

Abstract
Nucleotide-binding oligomerization domain-containing protein 2 (Nod2) is an innate immune receptor. To investigate the role of Nod2 in susceptibility to the autoimmune disease, type 1 diabetes mellitus (T1DM), we generated Nod2−/−non-obese diabetic (NOD) mice. The Nod2−/−NOD mice had different composition of the gut microbiota compared to Nod2+/+NOD mice and were significantly protected from diabetes, but only when housed separately from Nod2+/+NOD mice. This suggested that T1DM susceptibility in Nod2−/−NOD mice is dependent on the alteration of gut microbiota, which modulated the frequency and function of IgA-secreting B-cells and IL-10 promoting T-regulatory cells. Finally, colonizing germ-free NOD mice with Nod2−/−NOD gut microbiota significantly reduced pro-inflammatory cytokinesecreting immune cells but increased T-regulatory cells. Thus, gut microbiota modulate the immune system and T1D susceptibility. Importantly, our study raises a critical question about the housing mode in the interpretation of the disease phenotype of genetically-modified mouse strains in T1DM studies.
Keywords: Type 1 diabetes mellitus, NOD, Nod2, Innate immunity, Gut microbiota
1. Introduction
Type 1 diabetes mellitus (T1DM) development is influenced by both genetic and environmental factors [1]. The incidence of T1DM has significantly increased worldwide [2–7] and environmental factors are thought to play a critical role in this. Innate immunity protects hosts from bacterial invasion by recognizing bacterial components through their pattern recognition receptors (PRRs). These PRRs include Toll-like receptors (TLRs) and nucleotidebinding oligomerization domain-like receptors (NOD-like receptors, NLRs). Interestingly, TLR deficiencies on the Non-Obese Diabetic (NOD) mouse background both increase susceptibility to and protect from T1DM [8–13]. Deficiency in MyD88, an adaptor protein that mediates most TLR signaling, induced complete protection from T1DM, which was abolished when MyD88−/−NOD mice were housed in germ-free (GF) conditions [14]. Therefore, interactions between the TLR family and gut microbiota regulate T1DM development. However, much less is known about the role of NLR family members in T1DM susceptibility, in humans and mice. We recently reported that NLRP3, an important inflammasome component, influences T1DM development by regulating chemokines and their receptors in both immune cells and islet beta cells, preventing immune cell migration to the targeted tissue [15].
Nucleotide-binding oligomerization domain-containing protein 2 (Nod2) is a cytosolic bacterial sensor of muramyl dipeptide that induces antimicrobial peptide release and inflammatory signaling required for maintenance of the homeostasis of gut microbiota [16–18]. Nod2 mutations are associated with high susceptibility to Crohn’s disease, a common inflammatory disorder of the bowel [19,20]. Furthermore, Nod2 deficiency in mice correlates with increased bacterial susceptibility [16,21]. To investigate the role of Nod2 in a T1DM model, we generated NOD mice deficient in Nod2 (Nod2−/−NOD). We hypothesized that Nod2 modulates T1DM development by regulating the composition of gut microbiota, which alters the immune cell function. Our results fully supported our hypothesis and raise an important point about alteration of immunological phenotype by housing conditions.
2. Materials and methods
2.1. Mice
NOD/Caj mice have been maintained at Yale University for approximately 30 years. Nod2−/−B6 mice [16] were obtained from the Jackson Laboratory. They were backcrossed to NOD mice for 10 generations and the purity of the NOD genetic background was further confirmed by mouse genome scan (DartMouse). The mice were housed in specific pathogen-free (SPF) or germ-free (GF) conditions in a 12-h dark/light cycle. All mice received irradiated food ad libitum and were housed in individually-ventilated filter cages (SPF) or open-topped cages within a gnotobiotic isolator (GF). Three types of experimental breeding and four types of experimental housing strategies were applied in this study as summarized in Supplementary Table 1. In the cohoused group (CH) equal numbers (2 + 2 or 3 + 3) of Nod2+/+NOD and Nod2−/−NOD mice were housed in the same cage, whereas non-cohoused (NCH) mice refers to Nod2+/+NOD and Nod2−/−NOD mice housed by genotype, i.e., Nod2+/+NOD mice were only housed together with Nod2+/+NOD and vice versa. The use of mice in this study was approved by the Institutional Animal Care and Use Committee at Yale University.
2.2. Natural history of diabetes development
Female Nod2−/−NOD and Nod2+/+NOD mice were assessed weekly for glycosuria for 30 weeks. Diabetes was confirmed by blood glucose of ≥250 mg/dL (≥13.9 mmol/L).
2.3. Short term and long term co-housing
Three to 4-week-old female Nod2−/−NOD or Nod2+/+NOD littermates were divided into groups, whereby Nod2+/+NOD and Nod2−/−NOD mice were housed in separate groups or co-housed in cages where the genotypes were mixed. Experiments were terminated when the mice were aged 9–10 weeks or 30 weeks old.
2.4. Histopathology and insulitis
Pancreata from 12-week old female mice were fixed in 10% buffered formalin and embedded in paraffin. Tissues were sectioned and stained with hematoxylin and eosin. Insulitis was scored under light microscopy with the following grading scale: 0, no insulitis; I, infiltration <25% of the islet; II, 25–75% infiltration; III, >75% infiltration. A range of 170–212 islets were scored for insulitis in each group (n = 7–8 mice). Significance was determined using a Chi square test. **p < 0.01.
2.5. Extraction of gut bacterial DNA
Fecal samples from female Nod2−/−NOD and Nod2+/+NOD mice were collected from 12-week old mice and resuspended in 300 μl TE buffer containing 0.5% SDS and 200 μg/ml Proteinase K. Bacterial DNA was extracted as previously described [22].
2.6. 16S rRNA sequencing and microbiota classification
The V4 region of the 16S rRNA gene was amplified from each DNA sample using a bar-coded broadly-conserved bacterial forward (5’-GTGCCAGCMGCCGCGGTAA-3’) and reverse primer (5’GGACTACHVGGGTWTCTAAT-3’). The PCR products were purified using a Qiagen gel extraction kit. The DNA concentration was quantified and equimolar amounts of each sample were pooled and used for pyrosequencing with the Ion Torrent PGM sequencing system (Life Technologies). The sequencing data were analyzed with the QIIME software package and UPARSE pipeline to pick operational taxonomic units (OTUs). Taxonomy assignment was performed using representative sequences of each OTU. Betadiversity was calculated to compare differences between microbial communities, shown as Principal Coordinate Analysis (PCoA).
2.7. Quantitative real-time PCR (qPCR)
Quantitative real-time PCR (qPCR) was performed using Bio-Rad iQ5 qPCR detection system according to the manufacturer’s instructions. The relative mRNA level of segmented filamentous bacteria (SFB) was determined using the 2-ΔΔCt method and normalization using the 16S rRNA values with SFB specific primers (forward primer: 5’-AGGAGGAGTCTGCGGCACATTAGC-3’ and reverse primer: 5’-TCCCCACTGCTGCCTCCCGTAG-3’).
2.8. Gut lumen IgA detection
Gut contents were collected from Nod2+/+NOD and Nod2−/−NOD mice at 12 weeks of age by flushing the small intestine with 10 mls of sterile PBS. The gut flush was then centrifuged (2000 rpm, 5 min, RT) and the supernatant was collected for IgA detection using ELISA (Southern Biotech) following the protocol described by Harriman et al. [23].
2.9. Interaction of gut bacteria and immune cells in co-culture with splenocytes
The large intestine was harvested from 2-month old female Nod2−/−NOD and Nod2+/+NOD mice and flushed with 5 mls of sterile PBS. The gut contents were vortexed for 2 min followed by centrifuging for 5 min at low speed (52 × g) to remove dietary residue. The supernatant was transferred to a new tube and spun. After washing the pellet twice more, the combined supernatant was centrifuged at 469 × g to remove mammalian cells. Bacteria in the supernatant were pelleted by centrifugation at high speed (1876 × g, 5 min) and resuspended in PBS. Bacterial concentration was measured with a spectrophotometer (Bio-Rad) and heat-inactivated at 90 °C for 20 min. 108 heat-inactivated bacteria were co-cultured with splenocytes (2 × 106/ml) from Nod2−/−NOD or Nod2+/+NOD mice for 14 h prior to ICC.
2.10. Oral gavage
Fresh fecal pellets were harvested from 4 to 5 week old Nod2+/+NOD or Nod2−/−NOD mice housed separately. Pellets were homogenized and gently centrifuged to remove large particles (1 × g, 10 min). OD values were used to determine bacterial colony forming units (CFU). Germ-free WT NOD mice were then colonized with 2 × 108 CFU by oral gavage, and terminated for experiments 2–3 weeks after gavage. Successful colonization was evaluated by 16S rRNA bacterial sequencing of fecal pellets from the ex-GF mice.
2.11. Antibodies and reagents
Briefly, cells were incubated with an Fc-blocking antibody (clone 2.4G2) at 4° C for 15 min, prior to surface staining. Antibodies to IgA (eBioscience; clone mA-6E1), IgG1 (BioLegend; clone RMG11), IgG2b (BioLegend; clone RMG2b-1), CD4 (BioLegend; clone GK1.5), CD8α (BioLegend; clone 53–6.7), CD19 (BioLegend; clone 6D5), B220 (BioLegend; clone RA3–6B2), TCRβ (BioLegend; clone H57–597), CD25 (BioLegend; clone 3C5) were used with viability determined using a Zombie Aqua™ Fixable Viability Kit (Biolegend). Treg cell staining was conducted using Tonbo Biosciences buffers (Foxp3/Transcription Factor Staining Buffer Kit; cat no: TNB0607-KIT). Cells were stained as above, then fixed for 1 h at RT prior to permeabilzation. The cells were then incubated with the 2.4G2 Fc-blocking antibody at 4° C for 15 min prior to staining with antiFoxP3 PE (eBioscience; clone NRRF-30) for 30 min at 4° C. Hybridoma supernatants containing mAbs used for cell purification or stimulation, were generously provided by the late Charles Janeway Jr. (Yale University). RPMI-1640 media and heat-inactivated FCS were purchased from Invitrogen and Gemini, respectively.
2.12. Cell surface and intracellular cytokine (ICC) staining
Cells were stimulated with PMA (50 ng/ml, Sigma) and ionomycin (500 ng/ml, Sigma) in the presence of 1 μl GolgiPlug™ (BD) at 5 × 106/ml in cell culture medium for 4.5 h. Fc receptors were blocked using the Fc-blocking antibody (incubated at 4° C for 15 min), then the cells were stained (30 min at 4° C) with surface markers and viability dye. The cells were then washed prior to fixation (20 min at RT) and permeabilization, using eBioscience™ Intracellular fixation and permeabilization buffer set. The cells were then washed, blocked with the Fc-blocking antibody and then stained with anti-IFNγ (BioLegend; clone XMG1.2), anti-IL17a (BioLegend; clone TC11–18H10.1) and anti-TNFα (BioLegend; clone MP6-XT22) for 30 min at 4° C.
2.13. Treg suppression assays
Splenic and pancreatic lymph node (PLN)-derived Treg cells were isolated using Treg magnetic isolation kits (Stem cell technology, positive selection of CD4+CD25+ T-cells) and mitomycin-c-treated. Total NOD splenic APCs (post T-cell depletion and mitomycin-c-treatment) were used as alloantigen-stimulators. Purified splenic T cells from C57BL/6 mice were used as responders. Treg function was examined by suppression of the mixed lymphocyte reaction (MLR). Culture supernatants (to test for secreted cytokines) were collected on day 4 prior to 3H-thymidine addition for the last 18 h of the 5-day culture to assess proliferation.
2.14. Statistical analysis
Statistical analysis was performed using GraphPad Prism 7 software. Diabetes incidence was compared using log-rank test. In vitro assays were analyzed with Student’s t-test or ANOVA and P < 0.05 was considered significant.
3. Results
3.1. T1DM incidence in Nod2-deficient NOD mice was dependent on housing status
We set up three independent observation groups (summarized in Supplemental Table 1). Firstly, we investigated spontaneous diabetes in Nod2−/−NOD mice and their co-housed (CH) Nod2+/+NOD littermates (all females) over 30 weeks, from Nod2+/−breeding. We found that Nod2−/−NOD mice developed diabetes similar to their WT littermates (Fig. 1A). Secondly, to address the role of the gut microbiota (as Nod2 recognizes the bacterial component muramyl dipeptide) we set up another cohort of mice, in which Nod2−/−NOD and their Nod2+/+NOD female littermates from Nod2+/−breeding were separately housed (i.e. multiple mice of the same genotype were housed together) after weaning (non-cohoused, NCH). Interestingly, non-cohoused Nod2−/−NOD mice showed significantly reduced T1DM development (31% vs. 71%, p = 0.003, Fig. 1B). To confirm that the different diabetes phenotype was due to the housing conditions, in a third set of experiments, the offspring from homozygous Nod2−/−NOD breeding and homozygous Nod2+/+NOD breeding were then co-housed after weaning at 3–4 weeks until 30 weeks old. Nod2−/−NOD and Nod2+/+NOD mice were housed separately as controls. Similar to the results shown in Fig. 1B, the non-cohoused Nod2−/−NOD mice had significantly reduced T1DM incidence compared to non-cohoused Nod2+/+NOD mice (33.3% vs. 86.7%, p = 0.002, Fig. 1C). However, when the Nod2/NOD and Nod2+/+NOD mice were cohoused, the incidence of TIDM was increased in Nod2−/−NOD mice and indistinguishable from the Nod2+/+NOD counterparts, which had reduced incidence of diabetes (Fig. 1C). To assess disease severity, we also investigated the islet infiltration in 12-week old mice and found that Nod2−/−NOD mice had exacerbated islet infiltration when cohoused with WT Nod2+/+NOD mice (Fig. 1D). These results suggested that the gut microbiota affected T1DM development.
Fig. 1. Diabetes development in Nod2−/−NOD mice is dependent on housing status.
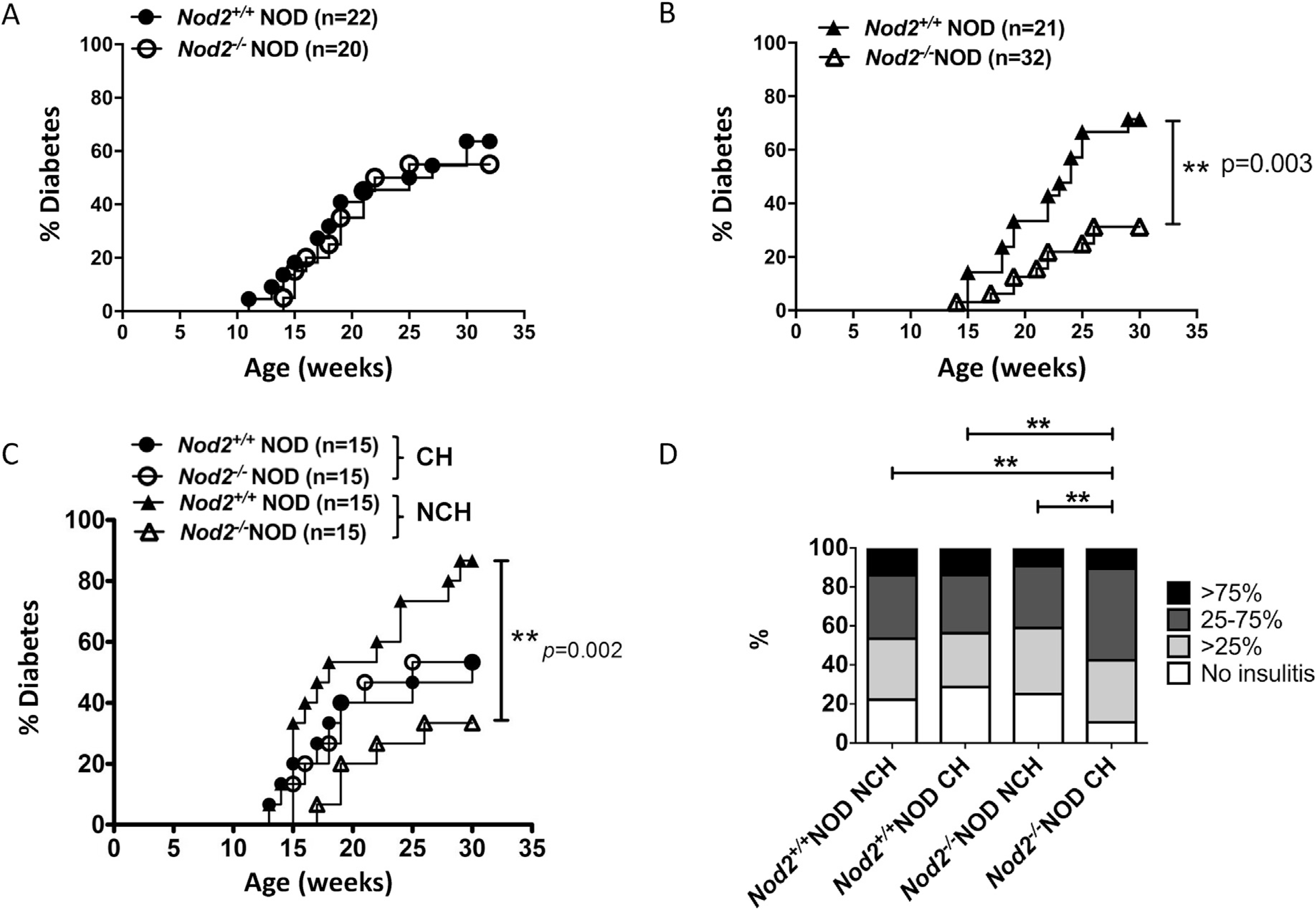
(A), Diabetes development was observed in female Nod2−/−NOD and Nod2+/+NOD mice that were reared together (CH, co-housed) or (B) separated by genotype (NCH, non-cohoused). (C), Diabetes development was observed in female Nod2−/−NOD and Nod2+/+NOD mice that were born from different mothers and either the strains were cohoused post-weaning (CH) or housed with littermates of their own genotype (NCH). Mice were screened weekly for glycosuria and diabetes confirmed by blood glucose >250 mg/dL (>13.9 mmol/L). The observation was terminated when the mice were 30 weeks old. Statistical analysis was performed by Log-rank test.
3.2. Diabetes susceptibility is modulated by altered gut microbiota in Nod2-deficient NOD mice
We conducted further studies using non-littermate mice (from Nod2−/−NOD breeding or Nod2+/+NOD breeding) that were housed together (cohoused) or separately (non-cohoused) at weaning. We specifically did not use Nod2−/−and Nod2+/+ littermates in order to study how distinct microbiota, shaped by Nod2, influence the immune responses. To verify whether gut microbiota were responsible for modifying T1DM susceptibility, we collected fresh fecal samples from pre-diabetic female Nod2−/−NOD and Nod2+/+NOD mice and conducted 16S rRNA sequencing. Principal component analysis (PCA) revealed that the composition of gut microbiota from Nod2−/−NOD mice was very different from Nod2−/−NOD mice when they were not cohoused (Fig. 2A); however, this difference was eliminated upon cohousing the mice (Fig. 2B). We further evaluated changes of the microbiota composition at 3 weeks (before housing changes) and at 8–9 weeks of age (non-cohoused and cohoused). Interestingly, the gut microbiota composition was dramatically altered in Nod2−/−NOD mice when cohoused compared to non-cohoused Nod2−/−NOD mice, with the latter similar to the microbiota composition they had at 3 weeks whereas the former were indistinguishable from the microbiota composition of Nod2+/+NOD mice (Supplemental Fig. 1A). Interestingly, Nod2+/+NOD mice also had a change in their microbiota when they were cohoused or non-cohoused, although the change was much more subtle. This, however, suggests that the subtle changes in the microbiota of Nod2-sufficient mice over time may protect the mice from developing diabetes (Fig. 1C).
Fig. 2. The composition of gut microbiota in Nod2−/−NOD mice is dependent on housing status.
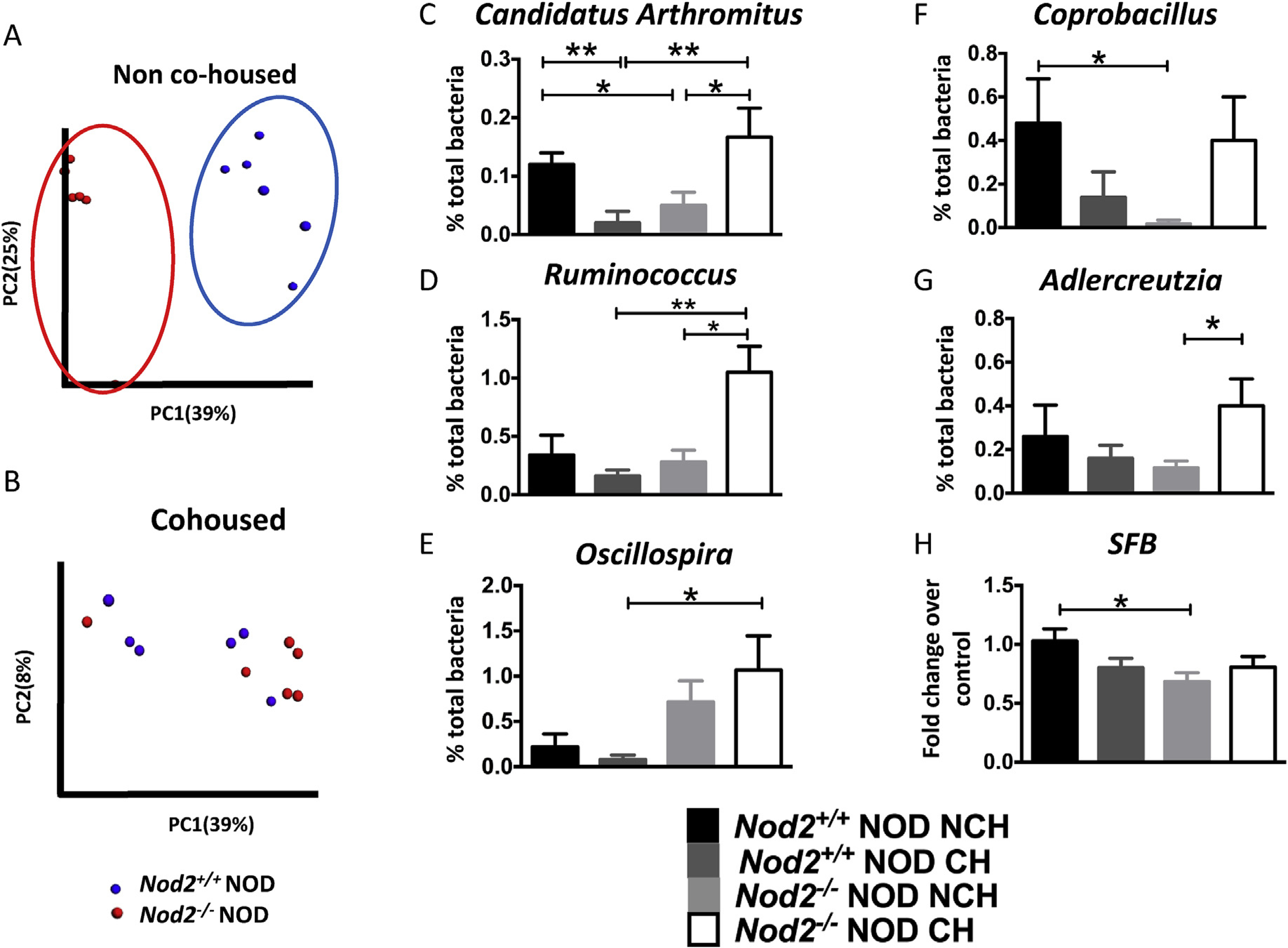
Bacterial DNA was extracted from fecal pellets from non-cohoused and cohoused 12-week old Nod2−/−NOD and Nod2+/+NOD female mice (n = 5–6/group, randomly selected from the mice in Fig. 1C) and used for pyrosequencing. Principal component analysis (unweighted) of beta diversity of the gut microbiota in Nod2+/+NOD and Nod2−/−NOD female mice is shown for non-cohoused (Fig. 2A) and cohoused (Fig. 2B) mice. The composition of gut microbiota was compared at genus level between Nod2−/−NOD and Nod2+/+NOD mice (n = 6 each) which were cohoused (CH) or non-cohoused (NCH) (Fig. 2C–G). The abundance of SFB in the gut microbiota was assessed by qPCR with SFB specific primers (Fig. 2H). The level of SFB expression was calculated by the delta-delta threshold cycle method after normalization with bacterial 16S rRNA. Each sample was plated in triplicate and the experiments were repeated twice. Student’s t-test was used for statistical analysis (*P < 0.05; **P < 0.001). Error bars represent SEM.
We identified that Candidatus Arthromitus and Ruminococcus, which are of the phylum Firmicutes, and Adlercreutzia, of the phylum Actinobacteria, were significantly increased in cohoused Nod2−/−NOD mice compared with non-cohoused Nod2−/−NOD mice (Fig. 2C, D, G). However, in the co-housed Nod2+/+NOD mice there was a significantly decreased frequency of Candidatus Arthromitus compared to non-cohoused Nod2+/+NOD mice (Fig. 2C). A similar trend was also noted in other genera including Coprobacillus (Fig. 2F). Interestingly, the effect of housing the mice had a stronger impact on the microbiota compared with the Nod2 gene effect, where only Oscillospira showed a trend towards a gene effect, being increased in Nod2−/−NOD mice (Fig. 2E).
To test if SFB contribute tothe disease protection in Nod2−/−NOD, we examined the abundance of SFB by qPCR in the fecal samples of Nod2-deficient or Nod2-sufficient NOD mice both co-housed and housed separately. While there were no significant differences between non-cohoused and cohoused mice of the same Nod2 genotype, there was a significant difference between non-cohoused Nod2+/+NOD and Nod2−/−NOD mice (Fig. 2H, p < 0.05), with higher levels of SFB in non-cohoused Nod2+/+NOD mice. Together, our data suggest that the diabetes-protected phenotype found in Nod2−/−NOD mice is mediated by gut microbiota and co-housing Nod2−/−NOD and Nod2+/+NOD mice can alter the composition of gut microbiota, leading to a change in diabetesphenotype. However, our data show a reduction of SFB in the protected mice, and thus do notsupport a protective roleof SFB in diabetes development in these mice.
Nod2-deficiency has been shown to reduce the level of antimicrobial peptides (AMPs) and thus alter the gut microbiota in other models [16,18]. Therefore, we investigated whether AMPs contribute to the altered gut microbiota composition in Nod2−/−NOD and Nod2+/+NOD mice, both cohoused and non-cohoused. As expected, non-cohoused Nod2−/−NOD mice had reduced levels of AMPs (particularly Reg3b) compared to non-cohoused Nod2+/+NOD mice (Supplemental Fig. 1B–D). Further, cohousing significantly increased AMP levels, as others have previously reported [24]. This suggests that alterations in AMP expression were at least partly responsible for the altered microbiota.
3.3. Effect of Nod2 on macrophages (MΦ) and dendritic cells (DCs)
We next examined the phenotype of the immune cells from cohoused and non-cohoused mice. As antigen presenting cells (APCs), especially MΦ and DCs, are able to take up and present antigens from the gut lumen [25,26], we hypothesized that APCs contributed to the modulation of T1DM susceptibility associated with Nod2. However, the frequency and phenotype of CD11b+ MΦ or CD11c+ DCs in WT and Nod2-deficient NOD mice were comparable, regardless of the housing conditions (data not shown). We then tested the function of MΦ and DCs using purified CD11b+ and CD11c+ populations from Nod2+/+NOD and Nod2−/−NOD mice. There were no obvious differences in APC function, assessed by stimulation of diabetogenic CD4+ BDC2.5 T-cells or CD8+ NY8.3 T-cells in vitro between Nod2−/−NOD or Nod2+/+NOD mice (Supplementary Fig. 2A and B). Finally, we tested the Mf and DCs in vivo, in adoptive transfer experiments in immune-deficient hosts. We transferred diabetogenic T-cells, from diabetic Nod2+/+NOD mice, to Nod2−/−and Nod2+/+ NOD. scid mice and evaluated diabetes development in the recipients. In line with our in vitro results, endogenous MΦ and DCs in Nod2-sufficient or deficient NOD. scid mice did not affect the incidence of diabetes in the recipients (Supplementary Fig. 2C). These results suggest that the MΦ and DCs are not responsible for altered Nod2-mediated spontaneous diabetes susceptibility, unlike the streptozotocin-induced model [27].
3.4. Effect of Nod2 on mucosal B-cells
We hypothesized thatNod2 may affect B-cell function, particularly IgA-producing B-cells. Therefore, we investigated the proportion of IgA+ B-cells in the Peyer’s patches (PPs). While there was a comparable proportion of IgA-producing B-cells in PPs of Nod2−/−NOD and Nod2+/+NOD mice when housed separately (Fig. 3A), co-housing led to a significant increase in IgA-producing B-cells in PPs of Nod2−/−NOD mice. A similar trend was also seen in cohoused Nod2+/+NOD mice (Fig. 3A). An increase in secreted IgA was also detected in the small intestine (Fig. 3B). This suggests that the cohousing induced altered gut microbiota, which in turn promoted mucosal B-cell responses, whereas in the steady state of gut microbiota, IgA-producing mucosal B-cells were not affected by Nod2. PP-residing IgG2b-expressing B-cells were also influenced by cohousing, as non co-housed Nod2−/−NOD mice had lower levels of IgG2b-expressing B-cells compared to co-housed Nod2−/−NOD mice (Fig. 3C). Further, IgG1-expressing B-cells were increased in the PP in CH WT Nod2+/+NOD mice when compared to CH Nod2−/−NOD mice (Fig. 3D). Together, these data suggest that the altered microbiota induced by cohousing affect the mucosal antibody responses.
Fig. 3. Cohousing affects IgA, IgG2b and IgG1 secreting B-cells in the PP and gut lumen.
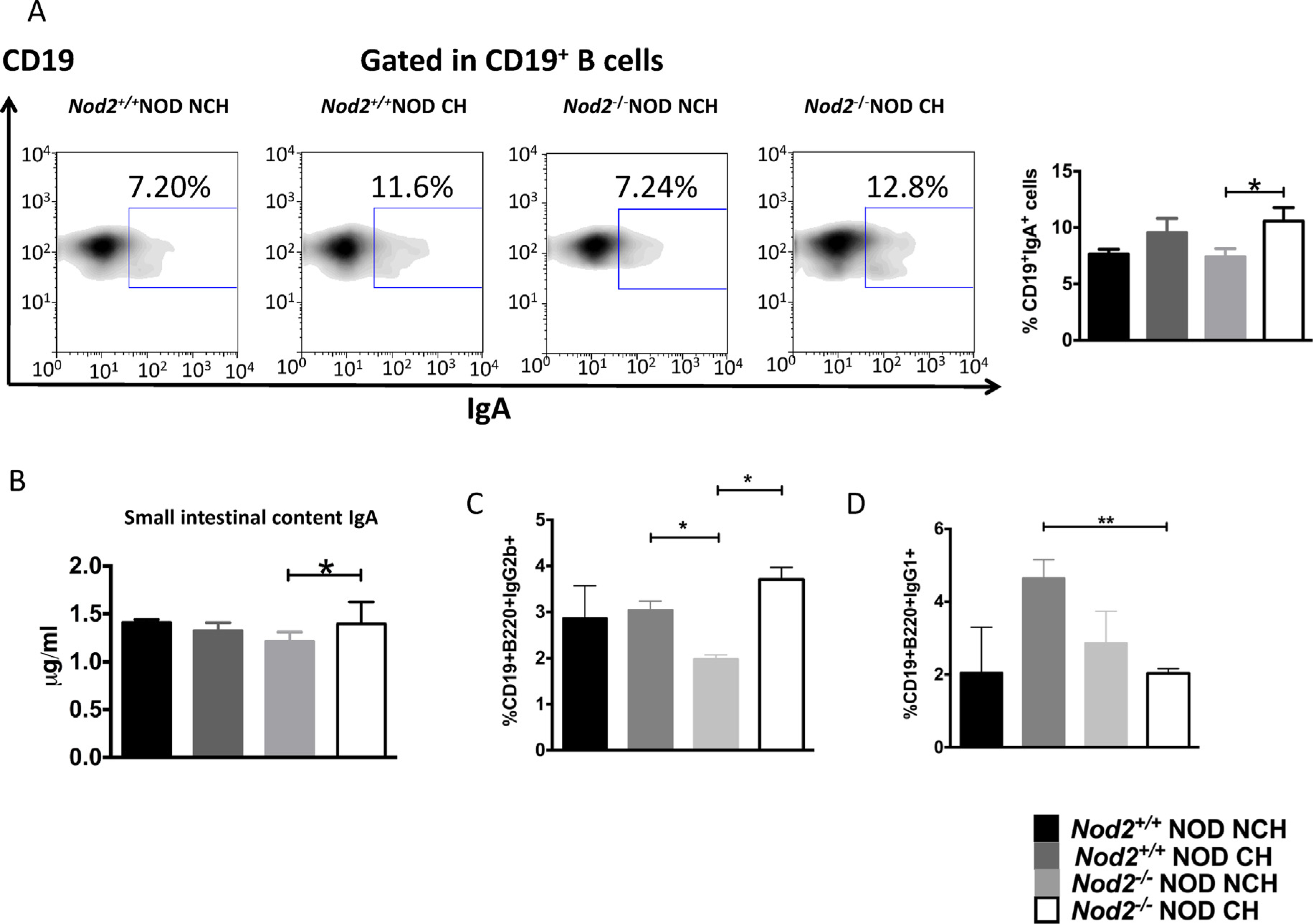
(A), IgA secreting B-cells (CD19+IgA+ B-cells) in the Peyer’s patches were investigated in 12-week old female Nod2−/−NOD and Nod2+/+NOD mice using flow cytometry. Left: A set of representative FACS plots. Right: The graphical summary of the IgA-secreting B-cells in the 4 groups (n = 8, pooled from two independent experiments). (B), IgA concentration in the small intestine of Nod2−/−NOD and Nod2+/+NOD mice cohoused (CH) or non-cohoused (NCH) was measured by ELISA. Data for (B) are from 3 individual experiments (n = 7–8). (C) and (D) IgG2b-secreting B-cells and IgG1-secreting B-cells in the Peyer’s patches were investigated in 12-week old female Nod2−/−NOD and Nod2+/+NOD mice using flow cytometry (n = 4). Student’s t-test was used for statistical analysis. *P < 0.05, **P < 0.01. Error bars represent SEM.
3.5. Effect of Nod2 on T-cells
To investigate if Nod2 affects T-cell function, we examined cytokine expression of T-cells from spleen, PLN and PPs. We found that, in the absence of Nod2, co-housing led to a significant increase of IFNγ-producing CD8+ T-cells in PPs of Nod2−/−NOD mice compared with non-cohoused Nod2−/−NOD or cohoused Nod2+/+NOD mice (Fig. 4A). Interestingly, a lower percentage of splenic IL17A-producing CD4+ T-cells was seen in Nod2−/−NOD mice whereas co-housing significantly increased the percentage of IL17A-producing CD4+ T-cells in PLN and PPs of Nod2−/−NOD mice compared with non-cohoused Nod2−/−NOD or Nod2+/+NOD mice (Fig. 4B). SFB have been shown to induce IL17-expressing T cells [28]. Interestingly, the CD4+IL17a+ T cells showed a similar pattern to the level of SFB detected (Fig. 2H), particularly splenic CD4+IL17a+ T cells (Fig. 4B). The increased proportion of IL17-expressing CD4+ T cells in both the PLN and PP from cohoused Nod2−/−NOD mice that had increased diabetes suggests that other microbiota are capable of inducing IL17 in CD4 T cells.
Fig. 4. Cytokine-producing CD4+ and CD8+ T-cells are altered by the absence of Nod2 and the cohousing conditions.
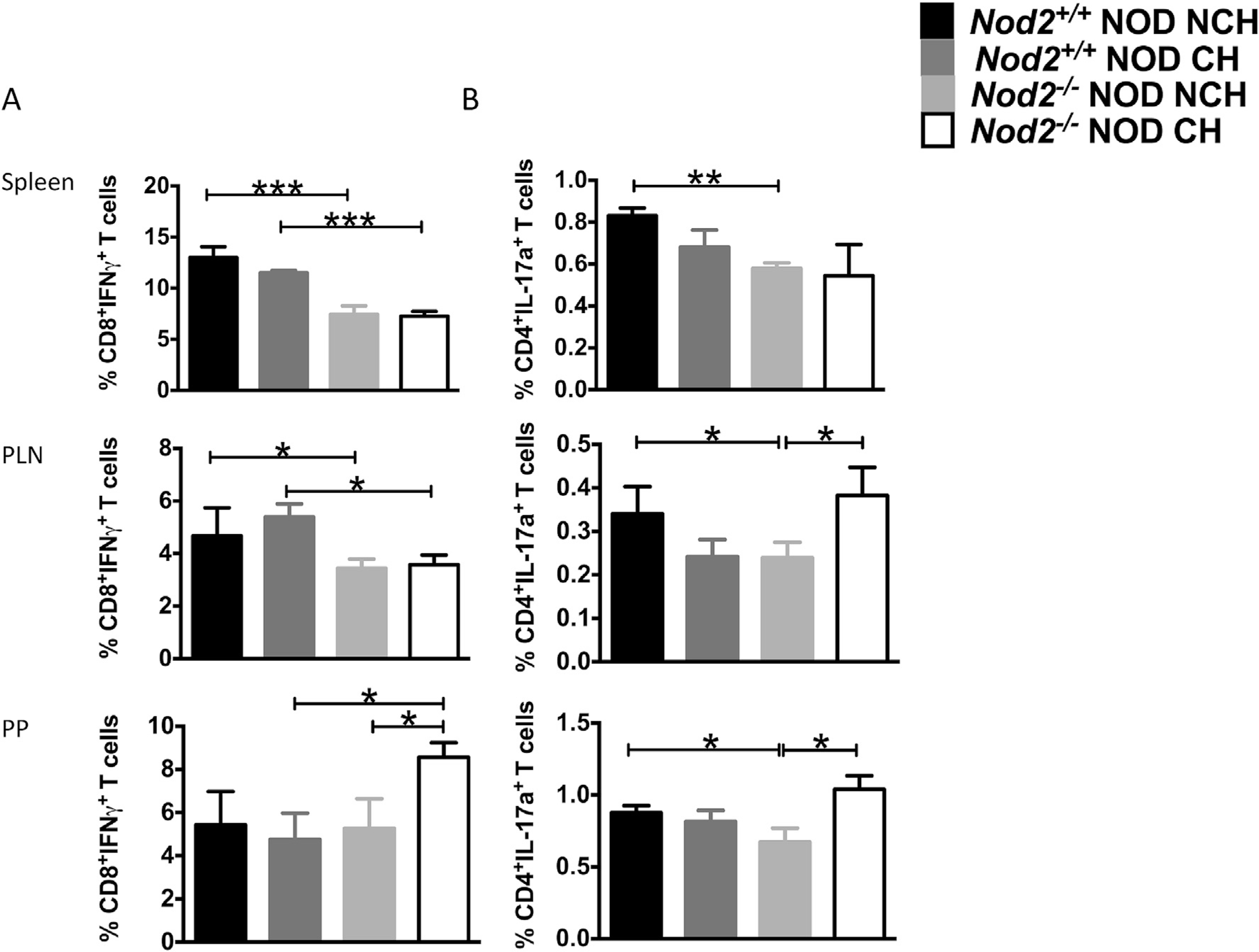
Intracellular staining was conducted on T cells from the spleen, pancreatic lymph nodes (PLN) and Peyer’s patches (PP) from 12-week old female Nod2−/−NOD (KO) and Nod2+/+NOD mice in both cohoused (CH) and non-cohoused (NCH) conditions. (A), The percentages of IFNγ-producing CD8+ T-cells and (B), IL-17a-producing CD4+ T-cells. The data represent at least 3 independent experiments (n = 3–4/group/ experiment). Student’s t-test was used for statistical analysis. *P < 0.05; **P < 0.001; ***P < 0.0001. Error bars represent SEM.
Next we investigated whether housing conditions affect Tregs. We found proportional changes in CD4+CD25+FoxP3+ T-cells predominantly in the PLNs (Fig. 5A and B). Interestingly, the Nod2+/+NOD mice housed separately, which have the highest incidence of diabetes, had the lowest proportion of Tregs. Further, the Nod2+/+NOD co-housed with Nod2−/−NOD mice (which reduced diabetes onset) or the Nod2−/−NOD mice housed separately (which have the lowest incidence of diabetes), had the highest proportions of Tregs. Our results suggested a role of the gut microbiota in inducing Tregs (Fig. 5A). To assess Treg function, we performed suppression assays using MLRs, in which T-cells from C57BL/6 mice (responders) were co-cultured with mitomycin-c-treated NOD APCs (stimulators) in the presence of purified Treg cells (suppressors) from Nod2+/+NOD and Nod2−/−NOD mice (housed together or separately). We found significant suppression of the proliferation of responder cells in the presence of Treg cells at 1:3 Treg:Tresponder ratio (Fig. 5C), although no suppression was observed at a 1:10 ratio (data not shown). Thus, housing status, which altered gut microbiota, affected both the number and function of the Treg cells. Secreted cytokines in the supernatants of the Treg suppression assays revealed a significant increase in anti-inflammatory IL-10, associated with Nod2 deficiency (Fig. 5D). Similar results were seen in an autoantigen-specific Treg suppression assay (data not shown).
Fig. 5. Treg proportion and function is affected by cohousing.
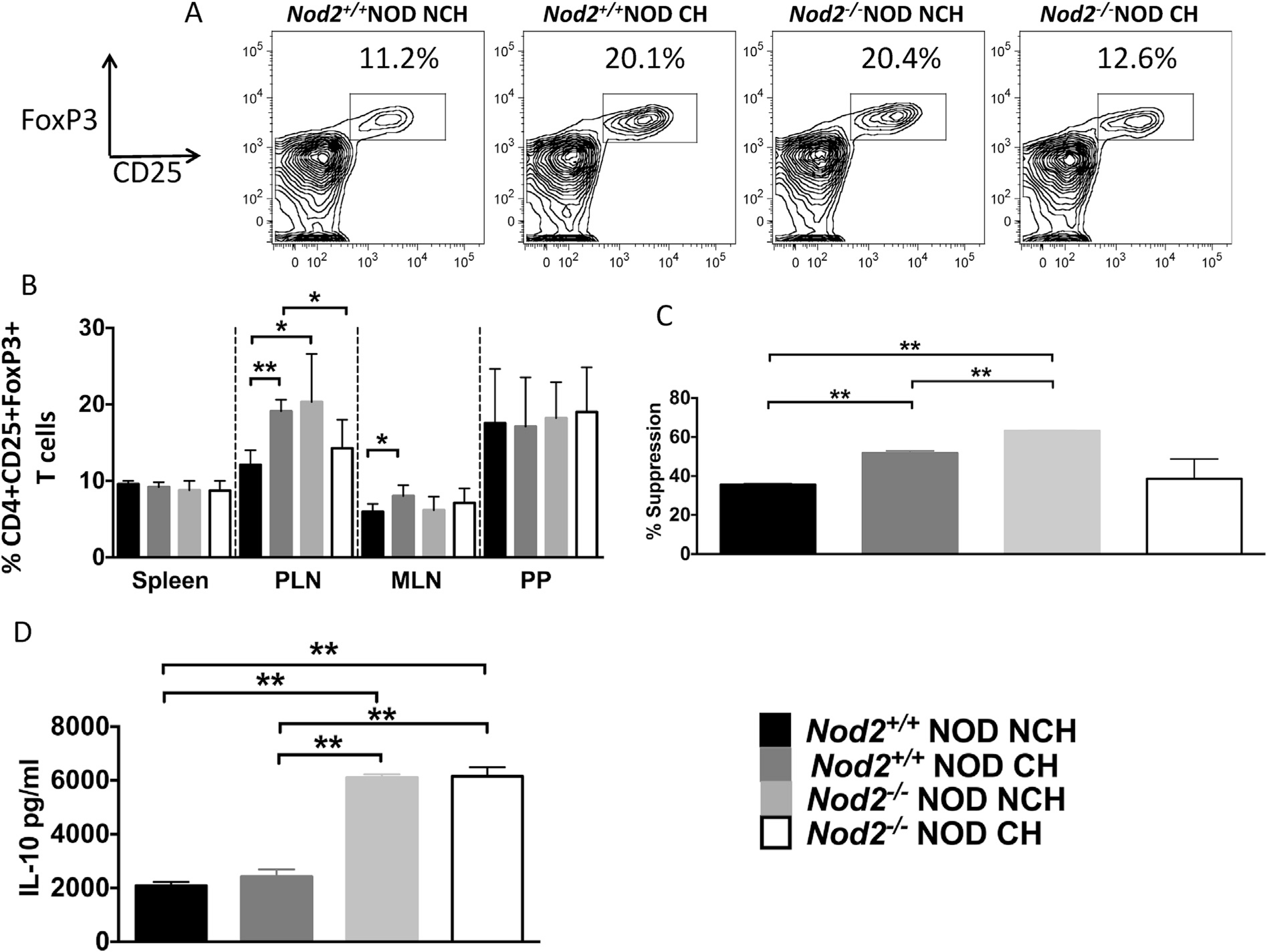
Intracellular staining was conducted on T-cells from the spleen, pancreatic lymph nodes (PLN), mesenteric lymph nodes (MLN) and Peyer’s patches (PP) from 12-week old female Nod2−/−NOD (KO) and Nod2+/+NOD mice in both cohoused (CH) and non-cohoused (NCH) conditions. (A), Representative Treg FACS plots shown for each group (gated on Live single B220−TCRbeta+ CD8a−CD4+CD25+ FoxP3+ cells). (B), Compiled data from A, (n =6–8). Pancreatic lymph node CD4+CD25 + Tregs were isolated using magnetic beads from 12-week old Nod2+/+NOD and Nod2−/−NOD mice in both cohoused and non-cohoused conditions and mitomycinc-treated (20 min, 37° C). NOD Splenic T cells were depleted using complement to enrich APCs, which were then mitomycin-c-treated (100,000/well). Isolated splenic T-cells from 12 week-old C57BL/6 mice were isolated and co-cultured with Tregs at a ratio of 1:3 (Treg:Tresponder) for 5 days, prior to supernatant collection and thymidine addition for the last 18 h (C), Proliferation suppression was calculated by (100-proliferation)/control × 100. Control was defined as the proliferation when no Tregs were added. (D), Supernatants were tested for the concentration of IL-10 by ELISA. Data shown are pooled from two independent experiments. Statistical analysis was performed using Student t-test, *P < 0.05; **P < 0.0001.
3.6. Direct effect of gut microbiota on T-cell function
To confirm if the housing-dependent change in gut microbiota was responsible for the different immune responses in the Nod2−/−NOD mice, we stimulated splenocytes from 2-month old female Nod2−/−NOD and Nod2+/+NOD mice with heat-inactivated colonic bacteria of Nod2−/−NOD or Nod2+/+NOD mice. Similar to the ICC staining shown (Fig. 4), there was a significant reduction in IFN-g producing CD8+ T-cells from Nod2−/−NOD mice when compared to Nod2+/+NOD mice, in response to gut bacteria from either Nod2-deficient or sufficient mice (Fig. 6A). However, gut bacteria from Nod2+/+NOD mice induced greater IFN-g production from CD8+ T-cells from Nod2−/−NOD or Nod2+/+NOD mice (Fig. 6A), indicating that gut bacteria from Nod2+/+NOD mice are more inflammatory. Interestingly, the gut bacteria, regardless of the donor, appeared to suppress IL-17A-producing CD4+ T-cells (Fig. 6B) and we did not find direct effects of gut bacteria on TNF-α or IL-10-producing T-cells in vitro (data not shown).
Fig. 6. The direct effect of gut microbiota on immune cells.
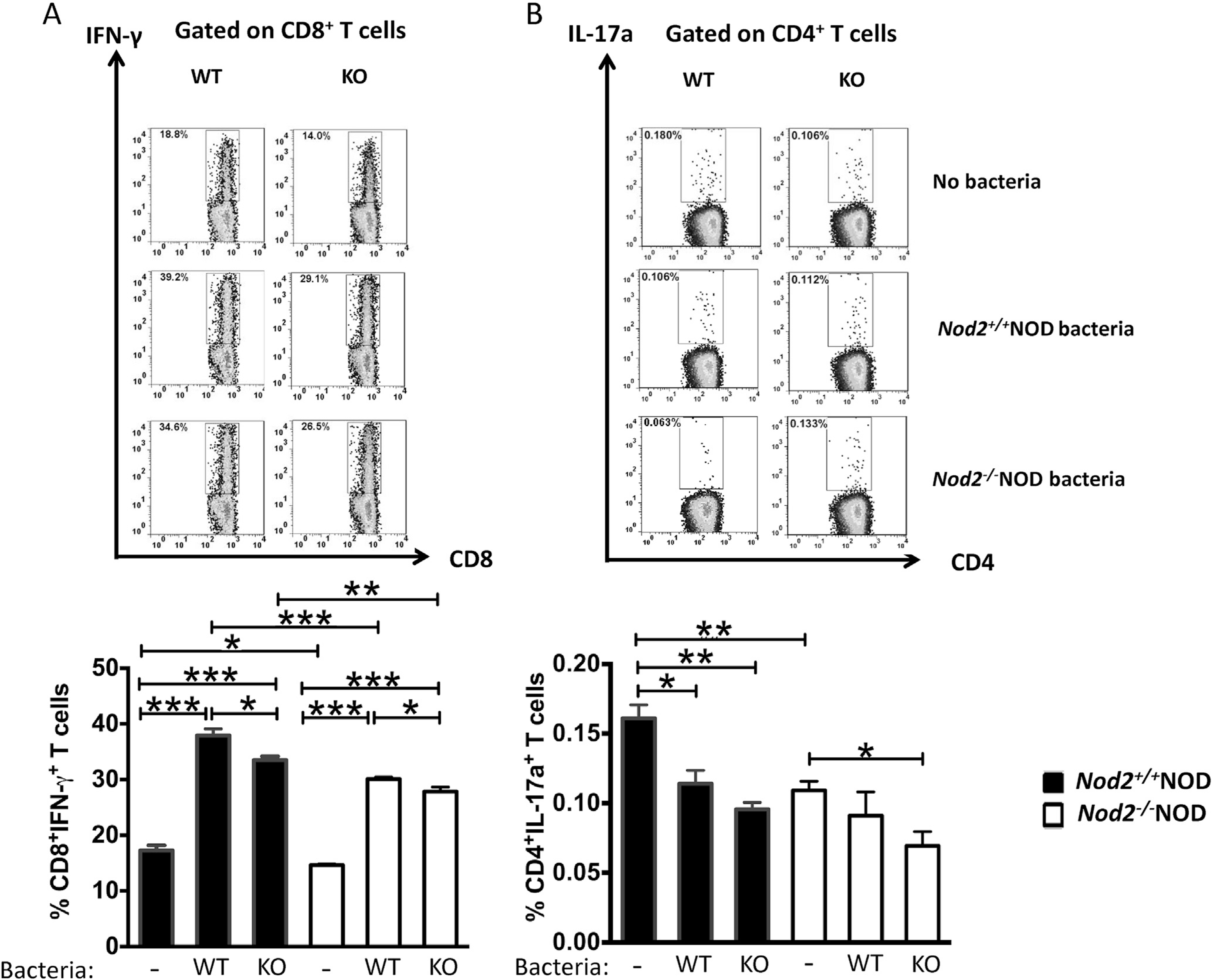
Gut bacteria from the large intestine of Nod2−/−NOD or Nod2+/+NOD mice were harvested and used to stimulate splenocytes (5 × 106/ml) from Nod2−/−NOD or Nod2+/+NOD mice with or without heat-inactivated gut bacteria (108 cfu) for 14 h. The expression of IFN-γ in CD8+ T-cells (A) and IL17A (B) in CD4+ T-cells was assessed by intracellular cytokine staining. Representative FACS plots are shown on the top. The results are pooled from three independent experiments. Student’s t-test was used for statistical analysis. *P < 0.05; **, P < 0.001; ***, P < 0.0001. * Data comparison was between cells incubated with no bacteria (control) and either Nod2−/−NOD or Nod2+/+NOD derived bacteria (A and B) or between Nod2−/−NOD or Nod2+/+NOD derived bacteria stimulation (A).
Taken together, our results suggest Nod2−/−NOD mice had a distinct gut microbiota composition compared with Nod2+/+NOD mice, and that the variance in components of gut microbiota altered the immune cell function which modulates T1DM susceptibility.
To further prove the concept, we colonized germ-free (GF) NOD mice with feces from 4 to 5 week old Nod2−/−NOD or Nod2+/+NOD mice that were housed separately. We studied the ex-GF mice 2–3 weeks after introduction of gut bacteria. 16S rRNA sequencing results showed a clear separation in the recipients depending on the donor microbiota they received (Fig. 7A). Interestingly, we found increased cecal weight in the ex-GF mice given Nod2−/−NOD feces (Supplemental Fig. 3A) and the small intestine was also longer (data not shown), neither of which were observed in SPF Nod2−/−NOD mice (data not shown). We also found reduced inflammatory cytokine-producing B cells, T cells in PLNs of ex-GF mice colonized with Nod2−/−NOD fecal bacteria (Fig. 7B). In line with the reduction of inflammatory cytokine production by immune cells, these mice also had a higher frequency and absolute number of CD4+CD25+FoxP3+ Treg cells in the mesenteric lymph nodes (MLNs) and PPs compared to their counterparts colonized with Nod2+/+NOD fecal bacteria (Fig. 7C–E).
Fig. 7. Colonization of GF NOD with bacteria alters immune cell cytokine secretion and function.
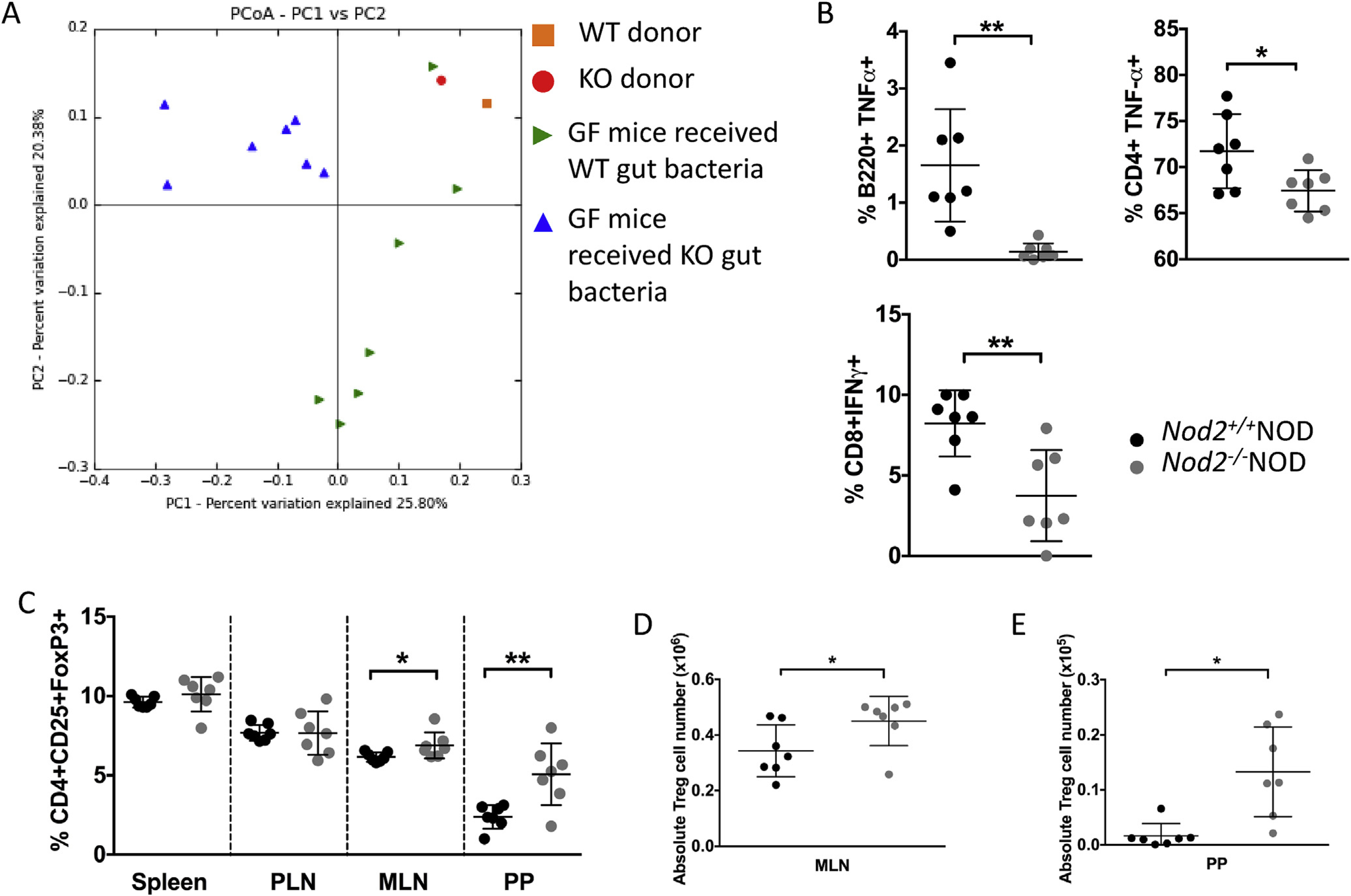
GF NOD mice were gavaged once with 2 × 108 CFU fecal bacteria from 4 to 5 week old Nod2−/−NOD KO or Nod2+/+NOD WT mice. Bacterial DNA was extracted from fecal pellets from WT or KO gavaged ex-GF NOD mice (n = 7/group) and the original bacteria donors. Pyrosequencing was conducted using the Ion Torrent PGM sequencing system. The sequencing data were analyzed with QIIME software package and UPARSE pipeline to pick operational taxonomic units (OTUs). Taxonomy assignment was performed at phylum level using representative sequences of each OTU. Principal component analysis (unweighted) of beta diversity of the gut microbiota from WT gavaged and KO gavaged mice is shown (A). 2–3 weeks later mice were sacrificed for immunophenotyping. Intracellular staining was conducted on B-cells (gated on live single TCRbeta-CD11b−CD11c−B220+) and T-cells (gated on live single TCRbeta+B22− cells then CD4 or CD8) from the pancreatic lymph nodes (B). FoxP3 staining was conducted following the instructions from the eBioscience staining kit for cells from spleen, pancreatic lymph nodes (PLN), mesenteric lymph nodes (MLN) and Peyer’s patches (PP). Cells were gated on live single B220−TCRbeta+CD8a−CD4+CD25+FoxP3+ cells (C). Absolute numbers were calculated for MLN (D) and PP (E) Treg cells. All data shown are pooled from two independent experiments (n = 3–4/experiment). Statistical analysis was performed using Student’s t-test, *P < 0.05 or **P < 0.0001. Lines represent the mean and the standard deviation.
There were no changes in phenotype and cytokine production in DCs or MΦ in the ex-GF mice (data not shown). However, given the results of the changes in IgA+ B-cells in cohoused SPF Nod2−/−NOD mice and altered cytokine production in the ex-GF mice colonized with Nod2−/−NOD fecal bacteria (Figs. 3 and 7B), we tested islet-specific APC function of the B-cells in these conventionalized GF mice. Supporting the immune tolerant status of these B-cells, B-cells from the ex-GF mice colonized with Nod2−/−NOD fecal bacteria showed attenuated antigen presentation to both BDC2.5 CD4+ T-cells and NY8.3 CD8+ T-cells compared to the B-cells from the ex-GF mice colonized with Nod2+/+NOD bacteria (Supplementary Fig. 3B–C). Taken together, we provide evidence that Nod2 alters the composition of gut microbiota, which in turn modulate the adaptive immune system and influence diabetes susceptibility.
4. Discussion
Our study sought to understand the role Nod2 plays in the development of spontaneous T1DM susceptibility. Our key finding was that Nod2-mediated T1DM susceptibility was dependent on the composition of the gut microbiota, which could be influenced by the housing of Nod2-deficient or sufficient NOD mice. The Nod2-deficient NOD mice were protected from T1DM development, if they were housed only with Nod2-deficient NOD mice. However, the disease protection was diminished if they were housed with Nod2-sufficient littermates. Conversely, the higher incidence of the Nod2-sufficient NOD mice was reduced when these mice were housed with their Nod2-deficient NOD littermates, emphasizing the importance of gut bacteria in influencing diabetes incidence. This was further confirmed by housing Nod2-deficient mice with Nod2-sufficient non-littermate mice. Although Nod2 deficiency led to a distinctive community of gut microbiota in NOD mice, its effect on diabetes was masked by the dominant community from Nod2-sufficient mice when cohoused. Thus, one of the roles of the Nod2 gene in T1DM development was to alter the gut microbiota. While macrophages and DCs were unaffected by Nod2 deficiency, the phenotype and functions of T- and B-cells were altered, some of which was likely to be related to the gut microbiota composition. This suggests that Nod2 activation is important for mediating changes in the gut microbiota, which affects the adaptive immune cells and subsequently diabetes development.
We recently showed that NLRP3 alters T1DM development by limiting the recruitment and infiltration of autoreactive CD4+ T-cells into the pancreas [15]. Our current study provides evidence that NLR signaling is important for modulating diabetes susceptibility by alteration of the microbial composition, which in turn affects the phenotype and functions of immune cells. Furthermore, this study demonstrates that the Nod2 gene effect on T1DM susceptibility is due to altering the composition of gut microbiota, and diabetes protection in Nod2-deficient mice is dependent on the gut microbial conditioning. This was supported by the results from three sets of experiments: i) cohousing Nod2−/−NOD with Nod2+/+NOD mice, littermates or non-littermates, ii) investigating the role of Nod2 in non-cohoused Nod2−/−NOD mice, in which diabetes development was significantly reduced; iii) reconstitution of gut microbiota from Nod2−/−NOD or Nod2+/+NOD to germ-free NOD mice.
The diabetes incidence differences in Nod2−/−NOD mice, when living with different housing partners, strongly supports the role of gut microbiota. We found a distinct profile of the gut microbiota between Nod2−/−NOD and Nod2+/+NOD mice when they are housed separately, which correlated with diabetes development. It is known that Nod2 is important for the expression of antimicrobial peptides and in controlling the expansion of intestinal bacteria [16,18]. In line with those reports, we found Nod2-deficient NOD mice expressed reduced levels of antimicrobial peptides; however, upon cohousing with Nod2-sufficient NOD mice, the levels of AMPs were increased. This suggests that the secretion of antimicrobial peptides is gut microbiota-dependent but can be independent of Nod2, as others have reported [24]. Therefore, the alterations of gut microbiota composition in the Nod2−/−NOD mice, caused by cohousing with Nod2+/+NOD mice, induce a large increase in AMP synthesis to similar levels seen in the Nod2-sufficient mice, thereby, diminishing diabetes protection in Nod2-deficient mice.
Interestingly, Nod2 deficiency had no effect on professional APCs, such as macrophages or DCs. However, the APC function of B-cells was attenuated in the absence of Nod2 or in the presence of Nod2−/−NOD gut microbiota. Nod2 deficiency also altered IgA-producing B-cells. It is well documented that a large amount of IgA is secreted into the gut lumen to protect the host from microbial invasion [29–31]. In Nod2 deficiency, a mild reduction of secreted IgA in gut lumen was observed; however, cohousing with Nod2+/+NOD mice significantly promoted IgA production into the gut lumen. This indicated that the gut microbiota tightly regulate the intestinal immune system in response to the newly-introduced gut bacteria by cohousing. Cohousing of Nod2−/−NOD mice with Nod2+/+NOD mice also affected pro-inflammatory cytokine-producing T-cells and Treg cells in the lymphoid tissue outside the intestine including PLN. This indicates that PLN have an intimate relationship with gut mucosal immunity. Turley has reported that PLNs drain antigens from both pancreas and intestine [32]. In line with this observation, we recently found a commensal microbial antigen that can be recognized by diabetogenic T cells, which in turn attack islet beta cells [33]. Similar results were also observed in an autoimmune uveitis model [34]. It is conceivable that Nod2−/−NOD mice harbor less diabetogenic gut microbiota, and thereby have a lower frequency of proinflammatory cytokine-producing T-cells but a higher frequency of Treg cells in PLNs. However, more immunostimulatory gut microbiota were present in Nod2−/−NOD mice after cohousing with WT mice, which led to a higher frequency of proinflammatory cytokine-producing T-cells and increased disease onset. The proof of concept using GF mice confirmed our hypothesis.
Costa reported recently that Nod2 is important in streptozotocin-induced diabetes in C57BL/6 mice [27]. Although both our study and theirs demonstrated increased Tregs in response to Nod2−/−microbiota, the cause of the Treg increase in the two model systems was different. In our spontaneous T1DM model, gut microbiota affected by housing status, regulated the change of Tregs; however, the anti-inflammatory cytokine IL-10 production was regulated by Nod2 gene expression (or lack of expression) but independent of housing conditions. While Costa and colleagues showed that CD11b+CD11c+ cells were important for mediating suppression and diabetes protection in their streptozotocin-induced disease model, we did not observe any changes in both phenotype and function of DCs or macrophages in vitro and in vivo. The changes we observed were in the B and T-cells in our spontaneous diabetes model. The discrepancy between the two studies was most likely due to the two very different model systems, streptozotocin-induced diabetes and spontaneous T1DM. More importantly, the two studies also used two very different mouse strains with different genetic backgrounds, C57BL/6 vs NOD.
Our study also raises a very important issue of housing conditions in interpretation of the disease phenotype in a geneticallytargeted mouse strain. We propose that the phenotype should be tested in two housing conditions – cohousing with gene-sufficient littermates and housing separately from gene-sufficient mice. If the disease phenotype is affected by the housing conditions when housed separately, as in our study, but not when cohoused, it suggests that the change in the gut microbiota is an integral part of the effect exerted by the specific gene.
Although the mouse Nod2 protein shares high homology with human Nod2 protein [35], it is not known if Nod2 has genetic associations with human T1D. However, a recent study suggests a high prevalence of inflammatory bowel disease (IBD) in patients with T1D [36]. It is not clear whether the patients developed IBD first or T1D first; however, Nod2 is a major susceptibility gene for IBD [19,20] and Nod2 plays an important role in mediating microbial signaling responses. It is possible that activation of Nod2 is possible that activation of Nod2 pathway may be context-dependent in individuals with T1D, i.e., dependent on microbial exposure, as we found in NOD mice.
In conclusion, our data suggest that Nod2 plays a role in T1DM development. Unlike inflammatory bowel disease, where the Nod2 gene plays a dominant role in the disease development, in T1DM, the role of the Nod2 gene contributes to alteration of the gut microbiota, but this effect can be over-ridden by altering the housing conditions. Thus, Nod2 acts as a recessive modulator in T1DM susceptibility. Finally, as human T1D incidence is increasing, this adds further weight to the increased importance of environmental factors in human studies.
Supplementary Material
Acknowledgements
We thank Karl Hager (Lab Medicine, Yale) for assistance with 16S rRNA sequencing and all the lab members for their kind technical help and critical scientific comments during the study.
Funding
This work was supported by NIH (DK092882, DK100500, and P30 DK945735 to LW), by ADA (1-14-BS-222) to LW and a Fulbright-Diabetes UK Research scholarship and a JDRF postdoctoral fellowship to JAP There are no conflicts of interest to disclose.
Footnotes
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.jaut.2017.05.007.
References
- [1].Tuomi T, Santoro N, Caprio S, Cai M, Weng J, Groop L, The many faces of diabetes: a disease with increasing heterogeneity, Lancet 383 (2014) 1084–1094. [DOI] [PubMed] [Google Scholar]
- [2].Variation and trends in incidence of childhood diabetes in Europe. EURODIAB ACE study group, Lancet 355 (2000) 873–876. [PubMed] [Google Scholar]
- [3].Gillespie KM, Bain SC, Barnett AH, Bingley PJ, Christie MR, Gill GV, et al. , The rising incidence of childhood type 1 diabetes and reduced contribution of high-risk HLA haplotypes, Lancet 364 (2004) 1699–1700. [DOI] [PubMed] [Google Scholar]
- [4].Bessaoud K, Boudraa G, Molinero de Ropolo M, de Sereday M, Marti ML, Moser M, et al. , Incidence and trends of childhood Type 1 diabetes worldwide 1990–1999, Diabet. Med. 23 (2006) 857–866. [DOI] [PubMed] [Google Scholar]
- [5].Harjutsalo V, Sjöberg L, Tuomilehto J, Time trends in the incidence of type 1 diabetes in Finnish children: a cohort study, Lancet 371 (2008) 1777–1782. [DOI] [PubMed] [Google Scholar]
- [6].Evertsen J, Alemzadeh R, Wang X, Increasing incidence of pediatric type 1 diabetes mellitus in southeastern Wisconsin: relationship with body weight at diagnosis, Plos One 4 (2009) e6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ehehalt S, Dietz K, Willasch AM, Neu A, Baden-Wuerttemberg Daibet I, Epidemiological perspectives on type 1 diabetes in childhood and adolescence in Germany - 20 years of the baden-wurttemberg diabetes incidence registry (DIARY), Diabetes Care 33 (2010) 338–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kim HS, Han MS, Chung KW, Kim S, Kim E, Kim MJ, et al. , Toll-like receptor 2 senses beta-cell death and contributes to the initiation of autoimmune diabetes, Immunity 27 (2007) 321–333. [DOI] [PubMed] [Google Scholar]
- [9].Wong FS, Hu C, Zhang L, Du W, Alexopoulou L, Flavell RA, et al. , The role of Toll-like receptors 3 and 9 in the development of autoimmune diabetes in NOD mice, Ann. N. Y. Acad. Sci. 1150 (2008) 146–148. [DOI] [PubMed] [Google Scholar]
- [10].Zhang Y, Lee AS, Shameli A, Geng X, Finegood D, Santamaria P, et al. , TLR9 blockade inhibits activation of diabetogenic CD8+ T cells and delays autoimmune diabetes, J. Immunol. 184 (2010) 5645–5653. [DOI] [PubMed] [Google Scholar]
- [11].Kim DH, Lee JC, Lee MK, Kim KW, Lee MS, Treatment of autoimmune diabetes in NOD mice by Toll-like receptor 2 tolerance in conjunction with dipeptidyl peptidase 4 inhibition, Diabetologia 55 (2012) 3308–3317. [DOI] [PubMed] [Google Scholar]
- [12].Gülden E, Ihira M, Ohashi A, Reinbeck AL, Freudenberg MA, Kolb H, et al. , Toll-like receptor 4 deficiency accelerates the development of insulindeficient diabetes in non-obese diabetic mice, PLoS One 8 (2013) e75385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tai N, Wong FS, Wen L, TLR9 deficiency promotes CD73 expression in T cells and diabetes protection in nonobese diabetic mice, J. Immunol 191 (2013) 2926–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. , Innate immunity and intestinal microbiota in the development of Type 1 diabetes, Nature 455 (2008) 1109–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hu C, Ding H, Li Y, Pearson JA, Zhang X, Flavell RA, et al. , NLRP3 deficiency protects from type 1 diabetes through the regulation of chemotaxis into the pancreatic islets, Proc. Natl. Acad. Sci. U. S. A 112 (2015) 11318–11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nuñez G, et al. , Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract, Science 307 (2005) 731–734. [DOI] [PubMed] [Google Scholar]
- [17].Biswas A, Liu YJ, Hao L, Mizoguchi A, Salzman NH, Bevins CL, et al. , Induction and rescue of Nod2-dependent Th1-driven granulomatous inflammation of the ileum, Proc. Natl. Acad. Sci. U. S. A 107 (2010) 14739–14744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ramanan D, Tang MS, Bowcutt R, Loke P, Cadwell K, Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal Bacteroides vulgatus, Immunity 41 (2014) 311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. , A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease, Nature 411 (2001) 603–606. [DOI] [PubMed] [Google Scholar]
- [20].Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, et al. , Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease, Nature 411 (2001) 599–603. [DOI] [PubMed] [Google Scholar]
- [21].Divangahi M, Mostowy S, Coulombe F, Kozak R, Guillot L, Veyrier F, et al. , NOD2-deficient mice have impaired resistance to Mycobacterium tuberculosis infection through defective innate and adaptive immunity, J. Immunol 181 (2008) 7157–7165. [DOI] [PubMed] [Google Scholar]
- [22].Peng J, Narasimhan S, Marchesi JR, Benson A, Wong FS, Wen L, Long term effect of gut microbiota transfer on diabetes development, J. Autoimmun 53 (2014) 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Harriman GR, Kunimoto DY, Elliott JF, Paetkau V, Strober W, The role of IL-5 in IgA B cell differentiation, J. Immunol 140 (1988) 3033–3039. [PubMed] [Google Scholar]
- [24].Shanahan MT, Carroll IM, Grossniklaus E, White A, von Furstenberg RJ, Barner R, et al. , Mouse Paneth cell antimicrobial function is independent of Nod2, Gut 63 (2014) 903–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].McDole JR, Wheeler LW, McDonald KG, Wang B, Konjufca V, Knoop KA, et al. , Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine, Nature 483 (2012) 345–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Farache J, Koren I, Milo I, Gurevich I, Kim KW, Zigmond E, et al. , Luminal bacteria recruit CD103+ dendritic cells into the intestinal epithelium to sample bacterial antigens for presentation, Immunity 38 (2013) 581–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Costa FR, Françozo MC, de Oliveira GG, Ignacio A, Castoldi A, Zamboni DS, et al. , Gut microbiota translocation to the pancreatic lymph nodes triggers NOD2 activation and contributes to T1D onset, J. Exp. Med 213 (2016) 1223–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. , Induction of intestinal Th17 cells by segmented filamentous bacteria, Cell 139 (2009) 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Russell MW, Brown TA, Claflin JL, Schroer K, Mestecky J, Immunoglobulin A-mediated hepatobiliary transport constitutes a natural pathway for disposing of bacterial antigens, Infect. Immun 42 (1983) 1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jonard PP, Rambaud JC, Dive C, Vaerman JP, Galian A, Delacroix DL, Secretion of immunoglobulins and plasma proteins from the jejunal mucosa. Transport rate and origin of polymeric immunoglobulin A, J. Clin. Invest 74 (1984) 525–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Phalipon A, Cardona A, Kraehenbuhl JP, Edelman L, Sansonetti PJ, Corthésy B, Secretory component: a new role in secretory IgA-mediated immune exclusion in vivo, Immunity 17 (2002) 107–115. [DOI] [PubMed] [Google Scholar]
- [32].Turley SJ, Lee JW, Dutton-Swain N, Mathis D, Benoist C, Endocrine self and gut non-self intersect in the pancreatic lymph nodes, Proc. Natl. Acad. Sci. U. S. A 102 (2005) 17729–17733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tai N, Peng J, Liu F, Gulden E, Hu Y, Zhang X, et al. , Microbial antigen mimics activate diabetogenic CD8 T cells in NOD mice, J. Exp. Med 213 (2016) 2129–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Horai R, Zárate-Bladés CR, Dillenburg-Pilla P, Chen J, Kielczewski JL, Silver PB, et al. , Microbiota-dependent activation of an autoreactive T cell receptor provokes autoimmunity in an immunologically privileged site, Immunity 43 (2015) 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pauleau AL, Murray PJ, Role of nod2 in the response of macrophages to tolllike receptor agonists, Mol. Cell Biol 23 (2003) 7531–7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Penny HA, Leeds JS, Kurien M, Averginos A, Hopper AD, Hadjivassiliou M, et al. , The relationship between inflammatory bowel disease and type 1 diabetes mellitus: a study of relative prevalence in comparison with population controls, J. Gastrointestin Liver Dis. 24 (2015) 125–126. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.