

Abstract
Even in the absence of coronary artery disease and hypertension, diabetes mellitus (DM) may increase the risk for heart failure development. This risk evolves from functional and structural alterations induced by diabetes in the heart, a cardiac entity termed diabetic cardiomyopathy (DbCM). Oxidative stress, defined as the imbalance of reactive oxygen species (ROS) has been increasingly proposed to contribute to the development of DbCM. There are several sources of ROS production including the mitochondria, NAD(P)H oxidase, xanthine oxidase, and uncoupled nitric oxide synthase. Overproduction of ROS in DbCM is thought to be counterbalanced by elevated antioxidant defense enzymes such as catalase and superoxide dismutase. Excess ROS in the cardiomyocyte results in further ROS production, mitochondrial DNA damage, lipid peroxidation, post-translational modifications of proteins and ultimately cell death and cardiac dysfunction. Furthermore, ROS modulates transcription factors responsible for expression of antioxidant enzymes. Lastly, evidence exists that several pharmacological agents may convey cardiovascular benefit by antioxidant mechanisms. As such, increasing our understanding of the pathways that lead to increased ROS production and impaired antioxidant defense may enable the development of therapeutic strategies against the progression of DbCM. Herein, we review the current knowledge about causes and consequences of ROS in DbCM, as well as the therapeutic potential and strategies of targeting oxidative stress in the diabetic heart.
Keywords: Diabetes, Diabetic cardiomyopathy, Diabetic heart, Oxidative stress, Reactive oxygen species, Mitochondria
1. Introduction
Diabetes mellitus (DM) has evolved as an pandemic that currently affects 463 million people worldwide and is projected to increase to 700 million by 2045 [1]. Estimates suggest that over half of people living with DM are even undiagnosed [1]. Despite numerous advances in care and intervention in subjects with DM, cardiovascular (CV) disease remains the leading cause of morbidity and mortality in these patients, mainly related to increased incidence and severity of coronary artery disease (CAD) and myocardial infarction, and consequently increased development of heart failure (HF). Patients with DM have approximately 75% increased risk of CV disease-related death or hospitalization for HF compared to patients without DM [2]. The risk for HF development in DM subjects is increased by two-fold [3,4], and the 1-year mortality due to HF in patients with DM is approximately 1.5-fold greater compared to those without DM [5]. Conversely, the prevalence of DM is four-fold higher in patients with HF compared to those without HF [3,6]. Thus, there appears to be a bidirectional association between HF and DM, where one increases and worsens the prognosis of the other [7,8].
2. Diabetic cardiomyopathy
Although CAD and hypertension are increased in patients suffering from DM, population-based studies have demonstrated that the increased HF risk in DM cannot be solely attributed to these comorbidities [34]. Thus, the term diabetic cardiomyopathy (DbCM) has been coined. DbCM has been defined as ventricular dysfunction in DM subjects in the absence of CAD, hypertension, valvular heart disease, congenital heart disease, and classical causes of cardiomyopathy, or the increased vulnerability of diabetic hearts to fail in the presence of concomitant stress [8–11]. DbCM is characterized by concentric hypertrophy, increased left ventricular (LV) mass, increased ventricular stiffening and impaired relaxation, reflecting a clinical phenotype of diastolic dysfunction that precedes LV dysfunction. At this stage in its pathophysiology DbCM shares features of HF with preserved ejection fraction (HFpEF) [12–15]. It has yet to be confirmed whether DbCM as defined above inexorably progresses to heart failure with reduced ejection fraction (HFrEF) over time [8,10]. Given the resistance of rodents to the development of atherosclerosis and hypertension in the absence of predisposing genetic manipulations [16], studies in rodent models of DM reporting impaired systolic function support the concept that DbCM could potentially predispose to, or may cause HFrEF [17–19]. Both rodent models of type 2 diabetes (T2D), including leptin-deficient db/db mice, obese ob/ob mice and Zucker diabetic fatty rats, as well as models of type 1 diabetes (T1D), elicited systolic cardiac dysfunction in several studies. Even though some studies have failed to show major cardiac contractile defects as a result of DM in animal models [20–22], this could be related to differences in methodology, gender, age, disease duration and model of DM (reviewed in Ref. [23]). Of note though, despite the absence of systolic dysfunction, many of these models may nevertheless exhibit diastolic dysfunction [24–26].
3. Oxidative stress
The “redox state” is determined by the balance between production of ROS and their removal by the antioxidant defense system. The term ROS includes: the free radicals, superoxide and hydroxyl (OH•); the non-radical, hydrogen peroxide (H2O2); and the result of the reaction of superoxide and nitric oxide (NO), peroxynitrite (ONOO•−). Production and degradation of ROS reflects the normal physiology of many cells; however, when an imbalance occurs from excessive ROS generation and the inability for these to be degraded, this is defined as “oxidative stress”.
In the cardiomyocyte, ROS may be generated in the mitochondria at the electron transport chain (ETC), by monoamine oxidase (MAO) or calpain; by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) and xanthine oxidase (XO) and uncoupling of nitric oxide oxidase (NOS) (Fig. 1 A). This production of ROS is counterbalanced by a complex antioxidant system which detoxifies ROS in order to maintain homeostasis (Fig. 1 B). However, under conditions of pathological stress, excessive ROS production and/or inadequate ROS detoxification may result in ROS-induced damage to DNA, proteins and lipids, leading to irreversible cell damage and death, ultimately resulting in cardiac dysfunction. As such, generation and detoxification of ROS must be tightly regulated in order to prevent oxidative damage, or reductive stress if these pathways are hyperactivated. Importantly, the causal role of oxidative stress in diabetic heart disease is supported by antioxidant interventions to scavenge ROS, which limit or prevent cardiac dysfunction in animal models of DM [27–31].
Fig. 1. Simplified schematic of A) sources of ROS and B) ROS detoxification by the antioxidant defense system.
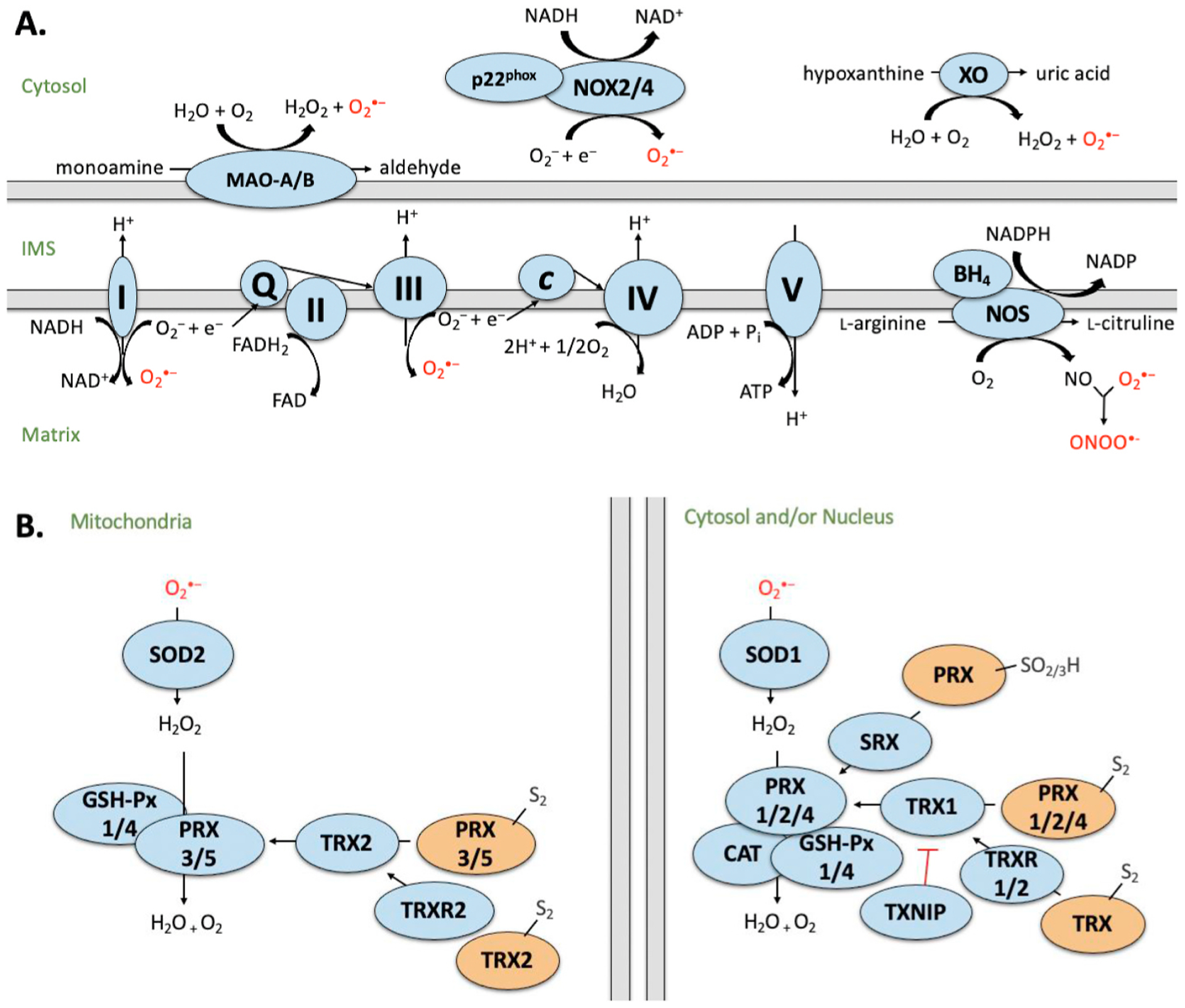
Sources of ROS generation in the cardiomyocyte including mitochondrial electron transport chain (ETC), monoamine oxidases (MAO), calpains, nicotinamide adenine dinucleotide (NADH) oxidases (NOX), xanthine oxidase (XO) and uncoupled nitric oxide synthase (NOS). Complex I (NADH dehydrogenase) transfers electrons from NADH and passes to ubiquinone (Q). Complex II (succinate dehydrogenase; SDH) donates electrons from succinate (as part of the Krebs cycle) to Q via FeS clusters. Complex III (cytochrome c oxidoreductase) transfers the electrons carried by ubiquinol (QH2) to cytochrome c (Cyt c). Complex IV (cytochrome c oxidase) transfers electrons (e−) from cytochrome c to O2 to generate water (H2O). Complex V (F1F0 ATP synthase) exchanges protons from the IMS to the matrix, which drives phosphorylation of ADP to form ATP. MAO generates H2O2 and reactive aldehydes as by-products during the oxidation of monoamines. NOX forms a heterodimer with p22phox and produces upon electron transfer from NADPH to molecular O2. XO reacts with molecular oxygen to produce and H2O2 during the production of uric acid. NOS consumes NADPH and O2 to convert L-arginine to L-citrulline, generating NO and NADP as by-products. I–V: Denotes ETC complex number. B) Generation of ROS is counterbalanced by the antioxidant defense system. Superoxide is converted to hydrogen peroxide (H2O2) by superoxide dismutase (SOD). H2O2 is converted to H2O by glutathione peroxidase (GSH-Px), peroxiredoxin (PRX) or catalase (CAT). Activity of PRX is regulated by sulfiredoxin (SRX) and thioredoxin (TRX), which is further regulated by TRX reductase (TRXR) and its endogenous inhibitor, thioredoxin interacting protein (TXNIP). Blue indicates active state. Orange indicates inactive state. Intermembrane space (IMS). Proteins have been compartmentalized according to their predominant intracellular localization.
The most well-studied markers of oxidative stress include the products of lipid peroxidation, malondialdehyde (MDA) and 4-hydroxynonenal (HNE), and the indicator of protein nitration, 3-nitrotyrosine (3-NT). MDA is generated via peroxidation of lysine residues by polyunsaturated fatty acids (FAs) and is commonly measured via calorimetric reaction with thiobarbituric acid (TBA) using a TBA reacting substances (TBARS) assay. HNE is a stable and reactive lipid peroxidation product, generated in a secondary reaction following initial peroxidation of polyunsaturated FAs. Interaction of HNE with macromolecules (e.g., proteins) is easily measured by immunoblotting or ELISA using specific antibodies. Protein nitration occurs when peroxynitrite or nitrogen dioxide (NO2) react with susceptible tyrosine residues and is most accurately measured by tandem mass spectrometry (MS/MS) in combination with high-performance liquid chromatography (HPLC) but can also be quantified by chromatography or immunohistochemical assays. Other indicators of oxidative stress include oxidized DNA molecules (i.e., modification of guanine [32]), protein modifications (e.g. carbonylation) [33] or modified polyunsaturated FAs (i.e., 8-isoprostane; 8-iso PGF2α) [34].
4. Oxidative stress in DbCM
Myocardial oxidative stress is a common observation in animal models of DM (Table 1). The most well-characterized animal model of T1DM is generated by intraperitoneal injection of the pancreatic betacell toxic glucosamine-nitrosourea antibiotic, streptozotocin (STZ), which induces hyperglycemia in mice, rats and guinea pigs. Both diastolic and systolic dysfunction occurs depending on the duration of diabetes and these functional changes have been correlated with increased cardiomyocyte ROS levels [35–38]. STZ-induced diabetes is characterized by increased lipid peroxidation [24,35,36,39], nitrotyrosine [24], carbonyl protein content [40] and reduced GSH [25,35, 41]. Elevated oxidative stress in STZ models is thought to be related to reduced ETC enzymatic activities [24,38,42], whereas the activity of specific detoxification enzymes has been reported to be increased [39, 43,44] or unaltered [35,39,44]. Other rodent models of T1D include the transgenic OVE26 mice, the insulin-dependent spontaneously diabetic BB Wistar rats (ISDBB), the Akita diabetic mouse, and alloxan (ALX)-induced diabetic mice. Of these, OVE26, ISDBB and ALX-induced T2D mice have been reported to display increased markers of lipid peroxidation, tyrosine nitration and protein carbonylation [45–47], in addition to increased expression of antioxidant defense systems [31, 48–50] in conjunction with impaired mitochondrial respiration [48]. Interestingly, others have observed reductions in lipid peroxidation in ALX-induced T1D [51] and in contrast to the other models, the Akita mouse model has decreased mitochondrial hydrogen peroxide generation and no induction of antioxidant defense pathways [52,53].
Table 1.
Overview of ROS, oxidative stress and cardiac structure and function in DbCM.
| Diabetes Model | ROS/Oxidative Stress | Cardiac Function/Structure | Author |
|---|---|---|---|
| Permeabilized atrial myofibers from T2D patients (LVEF >30%) | ↑ mitochondrial H2O2 emission ↓ GSH/GSSG ratio ↑HNE, 3-NT |
↔ systolic function | Anderson [74] |
| Obese T2D patients without HF, CAD and hypertension | ↑ glycogen ↑ HO-1, NQO1 |
↑ LV hypertrophy ↑ myocardial fibrosis |
Li [234] |
| Right atrial myocardium from diabetic patients without signs of CM | ↑ ROS levels ↑ MnSOD, CAT; ↔ Cu-ZnSOD activities |
↓ mitochondrial function ↑ contractile dysfunction of atrial without signs ↓ complex I activity in obese/diabetic individuals |
Montaigne [77] |
| Human diabetic hearts | ↓ NRF2 staining | Tan [233] | |
| STZ-induced T1D guinea pigs | ↑ ROS levels; ↔ GSH ↓ basal complex II state 4H2O2 emission ↔ SOD, GSH-Px, PRX, GSH, TRXR1/2, CAT, TRX1/2 ↑ oxidant-challenged H2O2 emission |
↓ complex II and IV state 3 VO2 ↓ ADP phosphorylation rates |
Tocchetti [38] |
| STZ-induced T1D rats | ↑ O2•− ; ↓ complex I H2O2 production; ↑/↓ H2O2 production; ↑ CM ROS, TBARS, MDA, lipid peroxide, protein carbonyl, 8-iso PGF2α, HNE-adduct of SDH subunit, NT of SCOT; ↓ mitochondrial MDA ↓ GSH; ↑ GSH conjugate efflux; ↔ γ-GSC mRNA, GSR activity; ↑ GSH/GSSG ratio ↑ p22phox, NOX4 mRNA ↑/↓ SOD content; ↑ CAT; ↔ CuZn-SOD, GSH-Px activities; MnSOD, GSH-Px, PRX3 protein |
↑/↓ cardiac hypertrophy ↓ systolic and diastolic function ↑ cardiac fibrosis, apoptosis ↑ mitochondrial dysfunction; ↓ mitochondrial transcription/translation, respiration ↑ respiratory uncoupling ↓ ETC proteins; complex I, SDH, SCOT activities |
Singh [37], Ghosh [35], Kanazawa [36] Kakkar [39], da Silva [40] Hamblin [41], Wohaieb [43], Herlein [44], Lashin [42], Turko [180], Turko [262], Guo [126] |
| STZ-induced T1D mice | ↑ ROS generation in IFM, NT, MDA, 4-HNE; AGE protein; ↔ protein carbonyl ↓ GSH/GSSG ratio ↑ NOX2 mRNA ↑ OGT, OGA mRNA; O-GlcNAc |
↔ cardiac hypertrophy ↓ diastolic function; contractility ↑ fibrosis ↑ DNA fragmentation ↑ Ca2+-handling dysfunction ↓ mitochondrial respiration |
Dabkowski [24], Ceylan-Isik [25], De Blasio [26] |
| ALX-induced T1D rats | ↑ intracellular ROS, MDA, protein carbonylation, lipid peroxide levels, ↑ NT of mitochondrial proteins; ↓ TBARS [51] ↓ GSH/GSSG ratio ↑ XO activity ↓ SOD, CAT, GST, GSR, GSH-Px activities |
↑ CM disorganization ↓ mitochondrial membrane potential; ↑ cytosolic cytochrome c translocation ↑ apoptosis ↓ stimulated SCOT, complex I activities |
Das [45], Genet [46], Parinandi [51], Turko [47] |
| OVE26 T1D mice | ↓ mitochondrial GSH; ↑ GSSG; ↔ GSH content ↑ CAT activity |
↑ CM morphological damage ↑ mitochondrial area, number, protein, mtDNA ↓ RCR, state 3 respiration, P/O, state 4 respiration |
Shen [48], Shen [31], Liang [50] |
| ISDBB T1D rats | ↑ CAT, GSSG reductase activities ↑ GSH content |
Wohaieb [49] | |
| Akita T1D mice | ↓ mitochondrial H2O2 production; ↔ ROS ↔ SOD2, PRDX3 mRNA/protein |
↔ O2 consumption | Bugger [52] |
| db/db obese/T2D mice | ↑ total ROS, O2•− , ONOO•-, 4-HNE, 8OHdG, MDA ↓ GSH, GSH/GSSG ratio ↑ gp91phox, NOX1 mRNA; ↑ PRX5; ↔ SOD2 proteins ↓ OGG-1 activity |
↑ cardiac remodeling ↓ systolic function ↑ apoptosis ↑ myocardial lipotoxicity ↓ complex III activity, ATP production, ATP/ADP ratio |
Mariappan [56], Das [58], Pan [59], Li [234] |
| ob/ob obese pre-T2D mice | ↓ GSH/GSSG; ↑ MDA, protein carbonyl ↑ p47phox, gp91phox protein |
Li [114] | |
| ZDF T2D rats | ↑ ROS, MDA, protein carbonylation, ↑ CAT, GSH-Px, Cu/Zn-SOD, Mn-SOD ↑ GSH, GST, GSR ↑ HO-1, iNOS protein |
↑ LV hypertrophy ↓ complex I, IV activities ↓ cytochrome c oxidase subunit 1 |
Raza [60], Conti [400] |
| fa/fa T2D rats | ↑ TBARS, lipid hydroperoxides, GSH; ↔ O2•− formation ↑ SOD activities | Vincent [61] | |
| HFD-fed T2D rats | ↑ TBARS, lipid hydroperoxides, GSH; ↔ O2•− formation ↑ iNOS synthase, NO ↑ SOD activities |
Vincent [61], Banerjee [62] |
|
| fructose-induced pre-T2D rats | ↔ MDA, 3-NT ↑ XO activity; ↑ PRX6; ↓ SOD2 mRNA |
↑/↓ ETC mRNA ↑ apoptosis |
Szűcs [63], Fan [129] |
| HFD-fed/STZ-induced pre-T2D [64] rats, mice [234] or miniswine [65] | ↑ mitochondrial ROS; total and NOX-stimulated O2•− production ↓ NO production; ↑ eNOS uncoupling ↑ NRF2 pathway activation |
↑ diastolic and systolic dysfunction ↑ LV mass/remodeling; CM size ↑ myocardial fibrosis, apoptosis ↑ myocardial lipotoxicity |
Koncsos [64], Heinonen [65], Li [234] |
| Goto-Kakizaki T2D rats | ↑ MDA ↓ GSH, CoQ9; ↔ α-tocopherol, CoQ10, Vitamin E |
↑ O2 consumption | Santos [68] |
Rodent models of T2D present with a range of phenotypes depending on the presence or absence of obesity, mode of diabetes induction, and duration of hyperglycemia. The db/db mouse is a severely hyperglycemic genetic model of obesity, insulin resistance and T2D, which presents with altered myocardial substrate use and reduced myocardial efficiency [54]. Similar to the db/db mouse, the male Zucker diabetic fatty (ZDF) rat spontaneously develops T2D and obesity. However, female ZDF rats require the addition of high-fat feeding to induce diabetes [55]. Db/db mice have elevated myocardial superoxide, hydrogen peroxide and peroxynitrite production [56,57]. Furthermore, both db/db mice and ZDF rats display increased lipid peroxidation and protein carbonylation levels [57–60]. Similar to T1D, these models also present with increased activity of antioxidant enzymes, such as CAT and MnSOD, and with impaired ETC activity and mitochondrial uncoupling [57,60]. High-fat or fructose feeding may also be used to induce obesity and/or diabetes and have been found to increase hydrogen peroxide, nitric oxide production [61,62], lipid peroxidation and nitrotyrosine [63], respectively. Studies using a combination of high-fat feeding and a single low-dose injection of STZ demonstrated an increase in mitochondrial ROS production before onset of impaired insulin secretion both in rodents [64] and a large animal model [65], suggesting that metabolic derangements that precede the onset of overt DM may already be sufficient to increase ROS and to contribute to the development of DbCM. Lastly, the Goto-Kakizaki (GK) rat is a unique model of insulin-resistance without obesity, characterized by mild diastolic dysfunction and susceptibility to oxidative stress [66–68]. However, based on the fact that lipid peroxidation is increased [68] while mitochondrial function is preserved in this model, it has been suggested that factors associated obesity and insulin resistance could be a critical mediator of the development of mitochondrial dysfunction, rather than hyperglycemia per se [69].
There exists a large body of literature on preclinical trials of antioxidants that report therapeutic benefit of ROS reduction on diabetic complications, implying that antioxidants may be a viable therapeutic option to reduce the consequences of chronic oxidative stress in diabetes. Importantly, antioxidant vitamin supplementation has been largely insufficient in showing positive outcomes in clinical trials of CVD, despite their success in preclinical studies [70–73]. As such, new research is focused on fine-tuning the administered dose, as well as optimizing the delivery. However, it should be noted that while evidence of oxidative stress has been reported in multiple organs in humans that exhibit well-characterized diabetes complications, only few data are available regarding oxidative stress in human DbCM (Table 1). Despite the limited number of studies, increased mitochondrial hydrogen peroxide emission, increased mitochondrial ROS levels, and increased levels of HNE-adducts have been consistently observed in atrial tissue of diabetic patients in these studies [74–77]. Although all of these data have been generated from atrial tissue, they are highly suggestive that oxidative stress could also occur in LV tissue of human DbCM. Nevertheless, in order to improve our understanding since clinical trials using antioxidant therapies have failed thus far, a more in-depth analyses of ROS generation and ROS-induced damage are needed, in particular from LV tissue of patients matching the definition of DbCM. Moreover, the development of reliable biomarkers for myocardial oxidative stress, would significantly advance the field.
5. Sources of ROS in diabetic cardiomyopathy
There are several principal sources of ROS generation within a cardiomyocyte that may contribute to oxidative stress in cardiac disease states. Several cytosolic enzymes, such as NADH oxidases (NOX), xanthine oxidase (XO) and uncoupled nitric oxide synthase (NOS), produce ROS during their catalytic activity and alterations in cytosolic ROS generation that may contribute to the pathophysiology myocardial dysfunction in diabetes has been described [78]. However, mitochondrial sources of ROS are thought to represent the major ROS burden in the context of diabetes. Most notably, the electron transport chain (ETC), p66Shc, and monoamine oxidase (MAO) are considered the major sources of ROS formation in mitochondria (Fig. 1A). However, given that direct comparison of cytosolic versus mitochondrial sources of ROS production in the diabetic heart are limited, it is difficult to evaluate the relative contributions of each ROS-producing enzyme. Furthermore, it should be noted that while mitochondrial ROS production is elevated in the T2D heart, this may not be the case for several models of T1D [44,79, 80], highlighting important differences that may contribute to the development of DbCM. Nonetheless, literally all of these have been shown or at least been implicated in the development of oxidative stress in DbCM, as schematically illustrated in Fig. 2. The underlying evidence (Table 2) will be presented and discussed in the following paragraphs.
Fig. 2. Targeting ROS-producing and antioxidant enzymes in the diabetic heart.
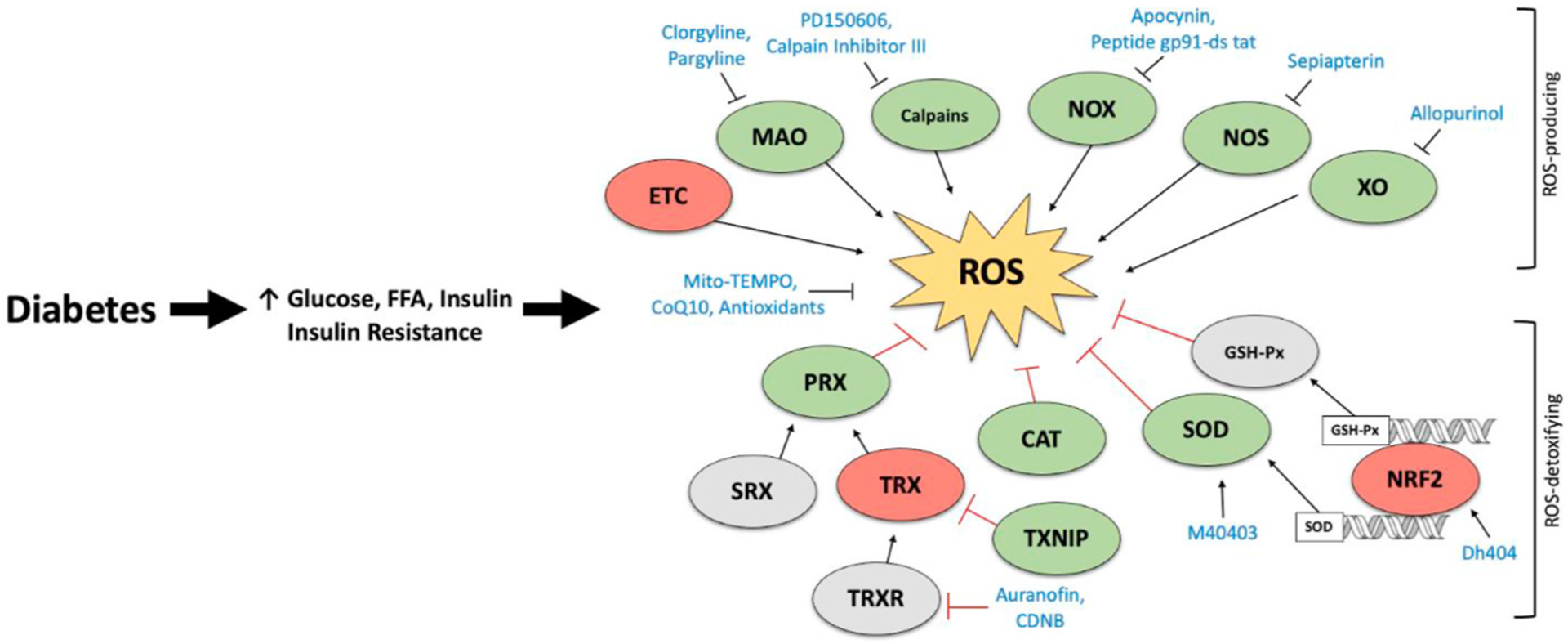
Enhanced expression/activity of reactive oxygen specifies (ROS)-producing enzymes induces ROS overproduction in the diabetic heart. Although production of ROS is counterbalanced by enhanced expression of the antioxidant defense system, insufficient ROS-detoxification may impair the “redox state” of the diabetic heart, thereby resulting in oxidative stress. Pharmacological inhibition of ROS-producing enzymes or activation of ROS-detoxifying enzymes offers a potential therapeutic approach for the treatment of diabetic cardiomyopathy (DbCM). Green indicates elevated expression/activity in DbCM. Red indicates reduced expression/activity in DbCM. Grey indicates unchanged or unknown expression/activity in DbCM. Blue indicates pharmacological activators/inhibitors. Catalase (CAT); 1-chloro-2,4-dinitrobenzene (CDNB); electron transport chain (ETC); free fatty acid (FFA); glutathione peroxidase (GSH-Px); monoamine oxidase (MAO); nitric oxide oxidase (NOS); nicotinamide adenine dinucleotide phosphate oxidase (NOX); peroxiredoxins (PRX); sulfiredoxin (SRX); superoxide dismutase (SOD); thioredoxin interacting protein (TXNIP); thioredoxin (TRX); TRX reductase (TRXR); xanthine oxidase (XO).
Table 2.
Studies investigating ROS-producing enzymes in DbCM.
| Intervention/Model | Diabetes Model | ROS/Oxidative Stress | Cardiac Structure/Function | Author |
|---|---|---|---|---|
| MAO inhibitor, Clorgyline [100] or Pargyline [101] | STZ-induced T1D rats | ↓ MDA, 4-HNE ↓ MAO-A activity; ↔ MAO-A protein ↓ SOD activity; UCP3 protein; ↑ GSH-Px activity; PRX-3 protein; ↔ CAT activity; PRX-5 protein |
↓ contractile dysfunction, diastolic stiffness, electrical abnormalities ↓ morphological changes, fibrosis, apoptosis ↓ mitochondrial depolarization |
Umbarkar [100], Deshwal [101] |
| CM-specific CAPN4 KO or calpastatin-TG | STZ-induced or OVE26 T1D mice | ↓ ROS, H2O2; ↔ TAC ↓ NOX, calpain activity |
↓ systolic and diastolic dysfunction ↓ cell death ↓ CM hypertrophy ↓ fibrosis, apoptosis |
Li [106], Li [107], Ni [109] |
| NOX inhibitor, Apocynin | STZ-induced T1D mice | ↓ O2•− , production, NT, NO ↔ p-eNOS (Ser1177) |
↑ systolic function ↔ cardiac remodeling ↑ CM contractility |
Roe [122] |
| NOX4 ASOs | STZ-induced T1D rats | ↓ NOX4; ↔ NOX1, NOX2 protein ↓ ROS (DHE), NOX-dependent O2•− production |
↓ systolic dysfunction ↓ cardiac fibrosis |
Maalouf [125] |
| CM-specific RAC1 KO or NOX inhibitor, Apocynin | STZ-induced T1D mice | ↓ O2•− , H2O2 production, total/mitochondrial ROS production ↓ Rac1 mRNA/protein/activity; membrane Rac1, p67phox protein; NOX activity; Rac1, gp91phox, p67phox, p47phox mRNA ↑ TRXR activity |
↓ cardiac dysfunction ↓ CM hypertrophy ↓ cardiac fibrosis, apoptosis |
Li [124], Shen [121] |
| XO inhibitor, Allopurinol | STZ-induced T1D mice/rats | ↓ ROS, 3-NT, 15-f2t-isoprostane ↓ SOD activity; MnSOD, iNOS protein, XO protein/activity; ↔ eNOS protein ↔ p22phox, p40phox, p47phox and gp91phox mRNA expression ↑ NRF2 mRNA/nuclear protein; ↓ Keap1 protein/mRNA |
↓ systolic & diastolic dysfunction ↓ cardiac remodeling & hypertrophy ↓ atrial CM disorder ↓ atrial & LV fibrosis, apoptosis |
Yang [128], Rajesh [130], Gao [131], Luo [132] |
| eNOS, iNOS and nNOS KO or BH4 precursor, Sepiapterin | STZ- induced T1D mice | iNOS−/−: ↓ MDA, 4-HNE, NT protein ↓ NOx bioavailability sepiapterin: ↓ MDA, 4-HNE, NT protein ↑ BH4 and the BH4/BH2 ratio ↓ NOx bioavailability |
sepiapterin: ↑ systolic function ↓ cardiac remodeling |
Jo [144] |
5.1. Mitochondrial electron transport chain (ETC)
The ETC, comprising protein complexes I-IV and the electron transfer carriers, ubiquinone and cytochrome c, is the main site of ATP production during oxidative phosphorylation (OXPHOS). Under physiological conditions, electrons are tightly coupled with ATP production, but ROS are nonetheless formed at the ETC from low levels of electron leak (0.2–2%), driven by the activity of ETC complexes, particularly at complex I and III [81]. Brownlee’s group provided strong evidence that under hyperglycemic conditions, ROS in vascular endothelial cells are derived from the mitochondrial ETC, as evidenced by an inhibition of increased ROS production by treatment with an uncoupling agent or a complex II inhibitor [82]. Numerous other studies demonstrated that mitochondrial ROS generation is also increased in cardiomyocytes under hyperglycemic or diabetic conditions, likely generated at the ETC, although the relative contribution of each OXPHOS complex remains a matter of debate [42,57,80]. ADP-stimulated, coupled respiration with electrons delivered via complex I or II is decreased in STZ-diabetic rats, however the amount of ROS generated per oxygen consumed was clearly increased, suggesting that impaired ETC complex activities are involved in increased ROS generation in cardiomyocyte mitochondria [83]. Similarly, succinate-driven ROS production in the presence of the complex V inhibitor, oligomycin, was increased in mitochondria isolated from db/db mouse hearts, and partial inhibition of ROS production following inhibition of complex I with rotenone suggested that ROS may be generated both at complex I but also at other ETC complexes [57]. It is widely speculated that decreased expression or altered composition of OXPHOS subunits observed in models of T1D [84,85] and T2D [57,60] may contribute to increased ETC-related ROS formation, since conditions that impair electron flow through the ETC may increase the likelihood that electrons slip non-specifically from the ETC to generate ROS, as occurs during ischemia reperfusion or heart failure [86].
Of note, complex I or II as a source of ROS-dependent cardiomyocyte dysfunction has also been observed following high glucose (HG)-stimulation of cardiomyocytes from diabetic OVE26 mice [31]. The observation that HG induces ROS production in cardiomyocytes from T2D but not wild-type mice implicates the diabetic milieu in predisposing cardiac mitochondria to ROS production [31]. In fact, a pronounced remodeling of mitochondrial proteins, including ETC subunits, has been observed in hearts of OVE26 mice using 2-D gel comparative proteomics, potentially revealing the biochemical basis for the predilection to ROS production [48]. Thus, it could be hypothesized that hyperglycemia may induce ROS production in diabetes as a result of acquired defects in the ETC. In addition, mice with cardiomyocyte-selective deletion of the insulin receptor demonstrated decreased expression of OXPHOS subunits, associated with increased mitochondrial ROS generation [57,85]. Importantly, the other systemic metabolic perturbations that typically accompany DM are absent in this model, including hyperglycemia. This observation suggests that impaired cardiomyocyte insulin signaling may induce mitochondrial proteomic remodeling, which may increase ROS generation even in the absence of hyperglycemia. Given that impaired cellular insulin signaling often precedes DM, it is possible that the onset of diabetes with concomitant hyperglycemia may further increase ROS generation in the diabetic heart. This hypothesis was partially supported by observations made by Montaigne et al., who observed decreased complex I activity in obese, diabetic individuals, but ROS production was only increased in diabetic hearts [77]. Niemann et al. observed decreased expression of complex I subunits and enzymatic activity of complex I, accompanied by increased protein carbonylation and oxidative DNA damage in right atrial tissue of obese and insulin resistant (but not diabetic) subjects, suggesting that mitochondrial proteomic remodeling during insulin resistance may be sufficient to induce increased ROS [87]. Partial inconsistencies among studies, suggest that a certain degree of ETC remodeling may be necessary to increase ROS generation. These changes are not only determined by obesity and impaired insulin signaling, but also by genetic predisposition and other, maybe unknown, mechanisms. Mechanisms that modulate OXPHOS expression during diabetes development may differ between subjects, but may include protein degradation, transcriptional regulation, altered protein composition, altered mitochondrial quality control, and others [88].
Given that the diabetic heart predominantly oxidizes FAs, it is also of interest that a cardiomyocyte-specific increase in FAs due to overexpression of ACSL1 has been shown to markedly increase mitochondrial ROS generation at the ETC, suggesting that DM-associated hyperlipidemia and/or the shift in substrate oxidation towards increased FA oxidation may contribute to increased mitochondrial ROS generation at the ETC [89]. Of note, this ROS generation occurred using succinate or palmitoyl-carnitine as a substrate, but not if glutamate was used, and ROS generation was completely blocked by rotenone, suggesting that ROS generation was predominantly driven via complex II. Thus, in addition to hyperglycemia and impaired insulin action, increased myocardial lipid utilization may also contribute to increased mitochondrial ROS generation in the diabetic heart.
5.2. p66Shc
Another important source of ROS production is a protein of the Src homology 2 domain and collagen-homology region (Shr) family, p66Shc. This cytosolic protein partially translocates to the mitochondria [90], where it catalyzes the electron transfer from cytochrome c to oxygen, thereby promoting production of hydrogen peroxide [91]. In addition, p66Shc can activate NOX and inhibit synthesis of antioxidant enzymes, thus further promoting the development of oxidative stress [92]. Importantly, it is believed that persistent transcription of p66Shc occurs as a result of epigenetic signals, such as DNA hypomethylation and H3 acetylation, regulated by miR-218 and miR-34a. Interestingly, mice lacking p66Shc are protected from the consequences of ROS on vasculature in T1D and T2D [93,94]. Of note, upregulation of p66Shc in the diabetic heart has been associated with oxidative stress and left ventricular dysfunction, which were reversed upon gene silencing of p66Shc [95]. Furthermore, ablation of p66Shc expression in cardiac progenitor cells (CPC) and myocytes prevented ROS-mediated tyrosine nitration and 8-OHdG, thereby preserving cardiac gross morphology and function following diabetes [96].
5.3. Mitochondrial monoamine oxidases (MAOs)
The mitochondrial flavoenzymes, monoamine oxidases (MAOs) generate hydrogen peroxide and reactive aldehydes as by-products during the oxidation of monoamines. Recently, MAOs have been implicated as a source of oxidative stress in the heart [97] and have gained traction as a possible target in the treatment of various cardiovascular diseases [98]. Both MAO-A, MAO-B are expressed in the rodent heart, but MAO-A, the major isoform [99], is significantly induced in the myocardium of T1D rats [100]. Under hyperglycemic conditions, enhanced mitochondrial MAO-dependent production of hydrogen peroxide precipitates mitochondrial permeability transition pore (MPTP) opening contributing to mitochondrial dysfunction and impaired endoplasmic reticulum (ER) homeostasis [101]. Pharmacological inhibition of MAO-A, using the specific inhibitor, clorgyline [100] or pargyline [101], attenuated STZ-induced oxidative stress and activation of the ER stress/unfolded protein response (UPR) [100,101]. Importantly, this was associated with reversal of diastolic dysfunction, normalization of cardiac electrical activity and reduced cardiac apoptosis and fibrosis. Altogether, these findings suggest that MAO-induced ROS formation leads to mitochondrial dysfunction and ER stress, that may contribute to the development of DbCM and may offer a promising target for therapeutic intervention. It should be noted that basal expression of MAO is relatively low in the human heart [102] which limits the translatability of findings drawn from studies in animal models. However, it has been reported that increased activity and expression of both MAO isoforms are elevated in the ventricles of end-stage ischemic failing hearts [103]. Therefore, although MAO expression is similar in right atrial appendages of coronary patients with/without diabetes [104], the effect of diabetes on MAO in the human ventricle is relatively underexplored.
5.4. Mitochondrial calpains
Calpains belong to a family of Ca2+-dependent thiolproteases that have been linked to oxidative stress [105]. The essential regulatory subunit is encoded by capn4. The most well-characterized isoforms include calpain-1 and −2, and are ubiquitously expressed in the normal heart. Hearts from T1D [106,107] and T2D [108] mice, display significantly increased calpain activity. Particularly, hyperglycemia [106, 107] and high fat feeding [108] increase calpain activity without altering protein expression of calpain-1 or −2. Importantly, calpain-1 has been directly implicated in superoxide generation via impaired ATP synthase activity [109] and its activation is thought to be mediated by the NOX subunit, gp91phox, in cardiomyocytes [106]. Pharmacological inhibition (PD150606, calpain inhibitor-I or III) of calpain activity or overexpression of the endogenous inhibitor, calpastatin, prevent apoptosis induced by hyperglycemia [106,107]. In addition, inhibition of calpain activity prevents HFD-induced apoptosis in vivo and in vitro. [108] The pro-apoptotic action of calpain may be mediated by stimulating mitochondrial ROS generation [109]. In support of this, reduction of calpain in both T1DM and T2DM prevents cardiac hypertrophy, systolic and diastolic dysfunction, and fibrosis [107,108]. Lastly, transgenic overexpression of calpastatin, which specifically inhibits calpain-1 and −2, has been found to decrease oxidative stress and thereby improve cardiac remodeling following ischemia-reperfusion injury in the diabetic heart [110]. Although increased calpain activity may contribute to oxidative stress in DbCM, additional studies in diverse models and humans will be required to confirm this.
5.5. NADPH oxidases (NOX)
NADPH oxidase (NOX) plays a crucial role in determining the redox state of the heart. Upon formation of a heterodimer with the lower-molecular-weight subunit p22phox, the catalytic unit of NOX becomes the site of formation upon electron transfer from NADPH to molecular O2. Of the seven known NOX isoforms, NOX2 and NOX4 have been well characterized in the heart [111]. Activation of NOX2, but not NOX4, depends on interaction with cytosolic subunits, p47phox, p67phox, p40phox, and Rac1. Importantly, NOX enzymes have been implicated in the pathophysiology of many cardiovascular diseases, including atherosclerosis, hypertension and heart failure [112]. Of note, NOX2 and/or its subunits are induced in both T1D [26,30,113] and T2D [56, 114]. Fatty acids may also activate NOX2 via a diacylglycerol – protein kinase A mechanism leading to ROS-induced inhibition of lysosomal activity that impairs autophagy in high-fat fed mice (PMID: 25529920). In addition, HG exposure enhances expression and activity of NOX2, and induces the expression and translocation of its catalytic subunits in cardiomyocytes [106,115–121]. Importantly, studies with the NOX inhibitor, apocynin, demonstrate beneficial effects against the development of diabetes-induced myocardial dysfunction [122]. Deletion or inhibition of NOX2 prevents palmitate-induced ROS production and cardiomyocyte mitochondrial dysfunction [123]. In addition, Rac1 was implicated in STZ-induced myocardial remodeling and dysfunction [124], which may be a result of hyperglycemia-induced cardiomyocyte apoptosis [121]. Furthermore, NOX4 expression is elevated in diabetic hearts [40,125,126], and inhibition by an antisense NOX4 oligonucleotide improved cardiac function in concert with decreased ROS production [125]. While most knowledge of NOX in DbCM pertains to T1D, evidence exists that attenuation of oxidative stress by the cholesterol-lowering drug, ezetimibe, reduced expression of gp91phox and NOX4 in db/db mice [127]. Thus, targeting ROS-producing enzymes such as NOX may have therapeutic potential in both T1D and T2D.
5.6. Xanthine oxidases (XO)
XO as a source of ROS reacts with molecular oxygen to produce superoxide and hydrogen peroxide leading to oxidative stress in diabetes. XO protein expression is significantly elevated in the atrium of STZ-induced T1D rats [128], and XO activity was increased in both ALX-induced T1D rats [45] and fructose-fed diabetic mice [129]. Importantly, increased XO enzymatic activity was accompanied by increased oxidative stress [45]. Of importance, inhibition of XO with allopurinol has been shown to reduce oxidative stress in pre-clinical models of T1D, thereby inhibiting diabetes-induced cardiac remodeling, left-ventricular dysfunction and atrial electrophysiological abnormalities [128,130,131]. Interestingly, attenuation of oxidative stress by allopurinol may be mediated by activation of the NRF2/HO-1 pathway [132]. Allopurinol also inhibits superoxide formation by aortic rings in rabbits with alloxan-induced T1D [133]. While the XO inhibitors, oxyopurinol and allopurinol, may provide CV benefits in patients with chronic heart failure or following myocardial infarction [134,135], their therapeutic potential in the setting of DbCM remains to be investigated. Given that allopurinol prevented an increase in lipid hydroperoxides in the plasma of diabetic patients and that it mediates the above mentioned CV benefits, allopurinol treatment may be a promising therapy to attenuate CV disease in particular in diabetes and deserves to be evaluated in clinical studies [133].
5.7. Uncoupling NO oxidases (NOS)
Under normal physiological conditions, nitric oxide synthases (endothelial, eNOS; inducible, iNOS; neuronal, nNOS) consume NADPH and O2 to convert L-arginine to L-citrulline, generating nitric oxide and NADP as by-products. However, depletion of the necessary cofactor, tetrahydrobiopterin (BH4) upon oxidation and/or reduced synthesis may lead to reduction of O2 to superoxide, a process called uncoupling of NOS [136,137]. Accumulating evidence suggests that under hyperglycemic conditions, the uncoupling of NOS generates more superoxide and less NO, thereby contributing to myocardial dysfunction [138]. Particularly, eNOS may promote oxidative stress by competing with the catalytic activity of SOD or by promoting formation of peroxynitrite, which generates protein nitrotyrosine [139]. Conversely, hydrogen peroxide was reported to modulate NOS, specifically in cardiomyocytes [140]. Reduced nitric oxide levels are not only detrimental for myocardial function [141,142], but further oxidative stress results upon the generation of peroxynitrite by the reaction of superoxide and NO. While nNOS is believed to be the primary nitric oxide producer in the diabetic cardiomyocyte [143], deletion of iNOS, but not nNOS or eNOS, was sufficient to repress markers of oxidative stress in the diabetic heart [144]. Importantly, the beneficial effects of sepiapterin, a precursor of BH4, on left-ventricular function in STZ-induced T1D is thought to be mediated via enhanced iNOS-derived nitric oxide [144]. Interestingly, both the unspecific NOS inhibitor, L-NAME, and the specific nNOS inhibitor, L-VNIO, partially restore the positive ionotropic effect observed in T1D hearts [143]. Studies with the nitroxyl donor, Angeli’s salt (AS), have highlighted the role of uncoupled NOS in hyperglycemia-induced cardiomyocyte damage both in vitro and in vivo. Specifically, treatment with AS significantly reduced myocardial ROS and improved cardiac function in STZ-induced T1D mice, without affecting hyperglycemia. [145] Importantly, this benefit was mediated by induction of the caveolin-3/eNOS complex to reduce cardiomyocyte hypertrophy and apoptosis [145].
6. Metabolic alterations driving ros-production in diabetic cardiomyopathy
DM is characterized by a perturbation of the systemic metabolic milieu that induces metabolic alterations as well as cellular injury in multiple tissues, including the heart. In T2D, obesity-associated systemic insulin resistance, mainly affecting skeletal muscle, adipose and liver tissue, contributes to increased secretion of insulin and hyperinsulinemia. Hyperlipidemia mainly results from increased lipolysis due to impaired insulin sensitivity of adipose tissue, as well as from hepatic overproduction of TG-rich lipoproteins. Hyperglycemia develops when insulin secretion by pancreatic beta cells is insufficient to offset impaired glucose uptake of insulin-sensitive tissues and increased hepatic gluconeogenesis. In contrast, T1D primarily results from destruction of pancreatic beta cells and thus insulin deficiency, mainly induced by environmental factors and genetic susceptibility. All of these metabolic alterations have been proposed to contribute to oxidative stress in DbCM, on the level of cardiomyocytes, endothelial cells, and the crosstalk between cardiomyocytes and endothelial cells [146,147].
6.1. Hyperglycemia
Glucotoxicity due to excess serum glucose levels has been implicated in many cardiac alterations in diabetes, including generation of AGE, fibrosis, maladaptive inflammatory signaling, O-Glc-NAcylation, and impaired Ca2+ handling. Importantly, hyperglycemia is also known to stimulate ROS production in the diabetic heart by diverse mechanisms, including through NOX, XO and NOS [132,145,148]. High concentrations of glucose have also been shown to trigger synthesis of superoxide, nitric oxide, peroxynitrite and AGE by mechanisms such as increased polyol pathway flux, increased hexosamine pathway flux, and activation of protein kinase [82,149–151]. Many of these pathological pathways may be activated by ROS stemming from mitochondria [82]. Furthermore, several interventions, including inhibition of the ETC complex II and overexpression of MnSOD, have been shown to prevent hyperglycemia-induced ROS production [82]. Moreover, a recent study showed that several long noncoding RNAs may play an essential role in high glucose-induced cardiomyocyte oxidative stress [152]. Altogether, these findings suggest that exacerbation of ROS production in the diabetic heart due to hyperglycemia may result in oxidative stress, which in turn may contribute to the development of cardiac abnormalities in diabetes.
6.2. Hyperlipidemia
The diabetic heart has increased preference for FA as a source of energy, mainly driven by systemic hyperlipidemia and decreased myocardial glucose utilization due to impaired myocardial insulin action or reduced glucose uptake [153,154]. Circulating FAs are taken up by the heart by simple diffusion across the plasma membrane via FA transporters. Inside the cell, FAs are utilized for membrane structure, energy metabolism, and as signalling molecules. However, overload of acyl-CoAs generated from an excess of FAs in the diabetic myocardium can lead to cytosolic and mitochondrial ROS overproduction [155]. Particularly, the delivery of reducing equivalents, NADH and FADH2 resulting from oxidation of fatty acids leads to generation of ROS in the ETC [156,157]. Of note, long-term exposure to the FA, palmitate, promotes ROS production and has been shown to cause increased mitochondrial fission [89]. In addition, accumulated acyl-CoA in the diabetic heart can be recycled to triglycerides or assimilated into complexes, such as ceramides. Several diabetic rodent models have implicated elevated ceramide content and myocardial ROS in the development of cardiac dysfunction [158]. Ceramides are also thought to directly inhibit complex III, thereby generating ROS and inflammation [159]. Lastly, FAs can also regulate lipid homeostasis and oxidation by interacting with members of the nuclear receptor family, such as peroxisome proliferator-activated receptors (PPARs), which may also modulate expression of NOX subunits and their role in contributing to oxidative stress [160].
6.3. Insulin resistance
In many models of T2D, increased ROS levels and evidence of myocardial oxidative stress are associated with impaired insulin-stimulated cardiomyocyte glucose utilization, raising the possibility that impaired cardiac insulin sensitivity could contribute to oxidative stress [161–164]. Myocardial insulin resistance is thought to contribute to functional and structural alterations in the heart and may result from effects that are secondary to systemic metabolic disturbances in insulin resistant states (hyperinsulinemia, hyperglycemia, hyperlipidemia), and/or from intrinsic perturbations of insulin signaling in myocardial tissue [154]. Impaired myocardial expression and translocation of GLUT4 are initial alterations that limit myocardial glucose uptake, and that may occur prior to any defect in the ability of insulin to increase PI3K and Akt signaling, both in animal models and humans with T2D [165,166]. While potential contributions due to accompanying metabolic abnormalities such as hyperglycemia, hyperlipidemia and hormonal changes cannot be clearly distinguished in many models of insulin resistance, obesity and T2D, mice with cardiomyocyte-restricted deletion of the insulin receptor strongly suggested that impaired glucose uptake, mitochondrial dysfunction, and contractile defects may be directly related to impaired cardiomyocyte insulin action [85,167,168]. Of note, these mice also exhibited increased mitochondrial ROS production, potentially related to repression of mitochondrial ETC proteins [85,168]. In addition, it has been proposed that superimposing diabetes may further increase mitochondrial ROS generation since induction of FAO capacity by diabetes may increase the delivery of reducing equivalent to the ETC, thus resulting in increased electron leakage and ROS generation [85]. Thus, the myocardial metabolic adaptations to impaired cardiomyocyte insulin signaling may predispose the heart to increase mitochondrial ROS generation. Furthermore, mice lacking GLUT4 display elevated expression of the NOX isoforms, NOX1 and NOX2 [169], and antioxidant treatment using TEMPOL reversed this upregulation.
6.4. Hyperinsulinemia
An underappreciated mechanism that may contribute to cardiac ROS production is hyperinsulinemia itself. In response to insulin resistance, the pancreas attempts to compensate by secreting additional insulin [170]. A well described observation is that prolonged hyperinsulinemia increases myocardial mass and decreases cardiac output [171,172]. In several models exhibiting hyperinsulinemia, an association with increased myocardial ROS has been observed, e.g. high sucrose diet [173]. While the mechanisms by which hyperinsulinemia may induce ROS remain largely to be elucidated, NOX is thought to be activated by insulin [174,175] and may thus represent a candidate mechanism that may induce ROS production in diabetic hearts.
7. Antioxidant defense system in diabetic cardiomyopathy
Generation of ROS during cellular metabolism is further controlled by the antioxidant defense system consisting of both enzymatic and non-enzymatic antioxidants [176]. This includes the enzymatic antioxidants, superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GSH-Px) and redoxins (Fig. 1 B) and the non-enzymatic antioxidants, vitamin E, β-carotene, and vitamin C. Based on the fact that antioxidant activities are low in the heart compared to other organs [177], the heart is rather susceptible to oxidative damage. Thus, it is not surprising that expression of many antioxidant enzymes is significantly altered in DbCM (Fig. 2; studies summarized in Table 3). Reports of the levels of antioxidant expression and/or activity in diabetic hearts are largely based on studies in rodent models, highlighting a need for a better understanding of ROS producing and ROS scavenging systems in human DbCM. Importantly, according to a study of human left ventricular tissue, the catalytic activity of GSH-Px is over 10-fold higher than that of CAT, MnSOD or Cu/ZnSOD, which may highlight the relative contribution of each component of antioxidant defense in the human myocardium [178].
Table 3.
Studies investigating the antioxidant defense system in DbCM.
| Intervention/Model | Diabetes Model | ROS/Oxidative Stress | Cardiac Structure/Function | Author |
|---|---|---|---|---|
| MnSOD-TG | OVE26 T1D mice genetic lipid overload-induced T2D mice | ↑ cardiac SOD activity ↑ mitochondrial MnSOD protein ↑ CAT protein/activity, GSH protein ↓ cardiomyocyte ROS ↓ mitochondrial H2O2, 4-HNE |
↑ RCR, state 3 respiration ↓ mitochondrial mass/number; ↑ mitochondrial function ↑ CM contractility ↓ mitochondrial size ↑ mitochondrial fission |
Shen [31] Tsushima [89] |
| CM-specific CAT-TG | STZ-induced T1D mice OVE26 T1D mice | ↓ ROS, H2O2, MPO, 3-NT of α-KGD, ATP-α and ATP-β ↓ iNOS protein; ↔ eNOS protein ↑ SIRT2 protein ↓ MDA; CM ROS |
↓ systolic & diastolic cardiac dysfunction ↓ CM contractile dysfunction ↓ LV hypertrophy ↓ myocardial disorder ↓ cardiac fibrosis, apoptosis ↓ mitochondrial damage ↑ CM contractility |
Cong [182], Turdi [183] Ye [29] |
| mPHGPx-TG | STZ-induced T1D mice | ↓ H2O2 production, 4-HNE, MDA in IFM | ↓ systolic dysfunction ↑ ATP synthase activity in IFM ↑ mitochondrial respiration ↓ mitochondrial dysfunction |
Baseler [185] |
| GSHPx-TG | STZ-induced T1D mice | ↓ TBARS, 4-HNE ↑ GSH-Px; ↔ SOD, CAT activities |
↓ CM hypertrophy ↓ fibrosis ↓ apoptosis |
Matsushima [186] |
| TRX2 activation or TRXR inhibitors, Auranofin & CDNB | STZ-induced T1D mice | ↓ ROS ↓ PRX-SO2/3H; ↔ TRX1 expression/localization |
↓ CM hypertrophy ↓ contractile dysfunction |
Li [190] |
| CM-specific TXNIP KO | STZ-induced T1D mice | ↔ ROS | ↔ LV hypertrophy, cardiac function ↑ inotropic reserve |
Myers [194] |
| GSH precursor, NAC | STZ-induced T1D mice | ↓ ROS, H2O2, TAS, NO ↑ GSH content |
↓ systolic & diastolic dysfunction ↓ LV hypertrophy ↓ fibrosis, apoptosis ↑ myofilament morphology |
Liu [199], Fiordaliso [200], Yildirim [201] |
| CM-specific MT-TG | OVE26 T1D mice STZ-induced T1D mice | ↓ GSSG; ↔ GSH ↓ , ROS, 3-NT; 3-NT of ATP synthase α ↓ Trp374/Tyr135 nitration of SCOT ↑ p47(phox) |
↓ myocardial injury ↓ CM contractile dysfunction; LV dysfunction/stiffness ↓ myocardial structural derangement ↓ fibrosis, necrosis ↑ ATP synthase, SCOT activity |
Liang [50] Wold [113], Cai [264], Cong [213], Cong [214] |
| SIRT3 KO mice | STZ-induced T1D mice | ↑ ROS | ↑ cardiac dysfunction ↑ necrosis ↓ ATP |
Song [223] |
| NRF2 KO | STZ-induced T1D mice HFD/STZ-induced T2D mice | ↑ 8-OHdG, MDA, 3-NT, 4-HNE ↓ NRF2, NQO1 mRNA ↑ ROS production ↓ NRF2, NQO1, HO1 mRNA/protein ↓ total, nuclear NRF2 protein ↑ 8-OHdG, MDA ↓ NQO-1, HO-1, MT mRNA; MT protein |
↓ cardiac dysfunction ↓ CM hypertrophy, myocardial structural damage ↓ CM contractility ↑ fibrosis, apoptosis ↑ CM hypertrophy ↓ systolic function ↑ fibrosis |
He [257], He [237], Zang [301] Gu [239] |
| CM-specific NRF2-TG mice | STZ-induced T1D mice | ↑ 4-HNE, 8-OHdG | ↑ cardiac dysfunction ↑ fibrosis, apoptosis |
Zang [301] |
| NRF2 activator, Dh404 | STZ-induced T1D mice | ↑ NRF2 protein ↓ 3-NT |
Tan [233] | |
| HO-1-TG mutHO-1-TG | STZ-induced T1D mice | HO-1-TG: ↓ p47phox, GSH-PX3 mRNA) mutHO-1-TG: ↑ p47phox, GSH-Px3 mRNA | HO-1-TG: ↓ systolic dysfunction mutHO-1-TG: ↑ apoptosis | Zhao [240] |
| ALDH2-TG | sucrose-fed pre-T2D mice | ↑ SIRT3, HO-1 protein ↓ ROS |
↓ myocardial dysfunction ↑ CM contractility ↑ intracellular Ca2+ handling ↑ NAD + activity |
Hu [252] |
7.1. Superoxide dismutase (SOD)
SODs, including MnSOD and CuSOD, are enzymes that accept an electron from superoxide and react with water to create hydrogen peroxide. The myocardial expression of SOD proteins has predominantly been shown to be elevated in models of obesity and T2D [57,61], T1D [39], and in a genetic model of lipid-overload, suggesting that increased SOD levels may protect from oxidative stress by detoxification of highly reactive superoxide [89]. MnSOD is thought to be an important antioxidant enzyme in the heart, given its relatively high levels of abundance within mitochondria, and based on the observation that overexpression of MnSOD in the heart is capable of maintaining mitochondrial morphology, enhancing mitochondrial respiration, and improving contractility in the T1D heart [31]. Furthermore, overexpression of MnSOD completely rescued superoxide production and lipid peroxidation in a genetic model of lipid-overload, thereby restoring mitochondrial cross-sectional area and number [89]. Furthermore, the SOD mimetic, M40403, attenuated lipid peroxidation and DNA damage induced by HG in the perfused heart, and normalized QT interval prolongation and coronary perfusion pressure [179]. Altogether, these findings highlight the critical role of in the development of HG-induced cardiac dysfunction and the therapeutic potential of targeting SOD enzymes in the diabetic heart. It should be kept in mind though that a severalfold overexpression of MnSOD (10–20-fold) in overexpression models, as well as application of SOD mimetics, might only indicate that intensive antioxidant treatment might be required to attenuate oxidative stress and damage, and that targeting SOD enzymes could be a useful approach. However, it is not possible to infer from these studies the pathophysiological significance of a lower levels of MnSOD induction by diabetes in the heart.
7.2. Catalase (CAT)
CAT enzymatically detoxifies hydrogen peroxide into oxygen and water, thus neutralizing it. Both increased expression [29,31,44,49,180] and activity [46,181] of CAT have been reported in rodent models of T1D, although little is known about CAT in the context of T2D. The argument that ROS are implicated in DbCM is further supported by the beneficial cardiac effects of CAT overexpression in diabetic models. Cardiomyocyte-specific overexpression of CAT was found to protect mouse hearts against STZ-induced DbCM, partially by suppressing NF-κB-dependent inflammatory responses and associated protein nitration [182]. This may occur as a result of reduced diabetes-induced cardiomyocyte mechanical abnormalities via altered AKT/Fox-O3a/SIRT2 signaling [183]. Interestingly, although overexpression of CAT preserved l cardiac morphology, prevented the contractile defects, and reduced MDA protein modification, it was unable to reverse the slowed Ca2+ decay induced by T1D [29]. In terms of T2D, CAT overexpression also protected against defects in cardiomyocyte contractility in agouti mice [29]. Similar to SOD enzymes, myocardial overexpression of CAT induced by diabetes may serve as an adaptive response to protect from the consequences of oxidative stress.
7.3. Glutathione peroxidase (GSH-Px)
Similar to CAT, GSH-Px is an enzymatic antioxidant that receives hydrogen peroxide from SOD and converts it to water, thereby preventing spontaneous formation of hydroxyl. In contrast, expression of several isoforms of GSH-Px appears to be unaltered in models of both T1D [39,46] and T2D [60,184]. As might be expected, forced overexpression of the mitochondria-targeted antioxidant enzyme, phospholipid hydroperoxide glutathione peroxidase 4 (GSH-Px4), improved ETC function and cardiac function in concert with attenuated hydrogen peroxide production and post-translational modifications (i.e. oxidation and deamination) in STZ-diabetic mice [185]. Furthermore, genetically-induced overexpression of GSH-Px1 attenuated diastolic dysfunction, myocyte hypertrophy, and interstitial fibrosis in STZ-diabetic mice [186]. Of note, mice with GSH-Px1 overexpression demonstrated development of hyperglycemia, hyperinsulinemia, and obesity in another study, which was however interpreted as an interference of GSH-Px1 activity with insulin signaling due to over quenching of physiological levels of ROS that are required for insulin signaling [187]. Thus, in DbCM, GSH-Px may play a less important role in balancing ROS homeostasis.
7.4. Peroxiredoxin (PRX)/Sulfiredoxin (SRX)/Thioredoxin (TRX)
Peroxiredoxins (PRX) are an antioxidant enzyme family which detoxify hydrogen peroxide and peroxynitrite. The expression of PRX3 is decreased and that of PRX5 is increased in the hearts of T1D rats [100, 181], whereas PRX6 was increased in the fructose-fed rats model of prediabetes [63]. However, the precise role of PRX in the development of DbCM remains unclear. Interestingly, induction of PRX3 expression by the flavonoid, quercetin, prevented T1D-induced oxidative stress and cardiac dysfunction [181]. Furthermore, HG-induced mitochondrial oxidative damage was reduced following overexpression PRX3 in cultured cardiomyocytes [181]. When insufficient amounts of reduced PRX are available to deal with peroxides, hyperoxidation of PRX may occur (Prx-SO2/3H), causing inactivation of the peroxidase activity [188]. Both HG-stimulated cardiomyocytes and T1D rats exhibit enhanced levels of hyperoxidized PRX [189,190]. However, this process may be reversed and prevented by the activity of sulfiredoxin (SRX), thereby restoring PRX activity. Importantly, overexpression of SRX prevents PRX hyperoxidation in T1D hearts and high-glucose stimulated cardiomyocytes, whereas SRX gene silencing induced ROS generation [189].
The antioxidant activity of PRX3 and PRX5 depend on recycling by the mitochondrial electron donor, thioredoxin (TRX) 2 [191]. Studies showed a decrease in the expression of TRX2 in HG-treated H9c2 cardiac cells and myocardium of STZ-induced T1D rats [190]. Of importance, overexpression of TRX2 reduced mitochondrial oxidative damage and restores ATP production in HG-treated H9c2 cells [190]. Furthermore, inhibition of TRX reductases (TRXR), thereby reducing TRX activity [192], may be sufficient to impair contractility in the isolated heart [190]. TRXR activity was significantly reduced in T1D hearts and activated by genetic deletion of RAC1, suggesting that NOX may regulate the activity of TRXR in diabetic hearts [121]. Lastly, the activity of the major thiol reducing TRX system is controlled by expression of its endogenous inhibitor, thioredoxin interacting protein (TXNIP), which is increased in diabetic human right atrial biopsies and hyperglycemic models [193,194]. The resulting reduction in TRX activity is further associated with increased ROS, all of which was reversed by downregulation of TXNIP in cultured neonatal cardiomyocytes [193]. Furthermore, diabetic mice with CM-specific deletion of TXNIP showed greater resistance to β-adrenergic stress than controls [194], although this was not attributed to reduced ROS in this study.
7.5. Glutathione (GSH)/glutathione disulfide (GSSG)
Glutathione (GSH) is a nonprotein thiol that defends against oxidative stress by donating a hydrogen molecule in the neutralization of hydrogen peroxide. As a result of this reaction, GSH is converted to the inactive glutathione disulfide (oxidized glutathione; GSSG) by glutaredoxins (GRX), which is reversed by the enzyme, glutathione reductase (GSR) [195]. Under conditions of excessive ROS production, the GSH system can become overwhelmed; therefore, the ratio of GSH to GSSG is considered an indication of oxidant levels. In fact, GSH depletion has been implicated directly in enhanced mitochondrial ROS production [192]. Particularly, GSH/GSSG ratio was found to be reduced in human permeabilized atrial myofibers from T2D patients with EF>30% [74]. This is further supported by rodent models of T2D which display both low GSH/GSSG [56,58,68,114] and high GSR expression. [60] Similar results were reported in most [25,31, 35,41,48,196] but not all T1D rodent models [35,49,51]. Particularly, the role of GRX, which is elevated in the serum of hyperglycemic patients, and LV myocardium of T2D rats [197], has not yet been explored. Nevertheless, free radical scavenging by GSH infusion has been shown to prevent the increase in coronary perfusion pressure induced by HG in isolated hearts [198]. Importantly, this was associated with reduced superoxide and nitrotyrosine levels, without any effect on nitric oxide content. GSH supplementation also prevented apoptosis in T1D rat hearts, suggesting that the GSH system may also regulate cell death in the diabetic heart [196]. The therapeutic potential of GSH is supported by studies using the GSH precursor, N-acetylcysteine (NAC), which showed that NAC treatment of STZ-induced T1D mice was sufficient to improve cardiac function and reduce cardiac fibrosis in conjunction with reduced ROS generation [199]. Importantly, NAC normalized ROS production and prevented glutathione loss in cardiomyocytes isolated from T1D rats, and this was associated with reduced apoptosis [200]. In addition, NAC treatment of diabetic rats resulted in a significant restoration in the expression levels of several microRNAs (miR-499, miR-1, miR-133a, and miR-133b) in the myocardium, which are otherwise markedly depressed in diabetic cardiomyocytes [201]. Furthermore, the ability of NAC to ameliorate hyperglycemia-induced cardiac injury may be mediated via enhanced endogenous CoQ levels [202]. Based on positive patient outcomes following NAC supplementation [203–205], improving antioxidant capacity may provide an opportunistic approach to treating oxidative stress-induced cardiac injury in diabetic patients [206]. Lastly, glutathione-S-transferases (GSTs), which catalyze the conjugation of GSH was reduced [45] and the multidrug resistance protein 1 (MRP1), a transporter implicated in the efflux of GSH was elevated [35], respectively, in T1D hearts, although their precise role in DbCM remains to be elucidated.
7.6. Metallothionine
Metallothionein (MT) is a cysteine-rich regulator of metal homeostasis that has been shown to have potent antioxidant properties against hydroxyl radical and peroxynitrite [207,208]. Of the four MT isoforms, MT-I and MT-II are the major isoforms in the human and animal heart [209]. Chronic overexpression of MT eliminated excess ROS production in diabetic cardiomyocytes [210], thereby alleviating hyperglycemia induced mitochondrial injury [50] and impairments in cardiomyocyte contractility [113]. In addition, elevated MT attenuated ischemic contractility seen in diabetic hearts [50]. MT exerts cardioprotective effects in diabetic hearts by inhibiting GSH depletion, lipid peroxidation and protein nitration, thereby reducing cardiomyocyte death [211]. Importantly, beneficial effects of MT overexpression are thought to occur without alterations in other antioxidants [212], but may be attributed to reductions in tyrosine nitration and resulting impairment of ATP synthase α subunit [213] and/or SCOT [214]. Interestingly, prevention of DbCM by zinc supplementation is believed to be mediated by elevated MT expression [215]. As such, MT could be a potential strategy for prevention of DbCM through suppression of nitrosative damage (further reviewed in Refs. [216–219]).
7.7. Sirtuins
Sirtuins (SIRTs) are NAD+-dependent deacylases that regulate protein function by removing posttranslational modifications from protein lysine residues. Impaired activity of sirtuins may contribute to mitochondrial defects in DbCM including oxidative stress. Particularly, impaired antioxidant function is a result of reduced levels of the mitochondrial SIRT3, which promotes increased acetylation and thereby inactivation of MnSOD [220]. Importantly, increased protein acetylation in db/db hearts is associated with decreased NAD+/NADH ratio and reduced expression of SIRT3 and its targets, LCAD and MnSOD [221, 222]. In support of this, deletion of SIRT3 elevates ROS production and enhances cardiomyocyte necrosis, thereby reducing mitochondrial membrane potential and aggravating cardiac dysfunction in T1D [223]. Moreover, overexpression of SIRT3 reduced HG-induced hydrogen peroxide production in human cardiomyocytes [224]. Interestingly, the antioxidant effects of the peptide, elabela, in diabetes-induced cardiac injury may be mediated by Foxo3a deacetylation by SIRT3 [225]. Although not well-characterized, reduced SIRT2 expression may also play an important role in DbCM and is reversed along with the beneficial cardiac effects of CAT overexpression [183]. In addition, the novel thiazolo[3,2-a]pyrimidine derivative, LF10, suppressed cardiac dysfunction in T1D upon restoring SIRT1 and preventing MAPK activation [226]. Nonetheless, the exact roles of SIRT1 and SIRT3 in the development of DbCM requires further investigation.
7.8. Nuclear factor erythroid 2-related factor 2 (NRF2)/Heme oxygenase (HO)-1
The nuclear factor erythroid 2-related factor 2 (NRF2) is a master transcriptional regulator that controls the expression of antioxidant genes in response to oxidative stress, thereby playing an important role in cell survival [227]. The activity of NRF2 is primarily regulated via its interaction with Kelch-like ECH-associated protein 1 (Keap1), which promotes ubiquitination/degradation of NRF2 [228]. Under conditions of oxidative stress, NRF2 dissociates from Keap1 and translocates to the nucleus, where it regulates gene expression upon binding to antioxidant response elements (AREs) located in the promoter of its target genes [229]. Cytoprotective genes upregulated by NRF2 include the antioxidant genes, GSH-Px and SODs, as well as the cytoplasmic detoxification enzyme, NAD(P)H quinone dehydrogenase (NQO)-1, and heme oxygenase (HO)-1 [230]. Importantly, evidence of oxidative stress in T1D has been attributed to impaired NRF2-induced antioxidant expression [132,231]. In support of this, nuclear expression of NRF2 and its downstream target, HO-1, are reduced following HG-stimulation of human cardiomyocytes [232]. It should be noted that while expression of NRF2 appears to be significantly downregulated in cardiac tissue from diabetic patients [233], HO-1 expression is increased in diabetic patients without heart failure, although this may be attributed to obesity rather than T2D [234]. Interestingly, mRNA content of HO-1 was also increased in STZ-induced diabetic rat hearts [235]. Thus, downregulation of NRF2 might not be an early response to hyperglycemia [236,237], however evidence exists that the NRF2 adaptive response to glucose may be impaired in T2D [238]. Importantly, deletion of NRF2 exacerbated ROS production and increased sensitivity to mitochondrial respiratory complex II inhibition in HG-stimulated cardiomyocytes, and this was associated with impaired cardiomyocyte contractility and increased cellular apoptosis [237]. Furthermore, deletion of NRF2 exacerbated HFD/STZ-induced cardiac damage, and this was partially reversed by supplementation with sulforaphane (SFN) [239].
Meanwhile, overexpression of HO-1 inhibited oxidative stress, inflammation and apoptosis and enhanced autophagy, thereby ameliorating the pathogenesis of cardiac dysfunction in T1D [240]. Furthermore, the protective myocardial effects of several natural compounds against hyperglycemia-induced oxidative stress have been shown to be dependent on NRF2/Keap1 in diabetes [241–245]. Notably, the beneficial effects of SFN in the diabetic heart appear to be mediated by NRF2-dependent induction of MT [236,239]. However, conflicting reports have also implied that cardiomyocyte-specific overexpression of NRF2 may worsen progression of DbCM, whereas global NRF2 knockout may prevent the progression of cardiac dysfunction in T1D [246]. Based on this, the potential of targeting NRF2 as a therapeutic strategy in diabetes requires further exploration (further reviewed in Refs. [247, 248]).
7.9. Aldehyde oxidases
Mitochondrial acetaldehyde dehydrogenase 2 (ALDH2) is a nuclear-encoded aldehyde oxidase responsible for the metabolism or detoxification of acetaldehyde and other toxic aldehydes. ALDH2 polymorphisms are associated with an increased risk of T2D in female CAD patients [249]. Furthermore, proteomics analysis of hearts from STZ-induced T1D rats revealed significantly decreased expression of ALDH2 [41]. Importantly, overexpression of ALDH2 attenuates HG-induced cardiac injury by suppressing mitochondrial ROS production and inhibiting NLRP3 inflammasome activation [250]. In addition, ALDH2 protects against myocardial dysfunction in diabetes through an AMPK-dependent regulation of autophagy [251]. Moreover, the cardioprotective effects of ALDH2 in insulin resistance-induced cardiomyopathy may involve SIRT3 [252]. These studies suggest potential therapeutic promise of targeting ALDH2 in DbCM, although further studies are required.
8. Consequences of ROS in diabetic cardiomyopathy
ROS has been well-documented to play a causative role in the development of DbCM [253,254]. Regardless of the source of ROS, increased ROS levels may result in damage to nucleic acids, proteins, and lipids, resulting in a plethora of cellular abnormalities that may culminate in cardiomyocyte dysfunction and death in diabetes (Fig. 3). Below, we discuss maladaptive cellular alterations evident in DbCM that have been causally linked to increased ROS and oxidative damage.
Fig. 3.
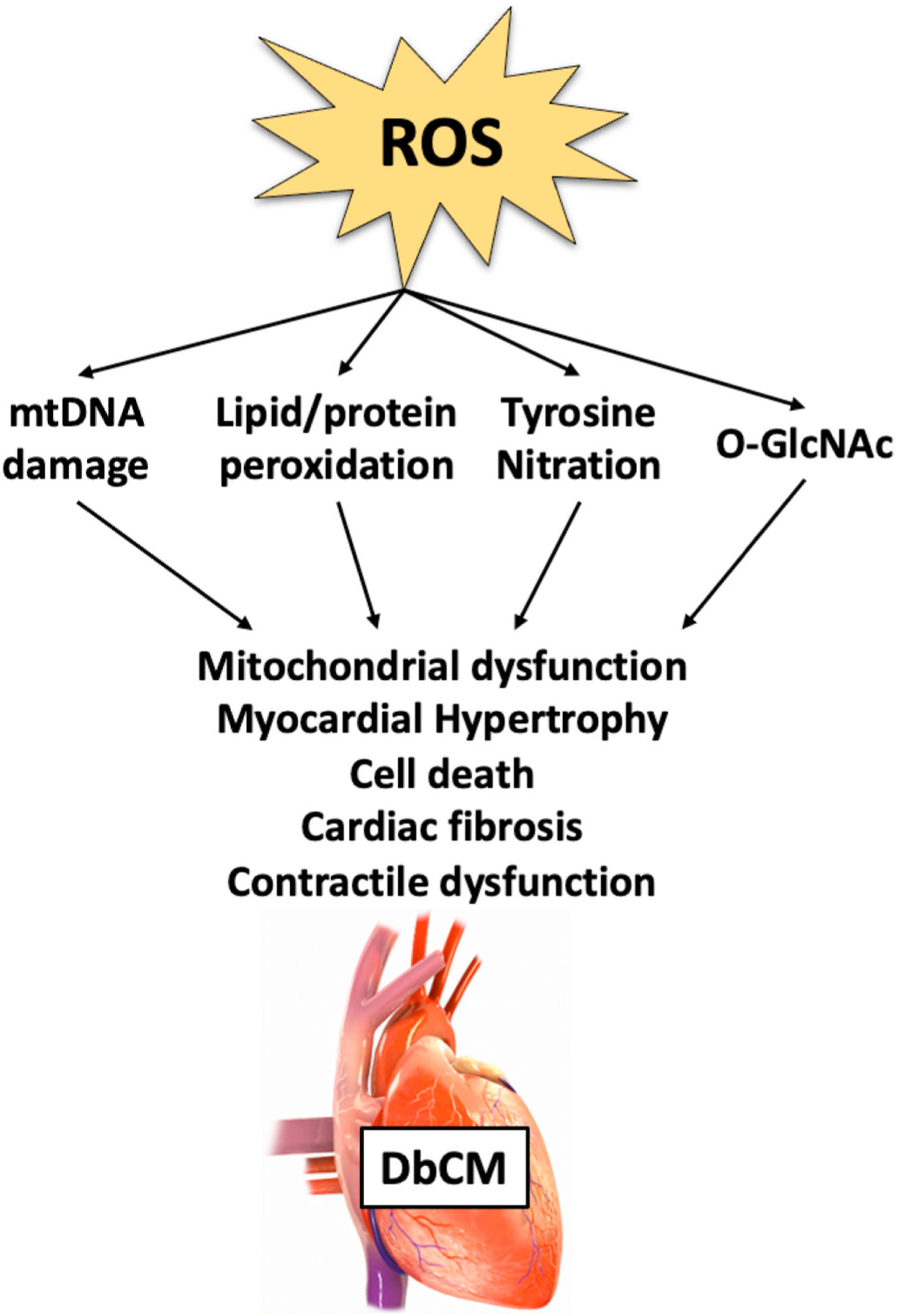
Consequences of ROS in the diabetic heart. Imbalance of reactive oxygen species (ROS) production and detoxification in the diabetic heart may result in damage to nucleic acids, proteins, and lipids, thereby resulting in mitochondrial dysfunction, myocardial hypertrophy, cell death. The convergence of these mechanisms leads to damage, which contributes to cardiac dysfunction observed in diabetic cardiomyopathy (DbCM). O-GlcNAcylation (O-GlcNAc); mitochondrial DNA (mtDNA).
8.1. DNA damage
Although there are relatively few proteins encoded by mitochondrial DNA (mtDNA), mtDNA appears to be particularly susceptible to oxidative damage [255]. MtDNA number, as well as expression of proteins that control replication and transcription of mtDNA are elevated in OVE26 T1D hearts [48], but reduced in HFD-induced T2D mice [256]. Oxidation of guanine residues results in the accumulation of 8-hydroxyguanine (8OHG), 8-hydroxydeoxyguanosine (8OHdG), and 8-oxo-7, 8-dihydro-2-deoxyguanosine (8-oxodG) [32,257]. Moreover, 8-oxoguanine glycosylase (OGG-1) is the primary enzyme responsible for reversing this reaction and may be inactivated in the diabetic heart by 4-HNE modification [59]. Importantly, overexpression of OGG-1 prevented fibrosis and improved contractile function the T1D heart [258]. In addition, HG-induced increases in 8-OHdG are attenuated by enhanced antioxidant capacity [179].
8.2. Lipid/protein peroxidation
In addition to DNA, unsaturated lipids are considered a vulnerable target of ROS. Malondialdehyde (MDA) is a product of lipid peroxidation (LPO) and is a commonly used marker of oxidative stress. Increased levels of lipid peroxides have been reported in the serum of diabetic patients [259–261] and in the heart of both T1D [24,35,39,45,196] and T2D [57,60,61,68,114,241] rodents, although this is not always the case [51,262]. In addition, hyperglycemia and/or hyperlipidemia both increase lipid peroxidation levels specifically in the heart or cardiomyocytes [179,232,240,241]. Importantly, the contribution of ROS to enhanced lipid peroxidation is evident by reduced MDA levels upon targeting ROS-producing enzymes [100,145], activating antioxidant defense systems [29,240], or improving ROS scavenging [27,226]. Of note, enhanced myocardial lipid peroxidation may be a direct consequence of myocardial lipid overload [61,89]. Proteins are also subject to peroxidation (marked by 4-HNE adducts) under conditions of persistent lipid peroxidation, as evidenced in atria of T2D patients [74]. 4-HNE modification of certain mitochondrial proteins, such as the FAD-containing subunit of succinate dehydrogenase [42], may contribute to mitochondrial defects and therefore myocardial contractile dysfunction in diabetes. In fact, inhibition of MAO [101] and induction of PRX-3 [181] were found to reduce cardiac levels of 4-HNE in association with reduced diastolic stiffness. Lastly, another modification shown to be a sensitive index of oxidative stress is 8-isoprostane prostaglandin (8-iso PGF2α) [34], which is also elevated in the heart of STZ-induced T1D rats [41].
8.3. Nitrotyrosine (3-NT)
In addition to peroxidation, nitration of proteins may also occur, particularly at tyrosine residues (3-nitrotyrosine; 3-NT). This is often used as a biomarker of cardiovascular risk [263], and may be implicated in the development of DbCM [264,265]. While 3-NT is increased in diabetic hearts, this damage can be reversed following inhibition of XO [130], by overexpressing antioxidant proteins [182,213,214], and by CoQ10 supplementation [27,28]. Indeed, reduced nitration of specific enzymes involved in metabolism [214,262,266], contraction and antioxidant defense [47] may be implicated in the impairments of the diabetic heart. Particularly, direct damage of ETC complexes have been characterized in hearts of STZ-induced diabetic rats [42,47]. Notably, enhanced antioxidative capacity by NRF2 may play a protective role in mediating against 3-NT in DbCM [257]. Interestingly, both lipid peroxidation and 3-NT are not increased in hearts of pre-diabetic mice [63], suggesting that this may be a consequence of prolonged metabolic disturbances in long-standing diabetes.
8.4. O-GlcNAcylation (O-GlcNAc)
O-GlcNAcylation (O-GlcNAc) is a specific posttranslational modification at Ser/Thr residues that alters the activity of the target protein. O-GlcNAc-transferase (OGT) and O-GlcNAcase (OGA) are the enzymes responsible for protein modification by O-GlcNAcylation [267,268]. Interestingly, expression of OGT and OGA appears to be increased in T1D in association with hyperglycemia-induced superoxide production [26]. Moreover, ROS-induced mitochondrial dysfunction in diabetes has been proposed to occur through O-GlcNAc [269]. In addition, subunits of the respiratory chain complexes I, II and IV undergo enhanced O-GlcNAc under HG conditions, in concert with impaired activity of specific proteins that regulate mitochondrial dynamics, optic atrophy 1 (OPA1) [270] and/or dynamin-related protein 1 (DRP1) [271]. Moreover, improper localization of OGT to the mitochondria prevents normal complex IV activity, thereby reducing mitochondrial membrane potential and impairing respiratory function [269,272]. O-GlcNAcylation of CaMKII during diabetes is also thought to impair Ca2+-handling and may be implicated in cardiac arrhythmias and dysfunction [273,274]. Altogether, these modifications induced by altered substrate metabolism and oxidative stress are thought to be toxic, particularly to the cardiomyocyte, and likely contribute to the onset of cardiac dysfunction in DbCM. Key myocardial proteins that are modified by O-GlcNAc and are involved in substrate metabolism and antioxidant defense (amongst other pathways) have been recently reviewed [254].
8.5. Advanced glycation end-products (AGE)
Advanced glycation end-products (AGEs) are the result of non-enzymatic glycation and oxidation of long-lived proteins and lipids, thereby disabling their functional properties [275,276]. Accumulation of AGE and its receptor (RAGE) have been noted to result from enhanced oxidative stress in hearts from pre-clinical models of diabetes [25,277], as well as cardiac tissue of diabetic HF [278] and aortic stenosis [279] patients. This is coupled with increased expression of NOX2 and its catalytic subunits [280]. Importantly, a causative role of AGE in oxidative stress and cardiac injury was recently demonstrated using engineered cardiac tissues (ECTs) as an in vitro model of DbCM [281]. Furthermore, AGE have been implicated in the progression of contractile dysfunction in DbCM by mechanisms that include increased Ca2+ leak in the heart [282]. Importantly, excess AGE formation is thought to contribute to myeloperoxidase (MPO) pathway induction [283], which will further exacerbate AGE formation [284], thus causing feed-forward-fueled pathological loops that amplify metabolic dysfunction. Furthermore, activation of the AGE/RAGE axis may impair activity of SIRT1 [285,286], although this phenomenon requires additional exploration in the diabetic heart.
8.6. Mitochondrial dysfunction
Since mitochondria are a source of ROS, they are also a major target of ROS damage. Importantly, mitochondrial dysfunction is a characteristic of obesity as well as diabetes/insulin resistance in the human heart [69,87]. Diabetic patients with both diastolic dysfunction [287] and normal [288] cardiac function display reduced cardiac phosphocreatine/ATP ratios, suggesting that mitochondrial energetics is compromised in the diabetic heart. Increased hydrogen peroxide emission is associated with impaired mitochondrial respiratory function in left atrial appendage of T2D patients with CAD [74]. Evidence of increased mitochondrial oxidative stress was associated with increased sensitivity to calcium-induced MPTP opening in atria from T2D patients [74,75]. Mitochondrial bioenergetic dysfunction in diabetes may also result from increased uncoupling [40], which negatively impacts mitochondrial energetics and Ca2+ uptake [289]. In addition, impaired removal of damaged, ROS-overproducing mitochondria through mitophagy within diabetic cardiomyocytes has been described [290]. Of note, MT overexpression restored mitochondrial structure which is otherwise disorganized in hearts of OVE26 mice [50]. In addition, overexpression of MnSOD improved respiration and normalized mass in diabetic mitochondria [31]. Furthermore, pharmacological and genetic inhibition of NOX2 prevents palmitate-induced ROS generation, depolarization of the mitochondrial inner membrane and Ca2+ overload in isolated cardiomyocytes [123].
8.7. Cell death
Enhanced cardiomyocyte apoptosis in both diabetic humans and rodent models may contribute to diabetic cardiac remodeling and dysfunction [200,291–293]. Evidence of myocardial apoptosis in both T1D [37,100,196] and T2D [294] models includes increased number of TUNEL-positive nuclei, reduced B-cell lymphoma 2 (BCL-2)/Bcl-2-associated X (BAX) ratio, and increased caspase (CASP) signaling (particularly CASP3, CASP9 expression and activity). In addition, in vitro exposure of cardiomyocytes to hyperglycemia [224,237,295,296] and increased fatty acids [297,298] will activate apoptosis. Increased cell death is associated with increased 3-NT [299,300], thereby implicating ROS in diabetes-related apoptosis [96,292]. Apoptosis may also result from GSH depletion linking oxidative stress with autophagy induction [196]. While the mechanisms by which hyperglycemia induces cardiomyocyte apoptosis are not fully understood, one proposed mechanism is via calpain-induced superoxide production [109]. Particularly, modulating the calpain/calpastatin system protects cardiomyocytes from apoptotic death in a model of T1D [106]. Interestingly, NOX has also been implicated in cardiomyocyte apoptosis since knockdown of Rac1 or treatment with the NOX inhibitor, NSC23766, significantly inhibited apoptosis and resulted in improved myocardial function of T2D mice [121]. There have been conflicting reports as to the potential role of Nrf2 in mediating diabetes-induced cardiomyocyte apoptosis. While some groups have reported that absence of NRF2 exacerbates [237,257] and overexpression of HO-1 prevents oxidative stress and apoptosis, others have suggested that deletion of NRF2 inhibited apoptosis and the progression of cardiac dysfunction in T1D [301]. Novel opportunities to ablate the formation of ROS and reduce apoptosis have been suggested by results of inhibition of several long-noncoding RNAs (lnRNA) [152]. Taken together, suppression of cardiomyocyte apoptosis by inhibiting upstream activation pathways induced by oxidative stress, may be beneficial in reducing the deleterious effects of diabetes on the heart.
8.8. Cardiac remodeling and dysfunction
Exacerbation of cardiac remodeling and dysfunction in diabetes may represent the clinically most relevant feature of DbCM, which can be considered as a common downstream consequence of numerous mechanisms that may contribute to DbCM, including oxidative stress. Cardiomyocyte loss precipitates replacement fibrosis by activated fibroblasts altering tissue composition and ventricular compliance leading to cardiac remodeling and impaired diastolic function [302]. Pre-diabetic rodents display early manifestation of a mild hypertrophy and diastolic dysfunction, without signs of fibrosis [63,64]. Importantly, mitochondrial oxidative stress is associated with the progression of diastolic dysfunction in a model HFD-induced T2D [303]. HG induces hemodynamic and electrical alterations characterized by QT interval prolongation and increased coronary perfusion pressure in the isolated working rat heart [179]. A causative role of ROS in the development of DbCM is evident by reversal of diabetes-induced electrical remodeling and prevention of diastolic stiffness by targeting ROS producing proteins or enzymes, such as calpains [107–109], NOX [121,122,124,125], MAOs [100,101], and XO [128,130,132]. Furthermore, enhancements of the antioxidant defense system appear to protect against diabetes-induced impairment in contractile function, as evidenced by overexpression of CAT [29,182], MT [50,113,210,264], mPHGPx [185], and genetic and pharmacological activation of SOD [31,179]. Reversal of impaired contractility and improved calcium handling as a result of these therapies, may be related to increased expression of contractile proteins [304], and reduced tyrosine nitration of metabolic proteins [213,214]. Additionally, loss of NRF2-induced expression of cytoprotective genes may contribute to myocardial structural damage and impaired response to isoproterenol stimulation of diabetic hearts [257]. Altogether, these findings, as well as improved cardiac morphology upon restoring antioxidant status by agents such as NAC [201] and quercetin [181], suggest that attenuating oxidative stress in diabetes may offer a strategic approach to limit cardiac functional abnormalities as a consequence of DbCM.
9. ROS scavengers
Preclinical studies showing beneficial effects of decreasing oxidative stress in the diabetic heart fostered studies in animals and humans to evaluate antioxidant treatment as a useful approach to attenuate diabetic heart disease (Table 4). These approaches include mitochondria-targeted therapies, given the crucial role of mitochondrial ROS in DbCM.
Table 4.
Effects of ROS scavengers and drugs recommended for diabetic subjects on myocardial ROS and oxidative stress.
| Target | Model | ROS/Oxidative Stress | Cardiac Structure/Function | Author |
|---|---|---|---|---|
| mito-TEMPO | STZ-induced T1D mice or db/db T2D mice | ↓ mitochondrial H2O2 production, intracellular ROS, protein carbonyl content, TAC ↓ gp91phox, p47phox mRNA |
↓ CM hypertrophy ↓ apoptosis ↓ systolic & diastolic dysfunction |
Ni [306] |
| Resveratrol | STZ-induced T1D mice STZ/nicotinamide-induced T1D rats Leprdb T2D mice fructose-induced T2D rat pre-diabetic ZDF rats | ↓ 8OHdG, MDA, 15-F2t-IsoP ↓ SIRT1, Rab7 protein, acetylated/total histone 3, acetylated/total FOXO1 ↓ TBARS, GSSG/GSH ↑ SOD ↓ nitrite/nitrate ↓ O2•− production, NT ↑ nitrite/nitrate levels; eNOS, iNOS protein ↓ NOX activity; gp91phox, gp91phox mRNA/protein ↓ ROS, AGE, protein carbonyl content, NO content ↓ NOX activity; NOX1, NOX4 mRNA ↑ SOD activity, GSH level ↑ SIRT-1 mRNA/protein/activity ↓ mitochondrial H2O2 emission ↔ GSH/GSSG; SOD2, CAT protein |
↓ cardiac dysfunction ↓ cardiac remodeling ↓ fibrosis, apoptosis ↓ apoptosis ↔ cardiac remodeling, systolic function ↓ diastolic dysfunction ↓ cardiac hypertrophy ↓ electrophysiological abnormalities ↑ NAD, NAD/NADH ratio ↓ apoptosis ↔ systolic, diastolic function ↓ cardiac fibrosis ↔ ETC subunits, ADP respiratory kinetics ↓ lipotoxicity |
Wang [318] Soufi [315] Zhang [314] Bagul [317] Beaudoin [316] |
| Piceatannol, a natural hydroxylated analog of Resveratrol | STZ-induced T1D rats | ↓ MDA, ↑ SOD activity ↑ NRF2, HO-1 mRNA |
↓ systolic dysfunction ↓ fibrosis, apoptosis |
Li [244] |
| Quercetin | high-cholesterol diet-induced T2D rats STZ-induced T1D rats | ↓ 8-isoprostane, TBARS ↑ GSH/GSSG; SOD, CAT, GSH-Px activities ↑ NRF2 nuclear translocation; HO-1 mRNA ↓ 4-HNE ↑ PRX3, TRX2 protein; TRXR2 activity; ↔ PRX5 protein; ↑ SOD activity; ↔ CAT, GSH-Px activities; ↓ UCP3 protein ↑ NRF2, NRF1 mRNA |
↓ diastolic dysfunction ↓ cardiac hypertrophy ↔ cardiac remodeling, systolic function ↓ CM density ↓ cardiac cholesterol content ↑ATP levels ↓ CM hypertrophy ↓ myocardial fibrosis ↑ cardiac conductivity & contractility ↓ apoptosis |
Castillo [320] Arkat [181] |
| Taxifolin, a dihydroquercetin | STZ-induced T1D mice | ↓ MDA ↑ SOD, GSH-Px, CAT activities ↓ NOX activities |
↓ cardiac hypertrophy/remodeling ↓ fibrosis, apoptosis |
Sun [321] |
| sodium selenate or omega-3/vitamin E supplementation | STZ-induced T1D rats | ↔ free/total SH, nitrite | ↓ cardiac dysfunction | Aydemir-Koksoy [324] |
| alpha-lipoic acid | STZ-induced T1D rats | ↓ MDA; ↑ GSH/GSSG ↑ MnSOD activity |
↓ cardiac dysfunction ↓ fibrosis |
Li [325] |
| vitamin E | STZ-induced T1D rats | ↓ 8-iso PGF2α ↓ GSSG |
↓ cardiac dysfunction | Hamblin [326] |
| ascorbic acid, dl α-tocopherol acetate, β-carotene, N acetyl cysteine, selenium | STZ-induced T1D rats | ↓ MDA ↓ XO mRNA/protein; MAO-A mRNA ↓ CAT ↓ HO-1 mRNA |
Kumar [401] | |
| coenzyme Q10 | STZ-induced T1D mice | ↓ 3-NT, NOX-derived O2•− production ↓ NOX2, p47phox mRNA; p22phox protein |
↓ diastolic dysfunction ↓ CM hypertrophy ↓ fibrosis, apoptosis |
Huynh [30], De Blasio [28] |
| coenzyme Q10 | db/db T2D mice | ↓ MDA; O2•− | ↓ cardiac hypertrophy ↓ diastolic dysfunction ↓ fibrosis, apoptosis |
Huynh [27] |
| SGLT2 inhibitor, Empagliflozin | STZ-induced T1D mice HFD-fed T2D mice ZDF obese/T2D rats db/db T2D mice KK-Ay T2D mice | ↓ cytosolic & mitochondrial ROS ↓ O2•− production ↑ mtDNA content ↑ p-eNOS, NRF2, HO-1, p-p65(Ser536) protein; ↑ NRF2, HO-1 mRNA ↓ H2O2, 3-NT, GSH, and lipid peroxide; ↑ NO bioavailability ↓ p-eNOSser1177, eNOS monomer ↓ ROS ↑ SOD2; ↓ NOX4; ↔ NOX2 ↓ lipid hydroperoxide, MDA ↑ GSH-Px, SOD; ↓ NOX4 protein ↑ NRF2, HO-1 protein; nuclear NRF2 |
↓ LV hypertrophy ↓ diastolic dysfunction ↓ perivascular/interstitial fibrosis ↓ mitochondrial injury ↑ passive force ↓ LV hypertrophy ↓ systolic &diastolic dysfunction ↓ fibrosis, apoptosis |
Lee [351] Sun [256] Kolijn [352] Xue [353] Li [355] |
| GLP-1RA, Exenatide | HFD-fed T2D mice & STZ-induced T1D mice [362] | ↑ MnSOD, CAT protein | ↓ systolic &diastolic dysfunction | Ding [362] |
| GLP-1RA, OHP2 & Semaglutide [354], or Exendin-4 [402] | HFD/STZ-induced T2D rats [354,402], | ↓ ROS production, MDA ↓ p22phox, p40phox ↑ GSH-Px1, HO-1; ↔ SOD1 mRNA |
↓ LV hypertrophy ↓ systolic & diastolic dysfunction ↓ fibrosis, apoptosis ↓ mitochondrial dysfunction, fission ↓ cardiac lipotoxicity ↑ cardiac ATP |
Qian [354], Wu [402] |
| GLP-1RA, Exendin-4 | KK-Ay or HFD-induced T2D mice [403] | ↓ ROS, ↓ NOX4; ↔ NOX2 mRNA ↑ SOD, GSH-Px |
↓ CM hypertrophy ↓ systolic & diastolic dysfunction ↓ fibrosis ↓ mitochondrial remodeling ↓ cardiac lipotoxicity |
Monji [403] |
| DPP-4 inhibitor, Sitagliptin [364,365,369], Vildagliptin [365,366] or Linagliptin [367,371] | STZ-induced T1D rats HFD-fed T2D mice/rats ZDF obese/T2D rats | ↓ MDA, NO, GSH content ↓ SOD, CAT activities ↓ TBARS, protein carbonylation, 3-NT, MDA, 4-HNE, ROS production ↑ CAT ↓ 3-NT; ↔ ROS, RNS ↓ iNOS |
↓ cardiac hypertrophy ↓ CM degeneration ↓ fibrosis ↓ cardiac hypertrophy ↓ diastolic dysfunction ↑ HR variability ↓ cardiac fibrosis ↓ mitochondrial depolarization, swelling ↓ systolic & diastolic dysfunction ↓ ultrastructural abnormalities ↑ autophagy |
Al-Rasheed [364] Apaijai [365], Nath [366], Aroor [367] Zhou [369], Aroor [371] |
| DPP-4 inhibitor, Alogliptin [368] or MK0626 [372] | ALX-induced T1D rabbits HFD-fed T2D mice | ↓ MDA, 8-OHdG, mitochondrial ROS ↔ SOD activity ↔ 3-NT |
↓ cardiac dysfunction ↓ fibrosis ↑ mitochondrial respiration ↑ mitochondrial biogenesis ↓ diastolic dysfunction ↓ fibrosis |
Zhang [368] Bostick [372] |
| ARBSs, Candesartan [37], Valsartan [375], Azilsartan [376], and Olmesartan [238]; ACI-Is, Benazepril [37] and Ramipril [30]; renin inhibitor, Aliskiren [37,375]; | STZ-induced T1D mice/rats db/db T2D mice obese OLETF T2D rats | ↓ ROS; NT; ↑ GSH ↑ mitochondrial SOD activity; ↔ CAT activity; ↑ cytosolic NRF2, acetylated NRF2; ↓ Keap1 mRNA ↓ 8-OHdG, NT ↓ NT; ↑ GSH ↑ mitochondrial SOD; ↔ CAT activities ↑ cytosolic/acetylated NRF2; ↓ Keap1 mRNA |
↓ LV hypertrophy ↓ systolic & diastolic dysfunction ↓ fibrosis ↓ apoptosis ↓ systolic & diastolic dysfunction ↓ cardiac hypertrophy ↓ fibrosis ↓ aconitase activity; ↔ complex I/II activities ↑ NAD+/NADH ratio |
Singh [37], Thomas [375], Huynh [30] Sukumaran [376] Thorwald [238] |
| ARNi, LCZ696 | STZ-induced T1D mice | ↑ GSH, GSH/GSSG ratio | ↓ cardiac hypertrophy ↓ cardiac dysfunction ↓ apoptosis & fibrosis |
Ge [377] |
9.1. Mito-TEMPO & MitoQ
Based on inconclusive evidence that antioxidants are sufficiently delivered to the mitochondria to therapeutically target ROS production, mitochondrial targeted antioxidants have been developed. In these compounds, the active antioxidant substance (e.g., TEMPO) is conjugated to the lipophilic triphenylphosphonium cation (TPP+), which in some cases facilitates an up to 1000-fold accumulation within mitochondria, driven by the negative intramitochondrial charge. As expected, mito-TEMPO passes through lipid bilayers, to scavenge ROS, and to eliminate the increase in ROS production induced by palmitate in cardiomyocytes [123,305] Interestingly, mito-TEMPO treatment improved diastolic function in db/db T2DM mice, and both diastolic and systolic function in STZ-induced T1DM mice [306]. Importantly, this was associated with inhibition of mitochondrial ROS production and oxidative stress, as well as reduced apoptosis. Another TPP+ conjugated compound is MitoQ (TPP+ conjugated to ubiquinone), which is reduced to ubiquinol within mitochondria and then acts as an antioxidant at the ETC [307]. Importantly, MitoQ reduces oxidative stress, enhances OXPHOS and prolongs cell survival under hyperglycemic and hyper-lipidemic conditions [308,309]. While protective effects of MitoQ have been established in the diabetic kidney [310] and in the heart following ischemia-reperfusion injury [311], experimental data on treatment effects of this potent antioxidant in models of DbCM are currently lacking. Given the safety of MitoQ in humans and its efficacy in attenuating ROS-induced vascular endothelial cell dysfunction in human subjects, studies of this promising compound in the setting of DbCM would be of utmost interest [312].
9.2. Flavonoids
Flavonoids are naturally occurring compounds acting upstream of mitochondria that have shown promise in models of diabetes, potentially as a consequence of their antioxidant properties. Resveratrol, a naturally occurring polyphenol found in grapes, has been shown to elicit protective effects in diabetes, particularly in the heart (extensively reviewed elsewhere [313]). Improved cardiac function following resveratrol treatment of T2D db/db mice was associated with reduced 3-NT and improved nitric oxide availability, likely upon reduced iNOS expression, enhanced eNOS-induced nitric oxide production and inhibition of ROS production by NOX [314]. Furthermore, resveratrol is believed to enhance endogenous antioxidant defense [296], thereby protecting cardiomyocytes from hyperglycemia-induced apoptosis [315]. Reducing mitochondrial ROS generation by resveratrol, attenuated cardiac fibrosis, accumulation of reactive lipids, and respiratory kinetics for palmitoyl-CoA in T2D ZDF rats [316]. Attenuation of cardiac oxidative stress by resveratrol has also been attributed to activation of SIRT1 and resulting deacetylation of NFkB-p65 and histone H3 [317]. These beneficial cardiac effects of SIRT1 activation by resveratrol in DbCM may depend on autophagic flux [318]. Furthermore, the natural hydroxylated analog of resveratrol, piceatannol, suppressed the pathological changes induced by oxidative stress in T1D hearts, in an NRF2 dependent manner [244]. The flavonoid quercetin, which is highly abundant in the human diet, accumulates in mitochondria [319] and is thought to have cardiac benefits via reducing oxidative stress. Quercetin reduced oxidative stress in hypercholesterolemic/hyperglycemic rats, thereby restoring cardiac bioenergetics [320]. The beneficial effects of quercetin may be mediated by induction of PRX3 expression [181]. In addition, the active quercetin glucoside, spiraeoside, prevented HG-induced oxidative stress and apoptosis in human cardiomyocytes upon activation of NRF2/HO-1 [232]. Lastly, taxifolin, also known as dihydroquercetin, reduced hyperglycemia-induced ROS and apoptosis both in vivo and in vitro, possibly by activation of JAK/STAT3 signaling [321]. Thus, modulation of diet composition towards increased flavonoid uptake, or adding flavonoids as a nutritional supplement to the diet, may represent a promising approach to attenuate diabetic heart disease.
9.3. Natural antioxidants
There is growing interest in the use of non-enzymatic antioxidants as a therapeutic approach for cardiovascular diseases. Vitamin E (α-tocopherol) acts as an antioxidant by interrupting formation of lipid radicals, thereby producing a much more stable vitamin E radical [322]. Vitamin C (ascorbic acid) is then responsible for reducing vitamin E back into a functional free radical scavenger [323]. Studies with these natural antioxidants and others have supported therapeutic potential in the context hyperglycemia-induced cardiac dysfunction [324,325]. Particularly, vitamin E supplementation was found to attenuate reduced LV contractility and slowing of LV relaxation upon reducing myocardial 8-iso PGF2α and GSSG accumulation in STZ-induced T1DM rats [326]. Treatment of cardiomyocytes with vitamin C prevented STZ-induced oxidative stress and contractile dysfunction [327]. Nonetheless, while these results appear to be promising, the effects of these agents in human studies suggest that vitamin/antioxidant supplementation is insufficient to provide a long-standing clinical benefit on CV outcomes [206,328, 329]. While short-term intra-arterial infusions of vitamin C have been shown to improve endothelial function in both T1D and T2D [330,331], administration of a combination of vitamin C and vitamin E for 6 months improved endothelial function only in patients with T1D but not T2D [332]. In another study, high doses of oral vitamin C did not improve endothelial function in patients with type 2 diabetes [333]. Vasodilatory function and LV function did also not improve in response to 12 months of high dose vitamin E supplementation both in T1D and T2D subjects [334]. In the HOPE trial, which investigated a two-by-two factorial design to receive either 400 IU of vitamin E daily or matching placebo and either ramipril or matching placebo, no beneficial effects were observed on CV events, CV death, or HF [328]. Extended follow-up of this study cohort even demonstrated an increased risk for heart failure or heart failure hospitalization [335]. Finally, lack of an effect of vitamin E supplementation in prevention of major CV events in diabetic subjects was also confirmed by a meta-analysis of 50 studies [329]. Interestingly, vitamin E supplementation was actually found to reduce, rather than increase, total antioxidant capacity [336]. Thus, long term antioxidant treatment with vitamin E and/or C does not seem to lead to any clinical benefit with respect to vascular outcomes or heart failure development, although the effects on the molecular biology of DbCM has not been evaluated. A differential effect between T1D and T2D cannot be ruled out. Explanation for a lack of efficiency may include that the administered dose and duration of supplementation with vitamins may be insufficient to reverse the pathological changes associated with T2D [337–340]. While it has been shown that vitamin C supplementation indeed may not reach similar serum levels in diabetic patients as it does in healthy subjects [333,341], the extended follow-up period in the HOPE-TOO trial with several years of observation argue against a treatment duration issue [335]. Another explanation for lack of efficiency may be that antioxidants, such as vitamin E, may not reach the target cell in humans as they do in animal models despite achieving appropriate serum concentrations. Novel studies have demonstrated the use of nanoparticle-based therapies that enable improved tissue delivery of vitamin E with reduced side effects in the treatment of animals in models of endotoxemia [342]. In addition, improved treatment efficacy due to nanoparticle-mediated drug delivery has been shown for a number of substances, such as cisplatin improving colon cancer chemotherapy or adenosine providing neuroprotection following stroke [343,344]. Although not yet explored in the setting of DbCM, these targeted treatments in general may offer a unique approach to combat the pitfalls experienced using antioxidant therapies in clinical trials to date. Finally, future trials might need to focus on those individuals with clear evidence of oxidative stress. Viability of this approach will require identification of robust biomarkers.
9.4. Coenzyme Q10 & Q9
Coenzyme Q10 (CoQ10) is a cofactor of mitochondrial respiration that acts as an endogenously synthesized lipid soluble antioxidant [345]. Supplementation with CoQ10 attenuates oxidative stress and cardiac dysfunction in rodent models of both T1D [30] and T2D [27] by preventing induction of ROS. Furthermore, chronic CoQ10 supplementation prevented diabetes-induced cardiomyopathy despite impaired cardiac PI3K signalling [28]. Reduced levels of coenzyme Q9 in the T1D heart is postulated to increase its susceptibility to mitochondrial oxidative stress [68], although its therapeutic potential in the treatment of DbCM has not yet been explored. Nevertheless, this compound holds promise of showing protective effects in human diabetic hearts, given that a number of small sized clinical trials reported potential prognostic advantages in subjects with HFrEF treated with CoQ10 supplementation (reviewed in Ref. [346]).
10. ROS scavenging properties of approved drugs used in diabetic individuals
10.1. Sodium/glucose co-transporter 2 inhibitors (SGLT2) inhibitors
Several classes of drugs in routine use in clinical practice might mediate some of their beneficial cardiovascular effects because of antioxidant properties (Table 4). Sodium/glucose co-transporter 2 inhibitors (SGLT2i) block 90% of glucose reabsorption in the kidney, thereby lowering serum glucose levels. In studies investigating the cardiovascular safety of the antidiabetic drugs, SGLT2i have shown considerable reduction in cardiovascular risk in T2D patients with high cardiovascular risk or known cardiovascular disease [347–349]. Although a number of mechanisms have been proposed to underlie these beneficial effects of SGLT2i, specific mechanisms of action remain elusive. Glucose-lowering effects of SGLT2i may be of less importance for these beneficial effects, given that the heart failure risk reduction was similar in heart failure patients, in the presence or absence of T2D [350]. Of note, several studies have highlighted the antioxidant properties of the SGLT2i, empagliflozin, in the heart of T1D [351] and T2D[256,352,353, 354] mice. Interestingly, cytosolic and mitochondrial hydrogen peroxide correlated with stiffness of cardiomyocytes from ZDF rats, and this was reversed following treatment with empagliflozin [352]. Importantly, these improvements are likely a result of improved NO bioavailability and attenuation of eNOS activation, thus resulting in improved endothelial function [352]. This is further supported by reduced cytosolic ROS production in ventricular cardiomyocytes associated with enhanced expression of Ca2+-handling proteins [351]. These beneficial effects of empagliflozin on myocardial structure and function in the diabetic heart have been linked to reduced myocardial oxidative stress mediated by enhanced nuclear translocation of NRF2 and subsequent expression of antioxidant proteins [355]. The attenuation of myocardial hypertrophy and fibrosis, and improvement of cardiac function observed in T2D hearts may be dependent on sGC-cGMP-PKG signaling [353] or Sestrin2-mediated AMPK-mTOR signaling [256]. While empagliflozin has been shown to reduce expression of key enzymes implicated in oxidative stress [256,353], this effect is most likely secondary to reductions in hyperglycemia or other metabolic improvements, rather than upon direct action on ROS-producing or ROS-scavenging proteins. Lastly, empagliflozin treatment reduced cardiomyocytes stiffness and attenuated oxidative stress parameters in human LV biopsies, although these were isolated from non-diabetic patients with diastolic dysfunction [352]. Of note, the ongoing EMPA-HEART trial will further determine the antioxidant potential of empagliflozin in diabetic patients by measuring plasma biomarkers of oxidative stress, such as myeloperoxidase (MPO) [356].
10.2. Glucagon-like peptide-1 (GLP-1) receptor agonists (GLP-1RAs)
Another class of antidiabetic drugs that showed beneficial cardiovascular effects in T2D is glucagon-like peptide-1 (GLP-1) receptor agonists (GLP-1RAs). Although some large scale clinical trials for GLP-1RAs [357–359] failed to show any cardiovascular benefit in patients with T2D and existing CVD, the LEADER [360] and SUSTAIN-6 [361] trials demonstrated a significantly lower rate of first occurrence of CV death and nonfatal MI and stroke upon treatment with liraglutide and semaglutide, respectively. Pre-clinical studies suggested that cardiac protective effects of GLP-1RAs may be driven by antioxidant effects. The GLP-1RA, exenatide, attenuated ROS production and enhanced antioxidant defense to protect against cardiac dysfunction in both T1D and T2D models [362]. Interestingly, the beneficial effects of exenatide against apoptosis in isolated cardiomyocytes was partially abolished in the presence of the CAT-inhibitor, 3-AT [362], suggesting that the antioxidant effects of this GLP-1RA may be dependent on CAT-mediated ROS-scavenging. Additionally, the GLP-1RA, liraglutide, was also recently shown to ameliorate interleukin (IL)-1β-induced cellular ROS production and NOX4 expression in cultured cardiomyocytes [363]. Furthermore, the novel GLP-1RA, oral hypoglycemic peptide 2 (OHP2), as well as both semaglutide reduced myocardial lipid accumulation, oxidative stress and mitochondrial dysfunction in diabetic hearts [354]. However, results of CV outcome trials of GLP1RAs have not revealed a signal of reduced HF hospitalization [357,358,360].
10.3. Dipeptidyl peptidase-4 (DPP-4) inhibitors
DPP-4 inhibitors which impair the degradation of GLP-1, reducing glucagon and blood glucose levels in diabetes, may also have antioxidant effects in the heart. For example, sitagliptin reduced lipid peroxidation and enhanced antioxidant defense in hearts of T1D rats and this was associated with a reduction in indices of cardiovascular risk [364]. Vildagliptin and sitagliptin both reduced lipid peroxidation, protein carbonylation and ROS production in rodent models of HFD-induced T2D which is associated with improved HR variability and reduced mitochondrial depolarization and swelling [365,366]. Furthermore, the cardiac improvements observed in female T2D mice upon treatment with DPP-4 inhibitor, linagliptin, are similarly associated with reduced markers of cardiac oxidative stress [367]. The beneficial anti-oxidative effects of DPP-4 inhibitors may be due to improved mitochondrial function and increased mitochondrial biogenesis [368] and related to improved autophagy [369]. Interestingly, studies in the kidney have demonstrated that the antioxidant effect of DPP-4 inhibitors may be dependent on an intact antioxidant system [370], although whether this is the case in the diabetic heart has yet to be confirmed. Interestingly, reduced oxidative stress parameters in the diabetic heart are not observed with DPP-4 inhibitors in all models of diabetes [371,372], suggesting the antioxidant potential of DPP-4 inhibition requires addition investigation. Unfortunately, while these preclinical data appear promising, none of the recently published large CV outcome trials demonstrated any significant cardiovascular benefit in patients with T2D.
10.4. Renin-angiotensin-aldosterone system (RAAS)
Angiotensin-converting enzyme inhibitors (ACEi) and angiotensin receptor blockers (ARB) are commonly used for the treatment of hypertension and to prevent or attenuate diabetic nephropathy in diabetic individuals. Activation of the RAAS has also been implicated in the pathogenesis of DbCM by promoting loss of CM and myocardial fibrosis [373,374]. ACEi and ARB decrease oxidative stress in diabetic hearts, resulting in reduced apoptosis [37]. Benazeprilat and valsartan, partially protected against STZ-induced cardiac dysfunction by reducing myocardial oxidative stress [375]. Azilsartan significantly reduced oxidative DNA damage and cardiac dysfunction in db/db mice [376]. Reduced oxidative stress by olmesartan may be mediated by enhanced expression and/or activation of cardiac NRF2 in T2D OLETF rats [238]. The ACE inhibitor ramipril ameliorates diabetes-related redox imbalance, as evidenced by reduced superoxide, 3-NT content, and NOX expression [30]. Furthermore, the first angiotensin receptor-neprilysin inhibitor (ARNi) to be used clinically, LCZ696 (valsartan/sacubitril), improved ventricular remodeling and cardiac function, ameliorated oxidative stress and inhibited apoptosis in STZ-induced diabetic hearts [377]. Together, these findings further implicate the RAAS in oxidative stress in diabetes, and suggest that ACEi and ARBs used to treat diabetic subjects may at least partially exert beneficial cardiovascular effects by attenuating oxidative stress in the heart.
10.5. Others
In addition to the aforementioned clinically used agents that may reduce cardiac oxidative stress, additional anti-diabetic or cholesterol lowering drugs have shown antioxidant properties in human trials. For example, the thiazolidinedione, rosiglitazone, reduced plasma peroxide levels in non-diabetic obese subjects [378]. Similarly, metformin restored markers of antioxidant status in patients with T2DM [379]. Lastly, reductions in cardiovascular events by statins have also been attributed to anti-inflammatory and antioxidant properties [380]. Nevertheless, the potential of these agents to prevent or reverse deleterious effects of diabetes-induced oxidative stress in the heart requires additional exploration.
11. Reductive stress in diabetic cardiomyopathy
While oxidative stress in the diabetic heart has been extensively documented, the role of reductive stress (RS), due to excess NADH, glutathione, and NADPH [381], has garnered less attention. Considerable evidence suggests that hyperglycaemia and other metabolic disturbances associated with diabetes result in the generation of ROS, ultimately leading to increased oxidative stress in a variety of tissues [382]. Glucose and fatty acid metabolism involves redox reactions that include electron extraction, storage, and transportation. Electrons are mainly stored in NADH, therefore, prolonged hyperglycemia for example could increase the NADH levels, which could tip the redox balance towards RS. The reductive power through nicotinamide adenine dinucleotides is crucial for several cellular processes. In the diabetic setting, alterations in the NAD+/NADH ratio due to increased NADH could generate reactive oxygen species, which induce the formation of intracellular advanced glycation end products [383,384]. Of interest, changes in α-hydroxybutyrate levels in liver that were associated with insulin resistance and mitochondrial disorders induced hepatic reductive stress (RS), which contributes to metabolic traits and cardiometabolic disease [385]. Thus, aberrant NADH redox balance may play an important role in the pathogenesis of diabetes and its complications.
An imbalance of NAD+/NADH often occurs as a result of impaired complex I function in concert with increased glucose flux via the sorbitol pathway [386] leading to ROS overproduction upon overwhelming the ETC and antioxidant defense capacity [387]. Notably, metabolites of the polyol pathway were found to be highly increased in the ventricles of diabetic rats [388]. In fact, this metabolic imbalance has been implicated in hemodynamic impairments observed in diabetic tissues [389, 390]. Particularly, aldose reductase (which utilizes NADPH in the conversion of glucose to sorbitol) is activated by glucose and is therefore thought to be involved in HG-driven complications of diabetes [391]. Importantly, inhibition of the key enzymes of the polyol pathway (aldose reductase or sorbitol dehydrogenase) protects against myocardial ischemia-reperfusion injury and prevents the decline in cardiac function in T2D rats [392]. Given that overproduction of ROS may develop as a result of NADH-imposed reductive stress, strategies to prevent reductive stress (RS) may provide novel therapeutic approaches for the treatment of DbCM.
Although the generation of ROS is a metabolic process, the impact of ROS and the extent of the resultant oxidative stress are often controlled by antioxidants that are transcriptionally regulated through Nrf2. Nrf2 depletion and downregulation of antioxidant genes results in dysfunctional myogenic tone in diabetes, which was reversed by sulforaphane, an Nrf2 activator [393]. Genetic overexpression of Nrf2 prevents the onset of T2D in mice and pharmacological activation of Nrf2 reduces oxidative stress, and a myriad of diabetic complications, including cardiovascular complications, nephropathy, and neuropathy [236,394, 395]. A recent study has shown that ChREBPα increases anabolism and mitochondrial mass through Nrf2 signaling [396]. Of note, activation of Nrf2 is essential for glucose-mediated proliferation in rodent and human β-cells [396].
Overall, these investigations indicate the positive impact of Nrf2 in mitigating diabetes and its associated complications. However, after the emergence of the “reductive stress” concept, a dual role of Nrf2, particularly if overactivated is being reported in diverse pathologies including diabetes [246,397,398]. Thus it is evident that both oxidative and reductive stress could play a significant role in the pathogenesis and progression of T2DM. Given that oxidative stress originates from NADH-imposed RS [383,399], attenuating hyperglycemia-triggered RS may provide a potential therapeutic opportunity for preventing the development of diabetes and diabetic complications.
12. Conclusions
In the current review, we present evidence supporting the role of oxidative stress as an important contributor to the development of DbCM. Defects in both ROS-producing enzymes, as well as ROS detoxification enzymes are considered to render the heart susceptible to oxidative damage leading to cardiac dysfunction. While several novel therapeutic targets have been explored in animal models of both T1D and T2D, confirming their potential in large animal models of diabetes in future studies will provide additional insight and increase the possibility of success in human trials. To date, translating antioxidant therapies into human treatment remains a major challenge, related to many factors such as dosing issues, questionable efficacy in human cardiac cells, or failure to target relevant molecular, functional and structural alterations in the diabetic heart. Thus, in addition to a more detailed understanding of the molecular LV changes and the clinical outcomes of subjects with diabetes, significantly more clinical trials need to be performed to close the gaps between preclinical findings and clinical outcomes, particular in those with preclinical signs of DbCM. In the interim, patients with diabetes may benefit from longstanding therapies such as ACEi or ARBs, and from the recent introduction of antidiabetic drugs such as SGLT2i or GLP-1RA that may partially protect the heart by exerting antioxidant properties.
Acknowledgement
H.B. received research funding from the Austrian Science Fund (FWF; P-33874-B). E.D.A. has been supported over the years by the following collaborative awards: U01 HL70525, RO1 HL73167, U01 HL087947, R24 DK092784, U54 HL112311, and R61HL141783 (NIH); and 16SFRN31810000, and 20SFRN35120123 (American Heart Association). NSR was supported by funding from NHLBI (1HL118067), NIA (AG042860), the American Heart Association (BGIA 0865015F), University of Utah Center for Aging Pilot grant (2009), the start-up funds from the Division of Cardiovascular Medicine/Department of Medicine, University of Utah and by Department of Pathology, the University of Alabama at Birmingham, AL and UAB-AMC21 grant by the University of Alabama at Birmingham, AL.
Abbreviations
- ATP
adenosine triphosphate
- AMVM
adult mouse ventricular myocytes
- ALX
alloxan
- AS
Angeli’s salt
- ACE
angiotensin-converting enzyme
- ARB
angiotensin receptor blockers
- ARNi
angiotensin receptor-neprilysin inhibitor
- ARE
antioxidant response elements
- ASOs
antisense oligonucleotides
- BCL-2
B-cell lymphoma 2
- BAX
BCL-2-associated X
- Ca2+
calcium
- CAPN4
calpain small subunit 1
- CM
cardiomyocyte
- CV
cardiovascular
- CASP
caspase
- CAT
catalase
- CoQ10
coenzyme Q10
- CAD
coronary artery disease
- DbCM
diabetic cardiomyopathy
- DM
diabetes mellitus
- DPP-4
dipeptidyl peptidase-4
- HF
heart failure
- ETC
electron transport chain
- eNOS
endothelial NOS
- GLP-1RAs
GLP-1 receptor agonists
- GLP-1
glucagon-like peptide-1
- GRX
glutaredoxins
- GSH
glutathione
- GSSG
glutathione disulfide
- GSH-Px
glutathione peroxidase
- GSR
glutathione reductase
- GK
Goto-Kakizaki
- HO
heme oxygenase
- HG
high glucose
- HPLC
high-performance liquid chromatography
- HFrEF
HF with reduced ejection fraction
- HFpEF
HF with preserved ejection fraction
- H2O2
hydrogen peroxide
- OH•
hydroxyl
- iNOS
inducible NOS
- IMM
inner mitochondrial membrane
- IFM
interfibrillar mitochondria
- ISDBB
insulin-dependent spontaneously diabetic BB Wistar rats
- IL
interleukin
- Keap1
Kelch-like ECH-associated protein 1
- KO
knockout
- LV
left ventricular
- lnRNA
long-noncoding RNAs
- LVDV
LV diastolic volume
- MDA
malondialdehyde
- MT
metallothionein
- MPTP
mitochondrial permeability transition pore
- mPHGPx
mitochondria phospholipid hydroperoxide glutathione peroxidase 4
- mtDNA
mitochondrial DNA
- MAO
monoamine oxidases
- MRP1
multidrug resistance protein 1
- MPO
myeloperoxidase
- NADPH
nicotinamide adenine dinucleotide phosphate
- NOX
NADPH oxidases
- NQO
NADPH quinone dehydrogenase
- NRCM
neonatal rat cardiomyocyte
- nNOS
neuronal NOS
- NO
nitric oxide
- NOx
NO and nitrogen dioxide
- NOS
NO oxidases
- NRF2
nuclear factor erythroid 2-related factor 2
- NAC
N-acetylcysteine
- OHP2
oral hypoglycemic peptide 2
- OLETF
Otsuka Long Evans Tokushima Fatty
- OXPHOS
oxidative phosphorylation
- O-GlcNAc
O-GlcNAcylation
- OGA
O-GlcNAcase
- OGT
O-GlcNAc-transferase
- PS
peak shortening
- PRX
peroxiredoxins
- ONOO•−
peroxynitrite
- SH
protein sulfhydryl
- mPHGPx
phospholipid hydroperoxide glutathione peroxidase 4
- RAAS
renin-angiotensin-aldosterone system
- ROS
reactive oxygen species
- SGLT2i
sodium/glucose co-transporter 2 inhibitors
- STZ
streptozotocin
- SSM
subsarcolemmal mitochondria
superoxide
- SOD
superoxide dismutase
- MS/MS
tandem mass spectrometry
- dUTP
terminal deoxynucleotidyl transferase
- TUNEL
nick end labeling
- TBARS
TBA reacting substances
- BH4
tetrahydrobiopterin
- TBA
thiobarbituric acid
- TG
transgenic
- TPP+
triphenylphosphonium cation
- T1D
type 1 diabetes
- T2D
type 2 diabetes
- UPR
unfolded protein response
- XO
xanthine oxidase
- CDNB
1-chloro-2,4-dinitrobenzene
- 3-NT
3-nitrotyrosine
- 4-HNE
4-hydroxynonenal
- OGG-1
8-oxoguanine glycosylase
- 15-F2t-IsoP
15-F2t-isoprostane
- γ-GCS
γ-glutamylcysteine synthetase
References
- [1].Saeedi P, et al. , Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the international diabetes federation diabetes atlas, 9th edition, Diabetes Res. Clin. Pract (2019), 10.1016/j.diabres.2019.107843. [DOI] [PubMed] [Google Scholar]
- [2].Kristensen SL, et al. , Clinical and echocardiographic characteristics and cardiovascular outcomes according to diabetes status in patients with heart failure and preserved ejection fraction: a report from the I-preserve trial (irbesartan in heart failure with preserved ejection, Circulation (2017), 10.1161/CIRCULATIONAHA.116.024593. [DOI] [PubMed] [Google Scholar]
- [3].Bertoni AG, et al. , Heart failure prevalence, incidence, and mortality in the elderly with diabetes, Diabetes Care (2004), 10.2337/diacare.27.3.699. [DOI] [PubMed] [Google Scholar]
- [4].Kannel WB, Hjortland M, Castelli WP, Role of diabetes in congestive heart failure: the Framingham study, Am. J. Cardiol (1974), 10.1016/0002-9149(74)90089-7. [DOI] [PubMed] [Google Scholar]
- [5].Gustafsson I, et al. , Influence of diabetes and diabetes-gender interaction on the risk of death in patients hospitalized with congestive heart failure, J. Am. Coll. Cardiol (2004), 10.1016/j.jacc.2003.11.024. [DOI] [PubMed] [Google Scholar]
- [6].Lombardi C, Spigoni V, Gorga E, Dei Cas A, Novel insight into the dangerous connection between diabetes and heart failure, Herz (2016), 10.1007/s00059-016-4415-7. [DOI] [PubMed] [Google Scholar]
- [7].Dei Cas A, Fonarow GC, Gheorghiade M, Butler J, Concomitant diabetes mellitus and heart failure, Curr. Probl. Cardiol (2015), 10.1016/j.cpcardiol.2014.09.002. [DOI] [PubMed] [Google Scholar]
- [8].Maack C, et al. , Heart failure and diabetes: metabolic alterations and therapeutic interventions: a state-of-The-Art review from the translational research committee of the heart failure association-European society of cardiology, Eur. Heart J (2018), 10.1093/eurheartj/ehy596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rubler S, et al. , New type of cardiomyopathy associated with diabetic glomerulosclerosis, Am. J. Cardiol (1972), 10.1016/0002-9149(72)90595-4. [DOI] [PubMed] [Google Scholar]
- [10].Seferovíc PM, Paulus WJ, Clinical diabetic cardiomyopathy: a two-faced disease with restrictive and dilated phenotypes, Eur. Heart J (2015), 10.1093/eurheartj/ehv134. [DOI] [PubMed] [Google Scholar]
- [11].Ritchie RH, Dale Abel E, Basic mechanisms of diabetic heart disease, Circ. Res (2020), 10.1161/CIRCRESAHA.120.315913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Levelt E, et al. , Relationship between left ventricular structural and metabolic remodeling in type 2 diabetes, Diabetes (2016), 10.2337/db15-0627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Devereux RB, et al. , Impact of diabetes on cardiac structure and function: the Strong Heart Study, Circulation (2000), 10.1161/01.CIR.101.19.2271. [DOI] [PubMed] [Google Scholar]
- [14].Boyer JK, Thanigaraj S, Schechtman KB, Pérez JE, Prevalence of ventricular diastolic dysfunction in asymptomatic, normotensive patients with diabetes mellitus, Am. J. Cardiol (2004), 10.1016/j.amjcard.2003.12.026. [DOI] [PubMed] [Google Scholar]
- [15].Lourenço AP, et al. , An integrative translational approach to study heart failure with preserved ejection fraction: a position paper from the Working Group on Myocardial Function of the European Society of Cardiology, Eur. J. Heart Fail (2018), 10.1002/ejhf.1059. [DOI] [PubMed] [Google Scholar]
- [16].Barter PJ, et al. , Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis, Arterioscler. Thromb. Vasc. Biol (2003), 10.1161/01.ATV.0000054658.91146.64. [DOI] [PubMed] [Google Scholar]
- [17].Nielsen LB, Bartels ED, Bollano E, Overexpression of apolipoprotein B in the heart impedes cardiac triglyceride accumulation and development of cardiac dysfunction in diabetic mice, J. Biol. Chem (2002), 10.1074/jbc.M203458200. [DOI] [PubMed] [Google Scholar]
- [18].Kajstura J, et al. , IGF-1 0verexpression inhibits the development of diabetic cardiomyopathy and angiotensin II-mediated oxidative stress, Diabetes (2001), 10.2337/diabetes.50.6.1414. [DOI] [PubMed] [Google Scholar]
- [19].van Linthout S, et al. , Reduced MMP-2 activity contributes to cardiac fibrosis in experimental diabetic cardiomyopathy, Basic Res. Cardiol (2008), 10.1007/s00395-008-0715-2. [DOI] [PubMed] [Google Scholar]
- [20].Qin F, et al. , The polyphenols resveratrol and S17834 prevent the structural and functional sequelae of diet-induced metabolic heart disease in mice, Circulation (2012), 10.1161/CIRCULATIONAHA.111.067801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Shao Q, et al. , Empagliflozin, a sodium glucose co-transporter-2 inhibitor, alleviates atrial remodeling and improves mitochondrial function in high-fat diet/streptozotocin-induced diabetic rats, Cardiovasc. Diabetol (2019), 10.1186/s12933-019-0964-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Christoffersen C, et al. , Cardiac lipid accumulation associated with diastolic dysfunction in obese mice, Endocrinology (2003), 10.1210/en.2003-0242. [DOI] [PubMed] [Google Scholar]
- [23].Bugger H, Abel ED, Rodent models of diabetic cardiomyopathy, DMM Disease Models and Mechanisms (2009), 10.1242/dmm.001941. [DOI] [PubMed] [Google Scholar]
- [24].Dabkowski ER, et al. , Diabetic cardiomyopathy-associated dysfunction in spatially distinct mitochondrial subpopulations, Am. J. Physiol. Heart Circ. Physiol (2009), 10.1152/ajpheart.00467.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ceylan-Isik AF, Wu S, Li Q, Li SY, Ren J High-dose benfotiamine rescues cardiomyocyte contractile dysfunction in streptozotocin-induced diabetes mellitus, J. Appl. Physiol (2006), 10.1152/japplphysiol.00988.2005. [DOI] [PubMed] [Google Scholar]
- [26].De Blasio MJ, et al. , Defining the progression of diabetic cardiomyopathy in a mouse model of type 1 diabetes, Front. Physiol 11 (2020) 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Huynh K, et al. , Coenzyme Q 10 attenuates diastolic dysfunction, cardiomyocyte hypertrophy and cardiac fibrosis in the db/db mouse model of type 2 diabetes, Diabetologia (2012), 10.1007/s00125-012-2495-3. [DOI] [PubMed] [Google Scholar]
- [28].De Blasio MJ, et al. , Therapeutic targeting of oxidative stress with coenzyme Q10 counteracts exaggerated diabetic cardiomyopathy in a mouse model of diabetes with diminished PI3K(p110α) signaling, Free Radic. Biol. Med (2015), 10.1016/j.freeradbiomed.2015.04.028. [DOI] [PubMed] [Google Scholar]
- [29].Ye G, et al. , Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes, Diabetes (2004), 10.2337/diabetes.53.5.1336. [DOI] [PubMed] [Google Scholar]
- [30].Huynh K, et al. , Targeting the upregulation of reactive oxygen species subsequent to hyperglycemia prevents type 1 diabetic cardiomyopathy in mice, Free Radic. Biol. Med (2013), 10.1016/j.freeradbiomed.2013.02.021. [DOI] [PubMed] [Google Scholar]
- [31].Shen X, Zheng S, Metreveli NS, Epstein PN, Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy, Diabetes (2006), 10.2337/diabetes.55.03.06.db05-1039. [DOI] [PubMed] [Google Scholar]
- [32].Valavanidis A, Vlachogianni T, Fiotakis C, 8-Hydroxy-2′ -deoxyguanosine (8-OHdG): a critical biomarker of oxidative stress and carcinogenesis, J. Environ. Sci. Health Part C Environ. Carcinog. Ecotoxicol. Rev (2009), 10.1080/10590500902885684. [DOI] [PubMed] [Google Scholar]
- [33].DayanandC D, Vegi P, Kutty AVM, Protein carbonyl content as a stable oxidative stress marker in type ii diabetes, 2012.
- [34].Delanty N, et al. , 8-epi PGF(2α) generation during coronary reperfusion: a potential quantitative marker of oxidant stress in vivo, Circulation (1997), 10.1161/01.CIR.95.11.2492. [DOI] [PubMed] [Google Scholar]
- [35].Ghosh S, et al. , Increased efflux of glutathione conjugate in acutely diabetic cardiomyocytes, Can. J. Physiol. Pharmacol (2004), 10.1139/y04-060. [DOI] [PubMed] [Google Scholar]
- [36].Kanazawa A, et al. , Reduced activity of mtTFA decreases the transcription in mitochondria isolated from diabetic rat heart, Am. J. Physiol. Endocrinol. Metab (2002), 10.1152/ajpendo.00255.2001. [DOI] [PubMed] [Google Scholar]
- [37].Singh VP, Le B, Khode R, Baker KM, Kumar R, Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis, Diabetes (2008), 10.2337/db08-0805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tocchetti CG, et al. , Impaired mitochondrial energy supply coupled to increased H2O2 emission under energy/redox stress leads to myocardial dysfunction during Type I diabetes, Clin. Sci (2015), 10.1042/CS20150204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kakkar R, Kalra J, Mantha SV, Prasad K, Lipid peroxidation and activity of antioxidant enzymes in diabetic rats, Mol. Cell. Biochem (1995), 10.1007/BF01322333. [DOI] [PubMed] [Google Scholar]
- [40].Da Silva MF, et al. , Attenuation of Ca2+ homeostasis, oxidative stress, and mitochondrial dysfunctions in diabetic rat heart: insulin therapy or aerobic exercise? J. Appl. Physiol (2015) 10.1152/japplphysiol.00915.2014. [DOI] [PubMed] [Google Scholar]
- [41].Hamblin M, et al. , Alterations in the diabetic myocardial proteome coupled with increased myocardial oxidative stress underlies diabetic cardiomyopathy, J. Mol. Cell. Cardiol (2007), 10.1016/j.yjmcc.2006.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lashin OM, Szweda PA, Szweda LI, Romani AMP, Decreased complex II respiration and HNE-modified SDH subunit in diabetic heart, Free Radic. Biol. Med (2006), 10.1016/j.freeradbiomed.2005.10.040. [DOI] [PubMed] [Google Scholar]
- [43].Wohaieb SA, Godin DV, Alterations in free radical tissue-defense mechanisms in streptozocin-induced diabetes in rat. Effects of insulin treatment, Diabetes (1987), 10.2337/diabetes.36.9.1014. [DOI] [PubMed] [Google Scholar]
- [44].Herlein JA, Fink BD, O’Malley Y, Sivitz WI, Superoxide and respiratory coupling in mitochondria of insulin-deficient diabetic rats, Endocrinology (2009), 10.1210/en.2008-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Das J, Vasan V, Sil PC, Taurine exerts hypoglycemic effect in alloxan-induced diabetic rats, improves insulin-mediated glucose transport signaling pathway in heart and ameliorates cardiac oxidative stress and apoptosis, Toxicol. Appl. Pharmacol (2012), 10.1016/j.taap.2011.11.009. [DOI] [PubMed] [Google Scholar]
- [46].Genet S, Kale RK, Baquer NZ, Alterations in antioxidant enzymes and oxidative damage in experimental diabetic rat tissues: effect of vanadate and fenugreek (Trigonella foenum graecum), Mol. Cell. Biochem (2002), 10.1023/A:1016103131408. [DOI] [PubMed] [Google Scholar]
- [47].Turko IV, et al. , Protein tyrosine nitration in the mitochondria from diabetic mouse heart: implications to dysfunctional mitochondria in diabetes, J. Biol. Chem (2003), 10.1074/jbc.M303734200. [DOI] [PubMed] [Google Scholar]
- [48].Shen X, et al. , Cardiac mitochondrial damage and biogenesis in a chronic model of type 1 diabetes, Am. J. Physiol. Endocrinol. Metab (2004), 10.1152/ajpendo.00047.2004. [DOI] [PubMed] [Google Scholar]
- [49].Wohaieb SA, Godin DV, Alterations in tissue antioxidant systems in the spontaneously diabetic (BB Wistar) rat, Can. J. Physiol. Pharmacol 65 (1987) 2191–2195. [DOI] [PubMed] [Google Scholar]
- [50].Liang Q, et al. , Overexpression of metallothionein reduces diabetic cardiomyopathy, Diabetes (2002), 10.2337/diabetes.51.1.174. [DOI] [PubMed] [Google Scholar]
- [51].Parinandi NL, Thompson EW, Schmid HHO, Diabetic heart and kidney exhibit increased resistance to lipid peroxidation, Biochim. Biophys. Acta Lipids Lipid. Metabol (1990), 10.1016/0005-2760(90)90261-U. [DOI] [PubMed] [Google Scholar]
- [52].Bugger H, et al. , Type 1 diabetic akita mouse hearts are insulin sensitive but manifest structurally abnormal mitochondria that remain coupled despite increased uncoupling protein 3, Diabetes (2008), 10.2337/db08-0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Bugger H, et al. , Tissue-specific remodeling of the mitochondrial proteome in type 1 diabetic akita mice, Diabetes (2009), 10.2337/db09-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Buchanan J, et al. , Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity, Endocrinology (2005), 10.1210/en.2005-0938. [DOI] [PubMed] [Google Scholar]
- [55].Corsetti JP, Sparks JD, Peterson RG, Smith RL, Sparks CE, Effect of dietary fat on the development of non-insulin dependent diabetes mellitus in obese Zucker diabetic fatty male and female rats, Atherosclerosis (2000), 10.1016/S0021-9150(99)00265-8. [DOI] [PubMed] [Google Scholar]
- [56].Mariappan N, et al. , NF-κB-induced oxidative stress contributes to mitochondrial and cardiac dysfunction in type II diabetes, Cardiovasc. Res (2010), 10.1093/cvr/cvp305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Boudina S, et al. , Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins, Diabetes (2007), 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
- [58].Das A, et al. , Mammalian target of rapamycin (mTOR) inhibition with rapamycin improves cardiac function in type 2 diabetic mice : potential role of attenuated oxidative stress and altered contractile protein expression, J. Biol. Chem (2014), 10.1074/jbc.M113.521062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Pan G, et al. , 4-Hydroxy-2-nonenal attenuates 8-oxoguanine DNA glycosylase 1 activity, J. Cell. Biochem (2020), 10.1002/jcb.29814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Raza H, John A, Howarth FC, Alterations in glutathione redox metabolism, oxidative stress, and mitochondrial function in the left ventricle of elderly zucker diabetic fatty rat heart, Int. J. Mol. Sci (2012), 10.3390/ijms131216241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Vincent HK, Powers SK, Dirks AJ, Scarpace PJ, Mechanism for obesity-induced increase in myocardial lipid peroxidation, Int. J. Obes (2001), 10.1038/sj.ijo.0801536. [DOI] [PubMed] [Google Scholar]
- [62].Banerjee A, et al. , Altered composition of high-lipid diet may generate reactive oxygen species by disturbing the balance of antioxidant and free radicals, J. Basic Clin. Physiol. Pharmacol (2020), 10.1515/jbcpp-2019-0141. [DOI] [PubMed] [Google Scholar]
- [63].Szűcs G, et al. , Prediabetes induced by fructose-enriched diet influences cardiac lipidome and proteome and leads to deterioration of cardiac function prior to the development of excessive oxidative stress and cell damage, Oxid. Med. Cell. Longev (2019), 10.1155/2019/3218275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Koncsos G, et al. , Diastolic dysfunction in prediabetic male rats: role of mitochondrial oxidative stress, Am. J. Physiol. Heart Circ. Physiol (2016), 10.1152/ajpheart.00049.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Heinonen I, et al. , Cellular, mitochondrial and molecular alterations associate with early left ventricular diastolic dysfunction in a porcine model of diabetic metabolic derangement, Sci. Rep (2020), 10.1038/s41598-020-68637-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].El-Omar MM, Yang ZK, Phillips AO, Shah AM, Cardiac dysfunction in the Goto-Kakizaki rat - a model of type II diabetes mellitus, Basic Res. Cardiol (2004), 10.1007/s00395-004-0440-4. [DOI] [PubMed] [Google Scholar]
- [67].Kuwabara WMT, et al. , Comparison of Goto-Kakizaki rats and high fat diet-induced obese rats: are they reliable models to study Type 2 Diabetes mellitus? PloS One (2017) 10.1371/journal.pone.0189622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Santos DL, et al. , Diabetes and mitochondrial oxidative stress: a study using heart mitochondria from the diabetic Goto-Kakizaki rat, Mol. Cell. Biochem (2003), 10.1023/A:1023475022025. [DOI] [PubMed] [Google Scholar]
- [69].Lai N, Kummitha CM, Loy F, Isola R, Hoppel CL, Bioenergetic functions in subpopulations of heart mitochondria are preserved in a non-obese type 2 diabetes rat model (Goto-Kakizaki), Sci. Rep (2020), 10.1038/s41598-020-62370-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Cook NR, et al. , A randomized factorial trial of vitamins C and E and beta carotene in the secondary prevention of cardiovascular events in women: results from the women’s antioxidant cardiovascular study, Arch. Intern. Med (2007), 10.1001/archinte.167.15.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Sesso HD, et al. , Vitamins E and C in the prevention of cardiovascular disease in men: the physicians’ health study II randomized controlled trial, JAMA, J. Am. Med. Assoc (2008), 10.1001/jama.2008.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Schwingshackl L, et al. , Dietary Supplements and risk of cause-specific death, cardiovascular disease, and cancer: a systematic review and meta-analysis of primary prevention trials, Advances in Nutrition (2017), 10.3945/an.116.013516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Miller ER, et al. , Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality, Ann. Intern. Med (2005), 10.7326/0003-4819-142-1-200501040-00110. [DOI] [PubMed] [Google Scholar]
- [74].Anderson EJ, et al. , Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart, J. Am. Coll. Cardiol (2009), 10.1016/j.jacc.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Anderson EJ, et al. , Increased propensity for cell death in diabetic human heart is mediated by mitochondrial-dependent pathways, Am. J. Physiol. Heart Circ. Physiol (2011), 10.1152/ajpheart.00932.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Katunga LA, et al. , Obesity in a model of gpx4 haploinsufficiency uncovers a causal role for lipid-derived aldehydes in human metabolic disease and cardiomyopathy, Mol. Metab (2015), 10.1016/j.molmet.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Montaigne D, et al. , Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients, Circulation (2014), 10.1161/CIRCULATIONAHA.113.008476. [DOI] [PubMed] [Google Scholar]
- [78].Jaishy B, et al. , Lipid-induced NOX2 activation inhibits autophagic flux by impairing lysosomal enzyme activity, J. Lipid Res (2015), 10.1194/jlr.M055152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Boudina S, Abel ED, Diabetic cardiomyopathy revisited, Circulation (2007), 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- [80].Bugger H, Abel ED, Molecular mechanisms for myocardial mitochondrial dysfunction in the metabolic syndrome, Clin. Sci (2008), 10.1042/CS20070166. [DOI] [PubMed] [Google Scholar]
- [81].Turrens JF, Mitochondrial formation of reactive oxygen species, J. Physiol (2003), 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Nishikawa T, et al. , Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage, Nature (2000), 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- [83].Pham T, Loiselle D, Power A, Hickey AJR, Mitochondrial inefficiencies and anoxic ATP hydrolysis capacities in diabetic rat heart, Am. J. Physiol. Cell Physiol (2014), 10.1152/ajpcell.00006.2014. [DOI] [PubMed] [Google Scholar]
- [84].Baseler WA, et al. , Proteomic alterations of distinct mitochondrial subpopulations in the type 1 diabetic heart: contribution of protein import dysfunction, Am. J. Physiol. Regul. Integr. Comp. Physiol (2011), 10.1152/ajpregu.00423.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Bugger H, et al. , Genetic loss of insulin receptors worsens cardiac efficiency in diabetes, J. Mol. Cell. Cardiol (2012), 10.1016/jyjmcc.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Bugger H, Pfeil K, Mitochondrial ROS in myocardial ischemia reperfusion and remodeling, Biochim. Biophys. Acta (BBA) - Mol. Basis Dis (2020), 10.1016/j.bbadis.2020.165768. [DOI] [PubMed] [Google Scholar]
- [87].Niemann B, et al. , Obesity induces signs of premature cardiac aging in younger patients: the role of mitochondria, J. Am. Coll. Cardiol (2011), 10.1016/j.jacc.2010.09.040. [DOI] [PubMed] [Google Scholar]
- [88].Sivitz WI, Yorek MA, Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities, Antioxidants Redox Signal (2010), 10.1089/ars.2009.2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Tsushima K, et al. , Mitochondrial reactive oxygen species in lipotoxic hearts induce post-translational modifications of AKAP121, DRP1, and OPA1 that promote mitochondrial fission, Circ. Res (2018), 10.1161/CIRCRESAHA.117.311307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Orsini F, et al. , The life span determinant p66Shc localizes to mitochondria where it associates with mitochondrial heat shock protein 70 and regulates transmembrane potential, J. Biol. Chem (2004), 10.1074/jbc.M401844200. [DOI] [PubMed] [Google Scholar]
- [91].Giorgio M, et al. , Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis, Cell (2005), 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- [92].Galimov ER, The role of p66shc in oxidative stress and apoptosis, Acta Naturae (2010), 10.32607/20758251-2010-2-4-44-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Camici GG, et al. , Genetic deletion of p66Shc adaptor protein prevents hyperglycemia-induced endothelial dysfunction and oxidative stress, Proc. Natl. Acad. Sci. U.S.A (2007), 10.1073/pnas.0609656104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Napoli C, et al. , Deletion of the p66Shc longevity gene reduces systemic and tissue oxidative stress, vascular cell apoptosis, and early atherogenesis in mice fed a high-fat diet, Proc. Natl. Acad. Sci. U.S.A (2003), 10.1073/pnas.0336359100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Costantino S, et al. , Hyperglycaemia-induced epigenetic changes drive persistent cardiac dysfunction via the adaptor p66 Shc, Int. J. Cardiol (2018), 10.1016/j.ijcard.2018.04.082. [DOI] [PubMed] [Google Scholar]
- [96].Rota M, et al. , Diabetes promotes cardiac stem cell aging and heart failure, which are prevented by deletion of the p66shc gene, Circ. Res (2006), 10.1161/01.RES.0000231289.63468.08. [DOI] [PubMed] [Google Scholar]
- [97].Kaludercic N, Mialet-Perez J, Paolocci N, Parini A, Di Lisa F, Monoamine oxidases as sources of oxidants in the heart, J. Mol. Cell. Cardiol (2014), 10.1016/j.yjmcc.2013.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Deshwal S, Di Sante M, Di Lisa F, Kaludercic N, Emerging role of monoamine oxidase as a therapeutic target for cardiovascular disease, Curr. Opin. Pharmacol (2017), 10.1016/j.coph.2017.04.003. [DOI] [PubMed] [Google Scholar]
- [99].Kaludercic N, Carpi A, Menabò R, Di Lisa F, Paolocci N, Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury, Biochim. Biophys. Acta Mol. Cell Res (2011), 10.1016/j.bbamcr.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Umbarkar P, et al. , Monoamine oxidase-A is an important source of oxidative stress and promotes cardiac dysfunction, apoptosis, and fibrosis in diabetic cardiomyopathy, Free Radic. Biol. Med 87 (2015) 263–273. [DOI] [PubMed] [Google Scholar]
- [101].Deshwal S, et al. , Monoamine oxidase-dependent endoplasmic reticulum-mitochondria dysfunction and mast cell degranulation lead to adverse cardiac remodeling in diabetes, Cell Death Differ (2018), 10.1038/s41418-018-0071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Eisenhofer G, The role of neuronal and extraneuronal plasma membrane transporters in the inactivation of peripheral catecholamines, Pharmacol. Therapeut (2001), 10.1016/S0163-7258(01)00144-9. [DOI] [PubMed] [Google Scholar]
- [103].Manni ME, et al. , Monoamine oxidase is overactivated in left and right ventricles from ischemic hearts: an intriguing therapeutic target, Oxid. Med. Cell. Longev (2016), 10.1155/2016/4375418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Duicu OM, et al. , Assessment of mitochondrial dysfunction and monoamine oxidase contribution to oxidative stress in human diabetic hearts, Oxid. Med. Cell. Longev (2016), 10.1155/2016/8470394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Goll DE, Thompson VF, Li H, Wei W, Cong J, The calpain system, Physiol. Rev (2003), 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- [106].Li Y, Li Y, Feng Q, Arnold M, Peng T, Calpain activation contributes to hyperglycaemia-induced apoptosis in cardiomyocytes, Cardiovasc. Res (2009), 10.1093/cvr/cvp189. [DOI] [PubMed] [Google Scholar]
- [107].Li Y, et al. , Targeted inhibition of calpain reduces myocardial hypertrophy and fibrosis in mouse models of type 1 diabetes, Diabetes (2011), 10.2337/db10-1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Li S, et al. , Disruption of calpain reduces lipotoxicity-induced cardiac injury by preventing endoplasmic reticulum stress, Biochim. Biophys. Acta - Mol. Basis Dis (2016), 10.1016/j.bbadis.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Ni R, et al. , Mitochondrial calpain-1 disrupts ATP synthase and induces superoxide generation in type 1 diabetic hearts: a novel mechanism contributing to diabetic cardiomyopathy, Diabetes 65 (2016) 255–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Zheng D, et al. , Targeted inhibition of calpain in mitochondria alleviates oxidative stress-induced myocardial injury, Acta Pharmacol. Sin (2020), 10.1038/s41401-020-00526-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Lassègue B, San Martín A, Griendling KK, Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system, Circ. Res (2012), 10.1161/CIRCRESAHA.111.243972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Brandes RP, Weissmann N, Schröder K, NADPH oxidases in cardiovascular disease, Free Radic. Biol. Med (2010), 10.1016/j.freeradbiomed.2010.04.030. [DOI] [PubMed] [Google Scholar]
- [113].Wold LE, et al. , Metallothionein alleviates cardiac dysfunction in streptozotocin-induced diabetes: role of Ca2+ cycling proteins, NADPH oxidase, poly(ADP-Ribose) polymerase and myosin heavy chain isozyme, Free Radic. Biol. Med (2006), 10.1016/j.freeradbiomed.2005.12.009. [DOI] [PubMed] [Google Scholar]
- [114].Li SY, et al. , Cardiac contractile dysfunction in Lep/Lep obesity is accompanied by NADPH oxidase activation, oxidative modification of sarco(endo)plasmic reticulum Ca2+-ATPase and myosin heavy chain isozyme switch, Diabetologia (2006), 10.1007/s00125-006-0229-0. [DOI] [PubMed] [Google Scholar]
- [115].Tsai KH, et al. , NADPH oxidase-derived superoxide Anion-induced apoptosis is mediated via the JNK-dependent activation of NF-κB in cardiomyocytes exposed to high glucose, J. Cell. Physiol (2012), 10.1002/jcp.22847. [DOI] [PubMed] [Google Scholar]
- [116].Privratsky JR, Wold LE, Sowers JR, Quinn MT, Ren J, AT1 blockade prevents glucose-induced cardiac dysfunction in ventricular myocytes: role of the AT1 receptor and NADPH oxidase, Hypertension (2003), 10.1161/01.HYP.0000082814.62655.85. [DOI] [PubMed] [Google Scholar]
- [117].Kuo WW, et al. , Diallyl trisufide (DATS) suppresses high glucose-induced cardiomyocyte apoptosis by inhibiting JNK/NFκB signaling via attenuating ROS generation, Int. J. Cardiol (2013), 10.1016/j.ijcard.2012.09.080. [DOI] [PubMed] [Google Scholar]
- [118].Balteau M, et al. , NADPH oxidase activation by hyperglycaemia in cardiomyocytes is independent of glucose metabolism but requires SGLT1, Cardiovasc. Res (2011), 10.1093/cvr/cvr230. [DOI] [PubMed] [Google Scholar]
- [119].Balteau M, et al. , AMPK activation by glucagon-like peptide-1 prevents NADPH oxidase activation induced by hyperglycemia in adult cardiomyocytes, Am. J. Physiol. Heart Circ. Physiol (2014), 10.1152/ajpheart.00210.2014. [DOI] [PubMed] [Google Scholar]
- [120].Joseph D, Kimar C, Symington B, Milne R, Faadiel Essop M, The detrimental effects of acute hyperglycemia on myocardial glucose uptake, Life Sci (2014), 10.1016/j.lfs.2014.04.009. [DOI] [PubMed] [Google Scholar]
- [121].Shen E, et al. , Rac1 is required for cardiomyocyte apoptosis during hyperglycemia, Diabetes (2009), 10.2337/db08-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Roe ND, Thomas DP, Ren J, Inhibition of NADPH oxidase alleviates experimental diabetes-induced myocardial contractile dysfunction, Diabetes Obes. Metabol (2011), 10.1111/j.1463-1326.2011.01369.x. [DOI] [PubMed] [Google Scholar]
- [123].Joseph LC, et al. , Inhibition of napdh oxidase 2 (nox2) prevents oxidative stress and mitochondrial abnormalities caused by saturated fat in cardiomyocytes, PloS One (2016), 10.1371/journal.pone.0145750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Li J, et al. , Deficiency of Rac1 blocks NADPH oxidase activation, inhibits endoplasmic reticulum stress, and reduces myocardial remodeling in a mouse model of type 1 diabetes, Diabetes (2010), 10.2337/db09-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Maalouf RM, et al. , Nox4-derived reactive oxygen species mediate cardiomyocyte injury in early type 1 diabetes, Am. J. Physiol. Cell Physiol (2012), 10.1152/ajpcell.00331.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Guo Z, et al. , Effect of exenatide on the cardiac expression of adiponectin receptor 1 and NADPH oxidase subunits and heart function in streptozotocin-induced diabetic rats, Diabetol. Metab. Syndrome (2014), 10.1186/1758-5996-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Fukuda M, et al. , Ezetimibe ameliorates cardiovascular complications and hepatic steatosis in obese and type 2 diabetic db/db mice, J. Pharmacol. Exp. Therapeut (2010), 10.1124/jpet.110.170373. [DOI] [PubMed] [Google Scholar]
- [128].Yang Y, et al. , Xanthine oxidase inhibitor allopurinol improves atrial electrical remodeling in diabetic rats by inhibiting CaMKII/NCX signaling, Life Sci 259 (2020) 118290. [DOI] [PubMed] [Google Scholar]
- [129].Di Fan X, Wan LL, Duan M, Lu S, HDAC11 deletion reduces fructose-induced cardiac dyslipidemia, apoptosis and inflammation by attenuating oxidative stress injury, Biochem. Biophys. Res. Commun (2018), 10.1016/j.bbrc.2018.04.090. [DOI] [PubMed] [Google Scholar]
- [130].Rajesh M, et al. , Xanthine oxidase inhibitor allopurinol attenuates the development of diabetic cardiomyopathy, J. Cell Mol. Med (2009), 10.1111/j.1582-4934.2008.00564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Gao X, et al. , Allopurinol attenuates left ventricular dysfunction in rats with early stages of streptozotocin-induced diabetes, Diabetes. Metab. Res. Rev (2012), 10.1002/dmrr.2295. [DOI] [PubMed] [Google Scholar]
- [132].Luo J, et al. , Allopurinol reduces oxidative stress and activates Nrf2/p62 to attenuate diabetic cardiomyopathy in rats, J. Cell Mol. Med 24 (2020) 1760–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Desco MC, et al. , Xanthine oxidase is involved in free radical production in type 1 diabetes: protection by allopurinol, Diabetes (2002), 10.2337/diabetes.51.4.1118. [DOI] [PubMed] [Google Scholar]
- [134].Pacher P, Nivorozhkin A, Szabó C, Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol, Pharmacol. Rev (2006), 10.1124/pr.58.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].D’Oria R, et al. , The role of oxidative stress in cardiac disease: from physiological response to injury factor, Oxidative Medicine and Cellular Longevity (2020), 10.1155/2020/5732956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Mayer B, Hemmens B, Biosynthesis and action of nitric oxide in mammalian cells, Trends Biochem. Sci (1997), 10.1016/S0968-0004(97)01147-X. [DOI] [PubMed] [Google Scholar]
- [137].Stuehr D, Pou S, Rosen GM, Oxygen reduction by nitric-oxide synthases, J. Biol. Chem (2001), 10.1074/jbc.R100011200. [DOI] [PubMed] [Google Scholar]
- [138].Magenta A, Greco S, Capogrossi MC, Gaetano C, Martelli F, Nitric oxide, oxidative stress, and p66Shc interplay in diabetic endothelial dysfunction, BioMed Res. Int (2014), 10.1155/2014/193095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].A. Nitrotyrosine Ceriello, New findings as a marker of postprandial oxidative stress, in: Int. J. Clin. Pract, 2002. Supplement. [PubMed] [Google Scholar]
- [140].Sartoretto JL, Kalwa H, Pluth MD, Lippard SJ, Michel T, Hydrogen peroxide differentially modulates cardiac myocyte nitric oxide synthesis, Proc. Natl. Acad. Sci. U.S.A (2011), 10.1073/pnas.1111331108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Pechánová O, et al. , Cardiac NO signalling in the metabolic syndrome, Br. J. Pharmacol (2015), 10.1111/bph.12960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Simon JN, Duglan D, Casadei B, Carnicer R, Nitric oxide synthase regulation of cardiac excitation-contraction coupling in health and disease, J. Mol. Cell. Cardiol (2014), 10.1016/j.yjmcc.2014.03.004. [DOI] [PubMed] [Google Scholar]
- [143].Amour J, et al. , Altered contractile response due to increased β3-adrenoceptor stimulation in diabetic cardiomyopathy: the role of nitric oxide synthase 1-derived nitric oxide, Anesthesiology (2007), 10.1097/01.anes.0000278909.40408.24. [DOI] [PubMed] [Google Scholar]
- [144].Jo H, et al. , Inhibition of nitric oxide synthase uncoupling by sepiapterin improves left ventricular function in streptozotocin-induced diabetic mice, Clin. Exp. Pharmacol. Physiol (2011), 10.1111/j.1440-1681.2011.05535.x. [DOI] [PubMed] [Google Scholar]
- [145].Sun HJ, et al. , Induction of caveolin-3/eNOS complex by nitroxyl (HNO) ameliorates diabetic cardiomyopathy, Redox Biol (2020), 10.1016/j.redox.2020.101493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].de Zeeuw P, Wong BW, Carmeliet P, Metabolic adaptations in diabetic endothelial cells, Circ. J (2015), 10.1253/circj.CJ-15-0230. [DOI] [PubMed] [Google Scholar]
- [147].Wan A, Rodrigues B, Endothelial cell-cardiomyocyte crosstalk in diabetic cardiomyopathy, Cardiovasc. Res (2016), 10.1093/cvr/cvw159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].N D R, D P T, J R, Inhibition of NADPH oxidase alleviates experimental diabetes-induced myocardial contractile dysfunction, Diabetes Obes. Metabol 13 (5) (2011) 465–473. [DOI] [PubMed] [Google Scholar]
- [149].Jay D, Hitomi H, Griendling KK, Oxidative stress and diabetic cardiovascular complications, Free Radic. Biol. Med (2006), 10.1016/j.freeradbiomed.2005.06.018. [DOI] [PubMed] [Google Scholar]
- [150].Brownlee M, The pathobiology of diabetic complications: a unifying mechanism, Diabetes (2005), 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- [151].Ceriello A, Acute hyperglycaemia: a ‘new’ risk factor during myocardial infarction, Eur. Heart J (2005), 10.1093/eurheartj/ehi049. [DOI] [PubMed] [Google Scholar]
- [152].Yu M, et al. , RNA-Seq analysis and functional characterization revealed lncRNA NONRATT007560.2 regulated cardiomyocytes oxidative stress and apoptosis induced by high glucose, J. Cell. Biochem (2019), 10.1002/jcb.29134. [DOI] [PubMed] [Google Scholar]
- [153].Zhi YF, Prins JB, Marwick TH, Diabetic cardiomyopathy: evidence, mechanisms, and therapeutic implications, Endocr. Rev (2004), 10.1210/er.2003-0012. [DOI] [PubMed] [Google Scholar]
- [154].Abel ED, O’Shea KM, Ramasamy R, Insulin resistance: metabolic mechanisms and consequences in the heart, Arterioscler. Thromb. Vasc. Biol (2012), 10.1161/ATVBAHA.111.241984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Patti ME, Corvera S, The role of mitochondria in the pathogenesis of type 2 diabetes, Endocr. Rev (2010), 10.1210/er.2009-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Lopaschuk GD, Ussher JR, Folmes CDL, Jaswal JS, Stanley WC, Myocardial fatty acid metabolism in health and disease, Physiol. Rev (2010), 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- [157].Kessler G, Friedman J, Metabolism of fatty acids and glucose, Circulation (1998), 10.1161/01.CIR.98.13.1350.a. [DOI] [PubMed] [Google Scholar]
- [158].Van De Weijer T, Schrauwen-Hinderling VB, Schrauwen P, Lipotoxicity in type 2 diabetic cardiomyopathy, Cardiovasc. Res (2011), 10.1093/cvr/cvr212. [DOI] [PubMed] [Google Scholar]
- [159].Summers SA, Ceramides in insulin resistance and lipotoxicity, Prog. Lipid Res (2006), 10.1016/j.plipres.2005.11.002. [DOI] [PubMed] [Google Scholar]
- [160].Quintela AM, et al. , Activation of peroxisome proliferator-activated receptor-β/-δ (PPARβ/δ) prevents endothelial dysfunction in type 1 diabetic rats, Free Radic. Biol. Med (2012), 10.1016/j.freeradbiomed.2012.05.045. [DOI] [PubMed] [Google Scholar]
- [161].Mazumder PK, et al. , Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts, Diabetes (2004), 10.2337/diabetes.53.9.2366. [DOI] [PubMed] [Google Scholar]
- [162].Boudina S, et al. , Mitochondrial energetics in the heart in obesity-related diabetes, Diabetes 56 (2007) 2457. LP–2466. [DOI] [PubMed] [Google Scholar]
- [163].Carroll R, Carley AN, Dyck JRB, Severson DL, Metabolic effects of insulin on cardiomyocytes from control and diabetic db/db mouse hearts, Am. J. Physiol. Endocrinol. Metab (2005), 10.1152/ajpendo.00491.2004. [DOI] [PubMed] [Google Scholar]
- [164].Hafstad AD, Solevåg GH, Severson DL, Larsen TS, Aasum E, Perfused hearts from Type 2 diabetic (db/db) mice show metabolic responsiveness to insulin, Am. J. Physiol. Heart Circ. Physiol (2006), 10.1152/ajpheart.01063.2005. [DOI] [PubMed] [Google Scholar]
- [165].Wright JJ, et al. , Mechanisms for increased myocardial fatty acid utilization following short-term high-fat feeding, Cardiovasc. Res (2009), 10.1093/cvr/cvp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Cook SA, et al. , Abnormal myocardial insulin signalling in type 2 diabetes and left-ventricular dysfunction, Eur. Heart J (2010), 10.1093/eurheartj/ehp396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Broderick TL, Driedzic WR, Gillis M, Jacob J, Belke T, Effects of chronic food restriction and exercise training on the recovery of cardiac function following ischemia, Journals Gerontol. - Ser. A Biol. Sci. Med. Sci (2001), 10.1093/gerona/56.1.B33. [DOI] [PubMed] [Google Scholar]
- [168].Boudina S, et al. , Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart, Circulation (2009), 10.1161/CIRCULATIONAHA.108.792101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Ritchie RH, et al. , The antioxidant tempol inhibits cardiac hypertrophy in the insulin-resistant GLUT4-deficient mouse in vivo, J. Mol. Cell. Cardiol (2007), 10.1016/j.yjmcc.2007.03.900. [DOI] [PubMed] [Google Scholar]
- [170].Reaven G, Insulin resistance and coronary heart disease in nondiabetic individuals, Arterioscler. Thromb. Vasc. Biol (2012), 10.1161/ATVBAHA.111.241885. [DOI] [PubMed] [Google Scholar]
- [171].Mohammed Yusof NL, Zainalabidin S, Mohd Fauzi N, Budin SB, Cardioprotective effects of roselle (Hibiscus sabdariffa linn.) polyphenol-rich extract in streptozotocin-induced diabetic rats, Int. J. Cardiol (2017), 10.1016/j.ijcard.2017.09.037. [DOI] [Google Scholar]
- [172].Shimizu I, Minamino T, Physiological and pathological cardiac hypertrophy, J. Mol. Cell. Cardiol (2016), 10.1016/j.yjmcc.2016.06.001. [DOI] [PubMed] [Google Scholar]
- [173].Balderas-Villalobos J, et al. , Oxidative stress in cardiomyocytes contributes to decreased SERCA2a activity in rats with metabolic syndrome, Am. J. Physiol. Heart Circ. Physiol (2013), 10.1152/ajpheart.00211.2013. [DOI] [PubMed] [Google Scholar]
- [174].Kaneto H, Katakami N, Matsuhisa M, Matsuoka TA, Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis, Mediat. Inflamm (2010), 10.1155/2010/453892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Mahmoud AM, et al. , Nox2 contributes to hyperinsulinemia-induced redox imbalance and impaired vascular function, Redox Biol (2017), 10.1016/j.redox.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Palace V, Kumar D, Hill MF, Khaper N, Singal PK, Regional differences in non-enzymatic antioxidants in the heart under control and oxidative stress conditions, J. Mol. Cell. Cardiol (1999), 10.1006/jmcc.1998.0859. [DOI] [PubMed] [Google Scholar]
- [177].Doroshow JH, Locker GY, Myers CE, Enzymatic defenses of the mouse heart against reactive oxygen metabolites. Alterations produced by doxorubicin, J. Clin. Invest (1980), 10.1172/JCI109642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Dieterich S, Bieligk U, Beulich K, Hasenfuss G, Prestle J, Gene expression of antioxidative enzymes in the human heart: increased expression of catalase in the end-stage failing heart, Circulation (2000), 10.1161/01.CIR.101.1.33. [DOI] [PubMed] [Google Scholar]
- [179].Di Filippo C, et al. , M40403 prevents myocardial injury induced by acute hyperglycaemia in perfused rat heart, Eur. J. Pharmacol (2004), 10.1016/j.ejphar.2004.06.037. [DOI] [PubMed] [Google Scholar]
- [180].Turko IV, Murad F, Quantitative protein profiling in heart mitochondria from diabetic rats, J. Biol. Chem (2003), 10.1074/jbc.M303139200. [DOI] [PubMed] [Google Scholar]
- [181].Arkat S, Umbarkar P, Singh S, Sitasawad SL, Mitochondrial Peroxiredoxin-3 protects against hyperglycemia induced myocardial damage in Diabetic cardiomyopathy, Free Radic. Biol. Med (2016), 10.1016/j.freeradbiomed.2016.06.019. [DOI] [PubMed] [Google Scholar]
- [182].Cong W, et al. , Cardiac-specific overexpression of catalase prevents diabetes-induced pathological changes by inhibiting NF-κB signaling activation in the heart, J. Mol. Cell. Cardiol (2015), 10.1016/j.yjmcc.2015.10.010. [DOI] [PubMed] [Google Scholar]
- [183].Turdi S, Li Q, Lopez FL, Ren J, Catalase alleviates cardiomyocyte dysfunction in diabetes: role of Akt, Forkhead transcriptional factor and silent information regulator 2, Life Sci 81 (2007) 895–905. [DOI] [PubMed] [Google Scholar]
- [184].Bhatt NM, et al. , Restoring redox balance enhances contractility in heart trabeculae from type 2 diabetic rats exposed to high glucose, Am. J. Physiol. Heart Circ. Physiol (2015), 10.1152/ajpheart.00378.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Baseler WA, et al. , Reversal of mitochondrial proteomic loss in type 1 diabetic heart with overexpression of phospholipid hydroperoxide glutathione peroxidase, Am. J. Physiol. Regul. Integr. Comp. Physiol (2013), 10.1152/ajpregu.00249.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Matsushima S, et al. , Overexpression of glutathione peroxidase attenuates myocardial remodeling and preserves diastolic function in diabetic heart, Am. J. Physiol. Heart Circ. Physiol (2006), 10.1152/ajpheart.00427.2006. [DOI] [PubMed] [Google Scholar]
- [187].McClung JP, et al. , Development of insulin resistance and obesity in mice overexpressing cellular glutathione peroxidase, Proc. Natl. Acad. Sci. U.S.A (2004), 10.1073/pnas.0308096101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Rhee SG, Jeong W, Chang TS, Woo HA, Sulfiredoxin, the cysteine sulfinic acid reductase specific to 2-Cys peroxiredoxin: its discovery, mechanism of action, and biological significance, Kidney Int (2007), 10.1038/sj.ki.5002380. [DOI] [PubMed] [Google Scholar]
- [189].Shi S, et al. , Sulfiredoxin involved in the protection of peroxiredoxins against hyperoxidation in the early hyperglycaemia, Exp. Cell Res (2017), 10.1016/j.yexcr.2017.02.015. [DOI] [PubMed] [Google Scholar]
- [190].Li H, et al. , Thioredoxin 2 offers protection against mitochondrial oxidative stress in H9c2 cells and against myocardial hypertrophy induced by hyperglycemia, Int. J. Mol. Sci (2017), 10.3390/ijms18091958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Cox AG, Winterbourn CC, Hampton MB, Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling, Biochem. J (2010), 10.1042/BJ20091541. [DOI] [PubMed] [Google Scholar]
- [192].Stanley BA, et al. , Thioredoxin reductase-2 is essential for keeping low levels of H 2O 2 emission from isolated heart mitochondria, J. Biol. Chem (2011), 10.1074/jbc.M111.284612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Connelly KA, et al. , Impaired cardiac anti-oxidant activity in diabetes: human and correlative experimental studies, Acta Diabetol (2014), 10.1007/s00592-014-0608-9. [DOI] [PubMed] [Google Scholar]
- [194].Myers RB, et al. , Deletion of thioredoxin-interacting protein improves cardiac inotropic reserve in the streptozotocin-induced diabetic heart, Am. J. Physiol. Heart Circ. Physiol (2016), 10.1152/ajpheart.00051.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Li S, Li X, Rozanski GJ, Regulation of glutathione in cardiac myocytes, J. Mol. Cell. Cardiol (2003), 10.1016/S0022-2828(03)00230-X. [DOI] [PubMed] [Google Scholar]
- [196].Ghosh S, et al. , Cardiomyocyte apoptosis induced by short-term diabetes requires mitochondrial GSH depletion, Am. J. Physiol. Heart Circ. Physiol (2005), 10.1152/ajpheart.00038.2005. [DOI] [PubMed] [Google Scholar]
- [197].Qi X, et al. , Cardiac damage and dysfunction in diabetic cardiomyopathy are ameliorated by Grx1, Genet. Mol. Res (2016), 10.4238/gmr.15039000. [DOI] [PubMed] [Google Scholar]
- [198].Ceriello A, et al. , Acute hyperglycemia induces nitrotyrosine formation and apoptosis in perfused heart from rat, Diabetes (2002), 10.2337/diabetes.51.4.1076. [DOI] [PubMed] [Google Scholar]
- [199].Liu C, et al. , N-Acetyl Cysteine improves the diabetic cardiac function: possible role of fibrosis inhibition, BMC Cardiovasc. Disord (2015), 10.1186/s12872-015-0076-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Fiordaliso F, et al. , Antioxidant treatment attenuates hyperglycemia-induced cardiomyocyte death in rats, J. Mol. Cell. Cardiol (2004), 10.1016/j.yjmcc.2004.07.008. [DOI] [PubMed] [Google Scholar]
- [201].Yildirim SS, Akman D, Catalucci D, Turan B, Relationship between downregulation of miRNAs and increase of oxidative stress in the development of diabetic cardiac dysfunction: junctin as a target protein of miR-1, Cell Biochem. Biophys (2013), 10.1007/s12013-013-9672-y. [DOI] [PubMed] [Google Scholar]
- [202].Dludla PV, et al. , N-Acetyl cysteine ameliorates hyperglycemia-induced cardiomyocyte toxicity by improving mitochondrial energetics and enhancing endogenous Coenzyme Q9/10 levels, Toxicol. Reports 6 (2019) 1240–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Tossios P, et al. , N-acetylcysteine prevents reactive oxygen species-mediated myocardial stress in patients undergoing cardiac surgery: results of a randomized, double-blind, placebo-controlled clinical trial, J. Thorac. Cardiovasc. Surg (2003), 10.1016/S0022-5223(03)00968-1. [DOI] [PubMed] [Google Scholar]
- [204].Rodrigues AJ, et al. , Blood cardioplegia with N-acetylcysteine may reduce coronary endothelial activation and myocardial oxidative stress, Heart Surg. Forum (2009), 10.1532/HSF98.20081134. [DOI] [PubMed] [Google Scholar]
- [205].Mahmoud KM, Ammar AS, Effect of N-acetylcysteine on cardiac injury and oxidative stress after abdominal aortic aneurysm repair: a randomized controlled trial, Acta Anaesthesiol. Scand (2011), 10.1111/j.1399-6576.2011.02492.x. [DOI] [PubMed] [Google Scholar]
- [206].van der Pol A, van Gilst WH, Voors AA, van der Meer P, Treating oxidative stress in heart failure: past, present and future, Eur. J. Heart Fail (2019), 10.1002/ejhf.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Thornalley PJ. Vašák M, Possible role for metallothionein in protection against radiation-induced oxidative stress. Kinetics and mechanism of its reaction with superoxide and hydroxyl radicals, Biochim. Biophys. Acta (BBA)/Protein Struct. Mol (1985), 10.1016/0167-4838(85)90098-6. [DOI] [PubMed] [Google Scholar]
- [208].Miura T, Muraoka S, Ogiso T, Antioxidant activity of metallothionein compared with reduced glutathione, Life Sci (1997), 10.1016/S0024-3205(97)00156-2. [DOI] [PubMed] [Google Scholar]
- [209].Cai L, Satoh M, Tohyama C, Cherian MG, Metallothionein in radiation exposure: its induction and protective role, Toxicology (1999), 10.1016/S0300-483X(98)00150-4. [DOI] [PubMed] [Google Scholar]
- [210].Ye G, Metreveli NS, Ren J, Epstein PN, Metallothionein prevents diabetes-induced deficits in cardiomyocytes by inhibiting reactive oxygen species production, Diabetes 52 (2003) 777. LP–783. [DOI] [PubMed] [Google Scholar]
- [211].Cai L, et al. , Attenuation by metallothionein of early cardiac cell death via suppression of mitochondrial oxidative stress results in a prevention of diabetic cardiomyopathy, J. Am. Coll. Cardiol (2006), 10.1016/j.jacc.2006.07.022. [DOI] [PubMed] [Google Scholar]
- [212].Kang YJ, Chen V, Yu A, Voss-McCowan M, Epstein PN, Overexpression of metallothionein in the heart of transgenic mice suppresses doxorubicin cardiotoxicity, J. Clin. Invest (1997), 10.1172/JCI119672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Cong W, et al. , Metallothionein prevents cardiac pathological changes in diabetes by modulating nitration and inactivation of cardiac ATP synthase, J. Nutr. Biochem (2014), 10.1016/j.jnutbio.2013.12.007. [DOI] [PubMed] [Google Scholar]
- [214].Cong W, et al. , Metallothionein prevents diabetes-induced cardiac pathological changes, likely via the inhibition of succinyl-CoA:3-ketoacid coenzyme A transferase-1 nitration at Trp374, Am. J. Physiol. Metab 304 (2013) E826–E835. [DOI] [PubMed] [Google Scholar]
- [215].Wang J, et al. , Cardiac metallothionein induction plays the major role in the prevention of diabetic cardiomyopathy by zinc supplementation, Circulation (2006), 10.1161/CIRCULATIONAHA.105.537894. [DOI] [PubMed] [Google Scholar]
- [216].Cai L, Suppression of nitrative damage by metallothionein in diabetic heart contributes to the prevention of cardiomyopathy, Free Radic. Biol. Med (2006), 10.1016/j.freeradbiomed.2006.06.007. [DOI] [PubMed] [Google Scholar]
- [217].Cai L, Metallothionein as an adaptive protein prevents diabetes and its toxicity, Nonlinearity Biol. Toxicol. Med (2004), 10.1080/15401420490464367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Lu Cai, Diabetic cardiomyopathy and its prevention by metallothionein: experimental evidence, possible mechanisms and clinical implications, Curr. Med. Chem (2007), 10.2174/092986707781389646. [DOI] [PubMed] [Google Scholar]
- [219].Liu Q, Wang S, Cai L, Diabetic cardiomyopathy and its mechanisms: role of oxidative stress and damage, Journal of Diabetes Investigation (2014), 10.1111/jdi.12250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Sultana MR, et al. , Garlic activates SIRT-3 to prevent cardiac oxidative stress and mitochondrial dysfunction in diabetes, Life Sci (2016), 10.1016/j.lfs.2016.08.030. [DOI] [PubMed] [Google Scholar]
- [221].Sun Y, et al. , Exogenous H2S switches cardiac energy substrate metabolism by regulating SIRT3 expression in db/db mice, J. Mol. Med (2018), 10.1007/s00109-017-1616-3. [DOI] [PubMed] [Google Scholar]
- [222].Sun Y, et al. , Exogenous H2S reduces the acetylation levels of mitochondrial respiratory enzymes via regulating the NAD+-SIRT3 pathway in cardiac tissues of db/db mice, Am. J. Physiol. Endocrinol. Metab (2019), 10.1152/ajpendo.00326.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Song S, et al. , Sirtuin 3 deficiency exacerbates diabetic cardiomyopathy via necroptosis enhancement and NLRP3 activation, Acta Pharmacol. Sin (2020), 10.1038/s41401-020-0490-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Zong X, et al. , SIRT3 is a downstream target of PPAR-α implicated in high glucose-induced cardiomyocyte injury in AC16 cells, Exp. Ther. Med (2020), 10.3892/etm.2020.8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Li C, et al. , Elabela may regulate SIRT3-mediated inhibition of oxidative stress through Foxo3a deacetylation preventing diabetic-induced myocardial injury, J. Cell. Mol. Med 25 (1) (2020) 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Luo W, et al. , Blockage of ROS and MAPKs-mediated inflammation via restoring SIRT1 by a new compound LF10 prevents type 1 diabetic cardiomyopathy, Toxicol. Appl. Pharmacol (2019), 10.1016/j.taap.2019.03.005. [DOI] [PubMed] [Google Scholar]
- [227].Motohashi H, Yamamoto M, Nrf2-Keap1 defines a physiologically important stress response mechanism, Trends Mol. Med (2004), 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- [228].Chen QM, Maltagliati AJ, Nrf2 at the heart of oxidative stress and cardiac protection, Physiol. Genom (2018), 10.1152/physiolgenomics.00041.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Chen J, Zhang Z, Cai L, Diabetic cardiomyopathy and its prevention by Nrf2: current status, Diabetes and Metabolism Journal (2014), 10.4093/dmj.2014.38.5.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Kaspar JW, Niture SK, Jaiswal AK, Nrf2:INrf2 (Keap1) signaling in oxidative stress, Free Radic. Biol. Med (2009), 10.1016/j.freeradbiomed.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Qiang Ma XH, Disruption of Nrf2 synergizes with high glucose to cause heightened myocardial oxidative stress and severe cardiomyopathy in diabetic mice, J. Diabetes Metabol (2013), 10.4172/2155-6156.s7-002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Liu H et al. Spiraeoside protects human cardiomyocytes against high glucose-induced injury, oxidative stress, and apoptosis by activation of PI3K/Akt/Nrf2 pathway. J. Biochem. Mol. Toxicol n/a, e22548. [DOI] [PubMed] [Google Scholar]
- [233].Tan Y, et al. , Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo, Diabetes (2011), 10.2337/db10-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Li X, et al. , Distinct cardiac energy metabolism and oxidative stress adaptations between obese and non-obese type 2 diabetes mellitus, Theranostics 10 (2020) 2675–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Nishio Y, et al. , Altered activities of transcription factors and their related gene expression in cardiac tissues of diabetic rats, Diabetes (1998), 10.2337/diab.47.8.1318. [DOI] [PubMed] [Google Scholar]
- [236].Bai Y, et al. , Prevention by sulforaphane of diabetic cardiomyopathy is associated with up-regulation of Nrf2 expression and transcription activation, J. Mol. Cell. Cardiol (2013), 10.1016/j.yjmcc.2013.01.008. [DOI] [PubMed] [Google Scholar]
- [237].He X, Kan H, Cai L, Ma Q, Nrf2 is critical in defense against high glucose-induced oxidative damage in cardiomyocytes, J. Mol. Cell. Cardiol 46 (2009) 47–58. [DOI] [PubMed] [Google Scholar]
- [238].Thorwald MA, et al. , Nrf2-related gene expression is impaired during a glucose challenge in type II diabetic rat hearts, Free Radic. Biol. Med (2019), 10.1016/j.freeradbiomed.2018.10.405. [DOI] [PubMed] [Google Scholar]
- [239].Gu J, et al. , Metallothionein is downstream of Nrf2 and partially mediates sulforaphane prevention of diabetic cardiomyopathy, Diabetes (2017), 10.2337/db15-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Zhao Y, et al. , Heme oxygenase-1 prevents cardiac dysfunction in streptozotocin-diabetic mice by reducing inflammation, oxidative stress, apoptosis and enhancing autophagy, PloS One 8 (2013) e75927–e75927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Li R, et al. , Bailcalin protects against diabetic cardiomyopathy through Keap1/Nrf2/AMPK-mediated antioxidative and lipid-lowering effects, Oxid. Med. Cell. Longev (2019), 10.1155/2019/3206542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Lian Y, Xia X, Zhao H, Zhu Y, The potential of chrysophanol in protecting against high fat-induced cardiac injury through Nrf2-regulated anti-inflammation, anti-oxidant and anti-fibrosis in Nrf2 knockout mice, Biomed. Pharmacother 93 (2017) 1175–1189. [DOI] [PubMed] [Google Scholar]
- [243].Dludla PV, et al. , Aspalathin protects the heart against hyperglycemia-induced oxidative damage by up-regulating Nrf2 expression, Molecules (2017), 10.3390/molecules22010129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Li H, et al. , Piceatannol alleviates inflammation and oxidative stress via modulation of the Nrf2/HO-1 and NF-κB pathways in diabetic cardiomyopathy, Chem. Biol. Interact (2019), 10.1016/j.cbi.2019.108754. [DOI] [PubMed] [Google Scholar]
- [245].Ying Y, et al. , Phloretin prevents diabetic cardiomyopathy by dissociating Keap1/Nrf2 complex and inhibiting oxidative stress, Front. Endocrinol. (Lausanne) (2018), 10.3389/fendo.2018.00774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Zang H, et al. , Autophagy inhibition enables Nrf2 to exaggerate the progression of diabetic cardiomyopathy in mice, Diabetes (2020), 10.2337/db19-1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Li B, Liu S, Miao L, Cai L, Prevention of diabetic complications by activation of Nrf2: diabetic cardiomyopathy and nephropathy, Exp. Diabetes Res (2012), 10.1155/2012/216512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Chartoumpekis D, Kensler T, New player on an old field; the Keap1/Nrf2 pathway as a target for treatment of type 2 diabetes and metabolic syndrome, Curr. Diabetes Rev (2013), 10.2174/1573399811309020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Xu F, et al. , ALDH2 genetic polymorphism and the risk of type II diabetes mellitus in CAD patients, Hypertens. Res (2010), 10.1038/hr.2009.178. [DOI] [PubMed] [Google Scholar]
- [250].Cao R, et al. , ALDH2 overexpression alleviates high glucose-induced cardiotoxicity by inhibiting NLRP3 inflammasome activation, J. Diabetes Res (2019), 10.1155/2019/4857921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Guo Y, et al. , A novel protective mechanism for mitochondrial aldehyde dehydrogenase (ALDH2) in type i diabetes-induced cardiac dysfunction: role of AMPK-regulated autophagy, Biochim. Biophys. Acta - Mol. Basis Dis (2015), 10.1016/j.bbadis.2014.05.017. [DOI] [PubMed] [Google Scholar]
- [252].Hu N, Ren J, Zhang Y, Mitochondrial aldehyde dehydrogenase obliterates insulin resistance-induced cardiac dysfunction through deacetylation of PGC-1α, Oncotarget (2016) 10.18632/oncotarget.11977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Cai L, Kang YJ, Oxidative stress and diabetic cardiomyopathy: a brief review, Cardiovasc. Toxicol (2001), 10.1385/CT:1:3:181. [DOI] [PubMed] [Google Scholar]
- [254].Varga ZV, et al. , Interplay of oxidative, nitrosative/nitrative stress, inflammation, cell death and autophagy in diabetic cardiomyopathy, Biochim. Biophys. Acta (BBA) - Mol. Basis Dis (2015), 10.1016/j.bbadis.2014.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Wallace DC, A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine, Annu. Rev. Genet (2005), 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Sun X, et al. , Empagliflozin ameliorates obesity-related cardiac dysfunction by regulating sestrin2-mediated AMPK-mTOR signaling and redox homeostasis in high-fat diet-induced obese mice, Diabetes (2020), 10.2337/db19-0991. [DOI] [PubMed] [Google Scholar]
- [257].He X, Ma Q, Disruption of Nrf2 synergizes with high glucose to cause heightened myocardial oxidative stress and severe cardiomyopathy in diabetic mice, J. Diabetes Metab Suppl 7 (2012) 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Wang J, Wang Q, Watson LJ, Jones SP, Epstein PN, Cardiac overexpression of 8-oxoguanine dna glycosylase 1 protects mitochondrial dna and reduces cardiac fibrosis following transaortic constriction, Am. J. Physiol. Heart Circ. Physiol (2011), 10.1152/ajpheart.00157.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Kaji H, et al. , Increased lipoperoxide value and glutathione peroxidase activity in blood plasma of Type 2 (non-insulin-dependent) diabetic women, Klin. Wochenschr (1985), 10.1007/BF01733829. [DOI] [PubMed] [Google Scholar]
- [260].Griesmacher A, et al. , Enhanced serum levels of thiobarbituric-acid-reactive substances in diabetes mellitus, Am. J. Med (1995), 10.1016/S0002-9343(99)80347-7. [DOI] [PubMed] [Google Scholar]
- [261].Kesavulu MM, et al. , Lipid peroxidation and antioxidant enzyme status in Type 2 diabetics with coronary heart disease, Diabetes Res. Clin. Pract (2001), 10.1016/S0168-8227(01)00238-8. [DOI] [PubMed] [Google Scholar]
- [262].Turko IV, Marcondes S, Murad F, Diabetes-associated nitration of tyrosine and inactivation of succinyl-CoA:3-oxoacid CoA-transferase, Am. J. Physiol. Heart Circ. Physiol (2001), 10.1152/ajpheart.2001.281.6.h2289. [DOI] [PubMed] [Google Scholar]
- [263].Shishehbor MH, et al. , Association of nitrotyrosine levels with cardiovascular disease and modulation by statin therapy, J. Am. Med. Assoc (2003), 10.1001/jama.289.13.1675. [DOI] [PubMed] [Google Scholar]
- [264].Cai L, et al. , Inhibition of superoxide generation and associated nitrosative damage is involved in metallothionein prevention of diabetic cardiomyopathy, Diabetes (2005), 10.2337/diabetes.54.6.1829. [DOI] [PubMed] [Google Scholar]
- [265].Turko IV, Murad F, Protein nitration in cardiovascular diseases, Pharmacol. Rev (2002), 10.1124/pr.54.4.619. [DOI] [PubMed] [Google Scholar]
- [266].Lu N, Zhang Y, Li H, Gao Z, Oxidative and nitrative modifications of α-enolase in cardiac proteins from diabetic rats, Free Radic. Biol. Med (2010), 10.1016/j.freeradbiomed.2010.01.010. [DOI] [PubMed] [Google Scholar]
- [267].Chatham J, Marchase R, Protein O-GlcNAcylation: a critical regulator of the cellular response to stress, Curr. Signal Transduct. Ther (2010), 10.2174/157436210790226492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Qin CX, et al. , Insights into the role of maladaptive hexosamine biosynthesis and O-GlcNAcylation in development of diabetic cardiac complications, Pharmacol. Res (2017), 10.1016/j.phrs.2016.12.016. [DOI] [PubMed] [Google Scholar]
- [269].Hu Y, et al. , Increased enzymatic O-GlcNAcylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose, J. Biol. Chem (2009), 10.1074/jbc.M808518200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [270].Makino A, et al. , Regulation of mitochondrial morphology and function by O-GlcNAcylation in neonatal cardiac myocytes, Am. J. Physiol. Regul. Integr. Comp. Physiol (2011), 10.1152/ajpregu.00437.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].Gawlowski T, et al. , Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-β-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes, J. Biol. Chem (2012), 10.1074/jbc.M112.390682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [272].Banerjee PS, Ma J, Hart GW, Diabetes-associated dysregulation of O-GlcNAcylation in rat cardiac mitochondria, Proc. Natl. Acad. Sci. U.S.A (2015), 10.1073/pnas.1424017112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Erickson JR, et al. , Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation, Nature (2013), 10.1038/nature12537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [274].Bias WB, Cheung WD, Wang Z, Hart GW, Regulation of calcium/calmodulin-dependent kinase IV by O-GlcNAc modification, J. Biol. Chem (2009), 10.1074/jbc.M109.007310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [275].Goldin A, Beckman JA, Schmidt AM, Creager MA, Advanced glycation end products: sparking the development of diabetic vascular injury, Circulation (2006), 10.1161/CIRCULATIONAHA.106.621854. [DOI] [PubMed] [Google Scholar]
- [276].Wang J, Song Y, Wang Q, Kralik PM, Epstein PN, Causes and characteristics of diabetic cardiomyopathy, Rev. Diabet. Stud (2006), 10.1900/rds.2006.3.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Aragno M, et al. , Oxidative stress-dependent impairment of cardiac-specific transcription factors in experimental diabetes, Endocrinology (2006), 10.1210/en.2006-0728. [DOI] [PubMed] [Google Scholar]
- [278].Willemsen S, et al. , Tissue advanced glycation end products are associated with diastolic function and aerobic exercise capacity in diabetic heart failure patients, Eur. J. Heart Fail (2011), 10.1093/eurjhf/hfq168. [DOI] [PubMed] [Google Scholar]
- [279].Kopytek M, Ząbczyk M, Mazur P, Undas A, Natorska J, Accumulation of advanced glycation end products (AGEs) is associated with the severity of aortic stenosis in patients with concomitant type 2 diabetes, Cardiovasc. Diabetol 19 (2020) 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [280].Yu W, et al. , Curcumin alleviates diabetic cardiomyopathy in experimental diabetic rats, PloS One (2012), 10.1371/journal.pone.0052013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [281].Wang X, et al. , Engineered cardiac tissues: a novel in vitro model to investigate the pathophysiology of mouse diabetic cardiomyopathy, Acta Pharmacol. Sin (2020), 10.1038/s41401-020-00538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Yan D, et al. , Effects of advanced glycation end products on calcium handling in cardiomyocytes, Cardiol (2014), 10.1159/000364779. [DOI] [PubMed] [Google Scholar]
- [283].Sayej WN, et al. , Advanced glycation end products induce obesity and hepatosteatosis in CD-1 wild-type mice, BioMed Res. Int (2016), 10.1155/2016/7867852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [284].Anderson MM, Requena JR, Crowley JR, Thorpe SR, Heinecke JW, The myeloperoxidase system of human phagocytes generates N(ε)-(carboxymethyl) lysine on proteins: a mechanism for producing advanced glycation end products at sites of inflammation, J. Clin. Invest (1999), 10.1172/JCI3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [285].Cai W, et al. , Oral advanced glycation endproducts (AGEs) promote insulin resistance and diabetes by depleting the antioxidant defenses AGE receptor-1 and sirtuin 1, Proc. Natl. Acad. Sci. U.S.A (2012), 10.1073/pnas.1205847109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [286].Grossin N, et al. , Dietary CML-enriched protein induces functional arterial aging in a RAGE-dependent manner in mice, Mol. Nutr. Food Res (2015), 10.1002/mnfr.201400643. [DOI] [PubMed] [Google Scholar]
- [287].Diamant M, et al. , Diastolic dysfunction is associated with altered myocardial metabolism in asymptomatic normotensive patients with well-controlled type 2 diabetes mellitus, J. Am. Coll. Cardiol (2003), 10.1016/S0735-1097(03)00625-9. [DOI] [PubMed] [Google Scholar]
- [288].Scheuermann-Freestone M, et al. , Abnormal cardiac and skeletal muscle energy metabolism in patients with type 2 diabetes, Circulation (2003), 10.1161/01.CIR.0000072789.89096.10. [DOI] [PubMed] [Google Scholar]
- [289].Szabadkai G, Duchen MR, Mitochondria: the hub of cellular Ca2+ signaling, Physiology (2008), 10.1152/physiol.00046.2007. [DOI] [PubMed] [Google Scholar]
- [290].Kobayashi S, Liang Q, Autophagy and mitophagy in diabetic cardiomyopathy, Biochim. Biophys. Acta (BBA) - Mol. Basis Dis (2015), 10.1016/j.bbadis.2014.05.020. [DOI] [PubMed] [Google Scholar]
- [291].Frustaci A, et al. , Myocardial cell death in human diabetes, Circ. Res (2000), 10.1161/01.RES.87.12.1123. [DOI] [PubMed] [Google Scholar]
- [292].Cai L, et al. , Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome c-mediated caspase-3 activation pathway, Diabetes (2002), 10.2337/diabetes.51.6.1938. [DOI] [PubMed] [Google Scholar]
- [293].Cai L, Kang Y, J. Cell death and diabetic cardiomyopathy, Cardiovasc. Toxicol (2003), 10.1385/CT:3:3:219. [DOI] [PubMed] [Google Scholar]
- [294].Wu X, Huang LT, Zhou XL, Liu JC, Curcumin protects cardiomyopathy damage through inhibiting the production of reactive oxygen species in type 2 diabetic mice, Biochem. Biophys. Res. Commun (2020), 10.1016/j.bbrc.2020.05.053. [DOI] [PubMed] [Google Scholar]
- [295].Li J, et al. , Exogenous hydrogen sulfide protects against high glucose-induced apoptosis and oxidative stress by inhibiting the STAT3/HIF-1α pathway in H9c2 cardiomyocytes, Exp. Ther. Med (2019), 10.3892/etm.2019.8036. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [296].Guo S, et al. , Resveratrol attenuates high glucose-induced oxidative stress and cardiomyocyte apoptosis through AMPK, Mol. Cell. Endocrinol 412 (2015) 85–94. [DOI] [PubMed] [Google Scholar]
- [297].Wei CD, Li Y, Zheng HY, Tong YQ, Dai W, Palmitate induces H9c2 cell apoptosis by increasing reactive oxygen species generation and activation of the ERK1/2 signaling pathway, Mol. Med. Rep (2013), 10.3892/mmr.2013.1276. [DOI] [PubMed] [Google Scholar]
- [298].Oh CC, et al. , P38α mitogen-activated kinase mediates cardiomyocyte apoptosis induced by palmitate, Biochem. Biophys. Res. Commun (2014), 10.1016/j.bbrc.2014.06.023. [DOI] [PubMed] [Google Scholar]
- [299].Andrea F, et al. , Myocardial cell death in human diabetes, Circ. Res 87 (2000) 1123–1132. [DOI] [PubMed] [Google Scholar]
- [300].Munasinghe PE, et al. , Type-2 diabetes increases autophagy in the human heart through promotion of Beclin-1 mediated pathway, Int. J. Cardiol (2016), 10.1016/j.ijcard.2015.08.111. [DOI] [PubMed] [Google Scholar]
- [301].Zang H, et al. , Autophagy inhibition enables Nrf2 to exaggerate the progression of diabetic cardiomyopathy in mice, Diabetes (2020) db191176, 10.2337/db19-1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [302].Flarsheim CE, Grupp IL, Matlib MA, Mitochondrial dysfunction accompanies diastolic dysfunction in diabetic rat heart, Am. J. Physiol. Heart Circ. Physiol (1996), 10.1152/ajpheart.1996.271.1.h192. [DOI] [PubMed] [Google Scholar]
- [303].Chung J, et al. , MITOCHONDRIA-TARGETED antioxidant ameliorates diet-induced diabetes and diastolic dysfunction, J. Am. Coll. Cardiol (2013), 10.1016/s0735-1097(13)60597-5. [DOI] [Google Scholar]
- [304].Kralik PM, Ye G, Metreveli NS, Shen X, Epstein PN, Cardiomyocyte dysfunction in models of type 1 and type 2 diabetes, Cardiovasc. Toxicol 5 (2005) 285–292. [DOI] [PubMed] [Google Scholar]
- [305].Dikalova AE, et al. , Therapeutic targeting of mitochondrial superoxide in hypertension, Circ. Res (2010), 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [306].Ni R, et al. , Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy, Free Radic. Biol. Med (2016), 10.1016/j.freeradbiomed.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [307].Kelso GF, et al. , Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties, J. Biol. Chem (2001), 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- [308].Lim S, et al. , Mitochondria-targeted antioxidants protect pancreatic β-cells against oxidative stress and improve insulin secretion in glucotoxicity and glucolipotoxicity, Cell. Physiol. Biochem (2011), 10.1159/000335802. [DOI] [PubMed] [Google Scholar]
- [309].Ng LF, et al. , The mitochondria-targeted antioxidant MitoQ extends lifespan and improves healthspan of a transgenic Caenorhabditis elegans model of Alzheimer disease, Free Radic. Biol. Med (2014), 10.1016/j.freeradbiomed.2014.03.003. [DOI] [PubMed] [Google Scholar]
- [310].Chacko BK, et al. , Prevention of diabetic nephropathy in Ins2+/−AkitaJ mice by the mitochondria-targeted therapy MitoQ, Biochem. J (2010), 10.1042/BJ20100308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [311].Adlam VJ, et al. , Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury, Faseb. J (2005), 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- [312].Rossman MJ, et al. , Chronic supplementation with a mitochondrial antioxidant (MitoQ) improves vascular function in healthy older adults, Hypertension (2018), 10.1161/HYPERTENSIONAHA.117.10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [313].Ahmad I, Hoda M, Molecular mechanisms of action of resveratrol in modulation of diabetic and non-diabetic cardiomyopathy, Pharmacol. Res 161 (2020) 105112. [DOI] [PubMed] [Google Scholar]
- [314].Zhang H, et al. , Resveratrol improves left ventricular diastolic relaxation in type 2 diabetes by inhibiting oxidative/nitrative stress: in vivo demonstration with magnetic resonance imaging, Am. J. Physiol. Cell Physiol 299 (2010) H985–H994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [315].Soufi FG, Vardyani M, Sheervalilou R, Mohammadi M, Somi MH, Long-term treatment with resveratrol attenuates oxidative stress pro-inflammatory mediators and apoptosis in streptozotocin-nicotinamide-induced diabetic rats, Gen. Physiol. Biophys (2012), 10.4149/gpb_2012_039. [DOI] [PubMed] [Google Scholar]
- [316].Beaudoin MS, et al. , Impairments in mitochondrial palmitoyl-CoA respiratory kinetics that precede development of diabetic cardiomyopathy are prevented by resveratrol in ZDF rats, J. Physiol (2014), 10.1113/jphysiol.2013.270538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [317].Bagul PK, Deepthi N, Sultana R, Banerjee SK, Resveratrol ameliorates cardiac oxidative stress in diabetes through deacetylation of NFkB-p65 and histone 3, J. Nutr. Biochem 26 (2015) 1298–1307. [DOI] [PubMed] [Google Scholar]
- [318].Wang B, et al. , Resveratrol-enhanced autophagic flux ameliorates myocardial oxidative stress injury in diabetic mice, J. Cell Mol. Med (2014), 10.1111/jcmm.12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [319].Fiorani M, et al. , Mitochondria accumulate large amounts of quercetin: prevention of mitochondrial damage and release upon oxidation of the extramitochondrial fraction of the flavonoid, J. Nutr. Biochem (2010), 10.1016/j.jnutbio.2009.01.014. [DOI] [PubMed] [Google Scholar]
- [320].Castillo RL, et al. , Quercetin prevents diastolic dysfunction induced by a high-cholesterol diet: role of oxidative stress and bioenergetics in hyperglycemic rats, Oxid. Med. Cell. Longev (2018), 10.1155/2018/7239123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [321].Sun X, et al. , Taxifolin prevents diabetic cardiomyopathy in vivo and in vitro by inhibition of oxidative stress and cell apoptosis, Food Chem. Toxicol (2014), 10.1016/j.fct.2013.11.013. [DOI] [PubMed] [Google Scholar]
- [322].Packer L, Vitamin E is Nature’s master antioxidant, Sci. Med 1 (1994) 54–63. [Google Scholar]
- [323].Kaul N, Siveski-Iliskovic N, Hill M, Slezak J, Singal PK, Free radicals and the heart, J. Pharmacol. Toxicol. Methods (1993), 10.1016/1056-8719(93)90008-3. [DOI] [PubMed] [Google Scholar]
- [324].Aydemir-Koksoy A, Bilginoglu A, Sariahmetoglu M, Schulz R, Turan B, Antioxidant treatment protects diabetic rats from cardiac dysfunction by preserving contractile protein targets of oxidative stress, J. Nutr. Biochem (2010), 10.1016/j.jnutbio.2009.06.006. [DOI] [PubMed] [Google Scholar]
- [325].Li C, jun Lv L., Li H, Yu D. min, Cardiac fibrosis and dysfunction in experimental diabetic cardiomyopathy are ameliorated by alpha-lipoic acid, Cardiovasc. Diabetol (2012), 10.1186/1475-2840-11-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [326].Hamblin M, Smith HM, Hill MF, Dietary supplementation with vitamin E ameliorates cardiac failure in type I diabetic cardiomyopathy by suppressing myocardial generation of 8-iso-Prostaglandin F2α and oxidized glutathione, J. Card. Fail (2007), 10.1016/j.cardfail.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [327].Wold L, Streptozotocin directly impairs cardiac contractile function in isolated ventricular myocytes via a p38 map kinase-dependent oxidative stress mechanism, Biochem. Biophys. Res. Commun (2004), 10.1016/s0006-291x(04)00908-8. [DOI] [PubMed] [Google Scholar]
- [328].Yusuf S, Vitamin E supplementation and cardiovascular events in high-risk patients, N. Engl. J. Med (2000), 10.1056/NEJM200001203420302. [DOI] [PubMed] [Google Scholar]
- [329].Myung SK, et al. , Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: systematic review and meta-analysis of randomised controlled trials, BMJ (2013), 10.1136/bmj.f10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [330].Timimi FK, et al. , Vitamin C improves endothelium-dependent vasodilation in patients with insulin-dependent diabetes mellitus, J. Am. Coll. Cardiol (1998), 10.1016/S0735-1097(97)00536-6. [DOI] [PubMed] [Google Scholar]
- [331].Ting HH, et al. , Vitamin C improves endothelium-dependent vasodilation in patients with non-insulin-dependent diabetes mellitus, J. Clin. Invest (1996), 10.1172/JCI118394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [332].Beckman JA, et al. , Oral antioxidant therapy improves endothelial function in Type 1 but not Type 2 diabetes mellitus, Am. J. Physiol. Heart Circ. Physiol (2003), 10.1152/ajpheart.00403.2003. [DOI] [PubMed] [Google Scholar]
- [333].Chen H, et al. , High-dose oral vitamin C partially replenishes vitamin C levels in patients with Type 2 diabetes and low vitamin C levels but does not improve endothelial dysfunction or insulin resistance, Am. J. Physiol. Heart Circ. Physiol (2006), 10.1152/ajpheart.00768.2005. [DOI] [PubMed] [Google Scholar]
- [334].Economides PA, et al. , The effect of vitamin E on endothelial function of micro- and macrocirculation and left ventricular function in type 1 and type 2 diabetic patients, Diabetes (2005), 10.2337/diabetes.54.1.204. [DOI] [PubMed] [Google Scholar]
- [335].Lonn E, Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial, J. Am. Med. Assoc (2005), 10.1001/jama.293.11.1338. [DOI] [PubMed] [Google Scholar]
- [336].Gutierrez AD, de Serna DG, Robinson I, Schade DS, The response of γ vitamin E to varying dosages of α vitamin E plus vitamin C, Metabolism (2009), 10.1016/j.metabol.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [337].Dakhale GN, Chaudhari HV, Shrivastava M, Supplementation of vitamin C reduces blood glucose and improves glycosylated hemoglobin in type 2 diabetes mellitus: a randomized, double-blind study, Adv. Pharmacol. Sci (2011), 10.1155/2011/195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [338].Mazloom Z, et al. , Effect of vitamin C supplementation on postprandial oxidative stress and lipid profile in type 2 diabetic patients, Pakistan J. Biol. Sci (2011), 10.3923/pjbs.2011.900.904. [DOI] [PubMed] [Google Scholar]
- [339].Illison VK, Rondó PHC, de Oliveira AM, D’Abronzo FH, Campos KF, The relationship between plasma α-tocopherol concentration and vitamin E intake in patients with type 2 diabetes mellitus, Int. J. Vitam. Nutr. Res (2011), 10.1024/0300-9831/a000046. [DOI] [PubMed] [Google Scholar]
- [340].El-Aal AA, El-Ghffar EAA, Ghali AA, Zughbur MR, Sirdah MM, The effect of vitamin C and/or E supplementations on type 2 diabetic adult males under metformin treatment: a single-blinded randomized controlled clinical trial, Diabetes Metab. Syndr. Clin. Res. Rev (2018), 10.1016/j.dsx.2018.03.013. [DOI] [PubMed] [Google Scholar]
- [341].Jackson TS, Xu A, Vita JA, Keaney JF, Ascorbate prevents the interaction of superoxide and nitric oxide only at very high physiological concentrations, Circ. Res (1998), 10.1161/01.RES.83.9.916. [DOI] [PubMed] [Google Scholar]
- [342].Dormont F, et al. , Squalene-based multidrug nanoparticles for improved mitigation of uncontrolled inflammation in rodents, Sci. Adv (2020), 10.1126/sciadv.aaz5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [343].Gaudin A, et al. , Squalenoyl adenosine nanoparticles provide neuroprotection after stroke and spinal cord injury, Nat. Nanotechnol (2014), 10.1038/nnano.2014.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [344].Kotelevets L, et al. , A squalene-based nanomedicine for oral treatment of colon cancer, Canc. Res (2017), 10.1158/0008-5472.CAN-16-1741. [DOI] [PubMed] [Google Scholar]
- [345].Bentinger M, Brismar K, Dallner G, The antioxidant role of coenzyme Q, Mitochondrion (2007), 10.1016/j.mito.2007.02.006. [DOI] [PubMed] [Google Scholar]
- [346].Di Lorenzo A, et al. , Clinical evidence for Q10 coenzyme supplementation in heart failure: from energetics to functional improvement, J. Clin. Med (2020), 10.3390/jcm9051266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [347].Neal B, et al. , Canagliflozin and cardiovascular and renal events in type 2 diabetes, N. Engl. J. Med (2017), 10.1056/NEJMoa1611925. [DOI] [PubMed] [Google Scholar]
- [348].Zinman B, et al. , Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes, N. Engl. J. Med (2015), 10.1056/NEJMoa1504720. [DOI] [PubMed] [Google Scholar]
- [349].Wiviott SD, et al. , Dapagliflozin and cardiovascular outcomes in type 2 diabetes, N. Engl. J. Med (2019), 10.1056/NEJMoa1812389. [DOI] [PubMed] [Google Scholar]
- [350].Packer M, et al. , Cardiovascular and renal outcomes with empagliflozin in heart failure, N. Engl. J. Med (2020), 10.1056/nejmoa2022190. [DOI] [PubMed] [Google Scholar]
- [351].Lee TI, et al. , Empagliflozin attenuates myocardial sodium and calcium dysregulation and reverses cardiac remodeling in streptozotocin-induced diabetic rats, Int. J. Mol. Sci (2019), 10.3390/ijms20071680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [352].Kolijn D, et al. , Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase Gα oxidation, Cardiovasc. Res (2020), 10.1093/cvr/cvaa123. [DOI] [PubMed] [Google Scholar]
- [353].Xue M, et al. , Empagliflozin prevents cardiomyopathy via sGC-cGMP-PKG pathway in type 2 diabetes mice, Clin. Sci (2019), 10.1042/CS20190585. [DOI] [PubMed] [Google Scholar]
- [354].Qian P, et al. , A novel oral glucagon-like peptide 1 receptor agonist protects against diabetic cardiomyopathy via alleviating cardiac lipotoxicity induced mitochondria dysfunction, Biochem. Pharmacol (2020), 10.1016/j.bcp.2020.114209. [DOI] [PubMed] [Google Scholar]
- [355].Li C, et al. , SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart, Cardiovasc. Diabetol (2019), 10.1186/s12933-019-0816-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [356].Natali A, Nesti L, Fabiani I, Calogero E, Di Bello V, Impact of empagliflozin on subclinical left ventricular dysfunctions and on the mechanisms involved in myocardial disease progression in type 2 diabetes: rationale and design of the EMPA-HEART trial, Cardiovasc. Diabetol (2017), 10.1186/s12933-017-0615-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [357].Pfeffer MA, et al. , Lixisenatide in patients with type 2 diabetes and acute coronary syndrome, N. Engl. J. Med (2015), 10.1056/NEJMoa1509225. [DOI] [PubMed] [Google Scholar]
- [358].Holman RR, et al. , Effects of once-weekly exenatide on cardiovascular outcomes in type 2 diabetes, N. Engl. J. Med (2017), 10.1056/NEJMoa1612917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [359].Husain M, et al. , Oral semaglutide and cardiovascular outcomes in patients with type 2 diabetes, N. Engl. J. Med (2019), 10.1056/NEJMoa1901118. [DOI] [PubMed] [Google Scholar]
- [360].Marso SP, et al. , Liraglutide and cardiovascular outcomes in type 2 diabetes, N. Engl. J. Med (2016), 10.1056/NEJMoa1603827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [361].Marso SP, et al. , Semaglutide and cardiovascular outcomes in patients with type 2 diabetes, N. Engl. J. Med (2016), 10.1056/NEJMoa1607141. [DOI] [PubMed] [Google Scholar]
- [362].Ding W, et al. , Exenatide protects against cardiac dysfunction by attenuating oxidative stress in the diabetic mouse heart, Front. Endocrinol. (Lausanne) (2019), 10.3389/fendo.2019.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [363].Zhang L, et al. , GLP-1 receptor agonist liraglutide protects cardiomyocytes from IL-1β-induced metabolic disturbance and mitochondrial dysfunction, Chem. Biol. Interact 332 (2020) 109252, 10.1016/j.cbi.2020.109252. [DOI] [PubMed] [Google Scholar]
- [364].Al-Rasheed NM, et al. , Sitagliptin attenuates cardiomyopathy by modulating the JAK/STAT signaling pathway in experimental diabetic rats, Drug Des. Dev. Ther (2016), 10.2147/DDDT.S109287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [365].Apaijai N, Pintana H, Chattipakorn SC, Chattipakorn N, Effects of vildagliptin versus sitagliptin, on cardiac function, heart rate variability and mitochondrial function in obese insulin-resistant rats, Br. J. Pharmacol (2013), 10.1111/bph.12176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [366].Nath S, Ghosh SK, Choudhury Y, A murine model of type 2 diabetes mellitus developed using a combination of high fat diet and multiple low doses of streptozotocin treatment mimics the metabolic characteristics of type 2 diabetes mellitus in humans, J. Pharmacol. Toxicol. Methods (2017), 10.1016/j.vascn.2016.10.007. [DOI] [PubMed] [Google Scholar]
- [367].Aroor AR, et al. , Dipeptidyl peptidase-4 (DPP-4) inhibition with linagliptin reduces western diet-induced myocardial TRAF3IP2 expression, inflammation and fibrosis in female mice, Cardiovasc. Diabetol (2017), 10.1186/s12933-017-0544-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [368].Zhang X, et al. , Alogliptin prevents diastolic dysfunction and preserves left ventricular mitochondrial function in diabetic rabbits, Cardiovasc. Diabetol (2018), 10.1186/s12933-018-0803-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [369].Zhou Y, et al. , Sitagliptin protects cardiac function by reducing Nitroxidative stress and promoting autophagy in zucker diabetic fatty (ZDF) rats, Cardiovasc. Drugs Ther (2018), 10.1007/s10557-018-6831-9. [DOI] [PubMed] [Google Scholar]
- [370].Spencer NY, Yang Z, Sullivan JC, Klein T, Stanton RC, Linagliptin unmasks specific antioxidant pathways protective against albuminuria and kidney hypertrophy in a mouse model of diabetes, PloS One (2018), 10.1371/journal.pone.0200249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [371].Aroor AR, et al. , Dipeptidylpeptidase inhibition is associated with improvement in blood pressure and diastolic function in insulin-resistant male zucker obese rats, Endocrinology (2013), 10.1210/en.2013-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [372].Bostick B, et al. , Dipeptidyl peptidase inhibition prevents diastolic dysfunction and reduces myocardial fibrosis in a mouse model of Western diet induced obesity, Metabolism (2014), 10.1016/j.metabol.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [373].Miller JA, Floras JS, Zinman B, Skorecki KL, Logan AG, Effect of hyperglycaemia on arterial pressure, plasma renin activity and renal function in early diabetes, Clin. Sci (1996), 10.1042/cs0900189. [DOI] [PubMed] [Google Scholar]
- [374].Miller JA, Impact of hyperglycemia on the renin angiotensin system in early human type 1 diabetes mellitus, J. Am. Soc. Nephrol 10 (8) (1999) 1778–1785. [DOI] [PubMed] [Google Scholar]
- [375].Thomas CM, et al. , Direct renin inhibition prevents cardiac dysfunction in a diabetic mouse model: comparison with an angiotensin receptor antagonist and angiotensin-converting enzyme inhibitor, Clin. Sci (2013), 10.1042/CS20120448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [376].Sukumaran V, Tsuchimochi H, Tatsumi E, Shirai M, Pearson JT, Azilsartan ameliorates diabetic cardiomyopathy in young db/db mice through the modulation of ACE-2/ANG 1–7/Mas receptor cascade, Biochem. Pharmacol (2017), 10.1016/j.bcp.2017.07.022. [DOI] [PubMed] [Google Scholar]
- [377].Ge Q, Zhao L, Ren XM, Ye P, Hu ZY, Feature article: LCZ696, an angiotensin receptor-neprilysin inhibitor, ameliorates diabetic cardiomyopathy by inhibiting inflammation, oxidative stress and apoptosis, Exp. Biol. Med (2019), 10.1177/1535370219861283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [378].Manning PJ, et al. , The effect of rosiglitazone on oxidative stress and insulin resistance in overweight individuals, Diabetes Res. Clin. Pract (2008), 10.1016/j.diabres.2008.04.015. [DOI] [PubMed] [Google Scholar]
- [379].Chakraborty A, Chowdhury S, Bhattacharyya M, Effect of metformin on oxidative stress, nitrosative stress and inflammatory biomarkers in type 2 diabetes patients, Diabetes Res. Clin. Pract (2011), 10.1016/j.diabres.2010.11.030. [DOI] [PubMed] [Google Scholar]
- [380].Liakopoulos OJ, Kuhn EW, Slottosch I, Wassmer G, Wahlers T, Preoperative statin therapy for patients undergoing cardiac surgery, Cochrane Database Syst. Rev (2012), 10.1002/14651858.CD008493.pub2. [DOI] [PubMed] [Google Scholar]
- [381].Williamson JR, Kilo C, Ido Y, The role of cytosolic reductive stress in oxidant formation and diabetic complications, Diabetes Res. Clin. Pract (1999), 10.1016/S0168-8227(99)00034-0. [DOI] [PubMed] [Google Scholar]
- [382].Evans JL, Goldfine ID, Maddux BA, Grodsky GM, Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes, Endocr. Rev (2002), 10.1210/er.2001-0039. [DOI] [PubMed] [Google Scholar]
- [383].Teodoro JS, Rolo AP, Palmeira CM, The NAD ratio redox paradox: why does too much reductive power cause oxidative stress? Toxicol. Mech. Methods (2013) 10.3109/15376516.2012.759305. [DOI] [PubMed] [Google Scholar]
- [384].Tilton RG, et al. , Diabetes-induced glomerular dysfunction: links to a more reduced cytosolic ratio of NADH/NAD+, Kidney Int (1992) 10.1038/ki.1992.121. [DOI] [PubMed] [Google Scholar]
- [385].Goodman RP, et al. , Hepatic NADH reductive stress underlies common variation in metabolic traits, Nature (2020), 10.1038/s41586-020-2337-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [386].Wu J, Jin Z, Zheng H, Yan LJ, Sources and implications of NADH/NAD+ redox imbalance in diabetes and its complications, in: Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy, 2016, 10.2147/DMSO.S106087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [387].Giacco F, Brownlee M, Oxidative stress and diabetic complications, Circ. Res (2010), 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [388].Cotter MA, Cameron NE, Robertson S, Polyol pathway-mediated changes in cardiac muscle contractile properties: studies in streptozotocin-diabetic and galactose-fed rats, Exp. Physiol (1992), 10.1113/expphysiol.1992.sp003649. [DOI] [PubMed] [Google Scholar]
- [389].Williamson JR, et al. , Hyperglycemic pseudohypoxia and diabetic complications, Diabetes (1993), 10.2337/diab.42.6.801. [DOI] [PubMed] [Google Scholar]
- [390].Ido Y, Kilo C, Williamson JR, Interactions between the sorbitol pathway, non-enzymatic glycation, and diabetic vascular dysfunction, in: Nephrology Dialysis Transplantation, 1996, 10.1093/ndt/11.supp5.72. [DOI] [PubMed] [Google Scholar]
- [391].Steinmetz PR, Balko C, Gabbay KH, The sorbitol pathway and the complications of diabetes, N. Engl. J. Med (1973), 10.1056/nejm197304192881609. [DOI] [PubMed] [Google Scholar]
- [392].Li Q, et al. , Polyol pathway and modulation of ischemia-reperfusion injury in Type 2 diabetic BBZ rat hearts, Cardiovasc. Diabetol (2008), 10.1186/1475-2840-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [393].Velmurugan GV, Sundaresan NR, Gupta MP, White C, Defective Nrf2-dependent redox signalling contributes to microvascular dysfunction in type 2 diabetes, Cardiovasc. Res (2013), 10.1093/cvr/cvt125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [394].Cheng AS, Cheng YH, Chiou CH, Chang TL, Resveratrol upregulates Nrf2 expression to attenuate methylglyoxal-induced insulin resistance in hep G2 cells, J. Agric. Food Chem (2012), 10.1021/jf302831d. [DOI] [PubMed] [Google Scholar]
- [395].Uruno A, et al. , The Keap1-Nrf2 system prevents onset of diabetes mellitus, Mol. Cell Biol (2013), 10.1128/mcb.00225-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [396].Kumar A, et al. , Activation of Nrf2 is required for normal and ChREBPA-augmented glucose-stimulated B-cell proliferation, Diabetes (2018), 10.2337/db17-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [397].Zang HM, et al. , Nrf2 exaggerates cardiomyopathy associated with type 1 diabetes in mice, Diabetes (2018), 10.2337/db18-475-p. [DOI] [Google Scholar]
- [398].Zhao Y, Dong D, Reece EA, Wang AR, Yang P, Oxidative stress-induced miR-27a targets the redox gene nuclear factor erythroid 2-related factor 2 in diabetic embryopathy, Am. J. Obstet. Gynecol (2018), 10.1016/j.ajog.2017.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [399].Yan LJ, Pathogenesis of chronic hyperglycemia: from reductive stress to oxidative stress, Journal of Diabetes Research (2014), 10.1155/2014/137919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [400].Conti M, et al. , High levels of myocardial antioxidant defense in aging nondiabetic normotensive Zucker obese rats, Am. J. Physiol. Regul. Integr. Comp. Physiol (2004), 10.1152/ajpregu.00521.2002. [DOI] [PubMed] [Google Scholar]
- [401].Kumar S, Prasad S, Sitasawad SL, Multiple antioxidants improve cardiac complications and inhibit cardiac cell death in streptozotocin-induced diabetic rats, PloS One (2013), 10.1371/journal.pone.0067009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [402].Wu L, et al. , Glucagon-like peptide-1 ameliorates cardiac lipotoxicity in diabetic cardiomyopathy via the PPARα pathway, Aging Cell (2018), 10.1111/acel.12763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [403].Monji A, et al. , Glucagon-like peptide-1 receptor activation reverses cardiac remodeling via normalizing cardiac steatosis and oxidative stress in type 2 diabetes, Am. J. Physiol. Heart Circ. Physiol (2013), 10.1152/ajpheart.00990.2012. [DOI] [PubMed] [Google Scholar]