

Abstract
Background
Emerging evidence suggests that long noncoding RNAs (lncRNAs) play crucial roles in various cancers. In the present study, we aim to investigate the function and molecular mechanism of an up‐regulated and survival‐associated lncRNA, LINC00525, in lung adenocarcinoma (LUAD).
Methods
The expression level of LINC00525 in tissues was determined by quantitative reverse transcription polymerase chain reaction (RT‐qPCR) and in situ hybridization (ISH). The functional role of LINC00525 in LUAD was investigated using gain‐and loss‐of‐function approaches, both in vivo and in vitro. RNA pull‐down, RNA immunoprecipitation (RIP), chromatin immunoprecipitation (ChIP), triplex‐capture assay, dual‐luciferase assay, gene expression microarray, and bioinformatics analysis were used to investigate the potential underlying mechanisms involved.
Results
LINC00525 is highly expressed in LUAD cells and tissues. Survival analysis indicated that upregulation of LINC00525 was associated with poor prognosis in patients with LUAD patients. Knockdown of LINC00525 inhibited cell proliferation and cell cycle progression in vitro. In xenograft models, LINC00525 knockdown suppressed tumor growth and tumorigenesis of tumor‐bearing mice. Mechanistically, LINC00525 epigenetically suppressed p21 transcription by guiding Enhancer Of Zeste 2 Polycomb Repressive Complex 2 Subunit (EZH2) to the p21 promoter through an formation of RNA‐DNA triplex with the p21 promoter, leading to increased trimethylation of lysine 27 on histone 3 (H3K27me3) of the p21 promoter. In addition, LINC00525 repressed p21 expression post‐transcriptionally by enhancing p21 mRNA decay. LINC00525 promoted p21 mRNA decay by competitively binding to RNA Binding Motif Single Stranded Interacting Protein 2 (RBMS2).
Conclusion
Our findings demonstrate that LINC00525 promotes the progression of LUAD by reducing the transcription and stability of p21 mRNA in concert with EZH2 and RBMS2, thus suggesting that LINC00525 may be a potential therapeutic target for clinical intervention in LUAD.
Keywords: lung adenocarcinoma, LINC00525, p21, mRNA decay, RNA‐DNA triplex
We reported the dual function of LINC00525 in fine‐tuning p21 production at both the transcriptional and post‐transcriptional levels. We suggest that LINC00525 may represent a potential therapeutic target for clinical intervention in lung adenocarcinoma.
Abbreviations
- ActD
actinomycin D
- ARE
AU‐rich element
- ChIP
chromatin immunoprecipitation
- CHX
cycloheximide
- CI
confidence interval
- CISH
chromogenic in situ hybridization
- DEG
differentially expressed gene
- DMEM
Dulbecco's modified Eagle's medium
- EdU
5‐ethynyl‐20‐deoxyuridine
- FISH
fluorescence in situ hybridization
- GSEA
gene set enrichment analysis
- H3K27me3
trimethylation of lysine 27 of histone 3
- HR
hazard ratio
- IHC
immunohistochemical
- lncRNA
long noncoding RNA
- LUAD
lung adenocarcinoma
- NBT/BCIP
nitro blue tetrazolium/5‐bromo‐4‐chloro‐3‐indolylphosphate
- NSCLC
non‐small cell lung cancer
- nt
nucleotide
- OS
overall survival
- PBS
phosphate buffered saline
- PI
propidium iodide
- PRC2
polycomb repressive complex 2
- PVDF
polyvinylidene difluoride
- qPCR
quantitative PCR
- RIP
RNA immunoprecipitation
- RT‐qPCR
reverses transcription quantitative PCR
- RTCA
real‐time xCELLLigence analysis
- SDS‐PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- shRNA
short hairpin RNA
- TCGA
the Cancer Genome Atlas
- TFO
triplex‐forming oligonucleotide
- TMA
tissue microarray
- TrT
triple target site
1. BACKGROUND
Lung cancer is a leading cause of cancer‐related deaths worldwide [1]. Lung adenocarcinoma (LUAD) is currently the most prevalent histological type of lung cancer, especially in East Asia [2, 3]. Although considerable progress has been made in the diagnosis and treatment of LUAD, patients with advanced LUAD have a poor prognosis. Recurrence, metastasis, and drug resistance are the common causes of poor prognosis in patients with LUAD [4]. Thus, a better understanding of the molecular mechanisms underlying LUAD progression is the key for the development of novel biomarkers and effective therapeutic agents for LUAD.
Long noncoding RNAs (lncRNAs) are defined as transcribed RNA molecules greater than 200 nucleotides (nt) in length that lack protein‐coding capacity [5]. Accumulating evidence suggests that lncRNAs are critical regulators of gene expression at both the transcriptional and post‐transcriptional levels [6]. They influence various physiological and pathological processes, including the progression of tumors [7, 8, 9]. LncRNAs perform their functions by interacting with RNAs, DNAs, and proteins [5]. Cytoplasmic lncRNAs are capable of assembling cytoplasmic complexes and sequestering various cytosolic regulatory factors [6]. Nuclear lncRNAs regulate chromosome architecture and recruit transcriptional regulatory factors, such as polycomb repressive complex 2 (PRC2), to different chromosomal loci [10, 11]. PRC2 and its core components are responsible for the trimethylation of lysine 27 of histone 3 (H3K27me3) to silence gene transcription [12]. However, little attention has been paid to the mechanisms by which specific chromatin‐modifying enzymes are guided by lncRNAs to distinct genomic sites, and how a given lncRNA associates with specific genomic regions to alter the chromatin structure. Recent studies have demonstrated that lncRNAs are capable of generating structures, such as RNA‐DNA duplexes (also called R loops) and RNA‐DNA triplexes, to guide chromatin‐modifying enzymes to specific genomic positions [7, 12‐15]. The functions of various lncRNAs in LUAD have been reported in recent decades [16]. For example, Pan et al.[17] reported that lncRNA JPX promotes tumorigenesis and metastasis by activating Wnt/β‐catenin signaling in LUAD. Peng et al. [18] found that LINC00312 induces LUAD migration and vasculogenic mimicry by interacting with YBX1. However, the specific roles of lncRNAs in LUAD progression, and the involvement of endogenous lncRNAs in the formation of RNA‐DNA triplexes need to be investigated further.
LINC00525, a discovery lncRNA, has been found to be upregulated in a several cancers, including colorectal [19] and non‐small cell lung cancer (NSCLC) [20, 21]. These previous studies focused on the relationship between LINC00525 and miRNAs [19, 20]. However, the biological roles and mechanisms of LINC00525 in addition to sequestering microRNAs remain unknown.
In this study, we aimed to fully assess the biological functions of LINC00525 in LUAD and investigate the underlying mechanism in addition to sponging miRNAs. We also dissected the potential mechanisms of promoting LUAD progression at transcriptional as well as post‐transcriptional level. Our present findings uncover the clinical impact, biological roles, and underlying mechanisms of LINC00525 in LUAD.
2. MATERIALS AND METHODS
2.1. Cell lines, cell culture, and treatments
NSCLC cell lines (A549, H1299, PC9, and SPC‐A1) and human bronchial epithelial cell line (HBE) were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and maintained in RPMI‐1640 (Gibco, Carlsbad, CA, USA) or Dulbecco's modified Eagle's medium (DMEM; Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Gibco, Carlsbad, CA, USA) at 37°C with 5% CO2. All cell lines were authenticated and tested routinely for their authenticity and were free of mycoplasma contamination.
For p21 mRNA stability analysis, A549 cells were incubated with 5 μg/mL actinomycin D (ActD; Sigma‐Aldrich, St. Louis, MO, USA) for the indicated time periods (2, 4, 8 and 12 h). For P21 protein stability analysis, A549 cells were incubated with 50 μg/mL cycloheximide (CHX) (Sigma‐Aldrich, St. Louis, MO, USA) for the indicated time periods (2, 4, 8 and 12 h).
2.2. Tissue samples
Human LUAD tissues and paired adjacent normal tissues were collected from the Jiangsu Cancer Hospital (Jiangsu Institute of Cancer Research, Nanjing Medical University affiliated Cancer Hospital Nanjing, Jiangsu, China). All samples were obtained from surgical resection of patients with LUAD (stage I‐ IV) and reviewed by experienced pathologists at the Jiangsu Cancer Hospital. Specimens were collected, immediately frozen in liquid nitrogen after surgery, and stored at ‐80°C until use. A total of 92 pairs of LUAD and adjacent normal tissues were used to construct a tissue microarray (TMA) as described previously [22]. Thirty pairs of LUAD tissues and adjacent normal tissues were used to extract RNA. Written informed consent was obtained from all the subjects. The study protocol was approved by the Ethics Committee of the Nanjing Medical University, China.
2.3. RNA chromogenic in situ hybridization (CISH)
RNA CISH was performed to analyze LINC00525 expression in TMA using a digoxigenin‐labeled probe (5'‐GCCAAGGACCGAAGAGGAAATTGAACGA‐3'). Briefly, the sections were dewaxed and rehydrated, digested with proteinase K, fixed in 4% paraformaldehyde, and hybridized with the digoxin‐labeled probe overnight at 55°C. The samples were then incubated at 4°C overnight with an anti‐digoxin mAb (Roche, St Louis, MO, USA). Sections were stained with nitro blue tetrazolium/5‐bromo‐4‐chloro‐3‐indolylphosphate (NBT/BCIP) in the dark, mounted, and observed.
2.4. Immunohistochemical (IHC)
Serial paraffin‐embedded tissues (4 μm thick) were de‐waxed and rehydrated. Antigen retrieval was conducted in a pressure cooker for 5 min in 10 mM Tris containing 1 mM EDTA (pH 9). The sections were incubated with antibodies specific for Ki 67 (1:200, Servicebio, Wuhan, Hubei, China), P21 (1:200; Cell Signaling Technology, Danvers, Massachusetts), Cylcin D1 (1:200; Cell Signaling Technology, Danvers, Massachusetts) at 4°C, the immunodetection was then performed with DAB (diaminobenzidine) on the following day.
2.5. Fluorescence in situ hybridization (FISH)
Fluorescence in situ hybridization (FISH) assay was performed using a fluorescent in situ hybridization kit (RiboBio, Guangzhou, Guangdong, China) according to the manufacture's protocol. Briefly, A549 cells were fixed in 4% formaldehyde for 20 min and washed with PBS. The cells were then incubated with FISH probe in hybridization buffer. DAPI was used for counterstaining the nuclei, and images were obtained with microscopy.
2.6. Plasmid construction and transfection
The full‐length and antisense cDNA of human LINC00525 and short hairpin RNAs (shRNAs) targeting LINC00525 were synthesized and cloned into the pcDNA3.1 expression vector (Realgene, Nanjing, Jiangsu, China). Cells were transfected with in vitro‐synthesized siRNAs (Realgene, Nanjing, Jiangsu, China) or expression plasmids using Lipofectamine RNAi‐MAX (Invitrogen, Carlsbad, CA, USA) or Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. The sequences of the siRNAs and shRNAs used in this study are listed in Supplementary Table S1.
2.7. Real‐time xCELLLigence analysis (RTCA)
Cell proliferation was analyzed using a real‐time xCELLigence® analysis system (ACEA Biosciences, San Diego, CA, USA) according to the manufacturer's instructions. Briefly, after transfection, 5 × 103 cells were plated onto E‐Plates and incubated at 37 °C with 5% CO2, and proliferation was monitored every 15 min for at least 90 h.
2.8. Colony formation assay
Colony formation assay was used to monitor cellular clonogenic potential. Briefly, following transfection, 1.5 × 103 treated cells were plated in 6‐well plates in in triplicates. After 14 days of incubation, the cells were washed twice with phosphate buffered saline (PBS), fixed with methanol for 10 min, and stained with 0.1% crystal violet solution for 10 min prior to analysis.
2.9. 5‐ethynyl‐20‐deoxyuridine (EdU) assay
EdU assays were used to monitor cell proliferation. Briefly, LUAD cells were cultured in 96‐well plates in complete media until 80%‐90% confluent, and then treated with 50 μM EdU for 6 h to measure cell proliferation using an EdU DNA Cell Proliferation Kit (RiboBio, Guangzhou, Guangdong, China) according to the manufacturer's instructions.
2.10. Cell cycle analysis using flow cytometry
For cell cycle distribution analysis, 1 × 105 LUAD cells were fixed in ice‐cold 70% ethanol before staining with propidium iodide (PI), and analyzed on a flow cytometer (FACScan; BD Biosciences, Franklin Lake, NJ, USA) equipped with CellQuest software (BD Biosciences).
2.11. RNA extraction, reverses transcription quantitative PCR (RT‐qPCR), and quantitative PCR (qPCR)
Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacture's protocol. For the qRT‐PCR, cDNA was synthesized using a PrimeScript RT reagent kit (Takara, Beijing, China). The reaction was carried out for 15 min at 37°C and, 5 min at 85°C, and was then held at 4°C.
For the qPCR, the expression of genes was measured using PowerUpTM SYBR Green Master Mix (Vazyme, Nanjing, China) in triplicate using an Applied Biosystem Prism 7500 Fast Sequence Detection System (Applied Biosystems, Foster City, CA, USA). GAPDH, ACTB, and snRNA U6 were used as internal controls. The relative RNA amount was calculated using the 2ΔΔCt method and normalized to GAPDH. The primer sequences used for qRT‐PCR and qPCR are listed in Supplementary Table S2.
2.12. Luciferase assay
The wild‐type p21 promoter (‐1 to ‐2000) and its mutants (‐1 to ‐299), (‐300 to ‐999), and (‐1000 to ‐2000), were synthesized and cloned into the PGL3‐basic luciferase reporter plasmid (Realgene, Nanjing, Jiangsu, China). To construct the pcFLuc‐p21 3'UTR reporter plasmid, the p21 3'UTR region or AU‐rich element (ARE) mutant region of p21 were inserted into the PGL3‐basic luciferase reporter vector (Realgene, Nanjing, Jiangsu, China). To construct the ARE mutant reporter, the AUUUA motif in the p21 3'‐UTR was changed to AGGGA. Cells were transfected with a mixture of Renilla luciferase and the indicated luciferase reporter (Realgene, Nanjing, Jiangsu, China). Forty‐eight hours after transfection, the cells were harvested and luciferase activity was evaluated using the Dual‐Luciferase Assay Kit (Promega, Madison, USA) and GLOMA 96 Microplate Luminometer (Promega, Madison, Wisconsin, USA). The relative luciferase activity was normalized to Renilla luciferase activity.
2.13. Western blotting
Western blotting was performed as described previously [22]. Briefly, whole cell lysates were electrophoresed using 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) gels and were transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was then incubated with primary antibodies against P21 antibody (1:2000; Cell Signaling Technology, Danvers, Massachusetts, USA), cyclin D1 antibody (1:2000; Cell Signaling Technology, Danvers, Massachusetts, USA), cyclin E1 (1:2000; Cell Signaling Technology, Danvers, Massachusetts, USA), CDK4 antibody (1:1000; Cell Signaling Technology, Danvers, Massachusetts, USA), CDK6 antibody (1:1000; Cell Signaling Technology, Danvers, Massachusetts, USA), KI67 antibody (1:3000; Servicebio, Wuhan, Hubei, China) and EZH2 antibody (1:2000; Cell Signaling Technology, Danvers, Massachusetts, USA). The membranes were then incubated with the corresponding secondary antibodies (Cell Signaling Technology, Boston, MA, USA) and visualized using an enhanced chemiluminescence kit (Vazyme, Nanjing, Jiangsu, China).
2.14. Triplex‐capture assay
A549 cells were suspended in 1× nuclei isolation buffer (40 mM Tris‐HCl pH 7.5, 20 mM MgCl2, 4% Triton‐X100, and 1.28 M sucrose) and incubated on ice for 20 min. A549 nuclei (3 × 106 nuclei/reaction) were incubated with 2 μg biotinylated triplex‐forming oligonucleotides (TFOs) for 1 h at 30°C in 1× triplex‐forming buffer (10 mM Tris pH 7.5, 25 mM NaCl, and MgCl2). After UV (365 nm) irradiation and sonication (10 cycles, 30s on and 30 s off [Covaris, Woburn, MA USA] treatments the nuclei were centrifuged at 10,000 rpm and the supernatant was collected. The supernatants were then incubated with 50 μL streptavidin‐magnetic beads (Thermo Fisher Scientific, Waltham, Massachusetts USA) at 4°C with rotation for 1 h. The beads were washed five times with 1× Triplex‐forming buffer and then resuspended in 100 μL DNA isolation buffer (50 mM NaHCO3, 1% SDS, 200 mM NaCl, 20 mg/ml RNase A, or RNase H) for 30 min at 37°C. DNA was purified using phenol/chloroform extraction and ethanol precipitation and, qRT‐PCR was performed. The PCR primer sequences complementary to the different regions of p21 are listed in Supplementary Table S2.
2.15. Chromatin immunoprecipitation (ChIP)
Briefly, 1 × 107 cells were incubated in lysis buffer A (5 mM PIPES, 85 mM KCl, and 0.5% NP‐40) and B (1% SDS, 10 mM EDTA, and 50 mM Tris‐HCl) supplemented with protease inhibitor cocktail (Invitrogen, Carlsbad, CA, USA) and sonicated to generate chromatin fragments (100‐500 bp) using Covaris (Woburn, MA,USA). Cell lysates were treated with anti‐EZH2 (1:1000; Cell Signaling Technology, Boston, MA, USA), anti‐H3K27me3 (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), or anti‐IgG antibody (Invitrogen, Carlsbad, CA, USA) at 4℃ overnight. The supernatants were then incubated with Protein A/G magnetic beads (Invitrogen, Carlsbad, CA, USA) to pulldown the antibody‐chromatin complexes. After washing f times, the immunoprecipitated DNA was eluted, purified, and analyzed by qRT‐PCR using the Applied Biosystem Prism 7500 Fast Sequence Detection System (Applied Biosystems Inc, CA, USA).
2.16. RNA pull down and RNA immunoprecipitation (RIP)
Biotin‐labeled full‐length LINC00525 and antisense (Roche, St Louis, MO, USA) were treated with RNase‐free DNase I (Thermo Fisher Scientific, USA) and purified with the GeneJET RNA purification kit (Thermo Fisher Scientific, USA). Then the biotin‐labeled RNA was heated at 95 ℃ for 2 min and cooled on ice for 3 min followed by incubation with streptavidin agarose beads (Invitrogen, Carlsbad, CA,USA) overnight. A549 cell lysates were collected and incubated with the RNA‐capture beads (Invitrogen, Carlsbad, CA, USA) at 4 ℃ for 1 h. The beads were then washed five times with wash buffer (50 mM Tris‐HCl pH 7.4, 150 mM NaCl, 1 mM gCl2, and 0.05% NP‐40). After washing, the beads were boiled for 5 min at 95 ℃ in SDS loading buffer (Beyotime Biotechnology, Shanghai, China), separated by polyacrylamide gel electrophoresis (PAGE), and then visualized with a silver staining kit (Thermo Fisher Scientific, USA) or subjected to western blot analysis.
RIP assay was conducted using an EZ‐Magna RIP kit (Millipore, MA, USA). Briefly, 1 × 107 A549 cells were harvested and lysed with RIP lysis buffer (Millipore, MA, USA). Cell extracts were incubated with magnetic beads (Millipore, MA, USA) conjugated with anti‐EZH2 (Cell Signaling Technology, Boston, MA, USA), anti‐RBMS2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), or control anti‐IgG antibody (Millipore, MA, USA) at 4 ℃ overnight. The retrieved RNA was then subjected to qRT‐PCR analysis.
2.17. Isolation of RNA from nuclear and cytoplasmic fractions
The subcellular localization of LINC00525 was examined using the PARIS Kit (Ambion, Life Technologies, Carlsbad, CA, USA) according to the manufacture's protocol. Briefly, 1 × 107 A549 cells were resuspended in ice‐cold cell fractionation buffer and incubated for 5 min on ice. The cell suspension was then centrifuged at 4℃ for 3 min at 500 ×g. The cytoplasmic fraction was carefully aspirated from the nuclear pellets. The nuclear pellet was lysed in a cell disruption buffer. Finally, RNA was extracted from the cytoplasmic and nuclear fractions using TRIzol (Invitrogen, Carlsbad, CA, USA).
2.18. Mice and tumor formation assay
BALB/c nude female mice (4‐weeks‐old) were purchased from Vital River Laboratories (Beijing, China) and maintained according to protocols approved by the Nanjing Medical Experimental Animal Care Commission. For the tumor formation assay, control shRNA (sh‐ctrl) and sh‐LINC00525 transfected A549 cells (5 × 106 cells in 200 μL medium/PBS) were subcutaneously injected into the flank of each mouse. Tumor growth was evaluated using calipers every week following the injection. Tumor volume was calculated as (length × width2)/2. The mice were sacrificed by cervical dislocation after one month, and the tumors were excised and snap‐frozen or paraffin‐ embedded for further analysis.
2.19. Bioinformatics analysis
Clinical and RNA sequencing data pertaining to LUAD dataset consisting of 585 LUAD patients, including 56 adjacent normal tissue samples, were downloaded from the Cancer Genome Atlas (TCGA) data portal (https://portal.gdc.cancer.gov). A549 cells transfected with sh‐ctrl or sh‐LINC00525 were used for microarray analysis. The gene expression microarray experiments were performed by Capital Bio‐tech Inc (Beijing, China). The microarray data were submitted to the Gene Expression Omnibus, and can be accessed with the accession number GSE171460. The parameters Log2FC > 2 or Log2FC < ‐2 and FDR ≤ 0.05 were used to identify the differentially expressed genes (DEGs). Pathway enrichment analysis and gene set enrichment analysis (GSEA) were performed to analyze the DEGs between sh‐ctrl and sh‐LINC00525 transfected A549 cells. GSEA was performed using the gene set (v.7.1) from MSigDB (https://www.gsea‐msigdb.org/gsea/) LongTarget, a web‐based tool designed to predict DNA‐binding motifs in lncRNA sequence via Hoogsteen and reverse Hoogsteen interactions, was used to predict the potential TFOs within LINC00525 and the triple target sites (TrTs) in the p21 promoter (https://www.smu.edu.cn). The parameters used for prediction were a maximum error rate of 20, a minimum triplex length of 15 bp, and a maximum triplex length of 100 bp [23].
2.20. Statistical analysis
All data are represented as the mean ± standard deviation from at least three independent experiments. Student's t‐test or one‐way analysis of variance (ANOVA) was used to analyze parametric variables. Multivariate Cox regression was used to assess factors associated with LUAD survival. The Kaplan‐Meier method was used to evaluate the overall survival (OS). All statistical analyses were performed using SPSS 20 software (International Business Machines Corporation, Armonk, New York, USA), and statistical significance was set at P < 0.05.
3. RESULTS
3.1. Correlation between LINC00525 expression and clinical characteristics of LUAD
The expression of LINC00525 was analyzed in the TMA containing 92 pairs of LUAD and normal tissues by CISH (Figure 1A). LINC00525 was found to be significantly overexpressed in LUAD (Figure 1B). Statistical analysis revealed that LINC00525 expression was positively correlated with the TNM stage (Figure 1C‐E). Kaplan‐Meier survival curves showed that high LINC00525 expression was associated with poor OS (hazard ratio [HR] = 0.33; 95% confidence interval [CI] = 0.15‐0.74; P = 0.008; Figure 1F). Univariate and multivariate Cox proportional hazards analyses showed that high LINC00525 level was an independent prognostic factor for patients with LUAD (Figure 1G).
FIGURE 1.
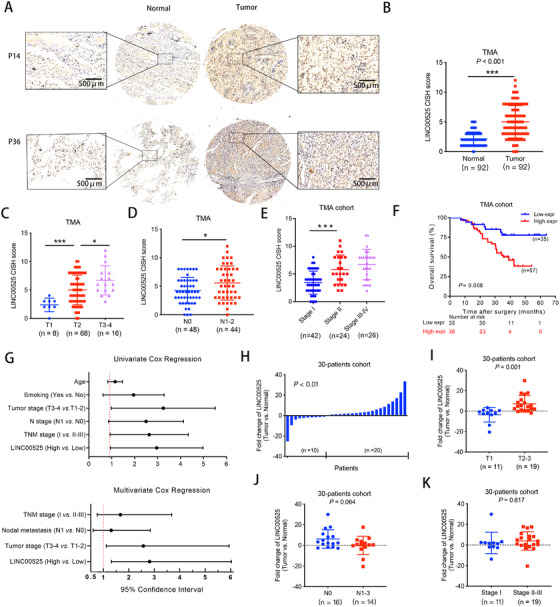
LINC00525 overexpression is associated with poor prognosis in LUAD. A. Expression of LINC00525 was analyzed by chromogenic in situ hybridization in a TMA cohort containing 92 paired LUAD tissues and adjacent normal tissues. Schematic representation of LINC00525 upregulation in LUAD and normal tissues is shown. B. The CISH results of the TMA. C‐E. Expression of LINC00525 was positively associated with tumor T stage (C), N stage (D), and overall TNM stage (E) in the TMA cohort. F. Kaplan‐Meier curves for overall survival of LUAD in the TMA cohort. Patients with high levels of LINC00525 had significantly shorter survival rates. G. Univariate and multivariate Cox regression analyses indicated that high expression of LINC00525 was an independent prognostic factor for poor survival. H. qRT‐PCR indicated that LINC00525 was upregulated in 20 pairs (out of 30 pairs) of LUAD tissues compared to adjacent normal tissues. I‐K. Correlation of LINC00525 expression in LUAD tissues (n = 30) assessed by qRT‐PCR with tumor T stage (I), N stage (J), and overall TNM stage (K). *P < 0.05; **P < 0.01; ***P < 0.001. Error bars, standard error of mean (SEM). Abbreviations: CISH, chromogenic in situ; LUAD, lung adenocarcinoma; qRT‐PCR, quantitative reverse transcription polymerase chain reaction; TMA, tissue microarray
The expression of LINC00525 was also analyzed in 30 pairs of LUAD and adjacent normal tissues by qRT‐PCR. The results showed that LINC00525 expression was significantly upregulated in LUAD tissues, consistent with the results obtained by CISH in the TMA (Figure 1H). Next, we evaluated the association between LINC00525 expression and clinical parameters. Patients with larger tumor size showed higher expression of LINC00525 (Figure 1I), but the expression of LINC00525 in N1‐3 patients and TNM stage II‐III patients was not different from that in patients with N0 (Figure 1J) and TNM stage I disease (Figure 1K). These data suggest that LINC00525 is overexpressed in LUAD tissues and positively associated with tumor size and poor prognosis.
Moreover, statistics from TCGA database indicated that LINC00525 was upregulated in various types of human cancers, such as lung, bladder, colon, rectal, and pancreatic cancers (Supplementary Figure S1A‐B), and LINC00525 overexpression was correlated with poor prognosis in LUAD (Supplementary Figure S1C‐E), which further indicated that LINC00525 may play an oncogenic role in the development and progression of various types of human cancers.
3.2. LINC00525 promoted the proliferation and cell cycle progression of LUAD cells
We examined the expression of LINC00525 in LUAD cell lines and found that compared to normal airway epithelial cells (HBE), LINC00525 was overexpressed in multiple LUAD cell lines, especially in A549 and PC9 cells (Supplementary Figure S2A). shRNAs and an expression vector of LINC00525 were used to knock down and overexpress LINC00525 in A549 and PC9 cell lines, respectively. We designed two shLINC00525 sequences (shLINC00525 #1 and #2) and found that shLINC00525 #2 exhibited a better efficacy in both A549 and PC9 cell lines. Therefore, shLINC00525 #2 was used in the subsequent assays (Figure 2A). As shown in Figure 2B, LINC00525 overexpression significantly upregulated the expression of LINC00525 in both A549 and PC9 cell lines. Gene expression microarray was performed to identify the DEG profile following LINC00525 knockdown in A549 cells. Pathway enrichment and GSEA analysis revealed that the genes that were highly co‐expressed with LINC00525 were involved in “cell cycle” and “cell proliferation” (Figure 2C‐D). Using RTCA system proliferation assay, EdU proliferation assay and colony formation assays, we determined that LINC00525 knockdown significantly impaired the proliferation ability of A549 (Figure 2E‐H) and PC9 (Supplementary Figure S2B‐E) cells, whereas ectopic expression of LINC00525 markedly enhanced cell proliferation. Flow cytometric analysis showed that knockdown of LINC00525 led to G1 phase cell cycle arrest in both A549 (Figure 2I) and PC9 cells (Supplementary Figure S2F), whereas ectopic expression of LINC00525 promoted cell cycle progression in A549 (Figure 2J) and PC9 cells (Supplementary Figure S2G). In vitro loss‐ and gain‐of‐function experiments suggested that LINC00525 promotes cell cycle progression and cell proliferation in LUAD.
FIGURE 2.
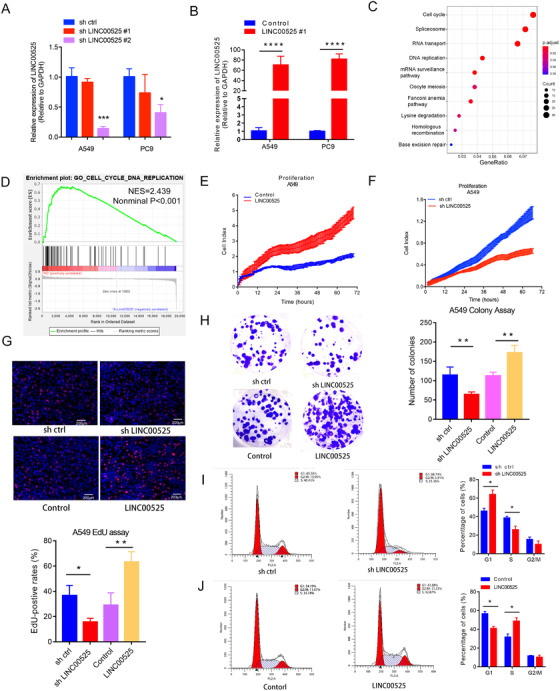
LINC00525 overexpression promotes LUAD cell proliferation. A. RNAi (shLINC00525) treatment reduced the expression of LINC00525 in both A549 and PC9 cells. B. LINC00525 overexpression increased LINC00525 expression in both A549 and PC9 cells. C‐D. KEGG pathway enrichment (C), and GSEA analysis (D) showed that the deregulated genes following LINC00525 knockdown were associated with the cell cycle. E‐F. RTCA was used to detect proliferation of A549 cells. Knockdown of LINC00525 markedly impaired, whereas overexpression of LINC00525 promoted the proliferation of A549 cells. G. Knockdown and overexpression of LINC00525 suppressed and induced the proliferation of A549 cells, respectively, as measured by EdU assay. H. Knockdown and overexpression of LINC00525 suppressed and induced clonogenicity of A549 cells, respectively. I‐J. Flow cytometric analyses were performed to analyze cell cycle progression in A549 cells. Overexpression and knockdown of LINC00525 inhibited and facilitated G1 to S transition in A549 cells, respectively. *P < 0.05; **P < 0.01; ***P < 0.001. Error bars, SEM. Abbreviations: EdU, 5‐Ethynyl‐20‐deoxyuridine; GSEA, Gene Set Enrichment Analysis; LUAD, lung adenocarcinoma; qRT‐PCR, quantitative reverse transcription polymerase chain reaction; RTCA, real‐time xCELLLigence analysis system
3.3. LINC00525 formed an RNA‐DNA triplex with p21 promoter to inhibit p21 transcription
To investigate how LINC00525 regulates cell cycle progression, we analyzed the mRNA expression of G1/S state‐related genes (p27, p21, cyclin D1, cyclin E1, Cyclin Dependent Kinase 2 (CDK2), Cyclin Dependent Kinase 4 (CDK4), and Cyclin Dependent Kinase 6 (CDK6)). As shown in Figure 3A and B, knockdown of LINC00525 led to the upregulation of p21, while ectopic expression of LINC00525 significantly downregulated p21 expression. Consistently, the expression of p21 mRNA was also significantly upregulated in the microarray analysis following LINC00525 silencing in A549 cells. Combined with the previous data, this suggests that LINC00525 may participate in the regulation of G1/S cell cycle progression. Therefore, next we determined whether LINC00525 modulates the expression of G1‐related Cyclin and CDK proteins. Western blotting revealed that the expression of P21 protein was enhanced after LINC00525 silencing, whereas ectopic expression of LINC00525 decreased the expression of P21 protein. In contrast, the expression levels of cyclin D1, cyclin E1, CDK4, and CDK6 were decreased after LINC00525 silencing, but increased after LINC00525 overexpression (Figure 3C). Furthermore, we found that the expression of LINC00525 was negatively correlated with the expression of p21 mRNA in a 50‐patient cohort from the Jiangsu Cancer Hospital (Figure 3D). Thus, we reasoned that LINC00525 may promote cell cycle by inhibiting p21expression. To further investigate the mechanisms underlying LINC00525‐induced p21 expression, we conducted fluorescence in situ hybridization (FISH) and subcellular fractionation assays and found that LINC00525 was localized both in the nucleus and cytoplasm (Figure 3E), suggesting that LINC00525 may exert its biological function at both the transcriptional and post‐transcriptional levels. To identify whether LINC00525 stimulates the transcription of p21 in the nucleus, we constructed p21‐luc promoter plasmids. The promoter luciferase assay showed an obvious increase in the transcriptional activity of the p21 promoter following LINC00525 knockdown, whereas ectopic LINC00525 expression significantly suppressed the activity of the p21 promoter fused to the luciferase reporter (Figure 3F and G).
FIGURE 3.
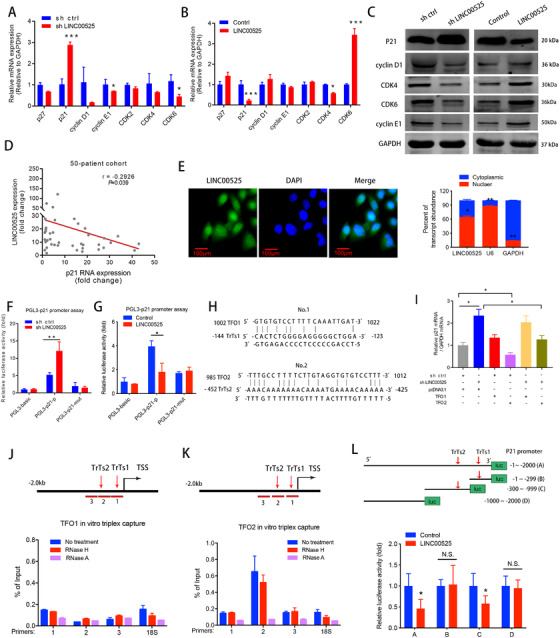
LINC00525 forms a triplex with the p21 promoter to inhibits p21 gene transcription. A‐B. qRT‐PCR analysis of the expression of G1 to S phase transition‐related genes in A549 cells following knockdown (A), or overexpression (B) of LINC00525. C. Western blotting demonstrated that knockdown of LINC00525 increased the expression of P21 protein and decreased the expressions of cyclin D1, cyclin E1, CDK4 and CDK6, whereas overexpression of LINC00525 had an opposite result. D. The expression of LINC00525 was negatively correlated with p21 mRNA as determined by qRT‐PCR in a 50‐patient cohort from Jiangsu Cancer Hospital. E, Florescence in situ hybridization (FISH), and the nuclear mass separation assays suggest that LINC00525 is distributed both in the nucleus and cytoplasm. F‐G. Dual‐luciferase reporter assays showed that LINC00525 inhibited the transcriptional activity of the p21 promoter. H. The potential binding sites of triplex‐forming oligonucleotides (TFOs) within LINC00525 and triplex target site (TrTs) in the promoter of p21 were predicted using the LongTarget program. I qRT‐PCR analysis indicated that overexpression of TFO2, but not TFO1, rescued the increase in p21 expression caused by LINC00525 knockdown. J‐K. Triplex‐capture assay was used to examine the binding of biotin‐labeled TFO1 (J) and TFO2 (K) and three diverse p21 promoter fragments in A549 cells. Triplex‐qPCR suggested that TFO2 formed a triplex structure (presented as a percentage of input) within the p21 promoter. Chromatin was pretreated with RNase A or RNase H. L. Luciferase activities of wildtype and truncated p21 promoter luciferase activities were evaluated by luciferase promoter assay in LINC00525‐overexpressing A549 cells. *, P <0.05; **P <0.01; ***P < 0.001. Error bars, SEM. Abbreviations: N.S, not significant. TFOs, triplex‐forming oligonucleotides; TrTs, triplex target sites; TSS: transcription start site; FISH, florescence in situ hybridization; qRT‐PCR, quantitative reverse transcription polymerase chain reaction
Recent studies have shown that nuclear RNA may bind to specific genomic loci to form RNA‐DNA triplex structures, which can regulate gene transcription [7, 12, 13]. To this end, the LongTarget program that predicts lncRNA‐DNA binding sites via Hoogsteen and reverse Hoogsteen interactions [23] was applied to LINC00525 and the p21 promoter sequences (‐1 to ‐2000 upstream of the transcription start site). Interestingly, two potential TFOs within LINC00525 and the corresponding TrTs in the p21 promoter were predicted (Figure 3H). Next, we subcloned the predicted TFO sequences into pcDNA3.1 vector and transfected them into A549 cells. Following LINC00525 knockdown, the expression of p21 increased (Figure 3I). Overexpression of TFO2, but not TFO1, attenuated the increase in p21 expression caused by LINC00525 knockdown (Figure 3I), suggesting that TFO2 rescues the effects of LINC00525 silencing. To further confirm the binding motifs, we performed a triplex‐capture assay using a series of primers spanning the p21 promoter (Figure 3J and K). TFO2 rather than TFO1, formed a triplex structure within the p21 promoter region. Moreover, the triplex structure was sensitive to RNase A treatment, but resistant to RNase H digestion. The former enzyme cleaves base‐paired nucleotides, whereas the latter cuts RNA in DNA‐RNA heteroduplexes [12]. These results suggest that LINC00525 TFO2 binds to the p21 promoter and the major binding regions can be mapped to fragment 2, which contains TrTs2. However, no obvious association with TrTs1 (fragment 1) was noted (Figure 3J and K). Furthermore, a mutant p21 promoter luciferase lacking TrTs2 failed to respond to ectopic LINC00525 expression (Figure 3L). Collectively, these data demonstrate that LINC00525, through the TFO2 sequence, forms a triplex with the p21 promoter via the predicted TrTs2.
3.4. LINC00525 directly interacted with EZH2 and RBMS2
Next, we performed an RNA pull‐down assay to identify LINC00525‐interacting proteins in A549 cells. Two obvious bands at 47 and 90 kDa were specifically enriched in the LINC00525 pull‐down cells (Figure 4A). EZH2 and RBMS2 were identified as two LINC00525‐interacting proteins via mass spectrometry and were detected in the anti‐biotinylated LINC00525 immunoprecipitates (Figure 4B). Consistently, the binding of LINC00525 with EZH2 and RBMS2 was further confirmed by RIP assays (Figure 4C and D). These data indicate that LINC00525 physically interacts with EZH2 and RBMS2.
FIGURE 4.
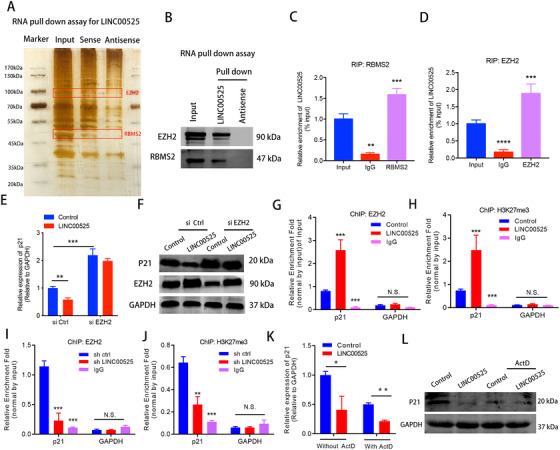
LINC00525 inhibits p21 mRNA transcription in an EZH2‐dependent manner. A. RNA pulldown assay followed by silver staining revealed proteins associated with biotinylated LINC00525. The band corresponding to LINC00525‐interacting proteins were analyzed by mass spectrometry and identified as EZH2 and RBMS2. B. EZH2, and RBMS2 recovered from the LINC00525 pulldown assay were analyzed by western blotting using the indicated antibodies. C‐D. RIP analysis using anti‐RBMS2 (C), and anti‐EZH2 (D) antibodies revealed interaction of the proteins with endogenous LINC00525 in A549 cells. E‐F. EZH2 silencing abrogated the suppressive effects of LINC00525 on p21 mRNA (E), and protein (F) levels, as shown by qRT‐PCR and western blotting. G‐H. ChIP‐qPCR showed that LINC00525 overexpression enhanced the occupancy of EZH2 and its substrate, H3K27me3, on the p21 promoter. IgG was used as the negative control. I‐J. LINC00525 silencing decreased the occupancy of EZH2 and H3K27me3 on the p21 promoter, as shown by ChIP‐qPCR. IgG was used as the negative control. K‐L. The mRNA (K) and protein (L) levels of p21 were significantly reduced in the presence of ActD as determined by qRT‐PCR and western blotting in LINC00525 overexpressing A549 cells. *P < 0.05; **P < 0.01; ***P < 0.001. Error bars, SEM. Abbreviations: RIP, RNA immunoprecipitation; ChIP, chromatin immunoprecipitation; ActD, actinomycin
3.5. EZH2 was recruited to the p21 promoter via LINC00525‐p21 triplex
Given the functional association between LINC00525 and EZH2, we reasoned that LINC00525 guides EZH2 to the promoter of p21 via the LINC00525‐p21 triplex. As shown by qRT‐PCR and western blotting, the expression of P21 was downregulated following the overexpression of LINC00525; however, EZH2 knockdown largely diminished the impact of LINC00525 on P21 expression (Figure 4E and F), suggesting that the LINC00525‐mediated p21 transcriptional inhibition is dependent on its interaction with EZH2.
EZH2, a PRC2 core component, has been reported to epigenetically regulate target gene expression through H3K27me3 histone modification [24]. To further confirm that LINC00525 inhibits p21 expression by associating with EZH2 and mediating H3K27 trimethylation, ChIP analysis was performed, which showed that LINC00525 overexpression significantly enhanced the occupancy of EZH2 and its substrate H3K27me3 on the p21 promoter (Figure 4G and H). Conversely, LINC00525 silencing decreased the occupancy of EZH2 and H3K27me3 on the p21 promoter (Figure 4I and J). Therefore, LINC00525 inhibits p21 mRNA transcription by physically bridging EZH2 and the p21 promoters. Thus, both are necessary for the accomplishment of LINC00525 activity
p21 performs its biological functions via its protein product. To investigate the contribution of LINC00525‐mediated inhibition of p21 mRNA on its protein level, we compared the effect of LINC00525 on P21 protein in the presence or absence of ActD. Interestingly, the expression of p21 mRNA and protein was significantly reduced when the ectopic expression of LINC00525 was observed in the presence of ActD (Figure 4K and L). These results suggest that in addition to transcriptional inhibition, LINC00525 may also regulate p21 mRNA stability, contributing to the reduction of P21 protein.
3.6. LINC00525 perturbed the binding of RBMS2 with p21 3'UTR to downregulate p21 mRNA stability
As shown in Figure 5A and B, the half‐life of the p21 mRNA was significantly longer following LINC00525 was silencing, whereas the half‐life of the p21 mRNA was significantly shorter in LINC00525 overexpressing A549 cells. However, the P21 protein stability was not affected following treatment with the translational inhibitor, cycloheximide (CHX) (Figure 5C). These results suggest that LINC00525 downregulates the stability of p21 mRNA, and consequently, the P21 protein synthesis inhibition. We further confirmed that the p21 mRNA stability was downregulated by LINC00525 using the p21 3'‐UTR reporter construct (Figure 5D). It has been reported that RBMS2 enhances p21 mRNA stability by directly binding to the AREs in the 3'UTR of p21 mRNA [25]. To investigate whether LINC00525‐interacted RBMS2 is involved in the regulation of p21 mRNA at a post‐transcriptional level, an RIP assay was performed in A549 cells. It was shown that RBMS2 directly bound to p21 mRNA (Figure 5E) and enhanced the stability of p21 mRNA at a post‐transcriptional level (Figure 5F). The interaction of RBMS2 with the p21 3'‐UTR was further confirmed using the p21 3'UTR or ARE mutant p21 3'‐UTR reporter constructs. Further analysis revealed that LINC00525‐mediated p21 mRNA degradation was rescued by RBMS2 overexpression (Figure 5G).
FIGURE 5.
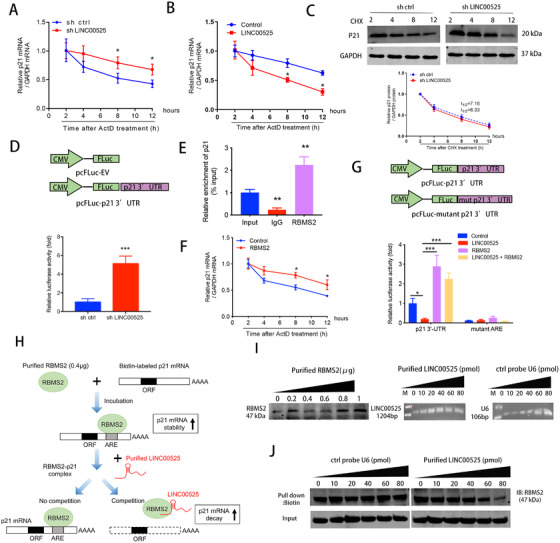
LINC00525 promotes p21 mRNA decay by competitively associating with RBMS2. A‐B. LINC00525 knockdown, LINC00525 overexpressing, and control A549 cells were incubated for the indicated times with 5 μg/mL ActD. RNA was purified and subjected to qRT‐PCR. Knockdown (A) and overexpression (B) of LINC00525 increased and reduced the half‐life of p21 mRNA, respectively. C. LINC00525 knockdown cells were treated with the translation inhibitor, CHX, after which lysates were prepared at the indicated times for western blotting (top). The band intensities were normalized using ImageJ software (bottom). D. Effect of LINC00525 on p21 3'‐UTR reporter activity. A549 cells were transfected with sh LINC00525 prior to pFL‐p21 3'‐UTR transfection, followed by luciferase reporter assay. A549 cells with LINC00525 knockdown showed increased p21 mRNA stability, as indicated by increased p21 3'UTR reporter activity. E. RIP evaluation of the interaction between RBMS2 and p21 3'UTR using an anti‐RBMS2 antibody (5 μg); IgG (5 μg) served as a negative control. F. RBMS2 overexpressing and control A549 cells were incubated with ActD for the indicated times, followed by qRT‐PCR. G. Luciferase reporters containing the p21 3'UTR region and ARE mutant region were constructed. Relative luciferase activity was measured and normalized to Renilla luciferase activity. H. Flow diagram showing the experimental design of purified LINC00525 competitively binding with purified RBMS2 at the 3'UTR of p21 mRNA. I. Amounts of purified RBMS2 protein, purified LINC00525, and control probe U6. J. Different amounts of purified LINC00525 competed with the RBMS2‐P21 complex in a dose dependent manner. *P < 0.05; **P < 0.01; ***P < 0.001. Error bars, SEM. Abbreviations: ORF, open reading frame; 3'UTR, 3'‐untranslated region; ARE, AU‐rich element; CHX, cycloheximide
To determine the interaction between p21 and LINC00525, we performed a competition assay. The biotin‐labeled p21 mRNA (1 μg) was synthesized and incubated with the purified RBMS2 protein. Purified LINC00525 produced by RNA synthesis was then added to the mixture (Figure 5H). The amount of purified RBMS2 protein, LINC00525, and control probe U6 were evaluated prior to the assays (Figure 5I). The assessment revealed that LINC00525 could abolish the binding of RBMS2 protein to the p21 3'UTR in a dose‐dependent manner (Figure 5J). Taken together, these data indicate that LINC00525 reduces p21 mRNA stability by competitively binding to RBMS2.
3.7. LINC00525 promoted LUAD proliferation by binding to EZH2 and RBMS2
To validate whether LINC00525 promotes cell proliferation by binding to EZH2 and RBMS2, we designed rescue experiments. As revealed by RTCA, EdU, and colony formation assays, EZH2 silencing or RBMS2 overexpression rescued the proliferation‐promoting effect produced by LINC00525, and the combination of EZH2 silencing and RBMS2 overexpression showed a stronger rescue effect (Figure 6A‐D). More importantly, knockdown of EZH2 or overexpression of RBMS2 partially reversed the effect of LINC00525 on p21 at both the mRNA and protein levels (Figure 6E‐F).
FIGURE 6.
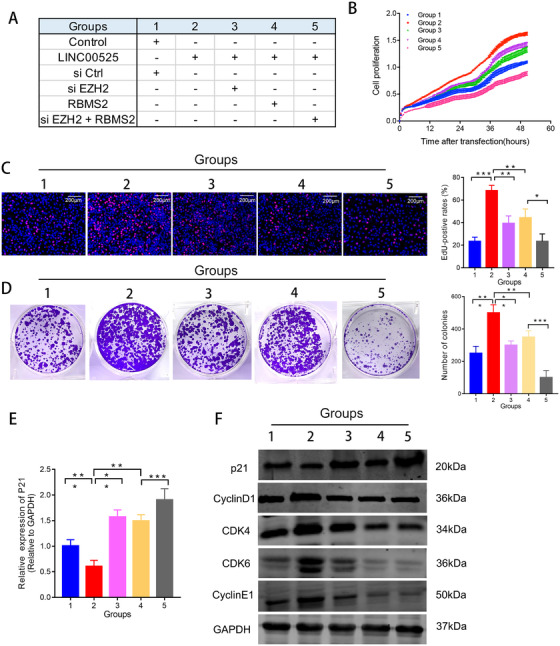
LINC00525 promotes cell proliferation by binding to EZH2 and RBMS2. A. Schematic diagram of the study design for the rescue experiments. B‐D. Knockdown of EZH2 and overexpression of RBMS2 abrogated the effects of LINC00525 on cell growth, as shown by RTCA assay (B), EdU (C), and colony formation (D) assays, respectively. E. p21 mRNA levels were determined by qRT‐PCR in A549 cells following the treatments indicated.. F. P21, cyclin D1, cyclin E1, CDK4 and CDK6 expression was evaluated by western blotting in A549 cells following the treatments indicated. *P < 0.05; **P < 0.01; ***P < 0.001. Error bars, SEM. Abbreviations: EdU, 5‐Ethynyl‐20‐deoxyuridine; qRT‐PCR, quantitative reverse transcription polymerase chain reaction; RTCA, real‐time xCELLLigence analysis system
3.8. LINC00525 promoted LUAD tumorigenesis in vivo
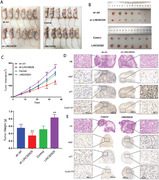
Given its role in cell proliferation, we examined the potential role of LINC00525 in vivo. In vivo xenograft model showed that LINC00525 silencing inhibited, whereas LINC00525 overexpression promoted lung cancer growth (Figure 7A‐C). Immunohistochemical (IHC) analyses revealed that tumors derived from the sh‐LINC005525 group had fewer Ki67‐ and cyclin D1‐positive cells, but more P21‐positive cells compared to the control group (Figure 7D). In contrast, tumors derived from LINC005525 overexpression group showed more Ki67‐ and cyclin D1‐positive cells, but fewer P21‐poistive cells compared to the control group (Figure 7E). Moreover, we performed a rescue experiment of P21 knockdown with LINC00525 overexpression in vivo. As shown in Supplementary Figure S3, P21 overexpression significantly abolished the tumorigenesis role of LINC00525 in vivo. These data suggested that LINC00525 may be a potential therapeutic target for lung cancer.
FIGURE 7.
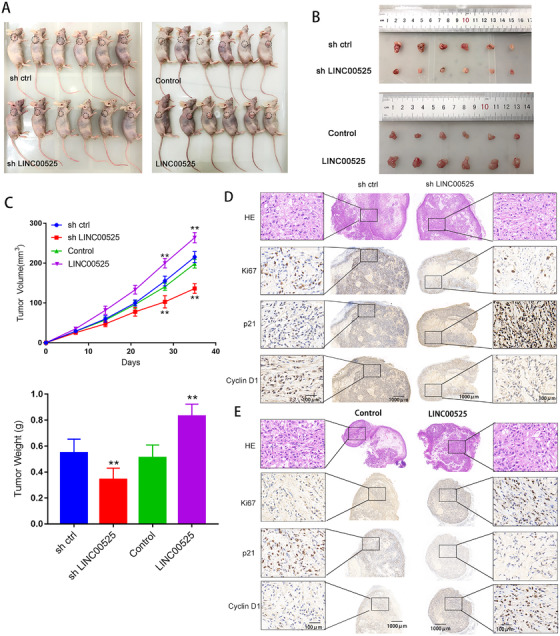
LINC00525 promotes LUAD tumorigenesis in vivo. A‐C. Xenograft tumor models showing that LINC00525 knockdown inhibited, whereas LINC00525 overexpression promoted LUADgrowth in vivo. D. Hematoxylin and Eosin and IHC stanning of xenograft tumor tissues showing that tumors derived from sh‐LINC00525 group with fewer Ki67 and cyclin D1‐postive cells, but more P21‐positvie cells. E. Hematoxylin and Eosin and IHC stanning of xenograft tumor tissues showing that tumors derived from LINC00525 group with more Ki67 and cyclin D1‐postive cells but fewer P21‐positvie cells. *P < 0.05; **P < 0.01; ***P < 0.001. Error bars, SEM. Abbreviations: ORF, open reading frame; 3'UTR, 3'‐untranslated region; IHC, Immunohistochemical
4. DISCUSSION
In this study, we found that LINC00525 promoted cell proliferation and cell cycle progression by downregulating p21 expression both at transcriptional and posttranscriptional levels. Specifically, LINC00525 knockdown suppressed tumor growth and tumorigenesis of tumor‐bearing mice, and P21 overexpression significantly abolished the tumorigenesis role of LINC00525 in vivo. LINC00525 may represent a potential therapeutic target for clinical intervention in lung adenocarcinoma.
Previous studies have reported that LINC00525 was overexpressed in CRC and NSCLC [19, 20, 21]. TCGA data demonstrated that LINC00525 was overexpressed in bladder, colon, rectum, pancreatic, and lung cancers, suggesting that LINC00525 may be a promising biomarker for pan‐cancer. However, the previous studies have focused on its interaction with miRNAs, and the biological roles and underlying mechanisms besides sequestering miRNAs are largely unknown. Here, we reported that LINC00525 promotes cell cycle progression in the G1 to S phases and plays an important role in the progression of LUAD. LINC00525 inhibited p21 gene transcription by recruiting EZH2 to the p21 promoter. Mechanistically, this suppression was achieved via the direct binding of LINC00525 with the DNA upstream of the transcription start site of p21, forming an RNA‐DNA triplex that anchored the LINC00525 and its associated EZH2 to the p21 promoter. Moreover, LINC00525 reduced p21 mRNA stability through competitive binding with RBMS2 in the cytoplasm.
FISH and subcellular fractionation assays showed that LINC00525 was localized to both the nucleus and cytoplasm. Many nuclear lncRNAs regulate gene expression via histone or DNA modifications. We found that LINC00525 physically associated with EZH2, a core component of the PRC2 histone methyltransferase complex, which mediates histone modification (H3K27me3) and represses transcription [26, 27]. Previous studies have revealed that many lncRNAs directly interact with EZH2 to silence the expression of tumor‐suppressive genes. For example, Huang et al. [28] found that lncRNA TUG1 interacts with EZH2 to silence KLF2 in hepatocellular carcinoma. Chen et al. [29] revealed that lncRNA SNHG20 associates with EZH2 to epigenetically silence p21 by mediating H3K27me3 enrichment at its promoter region. Zhu et al. [30] reported that lnc‐β‐Catm serves as a scaffold for EZH2 and β‐catenin and recruits EZH2 to the promoter region of β‐catenin. Notably, previous studies have shown that lncRNAs guide EZH2 to its target regions by interacting with sequence‐specific transcriptional factors [31, 32]. Here, we found that LINC00525 regulates the selectivity of EZH2 by forming an RNA‐DNA triplex with the promoter region of p21. In this study, triplex‐capture assays showed that LINC00525 directly interacts with TrTs2, a purine‐rich sequence located 462‐423 nt upstream of the transcription start site of p21, forming an RNA‐DNA triplex with double‐stranded DNA. As such, the LINC00525‐p21 promoter triplex guides EZH2 to its target site, contributing to the transcriptional repression of p21.
Interestingly, bioinformatics analysis has revealed that numerous triplex‐forming motifs are present across the human genome. A large number of these motifs accumulate in gene‐regulatory regions, particularly, the promoter regions, thus regulating gene expression [33]. More recently, a few lncRNAs have been identified to exhibit biological functions by forming RNA‐DNA triplexes. LncRNA Khps1 has been reported to activate the expression of the proto‐oncogene SPHK1 via triplex‐mediated changes in chromatin structure [7]. Another lncRNA, MEG3, regulates the TGF‐β pathway genes via triplex formation in GA‐rich sequences [13]. Wang et al. [12] recently reported that lncRNA HITT guided EZH2 to the promoter of HIF‐1α through RNA‐DNA triplex formation with the HIF‐1α promoter. Additionally, Chen et al. [14] found that lncRNA LNMAT1 forms an RNA‐DNA triplex that anchors its associated effector proteins to the CCL2 promoter. Collectively, these findings illustrate a prototypical mechanism that may be utilized by various lncRNAs to specifically target effector proteins.
Another important finding of our study is that LINC00525 regulates p21 mRNA stability by competitively binding to RBMS2 in the cytoplasm. Many RNA‐binding proteins have been reported to regulate p21 mRNA stability in cancer [34, 35]. RBMS2 is an RBM family member, which is downregulated in breast cancer and has been shown to inhibit the proliferation of breast cancer [25]. RBMS2 stabilizes p21 mRNA by binding to AREs of the p21 3'UTR. In our study, we determined that LINC00525 triggered p21 mRNA decay via competitive interaction with the RBMS2 protein. As shown, we demonstrated that RBMS2 protein interacts with the AREs of p21 mRNA 3'UTR to increase its stability.
Finally, we investigated the clinical relevance and the tumorigenic role of LINC00525 in vivo. RT‐PCR and CISH results showed that LINC00525 was highly expressed in LUAD tissues. High expression of LINC00525 correlates with a higher tumor grade and poor prognosis in LUAD. Depletion of LINC00525 efficiently reduces tumor size in vivo. In addition, we found a negative correlation between LINC00525 levels and p21 mRNA levels in the tumor tissues of patients with LUAD. These data indicate that LINC00525 serve as a biomarker and a promising therapeutic target for LUAD. However, there are a few limitations to the current study. Firstly, we did not identify a specific LINC00525‐interacting region with RBMS2 and EZH2. Secondly, the mechanisms underlying LINC00525 overexpression in LUAD remain unclear. Further studies are needed to identify the precise binding region of LINC00525 with the associated proteins and the upstream mechanisms of LINC00525 overexpression in LUAD.
5. CONCLUSIONS
In summary, our study provides evidence supporting the hypothesis that LINC00525 overexpression is clinically and functionally correlated with higher tumor grade and poor prognosis in LUAD. Here, we report the dual function of LINC00525 in fine‐tuning p21 expression at both the transcriptional and post‐transcriptional levels (Figure 8). In the nucleus, LINC00525 guides EZH2 to the p21 promoter via the formation of an RNA‐DNA triplex with the p21 gene promoter to suppress p21 gene transcription. In the cytoplasm, LINC00525 disturbs the binding of RBMS2 to the p21 3'UTR to downregulate p21 mRNA stability. Given the role of LINC00525 as potent, multilevel inhibitor of p21 expression in LUAD, we propose that LINC00525 may be a novel biomarker and a promising therapeutic target in LUAD.
FIGURE 8.
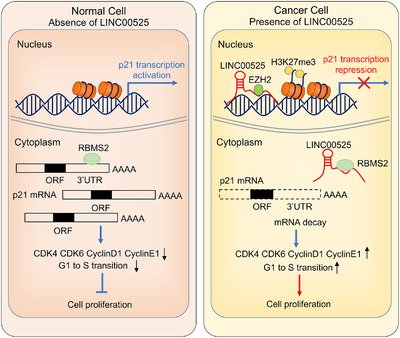
Schematic diagram showing the proposed mechanism by which LINC00525 promotes tumorigenesis of LUAD. F. LINC00525 inhibits p21 expression in two ways; it physiologically associates with EZH2 and guides it to the promoter region of p21 through the formation of a triplex with the p21 promoter in the nucleus, and it reduces p21 mRNA stability by competitively binding to RBMS2 in the cytoplasm. The downregulated P21 then activates Cyclin‐CDK complex and accelerates G1/S cell cycle transition
DECLARATIONS
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This study was approved by the Research Ethics Committee of the Nanjing Medical University (Nanjing, JiangSu, China). Written informed consent was obtained from all patients. All animal experiments were performed in accordance with the relevant guidelines and regulations.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The gene microarray data have been submitted to the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/), and the data can be accessed by the accession number GSE171460. The data that support the findings of this study are available from the corresponding author upon reasonable request.
COMPETING INTERESTS
The authors declare that they have no competing interests.
FUNDING
This work was supported by the grants from the National Natural Science Foundation of China (81802277, 81872378, and 81802907), China Postdoctoral Science Foundation (2018M642198), and Project of Jiangsu Provincial Medical Talent (ZDRCA2016033).
AUTHORS' CONTRIBUTIONS
Conception and design: TL and RY. Development of methodology: PF, HC, and ZM. Acquisition of data: PF, HC, ZM, CH, HZ, and SW. Analysis and interpretation of data: TL, PF, and HC. Writing, review, and/or revision of the manuscript: TL, and RY. Administrative, technical, or material support: WY, WX, JW, and LX. Study supervision: TL and RY. All authors read and approved the final manuscript.
Supporting information
SUPPORTING INFORMATION
SUPPORTING INFORMATION
SUPPORTING INFORMATION
SUPPORTING INFORMATION
ACKNOWLEDGMENTS
Not applicable.
Fang P, Chen H, Ma Z, Han C, Yin W, Wang S, et al. LncRNA LINC00525 suppresses p21 expression via mRNA decay and triplex‐mediated changes in chromatin structure in lung adenocarcinoma. Cancer Commun. 2021;41:596–614. 10.1002/cac2.12181
Contributor Information
Panqi Fang, Email: 1065944210@qq.com.
Hao Chen, Email: chenhaonick@163.com.
Zhifei Ma, Email: 626259564@163.com.
Chencheng Han, Email: 669707540@qq.com.
Tongyan Liu, Email: aprilliu.njmu@163.com.
Rong Yin, Email: rong_yin@njmu.edu.cn.
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. [DOI] [PubMed] [Google Scholar]
- 2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–32. [DOI] [PubMed] [Google Scholar]
- 3. Feng RM, Zong YN, Cao SM, Xu RH. Current cancer situation in China: good or bad news from the 2018 Global Cancer Statistics? Cancer Commun (Lond). 2019;39(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pompano RR, Chiang AH, Kastrup CJ, Ismagilov RF. Conceptual and experimental tools to understand spatial effects and transport phenomena in nonlinear biochemical networks illustrated with patchy switching. Annu Rev Biochem. 2017;86:333–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gil N, Ulitsky I. Regulation of gene expression by cis‐acting long non‐coding RNAs. Nat Rev Genet. 2020;21(2):102–17. [DOI] [PubMed] [Google Scholar]
- 6. Lee HC, Kang D, Han N, Lee Y, Hwang HJ, Lee SB, et al. A novel long noncoding RNA Linc‐ASEN represses cellular senescence through multileveled reduction of p21 expression. Cell Death Differ. 2020;27(6):1844–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Postepska‐Igielska A, Giwojna A, Gasri‐Plotnitsky L, Schmitt N, Dold A, Ginsberg D, et al. LncRNA Khps1 regulates expression of the proto‐oncogene SPHK1 via triplex‐mediated changes in chromatin structure. Mol Cell. 2015;60(4):626–36. [DOI] [PubMed] [Google Scholar]
- 8. Tan YT, Lin JF, Li T, Li JJ, Xu RH, Ju HQ. LncRNA‐mediated posttranslational modifications and reprogramming of energy metabolism in cancer. Cancer Commun (Lond). 2021;41(2):109–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Luo J, Langer LF, Liu J. A novel role of LncRNA in regulating tumor metabolism and angiogenesis under hypoxia. Cancer Commun (Lond). 2019;39(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. da Rocha ST, Boeva V, Escamilla‐Del‐Arenal M, Ancelin K, Granier C, Matias NR, et al. Jarid2 is implicated in the initial Xist‐induced targeting of PRC2 to the inactive X chromosome. Mol Cell. 2014;53(2):301–16. [DOI] [PubMed] [Google Scholar]
- 11. Xu M, Chen X, Lin K, Zeng K, Liu X, Pan B, et al. The long noncoding RNA SNHG1 regulates colorectal cancer cell growth through interactions with EZH2 and miR‐154‐5p. Mol Cancer. 2018;17(1):141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang X, Wang Y, Li L, Xue X, Xie H, Shi H, et al. A lncRNA coordinates with Ezh2 to inhibit HIF‐1alpha transcription and suppress cancer cell adaption to hypoxia. Oncogene. 2020;39(9):1860–74. [DOI] [PubMed] [Google Scholar]
- 13. Mondal T, Subhash S, Vaid R, Enroth S, Uday S, Reinius B, et al. MEG3 long noncoding RNA regulates the TGF‐beta pathway genes through formation of RNA‐DNA triplex structures. Nat Commun. 2015;6:7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen C, He W, Huang J, Wang B, Li H, Cai Q, et al. LNMAT1 promotes lymphatic metastasis of bladder cancer via CCL2 dependent macrophage recruitment. Nat Commun. 2018;9(1):3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang S, Ke H, Zhang H, Ma Y, Ao L, Zou L, et al. LncRNA MIR100HG promotes cell proliferation in triple‐negative breast cancer through triplex formation with p27 loci. Cell Death Dis. 2018;9(8):805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Donlic A, Zafferani M, Padroni G, Puri M, Hargrove AE. Regulation of MALAT1 triple helix stability and in vitro degradation by diphenylfurans. Nucleic Acids Res. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pan J, Fang S, Tian H, Zhou C, Zhao X, Tian H, et al. lncRNA JPX/miR‐33a‐5p/Twist1 axis regulates tumorigenesis and metastasis of lung cancer by activating Wnt/beta‐catenin signaling. Mol Cancer. 2020;19(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Peng Z, Wang J, Shan B, Li B, Peng W, Dong Y, et al. The long noncoding RNA LINC00312 induces lung adenocarcinoma migration and vasculogenic mimicry through directly binding YBX1. Mol Cancer. 2018;17(1):167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang S, Li J, Yang X. Long non‐coding RNA LINC00525 promotes the stemness and chemoresistance of colorectal cancer by targeting miR‐507/ELK3 axis. Int J Stem Cells. 2019;12(2):347–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang Z, Lin X, Zhang P, Liu Y, Liu Z, Qian B, et al. Long non‐coding RNA LINC00525 promotes the non‐small cell lung cancer progression by targeting miR‐338‐3p/IRS2 axis. Biomed Pharmacother. 2020;124:109858. [DOI] [PubMed] [Google Scholar]
- 21. Wang XW, Guo QQ, Wei Y, Ren KM, Zheng FS, Tang J, et al. Construction of a competing endogenous RNA network using differentially expressed lncRNAs, miRNAs and mRNAs in nonsmall cell lung cancer. Oncol Rep. 2019;42(6):2402–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qiu M, Xia W, Chen R, Wang S, Xu Y, Ma Z, et al. The circular RNA circPRKCI promotes tumor growth in lung adenocarcinoma. Cancer Res. 2018;78(11):2839–51. [DOI] [PubMed] [Google Scholar]
- 23. He S, Zhang H, Liu H, Zhu H. LongTarget: a tool to predict lncRNA DNA‐binding motifs and binding sites via Hoogsteen base‐pairing analysis. Bioinformatics. 2015;31(2):178–86. [DOI] [PubMed] [Google Scholar]
- 24. Laugesen A, Hojfeldt JW, Helin K. Molecular mechanisms directing PRC2 recruitment and H3K27 methylation. Mol Cell. 2019;74(1):8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sun X, Hu Y, Wu J, Shi L, Zhu L, Xi PW, et al. RBMS2 inhibits the proliferation by stabilizing P21 mRNA in breast cancer. J Exp Clin Cancer Res. 2018;37(1):298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jadhav U, Manieri E, Nalapareddy K, Madha S, Chakrabarti S, Wucherpfennig K, et al. Replicational dilution of H3K27me3 in mammalian cells and the role of poised promoters. Mol Cell. 2020;78(1):141‐51 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Long Y, Hwang T, Gooding AR, Goodrich KJ, Rinn JL, Cech TR. RNA is essential for PRC2 chromatin occupancy and function in human pluripotent stem cells. Nat Genet. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang MD, Chen WM, Qi FZ, Sun M, Xu TP, Ma P, et al. Long non‐coding RNA TUG1 is up‐regulated in hepatocellular carcinoma and promotes cell growth and apoptosis by epigenetically silencing of KLF2. Mol Cancer. 2015;14:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen Z, Chen X, Chen P, Yu S, Nie F, Lu B, et al. Long non‐coding RNA SNHG20 promotes non‐small cell lung cancer cell proliferation and migration by epigenetically silencing of P21 expression. Cell Death Dis. 2017;8(10):e3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhu P, Wang Y, Huang G, Ye B, Liu B, Wu J, et al. lnc‐beta‐Catm elicits EZH2‐dependent beta‐catenin stabilization and sustains liver CSC self‐renewal. Nat Struct Mol Biol. 2016;23(7):631–9. [DOI] [PubMed] [Google Scholar]
- 31. Battistelli C, Cicchini C, Santangelo L, Tramontano A, Grassi L, Gonzalez FJ, et al. The Snail repressor recruits EZH2 to specific genomic sites through the enrollment of the lncRNA HOTAIR in epithelial‐to‐mesenchymal transition. Oncogene. 2017;36(7):942–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pruszko M, Milano E, Forcato M, Donzelli S, Ganci F, Di Agostino S, et al. The mutant p53‐ID4 complex controls VEGFA isoforms by recruiting lncRNA MALAT1. EMBO Rep. 2017;18(8):1331–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buske FA, Bauer DC, Mattick JS, Bailey TL. Triplexator: detecting nucleic acid triple helices in genomic and transcriptomic data. Genome Res. 2012;22(7):1372–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Niu J, Zhao X, Liu Q, Yang J. Knockdown of MSI1 inhibited the cell proliferation of human osteosarcoma cells by targeting p21 and p27. Oncol Lett. 2017;14(5):5271–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu BQ, Jiang Y, Zhu F, Sun DL, He XZ. Long noncoding RNA PVT1 promotes EMT and cell proliferation and migration through downregulating p21 in pancreatic cancer cells. Technol Cancer Res Treat. 2017;16(6):819–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPORTING INFORMATION
SUPPORTING INFORMATION
SUPPORTING INFORMATION
SUPPORTING INFORMATION
Data Availability Statement
The gene microarray data have been submitted to the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/), and the data can be accessed by the accession number GSE171460. The data that support the findings of this study are available from the corresponding author upon reasonable request.