

Abstract
Although American men of European ancestry represent the largest population of prostate cancer (PCa) patients, men of African ancestry are disproportionately affected by PCa with higher prevalence and worse outcomes. These racial disparities in PCa are due to multiple factors, but variations in genomic susceptibility such as single nucleotide polymorphisms (SNP) may play an important role in determining cancer aggressiveness and treatment outcome. Using public databases, we have identified a PCa susceptibility SNP at an intronic enhancer of the NEDD9 gene, which is strongly associated with increased risk of patients with African ancestry. This genetic variation increased expression of NEDD9 by modulating the chromatin binding of certain transcription factors, including ERG and NANOG. Moreover, NEDD9 displayed oncogenic activity in PCa cells, promoting PCa tumor growth and metastasis in vitro and in vivo. Together, our study provides novel insights into the genetic mechanisms driving PCa racial disparities.
Keywords: NEDD9, susceptibility-associated genetic variation, prostate cancer, racial disparity, metastasis
INTRODUCTION
Prostate cancer (PCa) is one of the leading causes of cancer-related death in American men. The incidence and mortality of PCa are greater in men of African ancestry (AA) than European ancestry (EA) (1). While some of these disparities may be due to socioeconomic differences, the genetic differences between these racial subgroups also play an important role in determining the cancer aggressiveness and the outcome of the treatments (1–3). Through analyses of public databases, our previous study has identified a small panel of PCa risk-associated single nucleotide polymorphisms (SNPs) with significant differences of allele frequency in African men versus non-African men (4). The top-ranked SNP, rs4713266, located at an intronic region of the NEDD9 gene (Neural precursor Expressed, Developmentally Down-regulated 9) (5) was also previously identified in a large PCa GWAS analysis (6) and the frequency of the risk allele is significantly increased in AA men.
NEDD9 encodes a member of the Cas family of signaling transduction molecules, which is a non-catalytic focal adhesion protein phosphorylated by FAK and Src and functions as a signaling hub to regulate downstream signaling, such as the AKT pathway. In cancers, NEDD9 regulates cell migration, epithelial-mesenchymal transition (EMT), invasion, and metastasis (7–10). Importantly, gene amplification of NEDD9 is commonly found in the advanced metastatic castration-resistant PCa (~3%) and neuroendocrine PCa (~15%) (11,12), suggesting NEDD9 may play an important role in PCa progression.
The intronic region of NEDD9 that harbors rs4713266 is highly enriched for enhancer histone marks, indicating that this chromatin region may contain a putative enhancer. Using the CRISPR editing method to modify the nucleotide of rs4713266 in PCa cells, we show that NEDD9 expression was increased by converting the non-risk variant (T) to the risk variant (C). Mechanistically, we demonstrated that this genetic variation altered the binding and activity of multiple transcription factors, including ERG and NANOG. Although a NEDD9 germline variant has been reported being associated with PCa susceptibility (6), and NEDD9 is thought to regulate EMT and invasion in PCa cells (10), it remains unclear whether NEDD9 is involved in PCa development in vivo. Using VCaP and MDA-PCa-2b PCa models, we found that silencing NEDD9 repressed cell growth, migration, and invasion in vitro, and tumor growth and metastasis in vivo. Overall, our results indicate that increased expression of NEDD9 by the genetic variation of rs4713266 may drive PCa initiation and progression in AA men.
MATERIALS AND METHODS
Cell line and cell culture:
VCaP, CWR22-RV1, LNCaP, PC-3, and MDA-PCa-2b cell lines were purchased from ATCC and were authenticated every six months using short tandem repeat (STR) profiling and tested for mycoplasma contamination using MycoAlert mycoplasma detection kit (Lonza). VCaP and its derived stable cell lines were cultured in DMEM medium supplemented with 10% FBS (fetal bovine serum, Gibco). CWR22-RV1 and its isogenic stable lines and LNCaP derived stable cell lines were cultured in RPMI-1640 medium with 10% FBS. PC-3 cells were cultured in F-12K medium with 10% FBS. MDA-PCa-2b cells were cultured in BRFF-HPC1 medium with 20% FBS.
Chromatin immunoprecipitation (ChIP):
For the preparation of ChIP, dispensed cells were formalin-fixed, lysed, and sonicated to break the chromatin into 500–800 bp fragments, followed by immunoprecipitation with ChIP grade antibodies: anti-H3K4me1 (ab8895, Abcam), anti-H3K4me2 (07–030, Millipore), anti-H3K27ac (ab4729, Abcam), anti-p-Pol2-S5 (ab5131, Abcam), anti-ERG (ab110639, Abcam), anti-HA (ab9110, Abcam), and Rabbit/Mouse IgG (Millipore). The qPCR analysis was carried out using the SYBR Green method on QuantStudio 3 Real-time PCR system (Thermo Fisher Scientific). The primers for KLK3-Enh were previously listed (13) and other primers are NEDD9-Int1Enh Forward: 5’-AGGGTGAAATTGTTGTACTTAGAGA-3’, Reverse: 5’-TCCTGCTGTCTGTCTCCCAT-3’; NEDD9-S1 (ERG binding site) Forward: 5’-AAGCCTCACAGCCAGAGACT-3’, Reverse: 5’-CCGATCTGCGTCATTATCCT-3’; NEDD9-S2 (ERG binding site) Forward: 5’-ATGTCCAAGTCACTCAAGCC-3’, Reverse: 5’-TTTAAATAGGGAGGGGCAGGC-3’.
RT-PCR and immunoblotting:
The expression of mRNA was measured using real-time RT-PCR analyses with Taqman Fast one-step Mix RT-PCR reagents (Thermo Fisher Scientific) on QuantStudio 3 Real-time PCR system. The results were normalized to co-amplified GAPDH. The primer and probe sets for NEDD9, ERG, NANOG, and GAPDH were purchased as an inventoried mix from Applied Biosystems (Thermo Fisher Scientific). For immunoblotting, cells were lysed with RIPA buffer containing protease inhibitor cocktail (Thermo Fisher Scientific) and anti-ERG (ab110639, Abcam), anti-NEDD9 (ab18056, Abcam), anti-NANOG (3580, Cell Signaling), anti-p-Akt S473 (4060, Cell Signaling), anti-Akt (9272, Cell Signaling), anti-HA (H3663, Sigma), anti-GAPDH (ab8245, Abcam) were used. Immunoblotting results shown are representative of at least 3 independent experiments.
CRISPR/Cas9 Editing:
For the CRISPR gene activation approach, CWR22-RV1 cells were stably infected with lentiviral dCas9-VPR which expresses catalytically deactivated Cas9 fused to VP64, p65, and Rta transcription activator complex (Dharmacon), followed by blasticidin selection. The established stable cells were then infected with the lentiviral sgRNAs (52963, Addgene): sg-266 against the rs4713266 region (5’-AGTCACTCAAGCCTCACAGC-3’); sg-334 against the rs2018334 region (5’-GAAGGAACTTTAAACAATTA-3’); sg-NE against a nearby non-enhancer region (5’-CCGCACTGCTTAACACTCTC-3’), followed by puromycin selection.
For the CRISPR deletion approach, Cas9-expressing parental CWR22-RV1 cells were generated by stably infecting CWR22-RV1 cells with lentiviral Cas9-expressing vector (52962, Addgene) and selected with blasticidin. The cells were then stably infected with a pair of lentiviral sgRNAs (52963, Addgene) targeting the 5’ and 3’ boundary sequences of NEDD9-Int1Enh (sg-Int1Enh-5’: 5’-TAACAGCCAAATGAATTTCA-3’; sg-Int1Enh-3’: 5’-CCAACTCCTCCCTGCCTTTT-3’), followed by puromycin selection.
For the CRISPR nucleotide editing approach, Cas9-expressing CWR22-RV1 cells were first transfected with a single strand DNA template that contains a 60-nucleotide fragment of either the non-risk T allele or risk C allele for 24 hours (h) and then infected with a lentiviral sgRNA (Dharmacon) targeting rs4713266 site (5’-AGCCTCACAGCCAGAGACTA-3’), followed by puromycin selection. Colonies were then screened for genotyping of rs4713266. Cell clones with unsuccessful editing were selected as control lines (C/T heterozygous).
RNAi and transfection:
siRNAs against NEDD9, ERG, NANOG, and non-target control (NTC) were directly purchased (ON-TARGETplus, Dharmacon). The transfection reagent used in this study was lipofectamine 2000 (Thermo Fisher Scientific). VCaP-shNEDD9 and VCaP-shNTC cells were generated by stable infection of lentiviral shRNA against NEDD9 or NTC (Dharmacon), followed by puromycin selection and confirmation for NEDD9 expression.
RNA-Seq:
For RNA-seq analysis, VCaP-shNTC and VCaP-shNEDD9 cells were harvested for RNA extraction and followed by RNA-Seq library preparation with TruSeq Stranded RNA LT Kit (Illumina). Sequencing was performed on HiSeq 2500 Illumina Genome Analyzer. The single-end reads were processed by FastQC and aligned by STAR (version 2.5) (14) to the human Ensemble genome (Ensembl, GRCh38) with all default parameters. featureCounts (15) from Subread package was used to assign sequence reads to the genomic features. edgeR (16) was used for differential expression analysis and the list of differentially expressed genes was generated using 2-fold cut-off and P < 0.01 (FDR). The GEO accession for RNA-seq is GSE164531.
Cell proliferation assay:
Cells were stained with Muse Count & Viability Assay kit for 5 minutes and then counted by Muse® Cell Analyzer (EMD Millipore).
Luciferase reporter assay:
The reporters were created by inserting ~300bp or ~800bp DNA fragments around rs4713266 (C or T allele), or ~800bp DNA fragment around rs2018334 (G or A allele) into a pGL3 Firefly luciferase reporter vector containing a minimum promoter (E1761, Promega). PC-3 cells were then transfected with these reporters, ERG or NANOG expressing pcDNA3.1 plasmid, and a Renilla luciferase reporter. The activities of Firefly luciferase and Renilla luciferase were measured using the dual-luciferase reporter assay (Promega) and the results were normalized for Renilla activities.
Mouse xenograft:
VCaP-shNTC and VCaP-shNEDD9 xenograft tumors were established in the flanks of castrated male SCID mice (4–6 weeks) by injecting ~2 million cells mixed with 50% Matrigel. Tumor volume was measured by manual caliper using the formula V= (W2xL)/2. All animal experiments were approved by the UMass Boston Institutional Animal Care and Use Committee (IACUC) and were performed following institutional and national (USA) guidelines. The housing conditions were ambient temperatures of 65–75°F with 40–60% humidity and 12h light/12h dark cycle.
Invasion Assay:
Invasion assays were performed with Corning® BioCoat™ Matrigel® Invasion Chambers (354480, Corning). Per the manufacturer’s protocol, in brief, the same number of VCaP cells were seeded in the pre-moisturized upper chamber with serum free medium and the lower chamber was filled with medium containing 10% FBS as the chemoattractant. After three days, non-invading cells were removed by cotton swab and the invaded cells were fixed with 100% methanol and then stained with Giemsa staining solution (Fisher). All experiments were done in biological triplicates and images were taken by EVOS auto fluorescence microscope (Thermo Fisher Scientific).
Migration Assay:
Transwell migration assays were performed with Corning® FluoroBlok™ Inserts (351152, Corning). Per the manufacturer’s protocol, the same number of MDA-PCa-2b cells were seeded in the pre-moisturized upper chamber with serum free medium and the lower chamber was filled with medium containing 20% FBS as a chemoattractant. After two days, migrated cells were stained by Corning Calcein AM Fluorescent Dye (354217, Corning). All experiments were done in biological triplicates and images were taken by EVOS auto fluorescence microscope.
Zebrafish embryo metastasis assay:
Zebrafish embryos were generated from AB and TUE wild-type lines by natural spawning. All experiments were performed in 2–3-day post-fertilization embryos following an IACUC-approved protocol. Cell injections were performed as previously described (17,18). In brief, 2dpf embryos were dechorionated and anesthetized with 0.04 mg/ml tricaine and ~100 GFP-expressing cells were microinjected into the perivitelline space of each embryo using a borosilliac micropipette. After injection, embryos were washed to remove tricaine and then maintained in 96-well plates at 28°C. Embryos were imaged immediately after injection and then every hour till up to 24h.
Transcription factor binding prediction:
The database of TF binding motifs was downloaded from Jaspar. To identify potential TFs that may favor the binding to the risk allele of rs4713266 region, we used MotifDb R package. Each TF was scanned using their corresponding positions weight matrix to match the DNA region (including the complementary strand sequence) with a length of 4, 5, and 6 for T/C nucleotide (risk and non-risk alleles) and ranked by calculating the ratio of the matching score on the risk (cut-off >0.8) versus non-risk allele. The TFs that bind similarly to both risk and non-risk alleles were then removed.
Statistical analysis:
Data in bar graphs represent mean±SD of at least 3 biological repeats. Statistical analyses were generally performed using unpaired two-tailed Student’s t-test by comparing treatment versus vehicle control or otherwise as indicated. P-value<0.05 (*) was considered to be statistically significant. For animal studies, a two-tailed Student’s t-test was performed to determine the statistical difference of tumor growth at the final time point.
RESULTS
Identification of rs4713266 as the top-ranked AA-associated PCa risk SNP
Our previous analyses using several public databases including GWAS have identified a list of 38 SNPs (Supplementary Fig. 1) that are associated with increased PCa susceptibility and also have greater risk allele frequencies in African versus non-African populations (4). We then re-ranked these SNPs based on the correlation with PCa incidences in three racial groups from two studies (gnomAD and 1000Genomes) and identified rs4713266 (risk allele genotype C, non-risk allele genotype T) as the top-ranked AA-associated SNP (Fig. 1A). This SNP falls in a recently reported susceptibility locus of PCa at the chromatin region 6p24.2, which is located at an intronic region of the NEDD9 gene (6,19). The frequency of risk alleles (~80% in AA, ~50% in EA, and ~20% in East Asian men) significantly correlates with the PCa incidence and aggressiveness of racial subgroups. Susceptibility loci often contain multiple linkage disequilibrium (LD) variants. Using the HaploReg tool, we also identified a nearby rs2018334 (~1kb apart) as the LD variant of rs4713266 (r2=0.99), which is also highly correlated with the racial disparity in PCa incidence (20).
Figure 1. Identification of rs4713266 as the top-ranked AA-associated PCa risk SNP.
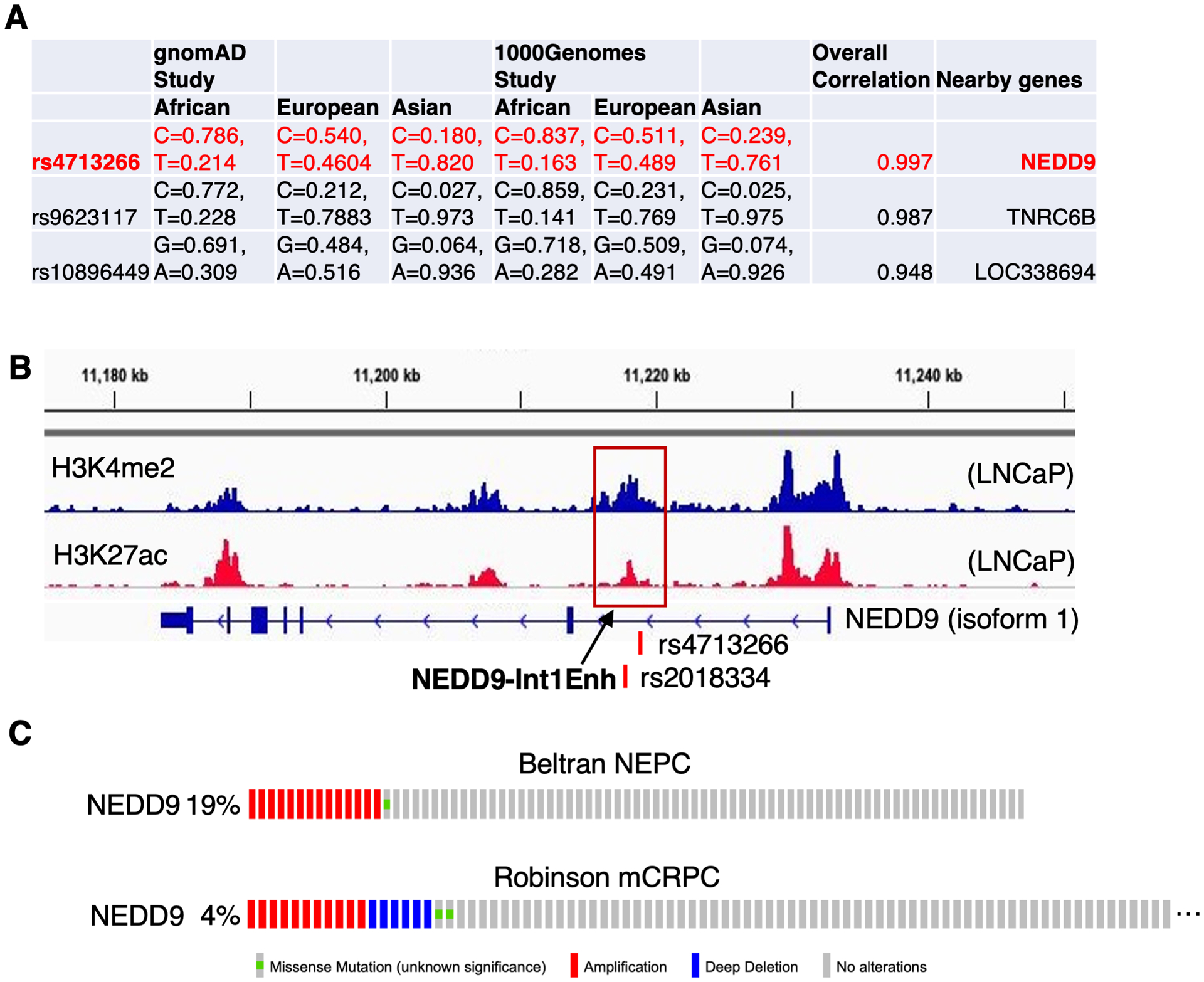
(A) Top three candidate SNPs identified from GWAS are associated with PCa risk and enriched in AA. (B) rs4713266 is mapped to a non-coding region of NEDD9 which is enriched for enhancer histone marks, H3K27ac and H3K4me2 (we named this enhancer NEDD9-Int1Enh). (C) NEDD9 amplification was found from two PCa cohorts.
The intronic region that contains these two SNPs is highly enriched for multiple enhancer marks, including H3K4me1/2, H3K27ac, and DNase hypersensitivity (20), suggesting it may be a putative enhancer of the NEDD9 gene. Searching public datasets of eQTL, we found that both SNPs are significantly associated with the expression of NEDD9 in peripheral blood samples (21), indicating that this susceptibility locus may be involved in the regulation of NEDD9 expression in PCa. Using our published ChIP-seq datasets in PCa cells (22), we show that the chromatin region overlapped with these variations were marked by high levels of H3K27ac and H3K4me2, suggesting that this intron region may contain a putative enhancer (named NEDD9-Int1Enh) and these two identified risk SNPs may play a role in differentially regulating the activity of this enhancer in PCa cells (Fig. 1B). Moreover, NEDD9 amplifications were also commonly found in the advanced metastatic castration-resistant PCa (mCRPC) and neuroendocrine PCa (NEPC) (11,23), suggesting increased NEDD9 expression may be required for PCa progression in a subset of patients (Fig. 1C).
Converting T to C at the rs4713266 site increases the expression of NEDD9 in PCa cells
To identify proper cell line models for the subsequent studies, we genotyped a panel of commonly used PCa cell line and xenograft models and found that two CRPC lines, VCaP and PC-3, have predominant risk alleles (C-allele) (Fig. 2A). Interestingly, VCaP cells always contain a low level of non-risk allele (T-allele) (Supplementary Fig. 2A and 2B). Examining a PCa cell line and patient sample dataset (24), we found that VCaP cells contain gene amplification of NEDD9 (Fig. 2B), which appeared to preferentially amplify the risk-allele. To further determine whether variations of these two SNPs may regulate NEDD9 expression, we selected CWR22-RV1 cell line (C/T heterozygous) for the subsequent gene editing studies using a series of CRISPR/Cas9 approaches. First, we made a genomic deletion of NEDD9-Int1Enh (~2kb) by using two lentiviral sgRNAs against the 5’ and 3’ boundaries of this enhancer in an established CWR22-RV1 cell line that expresses active Cas9 (25) (Supplementary Fig. 3A and 3B). As shown in Fig. 2C, the deletion decreased the mRNA expression of NEDD9 (~40%), indicating that this chromatin region is indeed an active enhancer regulating NEDD9 transcription. Second, to determine which SNP is the causal SNP we performed the CRISPR activation approach by stably expressing a catalytically dead Cas9 (dCas9) fused with an activator complex (VPR) and subsequently infecting cells with a lentiviral sgRNA against rs4713266 region (sg-266), rs2018334 region (sg-334), or a nearby non-enhancer region (sg-NE, negative control). As shown in Fig. 2D, only the activator complex bound at rs4713266 region (sg-266) increased the expression (~1.7 fold) of NEDD9, suggesting that rs4713266 but not rs2018334 is the causal SNP. Therefore, we only focused on rs4713266 for subsequent studies. Next, we directly alternated nucleotide sequence of rs4713266 in the Cas9-expressing CWR22-RV1 cell line by infecting cells with a lentiviral sgRNA against the SNP region and by cotransfecting a single strand template DNA oligo (containing T or C), followed by the selection for stable clones containing homozygous risk alleles (C/C) or non-risk alleles (T/T) (Fig. 2E and Supplementary Fig. 3C). As shown in Fig. 2F, the “C/C” line had a noticeable increase of H3K27ac and H3K4me2 at NEDD9-Int1Enh, indicating this enhancer is more active. Importantly, the alteration from the heterozygous C/T to the homozygous C/C resulted in a significant increase of NEDD9 mRNA expression and its coded protein expression (Fig. 2G and 2H). Together, these results demonstrated that T to C nucleotide editing at rs4713266 can increase NEDD9 expression.
Figure 2. Converting T to C at the rs4713266 site increases the expression of NEDD9 in PCa cells.
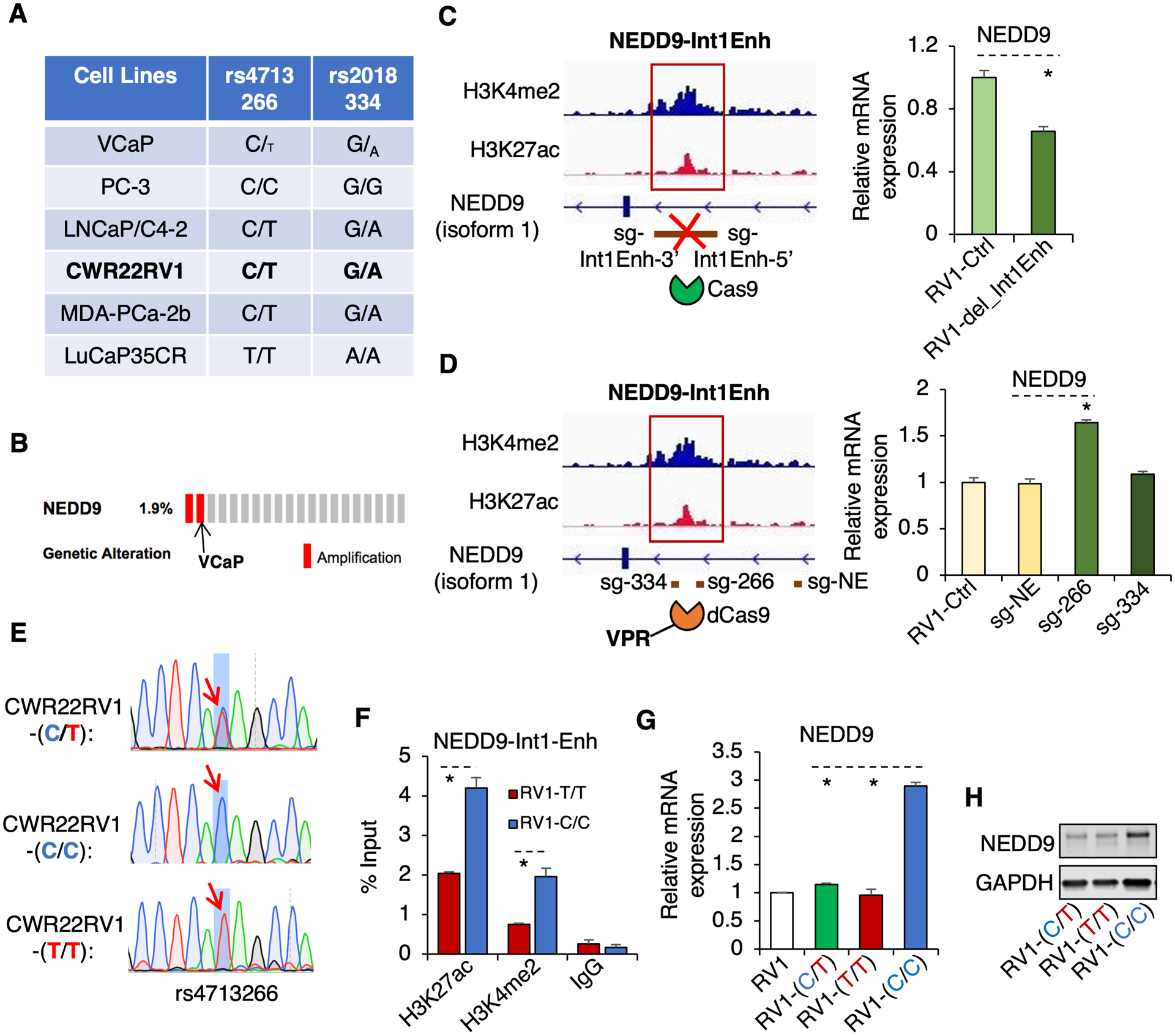
(A) Multiple PCa cell or xenograft lines were genotyped for rs4713266 and rs2018334 by Sanger sequencing. (B) Data acquired from cBioPortal indicated that the NEDD9 gene is amplified in VCaP cells. (C) Cas9-expressing CWR22-RV1 cells were stably infected with lentiviral sgRNAs against 5’ and 3’ boundary sites of NEDD9-Int1Enh. NEDD9 expression was then examined by qRT-PCR in these cells in comparison with parental cells. (D) CWR22-RV1 cells were stably infected with catalytically dead Cas9 fused with VPR, followed by the subsequent infection of lentiviral sgRNAs against the nearby sequences of rs4713266 (sg-266), rs2018334 (sg-334), or a non-enhancer region (sg-NE). NEDD9 expression was then examined by qRT-PCR in these cells in comparison with the parental cells. (E) CWR22-RV1 cells stably expressing Cas9 were established, followed by the infection of lentiviral sg-Int1Enh and the transfection of a single-strand DNA template that contains C or T at rs4713266. The C/C, T/T, or T/C isogenic cell lines were subsequently screened and selected. (F) ChIP-qPCR for H3K4me2 or H3K27ac at NEDD9-Int1Enh in the T/T cell line versus C/C cell line. (G, H) qRT-PCR for NEDD9 mRNA (G) and immunoblotting for NEDD9 protein (H) in these isogenic cell lines.
ERG preferentially activates the non-risk allele of NEDD9-Int1Enh
The above results suggest that NEDD9-Int1Enh is an enhancer involved in the regulation of NEDD9 expression. Therefore, the nucleotide variation at rs4713266 (C versus T) may result in differential recruitment and activity of transcription factors (TFs). Interestingly, searching public ChIP-seq databases for known TFs in PCa cells, we found that rs4713266 was overlapped with an ERG binding site (based on ERG ChIP-seq in VCaP cell line) (26) and a putative ERG binding motif was found to match the T-allele sequence near the SNP (Fig. 3A). ERG is a member of the ETS transcription factor family and the overexpression of ERG in PCa cells is primarily due to the chromosomal rearrangements that generate TMPRSS2-ERG fusion, which fuses the 5’ untranslated region of an androgen-regulated TMPRSS2 gene to the coding region of ERG gene (27).
Figure 3. ERG preferentially activates the non-risk allele of NEDD9-Int1Enh.
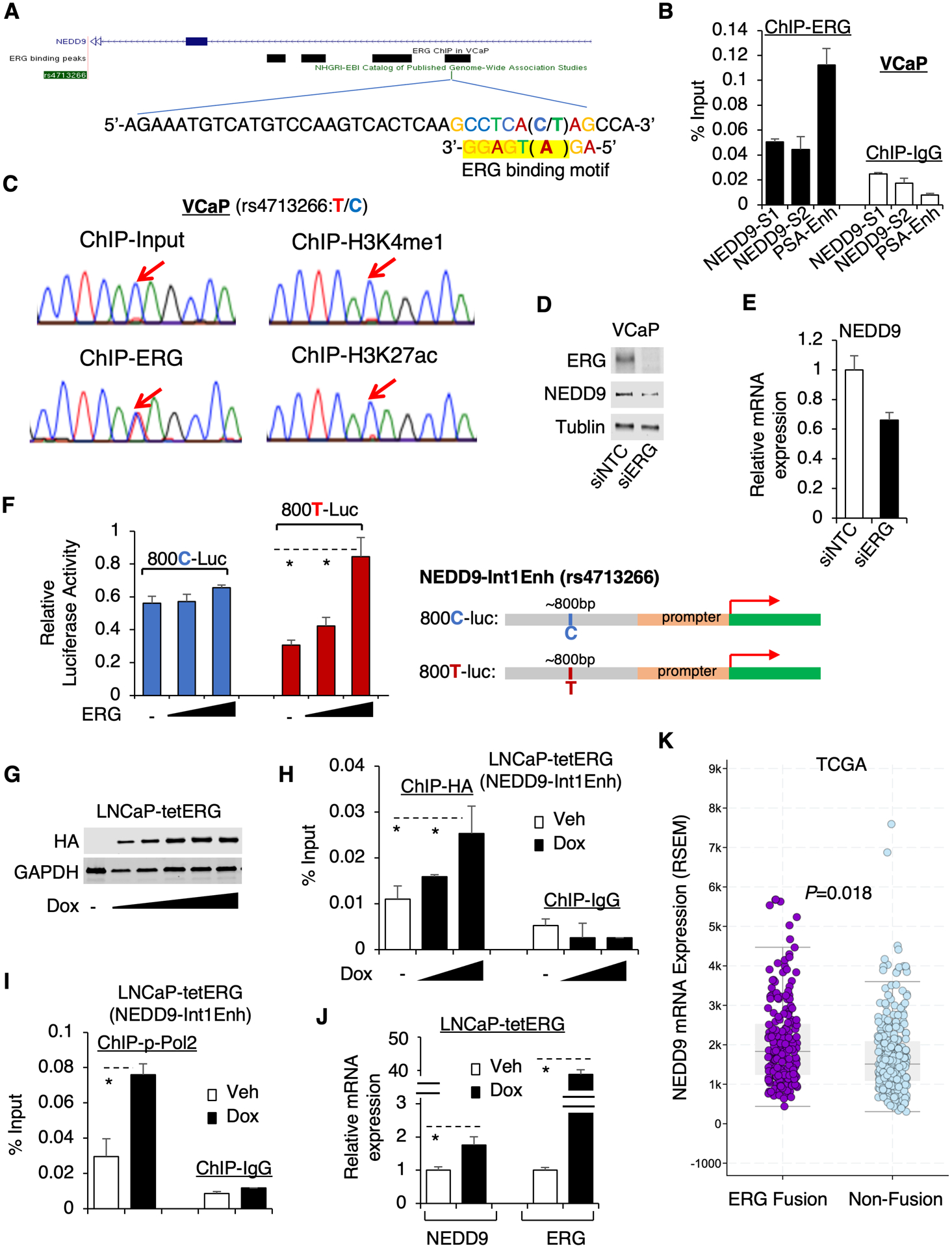
(A) ERG ChIP-seq in VCaP cells indicates ERG binding at NEDD9-Int1-Enh and a putative ERG binding motif at the SNP region was identified (matching sequence highlighted by yellow). (B) ChIP-qPCR for ERG binding at NEDD9-Int1Enh using two primer sets (NEDD9-S1 and S2) in VCaP cells. (C) The extracted DNA from ChIP-ERG, ChIP-H3K4me1, or ChIP-H3K27ac were PCR amplified and then sequenced. (D) Immunoblotting for ERG and NEDD9 in VCaP cells transfected with siRNA against ERG (siERG) or non-target control (siNTC). (E) qRT-PCR for NEDD9 mRNA expression in these cells. (F) DNA fragments (~800bp) containing C or T of rs4713266 were cloned into a luciferase reporter system containing a minimum promoter (800C-Luc and 800T-Luc). PC-3 cells were then transfected with ERG and 800C- or 800T-Luc and the luciferase activities were examined. (G) Immunoblotting for HA in LNCaP cells stably overexpressing doxycycline-inducible HA-tagged ERG (N-terminal 1–44aa truncated) (dox: 0, 0.05, 0.1, 0.2, 0.5, 1μM). (H, I) ChIP-qPCR for HA (dox: 0, 0.1, 0.5 μM) (H) or Ser5-phosphorylated RNA polymerase II (dox: 0, 0.1μM) (I) at NEDD9-Int1Enh site. (J) qRT-PCR for NEDD9 and ERG in this stable cell line treated with doxycycline for 48h. (K) NEDD9 expression in ERG fusion-positive versus fusion-negative PCa by examining the TCGA cohort (data acquired from cBioPortal).
To validate ERG binding, ChIP-qPCR of ERG was performed in VCaP cells (TMPRSS2-ERG positive) and ERG chromatin binding was detected at NEDD9-Int1Enh (Fig. 3B). However, this ERG binding was noticeably weaker than the previously reported ERG binding at the KLK3 enhancer. To determine whether ERG binding can be affected by T/C variation at rs4713266, we sequenced the DNA fragments that were immunoprecipitated with ERG at NEDD9-Int1Enh. As seen in Fig. 3C, while the risk allele C was predominantly found in the input DNA, the percentage of non-risk allele T was dramatically increased in the DNA fragments amplified from ChIP-ERG, indicating that the interaction of ERG with the non-risk allele is much stronger. However, the levels of active enhancer marks, H3K4me2 and H3K27ac, were not similarly enriched at the non-risk allele, suggesting the risk allele of the enhancer is also active for transcription and possibly driven by other TFs. As expected, silencing ERG in VCaP cells resulted in a modest decrease of NEDD9 expression (Fig. 3D and 3E).
To directly examine whether ERG binding at the T- versus C-allele could lead to differential activation of NEDD9 transcription, we cloned the DNA fragment (~800bp) of NEDD9-Int1Enh to contain only C or T at rs4713266 position into a luciferase reporter system (containing a minimum promoter) and then transfected the reporter together with the N-terminal truncated ERG (the primary protein product of TMPRSS2-ERG) in ERG-negative PC3 PCa cells. The basal activity of the C-allele driven reporter was over 2-fold higher than the T-allele driven reporter, consistent with the C-allele of the enhancer being more active (Fig. 3F). As a comparison, we also cloned the DNA fragment near rs2018334 (~800bp) into the reporter system, but the A to G alteration did not increase the reporter activity (Supplementary Fig. 4), further supporting that rs2018334 is not a causal SNP. Importantly, while the reporter containing the risk allele C of rs4713266 did not significantly respond to ERG expression, the reporter containing non-risk allele T displayed a significant dose-dependent increase of activity by expressing ERG, suggesting that ERG may preferentially bind to and activate the non-risk allele of NEDD9-Int1Enh. Moreover, we established a doxycycline-inducible HA-tagged ERG-expressing stable line (LNCaP-tetERG) in LNCaP cells (ERG-negative, T/C heterozygous) to further examine the ERG regulation on NEDD9 (Fig. 3G). The direct ERG binding at NEDD9-Int1Enh was detected by ChIP-HA when ERG expression was induced by doxycycline treatment, which also led to increased binding of Ser5 phosphorylated RNA polymerase II, an active marker for transcription initiation (Fig. 3H and 3I). More importantly, the expression of NEDD9 was increased ~2 fold by overexpression of ERG even though LNCaP cells only contain one copy of the non-risk allele. (Fig. 3J). Overall, these data highly suggest that ERG can regulate NEDD9 expression through binding to the non-risk T allele of NEDD9-Int1Enh.
We next analyzed a large PCa cohort (TCGA) (28) to determine whether NEDD9 expression could be associated with ERG fusion. In this cohort, the majority of patient samples are from men with EA and only very few samples are from AA men. As shown in Fig 3K, the expression of NEDD9 was significantly increased in TMPRSS2-ERG positive PCa, supporting that NEDD9 expression can be regulated by ERG. Similar results were also found from an analysis of another EA-dominant PCa cohort (MSKCC PCa, n=103) (24) (Supplementary Fig. 5). Together, these data indicate that NEDD9 can be directly regulated by ERG in TMPRSS2-ERG positive PCa through specific binding and activation on the non-risk T allele of NEDD9-Int1Enh.
NANOG preferentially activates the risk allele of NEDD9-Int1Enh
We next sought to identify TFs whose DNA binding may directly favor the risk C-allele of NEDD9-Int1Enh. To identify such TFs, we have conducted a motif prediction analysis using MotifDb tool and ranked TFs that may preferentially bind to the risk versus non-risk allele based on their differential motif matching scores (Fig. 4A). The top-ranked TF is NANOG (29) (Fig. 4B), which is a well-known stem cell factor and has been previously shown to regulate NEDD9 in other cancers and function to promote PCa progression (7,30,31). To determine if NANOG can specifically activate the risk allele of NEDD9-Int1Enh, we further cloned the core fragment (~300bp) of NEDD9-Int1Enh into a luciferase reporter system with a minimum promoter. As shown in Fig. 4C, NANOG only increased the activity of the reporter containing the risk-allele C, suggesting it can preferentially bind to and activate the risk allele of NEDD9-Int1Enh.
Figure 4. NANOG preferentially activates the risk allele of NEDD9-Int1Enh.
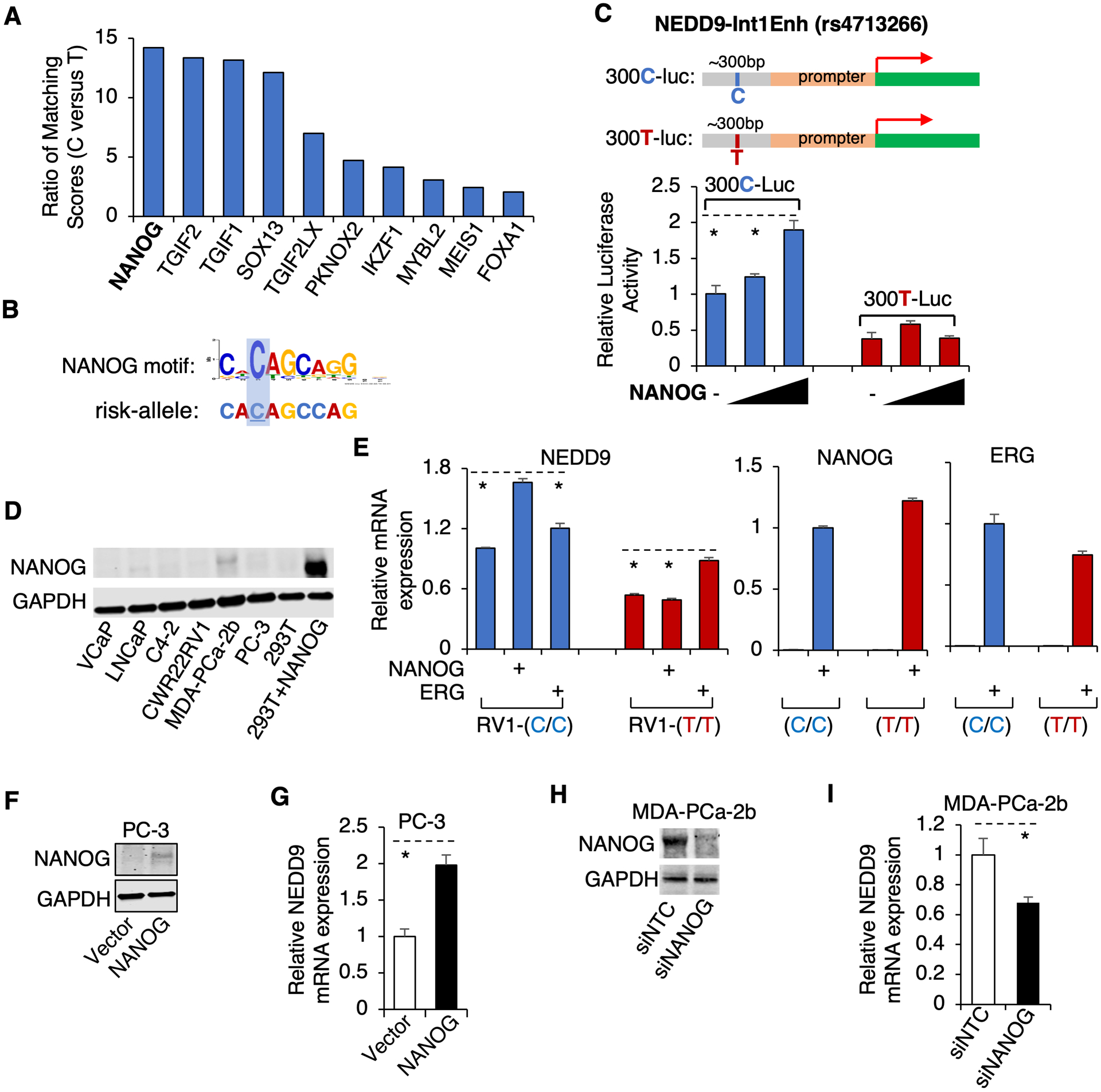
(A) Rank of potential risk-allele specific TFs based on the ratio of the matching score of risk-allele sequence versus the score of non-risk allele sequence. (B) NANOG binding motif specifically matches the risk allele DNA sequence. (C) DNA fragments (~300bp) containing C or T of rs4713266 were cloned into a luciferase reporter system containing a minimum promoter (300C-Luc and 300T-Luc). PC-3 cells were then transfected with ERG and 300C- or 300T-Luc and the luciferase activities were examined. (D) Immunoblotting for NANOG in the indicated cell lines. (E) CWR22-RV1-C/C and -T/T isogenic cell lines were transfected by NANOG or ERG expressing plasmids. NEDD9, NANOG, or ERG expression was examined by qRT-PCR. (F, G) PC-3 cells were transfected with NANOG, followed by immunoblotting for NANOG (F) and qRT-PCR for NEDD9 (G). (H) Immunoblotting for NANOG in MDA-PCa-2b cells transfected with siRNA against NANOG (siNANOG) or non-target control. (I) qRT-PCR for NANOG mRNA expression in these cells.
We then determined whether NANOG can promote the transcription of endogenous NEDD9. The protein expression of NANOG appeared to be weak in most PCa cell lines that we tested except for the MDA-PCa-2b cell line (Fig. 4D). Therefore, we transiently overexpressed NANOG in the CWR22-RV1-T/T line versus -C/C line (Supplementary Fig. 6A and 6B) to compare the effect on NEDD9 expression. As shown in Fig. 4E and 4F, NANOG expression increased NEDD9 mRNA level in the C/C line but had very little effect on NEDD9 in the T/T line, which was in sharp contrast to the effect of overexpressing ERG that only increased NEDD9 expression in the T/T line. Since the PC-3 cell line has both risk alleles (homozygous C/C) and expresses a very low level of NANOG (Fig. 4D), we also transfected PC-3 cells with the NANOG expressing vector. As shown in Fig. 4F and 4G, overexpression of NANOG increased NEDD9 mRNA expression for ~2 fold. Interestingly, PC-3 cells are heterozygous for another nearby SNP (rs72827133), but it is not overlapped with the NANOG motif (see Supplementary Fig. 2A). Furthermore, silencing NANOG in the MDA-PCa-2b cell line, which is derived from an AA PCa patient, also resulted in a reduction of NEDD9 expression (Fig. 4H and 4I). Overall, these results indicate that NANOG can specifically regulate NEDD9 expression through preferentially binding to the rs4713266 site and activating the risk allele of NEDD9-Int1Enh.
Functional characterization of NEDD9 in PCa cells
Through examining the expression of NEDD9 protein in available PCa cell lines, we found that NEDD9 expression was significantly higher in VCaP cells (Fig. 5A), consistent with the amplified NEDD9 gene found in this model (24,32). To further study the function of NEDD9 in vivo, we established VCaP stable cell lines infected by lentiviral shRNA against NEDD9 versus non-target control (Fig. 5B). The ErbB2-mediated AKT activation (S473-phosphorylation) was suppressed by NEDD9 depletion (Fig. 5C), likely due to the impairment of FAK/Src signaling as NEDD9 is a substrate of FAK/Src and the phosphorylation provides docking sites for the binding of downstream SH2-containing proteins (5). To further characterize the molecular functions and downstream signaling pathways of NEDD9 in PCa, we performed an RNA-seq analysis in VCaP-shNTC and VCaP-shNEDD9 cell lines. Gene set enrichment analysis (GSEA) using hallmark gene sets indicated that NEDD9 promotes Epithelial-to-Mesenchymal Transition (EMT), IL6/JAK/STAT3, KRAS, and IL2/STAT5 signaling pathways in PCa (Fig. 5D), which are consistent with the previous studies on NEDD9 functions in other cancers (8,33–35).
Figure 5. Functional characterization of NEDD9 in PCa cells.
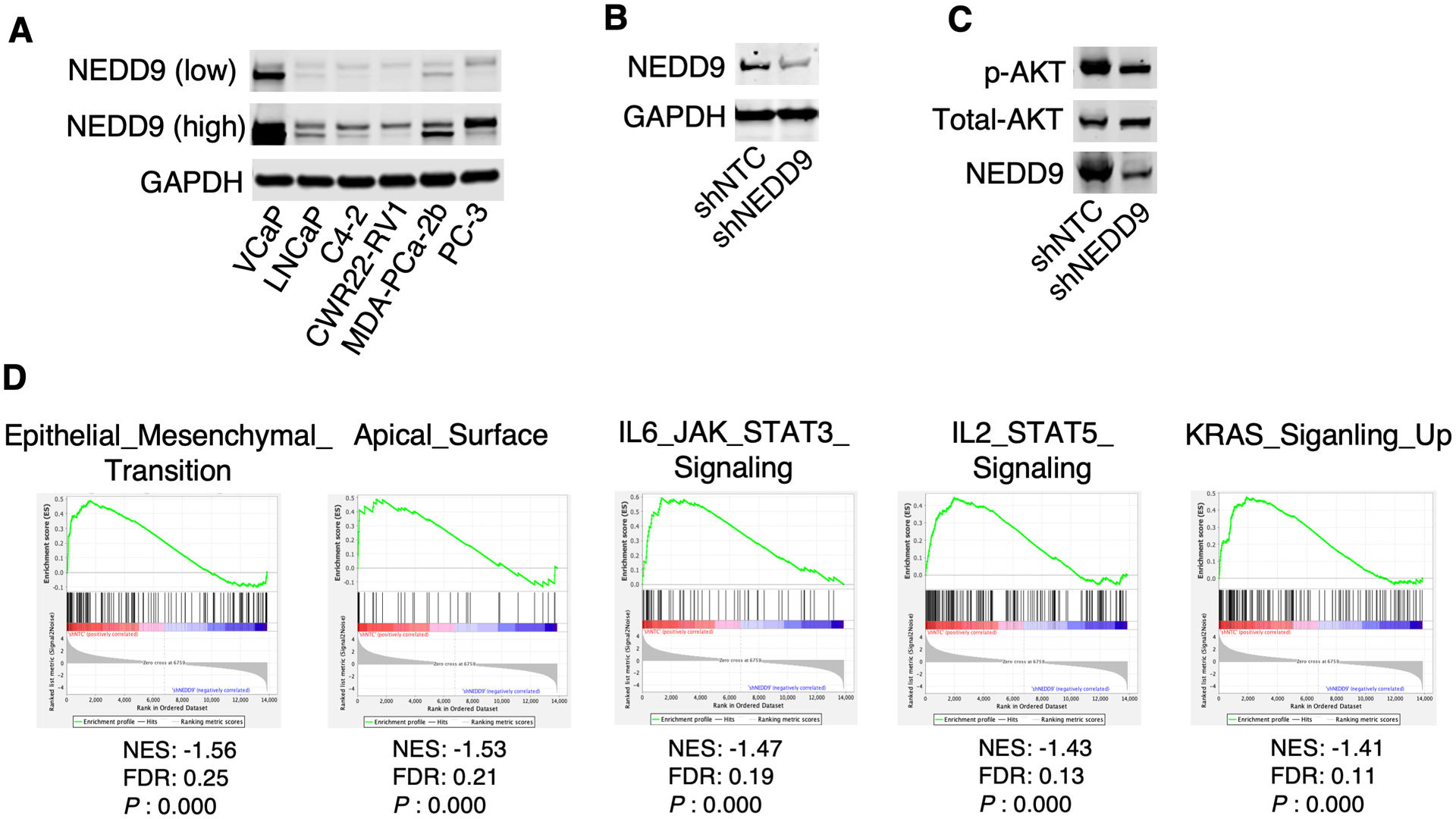
(A) NEDD9 protein expression (by immunoblotting) in PCa cell lines. (B) VCaP cells stably infected with lentiviral shRNA against NEDD9 or NTC were subjected to immunoblotting for NEDD9. (C) These stable cells were treated with 100 ng/ml heregulinβ to induce ErbB2/3 activity, followed by immunoblotting for S473-phosphorylated AKT. (D) RNA-seq analysis was done in VCaP-shNTC and VCaP-shNEDD9 cells and differentially expressed genes were identified. GSEA was then performed to identify enriched pathways that were associated with decreased expression of genes by NEDD9 depletion.
NEDD9 promotes PCa tumor growth and metastasis
We next sought to determine the role of NEDD9 in PCa initiation and progression. Using the transient RNAi approach, we found that silencing NEDD9 dramatically decreased VCaP cell proliferation (Fig. 6A and 6B). Importantly, stably silencing NEDD9 in VCaP cells markedly suppressed cell growth in vitro and xenograft tumor development in vivo (Fig. 6C and 6D). As several previous studies have demonstrated that NEDD9 may play a major role in tumor cell EMT, invasion, and metastasis (9,35–38), we next determined whether NEDD9 can induce PCa metastasis. As shown in Fig 6E and 6F, NEDD9 silencing decreased the invasion of VCaP cells in vitro. We then examined the in vivo metastasis using a zebrafish embryo model (zebrafish do not develop an adaptive immune system until 14d post-fertilization) by injecting VCaP-shNTC or shNEDD9 cells into the zebrafish embryos. As shown in Fig. 6G, while the control VCaP cells can quickly disseminate into the blood vessel within a few hours post-injection (8/10), cells with NEDD9 depletion stayed within the perivitelline space of each embryo (0/10 can metastasize), demonstrating that NEDD9 can strongly promote cancer cell intravasation, a key step for metastasis. Since the MDA-PCa-2b cell line also expresses significant levels of NEDD9 (see Fig. 5A), we next examined the function of NEDD9 using this model. Consistent with the effects found in the VCaP model, silencing NEDD9 in MDA-PCa-2b cells significantly decreased cell proliferation and migration (Fig. 6H–J). Overall, these results indicate that NEDD9 functions as an oncogene to promote PCa tumor growth, invasion, and metastasis.
Figure 6. NEDD9 promotes PCa tumor growth and metastasis.
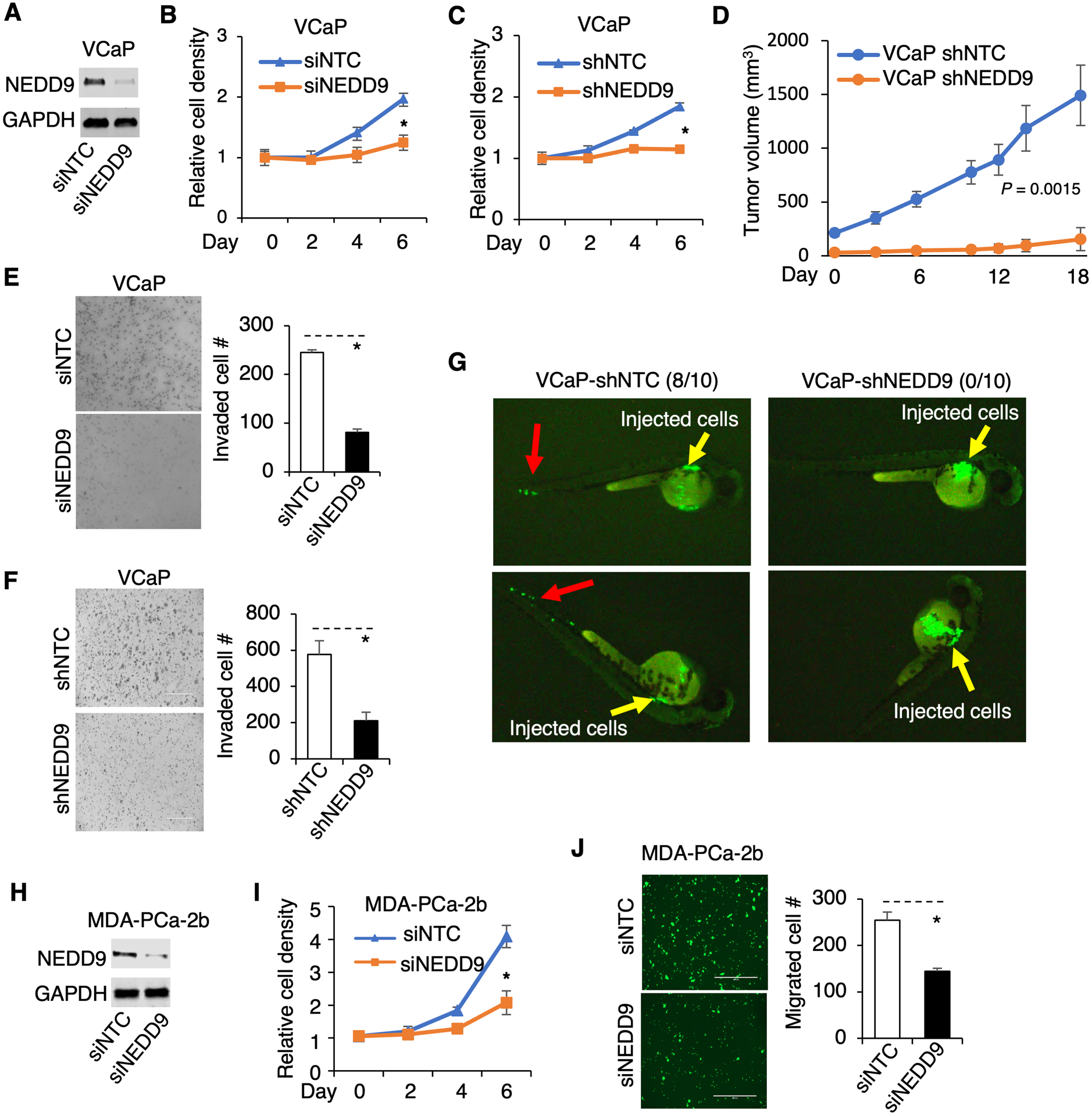
(A, B) VCaP cells transfected with siRNA against NTC or NEDD9 were subjected to immunoblotting for NEDD9 (A) and cell proliferation assay (B). (C) VCaP cells stably infected by shNTC versus shNEDD9 were subjected to cell proliferation assay. (D) These stable cells were subcutaneously injected into male SCID mice and the tumor volume was measured. (E, F) VCaP cells transfected with siNTC versus siNEDD9, or stably infected by shNTC versus shNEDD9 were subjected to Matrigel invasion assay. (G) VCaP cells stably infected by shNTC versus shNEDD9 (both GFP labeled) were injected into zebrafish embryos. The tumor cell metastasis was observed using a fluorescence microscope (yellow arrow: injected PCa cells; red arrow: disseminated PCa cells). (H, I, J) MDA-PCa-2b cells transfected with siRNA against NEDD9 or NTC were subjected to immunoblotting for NEDD9 (H), cell proliferation assay (I), and transwell migration assay (J).
DISCUSSION
Our study is an attempt to address one important molecular mechanism in driving or contributing to PCa initiation and progression and in part explain observed racial disparities in PCa. Although SNPs associated with PCa risk are commonly found within non-coding regions, the molecular functions of such SNPs are still largely unknown. This work focused on two highly related PCa susceptibility SNPs at 6p24.2 that map to a putative enhancer (NEDD9-Int1Enh) of NEDD9 proto-oncogene and aimed to determine the impact of these race-associated germline variations on NEDD9 expression and activity, and to identify potential TF(s) mediating NEDD9-Int1Enh activity, NEDD9 gene transcription, and PCa development. Our data strongly suggest that rs4713266 is the causal SNP that can alter NEDD9-Int1Enh activity and NEDD9 expression. Using bioinformatic analyses, we have identified a panel of candidate TFs, which have different preferences to activate the risk versus non-risk allele of NEDD9-Int1Enh. The identified and validated TF that preferentially activates the non-risk T allele is ERG, which is overexpressed in PCa due to the TMPRSS2-ERG fusion (27). Although ERG functions to promote PCa development (39,40), it is highly debated whether its expression is correlated with aggressiveness (such as Gleason score) or outcomes of PCa (41–43). Studies on ETS gene fusion in different racial groups have indicated that TMPRSS2-ERG rearrangement is significantly less frequent in AA men (i.e., ~10–20% in AA versus ~40–50% in EA) (3,44). This racially specific mutation frequency of ERG fusion is highly correlated with the non-risk allele frequency of rs4713266 in men with AA (~20%) versus EA (~50%) (see Fig. 1). Our findings strongly suggest that ERG can preferentially bind to and activate NEDD9-Int1Enh at the non-risk allele. We have also identified NANOG as a potential TF to preferentially activate the risk allele and NANOG has been recently reported to play an important role in driving PCa progression in SPOP mutated PCa (SPOP mutations are mutually exclusive to TMPRSS2-ERG fusions) (45). Interestingly, we found that the risk allele may also favor the binding of pioneer factors (MEIS1, FOXA1) that function to increase the accessibility of enhancers (46). This finding is consistent with the homozygous “C/C” line showing increased enhancer activity (see Fig. 2F). Nonetheless, our data indicate that future studies on characterizing the activities of such TFs are clearly needed. Overall, the working model we proposed in this study is that in EA patients NEDD9 expression is primarily driven by ERG through specifically activating the non-risk T allele of NEDD9-Int1Enh, while in AA patients, the risk C allele, potentially activated by NANOG and/or other TFs, is driving NEDD9 expression at a comparable or even higher level to mitigate the lack of ERG fusion. Importantly, ERG-driven NEDD9 overexpression may likely be suppressed by androgen deprivation therapies (ADTs) since TMPRSS2-ERG is a direct target of the androgen receptor. However, NANOG (or other TFs)-driven NEDD9 overexpression may be less affected with ADTs, which is consistent with the clinical observation that AA men, in general, do not respond well to ADTs.
Regardless of the genetic variation at NEDD9-Int1Enh, NEDD9 amplification was also commonly found in CRPC or NEPC but not in the primary PCa, indicating that NEDD9 may play an important role in PCa progression. Indeed, previous studies have suggested that NEDD9 functions as a major signaling molecule in driving tumor cell metastasis through regulating EMT, migration, and invasion. Using the VCaP PCa cell line in which NEDD9 is overexpressed due to the gene amplification, and in its derived mouse xenograft model, we found that silencing NEDD9 resulted in a significant reduction of tumor cell invasion in vitro, and suppression of the tumor growth in vivo. More importantly, using a zebrafish embryo metastasis model, we demonstrated a very strong activity of NEDD9 in promoting metastasis in vivo. These oncogenic activities of NEDD9 in PCa cells are presumably mediated by the downstream pathways identified from our RNA-seq analysis, such as JAK/STAT3, IL2/STAT5, and KRAS pathways. In addition, we also observed decreased AKT activation when NEDD9 was depleted. In particular, JAK/STAT3 signaling plays an essential role in regulating EMT and metastasis in cancer (47). Nonetheless, the findings of this study will have implications in clinical therapies: while directly targeting NEDD9 may be challenging, treatments targeting the identified risk SNP-associated TFs or NEDD9-mediated signaling pathways, such as inhibitors of FAK or JAK/STAT3, may be potentially used to treat PCa with African ancestry. Overall, these data represent an important advance in our mechanistic and epidemiological studies to address the PCa disparity in AA men.
Supplementary Material
SIGNIFICANCE.
A prostate cancer susceptibility genetic variation in NEDD9, which is strongly associated with the increased risk of patients with African ancestry, increases NEDD9 expression and promotes initiation and progression of prostate cancer.
ACKNOWLEDGMENTS
This work is supported by grants from NIH (R01 CA211350 to C Cai., P20 CA233255 to T Rebbeck, U54 CA156734 to JA Macoska) and DOD (W81XWH-16-1-0445 and W81XWH-19-1-0361 to C Cai, W81XWH-19-1-0777 to S Gao, W81XWH-15-1-0151 to X Yuan). M Liu and M Labaf were supported by the graduate fellowship from Integrative Biosciences Program at University of Massachusetts Boston.
Footnotes
The authors declare no potential conflict of interest.
REFERENCES
- 1.McGinley KF, Tay KJ, Moul JW. Prostate cancer in men of African origin. Nat Rev Urol 2016;13:99–107 [DOI] [PubMed] [Google Scholar]
- 2.Yamoah K, Deville C, Vapiwala N, Spangler E, Zeigler-Johnson CM, Malkowicz B, et al. African American men with low-grade prostate cancer have increased disease recurrence after prostatectomy compared with Caucasian men. Urol Oncol 2015;33:70 e15–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamoah K, Johnson MH, Choeurng V, Faisal FA, Yousefi K, Haddad Z, et al. Novel Biomarker Signature That May Predict Aggressive Disease in African American Men With Prostate Cancer. J Clin Oncol 2015;33:2789–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lachance J, Berens AJ, Hansen MEB, Teng AK, Tishkoff SA, Rebbeck TR. Genetic Hitchhiking and Population Bottlenecks Contribute to Prostate Cancer Disparities in Men of African Descent. Cancer Res 2018;78:2432–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shagisultanova E, Gaponova AV, Gabbasov R, Nicolas E, Golemis EA. Preclinical and clinical studies of the NEDD9 scaffold protein in cancer and other diseases. Gene 2015;567:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Al Olama AA, Kote-Jarai Z, Berndt SI, Conti DV, Schumacher F, Han Y, et al. A meta-analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nat Genet 2014;46:1103–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh M, Cowell L, Seo S, O’Neill G, Golemis E. Molecular basis for HEF1/NEDD9/Cas-L action as a multifunctional co-ordinator of invasion, apoptosis and cell cycle. Cell Biochem Biophys 2007;48:54–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Izumchenko E, Singh MK, Plotnikova OV, Tikhmyanova N, Little JL, Serebriiskii IG, et al. NEDD9 promotes oncogenic signaling in mammary tumor development. Cancer Res 2009;69:7198–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo W, Ren D, Chen X, Tu X, Huang S, Wang M, et al. HEF1 promotes epithelial mesenchymal transition and bone invasion in prostate cancer under the regulation of microRNA-145. J Cell Biochem 2013;114:1606–15 [DOI] [PubMed] [Google Scholar]
- 10.Morimoto K, Tanaka T, Nitta Y, Ohnishi K, Kawashima H, Nakatani T. NEDD9 crucially regulates TGF-beta-triggered epithelial-mesenchymal transition and cell invasion in prostate cancer cells: involvement in cancer progressiveness. Prostate 2014;74:901–10 [DOI] [PubMed] [Google Scholar]
- 11.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;161:1215–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar A, Coleman I, Morrissey C, Zhang X, True LD, Gulati R, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med 2016;22:369–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao S, Gao Y, He HH, Han D, Han W, Avery A, et al. Androgen Receptor Tumor Suppressor Function Is Mediated by Recruitment of Retinoblastoma Protein. Cell Rep 2016;17:966–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30:923–30 [DOI] [PubMed] [Google Scholar]
- 16.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parolia A, Cieslik M, Chu SC, Xiao L, Ouchi T, Zhang Y, et al. Distinct structural classes of activating FOXA1 alterations in advanced prostate cancer. Nature 2019;571:413–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teng Y, Xie X, Walker S, White DT, Mumm JS, Cowell JK. Evaluating human cancer cell metastasis in zebrafish. BMC Cancer 2013;13:453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Al Olama AA, Kote-Jarai Z, Berndt SI, Conti DV, Schumacher F, Han Y, et al. A meta-analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nat Genet 2014;46:1103–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res 2012;40:D930–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet 2013;45:1238–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao S, Chen S, Han D, Wang Z, Li M, Han W, et al. Chromatin binding of FOXA1 is promoted by LSD1-mediated demethylation in prostate cancer. Nat Genet 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med 2016;22:298–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010;18:11–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Z, Gao S, Han D, Han W, Li M, Cai C. LSD1 Activates PI3K/AKT Signaling Through Regulating p85 Expression in Prostate Cancer Cells. Front Oncol 2019;9:721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu J, Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 2010;17:443–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005;310:644–8 [DOI] [PubMed] [Google Scholar]
- 28.Cancer Genome Atlas Research N. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015;163:1011–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res 2012;22:1798–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kregel S, Szmulewitz RZ, Vander Griend DJ. The pluripotency factor Nanog is directly upregulated by the androgen receptor in prostate cancer cells. Prostate 2014;74:1530–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeter CR, Liu B, Lu Y, Chao HP, Zhang D, Liu X, et al. NANOG reprograms prostate cancer cells to castration resistance via dynamically repressing and engaging the AR/FOXA1 signaling axis. Cell Discov 2016;2:16041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Astier A, Manie SN, Law SF, Canty T, Haghayghi N, Druker BJ, et al. Association of the Cas-like molecule HEF1 with CrkL following integrin and antigen receptor signaling in human B-cells: potential relevance to neoplastic lymphohematopoietic cells. Leuk Lymphoma 1997;28:65–729498705 [Google Scholar]
- 34.Ota J, Kimura F, Sato K, Wakimoto N, Nakamura Y, Nagata N, et al. Association of CrkL with STAT5 in hematopoietic cells stimulated by granulocyte-macrophage colony-stimulating factor or erythropoietin. Biochem Biophys Res Commun 1998;252:779–86 [DOI] [PubMed] [Google Scholar]
- 35.Jin Y, Li F, Zheng C, Wang Y, Fang Z, Guo C, et al. NEDD9 promotes lung cancer metastasis through epithelial-mesenchymal transition. Int J Cancer 2014;134:2294–304 [DOI] [PubMed] [Google Scholar]
- 36.Kim M, Gans JD, Nogueira C, Wang A, Paik JH, Feng B, et al. Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell 2006;125:1269–81 [DOI] [PubMed] [Google Scholar]
- 37.Natarajan M, Stewart JE, Golemis EA, Pugacheva EN, Alexandropoulos K, Cox BD, et al. HEF1 is a necessary and specific downstream effector of FAK that promotes the migration of glioblastoma cells. Oncogene 2006;25:1721–32 [DOI] [PubMed] [Google Scholar]
- 38.Gabbasov R, Xiao F, Howe CG, Bickel LE, O’Brien SW, Benrubi D, et al. NEDD9 promotes oncogenic signaling, a stem/mesenchymal gene signature, and aggressive ovarian cancer growth in mice. Oncogene 2018;37:4854–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Magi-Galluzzi C, Tsusuki T, Elson P, Simmerman K, LaFargue C, Esgueva R, et al. TMPRSS2-ERG gene fusion prevalence and class are significantly different in prostate cancer of Caucasian, African-American and Japanese patients. Prostate 2011;71:489–97 [DOI] [PubMed] [Google Scholar]
- 40.Cai C, Wang H, He HH, Chen S, He L, Ma F, et al. ERG induces androgen receptor-mediated regulation of SOX9 in prostate cancer. J Clin Invest 2013;123:1109–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petrovics G, Liu A, Shaheduzzaman S, Furusato B, Sun C, Chen Y, et al. Frequent overexpression of ETS-related gene-1 (ERG1) in prostate cancer transcriptome. Oncogene 2005;24:3847–52 [DOI] [PubMed] [Google Scholar]
- 42.Attard G, Clark J, Ambroisine L, Fisher G, Kovacs G, Flohr P, et al. Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene 2008;27:253–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gopalan A, Leversha MA, Satagopan JM, Zhou Q, Al-Ahmadie HA, Fine SW, et al. TMPRSS2-ERG gene fusion is not associated with outcome in patients treated by prostatectomy. Cancer Res 2009;69:1400–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou CK, Young D, Yeboah ED, Coburn SB, Tettey Y, Biritwum RB, et al. TMPRSS2:ERG Gene Fusions in Prostate Cancer of West African Men and a Meta-Analysis of Racial Differences. Am J Epidemiol 2017;186:1352–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang J, Chen M, Zhu Y, Dai X, Dang F, Ren J, et al. SPOP Promotes Nanog Destruction to Suppress Stem Cell Traits and Prostate Cancer Progression. Dev Cell 2019;48:329–44 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev 2011;25:2227–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin W Role of JAK/STAT3 Signaling in the Regulation of Metastasis, the Transition of Cancer Stem Cells, and Chemoresistance of Cancer by Epithelial-Mesenchymal Transition. Cells 2020;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.