

Abstract
The p53 tumor suppressor is frequently inactivated by mutations in cancer. Most p53 mutations are located in the DNA binding domain, causing local disruption of DNA binding surface or global misfolding. Rescuing the structural defect of mutant p53 is an attractive therapeutic strategy, but its potential remains unproven due to a lack of drugs capable of efficiently rescuing misfolded p53. Although mutant p53 in tumors are inactive at 37°C, ~15% are temperature sensitive and regain DNA binding activity at 32–34°C (ts mutants). This temperature is achievable using a therapeutic hypothermia procedure established for resuscitated cardiac arrest patients. To test whether hypothermia can be used to target tumors with ts p53 mutations, the core temperature of tumor-bearing mice was lowered to 32°C using the adenosine A1 receptor agonist N6-cyclohexyladenoxine (CHA) that suppresses brain-regulated thermogenesis. Hypothermia treatment (32 hours at 32°C × 5 cycles) activated endogenous ts mutant p53 in xenograft tumors and inhibited tumor growth in a p53-dependent fashion. Tumor regression and durable remission in a ts p53 lymphoma model was achieved by combining hypothermia with chemotherapy. The results raise the possibility of treating tumors expressing ts p53 mutations with hypothermia.
Introduction
P53 is a transcription factor inducible by stress signals such as DNA damage, oncogene activation, and nutrient deprivation. P53 tetramer binds to a specific DNA sequence and activates genes involved in cell cycle, apoptosis, and energy metabolism (1). P53 is the most frequently mutated gene in human cancer (>50% overall mutation rate). Most p53 mutations (~80%) are amino acid substitutions in the DNA binding domain that cause misfolding or disrupt the DNA binding surface (2,3). As a result, mutant p53 does not bind DNA or activate target genes.
Mutant p53 is resistant to MDM2-mediated ubiquitination and accumulates to high levels in tumor cells (4,5). Restoring the DNA binding function of mutant p53 is an attractive strategy with significant therapeutic potential (6,7). However, currently there are no effective mutant p53-targeted drugs approved for clinical use. Wild type (wt) p53 has poor structural stability. The stability is further reduced in tumor-derived p53 mutants, rendering them unable to bind DNA at 37°C (8). The development of a yeast assay for detecting p53 mutations unexpectedly revealed that ~11–15% of tumor-derived p53 mutants are temperature-sensitive (ts) and behaved like wt p53 in the yeast at 32°C (9–11). Most ts p53 mutations are located in the hydrophobic β sandwich core of the DNA binding domain and destabilize the folding at 37°C (9). These mutants are denatured at 37°C but can regain wt conformation at 32–34°C.
Early work using the mouse p53 ts mutant A135V and the human counterpart A138V in cell culture demonstrated potent apoptosis or cell cycle arrest activities at 32°C (12,13). Therefore, activating ts p53 mutants may produce strong anti-tumor effects because of their high expression levels. Furthermore, ts p53 mutants are only expressed in tumor cells but not in normal tissues, making them attractive tumor-specific targets for treatment using hypothermia. There are ~1,700,000 new cancer diagnosis per year in the U.S. If 50% of the new patients have p53 missense mutations and 15% of these mutations are temperature-sensitive, then 7.5% of new cancer patients (127,500 per year) have ts p53 mutations. Therefore, approaches that specifically target tumors expressing ts p53 will have wide impact.
Several mouse models demonstrated that restoring wt p53 expression induced tumor regression. Using p53-ER fusion protein to control p53 activity in Myc-induced lymphomas, Martins and colleagues showed restoring p53 activity for 6 hours led to apoptosis of most tumor cells (14). Continuous p53-ER activation for 7 days extended survival of tumor-bearing mice. Irradiation cooperated with p53 reactivation to increase PUMA expression and further extended survival (14). Using Cre-LoxP to control endogenous p53 expression, Ventura et al showed that restoring wt p53 expression induced rapid apoptosis in lymphomas and senescence in sarcomas, causing tumor regression (15). Using inducible shRNA to regulate p53 expression, Xue et al showed that p53 restoration in liver cancer induced tumor regression through senescence and immune clearance (16). These observations support the hypothesis that restoring ts mutant p53 activity in tumors should also have therapeutic effects.
In principle, cooling tumors to 32°C will activate ts mutant p53. Due to the locations of most tumors, this requires cooling the body core. Therapeutic hypothermia is the standard of care for resuscitated cardiac arrest patients (17–19). The core temperature of sedated patients is lowered to 32–34°C using cold blankets for up to 24 hours to improve neurological outcome. Newborn infants with hypoxic-ischemic encephalopathy are cooled for 72 hours for neuro protection (20). There are also pre-clinical studies to develop drug-induced hypothermia for treating stroke and brain injury (21). A seminal study showed the A1 adenosine receptor (A1AR) agonist N6-cyclohexyladenoxine (CHA) triggers entry into hibernation in arctic ground squirrels (22). CHA induces transient hypothermia in non-hibernating species such as rats and mice by suppressing thermogenesis and shiver response (23,24). Certain anti-psychotic drugs cause hypothermia as a side effect (25,26). The anti-psychotic drug chlorpromazine was used in combination with physical cooling to maintain long duration (2–5 days) hypothermia in patients with brain injury (27). Therefore, hypothermia is an established procedure that can potentially be repurposed for cancer treatment if significant efficacy is demonstrated.
In this report, we tested the therapeutic potential of hypothermia in mice bearing tumor xenografts with ts mutant p53. Using CHA to induce hypothermia for multiple cycles, we observed stasis effects against tumors expressing endogenous ts mutant p53. Furthermore, hypothermia synergized with chemotherapy to induce tumor regression and durable remission in a lymphoma xenograft model. The results suggest hypothermia should be further investigated as a strategy against tumors expressing ts mutant p53.
Materials and methods
Cell Lines and plasmids.
Cell lines with ts mutant p53 [GA10 (P152L/I232N, ATCC Cat# CRL-2393), SU-DHL-6 (Y234C, ATCC Cat# CRL-2959), NCI-H441 (R158L, ATCC Cat# HTB-174), NCI-H1355 (E285K, ATCC Cat# CRL-5865), NCI-H1963 (H214R/V147D, ATCC Cat# CRL-5982)] were recently purchased from ATCC in 2020 and therefore not tested for mycoplasma or authenticated. Patu8988t (R282W) was provided by Dr. Lixin Wan of Moffitt Cancer Center. All ts p53 mutations in the cell lines were validated by RT-PCR and DNA sequencing. Other cell lines [A549 (wt), MCF7 (wt), U2OS (wt), NCI-H1299 (null), Saos2 (null), MDA-MB-468 (R273H), DU145 (P223L)] were cryopreserved lab stocks last authenticated and tested negative for mycoplasma contamination in 2018. The cells were passaged for <60 days between thawing and experiments. GA-10 and SU-DHL-6 were cultured in RPMI-1640 medium with 20% fetal bovine serum (FBS). All other cell lines were cultured in Dulbecco modified Eagle medium (DMEM) with 10% FBS. H1299 cell lines stably expressing p53 ts or non-ts mutants, were established by infection with pLenti-p53 mutant viruses followed by Zeocin selection (ViraPower T-REX lentiviral expression system, Invitrogen). Each p53 mutation was generated by site-directed mutagenesis on the wt pLenti-p53 plasmid. To knockout p53 in GA10, SU-DHL-6, Patu8988t and NCI-H1963 cells, p53gRNA3 (5’CACCGCCATTGTTCAATATCGTCCG annealed to 5’AAACCGGACGATATTGAACAATGGC) was cloned into LentiCRISPRv2 vector (Addgene). Cells were infected with lentivirus expressing gRNA, selected with puromycin for single cell clones, and analyzed of p53 expression. Three p53-negative clones were pooled for tumor xenograft analysis.
The profiles of p53 mutations in clinical cases were generated from the Catalogue Of Somatic Mutations In Cancer (COSMIC) database (https://cancer.sanger.ac.uk/cosmic). The tumor cell lines information for p53 ts mutations were acquired from the International Agency for Research on Cancer (IARC) database (https://p53.iarc.fr/CellLines.aspx).
Western blot.
Cells were lysed in lysis buffer (50 mM Tris–HCl pH 8.0, 5 mM EDTA, 150 mM NaCl, 0.5% Nonidet P-40, 1x protease inhibitor cocktail) and centrifuged for 10 minutes at 14,000 × g, and the insoluble debris was discarded. Cell lysate (10–50 μg of protein) was fractionated by SDS–PAGE and transferred to Immobilion P filters (Millipore). The filter was blocked for 1 hr with phosphate buffered saline (PBS) containing 5% non-fat dry milk and 0.1% Tween 20, incubated with primary and secondary antibodies, and the filter was developed using SuperSignal reagent (Thermo Scientific). MDM2 was detected using monoclonal antibody 3G9 produced in house. Other markers were detected using commercial antibodies: Actin (Sigma A5441), p53-DO1 (BD Pharmingen #554293), p21 (BD Pharmingen #556430), PUMA (CellSignaling #12450), PARP (BD Pharmingen #556362), p53 pSer15 (CellSignaling #9284), p53 acetyl-Lys382 (CellSignaling #2525).
RNA isolation and quantitative RT-PCR.
Total RNA was extracted using the RNeasy Mini kit (Qiagen). cDNAs were prepared by reverse transcription of total RNA using Applied Biosystems™ High-Capacity cDNA Reverse Transcription Kit. The products were used for SYBR Green real-time PCR using the following primers: GAPDH (5’TCACCACCATGGAGAAGGC and 5’GCTAAGCAGTTGGTGGTGCA), p14-ARF (5’TCTTGGTGACCCTCCGGATTCGG and 5’TCAGCCAGGTCCACGGGCAGA).
Luciferase reporter assay.
H1299 cells (50,000/well) were seeded in 24-well plate and transfected with 2 ng pLenti-p53 plasmid (wt or mutant), 5 ng CMV-lacZ plasmid, and 20 ng p53-responsive luciferase reporter plasmid driven by MDM2, p21, or PUMA promoters. Transfection was achieved using Lipofectamine 3000 (Invitrogen). Twenty-four hours after transfection at 37°C, cells were transferred to 32°C for 20 hrs, and analyzed for luciferase and beta-galactosidase activity. The transcriptional activity of p53 was indicated by the ratio of luciferase/beta-galactosidase activity.
Chromatin immunoprecipitation (ChIP).
ChIP assay was performed using standard procedure. Crosslinked p53-DNA complexes were immunoprecipitated with DO-1 antibody. Samples were subjected to SYBR Green real-time PCR analysis using forward and reverse primers for the p53 binding sites in the p21 promoter (5′AGGAAGGGGATGGTAGGAGA and 5′ACACAAGCACACATGCATCA), and PUMA promoter (5′CTGTGGCCTTGTGTCTGTGAGTAC and 5′CCTAGCCCAAGGCAAGGAGGAC).
Cell proliferation and viability assays.
DNA replication was analyzed using 3H-thymidine incorporation assay. Cells were plated in 12-well plate (5×105/well) and incubated at 37°C or 32°C for 48 hrs. 3H-thymidine (4 μCi/well, 25 Ci/mmol, PerkinElmer) was added to culture media and incubated at 37°C for 3 hrs. Cells were washed twice with cold PBS and treated with 1 ml of ice-cold 5% Trichloroacetic acid at 4°C for 30 min. After washing twice with PBS, the cells were lysed in 1 ml of 1 N NaOH at 37°C for 30 min. The incorporated 3H-thymidine was analyzed by liquid scintillation counting. MTS cell viability analysis was performed using Celltiter 96 AQueous One Solution reagent following manufacturer instructions (Fisher #PR-G3580).
In vitro p53 refolding and DNA binding assay.
XL1-Blue cotransformed with pBB540 (Addgene #27393, expressing GrpE, ClpB.) and pBB550 (Addgene #27396, expressing DnaK, DnaJ, GroESL.) was cultured in LB medium containing chloramphenicol (30 μg/ml) and spectinomycin (50 μg/ml) to OD600=0.6 at 37°C and induced with 0.1 mM IPTG at 18°C for 18 hrs. Cell pellet from 250 ml culture was suspended in 5 ml phosphate buffer (100 mM potassium phosphate pH7.8, 0.01% Triton X-100, 1 μM ZnCl2. 1 mM DTT), disrupted by sonication, and centrifuged at 14,000 × g for 10 min at 4°C to prepare supernatant containing chaperones (stored at −80°C with 5% glycerol). The p53 in vitro DNA binding assay contained ZF-Nluc (luciferase aa 1–437 fused to C-terminus of a zinc finger protein and cloned into pET28 vector), p53-Cluc (luciferase aa 398–550 fused to C-terminus of p53 or R282W and R175H mutants and cloned into pET28), ZPBS12 plasmid DNA [pUC57 vector with a 560 bp DNA insert containing 12 copies of ZF binding site (ATGTAGGGAAAAGCCCGG) and p53 binding site (GAACATGTCCCAACATGTTG) with various spacing (0, 2, 4, 6, 8, 10 bp)]. BL21DE3 transformed with p53-Cluc or ZF-Nluc were cultured to OD600=0.6 at 37°C in LB medium containing 150 μM ZnCl2, and induced with 0.1 mM IPTG for 20 hrs at 16°C. Pelleted cells were sonicated in lysis buffer [50 mM HEPES (pH 7.5), 150 mM NaCl, 0.1% Nonidet P-40, 5% glycerol, 10 μM ZnCl2, 1 mM DTT] and centrifuged at 14,000 x g for 10 min at 4°C. The lysate was diluted to ~10 ng/μl total protein with dilution buffer [4.25% (vol/vol) 0.2 M NaH2PO4, 45.75% (vol/vol) 0.2 M Na2HPO4, 5% glycerol, 1 μM ZnCl2, 1 mM DTT]. The diluted BL21DE3 extract (10 μl, ~200 ng protein) containing p53-Cluc or R282W-Cluc was incubated at 34°C for 20 min to inactivate the ts p53. The heat-treated R282W-Cluc was mixed with 10 μl XL1-Blue extract containing chaperones (or XL1-Blue control extract), 5 mM ATP, and incubated at 23°C for 2 hrs for refolding. The refolded p53-Cluc mixture was combined with 200 ng ZF1-Nluc extract and 50 ng ZPBS12 plasmid DNA in a 40 μl DNA binding reaction mixture and incubated for 30 min at 23°C. Luciferase substrate A (25 mM glycylglycin pH7.8, 15 mM potassium phosphate pH7.8, 15 mM MgSO4, 4 mM EGTA, 2 mM ATP, 1 mM DTT) was combined with equal volume of luciferase substrate B (0.4 mg/ml D-luciferin, 25 mM glycylglycin pH7.8, 2 mM DTT), and 30 μl substrate mixture was added to the DNA binding reaction mixture. After incubating for 10 min at 23°C, luciferase signal was measured using a microplate luminometer.
Animal experiment.
Animal experiments were reviewed and approved by the University of South Florida IACUC. Athymic female nude mice (6-week old, Athymic Nude-Foxn1nu, Envigo) were injected subcutaneously with 0.1 ml 1:1 slurry of Matrigel (VWR 47743-715) and 1×107 cells in PBS at each site. The inoculated mice were housed in a cabinet set to 28°C (ARIA BIO-C36 ventilated cabinet, TECNIPLAST) to ensure that tumors develop at ~37°C. Each mouse received injections at 2 sites, tumors formed at >80% of sites injected with GA10 cells. N6-cyclohexyladenosine (CHA, Sigma C9901) was dissolved at 20 mg/ml in 25% (2-Hydroxypropyl)-β-cyclodextrin (w/v, Sigma H107) and heated at 65°C for 10 min to ensure dissolution. Camptothecin (CPT, Santa Cruz Biotechnology sc-200871B) was freshly dissolved in 1 N NaOH, diluted to ~0.5 mg/ml with saline, and adjusted to pH10 using HCl. To induce hypothermia, 0.2 ml saline+10% glucose containing CHA and CPT was administered by intra peritoneal injection to provide a CHA dosage of 10 mg/kg body weight and CPT (when indicated) dosage of 1.5 mg/kg body weight. The mice were kept in the 28°C cabinet and monitored using infrared thermal imaging camera (FLIR ONE Pro, FLIR® Systems, Inc.) to verify that body temperature was maintained at ~32°C. A second injection of CHA (10 mg/kg) in 0.4 ml saline+10% glucose was given after 10 hours to maintain hypothermia for ~24 hours. A third injection of CHA (10 mg/kg) in 0.4 ml saline+10% glucose was given after 10 hours to maintain hypothermia for ~32 hours. After regaining normal body temperature, the mice were kept for 2–4 days in the 28°C cabinet to allow weight recovery (~10% weight loss occurred during hypothermia due to loss of mobility and food intake). The treatment was repeated 4–5 times (24 hr × 6 or 32 hr × 5 format). In each subsequent round CHA dosage was increased by 25% to compensate for the gradual loss of sensitivity.
Flow cytometry.
GA-10 control or p53-knockout cells were cultured at 37°C or 32°C respectively for 48 hrs. The cells were collected by centrifuging at 3000 × g for 5 min, washed with PBS, stained with Annexin V-PE and 7-AAD, and subjected to FACS analysis using FACSCalibur flow cytometer (BD BioSciences). The data were analyzed using FlowJo software (TreeStar).
Immunohistochemistry.
Mice bearing GA-10 xenograft tumors were treated with hypothermia (with or without CPT) for 24 hrs and euthanized for tumor collection. The tumor samples were fixed in formalin and paraffin sections were prepared. After de-paraffinization and rehydration with xylene and a graded ethanol series, tissue sections were analyzed with TUNEL staining using the In situ Apoptosis Detection Kit (Abcam ab206386).
Statistical analysis.
The experimental results were presented as mean ± standard deviation (SD), and Student’s t-test was used to evaluate differences between groups. P < 0.05 was considered statistically significant.
Results
Somatic p53 ts mutation frequency in cancer.
P53 A135V (mouse) and A138V (human) mutants were frequently used for controlling p53 activity in culture (12,28). The human mutant V143A is also a ts mutation (29). A138V and V143A mutations occur at relatively low frequency in cancer, thus have limited clinical significance. We recently observed that R282W also behaved as a ts mutant similar to A138V in inducing MDM2 and p21 at 32°C (Fig.S1A–D). As one of six p53 hotspot mutants, R282W alone accounts for 3.3% (820/24,679) of p53 missense mutations in cancer according to the COSMIC database (Fig.1A, Table S1). Therefore, we analyzed the p53 mutations curated by the COSMIC database to obtain an estimate of ts mutation frequency in cancer.
Figure 1. Somatic p53 ts mutation frequency and distribution.
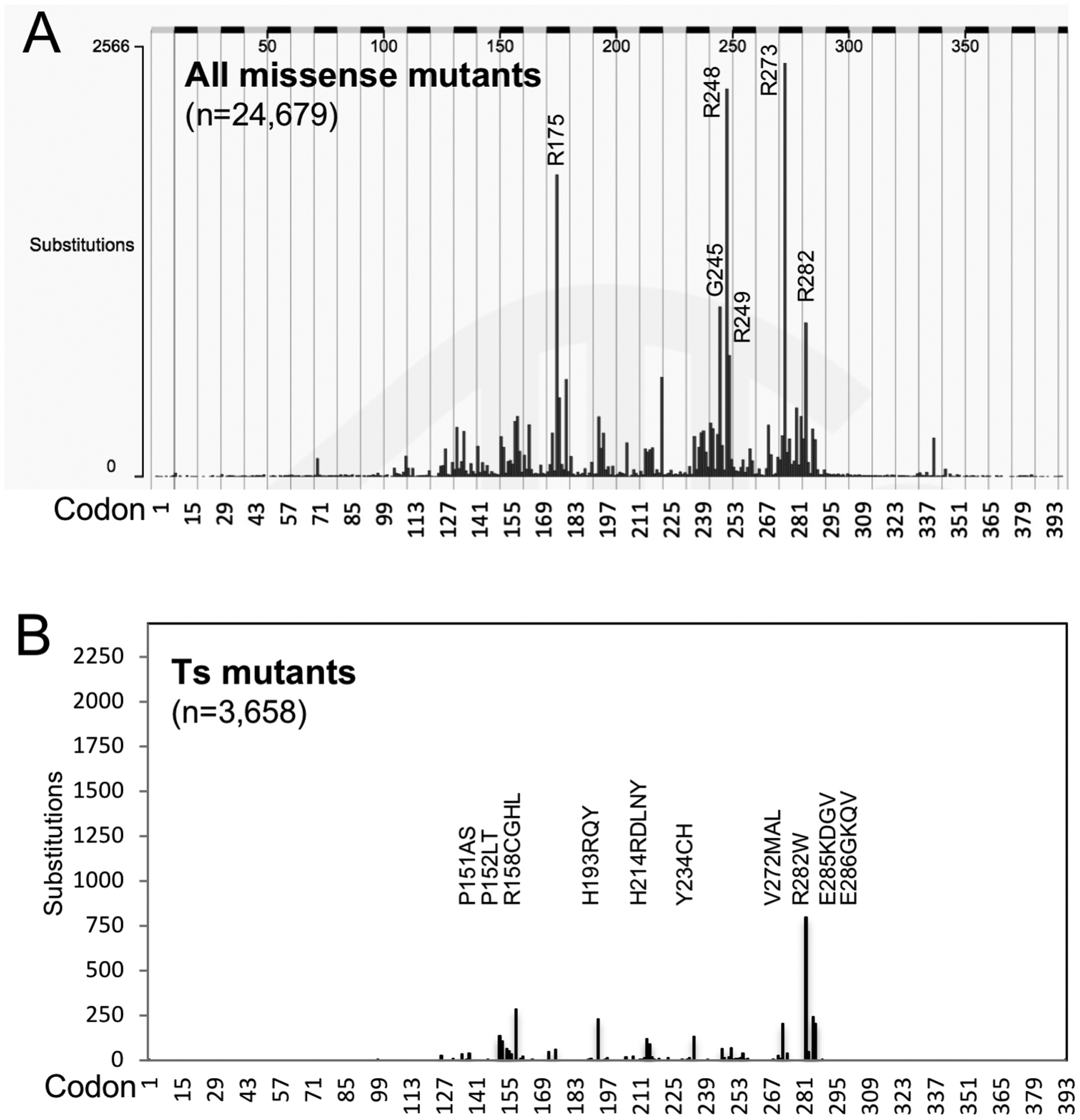
(A) COSMIC plot of p53 missense mutations detected in cancer. (B) P53 ts mutations from Table S1 plotted in the same scale as (A). Top-10 codons that generated the majority of ts mutants are marked.
A previous study by Shiraishi et al using a yeast transactivation assay at 30°C identified 142 p53 ts mutations out of a library of 2,314 mutants covering all single nucleotide mutations in the DNA binding domain (9). The 142 ts mutants were observed in 10.4% of 12,032 tumors with mutant p53 (9). However, the yeast assay did not identify several ts mutants validated in human cells such as R282W, A138V, and V143A. Inclusion of these ts mutants into the yeast-generated ts mutant list and analysis of the current COSMIC database showed that ~14.8% of tumors with p53 missense mutations express ts p53 (3,658/24,679, Table S1, Table S2). Lung and skin tumors had higher than average ts mutation frequency of 18.4% and 18.9% respectively (Table S2). The 3,658 ts mutations affected 62 amino acid residues in the 300-residue DNA binding domain (Table S1). The majority of ts mutations (68%, 2,484/3,658) occurred at 10 ts hotspot codons (Fig.1B).
Validation of ts p53 mutants from cancer.
We selected 17 most frequently observed ts mutants for validation in p53-null H1299 cells. These ts mutants together represent 9.6% (2,358/24,679) of tumors with p53 missense mutations (Table S3), or 64% (2,358/3,658) of tumors with ts p53 mutations. The ability of these mutants to activate the p21 and PUMA promoters at 32°C were confirmed in reporter assays after transient expression (Fig.2A, Fig.S2A–C). The ts mutants were also stably expressed in H1299 at 37°C and tested for induction of MDM2, p21 and PUMA after shifting to 32°C. Although all ts mutants activated the PUMA promoter-luc reporter in transient transfection assay, induction of endogenous PUMA in stably transfected H1299 was modest and in most cases could be stimulated by DNA-damaging agent camptothecin (CPT. Fig.2B, 2C, Fig.S3A–F). CPT appeared to synergize with ts p53 through several mechanisms. ChIP analysis showed that while 32°C alone modestly activated PUMA promoter binding by the ts mutants, CPT treatment further stimulated the binding (Fig.2D). In the absence of p53, CPT modestly stimulated PUMA expression (Fig.2B), suggesting it also primed the PUMA promoter for activation by p53. Furthermore, CPT stimulated ts p53 Ser15 phosphorylation and K382 acetylation, which may increase its DNA binding and transcriptional activity (Fig.2B, 2C).
Figure 2. Validation of p53 ts mutants in human cells.
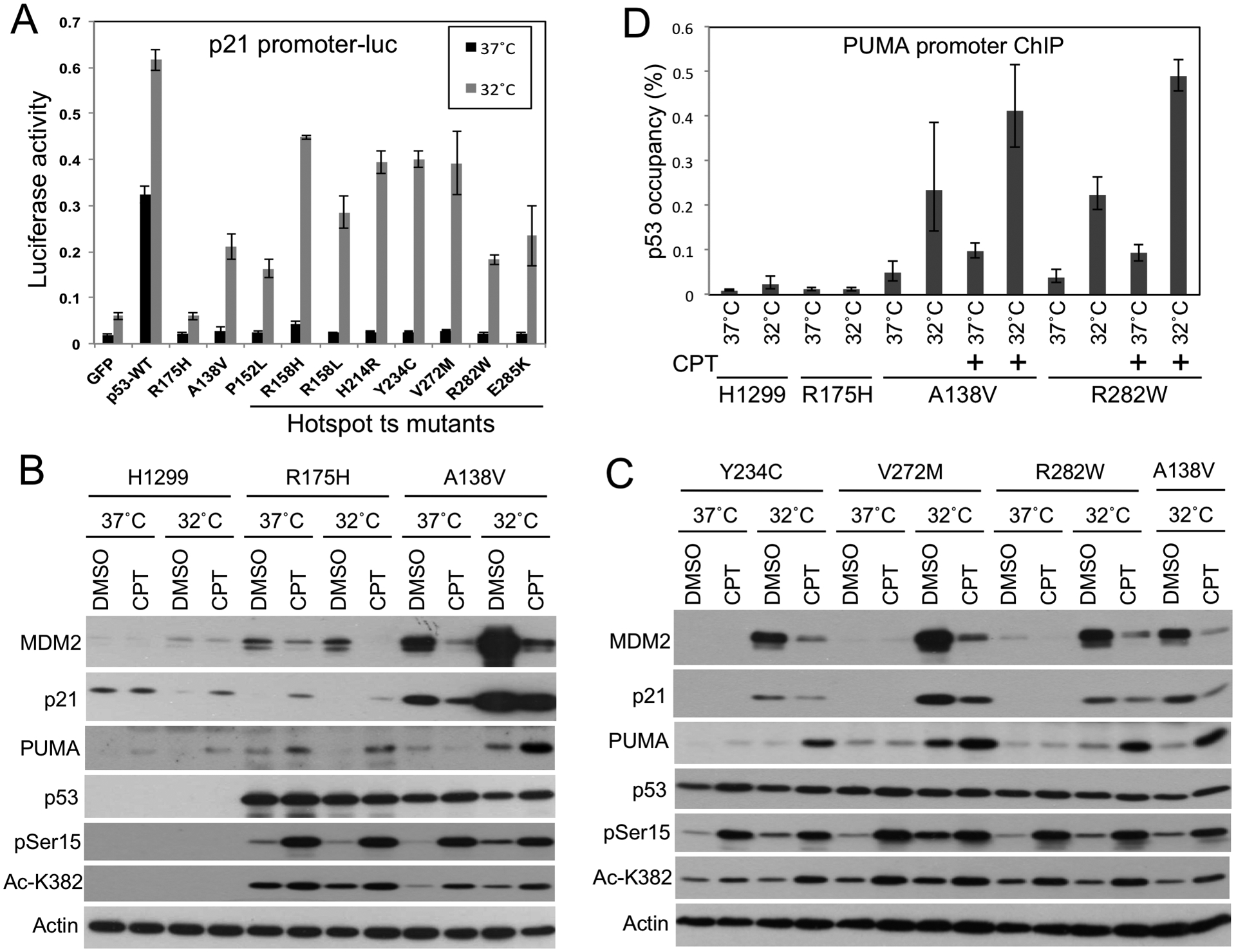
(A) P53 mutants transiently expressed using lentivirus vector in H1299 cells were tested for activation of p21-luc reporter at 37°C, or after shifting to 32°C for 18 hrs. (B,C) Representative p53 ts mutants stably expressed in H1299 cells were tested for induction of target gene expression after 20 hrs at 32°C. CPT (0.5 μM for 20 hrs) was added when cells were shifted to 32°C. (D) H1299 cells stably expressing p53 mutants where treated with 0.5 μM CPT at 32°C for 20 hrs. P53 binding to PUMA promoter was determined by ChIP. The results are average of 3 experiments (mean ± SD).
Activation of the ts mutants alone at 32°C induced H1299 cell cycle arrest as measured by 3H-thymidine incorporation assay (Fig.3A), but no apoptosis was detected (Fig.3B). However, the ts mutants cooperated with CPT to induce apoptosis at 32°C as indicated by PARP cleavage (Fig.3B), and loss of cell viability (Fig.3C, 3D). Apoptotic caspase activation may also explain the decrease of MDM2 in CPT-treated cells (Fig.2B, 2C), since MDM2 is a substrate for Caspase 2 and 3 (30,31). The results suggest the ts mutants regained some of the tumor suppressor functions at 32°C.
Figure 3. Induction of growth arrest and cell death by p53 ts mutants.
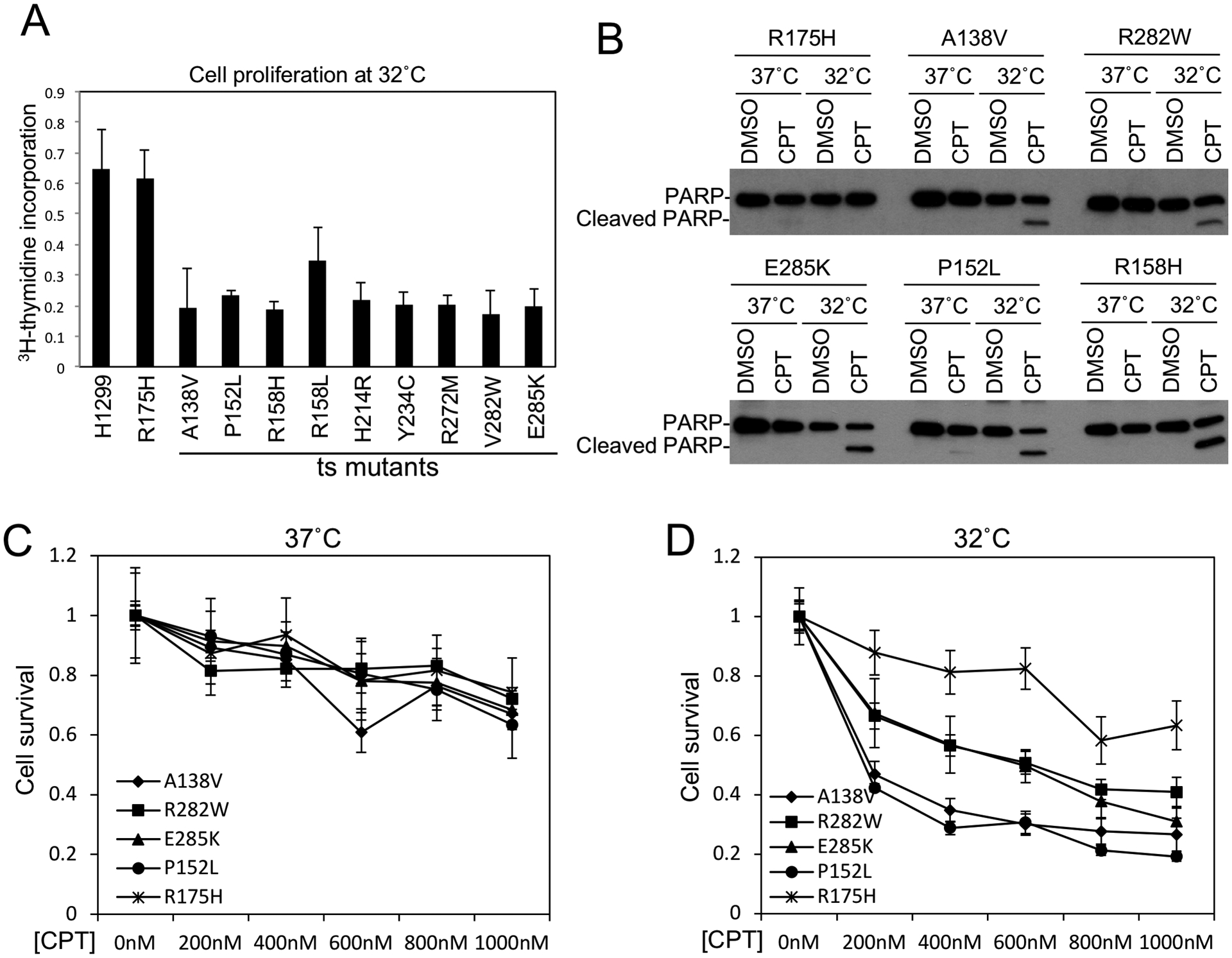
(A) H1299 cells stably infected with lentivirus expressing ts p53 mutants were shifted from 37°C to 32°C for 48 hrs and labeled with 3H-thymidine for 3 hrs. DNA replication rate was measured by scintillation counting and normalized to cell number. (B) H1299 stably expressing ts p53 mutants and non-ts R175H control were treated with 0.5 μM CPT for 48 hrs at 37°C or 32°C in culture medium with 1% FBS. PARP cleavage was determined by Western blot. (C, D) H1299 stably expressing ts p53 mutants were treated with CPT at indicated concentrations for 48 hrs at 37°C or 32°C. Cell viability was determined by MTS assay. The results are average of 3 experiments (mean ± SD).
Molecular chaperones mediate ts p53 refolding at low temperature.
Time course analysis using R158H and R282W showed that ts p53 induction of p21, MDM2 and PUMA were detectable 4 hours after shifting to 32°C and peaked after 12–24 hours (Fig.S4A). When the cells were shifted from 32°C back to 37°C, MDM2 and p21 decreased to background levels in 8–24 hours, whereas PUMA level remained elevated for 24 hours (Fig.S4B). ChIP analysis showed ts p53 DNA binding was activated and peaked 2–4 hours after shifting to 32°C and was completely inactivated if shifted back to 37°C for 1 hour, consistent with rapid heat inactivation (Fig.4A). Inhibition of new protein synthesis by cycloheximide (CHX) did not prevent the rescue of DNA binding at 32°C, indicating that refolding of stockpiled ts p53 contributed to most of the initial activation (Fig.4B). The hsp90 inhibitor 17-AAG blocked p53 activation at 32°C (Fig.4B), suggesting that ts p53 refolding in cells was chaperone-dependent.
Figure 4. Molecular chaperones mediate refolding of stockpiled ts p53 at permissive temperature.
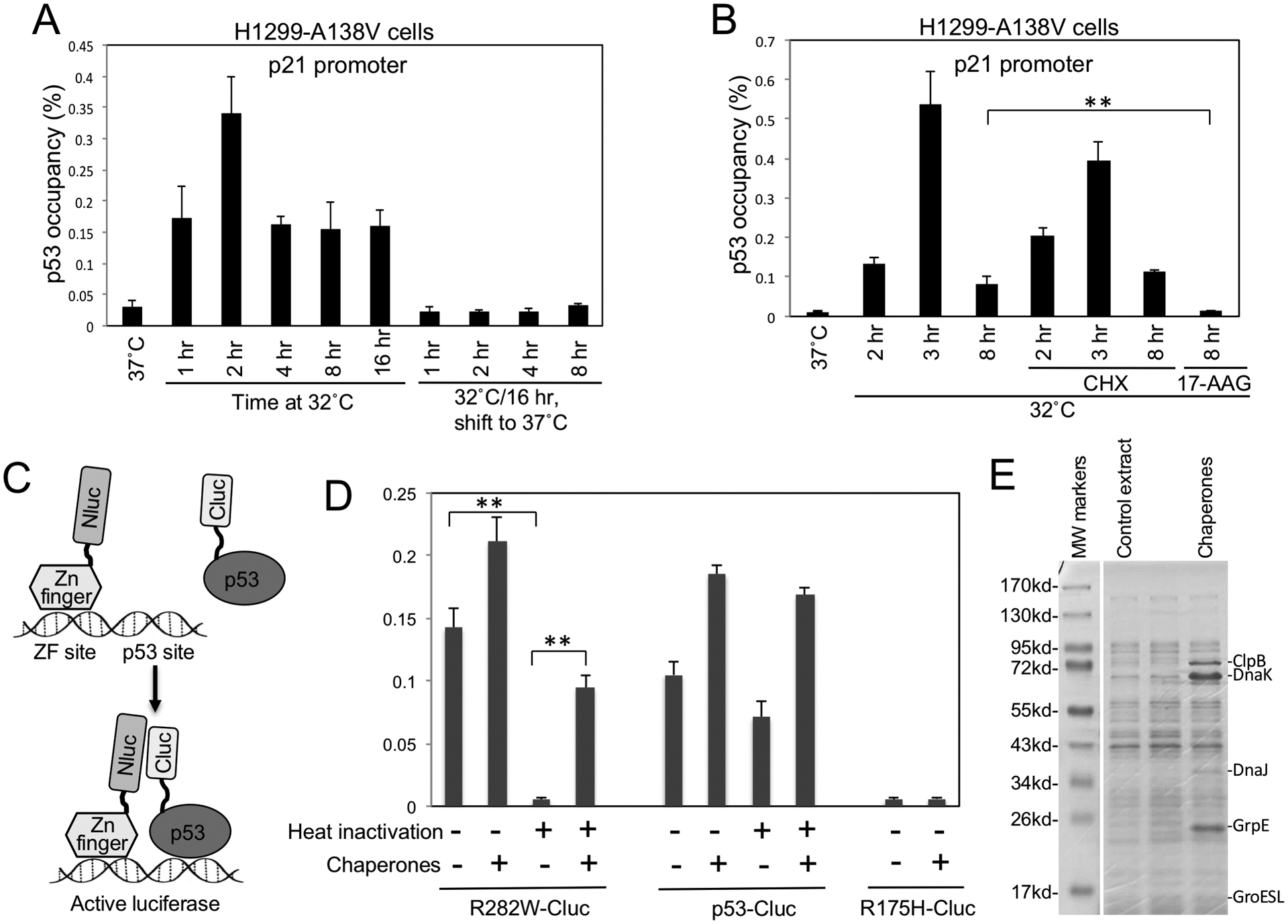
(A) H1299 expressing A138V was shifted from 37°C to 32°C for indicated durations, or pre-incubated at 32°C for 16 hrs and shifted back to 37°C for indicated durations. P53 binding to p21 promoter was determined by ChIP. (B) H1299 cells expressing A138V was shifted to 32°C in the absence or presence of 100 μg/ml cycloheximide (CHX) or 50 μM 17-AAG for indicated durations. P53 DNA binding was determined by ChIP. (C) Diagram of cell-free luciferase fragment complementation assay that detects the DNA binding of p53-Cluc fusion protein. ZF-Nluc and p53-Cluc binding to DNA containing ZF and p53 sites juxtapose Nluc and Cluc domains to restore luciferase activity. (D) P53-Cluc and R282W-Cluc were pre-treated for 30 min at 34°C to inactivate R282W. The heat-treated p53-Cluc proteins were mixed with ZF-Nluc, DNA, ATP, E. coli extract containing 5 chaperones and incubated for 3 hrs at 23°C. P53 DNA binding was detected by measuring luciferase activity. The results are average of 3 experiments (mean ± SD). **p<0.01. (E) Coomassie staining of E. coli extract expressing molecular chaperones.
To further test whether misfolded ts p53 can post-translationally refold at low temperature, we monitored the refolding using an in vitro system. We recently established a cell-free assay based on luciferase fragment complementation to detect DNA binding by p53-Cluc fusion protein (Fig.4C) (32). R282W-Cluc produced in 16°C E. coli culture was active in DNA binding at 23°C, but was inactivated after incubating at 34°C; in contrast, wt p53-Cluc remained active after heating at 34°C (Fig.4D). Incubation of heat-inactivated R282W-Cluc at 23°C for 3 hours did not restore DNA binding. However, co-incubation with a mixture of chaperones (DnaK, DnaJ, ClpB, GrpE, GroESL, Fig.4E) restored ~70% of R282W-Cluc DNA binding (Fig.4D) (33). The chaperones did not rescue the non-ts mutant R175H-Cluc (Fig.4D). Wt p53-Cluc was modestly resistant to heating at 34°C, its DNA binding was stimulated ~2-fold by the chaperones (Fig.4D), suggesting there was refolding of a spontaneously denatured sub-population. The results suggest that post-translational refolding of denatured ts p53 at low temperatures required the assistance of molecular chaperones.
Activation of endogenous ts p53 mutants in tumor cell lines.
The IARC TP53 Database showed that ts p53 mutations frequently detected in cancer are also present in many tumor cell lines (Table S3). The correlation suggests the ts mutations in the cell lines were originated from the tumors. Analysis of 7 tumor cell lines expressing ts p53 (6 of the 7 ts mutants were also tested in H1299 cells, Fig.2, Fig.S3) showed increased MDM2, p21, and PUMA expression at 32°C, suggesting the ts p53 mutants were activated in their natural context (Fig.5A). Cell lines with wt p53 or non-ts p53 mutant were not activated at 32° (Fig.S5A). Knockout of endogenous ts p53 by CRISPR/Cas9 gene editing in 3 cell lines abrogated the induction of MDM2/p21/PUMA and apoptosis at 32°C (Fig.5B, Fig.S5B/C), indicating the response was mediated by ts p53. Long incubation of GA10 lymphoma cells at 32°C led to cell death and population decline (Fig.5C), whereas GA10-p53KO cells continued to proliferate at 32°C (slower than 37°C as expected from less active metabolism). Therefore, endogenous ts p53 can be activated at 32°C to induce arrest or apoptosis. Analysis of DNA synthesis using 3H-thymidine labeling confirmed cell cycle arrest of lung tumor lines H441 (R158L) and H1355 (E285K) at 32°C (Fig.S6A). Annexin V staining also showed p53-mediated apoptosis in GA10 (P152L) cells at 32°C (Fig.S6B, S6C, S6D).
Figure 5. Activation of endogenous ts p53 mutants in tumor cell lines.
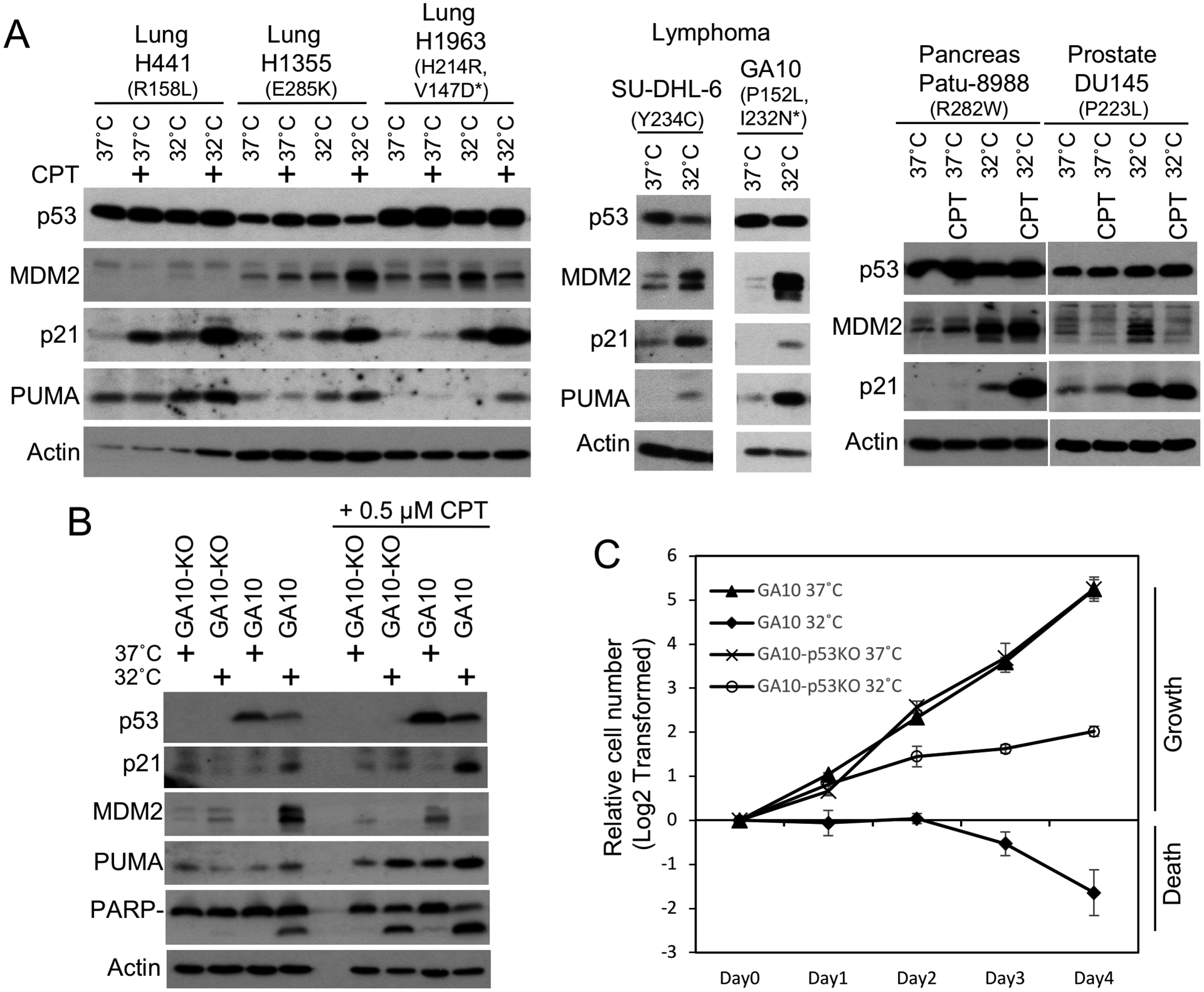
(A) Cell lines with endogenous ts mutant p53 (non-ts mutant alleles are marked by *) were cultured at 37°C or shifted to 32°C for 24 hrs and analyzed by Western blot. CPT was added to 0.5 μM where indicated. (B) GA10 cells were infected with lentivirus expressing Cas9 and p53 gRNA. A clonal cell line without p53 was analyzed by Western blot after culturing at 37°C or 32°C for 24 hrs. (C) GA10 cells with and without p53 knockout were seeded at identical starting numbers and cultured at 37°C or 32°C. Cell numbers were counted daily and plotted. The results are average of 3 experiments (mean ± SD).
Tumors with wt p53 often have silenced ARF expression that result in increased MDM2 activity, whereas tumors with mutant p53 generally retain ARF (34,35). The status of ARF in tumors with ts p53 have not been reported. RT-PCR analysis of 7 tumor cell lines with ts p53 showed they expressed various levels of ARF mRNA similar to non-ts mutant or p53-null cells (Fig.S6E). In contrast, 3 cell lines with wt p53 had no detectable ARF mRNA (Fig.S6E). The result suggests that ts p53 mutations also eliminate the selection pressure to silence ARF. The presence of ARF in the ts cell lines should limit MDM2 activity and facilitate p53 activation at 32°C. Consistent with this notion, despite increased MDM2 level at 32°C there was only modest or no down regulation of p53 in the ts cell lines (Fig.5A).
Pharmacological induction of hypothermia.
To test the effect of hypothermia on tumors with ts p53, the adenosine A1 receptor (A1AR) agonist CHA (N6-cyclohexyladenine) was used to lower mouse body temperature to 32°C for 24–48 hours. Thermal imaging of nude mice in standard ambient condition (23°C) showed anterior skin temperature of 37–38°C, providing a non-invasive measure of core temperature (Fig.6A). Intraperitoneal injection of CHA caused body temperature to decrease to ~3°C above the environment in ~10 minutes, consistent with the induction of hibernation-like response in rats and mice by A1AR agonists (36,37). Cell culture test suggested that 24 hours at 32°C was needed for optimal ts p53 activation (Fig.S4). This was achieved by keeping the CHA-treated mice in a 28°C environment (Fig.6A). Using multiple injections of CHA it was possible to maintain body temperature at 32°C for 24–32 hours. The procedure can be repeated 5–6 times with 2–4 days between treatments to allow recovery (referred to as 24 hr × 6 or 32 hr × 5 formats).
Figure 6. Hypothermia inhibits the growth of tumors expressing ts p53.
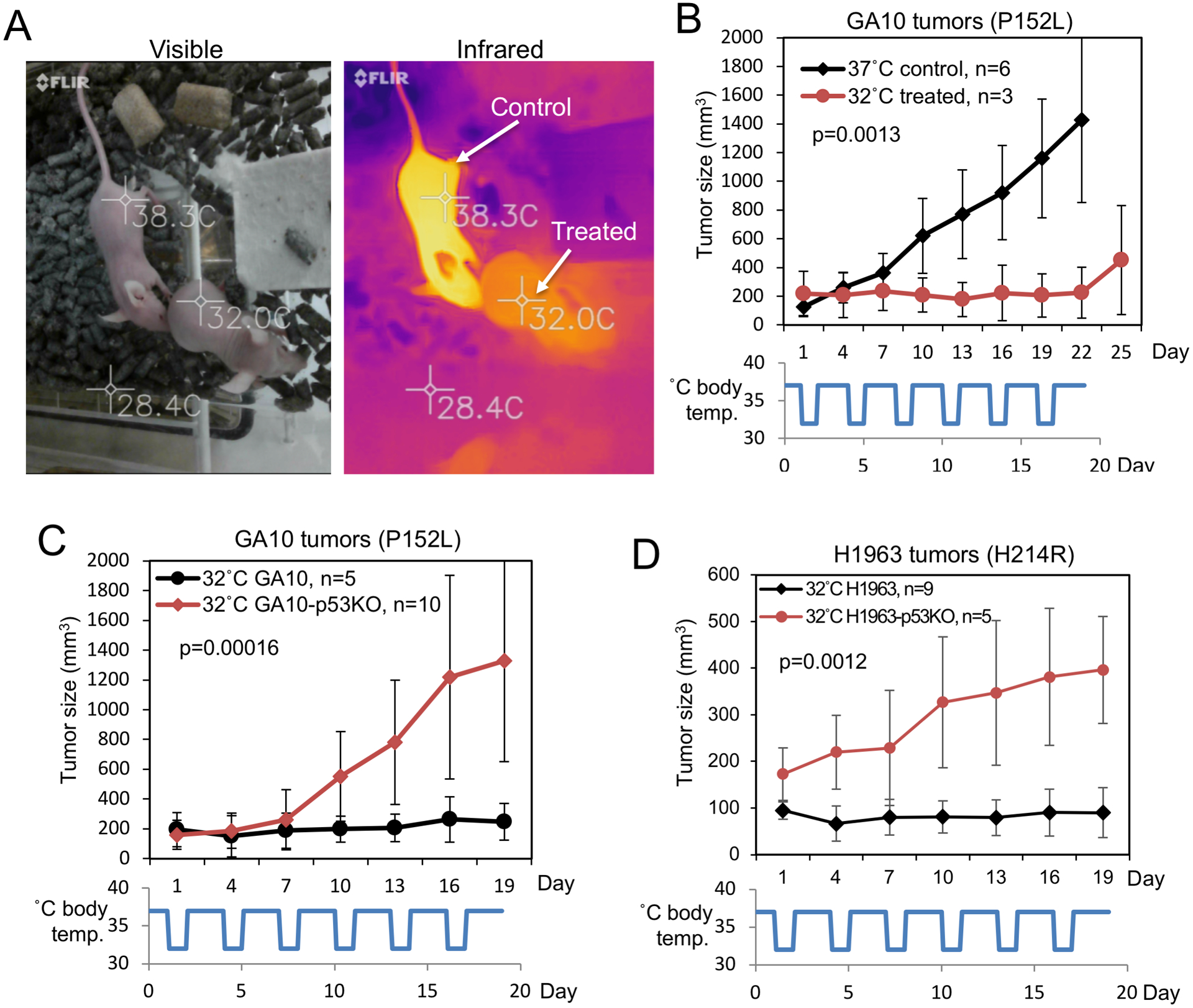
(A) Nude mice were injected with CHA (N6-cyclohexyladenine) and kept in a 28°C environment to maintain body temperature at 32°C. (B) Nude mice were inoculated subcutaneously with GA10 cells and tumors were allowed to grow to ~200 mm3. One group of mice was treated with 32°C hypothermia in 24 hr × 6 format. Control animals (37°C) were not treated. (C) GA10 and GA10-p53KO tumors were treated with 32°C hypothermia in 24 hr × 6 format and tumor growth was monitored. (D) H1963 and H1963-p53KO subcutaneous tumors were treated with 32°C hypothermia in 24 hr × 6 format and tumor growth was monitored. Tumor size (mean ± SD) was plotted over time.
Hypothermia inhibits the growth of tumors expressing ts p53.
To test the effect of ts p53 activation in vivo, nude mice bearing GA10 B-cell lymphoma subcutaneous xenografts were treated with 6 rounds of hypothermia, each round lowering body temperature to ~32°C for 24 hours (24 hr × 6 format). Tumor growth was attenuated during the treatments, but resumed after the treatments were stopped (Fig.6B). Untreated 37°C tumors grew rapidly (Fig.6B). GA10-p53KO tumors were not inhibited by hypothermia (Fig.6C), suggesting the effect of hypothermia was mediated by ts p53. Hypothermia also inhibited the growth of H1963 lung tumor xenografts, but did not stop H1963-p53KO tumors (Fig.6D). In 37°C control animals (no hypothermia treatment), p53 knockout did not affect the tumor growth rates of GA10/GA10-p53KO and H1963/H1963-p53KO in pair-wise comparisons (Fig.S7A, S7B). The results suggest that hypothermia activated the endogenous ts mutant p53 to inhibit tumor growth.
Hypothermia cooperates with chemotherapy to induce tumor regression.
To test whether ts p53 activation cooperates with chemotherapy, large GA10 and GA10-p53KO tumors (~300–1000 mm3) were treated with hypothermia in combination with CPT. CPT was given at a low dose (1.5 mg/kg) that modestly inhibited GA10 tumor growth at 37°C but did not cause shrinkage (Fig.S7C). At 37°C body temperature (p53 inactive), GA10 and GA10-p53KO tumors showed no difference in response to CPT (Fig.S7D). Hypothermia + CPT combination (in 32 hr × 5 format) induced significant regression of GA10 tumors (Fig.7A, 7B, 7C). Furthermore, 32% (12/37) of GA10 tumors did not relapse in the 50-day observation period after stopping treatment (Fig.7C). In contrast, GA10-p53KO tumors did not achieve complete response or durable remission, the p53-independent effects of CPT and hypothermia only led to modest shrinkage (Fig.7A, Fig.7C).
Figure 7. Hypothermia cooperates with chemotherapy to induce regression of tumors expressing ts p53.
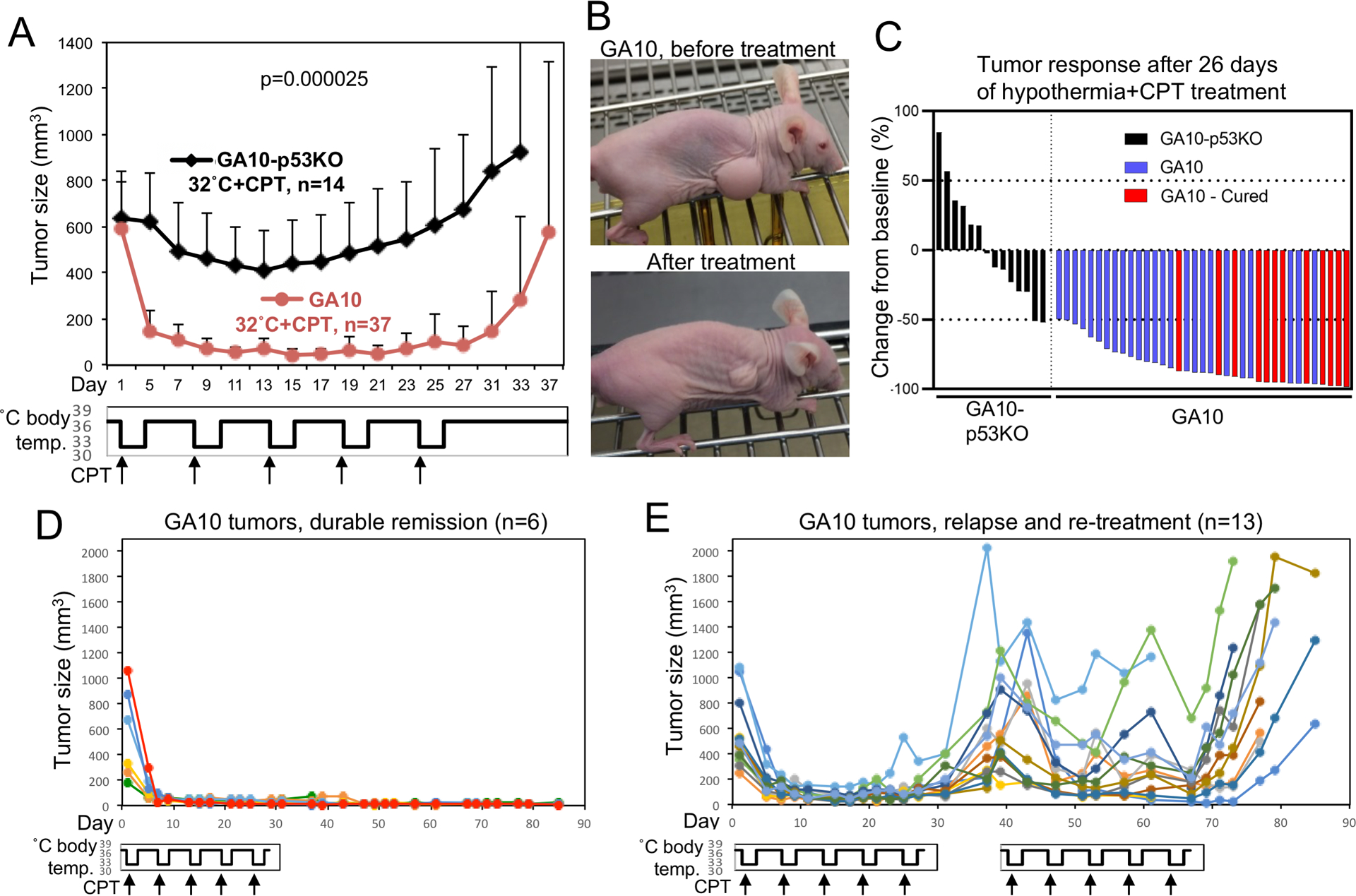
(A) Nude mice with subcutaneous GA10 and GA10-p53KO tumors were treated with combination of CPT and 32°C hypothermia in 32 hr × 5 format. CPT (1.5 mg/kg) was given at the beginning of each hypothermia cycle. Average size of tumors was plotted over time (mean ± SD). (B) Pictures of a GA10 partial response tumor before and after treatment. (C) Waterfall plot showing combined results of 3 experiments. Tumors with durable remission are marked as “cured”. (D) Individual growth curves of representative GA10 tumors from (A) that achieved durable remission after treatment. (E) Individual growth curves of GA10 tumors from (A) that relapsed and received re-treatment.
Growth curves of individual tumors with and without relapse showed that durable remissions occurred after achieving >80% shrinkage during the initial treatment (Fig.7D). Re-treatment of relapsed GA10 tumors resulted in weaker responses compared to the initial cycles, and did not produce new durable remission (Fig.7E). As expected, GA10-p53KO tumors responded poorly to both initial and repeat treatments (Fig.S8A). Western blot showed PARP cleavage and PUMA expression in GA10 tumors (but not GA10-p53KO) treated for 24 hours with the combination, consistent with ts p53-mediated apoptosis in vivo (Fig.S8B). The apoptosis in GA10 tumors was also confirmed by TUNEL staining (Fig.S9). The results showed that ts p53 activation cooperated with chemotherapy to induce tumor regression and durable remission.
Discussion
P53 inactivation in cancer occurs predominantly by missense mutations that result in accumulation of misfolded protein, providing a tumor-specific target (2,3). Conformational rescue of mutant p53 has been a long-standing challenge in drug development. Since ~15% of p53 missense mutants regain activity at 32°C, hypothermia should rescue this subclass of mutants without specific drugs. Physical cooling should activate ts p53 uniformly, bypassing common limitations in drug delivery. Furthermore, hypothermia does not damage normal tissues, whereas small molecules often have off-target toxicity. The results described in this study demonstrate that ts mutant p53 can be activated in tumors by inducing whole-body hypothermia, resulting in synergistic anti-tumor effects when combined with chemotherapy. The finding raises the possibility of repurposing therapeutic hypothermia for tumors with ts p53 mutations.
Tumor development selects for p53 mutations that inactivate DNA binding. Apparently many ts mutants are also selected because they are inactive at 37°C. The p53 ts mutations analyzed in the current work were defined by their re-activation at 32°C in yeast and mammalian cell culture (9). Therefore, the animal hypothermia experiments were also performed at 32°C in order to balance refolding efficiency, cellular metabolic activity, and clinical relevance. Analysis of 17 frequently observed p53 ts mutants (affecting ~64% of tumors expressing ts p53) showed they regained similar ability to induce target genes and growth arrest at 32°C, suggesting that similar results can be expected for other ts mutants.
Wt p53 is tightly regulated by the MDM2 feedback loop. A subset of tumors bypass the need to mutate p53 by silencing ARF or overexpressing MDM2 (34,35). Tumors with mutant p53 typically accumulate the p53 protein to high levels, partly due to inability to induce MDM2 and oncogene-activated expression of ARF. Activating ts p53 should in principle trigger its own degradation by MDM2. However, analysis of tumor cell lines expressing endogenous ts p53 showed no significant degradation at 32°C despite induction of MDM2. Ts p53 tumor cell lines retained ARF mRNA expression similar to non-ts mutant cell lines, possibly played a role in preventing p53 degradation at 32°C and sensitizing them to the cell cycle arrest or apoptotic effects of rescued p53.
If tumor development selects for complete loss of p53 activity, tissues that are not constantly maintained at 37°C (such as skin and breast) should have lower frequency of ts p53 mutations since they will be partially active and have less advantage. The COSMIC database shows that breast tumors have below-average ts p53 mutation frequency as expected, but surprisingly skin cancer has above-average frequency (Table S2). The reason for the discrepancy remains to be determined. It is possible that exposure to specific mutagens (i.e., UV irradiation) distorted the ts p53 mutation frequency in skin cancer. Alternatively, skin cancer may have more frequent ARF silencing or MDM2 overexpression that neutralize residual ts p53 activity at below-37°C temperatures.
Most ts p53 mutations are located at the hydrophobic β sandwich core of the DNA binding domain that reduce thermo stability (38). Our results suggest that ts p53 exists in a dynamic equilibrium between misfolded and wt conformations in the cell. Post-translational refolding of mutant protein contributes to rapid restoration of activity at 32°C. Furthermore, molecular chaperones are essential for rapid refolding at permissive temperature. Cell-free analysis showed that ts p53 cannot spontaneously regain wt conformation at low temperature without the help of molecular chaperones. The multi-domain organization and tetrameric status of p53 may lead to dependence on chaperones for accurate refolding. Previous work showed that optimal wt p53 activity in cells at 37°C required hsp90 (39). Hsp40/Hsp70 and Hsp90 exert opposing activities on the p53 DNA binding domain to maintain its conformation at a dynamic equilibrium (40,41).
Our results demonstrated that hypothermia can induce rapid regression of lymphomas and frequently achieved durable remission. Therefore, its potential in treating tumors with ts p53 warrants further investigation. The hypothermia procedure currently used in the clinic is performed under intensive care setting. Repurposing this procedure for cancer will require significant therapeutic benefits. Development of non-invasive methods to induce hypothermia will lower the threshold for translation to cancer treatment. Recent work identified specific neurons in the mouse hypothalamus that are sufficient to induce hypothermia upon stimulation (42,43). Hypothermia induction by anti-psychotic drugs is also well-documented (25). It is important to note that hypothermia affects energy metabolism, pharmacodynamics, immune functions, and physiology. Further studies will be needed to translate this strategy for cancer therapy.
Supplementary Material
Statement of Significance.
Pharmacological inhibition of brain-regulated thermogenesis and induction of 32°C whole-body hypothermia specifically targets tumors with temperature-sensitive p53 mutations, rescuing p53 transcriptional activity and inducing tumor regression.
Acknowledgements
The authors wish to thank the Moffitt Tissue Core and Flow Cytometry Core for their assistance. This work was supported by grants from the National Institutes of Health (CA141244, CA186917, GM115556) and Florida Department of Health (9BC09, 20B17) to J.Chen. H. Lee Moffitt Cancer Center & Research Institute is an NCI designated Comprehensive Cancer Center (P30-CA076292).
Footnotes
Conflict of interest: The authors declare they have no conflict of interests.
References
- 1.Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol 2007;8:275–83 [DOI] [PubMed] [Google Scholar]
- 2.Leroy B, Anderson M, Soussi T. TP53 mutations in human cancer: database reassessment and prospects for the next decade. Hum Mutat 2014;35:672–88 [DOI] [PubMed] [Google Scholar]
- 3.Joerger AC, Fersht AR. The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu Rev Biochem 2016;85:375–404 [DOI] [PubMed] [Google Scholar]
- 4.Peng Y, Chen L, Li C, Lu W, Chen J. Inhibition of MDM2 by hsp90 contributes to mutant p53 stabilization. J Biol Chem 2001;276:40583–90 [DOI] [PubMed] [Google Scholar]
- 5.Li D, Marchenko ND, Schulz R, Fischer V, Velasco-Hernandez T, Talos F, et al. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Molecular cancer research : MCR 2011;9:577–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nature reviews Drug discovery 2014;13:217–36 [DOI] [PubMed] [Google Scholar]
- 7.Bullock AN, Fersht AR. Rescuing the function of mutant p53. Nat Rev Cancer 2001;1:68–76 [DOI] [PubMed] [Google Scholar]
- 8.Bullock AN, Henckel J, DeDecker BS, Johnson CM, Nikolova PV, Proctor MR, et al. Thermodynamic stability of wild-type and mutant p53 core domain. Proc Natl Acad Sci U S A 1997;94:14338–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shiraishi K, Kato S, Han SY, Liu W, Otsuka K, Sakayori M, et al. Isolation of temperature-sensitive p53 mutations from a comprehensive missense mutation library. J Biol Chem 2004;279:348–55 [DOI] [PubMed] [Google Scholar]
- 10.Scharer E, Iggo R. Mammalian p53 can function as a transcription factor in yeast. Nucleic Acids Res 1992;20:1539–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Como CJ, Prives C. Human tumor-derived p53 proteins exhibit binding site selectivity and temperature sensitivity for transactivation in a yeast-based assay. Oncogene 1998;16:2527–39 [DOI] [PubMed] [Google Scholar]
- 12.Yonish-Rouach E, Resnitzky D, Lotem J, Sachs L, Kimchi A, Oren M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature 1991;352:345–7 [DOI] [PubMed] [Google Scholar]
- 13.Pochampally R, Fodera B, Chen L, Lu W, Chen J. Activation of an MDM2-specific caspase by p53 in the absence of apoptosis. J Biol Chem 1999;274:15271–7 [DOI] [PubMed] [Google Scholar]
- 14.Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 2006;127:1323–34 [DOI] [PubMed] [Google Scholar]
- 15.Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, et al. Restoration of p53 function leads to tumour regression in vivo. Nature 2007;445:661–5 [DOI] [PubMed] [Google Scholar]
- 16.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007;445:656–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gavrielatos G, Werner KD, Voridis E, Kremastinos DT. Contemporary practices in postcardiac arrest syndrome: the role of mild therapeutic hypothermia. Ther Adv Cardiovasc Dis 2010;4:325–33 [DOI] [PubMed] [Google Scholar]
- 18.Dietrich WD, Bramlett HM. Therapeutic hypothermia and targeted temperature management in traumatic brain injury: Clinical challenges for successful translation. Brain Res 2016;1640:94–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holzer M Targeted temperature management for comatose survivors of cardiac arrest. N Engl J Med 2010;363:1256–64 [DOI] [PubMed] [Google Scholar]
- 20.Shankaran S, Laptook AR, Ehrenkranz RA, Tyson JE, McDonald SA, Donovan EF, et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med 2005;353:1574–84 [DOI] [PubMed] [Google Scholar]
- 21.Zhang M, Wang H, Zhao J, Chen C, Leak RK, Xu Y, et al. Drug-induced hypothermia in stroke models: does it always protect? CNS Neurol Disord Drug Targets 2013;12:371–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jinka TR, Toien O, Drew KL. Season primes the brain in an arctic hibernator to facilitate entrance into torpor mediated by adenosine A(1) receptors. J Neurosci 2011;31:10752–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jinka TR, Combs VM, Drew KL. Translating drug-induced hibernation to therapeutic hypothermia. ACS Chem Neurosci 2015;6:899–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laughlin BW, Bailey IR, Rice SA, Barati Z, Bogren LK, Drew KL. Precise Control of Target Temperature Using N(6)-Cyclohexyladenosine and Real-Time Control of Surface Temperature. Ther Hypothermia Temp Manag 2018;8:108–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tarahovsky YS, Fadeeva IS, Komelina NP, Khrenov MO, Zakharova NM. Antipsychotic inductors of brain hypothermia and torpor-like states: perspectives of application. Psychopharmacology (Berl) 2017;234:173–84 [DOI] [PubMed] [Google Scholar]
- 26.Kreuzer P, Landgrebe M, Wittmann M, Schecklmann M, Poeppl TB, Hajak G, et al. Hypothermia associated with antipsychotic drug use: a clinical case series and review of current literature. J Clin Pharmacol 2012;52:1090–7 [DOI] [PubMed] [Google Scholar]
- 27.Jiang JY, Xu W, Li WP, Gao GY, Bao YH, Liang YM, et al. Effect of long-term mild hypothermia or short-term mild hypothermia on outcome of patients with severe traumatic brain injury. J Cereb Blood Flow Metab 2006;26:771–6 [DOI] [PubMed] [Google Scholar]
- 28.Yamato K, Yamamoto M, Hirano Y, Tsuchida N. A human temperature-sensitive p53 mutant p53Val-138: modulation of the cell cycle, viability and expression of p53-responsive genes. Oncogene 1995;11:1–6 [PubMed] [Google Scholar]
- 29.Friedlander P, Legros Y, Soussi T, Prives C. Regulation of mutant p53 temperature-sensitive DNA binding. J Biol Chem 1996;271:25468–78 [DOI] [PubMed] [Google Scholar]
- 30.Chen L, Marechal V, Moreau J, Levine AJ, Chen J. Proteolytic cleavage of the mdm2 oncoprotein during apoptosis. J Biol Chem 1997;272:22966–73 [DOI] [PubMed] [Google Scholar]
- 31.Oliver TG, Meylan E, Chang GP, Xue W, Burke JR, Humpton TJ, et al. Caspase-2-mediated cleavage of Mdm2 creates a p53-induced positive feedback loop. Mol Cell 2011;43:57–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He F, Borcherds W, Song T, Wei X, Das M, Chen L, et al. Interaction between p53 N terminus and core domain regulates specific and nonspecific DNA binding. Proc Natl Acad Sci U S A 2019;116:8859–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Marco A, Deuerling E, Mogk A, Tomoyasu T, Bukau B. Chaperone-based procedure to increase yields of soluble recombinant proteins produced in E. coli. BMC Biotechnol 2007;7:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stott FJ, Bates S, James MC, McConnell BB, Starborg M, Brookes S, et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. Embo J 1998;17:5001–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev 1999;13:2658–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tupone D, Madden CJ, Morrison SF. Central activation of the A1 adenosine receptor (A1AR) induces a hypothermic, torpor-like state in the rat. J Neurosci 2013;33:14512–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Futatsuki T, Yamashita A, Ikbar KN, Yamanaka A, Arita K, Kakihana Y, et al. Involvement of orexin neurons in fasting- and central adenosine-induced hypothermia. Sci Rep 2018;8:2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joerger AC, Ang HC, Fersht AR. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc Natl Acad Sci U S A 2006;103:15056–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walerych D, Kudla G, Gutkowska M, Wawrzynow B, Muller L, King FW, et al. Hsp90 chaperones wild-type p53 tumor suppressor protein. J Biol Chem 2004;279:48836–45 [DOI] [PubMed] [Google Scholar]
- 40.Boysen M, Kityk R, Mayer MP. Hsp70- and Hsp90-Mediated Regulation of the Conformation of p53 DNA Binding Domain and p53 Cancer Variants. Mol Cell 2019;74:831–43 e4 [DOI] [PubMed] [Google Scholar]
- 41.Dahiya V, Agam G, Lawatscheck J, Rutz DA, Lamb DC, Buchner J. Coordinated Conformational Processing of the Tumor Suppressor Protein p53 by the Hsp70 and Hsp90 Chaperone Machineries. Mol Cell 2019;74:816–30 e7 [DOI] [PubMed] [Google Scholar]
- 42.Takahashi TM, Sunagawa GA, Soya S, Abe M, Sakurai K, Ishikawa K, et al. A discrete neuronal circuit induces a hibernation-like state in rodents. Nature 2020;583:109–14 [DOI] [PubMed] [Google Scholar]
- 43.Hrvatin S, Sun S, Wilcox OF, Yao H, Lavin-Peter AJ, Cicconet M, et al. Neurons that regulate mouse torpor. Nature 2020;583:115–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.