

SUMMARY
Many long noncoding RNA (lncRNA) genes exist near cancer-associated loci, yet evidence connecting lncRNA functions to recurrent genetic alterations in cancer are lacking. Here, we report that DINO, the lncRNA transcribed from the cancer-associated DINO/CDKN1A locus, suppresses tumor formation independent of p21, the protein encoded at the locus. Loss of one or two alleles of Dino impairs p53 signaling and apoptosis, resulting in a haplo-insufficient tumor suppressor phenotype in genetically defined mouse models of tumorigenesis. A discrete region of the DINO/CDKN1A locus is recurrently hypermethylated in human cancers, silencing DINO but not CDKN1A, the gene encoding p21. Hypermethylation silences DINO, impairs p53 signaling pathway in trans, and is mutually exclusive with TP53 alterations, indicating that DINO and TP53 comprise a common tumor suppressor module. Therefore, DINO encodes a lncRNA essential for tumor suppression that is recurrently silenced in human cancers as a mechanism to escape p53-dependent tumor suppression.
Graphical Abstract
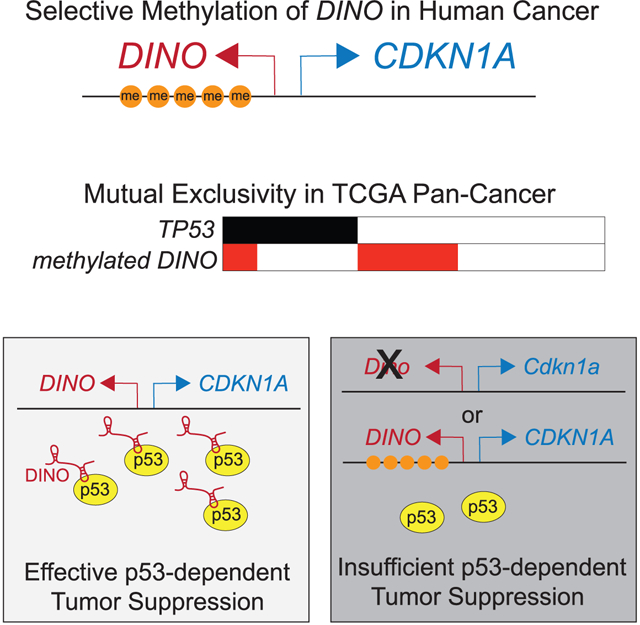
In brief
Tumorigenesis requires escape from the p53 tumor suppressor protein. How 50% of cancers develop despite an intact p53 pathway remains largely unknown. Marney et al. report here that loss of the lncRNA DINO provides a route to tumorigenesis in p53-intact cells, and DINO is recurrently silenced in human cancers.
INTRODUCTION
Genetic alterations and epigenomic CpG DNA methylation targeting tumor suppressor genes create a permissive state for tumorigenesis and are defining features of cancer (Hanahan and Weinberg, 2011; Flavahan et al., 2017). The tumor suppressor TP53 is the most frequently altered gene in human tumors, occurring in ~40% of cancers. MDM2, an E3 ubiquitin ligase that negative regulates p53 by targeting it for proteasomal degradation, is amplified in ~2% of human cancers. MDM2 amplification and TP53 mutations are generally mutually exclusive genetic alterations in human cancers. This suggested that MDM2 amplification may be sufficient for escape from p53-dependent tumor suppressor functions in TP53 intact tumors and was confirmed in genetically defined animal models. However, other components of the TP53 signaling pathway are rarely mutated in human cancers and few p53 target genes are individually required for the tumor suppressor phenotype. Indeed, putative tumor suppressor components of the p53 pathway, such as p21, lincRNA-p21, and miR-34, are not required for tumor suppression in rigorous, genetically defined animal models. Therefore, the mechanism by which human cancers can escape p53-dependent tumor suppression without altering TP53 or MDM2 remains a largely unanswered question.
Genome-wide association studies have identified numerous cancer-associated loci throughout the human genome, indicating that they may encode critical tumor suppressor functions. However, in many cases the identity of the genes disrupted at the loci and mechanism by which they contribute to cancer remain ambiguous. One of these, the CDKN1A locus, is a principal target of p53 during genotoxic and oncogenic stress, and genetic and epigenomic variation within the noncoding regulatory genome near the CDKN1A locus is associated with cancer (Dunlop et al., 2012; Béguelin et al., 2013; Hu et al., 2014; Roman-Gomez et al., 2002; Chambwe et al., 2014; Arribas et al., 2015). The protein coding gene at this locus, p21, is itself rarely mutated in human cancers, and genetically defined mouse models indicate that the p53 tumor suppressor pathway remains intact despite loss of p21 (Abbas and Dutta, 2009; Newbold et al., 2014; Valente et al., 2016). This suggests genetic information encoded at the CDKN1A locus other than the protein product p21 may be responsible for the loci’s tumor suppressor function.
Recent human transcriptome assemblies have identified nearly 59,000 long noncoding RNA (lncRNA) genes, many of which are located within or near protein-coding genes (Iyer et al., 2015). Many tumor suppressor loci contain both protein-coding and lncRNA genes, raising the possibility that lncRNA genes may contribute to the tumor suppressor functions of these loci. Although in vitro experimental data have indicated that lncRNAs participate in many essential cellular processes and regulate numerous cancer phenotypes, rigorous genetic analyses of lncRNA functions in cancer development are generally lacking, generating doubt about the role of these genes in human disease (Liu et al., 2017; Schmitt and Chang, 2016; Wang et al., 2018). We previously identified DINO as a lncRNA gene located within the CDKN1A locus that is required for p53 signaling in response to DNA damage (Schmitt et al., 2016). DINO binds to the p53 protein, increases p53 stability, and enhances p53 signaling. We set out to determine the functional contributions of DINO to the tumor suppressor phenotype encoded within the shared DINO/CDKN1A locus.
RESULTS
A genetic association in human cancers identifies a functional tumor suppressor network involving TP53 and the DINO/CDKN1A locus
We examined The Cancer Genome Atlas (TCGA) Pan-Cancer for recurrent genetic or epigenomic alterations of the DINO/CDKN1A locus and their relationship with TP53 alterations as indicator of potential functional association with the p53-dependent tumor suppressor pathway. Although somatic mutations and deep deletion of the DINO/CDKN1A locus are rare, epigenomic silencing has been reported in several types of human malignancies through mechanisms such as DNA methylation and repressive histone modifications (Béguelin et al., 2013; Roman-Gomez et al., 2002; Chambwe et al., 2014; Arribas et al., 2015). We found two distinct differentially methylated regions (DMRs) within the DINO/CDKN1A locus, one consisting of six CpGs downstream of the DINO transcription start site (TSS) (DMR1) and a second consisting of two CpGs downstream of the CDKN1A TSS (DMR2) (Figures S1A–S1D). We observed a striking, negative linear relationship between the average methylation of DMR1 and the frequency of TP53 mutations or deletions in tumor samples from the Pan-Cancer dataset (Figure 1A). Logistic regression confirmed the strong negative relationship between methylation within DMR1 and TP53 mutation or deletion (p < 10−36) but demonstrated no such association between TP53 mutation frequency and the methylation of DMR2 (p = 0.86). Because transcription factor occupancy can affect DNA methylation levels and DMR1 is within a p53 binding site at the DINO/CDKN1A locus, we examined whether TP53 status is associated with a differential methylation of p53 bound sites elsewhere in the genome (Younger et al., 2015). Logistic regression analyses reiterated that 6 of 8 CpGs within p53 bound site of the DINO/CDKN1A locus were individually, negatively associated with the probability of TP53 alterations, yet no similar association was observed for CpGs in the p53 binding sites at other canonical p53-regulated loci (Figures S1E and S1F; Table S3) (Knijnenburg et al., 2018). Therefore, increased methylation within the gene body of DINO is associated with reduced frequency of TP53 mutations in human tumors, a pattern distinct from methylation of the CDKN1A gene body and other p53-pathway genes.
Figure 1. DINO, a lncRNA gene recurrently methylated in human cancers, is a haplo-insufficient tumor suppressor.
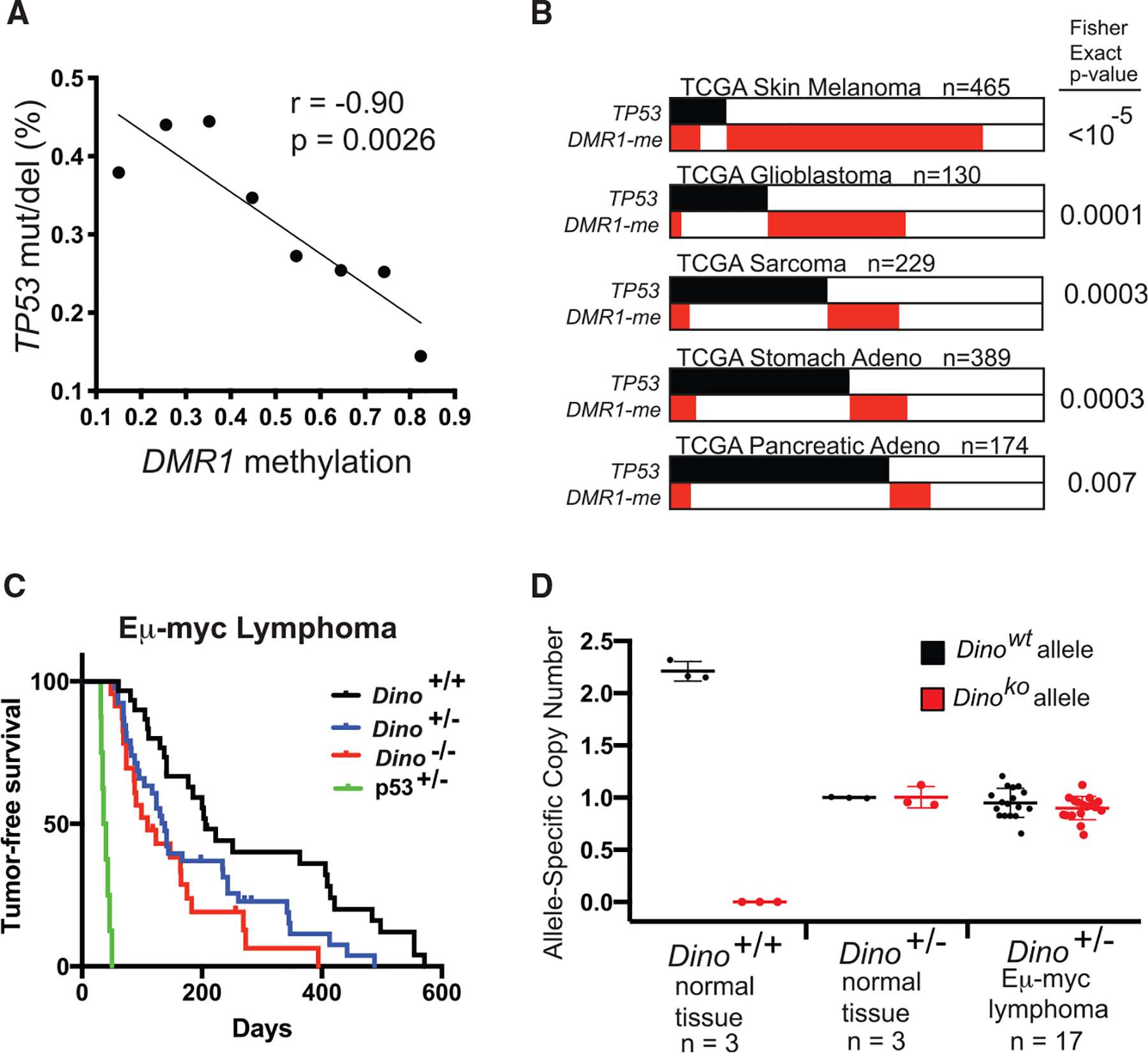
(A) Rate of TP53 somatic SNVs or deletions in 8,141 samples from the TCGA Pan-Cancer dataset in relation to methylation of DMR1. Samples are binned according to average DMR1 methylation in increments of 0.1 from <0.2 to ≥0.8, p value for correlation for 8 graphed data points.
(B) Mutual exclusivity analysis of DINO methylation (average methylation of DMR1 ≥0.5) and TP53 somatic SNVs or deletions in indicated TCGA datasets, Fisher exact test.
(C) Lymphoma-free survival of Eμ-myc mice of indicated genotype. Median lymphoma-free survival in Dino−/− mice occurs at 109 days (n = 23, p = 0.0007, log-rank test) and 138 days in Dino+/− mice (n = 40, p = 0.01, log-rank test) compared to 206 days in Dino+/+ mice (n = 30). Survival of p53+/− Eμ-myc mice are shown for comparison (median lymphoma-free survival 38 days, n = 8).
(D) Dino allele copy number in Eμ-myc Dino+/− lymphomas compared to indicated normal tissue controls as quantified by qPCR, mean ± SD.
See also Figures S1 and S2 and Tables S1, S2, and S3.
Mutual exclusivity analyses identify functionally related modules of genes involved in cancer pathways such as p53 tumor suppressor signaling (Ciriello et al., 2012). Functionally related genes are typically altered in a mutually exclusive manner because loss of one gene suffices to inactivate the tumor suppressor module, thus removing selective pressure to alter other components of the module. Similar to prior reports, we found no mutual exclusivity between CDKN1A alterations and TP53 alterations, again confirming that loss of CDKN1A is insufficient to inactivate the p53 tumor suppressor pathway (Figure S1G) (Cazier et al., 2014). However, as suggested by the logistic regression analysis, TP53 alterations were mutually exclusive with methylation of DINO (DMR1) regardless of the threshold value used to define DINO methylation status (Figure S1H; Table S2). K means clustering revealed two patterns of methylation at the DINO/CDKN1A locus in human cancers, predominately defined by average methylation of DINO greater thanor less than 0.5 (Figures S1C and S1D). Using this threshold, methylated DINO is mutually exclusive with TP53 alterations in the Pan-Cancer samples and individually in several TCGA cancer types (Figures 1B, S1I, and S1J; Table S1). Thus, we identified a focal region of differential methylation of DINO that is mutually exclusive with TP53 mutations, suggesting that DINO functions as a tumor suppressor gene in the p53 pathway.
The DINO/CDKN1A locus encodes a haplo-insufficient tumor suppressor function enforced exclusively by Dino
We used genetically modified mice to further examine the effect of each gene at the Dino/Cdkn1a locus on p53-associated tumor suppression. Cdkn1a−/− and Dino−/− mice provide two distinct alterations of the Dino/Cdkn1a locus that individually delete one gene at the locus while preserving the other (Schmitt et al., 2016; Brugarolas et al., 1995). Dino expression was fully intact in Cdkn1a−/− B cells, whereas Dino−/− B cells have a modest impairment in Cdkn1a expression relative to Dino+/+ B cells (Figures S2A and S2B). Therefore, studied together, these two alleles can dissect the individual contributions of each gene to tumor suppressor function. The Eμ-myc mouse model utilizes myc overexpression in B cells to induce B lymphoma and is one of the most widely utilized models for evaluation of the p53 tumor suppressor pathway. Although loss of members of the p53 pathway such as p19Arf and Trp53 accelerate B lymphoma formation in Eμ-myc mice, Cdkn1a−/− mice develop B cell lymphomas at a similar rate as Eμ-myc Cdkn1a+/+ mice, indicating that Cdkn1a/p21 does not contribute to the p53 tumor suppressor pathway in this model (Schmitt et al., 1999; Valente et al., 2016; Newbold et al., 2014). In contrast, we observed that loss of either one or two alleles of Dino significantly accelerated Eμ-myc lymphoma formation (Figure 1C). Furthermore, spleen mass, one measure of disease burden, was significantly elevated in Dino−/− mice compared to Dino+/+ mice and loss of one and two alleles of Dino resulted in lymphoma patterns progressively more similar to Eμ-myc p53+/− mice than Eμ-myc Dino+/+, p53+/+ (Figures S2C and S2D). Consistent with prior reports, Eμ-myc B cells isolated from mice prior to lymphoma development trended toward a modest activation of the p53 pathway in comparison to normal B cells, including Dino and other p53-pathway genes, although these results did not reach statistical significance due in part to high variability between replicates (Figure S2E). We found no evidence of LOH in Dino+/− lymphomas (Figure 1D), indicating that Dino suppresses tumor formation in a haplo-insufficient manner. Although CpGs within mouse Dino are methylated and little Dino expression was detected in lymphomas from Eμ-myc Dino+/+ and Eμ-myc Dino+/− (Figures S2F and S2G), CpG methylation at the Cdkn2a/Arf locus was generally low (Figure S2H). Furthermore, there were few genetic alterations of Cdkn2a/Arf and no alterations of Trp53 in Eμ-myc Dino+/+, Dino+/−, and Dino−/− lymphomas (Figure S2I), indicating that the p53 tumor suppressor pathway remains otherwise intact in these tumors. These data demonstrate that the Dino/Cdkn1a locus encodes a tumor suppressor program that is enforced by the lncRNA Dino independent of the protein product of the locus, p21. Although human cancer genetic data suggested that incremental increases in DINO methylation is associated with successively reduced selective pressure to mutate TP53, the Eμ-myc mouse genetic model definitively established that Dino suppresses tumorigenesis in a gene dose-dependent manner.
Progressively hypomorphic p53 in Dino+/− and Dino−/− cells impairs apoptosis and tumor suppression
p53 contributes to tumor suppression through both cell-autonomous and non-cell-autonomous mechanisms (Lujambio et al., 2013). To examine the mechanism by which Dino functions as a haplo-insufficient tumor suppressor, we next examined whether Dino suppresses tumor formation through cell-autonomous effects on p53 signaling. Because oncogene-expressing B cells were technically unsuitable for studying this question (Figure S2E), we turned to another well-characterized genetic model for p53-dependent tumor suppression, the mouse E1A, H-rasG12V fibrosarcoma model. Transformation of Dino+/− and Dino−/− mouse embryonic fibroblasts (MEFs) with E1A and H-rasG12V oncogenes increased in vitro clonogenic growth, similar to previous reports in p53−/− MEFs, and enhanced formation of fibrosarcoma in severe combined immunodeficiency (SCID) mice compared to Dino+/+ E1A, H-rasG12V MEFs, despite no difference in the proliferation rate of cells across these genotypes (Figures 2A, 2B, S3A, and S3B) (Lowe et al., 1994; Jiang et al., 2011). Importantly, following tumor formation in this model, p53 protein expression remained intact, the Trp53 allele remained wild-type, and Dino expression remained intact (Figures S3C and S3D). Thus, Dino’s haplo-insufficient tumor suppressor phenotype involves a cell autonomous mechanism in the E1A, H-rasG12V model of mouse fibrosarcoma.
Figure 2. Single and bi-allelic loss of Dino renders p53 progressively hypomorphic and impairs tumor suppression.
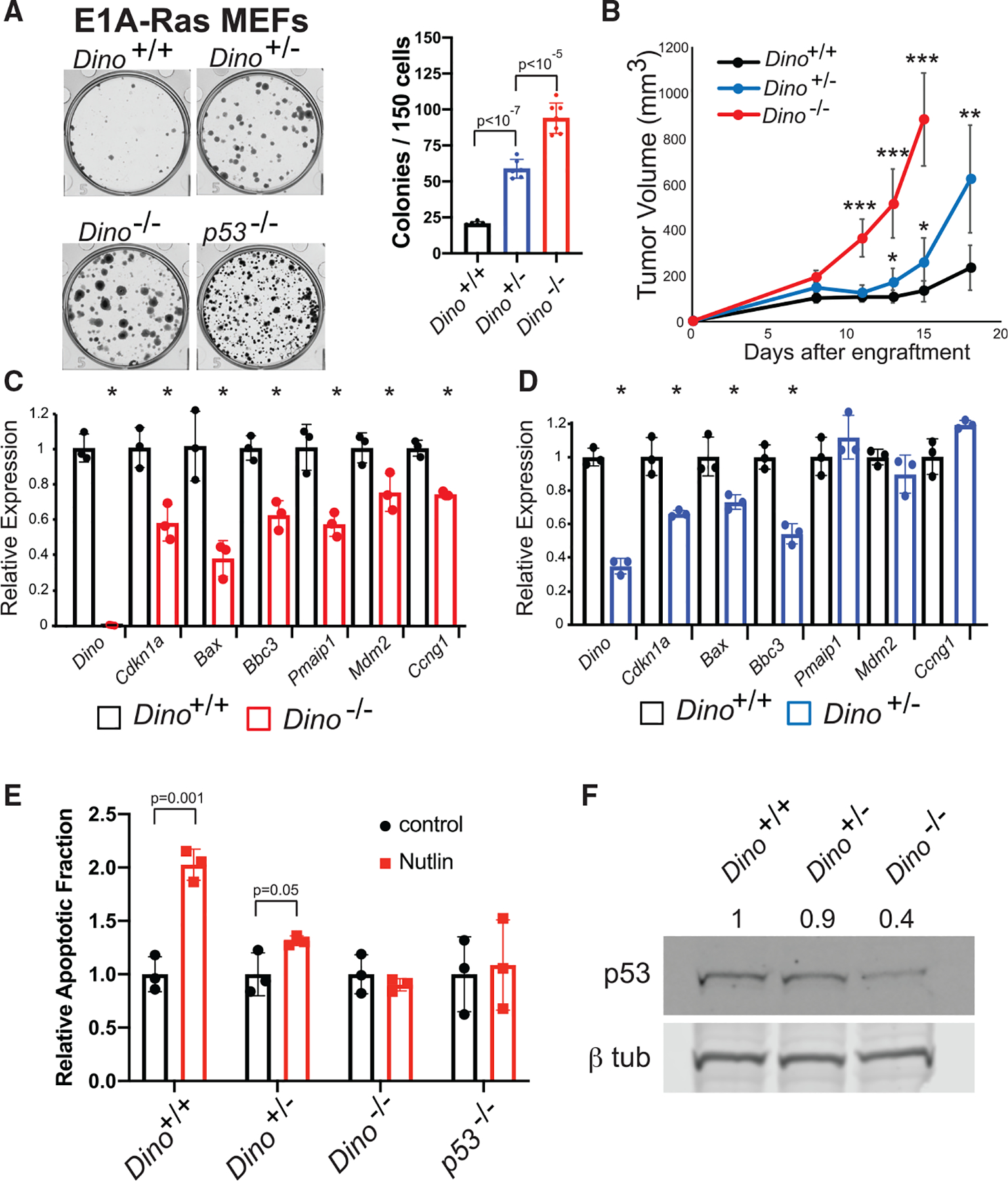
(A) In vitro clonogenic growth of Dino+/+, Dino+/−, and Dino−/− MEFs after transformation with E1A and HrasG12V retroviruses, mean ± SD, two-tailed t test.
(B) Fibrosarcoma formation in SCID mice after subcutaneous engraftment of 106 E1A-HrasG12V MEFs of indicated genotypes (mean ± SD, ***p < 10−7, **p = 0.001, *p < 0.01, two-tailed t test).
(C and D) The expression of p53-induced genes in Dino−/− E1A-HrasG12V cells (C) and Dino+/− E1A-HrasG12V cells (D) relative to Dino+/+ E1A-HrasG12V controls. (C) Mean ± SD, Dino, p = 2 × 10−5, Cdkn1a, p = 0.009, Bax, p = 0.008, Bbc3, p = 0.004, Pmaip1, p = 0.007, Mdm2, p = 0.03, Ccng1, p = 0.0007, two-tailed t test. (D) Mean ± SD, Dino, p = 10−4, Cdkn1a, p = 0.007, Bax, p = 0.02, Bbc3, p = 0.001, two-tailed t test.
(E) Apoptosis in Dino+/+, Dino+/−, Dino−/−, and p53−/−, E1A HrasG12V cells after 2.5 μM Nutlin-3a treatment, (mean ± SD, two-tailed t test).
(F) Western blot of p53 and β-tubulin in Dino+/+, Dino+/−, Dino−/− E1A HrasG12V cells with normalized p53 protein quantification.
See also Figure S3.
Even modestly hypomorphic p53 signaling is sufficient to impair p53-dependent tumor suppression despite intact Trp53 genes (Hemann et al., 2005). The expression of numerous p53-dependent genes are impaired in Dino−/− E1A, H-rasG12V MEFs, consistent with the requirement of Dino for activation of p53-dependent cellular responses to oncogenic stress (Figures 2C and S3E). Dino+/− E1A, H-rasG12V MEFs display an intermediate phenotype in the expression of p53 target genes, where the expression of a subset the p53-dependent genes are impaired relative to Dino+/+ controls (Figures 2D and S3F). Similarly, p53-dependent induction of apoptosis by the MDM2 inhibitor Nutlin-3a is increasingly impaired in Dino+/− and Dino−/− E1A, H-rasG12V MEFs compared to wild-type controls (Figure 2E). Consistent with our prior report that Dino stabilizes p53 protein, p53 protein is less abundant in Dino−/− E1A, H-rasG12V MEFs compared to wild-type controls, whereas p53 protein is minimally decreased Dino+/− E1A, H-rasG12V MEFs (Figure 2F). Therefore, cell-autonomous, hypomorphic p53 signaling in Dino+/− and Dino−/− MEFs is sufficient to impair p53-dependent cell fate decisions such as apoptosis, and thereby facilitate cellular transformation and tumorigenesis.
Reconstitution of Dino restores tumor suppression in Dino−/− cancer cells
Loss of potent tumor suppressor genes is typically sufficient to impair a tumor suppressor signaling pathway thereby removing selective pressure to alter other components of the pathway. For example, p53 null tumors typically retain wild-type copies of the other components of the p53 pathway (Klimovich et al., 2019), and as a result, restoration of p53 protein in p53 null tumors potently reactivates tumor suppression and leads to tumor regression or growth inhibition (Ventura et al., 2007). We next examined whether loss of Dino was sufficient to impair p53-dependent tumor suppression and remove selection pressure to alter other components of the signaling pathway in Dino−/− B lymphoma cells. We hypothesized that restoration of Dino in Dino−/− lymphoma cells would be sufficient to rescue tumor growth suppression if the p53 pathway was otherwise intact.
We generated cell lines from B lymphomas of three separate Eμ-myc Dino−/− mice. Lymphoma cells were infected with a dual promoter lentivirus expressing GFP and Dino (Dino-GFP), or an empty vector expressing GFP only (EV-GFP) as a control, and co-cultured in competition with Eμ-myc Dino−/− cells infected with mCherry expressing lentivirus (Figures 3A and S4A). We previously identified that DINO was induced >100-fold by DNA damage to ~1,000 copies per cell (Schmitt et al.,2016). Similarly, in normal B cells, Dino was expressed at a very low level in normal, unstimulated cells, but was induced by >100-fold by 2Gy irradiation (Figure S4B). Re-expression of Dino using a lentivirus in Eμ-myc Dino−/− cells resulted in Dino expression at levels similar to Dino expression levels following DNA damage in normal B cells (Figure S4B). Reintroduction of Dino by lentivirus resulted in impaired proliferation of Eμ-myc Dino−/− lymphoma cells relative to empty vector controls (Figure 3B), demonstrating that, similar to p53 restoration, tumor growth suppression can be rescued by the restoration of Dino expression into Dino−/− cancer cells. Each Eμ-myc Dino−/− B lymphoma cell line remained Trp53 wild-type by sequencing and demonstrated Cdkn1a induction following treatment with Nutlin-3a, confirming that the p53 signaling was otherwise intact in these Dino−/− lymphoma cells (Figures S4C–S4E). Engraftment of mice with Dino-GFP Eμ-myc Dino−/− lymphoma cells resulted in significantly fewer lymphomas than mice engrafted with EV-GFP Eμ-myc Dino−/− lymphoma cells infected with empty vector control (Figure 3C). Hence, myc-induced B cell transformation is facilitated by Dino loss in the absence of other p53-pathway alterations, and restoration of Dino expression is sufficient to re-establish tumor suppression.
Figure 3. Rescue of Dino expression restores tumor suppression in Dino−/− cells.
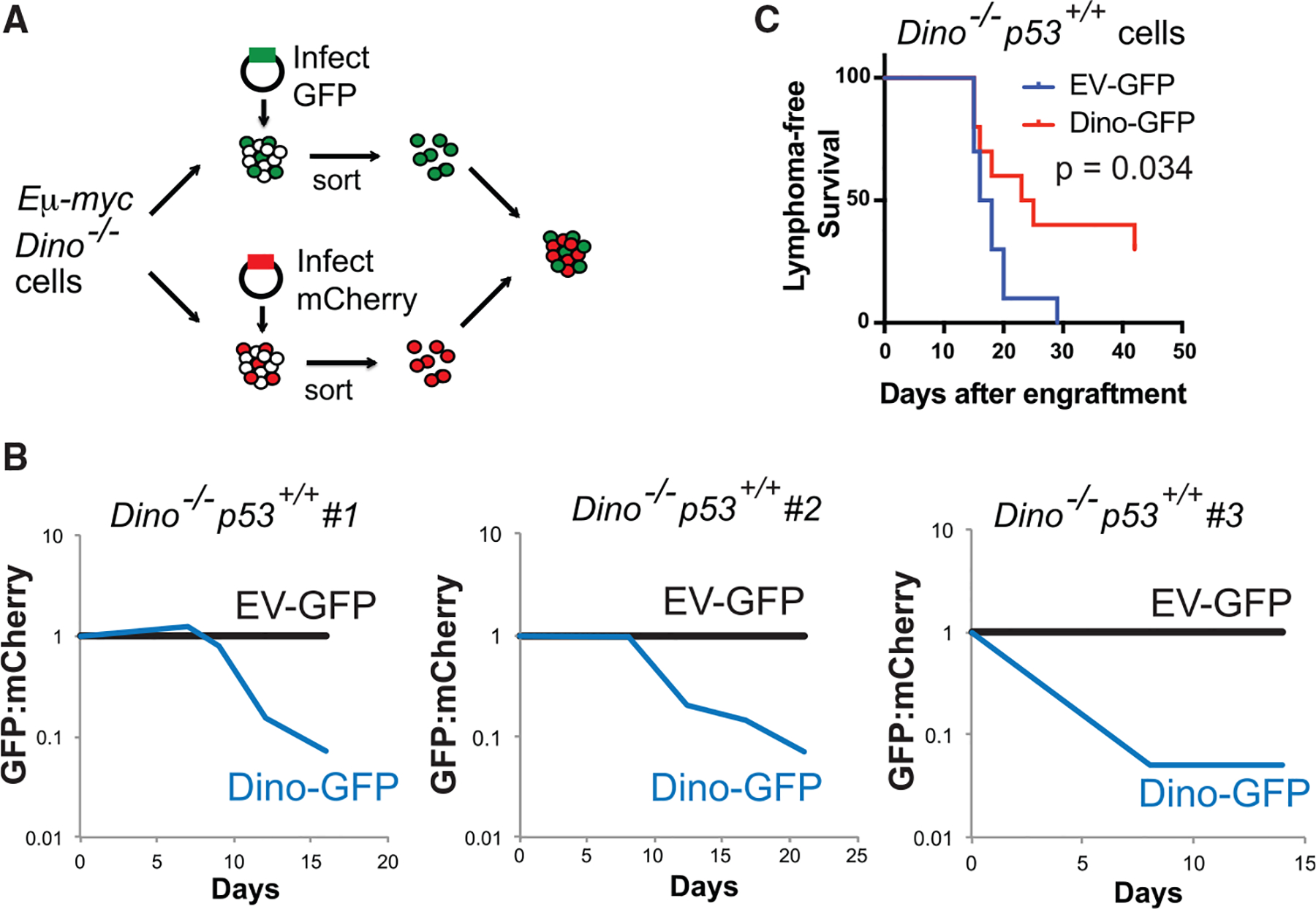
(A) Experimental strategy for rescue of Dino function in Dino−/− Eμ-myc lymphoma cells utilizing a two-promoter lentiviral construct expressing GFP and Dino (Dino-GFP), or GFP only (EV-GFP). After infection, GFP+ Eμ-myc lymphoma cells are co-cultured in competition with mCherry+ Eμ-myc cells from the same cell line.
(B) Growth competition assay for Dino−/−, p53+/+ Eμ-myc lymphoma cells infected with Dino-GFP or EV-GFP relative to competing mCherry+ Dino−/−, p53+/+ Eμ-myc lymphoma cells, two repeats were performed each in four independent cell lines totaling eight independent replicates of the experiment.
(C) Lymphoma-free survival after transplantation of in Dino−/−, p53+/+ Eμ-myc lymphoma cells infected with either EV-GFP or Dino-GFP lentivirus, n = 10, p = 0.034, log-rank test.
See also Figure S4.
Tumor suppression by Dino requires p53
Mutual exclusivity of methylated DINO and TP53 mutations in human tumors suggested a potential epistatic relationship between the two genes. In Eμ-myc Dino−/− B lymphoma cell line 1, restoration of Dino impaired lymphoma growth in vitro in two experiments performed between passage 5 and passage 21 after adaptation to cell culture. However, two separate experiments at late passages, 34 and 35, revealed that this cell line had subsequently evolved resistance to growth suppression by Dino (Figure 4A). Spontaneous inactivating p53 mutations have been noted to emerge in cell cultures at late passages. Sequencing of Trp53 in cell line 1 revealed that it remained p53+/+ until passage 22. However, by passage 35, a clonal population with an inactivating p53 mutation, p53R246Q, emerged contemporaneously with the resistance to growth suppression by Dino in the in vitro growth assay (Figures 4B, 4C, S4F, and S4G). To specifically isolate the effect of p53 on Dino’s tumor suppressor function, we introduced of Dino-GFP or EV-GFP lentivirus into p53 null Eμ-myc lymphoma cells. Similar to results in the in vitro growth suppression assay, Dino-GFP did not suppress lymphoma formation relative to EV-GFP infected cells, confirming that Dino requires p53 for its tumor suppressor function (Figure 4D). Therefore, consistent with mutual exclusivity analyses in human cancers, loss of Dino is sufficient for tumorigeneses in p53-intact cells but p53 is epistatic to Dino, and p53 loss abrogates the need to downregulate Dino.
Figure 4. p53 is epistatic to Dino in tumor suppression.
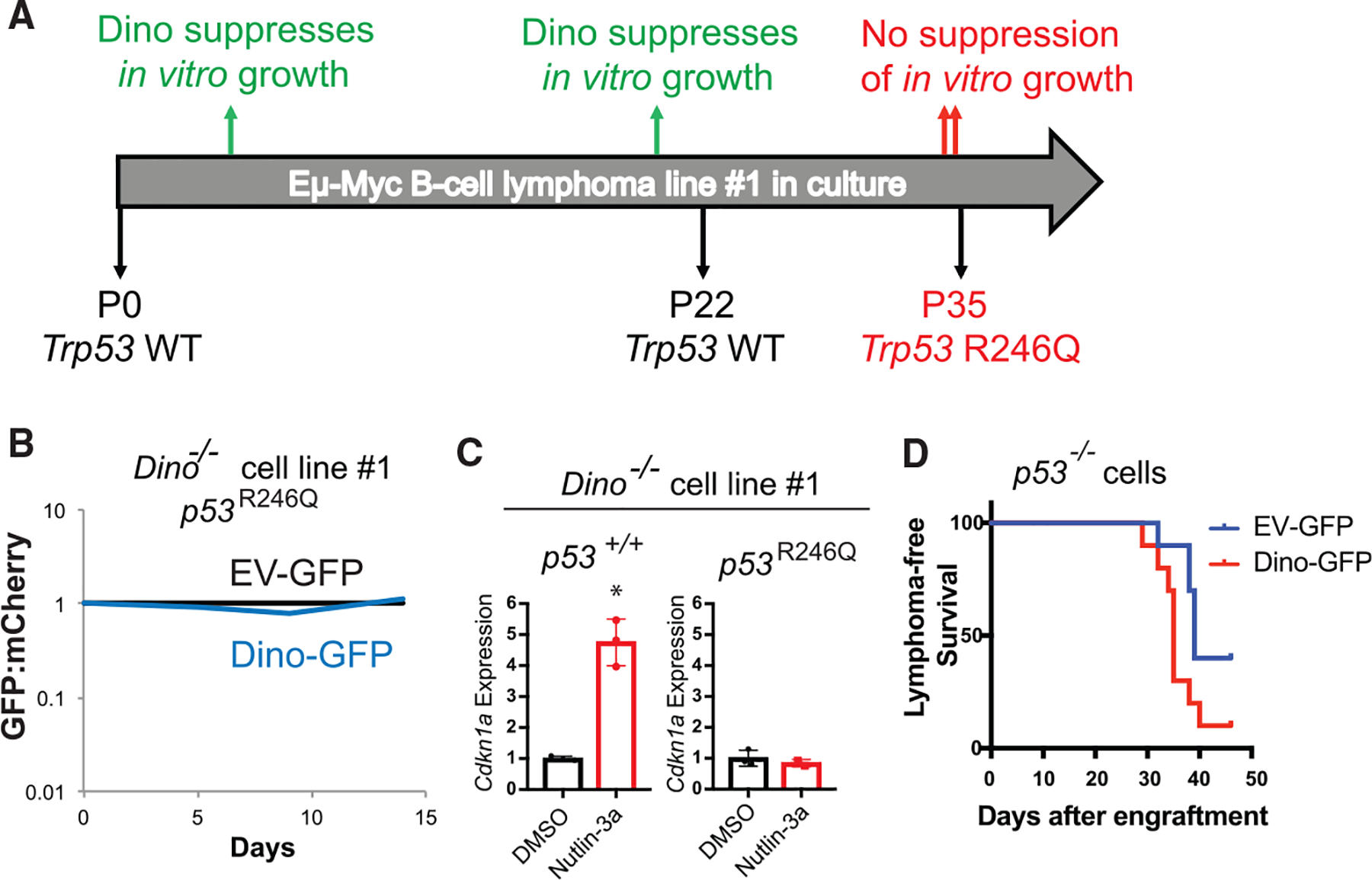
(A) Timeline of four independent in vitro growth competition assays in Eμ-myc lymphoma cell line 1 and the Trp53 status of the cell line during this time frame.
(B) Growth competition assay for passage 35 Dino−/− Eμ-myc lymphoma cell line 1 (p53R246Q) after infection with Dino-GFP or EV-GFP relative to competing mCherry+ Dino−/−, p53R246Q Eμ-myc lymphoma cells, two repeats of the experiment were performed before and after the cell line developed the Trp53 R246Q mutation.
(C) p53-dependent CDKN1A expression after 2uM Nutlin-3a treatment of in Dino−/− Eμ-myc lymphoma cell line 1 at passage 5 (p53+/+) and at passage 35 (p53R246Q), *p = 0.001, two-tailed t test.
(D) Lymphoma-free survival after transplantation of in p53 null Eμ-myc lymphoma cells after infection with either EV-GFP or Dino-GFP lentivirus n = 10, p = 0.033, log-rank test.
See also Figure S4.
Hypermethylation specifically silences DINO to impair p53 signaling and elicit cancer phenotypes similar to TP53 mutant cancers
Because our mouse genetic models define Dino as haplo-insufficient tumor suppressor within the Dino/Cdkn1a locus in sarcoma, we next used the TCGA sarcoma samples to examine how the methylation signature at the DINO/CDKN1A locus that is related to TP53 status regulates the expression of these genes. Similar to the Pan-Cancer dataset presented in Figure S1, the CpG island at the DINO/CDKN1A locus in The Cancer Genome Atlas Sarcoma (SARC) was unmethylated in nearly all samples but substantial differential methylation was observed in the CpG shore downstream of the DINO TSS (Figures S5A and S5B). Prior studies have demonstrated that CpG shores account for a large fraction of the DMRs that distinguish human cancers from normal tissues (Doi et al., 2009; Irizarry et al., 2009), and methylation within the first 2 kb downstream of TSSs is significantly, negatively correlated with gene expression in human cancer and normal tissues (Hovestadt et al., 2014; Schultz et al., 2015). In TCGA SARC, methylation values at each of the six CpGs that comprise the DINO DMR1 are significantly and negatively correlated with the expression of DINO, as is the average methylation within the DMR (Figures 5A, 5B, and S5C). In contrast, neither the individual CpG values within the DMR, nor the DMR average, correlate with the expression of CDKN1A (Figures 5A and 5C). Furthermore, tumor samples from TCGA SARC, Skin Cutaneous Melanoma (SKCM), and Glioblastoma Multi-forme (GBM) exhibit gain of DINO methylation relative to normal tissue controls (Figures S5D–S5F) (Benton et al., 2015; Do et al., 2016; Fujiwara et al., 2019; Marzese et al., 2014). Therefore, cancer-associated methylation gain in the DMR overlying DINO, and its relationship to TP53 mutations in human cancers, is specifically associated with the expression of DINO and unrelated to CDKN1A in human cancers.
Figure 5. Methylation silences DINO in human cancers.
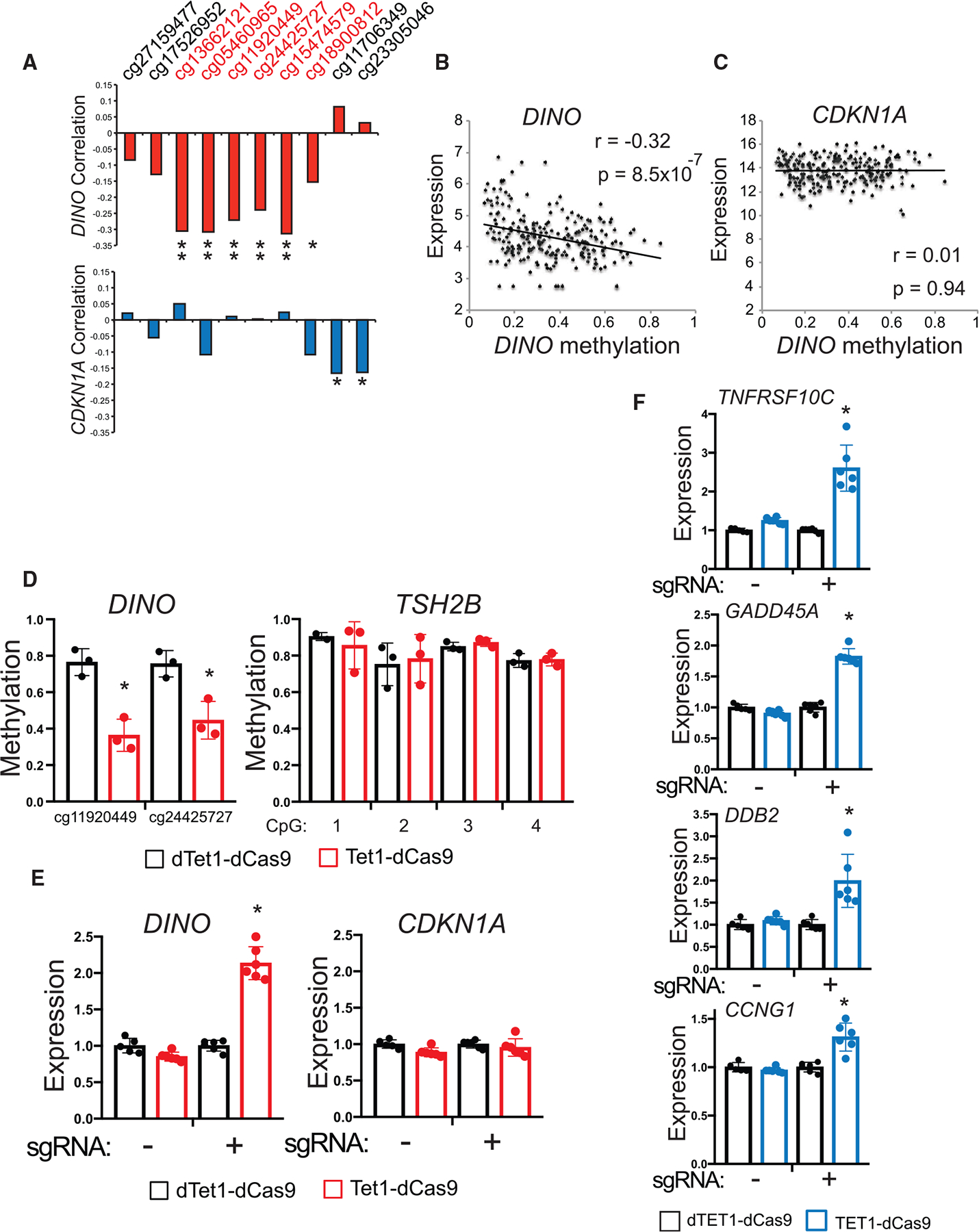
(A) Correlation coefficient for methylation at each CpG near the DINO DMR with DINO and CDKN1A expression in SARC samples, (DINO correlation: cg13662121 p = 3 × 10−6, cg05460965 p = 2 × 10−6, cg11920449 p = 3 × 10−5, cg24425727 p = 3 × 10−4, cg15474579 p = 1 × 10−6, cg18900812 p = 0.02; CDKN1A correlation: cg11706349 p = 0.01, cg23305046 p = 0.01). Pearson correlation two-tailed p value.
(B) Correlation of average DINO methylation with DINO expression in SARC (r = −0.32, p = 8.5 × 10−7). Pearson correlation two-tailed p value.
(C) Correlation of average DINO methylation with CDKN1A expression in SARC (r = 0.01, p = 0.94). Pearson correlation two-tailed p value.
(D) Methylation as measured by bisulfite sequencing at CpGs within the DINO DMR and TSH2B CpG island in HT-1080 cells. Cells were infected with lentivirus expressing either TET1-dCas9 or dead TET1-dCas9 (dTET1-dCas9) and a sgRNA targeting the DINO DMR. Above: map of the location of sgRNA within the DINO DMR. (mean ± SD, cg11920449, p = 0.004, cg24425727, p = 0.01, two-tailed t test).
(E) Expression of DINO and CDKN1A in HT-1080 cells stably expressing TET1-dCas9 or dTET1-dCas9 with or without a sgRNA targeting the DINO DMR. (mean ± SD, *p = 3.9 × 10−7, two-tailed t test).
(F) Expression of select p53-regulated genes located on different chromosomes than the DINO/CDKN1A locus. (mean ± SD, TNFRSF10C p = 6.4 × 10−5, GADD45A p = 8.7 × 10−8, DDB2 p = 0.0027, CCNG1 p = 0.0006, two-tailed t test).
See also Figure S5.
We next experimentally examined how methylation of this region regulates gene expression and p53 signaling in human sarcoma cells. In HT-1080 fibrosarcoma cells, in which DINO is methylated and TP53 is wild-type, treatment with demethylating agent 5-aza-2′-deoxycytidine significantly increased DINO expression, confirming that DNA methylation regulates DINO expression (Figures S5G and S5H). Furthermore, we confirmed that exogenous DINO expression alone in HT-1080 is sufficient to stabilize p53 protein (Figures S5I). Because demethylating agents broadly alter methylation levels throughout the cell, we used CRISPR methylation editing to specifically determine whether methylation within the DMR regulates the DINO/CDKN1A locus. Demethylation of the DINO DMR in HT-1080 cells using TET1-dCas9 and a single-guide RNA (sgRNA) targeting DINO achieved specific demethylation of DINO but not the control locus TSH2B (Figures 5D, 5E, and S5J) (Liu et al., 2016a). Targeted demethylation of DINO increased DINO gene expression but not CDKN1A, and also induced the expression of multiple p53-dependent genes in trans, similar to Nutlin-3a (Figure 5F, S5K and S5L). These results confirm that recurrent methylation of DINO in human cancers specifically silences DINO in order to impair p53 signaling in cancers that remain TP53 intact.
In some human cancers, TP53 mutations are not only a mechanism of escaping tumor suppression, but also result in distinct tumor phenotypes and clinical outcomes compared to TP53 intact tumors. As epigenomic silencing of DINO impaired p53 signaling in cancer cells, we examined whether silencing of DINO in TP53 intact tumors led to clinical outcomes similar to TP53 mutated cancers. Remarkably, clinical outcomes for DINO-methylated, TP53 wild-type tumors are similar to patients with TP53-altered tumors, and distinct from patients with DINO-un-methylated, TP53 wild-type tumors (Figures S5M–S5O). That patients with DINO-methylated, TP53 wild-type and TP53-altered GBM both survive better than the TP53-intact may be a result of TP53-pathway alterations occurring at a higher rate in GBM subtypes with slightly more favorable outcomes, as previously noted (Cantero et al., 2020). Although DINO hypermethylation and TP53 mutations were also mutually exclusive in TCGA SARC and Stomach Adenocarcinoma (STAD) samples, TP53 mutations were not associated with distinct clinical outcomes and so are not presented. Therefore, focal methylation within the DINO/CDKN1A locus specifically silences DINO in order to impair p53 signaling, resulting in tumors that are phenotypically similar to TP53 mutated cancers.
DISCUSSION
Although TP53 is the most frequently mutated gene in human cancers, the majority of human cancers develop while retaining intact TP53. The mechanism by which cancers escape p53-dependent tumor suppression in these cases have remained mostly unknown despite extensive examination of the protein-coding genes induced by p53 for function in tumor suppression. Our results indicate that the lncRNA gene DINO is a critical, haplo-insufficient effector of the p53-dependent tumor suppressor program, and provide one of the few rigorous genetic analyses definitively connecting a lncRNA to cancer phenotypes. Furthermore, we show that recurrent silencing of DINO is noted across multiple human cancer types in a manner mutually exclusive with TP53 mutations, and that DINO silencing downregulates p53 signaling, data indicating an important role for human DINO in tumorigenesis. Remarkably, these results indicate that epigenomic silencing of DINO may be among the most frequent alterations of the p53 tumor suppressor pathway in human cancer, after mutations of TP53 itself. Given that environmental factors, cellular differentiation, and stress can alter DNA methylation, DINO methylation may also titrate p53 signaling in other contexts in which p53 signaling play important roles such as development and aging.
Cancer genomic analyses have typically focused on the protein-coding genome. The contribution of the noncoding genome to cancer development has remained mostly unknown despite the fact that numerous cancer-associated loci reside in the noncoding genome, or within protein-coding loci with no known association with cancer biology. Our discovery that tumor suppressor function encoded at the cancer-associated DINO/CDKN1A locus arises from the function of the long noncoding RNA gene DINO, but not the protein product p21, highlights that alteration of the noncoding genome can initiate tumorigenesis. Although single nucleotide variants in protein-coding sequences unambiguously identify the affected gene, cancers contain numerous copy number variations and epigenomic alterations that can affect loci encoding numerous protein-coding genes and noncoding genetic sequences (Liu et al., 2016b; Barretina et al., 2010; Hovestadt et al., 2014). These results highlight that noncoding RNA genes should be carefully considered when examining the effects of structural alterations, mutations in the noncoding genome, or epigenomic regulation at work on cancer-associated loci.
Pharmacological reactivation of p53 signaling in cancer cells has been a long-standing goal for cancer therapy. Our results indicate that a potential novel therapeutic strategy to activate p53 signaling involves reversal of epigenomic silencing of DINO in TP53 intact cancers. That more than one-quarter of human cancers are DINO-methylated and TP53 wild-type shows this therapeutic opportunity may be relevant to a large population of cancer patients.
Limitations of the study
Our study utilized quantification of p53 protein by quantitative western blot in numerous panels. In experimental conditions using mouse fibroblasts with genetic perturbations of Dino, we observed clear disruption in p53 protein abundance (Figure 2F) following E1A-HrasG12V fibroblast transformation, similar to previous experiments (Schmitt et al., 2016). Furthermore, quantification of p53 protein abundance following overexpression of DINO by plasmid in HT-1080 cells revealed p53 protein stabilization, but which was less profound than noted in mouse fibroblasts. This may reflect the different efficacy of endogenous Dino versus exogenously expressed DINO, and/or differences in Dino-dependent p53 protein regulation in recently transformed primary cells compared to immortal human cancer cell lines. Future experiments to determine the mechanism by which DINO regulates p53 protein levels in normal and cancer cells will help clarify these questions.
Although we demonstrated that p53 protein abundance is highly correlated with Dino genotypes in E1A- and HrasG12V-transformed mouse fibroblasts in cell culture with highly controlled conditions prior to subcutaneous engraftment (Figure 2F), the role of Dino in p53 stabilization in fully formed mouse flank tumors was not assessed due to several confounding factors. These include the unknown degree of stromal inflitration from the host animal, as well as the effect of substantial difference in tumor size altering microenvironmental stressors such as nutrient availability and hypoxia, both of which can affect p53 signaling.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be addressed to and will be fulfilled by the Lead Contact, Adam Schmitt (schmitta@mskcc.org).
Materials availability
Plasmids generated in this study are available upon request.
Data and code availability
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the Key Resources Table. Original TCGA source data for bioinformatic analyses are available through the Genomics Data Commons and can be viewed through the UCSC Xena Browser.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-5-methylcytosine Antibody, clone 33D3 | Millipore Sigma | Cat# MABE146; RRID:AB_10863148 |
| anti-TurboGFP | Thermo Fisher Scientific | Cat# PA5–22688; RRID:AB_2540616 |
| Anti-p53 antibody, Mouse monoclonal (clone DO-1) | Sigma Aldrich | Cat# P6874; RRID:AB_1079545 |
| p53 (1C12) Mouse mAb | Cell Signaling | Cat# 2524; RRID:AB_331743 |
| beta Tubulin antibody | GeneTex | Cat# GTX101279; RRID:AB_1952434 |
| Anti-Actin Antibody, clone C4 | Millipore Sigma | Cat# MAB1501; RRID:AB_2223041 |
| IRDye® 800CW Goat anti-Mouse IgG Secondary Antibody | Li-Cor | Cat# 926–32210; RRID:AB_621842 |
| IRDye® 680LT Goat anti-Rabbit IgG Secondary Antibody | Li-Cor | Cat# 926–68021; RRID:AB_10706309 |
| Bacterial and virus strains | ||
| One Shot® Mach1 T1 Phage-Resistant Chemically Competent E. coli | Invitrogen | C862003 |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM high glucose | MSKCC Media Core | N/A |
| Fetal Bovine Serum | VWR | Cat# 97068–085 |
| Penicillin-Streptomycin | MSKCC Media Core | N/A |
| RPMI 1640 Medium | ThermoFisher Scientific | Cat# 11875093 |
| 2-mercaptoethanol | GIBCO | ThermoFisher Scientific cat# 21985023 |
| L-Asparagine | Sigma-Aldrich | Cat# A4159 |
| Purelink Genomic DNA Mini Kit | Invitrogen | Cat# K182002 |
| Brilliant II QRT-PCR SYBR® Green Low ROX Master Mix | Agilent Technologies | Cat# 600835 |
| Pan B Cell Isolation Kit II | Miltenyi Biotec | Cat# 130-104-443 |
| MACS BSA Stock Solution | Miltenyi Biotec | Cat# 130-091-376 |
| autoMACS Rinsing Solution | Miltenyi Biotec | Cat# 130-091-222 |
| MACS LS Columns | Miltenyi Biotec | Cat# 130-042-401 |
| RNeasy Plus Mini Kit | QIAGEN | Cat# 74136 |
| Phusion HF PCR Master Mix | NEB | Cat# M0531 |
| BamHI | NEB | Cat# R0136 |
| NotI-HF | NEB | Cat# R3189 |
| AarI | Thermo Scientific | Cat# ER1581 |
| Rapid DNA Dephos & Ligation Kit | Sigma-Aldrich | Cat# 4898117001 |
| Lenti-X Packaging Single Shots | Takara | Cat# 631282 |
| Lenti-X Concentrator | Takara | Cat# 631231 |
| Calphos Mammalian Transfection Kit | Clontech | Cat# 631312 |
| Puromycin 10mg/ml | Invivogen | Cat# ant-pr-1 |
| Hygromycin B (50 mg/mL) | ThermoFisher Scientific | Cat# 10687010 |
| Methanol | Fisher Scientific | Cat# A412P-4 |
| Crystal violet solution | Sigma-Aldrich | Cat# HT90132 |
| Nutlin-3 | Sigma-Aldrich | Cat# N6287 |
| DMSO | Fisher Scientific | Cat# BP231–100 |
| DAPI Staining Solution | Miltenyi Biotec | Cat# 130-111-570 |
| Dynabeada Protein G for Immunoprecipitation | Thermo Fisher Scientific | Cat# 10003D |
| Proteinase K | Thermo Fisher Scientific | Cat# AM2548 |
| Phenol:Chloroform Saturated Solution, Ph 6.7. ≥ 99.0% | MP Biomedicals | Cat# ICNA04802521 |
| 3M Sodium Acetate | Ambion | Cat# AM9740 |
| GlycoBlue | Ambion | Cat# AM9516 |
| 5-Aza-2-deoxycytidine | Sigma-Aldrich | Cat# A3656 |
| PureLink RNase A (20 mg/mL) | Fisher Scientific | Cat# 12091021 |
| EpiMark® Bisulfite Conversion Kit | New England Biolabs | Cat# E3318S |
| EpiMark® Hot Start Taq DNA Polymerase | New England Biolabs | Cat# M0490 |
| Cycloheximide | Sigma-Aldrich | Cat# C4859 |
| Bolt 4 to 12%, Bis-Tris, 1.0 mm Mini Protein Gel, 12-well | Thermo Fisher Scientific | Cat# NW04122BOX |
| 20X Bolt MOPS SDS Running Buffer | Thermo Fisher Scientific | Cat# B0001 |
| Invitrogen novex NuPAGE 4–12% Bis-Tris Midi Protein Gels, 20 well | Thermo Fisher Scientific | Cat# WG1402BOX |
| Bolt Transfer Buffer (20X) | Thermo Fisher Scientific | Cat# BT00061 |
| Nitrocellulose Membrane, 0.45 mm | BioRad | Cat# 1620115 |
| Revert 700 Total Protein Stain Kits for Western Blot Normalization | Li-Cor | Cat# 926–11010 |
| Critical commercial assays | ||
| Click-iT® Plus EdU Alexa Fluor® 488 Flow Cytometry Assay Kit | ThermoFisher Scientific | Cat# C10632 |
| ApoStat Apoptosis Detection Kit | R&D Systems | Cat# FMK012 |
| Qubit Protein Assay Kit | Invitrogen | Cat# Q33212 |
| Deposited data | ||
| Benign Adipose Methylation Array | Benton et al., 2015 | EMBL-EBI: E-MTAB-1866 |
| Benign Glia Methylation Array | Do et al., 2016 | GEO: GSE79144 |
| Benign Melanocyte Methylation | Fujiwara et al., 2019 | GEO: GSE122909 |
| Benign Melanocyte Methylation | Marzese et al., 2014 | GEO: GSE44662 |
| Experimental models: cell lines | ||
| Human: BJ Fibroblasts | ATCC | ATCC Cat# CRL-2522; RRID:CVCL_3653 |
| Human: HT-1080 | ATCC | ATCC Cat# CRL-7951; RRID:CVCL_0317 |
| Human: LentiX-293T | Takara | Cat# 632180 |
| Human: Pheonix-293T | Nolan Laboratory, Stanford University | N/A |
| Mouse Embryonic Fibroblasts | This paper | N/A |
| Mouse: Em-myc lymphoma cell lines | This paper | N/A |
| Experimental models: organisms/strains | ||
| Mouse: C57BL/6-Dinoegfp | Howard Chang, Stanford University | N/A |
| Mouse: C57BL/6 Eμ-myc | Scott Lowe, MSKCC | N/A |
| Mouse: C57BL/6 Cdkn1a−/− (B6.129S6(Cg)-Cdkn1atm1Led/J) | Jackson Labs | IMSR Cat# JAX:016565;RRID:IMSR_JAX:016565 |
| Mouse: CB17-Prkdc< scid > | Taconic | IMSR Cat# TAC:cb17sc; RRID:IMSR_TAC:cb17sc |
| Mouse: C57BL/6 | Jackson Labs | IMSR Cat# JAX:000664; RRID:IMSR_JAX:000664 |
| Oligonucleotides | ||
| PCR Primers | This paper (see Table S4) | N/A |
| Recombinant DNA | ||
| Plasmid: pCDH-EF1α-MCS-(PGK-GFP-T2A-Puro) | System Biosciences | Cat# CD813A-1 |
| Plasmid: pCDH-EF1α-Dino-(PGK-GFP-T2A-Puro) | This paper | N/A |
| Plasmid: pMSCV-IRES-mCherry FP | Addgene | RRID:Addgene_52114 |
| Plasmid: pBabe 12S E1A | Addgene | RRID:Addgene_18742 |
| Plasmid: pWZL hygro H-RasV12 | Addgene | RRID:Addgene_18749 |
| Plasmid: Fuw-dCas9-Tet1CD-P2A-BFP | Addgene | RRID:Addgene_108245 |
| Plasmid: Fuw-dCas9-dead Tet1CD-P2A-BFP | Addgene | RRID:Addgene_108246 |
| Plasmid: pgRNA-modified | Addgene | RRID:Addgene_84477 |
| Plasmid: pgRNA-modified_sgDINO | This paper | N/A |
| Software and algorithms | ||
| R | (R Core Team, 2020) | |
| featureCounts | CRAN | Liao et al., 2014 |
| DESeq2 | CRAN | Love et al., 2014 |
| XLSTAT | Addinsoft | N/A |
| Prism | GraphPad | v8 |
| ImageJ | NIH | Schneider et al., 2012 |
| FlowJo | BD Biosciences | N/A |
| FCS Express | De Novo Software | V6 |
| Custom Alt-R® CRISPR-Cas9 guide RNA tool | Integrated DNA Technologies | https://www.idtdna.com/site/order/designtool/index/CRISPR_CUSTOM |
| Image Studio | Li-Cor | N/A |
| Other | ||
| QuantStudio 6 Flex Real-Time PCR system | Applied Biosytems | N/A |
| MACS Multistand | Miltenyi Biotec | Cat# 130-042-303 |
| MidiMACS Separator | Miltenyi Biotec | Cat# 130-042-302 |
| LSR Fortessa | BD Biosciences | N/A |
| FACSAria | BD Biosciences | N/A |
| Biorupter Pico | Diagenode | B01060010 |
| Qubit 3.0 Fluorometer | Invitrogen | Cat# Q33216 |
| Odyssey CLx Imaging System | Li-Cor | Model# 9140 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
C57BL/6 Dino−/− mice (C57BL/6-Dinoegfp) mice were previously described (Schmitt et al., 2016), C57BL/6 Eμ-myc were kindly provided by Scott Lowe (Adams et al., 1985). C57BL/6 Cdkn1a−/− mice (B6.129S6(Cg)-Cdkn1atm1Led/J) were obtained from Jackson Laboratories (Deng et al., 1995).. E1A-HrasG12V MEFs were subcutaneously engrafted into 6-week old male CB17-Prkdc < scid > mice (Taconic). For tumor allograft studies, Eμ-myc B lymphoma cells were injected into the peritoneum of 6-week old male C57BL/6 mice (Jackson Laboratories), All experiments were performed in accordance with the MSKCC Institutional Animal Care and Use Committee (IACUC) protocol #1507011.
Cell lines
BJ fibroblasts (human, male origin; ATCC), HT-1080 (human, male origin; authenticated by the ATCC cell line authentication service), LentiX-293T (human; Takara Bio), Phoenix-293T (human; Nolan Lab Stanford University), and mouse embryonic fibroblasts were cultured in DMEM high glucose, 10% FBS (VWR) plus Penicillin-Streptomycin. Isolated Eμ-myc lymphoma cell lines were maintained in FMA media (RPMI-1640 (ThermoFisher Scientific) supplemented with 10% FBS, 50 μM 2-mercaptoethanol (GIBCO), 200 μM asparagine (Sigma-Aldrich), and Pen/Strep) (Corcoran et al., 1999). All cells were grown at 37°C in a humidified atmosphere of 5% CO2.
METHOD DETAILS
Computational analysis of TCGA samples
Genome-wide read counts were summarized using the R package featureCounts (Liao et al., 2014). Reads were mapped to the hg38 human gene set using Ensembl (Ensembl GRCh38.95 GTF), which included gene coordinates for DINO and CDKN1A. Raw read counts were normalized using variance stabilizing transformation from the R package DESeq2 (Love et al., 2014).
For DNA methylation, copy number alteration, and mutation analyses, TCGA Pan-Cancer data were downloaded through the UCSC Xena. A dataset consisting of 8141 tumor samples with complete TP53 mutations, TP53 copy number, and DINO/CDKN1A methylation data were used for downstream analyses. Average methylation within the DINO DMR was obtained by averaging the beta values of the six CpGs covered by the Illumina HM450 probes in this DMR: cg13662121, cg05460965, cg11920449, cg24425727, cg15474579, cg18900812. Methylated DINO was defined as an average beta value of the six CpGs ≥ 0.5. For mutual exclusivity analyses, TP53 was considered altered in any sample with deep deletion of TP53 (copy number ≤ −2) or any non-silent TP53 mutation. Fisher exact test was used to test for significant mutual exclusivity or co-alteration and logistic regression analyses for DNA methylation and TP53 alterations were performed using XLSTAT. TCGA data for normal tissue methylation were very limited for most cancer types in which DINO methylation and TP53 mutations/deletions were mutually exclusive. Illumina HM450 DINO/CDKN1A methylation data were available from prior studies and used to compare DINO methylation values of specific tumor types with benign samples from comparable tissues. Data accession numbers for benign adipose tissue (E-MTAB-1866), benign glia (GSE79144), and benign melanocytes (GSE122909, GSE44662).
For analyses of mutations associated with DINO methylation in DLBC, 48 tumor samples had both mutation and DNA methylation data, whereas the 11 samples were missing copy number data and were excluded from mutual exclusivity analysis of DINO methylation and CDKN2A deep deletion or TP53 mutation/deletion.
Survival analyses
Lymphoma-bearing mice were euthanized when showing evidence of lymphoma including immobility, rapid breathing, ascites or palpable tumors. Fibrosarcoma bearing mice were euthanized once tumor burden reached 1cm3. Kaplan-Meier curves were plotted using Prism (GraphPad), median survival calculated, and significance calculated using the Mantel-Cox Test.
B cell isolation
Spleens were dissected from three non-irradiated (control) wild-type mice, and three wild-type and three knockout animals 6 hours after 2Gy total body irradiation. Two male and 1 female 9-month old mice were used per condition. Untouched splenic B cells were isolated using the Pan B Cell Isolation Kit II (Miltenyi Biotec) as per manufacturer instructions. Briefly, spleens were dissociated by mashing though a 100μm filter into 1X PBS. 1×108 cells per condition were resuspended in 1X PBS, 0.5% bovine serum albumin and 2mM EDTA (MACS BSA Stock solution diluted 1:20 in MACS Rinsing Solution) and filtered through a 45 μm mesh. Cells were then magnetically labeled and separated using MACS LS Columns on a MACS separator following kit protocol. B cells were pelleted, snap frozen, and stored at −80C.
Quantitative RT-PCR
All RNA was isolated using RNeasy Plus Mini Kit (QIAGEN). RT-qPCR was performed using Brilliant II QRT-PCR SYBR® Green Low ROX Master Mix (Agilent Technologies) on an Applied Biosytems QuantStudio 6 Flex Real-Time PCR system using the indicated primers. Expression was normalized to Beta-actin (mouse) or GAPDH (human). Primer sequences are listed in Table S4. Significance calculated by t test, error bars show standard error of the mean.
Dino loss of heterozygosity
Splenic genomic DNA was prepared from 3-month-old Dino+/+, Dino+/− and Dino−/− (n = 3, 2 male and 1 female per genotype), and Dino+/−-Eμ-myc tumor bearing spleens dissected at time of death (Purelink Genomic DNA Mini Kit, Invitrogen). Q-PCR was carried out using primers specific to the endogenous Dino locus and EGFP (mutant allele, See Table S4), normalizing to L32 as a control (Brilliant II QRT-PCR SYBR® Green Low ROX Master Mix (Agilent Technologies), QuantStudio 6 Flex Real-Time PCR system (Applied Biosytems)). The relative abundance of Dino and EGFP in tumors is shown normalized to Dino+/− spleen.
Virus production
Full-length mouse Dino was PCR amplified (Phusion HF PCR Master Mix, NEB) using Fwd-BamHI and Rev-NotI encoding primers, and ligated into the multiple cloning site of pCDH-EF1α-MCS-(PGK-GFP-T2A-Puro) (System Biosciences CD813A-1). Empty vector was used to generate control copGFP positive virus, pMSCV-IRES-mCherry (Addgene plasmid # 52114) was used to make control mCherry positive virus. pBabe 12S E1A was a gift from Scott Lowe (Addgene plasmid # 18742). pWZL hygro H-Ras V12 was a gift from Scott Lowe (Addgene plasmid # 18749). Fuw-dCas9-Tet1CD-P2A-BFP (Addgene plasmid 108245), Fuw-dCas9-dead Tet1CD-P2A-BFP (Addgene plasmid # 108246) and pgRNA-modified (Addgene plasmid #84477) were a gift from Rudolf Jaenisch. All plasmids were propagated in single shot Mach I bacteria (Life Technologies), and Sanger sequenced prior to use. Lentiviral vectors were transfected into LentiX 293T cells (Takara) using Lenti-X Packaging Single Shots (Takara). Supernatants were filtered (0.45μM) and concentrated using Lenti-X concentrator (Takara) prior to use. Retroviral vectors were transfected into Pheonix-293T packaging cells using Calphos Mammalian Transfection Kit (Clontech).
Cell culture
MEFs were sequentially infected with E1A (puromycin selection, Invivogen) and RasG12V (hygromycin selection, ThermoFisher Scientific) viruses. For clonogenic cell survival assays, 150 cells were plated per well in triplicate in a 6 well dish and cultured for 8 days. Colonies were fixed with methanol, stained with crystal violet and counted using ImageJ. Edu incorporation was assayed by Click-iT® Plus EdU Alexa Fluor® 488 Flow Cytometry Assay Kit (Thermo Fisher Scientific). Apoptosis was assayed using the ApoStat Apoptosis Detection Kit (R&D Systems). Flow cytometry was carried out using a LSR Fortessa (BD Biosciences) and analyzed using FlowJo (BD Biosciences).
Single cell suspensions from lymphoma bearing spleens from Dino−/−-Eμ-myc mice were generated at time of death by mechanical disruption through 100μm mesh. Erythocytes were lysed with ACK Buffer (150mM NH4Cl, 10mM KHCO3, 0.1mM Na2EDTA, pH7.2–7.4). Isolated Eμ-myc lymphoma cell lines were maintained in FMA media and treated with 10mM Nutlin-3 (Sigma Aldrich #N6287) dissolved in DMSO (Fisher Scientific) for indicated cell culture treatments. For cryostorage, > 5×106 cells were pelleted and re-suspended in maintenance media + 10% additional FBS and 20% DMSO.
For competition assays primary mouse lymphoma cells were infected with the indicated virus and grown for 72 hours in culture. GFP positive and mCherry positive cells were FACS-sorted (FACSAria, BD Biosciences) and co-cultured in 96-well flat bottom plates for 14–21 days. Triplicate samples were taken at indicated time points for quantification of (DAPI-negative) fluorescent cells via flow cytometry (LSR Fortessa (BD Biosciences), FCS Express (De Novo Software)). Fluorescent cell ratios were calculated and normalized to empty vector-GFP:mCherry for each time-point for plotting purposes.
DNA methylation analyses
MeDIP was performed as described previously (Hsu et al., 2012). Briefly, genomic DNA was prepared by RNaseA (Fisher Scientific) and proteinase K digestion of cell pellets, followed by phenol:chloroform:isoamyl alcohol extraction and ethanol precipitation. DNA was sonicated (Biorupter-Pico, Diagenode) to produce random fragments from 300 to 1,000bp. Fragmented DNA was denatured for 10 min at 95°C, and immunoprecipitated overnight at 4°C 5-methylcytidine antibody (Millipore-Sigma) in IP buffer (10 mM sodium phosphate [pH 7.0], 140 mM NaCl, 0.05% Triton X-100). The mixture was incubated for 4 hours at 4°C with Protein G Dynabeads (Thermo Fisher Scientific) and washed three times with IP buffer. Beads were resuspended with digestion buffer (50 mM Tris [pH 8.0], 10 mM EDTA, 0.5% SDS) plus proteinase K (Ambion, 1μg/μl final concentration) and shaken overnight at 56°C. DNA was purified by phenol:chloroform:isoamyl extraction (MP Biomedicals) and ethanol precipitation, then analyzed by real-time PCR.
For genome-wide demethylation studies, HT-1080 cells were grown in the presence of 5-aza-2′-deoxycytidine (Millipore Sigma) for 72 hours before RNA purification and qRT-PCR. To assess the methylation status of specific loci both in cell culture and mouse tumor tissue, genomic DNA was prepared as above. Methylation was assayed by bisulfite conversion (Epimark Bisulfite conversion kit, NEB) using EpiMark Hotstart DNA polymerase (NEB) and bisulfite converted primer sequences (Table S4). PCR purified fragments were sanger sequenced a minimum of 3 times, and the average relative C:T ratio at each CpG calculated as previously described (Jiang et al., 2010). For targeted demethylation in HT1080 cells guide RNA was designed using the Custom Alt-R CRISPR Cas9 guide RNA tool (IDT). The sgRNA sequence ATTGTCACATGCTTCCGGGA was cloned into the AarI site of pgRNA-modified as previously reported (Liu et al., 2016a). HT-1080 cells were infected with either dCas9-deadTet1 or dCas9-Tet1 lentivirus, and FACS sorted 72 hours after infection using the viral BFP reporter protein. BFP-positive cells were infected with gRNA expressing virus and puromycin selected. Genomic DNA and RNA was isolated 10 days after sgRNA viral infection.
Trp53 mutation status
To assess Trp53 mutation status, genomic DNA was isolated from indicated cell lines and tissues (Purelink Genomic DNA Mini Kit, Invitrogen. PCR was performed using Trp53 primer pairs shown in Table S4 using Phusion polymerase (New England Biolabs #M0531L). Gel-purified DNA fragments were Sanger sequenced.
Tumor allografts
Single cell suspensions from lymphoma bearing spleens from Din−/− Eμ-myc and p53−/− Eμ-myc mice were generated at time of death by mechanical disruption through 100um mesh. Erythocytes were lysed with ACK Buffer (150mM NH4Cl, 10mM KHCO3, 0.1mM Na2EDTA, pH7.2–7.4). 200×105 cells suspended in 200ul sterile 1xPBS were IP injected into recipient 6-week old male C57BL/6 mice (Jackson Laboratories) to confirm the ability of the isolated cells to generate lymphoma in recipient animals. Lymphoma cells were isolated from the spleens of recipient mice with lymphoma, plated at 5×106/ml in FMA (Corcoran et al., 1999), and immediately spinfected with either Empty vector-GFP or mDino-GFP containing lentivirus (no polybrene). 48 hours post infection GFP-positive, live (DAPI-negative) cells were FACS-sorted and equal numbers of cells were IP injected into 10 recipient 6-week old male C57BL/6 mice per viral construct. Recipients were monitored for signs of lymphoma development and tumor bearing tissues were harvested at time of death. Viral infection of tumor cells was confirmed by direct fluorescence imaging of copGFP in recipient spleens and western blot (anti-TurboGFP, Invitrogen, data not shown).
For fibrosarcoma allograft studies, 1 × 106 cells were suspended in matrigel and were injected the flanks of CB17-Prkdc < scid > mice (Taconic), 5 animals and ten tumor engraftments per cell line. Tumor growth was monitored by three orthogonal measurements using digital calipers. Cell preparation, injection, and tumor monitoring was carried out by the Antitumor Assessment Core Facility at MSKCC.
Cycloheximide chase and western blotting
HT-1080 cells were treated with cycloheximide (Sigma, 25ug/ml) for the times indicated. For all Western Blots tissue was lysed in NET buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 5 mM EDTA, 0.1% NP40 substitute, protease inhibitors), sonicated and centrifuged to obtain the soluble protein fraction. Proteins were size separated by electrophoresis 4%–12% Bis-Tris protein gels (Invitrogen) and SDS-MOPS buffer (Invitrogen), then transferred to nitrocellulose (BioRad). Total protein was quantified using Qubit Protein Assay Kit (Invitrogen) prior to protein loading, and by total protein staining (Revert 700, Li-Cor) of the membrane. Blots were blocked in 5% Milk 1XPBS, and probed using antibodies against p53 (DO-1 (Sigma, human), 1C12 (Cell Signaling, mouse), B-tubulin (GeneTex) and Actin (MAB1501, EMD Millipore) in 5% milk 1X PBS-Tween. Bands were visualized using appropriate IRDye secondary antibodies and quantified using an Odyssey CLx Imaging System and ImageStudio (Li-Cor).
QUANTIFICATION AND STATISTICAL ANALYSIS
Genome-wide read counts were summarized using the R package featureCounts, mapped to the hg38 human gene set using Ensembl (Ensembl GRCh38.95 GTF), and normalized using variance stabilizing transformation from the R package DESeq2.
For DNA methylation, copy number alteration, and mutation analyses, average methylation within the DINO DMR was obtained by averaging the beta values of the six CpGs covered by the Illumina HM450 probes in this region. Methylated DINO was defined as an average beta value of the six CpGs ≥ 0.5. For mutual exclusivity analyses, TP53 was considered altered in any sample with deep deletion of TP53 (copy number ≤ −2) or any non-silent TP53 mutation. Fisher exact test was used to test for significant mutual exclusivity or co-alteration and logistic regression analyses for DNA methylation and TP53 alterations were performed using XLSTAT.
Kaplan-Meier curves were plotted using Prism (GraphPad) and mean median survival and significance were calculated using the Mantel-Cox Test. For quantitative PCR and RT-PCR a minimum of three biological replicates per condition were used, and significance calculated by t test. Error bars show standard error of the mean. Detailed descriptions of number and definition of replicates, plus the statistical methods used can be found in figure legends. All statistical analyses were performed using Excel, XLSTAT and Prism software version 8 (GraphPad).
Supplementary Material
Highlights.
DINO hypermethylation in human cancers is mutually exclusive with TP53 mutations
Dino is a haplo-insufficient tumor suppressor encoded in the Dino/Cdkn1a locus
Dino’s tumor suppressor mechanism requires p53
Hypermethylation silences DINO in human cancers to impair p53 signaling
ACKNOWLEDGMENTS
We thank Scott Lowe, Howard Chang, Simon Powell, Timothy Chan, and Azusa Tanaka for valuable discussions and feedback. Thanks to Scott Lowe for kindly providing the Eμ-myc mice. We thank Rachel Baum for able assistance. Animal husbandry was provided by the MSKCC/Weill Cornell animal facility. This study was supported by funding from the MSKCC Geoffrey Beene Cancer Research Center (to A.M.S. and N.W.), the American Cancer Society (RSG-19-158-01-RMC to A.M.S. and N.W.), NIH (R35GM124909 to A.M.S.), and the MSKCC Cancer Center Core (P30 CA008748).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109329.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPORTING CITATIONS
The following references appear in the Supplemental Information: Liu et al. (2018); Morgenstern and Land (1990); Samuelson and Lowe (1997); Serrano et al., (1997); Yosifov et al. (2020).
REFERENCES
- Abbas T, and Dutta A (2009). p21 in cancer: intricate networks and multiple activities. Nat. Rev. Cancer 9, 400–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, and Brinster RL (1985). The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 318, 533–538. [DOI] [PubMed] [Google Scholar]
- Arribas AJ, Rinaldi A, Mensah AA, Kwee I, Cascione L, Robles EF, Martinez-Climent JA, Oscier D, Arcaini L, Baldini L, et al. (2015). DNA methylation profiling identifies two splenic marginal zone lymphoma subgroups with different clinical and genetic features. Blood 125, 1922–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Taylor BS, Banerji S, Ramos AH, Lagos-Quintana M, Decarolis PL, Shah K, Socci ND, Weir BA, Ho A, et al. (2010). Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat. Genet 42, 715–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T, et al. (2013). EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 23, 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton MC, Johnstone A, Eccles D, Harmon B, Hayes MT, Lea RA, Griffiths L, Hoffman EP, Stubbs RS, and Macartney-Coxson D (2015). An analysis of DNA methylation in human adipose tissue reveals differential modification of obesity genes before and after gastric bypass and weight loss. Genome Biol. 16, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, and Hannon GJ (1995). Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature 377, 552–557. [DOI] [PubMed] [Google Scholar]
- Cantero D, Mollejo M, Sepúlveda JM, D’Haene N, Gutiérrez-Guamán MJ, Rodríguez de Lope Á, Fiaño C, Castresana JS, Lebrun L, Rey JA, et al. (2020). TP53, ATRX alterations, and low tumor mutation load feature IDH-wildtype giant cell glioblastoma despite exceptional ultra-mutated tumors. Neurooncol. Adv 2, vdz059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazier JB, Rao SR, McLean CM, Walker AK, Wright BJ, Jaeger EE, Kartsonaki C, Marsden L, Yau C, Camps C, et al. ; Oxford-Illumina WGS500 Consortium (2014). Whole-genome sequencing of bladder cancers reveals somatic CDKN1A mutations and clinicopathological associations with mutation burden. Nat. Commun 5, 3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambwe N, Kormaksson M, Geng H, De S, Michor F, Johnson NA, Morin RD, Scott DW, Godley LA, Gascoyne RD, et al. (2014). Variability in DNA methylation defines novel epigenetic subgroups of DLBCL associated with different clinical outcomes. Blood 123, 1699–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciriello G, Cerami E, Sander C, and Schultz N (2012). Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 22, 398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran LM, Tawfilis S, and Barlow LJ (1999). Generation of B lymphoma cell lines from knockout mice by transformation in vivo with an Emumyc transgene. J. Immunol. Methods 228, 131–138. [DOI] [PubMed] [Google Scholar]
- Deng C, Zhang P, Harper JW, Elledge SJ, and Leder P (1995). Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 82, 675–684. [DOI] [PubMed] [Google Scholar]
- Do C, Lang CF, Lin J, Darbary H, Krupska I, Gaba A, Petukhova L, Vonsattel JP, Gallagher MP, Goland RS, et al. (2016). Mechanisms and Disease Associations of Haplotype-Dependent Allele-Specific DNA Methylation. Am. J. Hum. Genet 98, 934–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S, et al. (2009). Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet 41, 1350–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop MG, Dobbins SE, Farrington SM, Jones AM, Palles C, Whiffin N, Tenesa A, Spain S, Broderick P, Ooi LY, et al. ; Colorectal Tumour Gene Identification (CORGI) Consortium; Swedish Low-Risk Colorectal Cancer Study Group; COIN Collaborative Group (2012). Common variation near CDKN1A, POLD3 and SHROOM2 influences colorectal cancer risk. Nat. Genet 44, 770–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan WA, Gaskell E, and Bernstein BE (2017). Epigenetic plasticity and the hallmarks of cancer. Science 357, eaal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara S, Nagai H, Jimbo H, Jimbo N, Tanaka T, Inoie M, and Nishigori C (2019). Gene Expression and Methylation Analysis in Melanomas and Melanocytes From the Same Patient: Loss of NPM2 Expression Is a Potential Immunohistochemical Marker for Melanoma. Front. Oncol 8, 675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hemann MT, Bric A, Teruya-Feldstein J, Herbst A, Nilsson JA, Cordon-Cardo C, Cleveland JL, Tansey WP, and Lowe SW (2005). Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature 436, 807–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovestadt V, Jones DT, Picelli S, Wang W, Kool M, Northcott PA, Sultan M, Stachurski K, Ryzhova M, Warnatz HJ, et al. (2014). Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature 510, 537–541. [DOI] [PubMed] [Google Scholar]
- Hsu CH, Peng KL, Kang ML, Chen YR, Yang YC, Tsai CH, Chu CS, Jeng YM, Chen YT, Lin FM, et al. (2012). TET1 suppresses cancer invasion by activating the tissue inhibitors of metalloproteinases. Cell Rep. 2, 568–579. [DOI] [PubMed] [Google Scholar]
- Hu X, Feng Y, Zhang D, Zhao SD, Hu Z, Greshock J, Zhang Y, Yang L, Zhong X, Wang LP, et al. (2014). A functional genomic approach identifies FAL1 as an oncogenic long noncoding RNA that associates with BMI1 and represses p21 expression in cancer. Cancer Cell 26, 344–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al. (2009). The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet 41, 178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, Barrette TR, Prensner JR, Evans JR, Zhao S, et al. (2015). The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet 47, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Zhang Y, Fei J, Chang X, Fan W, Qian X, Zhang T, and Lu D (2010). Rapid quantification of DNA methylation by measuring relative peak heights in direct bisulfite-PCR sequencing traces. Lab. Invest 90, 282–290. [DOI] [PubMed] [Google Scholar]
- Jiang D, Brady CA, Johnson TM, Lee EY, Park EJ, Scott MP, and Attardi LD (2011). Full p53 transcriptional activation potential is dispensable for tumor suppression in diverse lineages. Proc. Natl. Acad. Sci. USA 108, 17123–17128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimovich B, Mutlu S, Schneikert J, Elmshäuser S, Klimovich M, Nist A, Mernberger M, Timofeev O, and Stiewe T (2019). Loss of p53 function at late stages of tumorigenesis confers ARF-dependent vulnerability to p53 reactivation therapy. Proc. Natl. Acad. Sci. USA 116, 22288–22293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, Fan H, Shen H, Way GP, Greene CS, et al. ; Cancer Genome Atlas Research Network (2018). Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 23, 239–254.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, Shu J, Dadon D, Young RA, and Jaenisch R (2016a). Editing DNA Methylation in the Mammalian Genome. Cell 167, 233–247.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Chen C, Xu Z, Scuoppo C, Rillahan CD, Gao J, Spitzer B, Bos-bach B, Kastenhuber ER, Baslan T, et al. (2016b). Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature 531, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, Horlbeck MA, Cho SW, Birk HS, Malatesta M, He D, Attenello FJ, Villalta JE, Cho MY, Chen Y, et al. (2017). CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 355, aah7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XS, Wu H, Krzisch M, Wu X, Graef J, Muffat J, Hnisz D, Li CH, Yuan B, Xu C, et al. (2018). Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 172, 979–992.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe SW, Jacks T, Housman DE, and Ruley HE (1994). Abrogation of oncogene-associated apoptosis allows transformation of p53-deficient cells. Proc. Natl. Acad. Sci. USA 91, 2026–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA, Krizhanovsky V, and Lowe SW (2013). Non-cell-autonomous tumor suppression by p53. Cell 153, 449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzese DM, Scolyer RA, Huynh JL, Huang SK, Hirose H, Chong KK, Kiyohara E, Wang J, Kawas NP, Donovan NC, et al. (2014). Epigenome-wide DNA methylation landscape of melanoma progression to brain metastasis reveals aberrations on homeobox D cluster associated with prognosis. Hum. Mol. Genet 23, 226–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern JP, and Land H (1990). Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 18, 3587–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbold A, Salmon JM, Martin BP, Stanley K, and Johnstone RW (2014). The role of p21(waf1/cip1) and p27(Kip1) in HDACi-mediated tumor cell death and cell cycle arrest in the Eμ-myc model of B-cell lymphoma. Oncogene 33, 5415–5423. [DOI] [PubMed] [Google Scholar]
- R Core Team (2020). R: A language and environment for statistical computing (Vienna, Austria: R Foundation for Statistical Computing; ). [Google Scholar]
- Roman-Gomez J, Castillejo JA, Jimenez A, Gonzalez MG, Moreno F, Rodriguez Mdel.C., Barrios M, Maldonado J, and Torres A (2002). 5′ CpG island hypermethylation is associated with transcriptional silencing of the p21(CIP1/WAF1/SDI1) gene and confers poor prognosis in acute lymphoblastic leukemia. Blood 99, 2291–2296. [DOI] [PubMed] [Google Scholar]
- Samuelson AV, and Lowe SW (1997). Selective induction of p53 and chemosensitivity in RB-deficient cells by E1A mutants unable to bind the RB-related proteins. Proc. Natl. Acad. Sci. USA 94, 12094–12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt AM, and Chang HY (2016). Long Noncoding RNAs in Cancer Pathways. Cancer Cell 29, 452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt CA, McCurrach ME, de Stanchina E, Wallace-Brodeur RR, and Lowe SW (1999). INK4a/ARF mutations accelerate lymphomagenesis and promote chemoresistance by disabling p53. Genes Dev. 13, 2670–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt AM, Garcia JT, Hung T, Flynn RA, Shen Y, Qu K, Payumo AY, Peres-da-Silva A, Broz DK, Baum R, et al. (2016). An inducible long noncoding RNA amplifies DNA damage signaling. Nat. Genet 48, 1370–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz MD, He Y, Whitaker JW, Hariharan M, Mukamel EA, Leung D, Rajagopal N, Nery JR, Urich MA, Chen H, et al. (2015). Human body epigenome maps reveal noncanonical DNA methylation variation. Nature 523, 212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, and Lowe SW (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602. [DOI] [PubMed] [Google Scholar]
- Valente LJ, Grabow S, Vandenberg CJ, Strasser A, and Janic A (2016). Combined loss of PUMA and p21 accelerates c-MYC-driven lymphoma development considerably less than loss of one allele of p53. Oncogene 35, 3866–3871. [DOI] [PubMed] [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, and Jacks T (2007). Restoration of p53 function leads to tumour regression in vivo. Nature 445, 661–665. [DOI] [PubMed] [Google Scholar]
- Wang Z, Yang B, Zhang M, Guo W, Wu Z, Wang Y, Jia L, Li S, Cancer Genome Atlas Research Network; Xie W, and Yang D (2018). lncRNA Epigenetic Landscape Analysis Identifies EPIC1 as an Oncogenic lncRNA that Interacts with MYC and Promotes Cell-Cycle Progression in Cancer. Cancer Cell 33, 706–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosifov DY, Bloehdorn J, Döhner H, Lichter P, Stilgenbauer S, and Mertens D (2020). DNA methylation of chronic lymphocytic leukemia with differential response to chemotherapy. Sci. Data 7, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younger ST, Kenzelmann-Broz D, Jung H, Attardi LD, and Rinn JL (2015). Integrative genomic analysis reveals widespread enhancer regulation by p53 in response to DNA damage. Nucleic Acids Res. 43, 4447–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the Key Resources Table. Original TCGA source data for bioinformatic analyses are available through the Genomics Data Commons and can be viewed through the UCSC Xena Browser.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-5-methylcytosine Antibody, clone 33D3 | Millipore Sigma | Cat# MABE146; RRID:AB_10863148 |
| anti-TurboGFP | Thermo Fisher Scientific | Cat# PA5–22688; RRID:AB_2540616 |
| Anti-p53 antibody, Mouse monoclonal (clone DO-1) | Sigma Aldrich | Cat# P6874; RRID:AB_1079545 |
| p53 (1C12) Mouse mAb | Cell Signaling | Cat# 2524; RRID:AB_331743 |
| beta Tubulin antibody | GeneTex | Cat# GTX101279; RRID:AB_1952434 |
| Anti-Actin Antibody, clone C4 | Millipore Sigma | Cat# MAB1501; RRID:AB_2223041 |
| IRDye® 800CW Goat anti-Mouse IgG Secondary Antibody | Li-Cor | Cat# 926–32210; RRID:AB_621842 |
| IRDye® 680LT Goat anti-Rabbit IgG Secondary Antibody | Li-Cor | Cat# 926–68021; RRID:AB_10706309 |
| Bacterial and virus strains | ||
| One Shot® Mach1 T1 Phage-Resistant Chemically Competent E. coli | Invitrogen | C862003 |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM high glucose | MSKCC Media Core | N/A |
| Fetal Bovine Serum | VWR | Cat# 97068–085 |
| Penicillin-Streptomycin | MSKCC Media Core | N/A |
| RPMI 1640 Medium | ThermoFisher Scientific | Cat# 11875093 |
| 2-mercaptoethanol | GIBCO | ThermoFisher Scientific cat# 21985023 |
| L-Asparagine | Sigma-Aldrich | Cat# A4159 |
| Purelink Genomic DNA Mini Kit | Invitrogen | Cat# K182002 |
| Brilliant II QRT-PCR SYBR® Green Low ROX Master Mix | Agilent Technologies | Cat# 600835 |
| Pan B Cell Isolation Kit II | Miltenyi Biotec | Cat# 130-104-443 |
| MACS BSA Stock Solution | Miltenyi Biotec | Cat# 130-091-376 |
| autoMACS Rinsing Solution | Miltenyi Biotec | Cat# 130-091-222 |
| MACS LS Columns | Miltenyi Biotec | Cat# 130-042-401 |
| RNeasy Plus Mini Kit | QIAGEN | Cat# 74136 |
| Phusion HF PCR Master Mix | NEB | Cat# M0531 |
| BamHI | NEB | Cat# R0136 |
| NotI-HF | NEB | Cat# R3189 |
| AarI | Thermo Scientific | Cat# ER1581 |
| Rapid DNA Dephos & Ligation Kit | Sigma-Aldrich | Cat# 4898117001 |
| Lenti-X Packaging Single Shots | Takara | Cat# 631282 |
| Lenti-X Concentrator | Takara | Cat# 631231 |
| Calphos Mammalian Transfection Kit | Clontech | Cat# 631312 |
| Puromycin 10mg/ml | Invivogen | Cat# ant-pr-1 |
| Hygromycin B (50 mg/mL) | ThermoFisher Scientific | Cat# 10687010 |
| Methanol | Fisher Scientific | Cat# A412P-4 |
| Crystal violet solution | Sigma-Aldrich | Cat# HT90132 |
| Nutlin-3 | Sigma-Aldrich | Cat# N6287 |
| DMSO | Fisher Scientific | Cat# BP231–100 |
| DAPI Staining Solution | Miltenyi Biotec | Cat# 130-111-570 |
| Dynabeada Protein G for Immunoprecipitation | Thermo Fisher Scientific | Cat# 10003D |
| Proteinase K | Thermo Fisher Scientific | Cat# AM2548 |
| Phenol:Chloroform Saturated Solution, Ph 6.7. ≥ 99.0% | MP Biomedicals | Cat# ICNA04802521 |
| 3M Sodium Acetate | Ambion | Cat# AM9740 |
| GlycoBlue | Ambion | Cat# AM9516 |
| 5-Aza-2-deoxycytidine | Sigma-Aldrich | Cat# A3656 |
| PureLink RNase A (20 mg/mL) | Fisher Scientific | Cat# 12091021 |
| EpiMark® Bisulfite Conversion Kit | New England Biolabs | Cat# E3318S |
| EpiMark® Hot Start Taq DNA Polymerase | New England Biolabs | Cat# M0490 |
| Cycloheximide | Sigma-Aldrich | Cat# C4859 |
| Bolt 4 to 12%, Bis-Tris, 1.0 mm Mini Protein Gel, 12-well | Thermo Fisher Scientific | Cat# NW04122BOX |
| 20X Bolt MOPS SDS Running Buffer | Thermo Fisher Scientific | Cat# B0001 |
| Invitrogen novex NuPAGE 4–12% Bis-Tris Midi Protein Gels, 20 well | Thermo Fisher Scientific | Cat# WG1402BOX |
| Bolt Transfer Buffer (20X) | Thermo Fisher Scientific | Cat# BT00061 |
| Nitrocellulose Membrane, 0.45 mm | BioRad | Cat# 1620115 |
| Revert 700 Total Protein Stain Kits for Western Blot Normalization | Li-Cor | Cat# 926–11010 |
| Critical commercial assays | ||
| Click-iT® Plus EdU Alexa Fluor® 488 Flow Cytometry Assay Kit | ThermoFisher Scientific | Cat# C10632 |
| ApoStat Apoptosis Detection Kit | R&D Systems | Cat# FMK012 |
| Qubit Protein Assay Kit | Invitrogen | Cat# Q33212 |
| Deposited data | ||
| Benign Adipose Methylation Array | Benton et al., 2015 | EMBL-EBI: E-MTAB-1866 |
| Benign Glia Methylation Array | Do et al., 2016 | GEO: GSE79144 |
| Benign Melanocyte Methylation | Fujiwara et al., 2019 | GEO: GSE122909 |
| Benign Melanocyte Methylation | Marzese et al., 2014 | GEO: GSE44662 |
| Experimental models: cell lines | ||
| Human: BJ Fibroblasts | ATCC | ATCC Cat# CRL-2522; RRID:CVCL_3653 |
| Human: HT-1080 | ATCC | ATCC Cat# CRL-7951; RRID:CVCL_0317 |
| Human: LentiX-293T | Takara | Cat# 632180 |
| Human: Pheonix-293T | Nolan Laboratory, Stanford University | N/A |
| Mouse Embryonic Fibroblasts | This paper | N/A |
| Mouse: Em-myc lymphoma cell lines | This paper | N/A |
| Experimental models: organisms/strains | ||
| Mouse: C57BL/6-Dinoegfp | Howard Chang, Stanford University | N/A |
| Mouse: C57BL/6 Eμ-myc | Scott Lowe, MSKCC | N/A |
| Mouse: C57BL/6 Cdkn1a−/− (B6.129S6(Cg)-Cdkn1atm1Led/J) | Jackson Labs | IMSR Cat# JAX:016565;RRID:IMSR_JAX:016565 |
| Mouse: CB17-Prkdc< scid > | Taconic | IMSR Cat# TAC:cb17sc; RRID:IMSR_TAC:cb17sc |
| Mouse: C57BL/6 | Jackson Labs | IMSR Cat# JAX:000664; RRID:IMSR_JAX:000664 |
| Oligonucleotides | ||
| PCR Primers | This paper (see Table S4) | N/A |
| Recombinant DNA | ||
| Plasmid: pCDH-EF1α-MCS-(PGK-GFP-T2A-Puro) | System Biosciences | Cat# CD813A-1 |
| Plasmid: pCDH-EF1α-Dino-(PGK-GFP-T2A-Puro) | This paper | N/A |
| Plasmid: pMSCV-IRES-mCherry FP | Addgene | RRID:Addgene_52114 |
| Plasmid: pBabe 12S E1A | Addgene | RRID:Addgene_18742 |
| Plasmid: pWZL hygro H-RasV12 | Addgene | RRID:Addgene_18749 |
| Plasmid: Fuw-dCas9-Tet1CD-P2A-BFP | Addgene | RRID:Addgene_108245 |
| Plasmid: Fuw-dCas9-dead Tet1CD-P2A-BFP | Addgene | RRID:Addgene_108246 |
| Plasmid: pgRNA-modified | Addgene | RRID:Addgene_84477 |
| Plasmid: pgRNA-modified_sgDINO | This paper | N/A |
| Software and algorithms | ||
| R | (R Core Team, 2020) | |
| featureCounts | CRAN | Liao et al., 2014 |
| DESeq2 | CRAN | Love et al., 2014 |
| XLSTAT | Addinsoft | N/A |
| Prism | GraphPad | v8 |
| ImageJ | NIH | Schneider et al., 2012 |
| FlowJo | BD Biosciences | N/A |
| FCS Express | De Novo Software | V6 |
| Custom Alt-R® CRISPR-Cas9 guide RNA tool | Integrated DNA Technologies | https://www.idtdna.com/site/order/designtool/index/CRISPR_CUSTOM |
| Image Studio | Li-Cor | N/A |
| Other | ||
| QuantStudio 6 Flex Real-Time PCR system | Applied Biosytems | N/A |
| MACS Multistand | Miltenyi Biotec | Cat# 130-042-303 |
| MidiMACS Separator | Miltenyi Biotec | Cat# 130-042-302 |
| LSR Fortessa | BD Biosciences | N/A |
| FACSAria | BD Biosciences | N/A |
| Biorupter Pico | Diagenode | B01060010 |
| Qubit 3.0 Fluorometer | Invitrogen | Cat# Q33216 |
| Odyssey CLx Imaging System | Li-Cor | Model# 9140 |