


Abstract
Many type III CRISPR–Cas systems rely on the cyclic oligoadenylate (cOA) signaling pathway to exert immunization. However, LdCsm, a type III-A lactobacilli immune system mediates efficient plasmid clearance in spite of lacking cOA signaling. Thus, the system provides a good model for detailed characterization of the RNA-activated DNase in vitro and in vivo. We found ATP functions as a ligand to enhance the LdCsm ssDNase, and the ATP enhancement is essential for in vivo plasmid clearance. In vitro assays demonstrated LdCsm cleaved transcriptional bubbles at any positions in non-template strand, suggesting that DNA cleavage may occur for transcribing DNA. Destiny of target plasmid versus nontarget plasmid in Escherichia coli cells was investigated, and this revealed that the LdCsm effectors mediated co-transcriptional DNA cleavage to both target and nontarget plasmids, suggesting LdCsm effectors can mediate DNA cleavage to any transcriptional bubbles in close proximity upon activation. Subcellular locations of active LdCsm effectors were then manipulated by differential expression of LdCsm and CTR, and the data supported the hypothesis. Strikingly, stepwise induction experiments indicated allowing diffusion of LdCsm effector led to massive chromosomal DNA degradation, suggesting this unique IIIA system can facilitate infection abortion to eliminate virus-infected cells.
INTRODUCTION
CRISPR–Cas (Clustered Regularly Interspaced Short Palindromic Repeats, CRISPR-associated) systems provide the prokaryotic adaptive and heritable immunity that defends bacteria and archaea against invasion by mobile genetic elements in a small RNA-guided fashion (1–7). These antiviral systems fall into two broad classes (class 1 and 2) and six different types (types I–VI), which are further divided into a number of subtypes (8–10). Type III CRISPR–Cas systems are among the most interesting ones, and they possess three distinct activities: (a) target RNA cleavage via Csm3/Cmr4, the large backbone subunit (11–14), (b) target RNA-activated indiscriminate ssDNA cleavage from the HD nuclease domain of Csm1/Cmr2, the Cas10 subunit (15–19) and (c) cyclic oligoadenylate (cOA) generation by the Palm domains of Cas10 (20–25). The last activity produces cOA secondary signals that allosterically regulate activities of CARF (CRISPR-Associated Rossman Fold) domain nucleases (Csm6/Csx1/Can1/Can2/Card1), CRISPR-accessory enzymes (22,26–31) or NucC, an endonuclease that functions as the effector in the cyclic-oligonucleotide-based anti-phage signaling systems (CBASS) (32,33). The Cas10-hosted cOA synthesis and the CARF domain nucleases or the CBASS restriction enzyme constitute the cOA signaling pathway that is essential for efficiently protecting host against nucleic acid invasion and these enzyme effectors probably protect the cell population by selectively eliminating the infected cells (abortive infection) (34–41). In contrast, co-transcriptional DNA cleavage can facilitate clearance of invading DNA, allowing infected cells eventually to recover from virus infection (37).
Recently, we showed that LdCsm, a novel III-A CRISPR–Cas system present in Lactobacillus delbrueckii subsp. bulgaricus is capable of mediating plasmid clearance in a genetic assay in spite of lacking the cOA signaling pathway (42). This is very different from all other known type III immune systems, which primarily rely on the CRISPR signaling pathway to mediate antiviral defense, such as the Staphylococcus epidermidis Csm (SeCsm) system (36,37,40). Furthermore, it has been shown that the two Cas10-hosted activities function in concert to mediate anti-plasmid defense in which the Cas10 HD activity is essential for plasmid clearance as revealed for SeCsm (37). Nevertheless, there is a knowledge gap here: We have only had a rudimentary understanding of co-transcriptional nuclease activities of type III systems, which prevent us from clearly illustrating immunization mechanisms by type III CRISPR–Cas systems. An early work showed that the SeCsm effector performs co-transcriptional DNA cleavage at transcriptional bubbles, and the cleavage occurs for the 3′ flanking sequence of the target DNA in the non-template strand (43). However, contradictory results were reported more recently in which the SeCsm effector was only found to cleave co-transcriptional RNA as for the Thermus thermophilus Csm (TthCsm) (44). To this end, it remains unknown whether type III effector complexes can mediate co-transcriptional DNA cleavage.
Herein the LdCsm system was characterized in detail for the transcription-dependent DNA interference in vivo and the co-transcriptional DNA cleavage in vitro. We found that ATP functions as a ligand to enhance the LdCsm ssDNase to the optimum level that is essential for plasmid clearance by the unique immune system. Strikingly, while in vitro assays demonstrate LdCsm exhibits a strong DNA cleavage to the non-template strand DNA at any positions in the presence of multiple rounds of transcription, in vivo co-transcriptional DNA cleavage occurs for target plasmid, nontarget plasmid as well as in host chromosome. These results indicate the LdCsm co-transcriptional DNA cleavage also protects host cell populations by facilitating infection abortion to eliminate virus-infected cells, as for the CRISPR signaling pathway.
MATERIALS AND METHODS
Bacterial strains and growth conditions
Escherichia coli strains DH5α and BL21(DE3) were propagated in Luria-Bertani (LB) medium at 37°C with shaking at 200 rpm. If applicable, antibiotics were added to E. coli cultures as the following: 100 μg/ml ampicillin (Sigma-Aldrich), 25 μg/ml kanamycin (Sigma-Aldrich) and 10 μg/ml chloramphenicol (Sigma-Aldrich).
Construction of recombinant plasmids
Recombinant plasmids p15AIE-Cas, p15AIE-Cas-S1, pUCE, pUCE-S1, pET30a-Csm2, pBad-G, pBad-CTR and pBad-NTR were constructed previously (42). CRISPR plasmids were constructed for detection of 16S rRNAs of Lactobacillus plantarum and Lactobacillus casei. DNA fragments containing multiple copies of the 36-nt repeat-Lxx spacer (40 nt) were obtained from fusion PCR amplification using three primers, Re-Lxx-F, L-R1 and Re-L-R. The resulting amplification products were analyzed by agarose gel electrophoresis, and PCR products of ∼1 kb region were recovered from the agarose gel using an OMEGA gel-purification kit (OMEGA bio-tek). The purified DNA fragments were cloned into plasmid pJET1.2 (CloneJET PCR Cloning Kit, Thermo Scientific, USA). After confirming by DNA sequencing (GATC biotech), the CRISPR arrays were amplified individually from pJET clones by PCR. Insertion of each DNA fragment into plasmid pUCE at the BglII site yielded pUCE-xxx CRISPR plasmid (xxx represents the LPwt, LCwt, LPd or LCd spacers). These CRISPR plasmids were introduced into the E. coli strains, giving bacterial hosts for production of Csm RNPs carrying either the wild-type (WT) Csm3 or nuclease-dead Csm3 (dCsm3). Oligos and plasmids used in this study were listed in Supplementary Tables S1 and S2, respectively.
Purification of LdCsm effector complexes from E. coli
Bacterial strains carrying plasmids p15AIE-Cas, p15AIE-Cas-S1, pUCE, pUCE-S1, pET30a-Csm2, pBad-G, pBad-CTR were constructed previously. The wild-type (WT) and mutated LdCsm effectors were purified as described previously (42).
Labeling of DNA and RNA substrates
All DNA oligos, S10 RNA (nonhomologous RNA), S1–40 RNA (PTR), S1–46 RNA (CTR) and S1–48 RNA (NTR) oligonucleotides were purchased from Integrated DNA Technologies, other RNA oligonucleotides were generated by in vitro transcription using TranscriptAid T7 High Yield Transcription Kit (Thermo Scientific) (Supplementary Table S1). DNA and RNA oligonucleotides were 5′ labeled with [γ-32P]-ATP and T4 polynucleotide kinase (New England Biolabs) and gel-purified after denaturing polyacrylamide gel electrophoresis (PAGE).
Cleavage assay
Nucleic acid cleavage assays were conducted in 10 μl of reaction containing the indicated amount of effector complex and substrates in the cleavage buffer (50 mM Tris–Cl pH 6.8, 10 mM MgCl2, 50 mM KCl, 0.1 mg/ml BSA). In DNA cleavage assay, 500 nM (unless otherwise indicated) unlabeled RNA was supplemented to activate DNA cleavage activity. Samples were incubated at 37°C for indicated time periods and the reaction was stopped by addition of 2× RNA loading dye (New England Biolabs). For electrophoresis, samples were heated for 3 min at 95°C and analyzed on an 18% polyacrylamide denaturing gel. Results were recorded by phosphor imaging.
Fluorescence DNA cleavage assay
Each reaction mixture (20 μl in total) contains 50 nM complex, the indicated concentration target RNA, and/or the indicated concentration total RNA, and/or 100 nM ATP in the presence of 500 nM of FAM-poly-16T-BHQ1 ssDNA substrate (Tsingke biotechnology company, Wuhan, China). All reactions were set up with 384-well black plates (Thermo Fisher) and put in a fluorescence plate reader (FLUOstar Omega). Fluorescence DNA cleavage assay was conducted for up to 60 min at 37°C with fluorescence measurements taken every 1 min (λex: 485 nm; λem: 535 nm). Relative fluorescence units (RFU) were obtained by subtraction of individual fluorescence values of reference reactions (lacking CTR) from the fluorescence values of each test/assay reaction.
Electrophoretic mobility shift assay
Unspecific ssDNA and ATP binding assay was performed by incubating different amounts of Csm complex (specified in each experiment) with 5 nM 32P-5′-labeled S10–60 ssDNA or ∼2 nM [γ-32P]-ATP in the cleavage buffer. All reactions were incubated at 37°C for 3 min. Then, the same volume of 2× native loading buffer (0.1% bromophenol blue, 15% sucrose, w/v) was added, and the samples were immediately kept on ice until needed. Electrophoresis was carried out on an 8% nondenaturing polyacrylamide gel at 4°C using 40 mM Tris, 20 mM acetic acid (pH 8.4 at 25°C) as the running buffer. Gels were analyzed by phosphor imaging. Relative ATP binding of LdCsm was estimated by image quantification of bands on non-denaturing PAGE, using the accessory analysis tool equipped with a Typhoon FLA 7000. Results of average of three independent assays are shown with bars representing the mean standard deviation (± SD).
Plasmid interference assay
Plasmid interference assays were performed as previously described (42). Escherichia coli BL21(DE3) strain carrying p15AIE-Cas-S1 or p15AIE-Casmut-S1 (80 μl) was transformed with 100 ng plasmid DNA of each of the following plasmids, pBad-G, pBad-CTR or pBad-NTR by electroporation, using a Gene Pulser II Electroporation System (Bio-Rad). Then, 920 μl of SOC medium was immediately added to electroporated cells and incubated with shaking (200 rpm) at 37°C for 60 min. A series of dilutions were then made for each transformation, and 100 μl of each dilution was plated onto LB agar plates containing 0.05 mM isopropyl-β-d-thiogalactoside (IPTG), Ampicillin, Kanamycin and various concentrations of L-arabinose. Transformation experiments were repeated for three times.
In vitro assay for transcription-dependent DNA cleavage activity
Two pairs of DNA oligos, one for Target DNA, and the other for Nontarget DNA were ordered from Integrated DNA Technologies. Each has a template strand (TS) and non-template strand (NTS). All four oligos were then radioactively labeled. Annealing of a labeled TS and the corresponding unlabeled NTS yielded TS-labeled dsDNA and vice versa. The resulting four dsDNA substrates were purified by recovering the corresponding bands from an 8% native polyacrylamide gel. Co-transcriptional cleavage assay was conducted with 50 nM annealed dsDNA, 50 nM LdCsm, 2 U/μl T7 RNA polymerase (Thermo Fisher) and 50 ng/μl E. coli total RNA. RNA transcription was initiated with the addition of 2.5 mM rNTPs (Thermo Fisher) at 37°C, and samples were collected at timed intervals of 1, 2 or 3 h and quenched by 1 mg/ml RNase A (Thermo fisher) and 20 mM EDTA, and kept on ice for 10 min. Then, 1 μM TS or NTS oligos (TS oligo for TS cleavage assay, and NTS oligo for NTS cleavage assay) was added to unwind residual labeled oligos. After addition of 2X RNA loading dye, the mixtures were treated at 95°C for 5 min before loading onto an 18% denaturing gel. After gel electrophoresis, radio-active signals in the gels were revealed by phosphor imaging.
Analysis of in vivo DNA cleavage of target plasmid and nontarget plasmid by plasmid minipreparation from bacterial colonies and cultures
Simultaneous or stepwise induction of CTR and LdCsm synthesis was conducted as described below. Escherichia coli cells harboring pBad-CTR or pBad-NTR plus p15AIE-Cas-S1 were grown in the LB medium with ampicillin (Amp) and kanamycin (Kan) plus 0.5% glucose, the uninducible medium. When OD600 of the culture attained 0.6, cells were collected by centrifugation and used for the induction experiments. For simultaneous induction, the harvested cells were suspended in the LB broth containing both inducers, i.e. 0.3 mM IPTG and 0.1% l-arabinose to induce the synthesis of LdCsm effectors and CTR/NTR, respectively. For stepwise induction, cells were resuspended in the LB broth containing 0.3 mM IPTG for LdCsm synthesis. After 30 min incubation, 0.1% arabinose was added to induce transcription of CTR/NTR. Replacement of L-arabinose with glucose gave the corresponding references. All cultures were then incubated for 2 h during which 100 μl of cell samples were taken out every 15 min. Plasmid DNAs were prepared from the cell samples and analyzed by agarose gel electrophoresis.
Minipreparation of plasmid DNAs from bacterial colonies were performed as previously described (45) with minor modification. Briefly, bacterial colonies were transferred to a microfuge tube with 50 μl sterile water containing 50 μg/ml RNase A, 50 μl of the freshly made solution of NSS (0.2 M NaOH, 1% SDS and 20% sucrose) was added. After pipetting up and down for six times to mix the cells and the lysis buffer, the tube was kept at room temperature for 3 min. Then, 5 μl of 4 M KCl was added, and the tube was vortexed for 10 s and kept on ice for 5 min. Bacterial debris were removed by centrifugation at maximum speed for 5 min, 10 μl of the supernatant was analyzed by 1% agarose gel electrophoresis with Gel-Red® Nucleic Acid Gel Stain (Biotium, USA) in 1× TAE buffer. Examine the gel under UV illumination. For cell culture, 100 μl culture was centrifugated and resuspended in 50 μl of water with 50 μg/ml RNase A, the following steps were same with bacterial colonies.
Southern blotting analysis was conducted as described previously (46), using radio-labeled oligos of a repeat-probe (for plasmid p15AIE-Cas-S1) and a pBR322-probe and a Kan-probe (for plasmid pBad-CTR), individually (Supplementary Table S1).
RESULTS
ATP functions as a ligand to enhance the ssDNA degradation by LdCsm
Type III effector complexes possess two ATP-binding sites located in the Palm 1 and Palm 2 domains of their Cas10 subunit (47,48). The Palm 2 domain is well-conserved in LdCsm1, the Cas10 protein of this unique III-A CRISPR–Cas system although it does not catalyze synthesis of cOA. To test if LdCsm could bind ATP, wild-type LdCsm ribonucleoprotein complexes were mixed with ∼2 nM [γ-32P]-ATP in the presence or absence of a cognate target RNA (CTR), the activator RNA that carries a 3′ anti-tag sequence showing mismatches to the repeat tag of crRNA. After incubation at 37°C for 3 min, these samples were analyzed by non-denaturing PAGE. We found that both the binary effector and the CTR-bound ternary effector formed ATP-LdCsm complexes, and the intensity of the complex increased along with the increase of the LdCsm concentration (Figure 1A). Since ATP has not been hydrolyzed or chemically modified, the nucleotide binds to the effector complex as a ligand.
Figure 1.
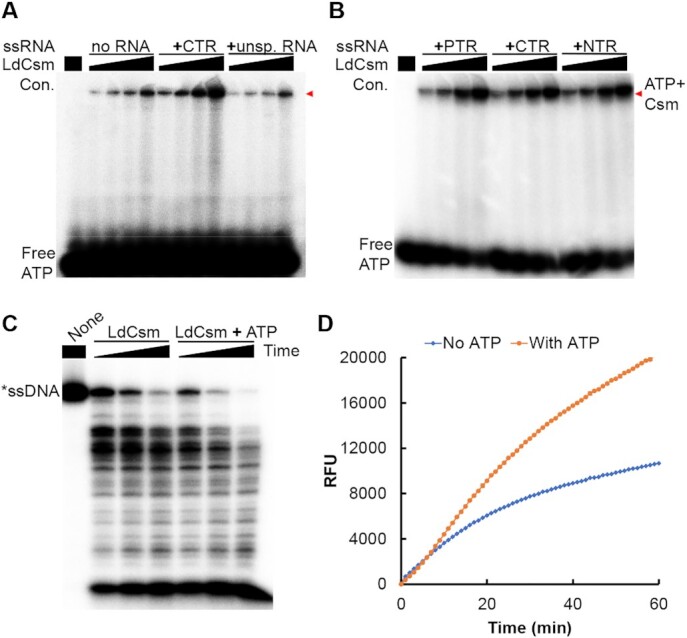
ATP stimulates the efficiency of the LdCsm RNA-activated DNA cleavage. (A) LdCsm binds ATP (B) Formation of LdCsm ternary complexes facilitates the ATP binding. Binding assay was conducted with ∼2 nM [γ-32P]-ATP, 50 nM of indicated RNA and 25, 50, 75 or 100 nM of LdCsm. PTR: Protospacer Target RNA; NTR: noncognate target RNA; CTR: cognate target RNA. Red arrowheads indicate the Csm–ATP complex. (C) ATP enhances the LdCsm RNA-activated ssDNA cleavage. Cleavage assay was conducted for 5, 10 and 20 min. (D) Reporter DNA assay for the RNA-activated ssDNase by LdCsm. Fluorophore quencher-labelled poly-16dT (FAM-poly-16T-BHQ1) was used as the reporter molecule. RFU: relative fluorescence units.
We noticed that the intensity of the radio-active LdCsm ternary complex was much higher than the binary one (Figure 1A), suggesting target RNA binding increased the capacity of the LdCsm ATP binding. Next, three target RNAs carrying different 3′ anti-tag sequences were tested for influence of the 3′ anti-tag sequence on the LdCsm ATP binding, including protospacer target RNA (PTR) lacking any 3′ anti-tag sequence, the CTR and non-cognate target RNA (NTR) carrying the 3′ anti-tag sequence fully matched to the repeat tag of crRNAs. We found that LdCsm binds ATP equally well in the presence of any target RNA type (Figure 1B), indicating that ternary LdCsm complexes possess higher affinity to the ATP ligand and anti-tag sequences of target RNAs do not play a role in the LdCsm ATP binding.
Since LdCsm is inactive in cOA synthesis, we tested if the ATP-binding could influence the LdCsm ssDNA cleavage activity. DNA cleavage reactions were then set up with the wild-type LdCsm effector complex, the CTR and the DNA substrate, in the presence or absence of 100 μM ATP. After incubation for 5, 10 or 20 min at 37°C, cleavage products were analyzed by denaturing PAGE. As shown in Figure 1C, intensities of cleavage products at 5 and 10 min in the presence of ATP were comparable with those at 10 and 20 min in the absence of ATP, respectively. These results indicated that ATP enhanced the LdCsm ssDNase activity.
To gain an insight into the possible mechanism of the ATP stimulation, a number of nucleotides were tested for their influence on the LdCsm DNase activity. These included ADP, AMP, GTP, CTP and UTP as well as AMP-PNP, 3′-dATP and 2′-dATP, the ATP analogues. We found that all ATP analogues, i.e. AMP-PNP, 3′-dATP and 2′-dATP, enhanced the LdCsm ssDNA degradation whereas other nucleotides did not exhibit any influence on the DNA cleavage (Supplementary Figure S1). Since the common moieties of ATP and its active analogues are the adenine base and the triphosphate moiety, we reasoned that these two chemical groups could be involved in the specific ligand-enzyme interaction to stimulate the LdCsm DNase activity.
To further characterize the ATP enhancement, we developed a quantitative DNA cleavage assay based on a DNA reporter, i.e. fluorophore quencher-labelled 16-nt poly-dT containing a 5′-carboxyfluorescein) and 3′-Black Hole Quencher-1 (FAM-poly-16T-BHQ1). The rationale is that cleavage of the polyT in the middle of the ssDNA reporter generates fluorescent FAM-polyT products, which can be quantitated by fluorophotometry. We found that, in the presence of 100 μM ATP, the fluorescence intensity derived from the DNA reporter increased proportionally with the elapse of time, attaining ca. 2-fold difference after incubation at 37°C for 60 min (Figure 1D).
The fluorescence assay was then employed to study the kinetics of the LdCsm DNase. Reactions were setup with microplates containing 10 nM LdCsm RNP and 10 nM to 50 μM of FAM-poly-16T-BHQ1 and placed in a FLUOstar Omega reader. The yield of cleavage products was estimated by continuously measuring the intensity of the FAM fluorescence generated for the entire reaction period. The resulting data were used for calculation of fluorescence production rates (V) for all tested substrate concentrations [S], which were then plotted against contents of cleavage products. The data could be fitted to the Michaelis-Menten model (Supplementary Figures S2A and B), yielding a Vmax of 142.1 ± 5.6 RFU/min with Km of 4969.4 ± 490.9 nM for the LdCsm DNA cleavage in the absence of ATP, and a Vmax of 189.9 ± 6.2 RFU/min with a Km value of 4675.7 ± 391.2 nM in the presence of ATP. These results suggested that ATP may facilitate the DNA cleavage by changing the kinetics of the reaction (Supplementary Figure S2C).
Conserved motifs in the LdCsm1 Palm2 domain are responsible for ATP binding and ATP stimulation
In the crystal structures of S. thermophilus Csm (StCsm) and Pyrococcus furiosus PfCmr3-PfCmr2dHD subcomplex, ATP binds to the Palm1 and Palm2 domains of Cas10 proteins (47,48). These analyses have implicated the following amino acid residues in the nucleotide binding, including H305 in the Palm1 domain, D519 and D522 in P-loop, as well as S549, and D577 in the GGDD motif of the Palm2 domain (48), all of which are conserved in LdCsm1 (Supplementary Figure S3A). To characterize these conserved amino acids, we constructed mutated ldcsm1 genes carrying individual alanine substitution. These include 3 Palm1 domain mutants, i.e., Csm1P1S274A, Csm1P1H307A and Csm1P1SH (S274A and H307A double substitutions), and six Palm 2 mutations, i.e. Csm1P2S571A, Csm1P2D541A, Csm1P2D543A, Csm1P2DxD (D541A and D543A double substitutions), Csm1P2DD (D599A and D600A double substitutions), and Csm1P2KK (K659A and K663A double substitutions). The WT ldcsm1 in the cas gene plasmid (p15AIE-cas-S1) was then replaced with each of the above mutants, giving new cas gene plasmids. The resulting cas gene plasmids were introduced into E. coli individually, yielding E. coli transformants that were used for expression and purification of mutated LdCsm effectors. Mutated LdCsm effector complexes of homogeneity were obtained (Supplementary Figure S3B).
Analysis of ATP binding by the wild-type and mutated effector complexes revealed that five mutations in the LdCsm1 P2 domain impaired the ATP binding. These included Csm1P2D541A, Csm1P2D543A, Csm1P2DxD, Csm1P2DD and Csm1P2KK, all of which are charged amino acids that are well conserved in Palm2 (DxD, GGDD and KxxxK, Figure 2A). Mutagenesis of other conserved amino acids of LdCsm1 including in S274 and H307 (single and double substitutions) in Palm1 as well as S571 in Palm 2 did not show any influence on the ATP binding (Figure 2A). These results indicated that these conserved charged amino acid residues in Palm 2 are involved in coordinating ATP binding.
Figure 2.

Effect of LdCsm1 mutations on ATP binding and ATP-stimulated ssDNA cleavage activity by LdCsm effector. (A) ATP binding assay with LdCsm1-mutated effectors. Red arrowheads indicate the Csm-ATP complex. (B) Reporter DNA assay on the ssDNase activity of LdCsm1-mutated effectors. The ssDNase activity of each LdCsm effector in the absence of ATP was set to 100% to which the relative activity was calculated for the same effector in the presence of 100 μM ATP. (C) RNA-activated ssDNA cleavage assay with the mutated LdCsm effectors. Incubation time was for 30 min except for Csm1P2D543A and Csm1P2DxD (120 min). (D) Anti-plasmid activity by the LdCsm system and LdCsm1 D541 mutants. Electroporated E. coli cells were plated on nutrient media containing either glucose or arabinose as the carbon source overnight at 37°C. Concentrations of the L-arabinose inducer are indicated on the top of the panel, with glucose (0.5% Glu) as a reference.
Then, we evaluated the role of these conserved amino acids in ATP stimulation of the LdCsm DNase activity using the fluorescence DNA cleavage assay. As shown in Figure 2B, addition of ATP increased the relative activity of each enzyme by 50–100% except for mutated RNPs carrying Csm1P2D541A or Csm1P2DxD substitutions, and these data are in good agreement with those obtained from the radio-labelled ssDNA cleavage assay of the corresponding mutated LdCsm effectors (Figure 2C).
Noticeably, mutagenetic analyses also revealed Csm1P2D541A substitution was the only mutation that abolished the ATP stimulation (Figure 2B). To confirm the finding, three additional mutants were constructed for Csm1 D541 residue, giving Csm1P2D541N, Csm1P2D541T and Csm1P2D541L. Mutated LdCsm effectors were obtained (Supplementary Figure S4A) and they exhibited similar target RNA cleavage activities (Supplementary Figure S4B). These mutated LdCsm RNPs were then tested for ATP stimulation. The effector carrying the Csm1P2D541N substitution still exhibited ATP-stimulated ssDNase activity, indicating that the aspartate residue can be replaced with asparagine of similar properties, but mutations of the Csm1P2D541T and Csm1P2D541L abolished the stimulation (Supplementary Figure S4C). These results are consistent with the essential role of D541 in ATP stimulation. Altogether, we found three motifs in the Palm2 domain of LdCsm1 contributed to the LdCsm ATP binding, which include the D599 D600 in the GGDD motif, D541 D543 in the DxD motif, and the K659 K663 in the KxxxK motif, whereas only one of these motifs, D541 is further involved in mediating ATP stimulation.
Plasmid clearance by LdCsm requires a robust ssDNase activity
Since mutated LdCsm effectors carrying the three D541 substitutions exhibited very similar ssDNase activities compared with the wild-type LdCsm effector in the absence of ATP (Supplementary Figure S4C), these mutants were chosen for investigating the importance of the ATP-stimulated ssDNase activity in LdCsm immunization. Four LdCsm-expressing plasmids were constructed, i.e. p15AIE-cas-S1, p15AIE-cas(Csm1P2D541N)-S1, p15AIE-cas(Csm1P2D541T)-S1 as well as p15AIE-cas(Csm1P2D541L)-S1. These plasmids were introduced into E. coli BL21 (DE3) by electroporation. Each strain was then transformed with the target plasmid pBad-CTR, which carries the CTR-S1 protospacer-GFP fusion gene under the control of an L-arabinose-inducible promoter (42). Electroporated E. coli cells were plated on LB media that were further supplemented either with glucose (for inhibition of target RNA expression) or with l-arabinose (for induction of target RNA expression). Furthermore, in the presence of different concentrations of l-arabinose, CTR RNA is to be expressed into different levels, and this would allow the determination of the minimal amounts of CTR RNA required for complete plasmid clearance for the WT and Csm1-mutated LdCsm systems.
We found that 0.01–0.04% of l-arabinose greatly reduced colony sizes of E. coli cells expressing the WT and Csm1P2D541N effector, indicating that the LdCsm interference already occurred at a low level of CTR induction (Supplementary Figure S4D). At higher levels of the inducer (≥0.1% of l-arabinose), the LdCsm immunity completely prevented colony formation of both transformants (WT and Csm1P2D541N). In comparison, E. coli cells hosting the mutated LdCsm systems with impaired ATP stimulation (due to Csm1P2D541T and Csm1P2D541L substitution) formed colonies under all tested concentrations of l-arabinose, the inducer of CTR synthesis (Figure 2D and Supplementary Figure S4D). These results indicated that these mutated LdCsm systems failed to attain the immunization level required for plasmid clearance.
This hypothesis was further tested with two other LdCsm1 mutants, namely Csm1P2D541A and Csm1P2KK. The former did not possess the ATP stimulation and the latter exhibited generally impaired DNase activity but retained the ATP stimulation (Figure 2C). Investigation of their in vivo plasmid clearance revealed that none of the mutated LdCsm systems mediated plasmid clearance since E. coli colonies appeared on plates in all tested concentrations of l-arabinose (Supplementary Figure S4E). These results reinforced the conclusion that the WT and Csm1P2D541N are capable of mediating plasmid clearance in host cells.
LdCsm performs DNA degradation at transcriptional bubbles indiscriminately for target vs. non-target dsDNA
A previous work reported that SeCsm performs the co-transcriptional DNA cleavage in the non-template strand of the transcriptional bubble formed in the target DNA region (43). More recently, in vitro co-transcriptional DNA cleavage was further examined for III-A CRISPR–Cas systems of T. thermophilus and S. epidermidis, and this revealed that DNA cleavage does not occur at the transcriptional bubbles in the target DNA region (44). For this reason, the authors have argued for a more important role of co-transcriptional RNA cleavage in the III-A CRISPR–Cas immunization (44). Nevertheless, our genetic analysis of the LdCsm system revealed DNA cleavage by the LdCsm effector is essential for immunization and inactivation of its RNA cleavage facilitates the immunity (42). These results suggested a more important role for the DNA cleavage in LdCsm immunization, and prompted us to further characterize the co-transcription DNA cleavage by this type III-A CRISPR–Cas system.
To do that, two 90 bp double-strand (ds) DNA substrates were generated by annealing of two complementary oligonucleotides, one was labeled for the template strand, while the other, for non-template strand. These DNA substrates contain a T7 promoter and a cognate target DNA sequence of the S1 crRNA and therefore named as the ‘Target dsDNA’ (Figure 3A). Co-transcriptional DNA cleavage assay was conducted with 50 nM radio-labeled Target dsDNA, 50 nM LdCsm, 2 U/μl T7 RNA polymerase in the presence of 50 ng/μl E. coli total RNA, and four different LdCsm effectors were tested, including WT, Csm1P2D541A, Csm1P2KK and LdCsmdCsm3. Transcription was initiated by supplementing 2.5 mM rNTPs and incubation was conducted at 37°C for 1, 2 or 3 h. Cleavage products were analyzed by denaturing PAGE and autoradiography. We found that, for all four tested LdCsm effectors, cleavage products accumulated for many different sites distributed in the entire non-template strand except for the promoter sequence (Figure 3B, upper panel), but the template strand remained intact (Figure 3B, bottom panel). These results indicated that the DNA cleavage by LdCsm is indeed transcription-dependent, and the cleavage occurred in the positions beyond in the transcriptional bubble containing the target DNA site. Intriguingly, this pattern of the transcription-dependent DNA cleavage is very different from the reported co-transcriptional model (44,49), and this is because our results have suggested that, once loaded with the CTR produced from transcription of Target dsDNA, the ternary LdCsm can mediate DNA cleavage to any accessible transcriptional bubbles.
Figure 3.

LdCsm performs indiscriminate DNA degradation at the transcriptional bubble of target and non-target in vitro. Schematic of the Target dsDNA (A) and the Nontarget dsDNA (C). The Target dsDNA substrate contains a T7 promoter and the target sequence (backbone of the protospacer is highlighted yellow and CTR is in red), and the direction of transcription is indicated with an arrow. Lines in the bottom indicate the positions of T7 promoter and target sequence in target dsDNA substrate as well as the size of the full length dsDNAs. The Nontarget dsDNA substrate contains the T7 promoter and the non-homologous sequence, lines in the bottom indicate the position of T7 promoter. Denaturing PAGE analysis of the cleavage products of the Target dsDNA (B) or the Nontarget dsDNA (D). Either non-template strand (Top panel) or template strand (bottom panel) was labeled. Cleavage assay was conducted for 1, 2 and 3 h. Reference reactions lacking one of assay components were conducted for 180 min. M: ssDNA size ladder.
To test the assumption, we designed a ‘Nontarget dsDNA’, which is not related to Target dsDNA (Supplementary Table S1). Nontarget dsDNA substrates were then generated as for Target dsDNAs and tested for transcription-dependent DNA cleavage alone, or together with a cold Target dsDNA (Figure 3C). We found that LdCsm did not show any detectable DNA cleavage to the Nontarget dsDNA (Figure 3D, reactions lacking cold Target dsDNA). However, when an equal amount of a cold Target dsDNA was added into the cleavage reaction containing labeled Nontarget dsDNA, LdCsm-mediated cleavage also occurred for Nontarget dsDNA. Again, cleavage products were solely derived from the non-template strand DNA, but not from the template strand, and cleavage products accumulated along with the increase of incubation time for all four tested LdCsm effector complexes (Figure 3D). These results indicated that, upon activation, the LdCsm effector mediates transcription-dependent DNA cleavage to the non-template strand of any accessible transcriptional bubbles, not only for those present in the target DNA substrate but also those in the nontarget DNA.
LdCsm also cleaves non-target plasmid and host chromosome
We were then very interested to test if this mode of DNA cleavage could occur in vivo. For this purpose, we investigate if LdCsm could mediate DNA cleavage to a non-target plasmid. In the in vivo DNA interference assay recently developed for LdCsm (42), two plasmids were used, pBad-CTR and p15AIE-Cas-S1. The former represented a target plasmid, and the latter, a nontarget plasmid. Thus, this assay could readily be adapted for the investigation of nontarget DNA cleavage by LdCsm. Nevertheless, there was prerequisite to this assay: the target plasmid versus the nontarget plasmid had to be distinguished from each other in plasmid preparations. To enable that, we isolated the supercoiled form of plasmid for pBad-CTR and p15AIE-Cas-S1, from which the corresponding linearized and nicked forms of plasmid were prepared. Then, plasmid DNAs were prepared from the E. coli cells carrying pBad-CTR and p15AIE-Cas-S1. These plasmid preparations were either run alone, or together with one of their topological standards. As shown in Supplementary Figure S5, different topological forms of both pBad-CTR and p15AIE-Cas-S1 present in plasmid preparations could be identified by running along with the standards of their topological forms, and their identities were further confirmed by Southern analysis.
The method was then employed to examine the influence of the CTR expression on LdCsm immunization of target vs. nontarget plasmids. E. coli strains containing pBad-G, or pBad-NTR or pBad-CTR (called the pBad-G, or pBad-NTR or pBad-CTR cells). All three strains were then transformed with p15AIE-Cas-S1 by electroporation. Electroporated cells were plated on selective LB media containing 0.5% glucose in which target RNAs synthesis is inhibited or 0.01%, 0.04% or 0.1% of L-arabinose in which target RNAs synthesis is induced, in addition to ampicillin and kanamycin to select for each of the plasmids individually. As shown in Figure 4A, introduction of p15AIE-Cas-S1 into pBad-G or pBad-NTR cells did not influence the cell growth as colonies of similar sizes formed on the selective plates for both transformation; electroporation of the non-target plasmid into pBad-CTR cells resulted in smaller colony sizes in the presence of 0.01%, and 0.04% l-arabinose, and further increase of l-arabinose to 0.1% completely inhibited the formation of single colonies. Nevertheless, complete growth stop did not occur for the transformed pBad-CTR cells on 0.1% arabinose plates since clusters of colonies or cell lawn appeared on the plate when 100-fold more cells were used for plating (Figure 4A). Plasmids were then extracted from colonies or cell mass and analyzed by agarose gel electrophoresis. In the negative reference (pBad-NTR), bands of both the NTR-protected target plasmid and the nontarget plasmid (p15AIE-Cas-S1) did not change upon a strong induction of NTR synthesis (Figure 4B). However, in the presence of pBad-CTR, the intensity of nontarget plasmid decreased along with the increase of the l-arabinose concentration, whereas the target plasmid, pBad-CTR remained at more or less the same level (Figure 4C and Supplementary Figure S6A). These results suggested that the LdCsm system preferably targeted the nontarget plasmid for degradation under this experimental condition.
Figure 4.

LdCsm cleaves Nontarget plasmid in vivo. (A) Schematic of the interference plasmid assay for determination of the LdCsm anti-plasmid immunity. Gene expression from these plasmids, pBad-G, pBad-CTR or pBad-NTR, is under the control from an L-arabinose-inducible promoter from which CTRs or NTRs are to be expressed in the presence of l-arabinose but their expression is to be repressed in glucose-containing media. (B) Minipreparation of plasmid DNAs from E. coli colonies electroporated with pBad-NTR. (C) Minipreparation of plasmid DNAs from E. coli colonies electroporated with pBad-CTR. Empty triangles indicate the plasmid p15AIE-Cas-S1, filled triangles indicate the plasmid pBad-CTR or pBad-NTR. Three topological forms of plasmids DNA were analyzed: SC: supercoiled form; Linear: linearized form; Nicked: opened circle (relaxed) form.
This was very surprising since it was expected that the immune system should exert immune response primarily to the target plasmid that mimics invading viruses. Nevertheless, the preferred interference to nontarget plasmid could reflect the fact that the system may indiscriminately target proximal transcriptional bubbles for degradation after activation by CTR. To test the hypothesis, experiments were designed to study the onset of co-transcriptional DNA cleavage by LdCsm in the cells where synthesis of target RNA and LdCsm was induced simultaneously or step-wisely (Figure 5A). Experiments were conducted as described in Materials and Methods. Cell samples were used for extracting plasmid DNAs, following the previous protocol (45), with the plasmid DNAs analyzed by agarose gel electrophoresis. We found, in the stepwise induction of LdCsm effector and CTR, massive DNA degradation occurred already at 30 min after CTR synthesis (CTR in L-arabinose, Figure 5C), which was in strict contrast to the preferred degradation of nontarget plasmid observed with the simultaneously induction of both LdCsm and CTR (CTR in l-arabinose, Figure 5B). These results suggested that, in the 30 min time window before the onset of CTR synthesis, the binary LdCsm effector complexes could have already diffused to different sublocations. Then, upon the arabinose induction, newly synthesized CTR immediately reached the pre-distributed binary LdCsm effector complexes and form the ternary effector complexes. Active LdCsm effectors were then tethered to nearby transcriptional bubbles and cleaved their non-template DNA strand without discriminating whether these were on plasmids or from the chromosome. Furthermore, analysis of cell survival by serial dilutions of cells and plating on LB agar showed only ca. 15% of pBad-CTR cells were viable, relative to the pBad-NTR cells, at 120 min after the second induction (Supplementary Figure S7), demonstrating that transformed pBad-CTR cells underwent massive degradation of chromosome DNA observed (CTR in l-arabinose, Figure 5C).
Figure 5.

LdCsm restricts the Nontarget plasmids and host chromosome. (A) Schematic of the assay for investigating the effect of plasmid-borne expression of CTR or LdCsm on the stability of the expressing plasmid (see Materials and Methods for a detailed description). (B) Stability of target vs. nontarget plasmids and host chromosome in E. coli cells upon simultaneous induction. Sample 0: 0 min after addition of 0.3 mM IPTG plus 0.5% glucose or 0.1% l-arabinose. Sample 1 – 8: 15, 30, 45, 60, 75, 90, 105 and 120 min after addition of glucose or L-arabinose. (C) Stability of target versus nontarget plasmids and host chromosome in E. coli cells upon stepwise induction. Sample 0, 30 min after addition of 0.3 mM IPTG and immediately before addition of 0.5% glucose or 0.1% l-arabinose; Sample 1 – 8: 15, 30, 45, 60, 75, 90, 105 and 120 min after addition of glucose or l-arabinose after addition of glucose or l-arabinose. Empty triangles indicate the plasmid p15AIE-Cas-S1, filled triangles indicate the plasmid pBad-CTR or pBad-NTR. Three topological forms of plasmids DNA were analyzed: SC: supercoiled form; linear: linearized form; nicked: opened circle (relaxed) form.
Taken all together, we demonstrate that, upon activation by CTR, LdCsm causes instability not only to the target plasmid but also to nontarget plasmid as well as to the host chromosome.
LdCsm provides a platform for RNA detection
To date, a few multi-domain Cas proteins have been exploited for nucleic acid detection such as Cas13a and Cas12a: the former is an RNA-guided RNase, and the latter a DNA-guided DNase (50–52). The capability of mediating target RNA-activated ssDNA cleavage by LdCsm renders it possible to harness the system for RNA detection using a DNA reporter as demonstrated with the fluorescence DNA cleavage assay developed in this work (Figure 6A). To explore the possibility, 100–500 ng/μl Sulfolobus islandicus E234 and E. coli BL21 total RNAs were mixed with different LdCsm effector complexes (the WT or LdCsmdCsm3) in the presence of 10 or 100 nM cognate target RNA. Fluorescence assay was conducted with a FLUOstar Omega, and the fluorescence was followed during incubation. We found that, while fluorescence in the RNPs in the presence of 100–500 ng/μl S. islandicus E234 or E. coli BL21 total RNAs showed little change, target RNA-dependent fluorescence increased proportionally during 60 min incubation for LdCsmdCsm3 RNP (Figure 6B), indicating that the effector mediates specific RNA detection.
Figure 6.

LdCsm is capable of sensitive RNA detection. (A) Schematic of LdCsm RNA detection approach using a quenched fluorescent ssDNA oligo. (B) Quantification of fluorescence generated after 60 min by LdCsmdCsm3 effector loaded with 0, 10 or 100 nM CTR in the presence of varying concentrations of S. islandicus E234 or E. coli total RNA, RFU: Relative fluorescence units. Relative ssDNase activities of WT LdCsm (C) and LdCsm LdCsmdCsm3 (E). The number of mismatches between RNA activators and the crRNA are indicated. Relative ssDNase activities are calculated with the linear region of the fluorescence curve for each sample with DNase activities of the LdCsm with the full matched target RNA (PT) as the reference (set to 1). Data are shown with averages of three independent experiments with indicated standard derivations. (D, F) LdCsm system detection can discriminate between two closely related Lactobacillus strains (L. plantarum and L. casei) by 16S rRNA. Top in both: Schematic of Lactobacillus strain target regions and the crRNA sequences used for detection. Mismatches in target sequences are highlighted in blue and pink. Specific detection of L. plantarum versus L. casei by WT LdCsm (D) and LdCsmdCsm3 (F). The transcriptional 16S RNA in vitro and 50 ng/μl total RNA were used as the activator for WT LdCsm and LdCsmdCsm3 effectors, respectively.
To gain an understanding of how mismatches could affect the RNA detection reaction by LdCsmdCsm3 RNP, a series of target RNAs were designed carrying mismatch(es) to the crRNA at different position(s). These RNAs were tested for their capability of activating the ssDNase activity of either WT or LdCsmdCsm3. This revealed that single base-pair (bp) mismatches showed different impact on the DNase activity of these effectors (Figure 6C). In the case of LdCsmdCsm3 RNP, 2 bp mismatches in position 3 and 4 of crRNA with target RNA has ∼10-fold inhibitory effect on fluorophore quencher-labelled reporter ssDNA, while both single mismatch in position 3 or position 4 only exhibit ∼30% inhibitory effects (Figure 6E), thus, single-molecule sensitivity could be achieved by comparing the single mismatch in position 3 or position 4 with dual mismatches.
To test the usefulness of the LdCsm-based RNA detection, we investigated whether WT or LdCsmdCsm3 RNPs could distinguish 16S rRNAs of two closely related Lactobacillus strains, L. plantarum and L. casei. Two LdCsm RNPs with LPwt and LCwt crRNA, and two LdCsmdCsm3 RNPs with LPwt and LCwt crRNA, targeting L. plantarum and L. casei respectively, were obtained. Fragments of ∼300 bp 16S rDNA were obtained by PCR from L. plantarum and L. casei, and used as templates for in vitro transcription using T7 RNA polymerase. The resulting RNA transcripts were the employed as target RNAs to test the usefulness of LdCsm effector in discrimination of closely related RNA targets. The results showed that the relative activities of WT or LdCsmdCsm3 RNPs catalyzes the fluorescent ssDNA cleavage in the presence of the corresponding targets were ∼10 folds higher than that in presence of the other RNA (Figure 6D, F). Furthermore, LdCsmdCsm3 RNP was capable of facilitating the fluorescent ssDNA cleavage in the presence of total RNA of the corresponding lactobacilli (Figure 6F). To this end, we concluded that target RNA-stimulated cleavage of ssDNA substrates by either WT LdCsm or LdCsmdCsm3 RNPs has the potential to be used for specific RNA detection.
WT LdCsm and LdCsmdCsm3 RNPs were also explored for specific RNA quantification. Various concentrations of CTR were tested for their capability of activating the ssDNase of each effector, and we found that, while it required 0.2 nM CTR to activate WT LdCsm RNP (Supplementary Figure S8A), 0.01 nM was sufficient to activate LdCsmdCsm3 RNP (Supplementary Figure S8B). Therefore, the mutated effector is 10–20 folds more sensitive than the WT effector in RNA detection. In addition, since RFU value accumulation was positively correlated with the increase of target RNA concentration (Supplementary Figure S8C), suggesting that the mutated LdCsmdCsm3 RNP could be useful for specific RNA quantification, too. Taken together, LdCsm exhibits a great potential for specific RNA detection and quantification.
DISCUSSION
Type III immune systems exhibit multiple activities, including target RNA cleavage (11–14), RNA-activated unspecific DNA cleavage (15–19) and cOA synthesis (20–25). However, it remains elusive how these activities have contributed to immune responses observed for individual type III systems. This is mainly because there is a knowledge gap in our understanding of how the in vitro RNA-activated ssDNA cleavage activity is related to the in vivo immune response mediated from the Cas10 HD domain. Herein we show that LdCsm mediates DNA cleavage to the non-template DNA segment at any transcriptional bubble in a close proximity. This finding not only provides the molecular mechanism for the final clearance of invading DNA by type III systems, but also supports the presence of abortive infection facilitated by the LdCsm co-transcriptional DNA cleavage. An updated understanding of the immunization mechanisms by type III CRISPR–Cas systems is presented in Figure 7, including the importance of ATP stimulation of ssDNA degradation from the HD domain and the immunization mechanisms based on co-transcriptional DNA cleavage.
Figure 7.

Mechanisms of immunization provided by Type III-A CRISPR–Cas systems. (A) The immunization mechanism of type III CRISPR–Cas systems. Four enzymatic activities were found, including (i) crRNA-guided backbone RNase, (ii) target RNA-activated ssDNase, (iii) target RNA-dependent and ATP-stimulated ssDNase, (iv) target RNA-activated synthetase, producing a cyclic oligoadenylate (cOAn) second messengers that activate CRISPR–Cas associated proteins such as NucC, Can1, Can2, Card1, Csx1 and Csm6. (B) A model for immunization by the LdCsm system. At a low level of target RNAs, active LdCsm effectors are formed in a close proximity to phage DNA. At a high level of target RNAs, active LdCsm effectors are formed in close proximity to chromosomal DNA. In the latter scenario, LdCsm effectors will mediate DNA cleavage to any nearby transcriptional bubbles, many of which are the chromosomal ones. Thus, LdCsm can facilitate cell death to infected cells.
We find in this work that LdCsm possesses the ATP-stimulated ssDNase activity and it is the only known III-A CRISPR–Cas system that relies on the ATP stimulation to attain the full immunity. Very recently, ATP was found to facilitate ssDNA cleavage by a unique S. islandicus Cmr complex (53), and the III-B system represents the only other type III CRISPR–Cas that can also be activated by ATP. Interestingly, the presence of the target RNA has strongly increased the affinity of LdCsm complex towards ATP, relative to the binary effector complex (Figure 1A and B). This suggests that the target RNA binding increases the capacity or the stability of the LdCsm ATP binding during the formation of the LdCsm ternary complex. As structures of LdCsm are not yet available, it remains elusive how ATP binding is facilitated by target RNA binding and how ATP binding enhances the immunization. Nevertheless, in the structure of StCsm, ATP stabilizes the 3′ anti-tag of the target RNA based on comparisons between structures of StCsm with or without ATP (48). It is thus tempting to assume that ATP binding to the ternary complex may stabilize the conformational change, and thereby enhancing the ssDNA cleavage from the HD domain of the Cas10 subunit. Furthermore, a few LdCsm1 mutants, such as Csm1P2D541T and Csm1P2D541L, lack the ATP-stimulated ssDNA cleavage and consequently fail to mediate plasmid clearance in E. coli (Figure 2D and Supplementary Figure S4). These mutated LdCsm systems could be compared with most known type III immune systems in which the effector complexes show a moderate ssDNase activity that is insufficient to yield plasmid clearance in their hosts. To this end, we speculate that LdCsm may have lost the cOA synthesis activity and in the meantime gained the ATP stimulation during evolution.
The most important finding presented in this work is probably the demonstration of the LdCsm-mediated co-transcriptional DNA cleavage in vitro and in vivo. Co-transcriptional DNA cleavage was first suggested for type III CRISPR–Cas systems from the investigation of the SeCsm system. In their study, the authors find SeCsm cleaves the non-template strand DNA at the transcriptional bubble containing the target site, and the cleavage occurs for the target and its 3′ flanking sequence (43). However, investigation of TthCsm by another group failed to detect such co-transcriptional DNA cleavage, and the authors further unravel that the type III effectors only cleave RNA transcripts in a co-transcriptional fashion, the cleavage products identified in the early work are due to the presence of an excessive amount of non-template DNA in the transcriptional assay (44). In our experiments of co-transcriptional cleavage, ssDNAs were removed from dsDNA templates by gel purification, and this is to ensure that all cleavage products, if observed, are derived from template dsDNAs. For this reason, we attribute the cleavage products observed in Figure 3B to the cleavage in the non-template strand DNA segments by LdCsm at any transcriptional bubbles.
We find that, when the expression of CTR and LdCsm is induced simultaneously, the LdCsm system preferably targets the nontarget plasmid for degradation, whereas the target plasmid has remained at the more or less same level (Figure 4C and Supplementary Figure S6A). These results do not fit with the expectation that type III immune systems mediate specific DNA interference at the target DNA region of invading genetic elements. There are two possible reasons to the observation. First, since CTR is to be obtained by transcription of a short DNA segment, the target RNA is readily obtained upon induction. In contrast, synthesis of binary LdCsm effectors requires a series of events, including crRNA synthesis and processing, transcription of a long cas gene locus and protein synthesis from the long mRNA as well as effector assembly with Cas proteins and mature crRNA. For this reason, newly synthesized CTR could have already diffused to a close proximity to the subcellular locations in which binary LdCsm effectors are being assembled. After CTR loading, the activated LdCsm would readily target its proximal transcriptional bubbles for degradation, many of which are located on the LdCsm expression plasmid, which is the nontarget plasmid. Second, the plasmid-borne expression would produce many more transcriptional bubbles in p15AIE-Cas-S1 than those in pBad-CTR, which are accessible for DNA cleavage by LdCsm before inactivation by target RNA cleavage in the spatiotemporal regulation (17). To this end, these results fit well to the model that LdCsm mediates co-transcriptional cleavage to its proximal transcriptional bubbles regardless if they are on invading genetic elements or host chromosomes.
Strikingly, we find that the indiscriminate DNA cleavage also occurs in vivo since step-wise expression of the inactive binary LdCsm effector and activator RNA yields robust degradation of cellular DNA probably from transcriptional bubbles (Figure 5B). In this scenario, massive DNA degradation by LdCsm would lead to cell death of infected bacterial cells, thereby blocking phage replication in the infected cells and reducing the release of new progeny particles to the environment. As a result, the immunization prevents phage spreading by an abortive infection mechanism. Currently, CRISPR-based suicidal mechanism proposed for several distinctive CRISPR–Cas variants, e.g. type III (20,21,37), type VI (54), and type I-E and I-F immunity against virulent phages (55,56). Possibly, abortive infection represents a more common mechanism of antiviral defense than currently appreciated. To this end, the ATP stimulation can be regarded a feature of type III systems which could have lost during evolution, probably along with a simultaneous gain of the CRISPR signaling pathway. In this scenario, the mutated LdCsm systems lacking ATP enhancement could be regarded as the equivalent of most type III immune systems in which co-transcriptional DNA cleavage cannot mediate immunization alone, as exemplified with several mutated LdCsm systems (Figure 3). Since these mutated LdCsm systems also target nontarget plasmids and chromosome in vivo (Supplementary Figures S6B, S6C and S7), this feature may be conserved in other type III CRISPR–Cas systems. Indeed, a very recent study with SeCsm has revealed the immune system can also damage the host chromosome, facilitating specific mutations in the host (57), suggesting chromosome targeting could be a general feature for type III CRISPR–Cas systems.
Compared with other type III effectors, LdCsm exhibits high specificity to distinguish one single mismatch (Figure 6C), suggesting the LdCsm effector has a better potential in the application of specific RNA detection than other known type III immune systems. There is another important difference between LdCsm and other type III effectors: while a majority of LdCsm crRNAs consist of an 8 nt 5′ tag and a 26 nt spacer region (42), crRNAs from other known III-A and III-B systems are usually longer in length, e.g. 29 or 35 nt spacer in SeCsm (36,58), 32 nt spacer in StCsm (14), ∼37 nt spacer in TtCsm (12), 30 nt spacer in ToCsm (59), ∼31 or 37 nt spacer in PfCmr (60), ∼32 or 38 nt spacer in TtCmr (61), 32 or 38 nt spacer in SisCmr-α (18). Compared with the well-known detection platform based on CRISPR–Cas13 systems, the LdCsm system has some advantages in RNA detection, too. For instance, LdCsm degrades non-homologous ssDNA, while CRISPR–Cas13 targets RNA (50,62). Since ssDNA is a more stable reporter than RNA, the LdCsm-based RNA detection is much cheaper to Cas13-based ones. In addition, since CRISPR–Cas13 targets RNA, of course, it can also degrade target RNA, but LdCsm does not. Taken together, further investigation of the LdCsm immune system will not only clarify the function of co-transcriptional DNA cleavage in abortive infection but also demonstrate its usefulness in RNA detection as well as RNA quantification.
Supplementary Material
ACKNOWLEDGEMENTS
We thank colleagues in the Danish Archaea Centre in Copenhagen and those in the CRISPR and Archaea Biology Research Center in Shandong University for stimulating discussions.
Contributor Information
Jinzhong Lin, CRISPR and Archaea Biology Research Center, State Key Laboratory of Microbial Technology, Shandong University, 72 Binhai Road, Jimo, Qingdao, Shandong 266237, P.R. China; Archaea Centre, Department of Biology, University of Copenhagen, Ole Maaløes Vej 5, DK-2200 Copenhagen N, Denmark.
Yulong Shen, CRISPR and Archaea Biology Research Center, State Key Laboratory of Microbial Technology, Shandong University, 72 Binhai Road, Jimo, Qingdao, Shandong 266237, P.R. China.
Jinfeng Ni, CRISPR and Archaea Biology Research Center, State Key Laboratory of Microbial Technology, Shandong University, 72 Binhai Road, Jimo, Qingdao, Shandong 266237, P.R. China.
Qunxin She, CRISPR and Archaea Biology Research Center, State Key Laboratory of Microbial Technology, Shandong University, 72 Binhai Road, Jimo, Qingdao, Shandong 266237, P.R. China; Archaea Centre, Department of Biology, University of Copenhagen, Ole Maaløes Vej 5, DK-2200 Copenhagen N, Denmark.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Chinese National Transgenic Science and Technology Program [2019ZX08010003 to Q.S.]; National Natural Science Foundation of China [31771380 to Q.S., 31670061 and 31970546 to Y.S., 31970119 to J.N.]; Danish Council for Independent Research [DFF-4181-00274 to Q.S.]. Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement. Q.S. has filed a patent application for RNA detection and quantification with LdCsm effectors.
REFERENCES
- 1. Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D.A., Horvath P.. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007; 315:1709–1712. [DOI] [PubMed] [Google Scholar]
- 2. Marraffini L.A. CRISPR–Cas immunity in prokaryotes. Nature. 2015; 526:55–61. [DOI] [PubMed] [Google Scholar]
- 3. Marraffini L.A., Sontheimer E.J.. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008; 322:1843–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Horvath P., Barrangou R.. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010; 327:167–170. [DOI] [PubMed] [Google Scholar]
- 5. van der Oost J., Westra E.R., Jackson R.N., Wiedenheft B.. Unravelling the structural and mechanistic basis of CRISPR–Cas systems. Nat. Rev. Microbiol. 2014; 12:479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hille F., Richter H., Wong S.P., Bratovic M., Ressel S., Charpentier E.. The biology of CRISPR–Cas: backward and forward. Cell. 2018; 172:1239–1259. [DOI] [PubMed] [Google Scholar]
- 7. Wright A.V., Nunez J.K., Doudna J.A.. Biology and applications of CRISPR systems: harnessing nature's toolbox for genome engineering. Cell. 2016; 164:29–44. [DOI] [PubMed] [Google Scholar]
- 8. Koonin E.V., Makarova K.S., Zhang F.. 2017) Diversity, classification and evolution of CRISPR–Cas systems. Curr. Opin. Microbiol. 37:67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mohanraju P., Makarova K.S., Zetsche B., Zhang F., Koonin E.V., van der Oost J.. Diverse evolutionary roots and mechanistic variations of the CRISPR–Cas systems. Science. 2016; 353:aad5147. [DOI] [PubMed] [Google Scholar]
- 10. Makarova K.S., Wolf Y.I., Alkhnbashi O.S., Costa F., Shah S.A., Saunders S.J., Barrangou R., Brouns S.J., Charpentier E., Haft D.H.et al.. An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Microbiol. 2015; 13:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Benda C., Ebert J., Scheltema R.A., Schiller H.B., Baumgartner M., Bonneau F., Mann M., Conti E.. Structural model of a CRISPR RNA-silencing complex reveals the RNA-target cleavage activity in Cmr4. Mol. Cell. 2014; 56:43–54. [DOI] [PubMed] [Google Scholar]
- 12. Staals R.H., Zhu Y., Taylor D.W., Kornfeld J.E., Sharma K., Barendregt A., Koehorst J.J., Vlot M., Neupane N., Varossieau K.et al.. RNA targeting by the type III-A CRISPR–Cas Csm complex of Thermus thermophilus. Mol. Cell. 2014; 56:518–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ramia N.F., Spilman M., Tang L., Shao Y.M., Elmore J., Hale C., Cocozaki A., Bhattacharya N., Terns R.M., Terns M.P.et al.. Essential structural and functional roles of the Cmr4 subunit in RNA cleavage by the Cmr CRISPR–Cas complex. Cell Rep. 2014; 9:1610–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tamulaitis G., Kazlauskiene M., Manakova E., Venclovas C., Nwokeoji A.O., Dickman M.J., Horvath P., Siksnys V.. Programmable RNA shredding by the type III-A CRISPR–Cas system of Streptococcus thermophilus. Mol. Cell. 2014; 56:506–517. [DOI] [PubMed] [Google Scholar]
- 15. Elmore J.R., Sheppard N.F., Ramia N., Deighan T., Li H., Terns R.M., Terns M.P.. Bipartite recognition of target RNAs activates DNA cleavage by the Type III-B CRISPR–Cas system. Genes Dev. 2016; 30:447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Estrella M.A., Kuo F.T., Bailey S.. RNA-activated DNA cleavage by the Type III-B CRISPR–Cas effector complex. Genes Dev. 2016; 30:460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kazlauskiene M., Tamulaitis G., Kostiuk G., Venclovas C., Siksnys V.. Spatiotemporal control of type III-A CRISPR–Cas immunity: coupling DNA degradation with the target RNA recognition. Mol. Cell. 2016; 62:295–306. [DOI] [PubMed] [Google Scholar]
- 18. Han W., Li Y., Deng L., Feng M., Peng W., Hallstrom S., Zhang J., Peng N., Liang Y.X., White M.F.et al.. A type III-B CRISPR–Cas effector complex mediating massive target DNA destruction. Nucleic Acids Res. 2017; 45:1983–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu T.Y., Iavarone A.T., Doudna J.A.. RNA and DNA targeting by a reconstituted thermus thermophilus Type III-A CRISPR–Cas system. PLoS One. 2017; 12:e0170552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kazlauskiene M., Kostiuk G., Venclovas C., Tamulaitis G., Siksnys V.. A cyclic oligonucleotide signaling pathway in type III CRISPR–Cas systems. Science. 2017; 357:605–609. [DOI] [PubMed] [Google Scholar]
- 21. Niewoehner O., Garcia-Doval C., Rostol J.T., Berk C., Schwede F., Bigler L., Hall J., Marraffini L.A., Jinek M.. Type III CRISPR–Cas systems produce cyclic oligoadenylate second messengers. Nature. 2017; 548:543–548. [DOI] [PubMed] [Google Scholar]
- 22. Han W., Pan S., Lopez-Mendez B., Montoya G., She Q.. Allosteric regulation of Csx1, a type IIIB-associated CARF domain ribonuclease by RNAs carrying a tetraadenylate tail. Nucleic Acids Res. 2017; 45:10740–10750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Han W., Stella S., Zhang Y., Guo T., Sulek K., Peng-Lundgren L., Montoya G., She Q.. A Type III-B Cmr effector complex catalyzes the synthesis of cyclic oligoadenylate second messengers by cooperative substrate binding. Nucleic Acids Res. 2018; 46:10319–10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rouillon C., Athukoralage J.S., Graham S., Gruschow S., White M.F.. Control of cyclic oligoadenylate synthesis in a type III CRISPR system. Elife. 2018; 7:e36734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jia N., Jones R., Sukenick G., Patel D.J.. Second messenger cA4 formation within the composite Csm1 palm pocket of Type III-A CRISPR–Cas Csm complex and its release path. Mol. Cell. 2019; 75:933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jia N., Jones R., Yang G., Ouerfelli O., Patel D.J.. CRISPR–Cas III-A Csm6 CARF domain is a ring nuclease triggering stepwise cA4 cleavage with ApA>p formation terminating RNase activity. Mol. Cell. 2019; 75:944–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Niewoehner O., Jinek M.. Structural basis for the endoribonuclease activity of the type III-A CRISPR-associated protein Csm6. RNA. 2016; 22:318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sheppard N.F., Glover C.V., Terns R.M., Terns M.P.. The CRISPR-associated Csx1 protein of Pyrococcus furiosus is an adenosine-specific endoribonuclease. RNA. 2016; 22:216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McMahon S.A., Zhu W., Graham S., Rambo R., White M.F., Gloster T.M.. Structure and mechanism of a Type III CRISPR defence DNA nuclease activated by cyclic oligoadenylate. Nat. Commun. 2020; 11:500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rostol J.T., Xie W., Kuryavyi V., Maguin P., Kao K., Froom R., Patel D.J., Marraffini L.A.. The Card1 nuclease provides defence during type III CRISPR immunity. Nature. 2021; 590:624–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhu W., McQuarrie S., Gruschow S., McMahon S.A., Graham S., Gloster T.M., White M.F.. The CRISPR ancillary effector Can2 is a dual-specificity nuclease potentiating type III CRISPR defence. Nucleic Acids Res. 2021; 49:2777–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Millman A., Melamed S., Amitai G., Sorek R.. Diversity and classification of cyclic-oligonucleotide-based anti-phage signalling systems. Nat. Microbiol. 2020; 5:1608–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lau R.K., Ye Q., Birkholz E.A., Berg K.R., Patel L., Mathews I.T., Watrous J.D., Ego K., Whiteley A.T., Lowey B.et al.. Structure and mechanism of a cyclic trinucleotide-activated bacterial endonuclease mediating bacteriophage immunity. Mol. Cell. 2020; 77:723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Deng L., Garrett R.A., Shah S.A., Peng X., She Q.. A novel interference mechanism by a type IIIB CRISPR-Cmr module in Sulfolobus. Mol. Microbiol. 2013; 87:1088–1099. [DOI] [PubMed] [Google Scholar]
- 35. Foster K., Kalter J., Woodside W., Terns R.M., Terns M.P.. The ribonuclease activity of Csm6 is required for anti-plasmid immunity by Type III-A CRISPR–Cas systems. RNA Biol. 2019; 16:449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hatoum-Aslan A., Maniv I., Samai P., Marraffini L.A.. Genetic characterization of antiplasmid immunity through a type III-A CRISPR–Cas system. J. Bacteriol. 2014; 196:310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rostol J.T., Marraffini L.A.. Non-specific degradation of transcripts promotes plasmid clearance during type III-A CRISPR–Cas immunity. Nat. Microbiol. 2019; 4:656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bhoobalan-Chitty Y., Johansen T.B., Di Cianni N., Peng X.. Inhibition of Type III CRISPR–Cas immunity by an archaeal virus-encoded anti-CRISPR protein. Cell. 2019; 179:448–458. [DOI] [PubMed] [Google Scholar]
- 39. Foster K., Gruschow S., Bailey S., White M.F., Terns M.P.. Regulation of the RNA and DNA nuclease activities required for Pyrococcus furiosus Type III-B CRISPR–Cas immunity. Nucleic Acids Res. 2020; 48:4418–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jiang W., Samai P., Marraffini L.A.. Degradation of phage transcripts by CRISPR-associated RNases enables type III CRISPR–Cas immunity. Cell. 2016; 164:710–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gruschow S., Athukoralage J.S., Graham S., Hoogeboom T., White M.F.. Cyclic oligoadenylate signalling mediates Mycobacterium tuberculosis CRISPR defence. Nucleic Acids Res. 2019; 47:9259–9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lin J., Feng M., Zhang H., She Q.. Characterization of a novel type III CRISPR–Cas effector provides new insights into the allosteric activation and suppression of the Cas10 DNase. Cell Discov. 2020; 6:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Samai P., Pyenson N., Jiang W., Goldberg G.W., Hatoum-Aslan A., Marraffini L.A.. Co-transcriptional DNA and RNA Cleavage during Type III CRISPR–Cas Immunity. Cell. 2015; 161:1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu T.Y., Liu J.J., Aditham A.J., Nogales E., Doudna J.A.. Target preference of Type III-A CRISPR–Cas complexes at the transcription bubble. Nat. Commun. 2019; 10:3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sambrook J., Russell W.D.. Molecular Cloning: A Laboratory Manual (3-Volume Set). 2001; Cold Spring Harbor Laboratory Press. [Google Scholar]
- 46. Southern E. Southern blotting. Nat. Protoc. 2006; 1:518–525. [DOI] [PubMed] [Google Scholar]
- 47. Osawa T., Inanaga H., Numata T.. Crystal structure of the Cmr2-Cmr3 subcomplex in the CRISPR–Cas RNA silencing effector complex. J. Mol. Biol. 2013; 425:3811–3823. [DOI] [PubMed] [Google Scholar]
- 48. You L., Ma J., Wang J., Artamonova D., Wang M., Liu L., Xiang H., Severinov K., Zhang X., Wang Y.. Structure Studies of the CRISPR-Csm Complex Reveal Mechanism of Co-transcriptional Interference. Cell. 2019; 176:239–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Samai P., Pyenson N., Jiang W., Goldberg G.W., Hatoum-Aslan A., Marraffini L.A.. Co-transcriptional DNA and RNA cleavage during Type III CRISPR–Cas Immunity. Cell. 2015; 161:1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. East-Seletsky A., O’Connell M.R., Knight S.C., Burstein D., Cate J.H., Tjian R., Doudna J.A.. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature. 2016; 538:270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gootenberg J.S., Abudayyeh O.O., Lee J.W., Essletzbichler P., Dy A.J., Joung J., Verdine V., Donghia N., Daringer N.M., Freije C.A.et al.. Nucleic acid detection with CRISPR–Cas13a/C2c2. Science. 2017; 356:438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen J.S., Ma E., Harrington L.B., Da Costa M., Tian X., Palefsky J.M., Doudna J.A.. CRISPR–Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science. 2018; 360:436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sofos N., Feng M., Stella S., Pape T., Fuglsang A., Lin J., Huang Q., Li Y., She Q., Montoya G.. Structures of the Cmr-beta complex reveal the regulation of the immunity mechanism of Type III-B CRISPR–Cas. Mol. Cell. 2020; 79:741–757. [DOI] [PubMed] [Google Scholar]
- 54. Meeske A.J., Nakandakari-Higa S., Marraffini L.A.. Cas13-induced cellular dormancy prevents the rise of CRISPR-resistant bacteriophage. Nature. 2019; 570:241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Strotskaya A., Savitskaya E., Metlitskaya A., Morozova N., Datsenko K.A., Semenova E., Severinov K.. The action of Escherichia coli CRISPR–Cas system on lytic bacteriophages with different lifestyles and development strategies. Nucleic Acids Res. 2017; 45:1946–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Watson B.N.J., Vercoe R.B., Salmond G.P.C., Westra E.R., Staals R.H.J., Fineran P.C.. Type I-F CRISPR–Cas resistance against virulent phages results in abortive infection and provides population-level immunity. Nat. Commun. 2019; 10:5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mo C.Y., Mathai J., Rostol J.T., Varble A., Banh D.V., Marraffini L.A.. Type III-A CRISPR immunity promotes mutagenesis of staphylococci. Nature. 2021; 592:611–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hatoum-Aslan A., Maniv I., Marraffini L.A.. Mature clustered, regularly interspaced, short palindromic repeats RNA (crRNA) length is measured by a ruler mechanism anchored at the precursor processing site. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:21218–21222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Park K.H., An Y., Jung T.Y., Baek I.Y., Noh H., Ahn W.C., Hebert H., Song J.J., Kim J.H., Oh B.H.et al.. RNA activation-independent DNA targeting of the Type III CRISPR–Cas system by a Csm complex. EMBO Rep. 2017; 18:826–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hale C.R., Zhao P., Olson S., Duff M.O., Graveley B.R., Wells L., Terns R.M., Terns M.P.. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009; 139:945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Staals R.H.J., Agari Y., Maki-Yonekura S., Zhu Y., Taylor D.W., van Duijn E., Barendregt A., Vlot M., Koehorst J.J., Sakamoto K.et al.. Structure and activity of the RNA-targeting Type III-B CRISPR–Cas complex of Thermus thermophilus. Mol. Cell. 2013; 52:135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Abudayyeh O.O., Gootenberg J.S., Konermann S., Joung J., Slaymaker I.M., Cox D.B., Shmakov S., Makarova K.S., Semenova E., Minakhin L.et al.. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016; 353:aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.