

Abstract
RNA interference (RNAi)-based therapeutics (miRNAs, siRNAs) have great potential for treating various human diseases through their ability to downregulate proteins associated with disease progression. However, the development of RNAi-based therapeutics is limited by lack of safe and specific delivery strategies. A great effort has been made to overcome some of these challenges resulting in development of N-acetylgalactosamine (GalNAc) ligands that are being used for delivery of siRNAs for the treatment of diseases that affect the liver. The successes achieved using GalNAc-siRNAs have paved the way for developing RNAi-based delivery strategies that can target extrahepatic diseases including cancer. This includes targeting survival signals directly in the cancer cells and indirectly through targeting cancer-associated immunosuppressive cells. To achieve targeting specificity, RNAi molecules are being directly conjugated to a targeting ligand or being packaged into a delivery vehicle engineered to overexpress a targeting ligand on its surface. In both cases, the ligand binds to a cell surface receptor that is highly upregulated by the target cells, while not expressed, or expressed at low levels on normal cells. In this review, we summarize the most recent RNAi delivery strategies, including extracellular vesicles, that use a ligand-mediated approach for targeting various oncological diseases.
Graphical Abstract
Graphical Abstract.
Ligands developed and tested for delivery of RNAi therapeutics.
INTRODUCTION
Several diseases including cancer are characterized by aberrant gene expression including upregulation, constitutive activation, or mutations that contribute to disease progression. Ideal therapeutic strategies for modulating these genes include RNA interference (RNAi)-based approaches, which include short interfering RNAs (siRNAs) and microRNAs (miRNAs). siRNAs and miRNAs can be easily designed and synthesized for modulating various genes, even those that are challenging to target with traditional small molecule inhibitors.
miRNAs are small non-coding RNAs that regulate gene expression through imperfect base pairing to various target messenger RNAs (mRNAs), most commonly resulting in mRNA cleavage or translation repression (1). The imperfect base pairing gives miRNAs the unique ability to bind to and regulate multiple target mRNAs. Thus, it is perhaps not surprising that miRNA dysregulation is a common occurrence in human diseases, including cancers where tumor suppressive miRNAs such as miR-34a or let-7 are downregulated and oncogenic miRNAs such as miR-21 and miR-155 are upregulated (2,3).The dysregulated miRNAs contribute to various hallmarks of cancer, such as sustained cancer cell proliferation, induction of angiogenesis, metastatic and invasion phenotypes, and resistance to cell death and anti-cancer agents (4,5) (Figure 1). Based on these intriguing properties of miRNAs, miRNA-based therapeutics, including restoring tumor suppressive miRNAs or antagonizing oncogenic miRNAs, have been developed and will continue to evolve. While miRNA restoration depends mainly on exogenous delivery of miRNA duplexes to restore the levels of downregulated tumor suppressive miRNAs, antagonizing oncogenic miRNAs aims to sequester or inhibit abundantly expressed oncogenic miRNAs through multiple approaches, including use of antisense oligonucleotides or small molecules inhibitors (6) (Figure 1).
Figure 1.

miRNA dysregulation in cancer and miRNA-based therapeutics strategies. In cancer, oncogenic miRNAs are upregulated while tumor suppressive miRNAs are downregulated, both of which contribute to disease progression (sustained proliferation, activation of metastasis and invasion, induction of angiogenesis, and resistance to death). To overcome miRNA dysregulation, two miRNA-based therapeutic strategies have been developed. Bottom left, when a tumor suppressive miRNA is downregulated, miRNA levels can be upregulated through exogenous delivery of synthetic miRNAs or through small molecules that can restore normal miRNA biogenesis. Restoring tumor suppressive miRNAs levels downregulates the expression of target cancer oncogenes, inhibiting disease progression. Bottom right, conversely, when an oncogenic miRNA is upregulated, the miRNA can be inhibited using antisense oligonucleotides, miRNA sponges, or small molecule inhibitors. Oncogenic miRNAs inhibition upregulates the expression of tumor suppressor proteins which helps to inhibit disease progression.
MiRNAs inhibit the expression of target genes through imperfect base pairing with target messenger RNAs (mRNAs), which allows a single miRNA the ability to regulate the expression of multiple genes, potentially acting as a multi-drug cocktail. For example, the tumor suppressive miRNA, miR-34 can downregulate genes involved in proliferation (c-MYC, Androgen Receptor), angiogenesis (VEGF), anti-apoptosis (BCL2) and immune response (PD-L1) resulting in a potent antitumor response (7–11). Indeed, several preclinical studies have validated that miR-34, or other tumor suppressive miRNAs such as let-7, can inhibit proliferation, survival, and metastasis of various tumors grown in vivo (12–19). In addition to tumor suppressive miRNAs, siRNAs have shown potential for treating various cancer types such as breast and prostate cancers, both of which have been treated with a PLK1 siRNA (20,21), and blood cancers that have been treated with a STAT3 siRNA (22,23). While miRNAs do not require perfect base pairing with their targets, siRNAs downregulate gene expression through perfect base pairing with a complementary mRNA sequence resulting in mRNA degradation. Various comprehensive reviews have been written over the years on the mechanisms involved in miRNA and siRNA targeting and their use as anti-cancer agents (see (24–27)). Here, we focus on the successes and challenges that still remain with attempting to achieve efficient and toxic-free in vivo delivery of therapeutically-relevant miRNAs and siRNAs.
The development of RNAi-based therapeutics has been limited by several challenges including delivery-associated toxicity, reduced stability, and immunogenic effects related to unmodified RNAi molecules (28). The sensitivity of unmodified RNAi molecules to degradation by nucleases results in decreased RNAi activity, requiring repetitive and high dosing to achieve the intended therapeutic response. Several chemical modifications can overcome these issues. For example, chemical modifications that include replacing the phosphodiester backbone with phosphorothioate (PS) bonds and introducing 2′-O-methyl (2′-OMe) and 2′-fluoro (2′-F) modifications in place of the unstable 2′-OH of the ribose sugar, reduce immunogenic effects, and enhance both stability and activity of siRNAs (29) (Figure 2A). Efforts are ongoing to identify additional modifications and to determine how the modifications affect targeting. Since chemical modifications in the 2′-position of the ribose sugar enhance the binding affinity of siRNA to its RNA target (30), they may induce unintended off-target effects. Although it has been shown that siRNA containing 2′-O-methyl modifications at various positions along the siRNA could reduce off-target effects (31,32), it is critical to determine how various modification patterns affect global gene targeting. Beyond the stability and immunogenicity issues, the main challenge that limits the development of RNAi-based cancer therapeutics is lack of safe, specific, and effective delivery strategies.
Figure 2.

Design of ligand-targeted miRNA or siRNA conjugates and the critical characteristics for each component. (A) Various targeting moieties and chemical modifications commonly used for miRNA or siRNA delivery. (B) Ligand-targeted miRNA or siRNA conjugates include a targeting ligand, linker, and miRNA or siRNA. The ligand should have high affinity and specificity for a receptor that is upregulated by the diseased cells but not normal cells. Rapid binding and uptake of the ligand is also important to avoid clearance from circulation. In the case of ligands with low affinity, multivalent designs can enhance binding of the ligand to target cells. Ligand size should be considered carefully, as small ligands often penetrate the dense architecture of the tumor to reach target cells, yet larger ligands are likely to be retained in circulation longer. Ligands can be attached to the RNAi molecule using a cleavable or non-cleavable linker. Optimizing the linker chemistry can enhance ligand binding affinity and can be used to attach an endosomal escape agent to the delivery system. In addition, the miRNA or siRNA should be designed to ensure stability and activity, low immunogenicity, and preferential loading of the guide strand to minimize any potential off-target effects.
A great effort has been made to overcome delivery issues and to ensure the selective targeting of RNAi molecules to diseased cells. For example, the development of N-acetylgalactosamine (GalNAc)-siRNA conjugates by Alnylam Pharmaceuticals is considered a breakthrough in RNAi delivery to the liver (33) (see Figure 4A for the chemical structure). GalNAc is a high affinity ligand that binds the asialoglycoprotein receptor (ASGPR) which is upregulated by hepatocytes resulting in ∼106 receptors per cell (34,35). Upon binding to the receptor, GalNAc-siRNA conjugates are rapidly internalized through receptor-mediated endocytosis. Due to endosomal acidification that causes disruption of ionic interactions, GalNAc-siRNAs are released from ASGPRs which rapidly recycle back to the cell surface every 10–15 min (34,35). The siRNAs are slowly released from the endosomes into the cytosol where loading into the RNA-induced silencing complex (RISC) for subsequent gene targeting takes place (34). The stability of the chemically modified siRNA in the acidic compartments and the slow release rate contribute to the robust and sustained activity of GalNAc-siRNA conjugates in vivo (34). In addition, the unique features of ASGPR in hepatocytes, including significant overexpression and rapid internalization and recycling rate, in addition to the ability of GalNAc to disengage from its receptor, are important factors that have resulted in the exceptional success of GalNAc–siRNA conjugates. Furthermore, GalNAc-siRNA conjugates are chemically modified using 2′-F, and 2′-OMe modifications, and include PS linkages at certain positions which enhances both stability and activity of the siRNA (30,33,36). For any receptor-targeted delivery approach, including GalNAc conjugates, the concentration of the receptor on the cell surface and the internalization kinetics are critical for achieving efficient uptake and targeting. Indeed, the overall level of ASGPR on the surface of hepatocytes is sufficient to achieve robust delivery and activity of GalNAc-siRNAs conjugates, as a reduction in ASGPR by 50% still supports a similar siRNA response (37). In contrast to the liver, delivery of RNAi molecules for targeting non-hepatic diseases is much more difficult. Ligand-RNAi conjugates must accumulate at a sufficient rate in target cells to achieve silencing. However, barriers including the endothelial barrier, renal clearance, inadequate receptor expression, and endosomal entrapment need to be overcome to achieve sufficient silencing (35,38).
Figure 4.

Examples of chemical structures of some common ligand-conjugates used to deliver miRNAs or siRNAs to diseased cells. The ligand structure is indicated in blue followed by the linker and the RNA. (A) Alnylam Pharmaceuticals: tri-GalNAc ligand conjugated to an siRNA to target the asialoglycoprotein receptor (ASGPR) on liver hepatocytes. (B) Kasinski group: folate conjugated to miR-34a through an unreleasable (top) or releasable (bottom, shown in red) linker for targeting folate-receptor (FR) expressing cancer cells (C) Desaulniers group: folate conjugated to the center of an siRNA for targeting FR expressing cancer cells. (D) Lieberman group: an EpCAM aptamer linked to an siRNA for targeting EpCAM+ epithelial breast cancers; U-U-U, linker. (E) Kortylewski group: CpG (D19) oligodeoxynucleotide (ODN) conjugated to miR-146a for treatment of inflammatory disorders and inhibition of leukemia progression; Asterisks, phosphorothioated bonds; x, C3 units of a carbon linker.
In addition to enhancing the stability and activity of siRNAs, considerations need to be made regarding unintended targeting at the molecular level, so-called off-target effects. Alnylam developed a design that limits the off-target effect of siRNAs (39). In the seed region of the siRNA antisense strand a thermally destabilizing modification, such as a glycol nucleic acid (GNA) is introduced. GNA modifications thermally destabilize non-specific interactions, while preserving on-target knockdown. Since sequence-specific RNA-based off-target effects might cause potential hepatotoxicity (39), the broad use of thermally destabilizing modifications will need to be explored in more detail, which could help overcome many of the off-target toxicities observed with other RNAi approaches.
After a great effort in the siRNA therapeutics field, Alnylam Pharmaceuticals has achieved US FDA approval for three siRNAs-based drugs – ONPATTRO® (Patisiran) in 2018, GIVLAARI® (Givosiran) in 2019, and most recently Oxlumo™ (Lumasiran) in 2020. Patisiran targets transthyretin (TTR) for the treatment of hereditary transthyretin amyloidosis (40), Givosiran targets aminolevulinic acid synthase 1 (ALAS1) for the treatment of acute hepatic porphyria (41), and Lumasiran treats primary hyperoxaluria type 1 (PH1) by targeting glycolate oxidase (GO) (42). While Patisiran uses lipid nanoparticles for delivery of siRNA, Givosiran and Lumasiran capitalize on direct conjugation of the siRNA to a tri-GalNAc ligand (40–42) (see Figure 4A for chemical structure). The success of these siRNA-based drugs offers the promise of developing other RNAi-based therapeutics for targeting extrahepatic diseases such as cancers. Even though targeting tissues beyond the liver is more complicated and additional effort is still needed, great progress has been made to overcome some of the limitations that stand against delivering RNAi molecules to these tissues. In this review, we summarize the strategies that have been developed to achieve specific delivery of miRNAs and siRNAs for targeting oncological and survival signaling in tumor cells as well as in tumor-associated immunosuppressive cells.
LIGAND-BASED STRATEGIES FOR RNAi DELIVERY
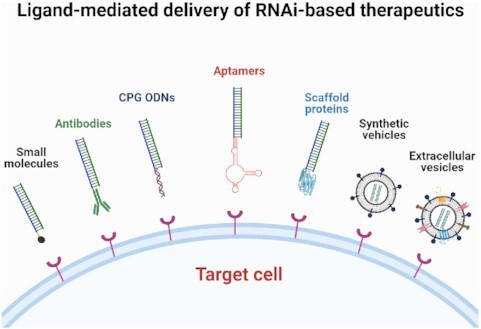
Several RNAi-targeted delivery strategies have been developed to achieve selective delivery of RNAi molecules to tumors. These strategies include direct conjugation of the RNAi molecule to a targeting ligand in the absence of a delivery vehicle or packaging the RNAi molecules into a delivery vehicle that is engineered to display a targeting ligand on its surface (Figure 3). Various ligands used for both delivery strategies are described in this review and are summarized in Table 1.
Figure 3.

Proposed internalization mechanisms of ligand-targeted miRNA or siRNA. Left panel: Internalization of ligand decorated synthetic lipids or natural extracellular vesicles (EVs) packaged with tumor suppressive miRNA/siRNA. After binding of the ligand to its receptor on the surface of cancer cells, ligand displaying delivery vehicles (synthetic vehicles or natural EVs) undergo receptor-mediated endocytosis or fusion with the cell membrane followed by release of miRNAs or siRNAs into the cytosol. Right panel: Internalization of vehicle free ligand-conjugated miRNA or siRNA. The ligand, conjugated to the miRNA or siRNA, binds to its receptor on the cell surface and is internalized via receptor-mediated endocytosis followed by release of the miRNA or siRNA into the cytosol while the receptor is recycled back to the cell surface. In the cytosol, tumor suppressive miRNAs or siRNAs engage with the RNA-induced silencing complex (RISC) and modulate gene expression resulting in downregulation of target genes and thus, reducing tumorigenesis.
Table 1.
Ligand-targeted miRNA or siRNA delivery systems for cancer therapy
| Ligand/receptor | miRNA/siRNA | RNA chemical modification | Condition | Outcome | Ref. |
|---|---|---|---|---|---|
| Ligand-targeted (vehicle-free) delivery | |||||
| Folate/FR | miR-34a | 2′-O-Methyl nucleotides | Breast and lung cancer | Inhibition of tumor growth | (71) |
| Folate-nigericin/FR | miR-34a | 2′-O-Methyl nucleotides | Breast cancer | Downregulation of cell proliferation | (73) |
| Folate/FR | siLuciferase | 2′-O-Methyl nucleotides | Breast cancer | Downregulation of Luciferase activity | (71) |
| Folate-ODN/ FR | siαV integrin, siSurvivin | Unmodified | FR+ HUVECs/ KB cells | Significant downregulation of target genes | (69) |
| Folate/FR | siLuciferase, siBCL-2 | Unmodified * | FR+ HeLa cells | Significant downregulation of target genes | (70) |
| DUPA-dsRBD/ PSMA | siPLK1 | 2′-O-Methyl or 2′-F nucleotides, PS bonds | Prostate cancer | Inhibition of tumor growth | (43) |
| Trivalent DUPA/ PSMA | siPLK1 | 2′-O-Methyl nucleotides, PS bonds | Prostate cancer | Inhibition of tumor growth | (78) |
| GL21.T aptamer/ AXL receptor | let-7g | 2′-F pyrimidines | Lung cancer | Inhibition of tumor growth | (83) |
| GL21.T aptamer/ AXL receptor | miR-212 | 2′-F pyrimidines | Lung cancer | Sensitization of lung cancer to TRAIL therapy | (84) |
| GL21.T aptamer/ AXL receptor | miR-34c | 2′-F pyrimidines | Lung cancer | Synergistic inhibition of cell proliferation by GL21.T-miR-34c and erlotinib | (86) |
| GL21.T aptamer/ AXL receptor | miR-148b | 2′-F pyrimidines | Breast cancer and melanoma | Apoptosis and necrosis in breast tumor, prevented tumor cell dissemination | (87) |
| anti-KIT aptamer/ KIT receptor | miR-26a | 2′-F uridines | Basal-like breast cancer cells and HSPCs | Inhibition of tumor growth and protection against chemotherapy induced myelosuppression | (88) |
| apt69.T aptamer/ BCMA | miR-137 | 2′-F pyrimidines | Multiple myeloma | Downregulation of cell viability | (89) |
| EpCAM aptamer/ EpCAM receptor | siPLK1 | 2′-F pyrimidines | Breast cancer | Inhibition of tumor growth | (21) |
| A10 aptamer/PSMA | siPLK1 and siBCL-2 | 2′-F pyrimidines | Prostate cancer | Inhibition of tumor growth by A10-siPLK1 | (20,90) |
| CpG-ODN/SR, TLR9 | miR-146a | 2′-O-Methyl nucleotide | Del(5q) MDS and AML | Inhibition of NF-κB inflammatory activity and disseminated leukemia progression | (104) |
| CpG-ODN/ SR, TLR9 | siSTAT3 | Deoxyribonucleotides | Melanoma, multiple myeloma, and AML | Induction of antitumor immunity and Inhibition of tumor growth | (22,23,108) |
| CpG-ODN/ SR, TLR9 | siSTAT3 | Deoxyribonucleotides | MDSCs from Prostate cancer patients | Abrogation of MDSC immunosuppressive activity | (109) |
| ScFv/Her2 | siPLK1 | 2′-O-Methyl nucleotides | Breast cancer | Inhibition of tumor growth and metastasis and prolonged survival | (113) |
| mAb/Transferrin receptor | siLuciferase | Unmodified | Brain tumor | Significant reduction of luciferase expression | (115) |
| mAb (Hu3S193) /Lewis-Y | siSTAT3 | Unmodified | Lewis-Y+ A431 cells | Significant silencing of STAT3 | (116) |
| mAb/EGFR | siKRAS | Not reported | Colon Cancer | Significant inhibition of tumor growth | (117) |
| ScFv/PSMA | siNotch1 | Unmodified | Prostate cancer | Significant inhibition of tumor growth | (118) |
| mAb/PSMA | siTRIM24 | 2′-O-Methyl nucleotide | Prostate cancer | Significant inhibition of tumor growth | (119) |
| Dual variable domain antibody/ BCMA, SLAMF7 or CD138 | siCTNNB1 | 2′-O-Methyl, 2′-F nucleotides, PS bonds | Multiple myeloma | Significant downregulation of CTNNB1 | (120) |
| DARPin/EpCAM | siBCL-2 | 2′-O-Methyl nucleotides | Breast cancer | Sensitization to doxorubicin treatment | (123) |
| Centyrins/EGFR or EpCAM | siCTNNB1 | 2′-O-Methyl, 2′-deoxy-2′-F nucleotides,PS bonds | A431 tumor or colorectal cancer cell lines | Significant downregulation of CTNNB1 and reduction of colorectal cancer cell viability in vitro | (124) |
| Targeted extracellular vesicles (EVs) | |||||
| GE11 peptide/ EGFR | let-7a | Unmodified | Breast cancer | Inhibition of tumor growth | (140) |
| AS1411 aptamer/ nucleolin | let-7a | Not reported | Breast cancer | Inhibition of tumor growth | (141) |
| A15/integrin αvβ3 | miR-159 | Cholesterol-modified | Breast cancer | Inhibition of tumor growth | (142) |
| IL3/IL3-R | siBCR-ABL | Not reported | Chronic myeloid leukemia | Inhibition of tumor growth | (145) |
| Folate/FR | siSurvivin | 2′-F nucleotides | Colorectal cancer (PDX-CRC) | Inhibition of tumor growth | (147) |
| EGFR aptamer/EGFR | siSurvivin | 2′-F nucleotides | Breast cancer | Inhibition of tumor growth | (147) |
| PSMA aptamer (A9g)/PSMA | siSurvivin | 2′-F nucleotides | Prostate cancer | Inhibition of tumor growth | (147) |
FR: folate receptor; PSMA: prostate specific membrane antigen; dsRBD: double stranded RNA binding domain; DUPA: (2- [3-(1,3-dicarboxy propyl) ureido] pentanedioic acid); BCMA: B cell maturation antigen; ODN: oligodeoxynucleotides; SR: scavenger receptor; TLR: Toll-like receptor; A15: disintegrin and metalloproteinase 15; IL3: interleukin 3; IL3-R: interleukin 3-receptor; EGFR: epidermal growth factor receptor; ScFv: single-chain fragmented antibody; mAb: Monoclonal antibody; Si: siRNA (short interfering RNA); miR: microRNA; Bcl-2: B-cell lymphoma 2; Plk1: polo-like kinase 1; STAT3: signal transducer and activator of transcription 3; TRIM24: Tripartite motif-containing protein 24; 2′-F: 2′- fluoro; PS: phosphorothioate; HSPCs: hematopoietic stem/progenitor cells; Del(5q) MDS: chromosome-5q deletion myelodysplastic syndrome; AML: acute myeloid leukemia; MDSCs: myeloid derived suppressor cells; NF-κB: nuclear factor-κB; PDX-CRC: patient-derived colorectal cancer xenograft; Asterisk *, siRNA with internal folic acid modification.
Vehicle-free delivery of RNAi molecules
Due to lack of specificity and poor cellular uptake of unconjugated miRNAs and siRNAs, as well as the potential toxicity of lipid transfecting agents, translating small RNAs into the clinic has been challenging (28). To overcome some of these disadvantages, while capitalizing on the power of small RNA therapeutics, various miRNA and siRNA delivery approaches have been developed and tested. A major advance includes direct conjugation of a targeting ligand to the RNA in the absence of a delivery vehicle. Several targeting ligands have been developed, including those based on small molecules, antibodies, aptamers, or synthetic CpG oligodeoxynucleotides (CpG-ODN) (Figure 2A). When using these so-called vehicle-free delivery approaches certain important features need to be considered (Figure 2B). Firstly, to obtain specificity, the ligand of choice needs to interact with a high affinity receptor that is upregulated on the surface of the intended target or diseased cells. The same receptor should not be expressed, or expressed at a relatively low level, on normal cells, or should be inconsequential for targeted delivery—for example, the location of the receptor on normal cells should be inaccessible to blood-born ligands. Secondly, to maximize internalization, binding and uptake of the ligand conjugates by the target cells must occur rapidly before clearance of the ligand conjugate from circulation. Of course, a high affinity ligand can support this feature; however, for certain ligands, engineering a divalent or multivalent ligand may further enhance both binding and uptake (43). Thirdly, to reduce the possibility of RNA degradation by serum nucleases and to increase intracellular half-life, chemical modification of the RNA is critical. However, modification positions within the RNA and the type of modification chemistry must be optimized carefully to prevent interference with the silencing activity of the RNA and/or unintended off-target effects. In fact, several recently developed chemically modified ligand-siRNA conjugates can enhanced both siRNA stability and silencing activity of the siRNA, resulting in long-lasting downregulation of target genes as discussed below (29,33). Fourthly, to promote the most robust interaction between the ligand and the receptor, inclusion of a linker between the ligand and the RNA should be considered. The linker itself can be modified to enhance binding to the receptor and the pharmacokinetics properties of the targeting ligand. This strategy has been employed for ligands that target Prostate-Specific Membrane Antigen (PSMA) (44,45). In general, the RNA can be linked to the ligand via a cleavable or non-cleavable linker depending on the stability of the conjugate and the activity of the conjugated RNA. Finally, since vehicle-free delivery approaches depend mainly on internalization of a targeting ligand by a specific receptor, they are subjected to endosomal entrapment, which is a rate-limiting factor in RNAi activity. Some targeting ligands have recently been modified to contain an endosomal escape agent to overcome this problem (43,46). This section summarizes some of the more commonly used, and recently developed vehicle-free delivery approaches, including the use of small molecule ligands, aptamers, CpG-ODNs, antibodies, and high-affinity scaffold proteins as effective ligands for delivery of miRNAs and siRNAs.
Small molecule-mediated delivery of RNAi
Several small molecule ligands have been developed for delivery of miRNAs or siRNAs specifically and robustly to diseased cells. Either naturally occurring or synthetic small molecules are identified, designed, and/or synthesized to bind to particular receptors with high affinity and specificity. After binding to the receptor on cell surface, the small molecule-RNA conjugates are internalized through receptor-mediated endocytosis, ultimately releasing some of the therapeutic RNA into the cytosol for target gene silencing (Figure 3, right panel). Small molecule ligands have many advantages that make them promising molecules for RNAi-mediated delivery which include low cost and feasibility of synthesis, and the ability to penetrate the dense architecture of the tumor microenvironment allowing them to effectively reach the intended cancer cells (47). Also, most small molecules contain derivatizable functional groups that make them amenable for facile conjugation to an RNA or for adding additional modifications (47).
Folate
Folate, vitamin B9, is an essential vitamin that is required by almost all cells and is a high affinity binding partner of the folate receptor (FR) which has four isoforms (α, β, γ and δ) (48–50). In non-diseased tissue, FR-α is expressed on the apical surface of epithelial cells in the lungs, mammary ducts, choroid plexus, and kidneys (50), whereas FR-β is expressed by immunosuppressive myeloid cells in the tumor microenvironment (51,52). Unlike other FR isomers, FR-γ is a secreted protein produced by certain hematopoietic cells (53) while the FR-δ isoform is expressed by transforming growth factor-beta (TGF-β) induced regulatory T-cells (Treg) as well as naturally arising Treg cells (54,55). After binding to the FR on the cell surface, folate molecules are rapidly internalized by the cells via receptor-mediated endocytosis (48). In addition to the high-affinity FRs, mammalian cells use other pathways to transport folate into cells, including the reduced folate carrier (RFC) and the proton-coupled folate transporter (PCFT). The RFC is ubiquitously expressed and is considered the major transporter of reduced folate in mammals (56). Importantly, the affinity of the RFC for folic acid is approximately 50-100-fold lower than the affinity for reduced folate, which is the major folate metabolite found in circulation (56). Thus, uptake of folate-conjugates for therapeutic intervention is not expected to occur in cells that only express the RFC. PCFT, the primary transporter system that facilitates folate absorption in the intestine, also has reduced affinity for folate in comparison to FR-α (57). Thus, due to the high affinity of FR-α and FR-β for folate, and their upregulation in cancer, it is unlikely that folate-conjugated drugs would be significantly taken up by normal cells.
While expression of the high-affinity FRs is limited on normal cells, FR expression is highly upregulated in numerous cancer types including tumors arising in the lung, breast, ovary, colon, and kidney (48,49,58–62). This high expression allows for substantial delivery of conjugated cargo to these cancerous tissues. For targeted agents, such as folate conjugates, typically a three-fold increase in therapeutic delivery between a normal and targeted cell is considered a major improvement in case of extrahepatic delivery over non-targeted agents, far less than the orders of magnitude observed when delivering GalNAc-siRNA conjugates to the liver. For ligands with a reduced delivery rate, incorporation of an endosomal escape agent is often required to achieve a therapeutic response. For FR expressing tumors the level of FR expression can easily be 10–100-fold higher than the level in normal tissues (60,62), which is more than adequate for delivering a biologically relevant concentration of RNAi molecules. While overexpression of the FR is essential, there are multiple attributes of the folate ligand that make it suitable for delivery as well. Folate is inexpensive and easy to synthesize, nonimmunogenic, and due to its small size, it is able to access solid tumors easily (47,63). These features make folate a promising ligand for the delivery of imaging and therapeutic agents specifically to cancer cells (64,65).
The path to using folate as a targeting ligand began in 1991 when Dr. Philip Low's group from Purdue University successfully used folate conjugates for delivery of macromolecules to KB cervical carcinoma cells (66). This work paved the way for several medical applications using folate conjugates. With regard to siRNA delivery, folate was conjugated to an siRNA labeled with a DY647 fluorophore to monitor its uptake and biodistribution (67). Folate-siRNA-DY647 conjugates were specifically internalized in FR-expressing cells in comparison to the non-targeted siRNA duplex. When folate-siRNA was injected into nude mice bearing KB tumor xenografts, folate accumulated specifically at the tumor relative to other organs indicating specific binding of folate–siRNA conjugates by FR-expressing cells in vivo. However, following the intracellular trafficking of the siRNA in cells, it was observed that folate-siRNA conjugates were entrapped in the endosomes, likely hindering siRNA target gene silencing.
Approximately ten years later, through intramolecular conjugation, the same group incorporated an endosomal escape agent, nigericin, into their folate-siRNA conjugates (folate–nigericin–siRNA) to overcome the endosomal entrapment issue (68). This strategy depends on the difference in solute concentration between the endosome which is rich in sodium ions (Na+) and the cytoplasm which is rich in potassium ions (K+). Following endosomal uptake of folate-nigericin-siRNA, nigericin is liberated from the rest of the compound and translocates into the endosomal membrane where it acts as a K+/H+ antiporter facilitating the exchange of osmotically active K+ for osmotically inactive H+ causing a build-up of osmotic pressure that leads to endosomal swelling and bursting. Folate-nigericin-siRNA activity was assessed in FR+ MDA-MB-231 cells that stably express a luciferase 2 reporter gene (Luc2). The Luc2 reporter was more robustly silenced following treatment with folate-nigericin-siluc2, indicating that inclusion of nigericin enhances target gene silencing.
Following the initial conjugation of folate to an siRNA, other strategies were evaluated including non-covalently tethering of folate to an siRNA through nucleic acid base pairing between the siRNA and a random oligodeoxynucleotide (F-ODN-siRNA) (69). In vitro treatment of human vein endothelial cells (HUVECs) with folate conjugated to an siRNA targeting ITGAV, an mRNA encoding αV integrin resulted in 80% inhibition of ITGAV mRNA in KB cells. Also, a reduction in the survivin transcript was observed in KB cells following treatment with F-ODN conjugated to an siRNA targeting survivin. This strategy has many advantages including simple synthesis and preparation that is cost-effective, and it has the potential to achieve a synergistic effect through the ability to deliver multiple siRNAs or miRNAs using a single oligodeoxynucleotide.
In addition to conjugating folate through a linker at the 3′ or 5′ ends of siRNA, a centrally-modified folic acid was recently reported for siRNA delivery (70) (Figure 4C). In this case, an siRNA was synthesized that allowed folic acid to be conjugated at various base positions along the length of the sense strand, including the central region that spans the Ago2 cleavage site. Indeed, siRNAs delivered with a centrally located folic acid modification resulted in enhanced silencing activity in comparison to siRNAs conjugated to folic acid at or near the 3′ end of the sense strand. Despite the potential of these newly developed centrally modified siRNAs, in vivo evaluation of safety and efficacy has yet to be conducted.
While delivery of siRNAs tends to be the benchmark, our group was the first to successfully conjugate folate directly to a miRNA, which we termed FolamiRs (71) (Figure 4B). Using the tumor suppressive miRNA, miR-34a, it was determined that FolamiR-34a was rapidly internalized only by FR-expressing cancer cells and that internalized conjugates were active and induced target gene silencing. The therapeutic safety and efficacy of FolamiR-34a were observed in breast cancer xenografts and in a genetically engineered model of non-small cell lung cancer. Efficacy was achieved at doses 10–150-fold lower than what was observed in the same models using MRX34, an encapsulated version of miR-34a that made its way into clinical trials (17,72). Although FolamiRs therapeutic effect is promising, like most ligand-conjugates, endosomal entrapment of the FolamiR molecules is a rate-limiting step. In a follow-up study, intramolecular conjugation of the FolamiR to nigericin enhanced the silencing activity of the delivered miRNA by facilitating endosomal escape (73). The cytosolic enrichment of miR-34a by nigericin induced downregulation of the miR-34a targets, oncogenic MET, and the programmed death-ligand 1 (PD-L1).
Although folate is a promising ligand for the delivery of miRNAs and siRNAs specifically to cancer cells. It would be beneficial to evaluate the effect of folate-conjugated miRNAs and siRNAs on cells in the tumor microenvironment such as tumor-associated macrophages and myeloid-derived suppressor cells (MDSCs) that are known to express FRβ (51).
DUPA (2- [3-(1,3-dicarboxy propyl) ureido] pentanedioic acid)
DUPA is a synthetic urea-based ligand that binds to PSMA with nanomolar affinity resulting in saturation of the receptor in a short period of time (74). PSMA, also known as glutamate carboxypeptidase II, is a type II membrane protein that is localized to the plasma membrane (75). PSMA is expressed at low levels in normal prostate tissue but is highly upregulated in prostate cancer (74,76). It is also expressed in the neovasculature of several solid tumors and is associated with the progression of prostate cancer (74,76). PSMA is constitutively internalized and the internalization rate increases after binding to anti-PSMA antibodies (77). After internalization, PSMA rapidly recycles back to the surface of the cell providing additional internalization rounds (74).
DUPA conjugates were first used to deliver siRNAs selectively to PSMA expressing prostate cancer cells in 2009 (67). In vitro treatment of LNCaP cells with a fluorescently tagged siRNA directly conjugated to DUPA (DUPA-siRNA-cy5) resulted in significant uptake 1-h post-treatment. Similarly, intravenous injection of DUPA-siRNA-cy5 into nude mice bearing LNCaP xenografts led to significant accumulation of DUPA-siRNA in LNCaP tumors. Although this approach provides a novel way to specifically deliver siRNAs to LNCaP cells, and delivery of the fluorescent siRNA was robust, target gene silencing using DUPA-siRNA conjugates needs to be evaluated in vitro and in vivo.
While still directly conjugated to DUPA, Tai et al. developed a novel DUPA-targeted siRNA delivery strategy (referred to as RNP8) that results in the formation of a DUPA-siRNA complex (43). In this case, DUPA-siRNA conjugates were mixed with a double strand RNA-binding domain (dsRBD) octamer which results in docking of the siRNA portion of the conjugate into the dsRBD while the DUPA ligands remained exposed on the surface. To facilitate endosomal escape of the siRNAs, an endosomolytic peptide (poly-histidine) was inserted into the C-terminus of the dsRBD. One important feature of this approach is the multivalency, which enhanced the binding affinity of DUPA to PSMA over one order of magnitude, from a KD of 82.1 nM for monovalent DUPA (DUPA-siRNA) to a KD of 0.728 nM for RNP8.
Both activity and biodistribution of the RNP8 complex were enhanced over DUPA-siRNA conjugates. Activity of the two complexes was assessed in vitro using LNCaP cells and an siRNA targeting Plk1. Treatment of LNCaP cells with RNP8 downregulated the Plk1 transcript and protein in comparison to DUPA-siRNA, which suppressed Plk1 only following transfection. The lack of silencing activity of DUPA-siRNA (without the dsRBD octamer) could be attributed to endosomal entrapment or the lower internalization rate of DUPA-siRNA conjugates. To evaluate in vivo biodistribution, Alexa Fluor 680-labeled siRNA was used in the preparation of DUPA-siRNA and RNP8. Following intravenous injection into mice bearing LNCaP xenografts, RNP8 accumulated at a higher rate in LNCaP tumors in comparison to DUPA-siRNA. Although this was attributed to lower binding affinity and rapid clearance of DUPA-siRNA from circulation relative to RNP8, other possibilities should also be considered. For example, for DUPA-siRNA, the siRNA was linked to DUPA through a cleavable disulfide bond which could have been reduced prematurely in the circulation leading to reduced signal in the tumor. A similar phenomenon was observed using folate as a delivery ligand (71). In this case, inclusion of a cleavable bond worked in vitro; however, in vivo the disulfide was reduced prematurely in circulation resulting in folate, but not the RNA, reaching the tumor tissue (71). The discrepancy in DUPA-siRNA accumulation within LNCaP tumors between this study and the study by the Low group is likely due to difference in linker chemistries resulting in different affinities (45,67). The KD of DUPA-siRNA used in this study was 82.1 nM while the KD of the DUPA ligand used in the study conducted by the Low group was 14 ± 1 nM.
The antitumor activity of RNP8 was assessed in vivo where RNP8 induced stronger inhibition of tumor growth in comparison to DUPA-siRNA. Despite that, protein-based delivery approaches require extensive evaluation for proper protein folding and could be expensive to synthesize (78). Recognizing this, the same group designed a new siRNA delivery approach that replaced the RNA-binding domains with an ethidium dimer (Et2) that binds to small RNAs. The polyhistidine peptide was also substituted with polyvinylimidazole (PVIm) to facilitate endosomal escape. A multivalent DUPA trimer (DUPA3) was conjugated to the sense strand of the siRNA and the resulting complex was mixed with Et2–PVIm to generate the DUPA3–siRNA/Et2–PVIm complex. Cellular binding and uptake studies indicated that DUPA3–siRNA with or without Et2–PVIm was taken up by LNCaP cells. Following intracellular trafficking using confocal microscopy, it was observed that DUPA3–siRNA/Et2–PVIm successfully escaped from the endosomes while DUPA3–siRNA (without Et2–PVIm) remained trapped. In vivo, DUPA3–siRNA/Et2–PVIm, but not DUPA3–siRNA, significantly inhibited LNCaP tumor growth. Despite the nice antitumor effect of DUPA3–siRNA/Et2–PVIm, the safety and efficacy of DUPA3–siRNA/Et2–PVIm needs to be evaluated in immunocompetent mice before considering clinical applications.
Aptamer-mediated delivery of RNAi molecules
Aptamers are short single-stranded oligonucleotide sequences that can be designed and developed to bind to any target protein, including surface receptors, with high affinity and specificity (79). In comparison to conventional antibodies, aptamers are easier to manufacture, have no or minimal toxicity and immunogenicity, and are smaller in size (80). Based on these properties, several aptamers have been developed for selective delivery of RNAi therapeutics to cancer cells.
Aptamer-mediated delivery of miRNAs
In addition to the use of aptamers for delivery of chemotherapeutics (81,82), a great effort has been made towards the use of aptamers for the delivery of miRNAs. For example, an aptamer, GL21.T that binds to the AXL tyrosine kinase receptor was used to restore tumor suppressive miRNAs in cancer cells that overexpress the AXL receptor (83). To accomplish this, a multifunctional therapeutic agent was developed that included covalent conjugation of the let-7g miRNA to GL21.T (referred to as GL21.T-let). In this case, the ribose sugars of the RNA were modified with 2′-fluoro pyrimidines (2′F-Py) to provide protection against nucleases, and to reduce potential immunogenicity. GL21.T-let selectively delivered let-7g to AXL+ A549 cells and downregulated HMGA2 and N-Ras, two let-7-g targets through a Dicer-mediated mechanism. In vivo biodistribution studies determined that GL21.T-let conjugates accumulated in A549 (Axl+) xenografts but not in MCF-7 (Axl–) tumors, with some targeting to the kidneys. Therapeutically, intravenous injection of GL21.T-let only inhibited tumor growth of Axl+ A549. Quantification of immune-related genes, 2′-5′ oligoadenylate synthetase 1, interferon-inducible IFIT1 (p56), and interleukin-8 in the spleen and the liver, suggested that GL21.T-let is well tolerated.
Multiple additional studies also validated use of the GL21.T aptamer for miRNA delivery. For example, miR-212 levels were restored in AXL+ cells through GL21.T-mediated delivery resulting in downregulation of the anti-apoptotic protein, PED, and restoration of TNF-related apoptosis-inducing ligand (TRAIL)-mediated cytotoxicity (84). Although the efficacy of this strategy needs to be validated in vivo, it provides a new approach for combinatorial therapy using aptamer-miR-212 to sensitize lung cancer to TRAIL therapy. The GL21.T aptamer was also used to overcome resistance to the commonly administered tyrosine kinase inhibitor, erlotinib. One mechanism of acquired resistance to erlotinib is through AXL overexpression and activation (85). Downregulation of AXL by a miR-34c mimic restored non-small cell lung cancer sensitivity to erlotinib (86). Thus, Russo et al. conjugated GL21.T to miR-34c (GL21.T/miR-34c) followed by co-treatment of erlotinib-resistant HCC827 cells with GL21.T/miR-34c and erlotinib (86). Significant inhibition of cell proliferation was observed in cells treated with GL21.T/miR-34c. Recently, the GL21.T aptamer was used to deliver miR-148b to AXL expressing breast cancer and melanoma cells, which also resulted in inhibition of tumor progression through downregulation of miR-148b targets, ALCAM and ITGA5 (87).
Additional aptamer-miRNA conjugates have also shown promise for cancer treatment. For example, a c-Kit targeting aptamer was used to deliver miR-26a (miR-26a chimera) to basal-like breast cancer cells and hematopoietic stem cells (HSCs), both expressing the KIT receptor (88). Delivery of the miR-26a chimera resulted in significant inhibition of cell growth in KIT receptor positive cells and had a striking combinatorial effect when combined with the chemotherapeutic agent, 5-fluorouracil (5-FU). Since chemotherapeutic drugs are associated with hematopoietic toxicity (88), the study also tested whether the miR-26a chimera could protect against chemotherapy-induced myelosuppression. Although mice treated with 5-FU only showed significant leukopenia and thrombocytopenia, the number of leukocytes and thrombocytes was nearly doubled in mice treated with the combination (5-FU and miR-26a chimera). Similar results were also obtained when the miR-26a chimera was used in combination with carboplatin. Although the study showed therapeutic potential of miR-26a chimera against basal-like breast cancer cells, additional support for using the miR-26a chimera could be gained by validating these effects in primary cells and other breast cancer cell types.
Targeting cancerous plasma cells has also been reported with aptamer-miRNAs (89). In this case, the RNA aptamer, apt69.T was designed to bind to B Cell Maturation Antigen (BCMA), which is highly upregulated by plasma cells in multiple myeloma (MM). Conjugating apt69.T to miR-137 (apt69.T-miR-137) and treating U266 cells resulted in a reduction in cell viability. Despite that, further verification that targeting occurs in bone marrow-accumulated malignant plasma cells in vivo would be beneficial.
Aptamer-mediated delivery of siRNAs
Several aptamers have been developed for the delivery of siRNAs to tumor cells. For example, an EpCAM aptamer was used by the Lieberman group to target epithelial breast cancers that highly upregulate EpCAM (21). In this case, the EpCAM aptamer-siRNA chimeras (AsiC) were synthesized by linking the 5′ end of the siRNA sense strand to the 3′ end of the aptamer via a U–U–U linker followed by annealing of the siRNA antisense strand (Figure 4D). The long strand of the AsiC (i.e. EpCAM aptamer + linker + siRNA sense strand) was synthesized with 2′-fluoropyrimidines, which enhances stability of the RNA in 50% serum (t1/2 >> 36 h).
Cellular uptake studies indicate that the EpCAM aptamer was taken up by EpCAM+ MDA-MB-468 cells. Importantly, the binding of EpCAM-AsiC was evaluated using normal tissue and breast tumor biopsies from breast cancer patients. EpCAM-AsiC significantly accumulated in the tumor biopsies in comparison to normal tissue samples further confirming the selectivity of EpCAM-AsiC to tumors that have upregulated EpCAM. The antitumor activity of PLK1 EpCAM-AsiC was assessed in vivo using an EpCAM+ MB468 reporter cell line that stably expresses luciferase. Treating these cells with PLK1 EpCAM-AsiC for 24 h before implanting them into nude mice, completely inhibited tumor formation. Subcutaneous injection of PLK1 EpCAM-AsiC into nude mice implanted with EpCAM+ MB468-luc cells in one flank and EpCAM– MB231 cells in the other induced regression of MB468-luc cells, but not MB231 cells. Similar results were also obtained in mice bearing Her2+ MCF10CA1a cells.
In addition to the use of aptamers to deliver small RNAs to breast cancer tissues, aptamers have been used to deliver siRNAs to prostate cancer cells with high PSMA expression. For example, the A10 RNA aptamer was used to deliver siRNAs targeting PLK1 or BCL-2 to PSMA expressing prostate cancer cells (90). In vitro studies indicated that A10-PLK1 and A10-BCL-2 bound specifically to PSMA expressing LNCaP cells and induced downregulation of PLK1 or BCL-2, respectively. Silencing of PLK1 or BCL-2 using A10-siRNAs reduced cell proliferation and induced apoptosis specifically in PSMA expressing cells. In vivo treatment resulted in a significant reduction in LNCaP tumor volume following A10-Plk1 administration. No effect was observed for PSMA– PC-3 tumors indicating specificity and efficacy of A10-Plk1 in targeting PSMA expressing tumors. As a preliminary evaluation of immune response, the level of IFN-β was measured in LNCaP cells, resulting in no IFN-β production following treatment with A10-siRNA chimeras.
A second-generation PSMA-PLK1 chimera was developed by the same group to enhance the silencing activity, specificity, and stability of the chimeras (20). In this design, several aspects of the A10-Plk1 chimera were modified. The aptamer was reduced to 39 nucleotides instead of the original 71 nucleotides to facilitate chemical synthesis. Also, to enhance the silencing activity and specificity of aptamer-siRNA chimera, an siRNA with a two nucleotide (UU)-overhang at the 3′ end of the siRNA was synthesized, and the duplex structure was further optimized to favor guide strand processing. Second-generation PSMA-PLK1 chimeras induced enhanced PLK1 silencing in comparison to the first-generation A10-Plk1 chimera. One possible explanation for the enhanced silencing by the second-generation chimeras is that the modified siRNAs are better substrates for Dicer, which resulted in more processed duplexes. In vivo therapeutic efficacy studies using the second-generation PSMA-PLK1 chimeras showed complete regression of tumor growth. Clearly, aptamer and siRNA modifications can greatly affect efficacy and need to be considered wisely for each target. This study also determined that addition of a polyethylene glycol (PEG) group to the PSMA-PLK1 chimera enhances the antitumor activity and circulation half-life. Overall, this strategy has several advantages including increased serum retention and stability, prolonged target gene silencing in vivo, and stronger inhibition of tumor growth at low siRNA doses.
Although aptamers bind their targets with high affinity and specificity and have been extensively used for delivery of RNAi molecules for targeting various cancers, there are some limitations with the use of aptamers including high cost involved in large-scale production and sensitivity of unmodified aptamers to nucleases (91). Developing shorter aptamers and inclusion of chemical modifications that do not affect affinity and specificity are important considerations for the future development of aptamers for delivery of RNAi-based therapeutics.
CpG oligodeoxynucleotide (ODN)-mediated delivery of RNAi molecules
Another prominent synthetic oligonucleotide used for delivery of RNAi-based therapeutics is the CpG oligodeoxynucleotides (ODN), a Toll-like receptor 9 (TLR9) ligand that is rapidly internalized by certain cells and induces immune responses (92,93). Under normal physiological conditions, human TLR9 is mainly expressed by plasmacytoid dendritic cells (DCs) and B cells. However, under inflammatory and tumor conditions, certain cells, including tumor-associated macrophages and polymorphonuclear myeloid-derived suppressor cells (PMN-MDSC) upregulate TLR9 (93). The expression of TLR9 is not restricted to immune cells as it is also upregulated by various tumors, including prostate cancer, glioma stem cells, acute myeloid leukemia (AML), multiple myeloma (MM), and B cell lymphoma (22,94–97). TLR9 is an intracellular receptor, therefore, it is not directly implicated in the uptake of CpG-ODN (93). Instead, surface-localized receptors have been involved in the uptake of CpG-ODN such as SR-A1 (93), RAGE (98), CXCL16 (99), CD205 (100), CD14 (101) or SR-BI (102). However, once internalized, TLR9 is required for effective siRNA-mediated silencing when siRNAs are conjugated to a CpG-ODN (22). One possible explanation is that TLR9 activation might be important for mediating rapid siRNA release from the endosomes (22,103). Based on these features, CpG-ODNs are considered a promising approach for delivery of therapeutics including miRNAs and siRNAs to cancer cells as well as immunosuppressive cells in the tumor microenvironment.
CpG oligodeoxynucleotide (ODN)-mediated miRNA delivery
Lately, CpG-ODNs have been used to deliver miRNA to non-malignant myeloid and malignant leukemic cells (104). For example, CpG-miR-146a conjugates (C-miR146a) were used to restore miR-146a levels in chromosome-5q deletion myelodysplastic syndrome [del(5q) MDS] and AML. In both of these diseases, miR-146a loss, through derepression of IRAK1 and TRAF6 leads to NF-κB activation (24,104–106). Thus, C-miR146a was developed to modulate NF-κB inflammatory and tumorigenic activity (104). In this study, C-miR146a was synthesized by conjugating the 5′ end of the miR-146a sense strand to the 3′ end of CpG-A/D19-ODN using a carbon linker followed by hybridization of the miR-146a guide strand (Figure 4E). The sense strand was minimally modified with a single 2′-O-methyl-modification at the 3′ end. In vitro, the C-miR146a duplex was rapidly taken up by multiple human immune cells, mouse RAW264.7 macrophages, human MDSL, HL-60 leukemia cells, and human Raji lymphoma cells. C-miR146a uptake was mediated by scavenger receptor A and was dependent on clathrin-mediated endocytosis. In vivo, intravenous injection of C-miR146a into mir-146a-deficient mice restored miR-146a in the bone marrow and spleen resulting in a reduction in Irak1 and Traf6 up to 24-h post-injection and corrected aberrant myeloproliferation in mir-146a-deficient mice. The ability of C-miR146a in dampening cytokine release syndrome (CRS) induced by CD19-specific chimeric antigen receptor (CAR) T-cells was also assessed in vivo (104). Intraperitoneal injection of C-miR146a three days prior to CAR T-cell transfer, upregulated miR-146a in peritoneal myeloid cells and reduced the level of CRS-related cytokines, IL-6, and granulocyte colony-stimulating factor (G-CSF). This suggests that C-miR146a could be used to overcome adverse effects associated with CD19-CAR T-cell therapy without compromising its antitumor activity.
The antitumor activity of C-miR146a in HL-60, MDSL del(5q) leukemia cells, and MV4-11 AML cells was also evaluated. Although, treatment with C-miR146a induced cell death in all the cells, C-miR146a induced more robust cell death in miR-146a-deficient HL-60, MDSL cells. In vivo therapeutic efficacy studies using C-miR146a (10 mg/kg) resulted in inhibition of disseminated HL-60 leukemia progression. The effect was likely due to targeting of NF-κB mediated survival signaling by miR-146a. This work suggests that the C-miR146a strategy has therapeutic potential to target myeloproliferative disorders as well as myeloid leukemia. These studies also set the stage for using the CpG strategy for delivery of other tumor suppressive miRNAs or anti-cancer antagomirs.
CpG oligodeoxynucleotide (ODN)-mediated siRNA delivery
Activation of Signal Transducer and Activator of Transcription 3 (STAT3) is associated with oncogenesis, survival, and proliferation of cancer cells (107). Activated STAT3 also promotes production of several angiogenic and immunosuppressive factors in myeloid cells, and inhibits expression of certain T-helper cell 1 (Th1) costimulatory molecules (107). Since pharmacological targeting of STAT3 is challenging, several studies have used siRNAs conjugated to CpG ligands to selectively inhibit STAT3 in tumor cells and tumor-associated immunosuppressive cells (93).
In 2009, Kortylewski et al. developed a strategy that links a Stat3 siRNA to a CpG oligonucleotide for targeting TLR9 expressing cells in the tumor microenvironment (108). In this case, the CpG oligonucleotide was linked to the antisense strand of a Stat3 siRNA followed by hybridization to the sense strand to generate a CpG-Stat3 siRNA duplex (CpG-Stat3). After confirming uptake and silencing activity using TLR9 expressing cells in vitro, the therapeutic efficacy of CpG-Stat3 siRNA was confirmed in vivo using B16 tumor-bearing mice. Efficient uptake and Stat3 gene silencing were observed in tumor-associated macrophages, DCs, and B cells. In addition, CpG-Stat3 siRNA conjugates induced stronger inhibition of B16 tumor growth in comparison to the negative control. This effect was mainly immune-mediated as treatment with CpG-Stat3 siRNA conjugates led to an increase in tumor-infiltrating neutrophils and CD8+T cells, and a reduction in immunosuppressive CD4+FoxP3+ regulatory T cells. These results indicated that combining STAT3 inhibition with TLR9 stimulation might enhance the antitumor immune response. Afterward, the CpG-Stat3 siRNA strategy was further optimized for targeting hematologic malignancies (23). In this study, the CpG-Stat3 siRNA induced regression in the syngeneic Cbfb/Myh11/Mpl (CMM) AML model (23). CpG-Stat3 conjugates induced potent antitumor immunity and eradicated disseminated AML cells in immunocompetent mice. Although the safety and efficacy of this strategy needs to be validated using humanized mouse models of AML, it provides a potential solution for targeting leukemia-initiating cells and for eradicating disseminated AML in vivo.
Based on the success of the CpG-Stat3 siRNA strategy in murine tumor models, the strategy was further optimized for targeting STAT3 in human TLR9+ immune cells and blood cancer cells (22). Human-specific CpG type A (CpG(A)/D19) was linked to the antisense strand of the STAT3 siRNA through a flexible carbon chain linker followed by annealing of the sense strand to generate CpG(A)-STAT3 siRNA conjugates. In vitro uptake and activity studies indicated that CpG(A)-STAT3 siRNA conjugates were internalized by human myeloid dendritic cells, plasmacytoid dendritic cells (pDCs), and B cells resulting in 60% knockdown of STAT3 in DCs. The CpG(A)-STAT3 siRNA induced stronger upregulation of the HLA-DR complex and the costimulatory molecule CD86 in DCs in comparison to control CpG(A)-Luc siRNA conjugates. In addition, treatment of pDCs with CpG(A)-STAT3 enhanced their ability to induce T cell proliferation. Overall, these results indicated that the CpG(A)-STAT3 siRNA has strong immunostimulatory properties in human immune cells. This study also evaluated the ability of CpG(A) to deliver STAT3 siRNA to TLR9+ hematologic malignancies, MM and AML. Although CpG(A)-STAT3 conjugates initially accumulated within early endosomes, CpG(A)-STAT3 downregulated STAT3 by approximately 50% suggesting that at least some of the siRNA escaped the endosome. In vivo gene silencing efficacy of CpG(A)-siRNAs in AML xenografts produced effective targeting of BCL-XL, an anti-apoptotic protein, and STAT3, and significantly inhibited tumor growth. Similar results were obtained using myeloma or leukemia xenografts.
The CpG-STAT3 siRNA strategy was further adapted for targeting tumor-associated immunosuppressive cells, such as MDSCs (109). MDSCs are a heterogeneous population of cells that expand during cancer progression and are associated with poor patients' survival (109–111). Hossain et al. found that TLR9 is overexpressed by a subset of granulocytic-MDSC (G-MDSC) that accumulates in the peripheral blood of prostate cancer patients during disease progression (109). This population of G-MDSC had elevated STAT3 activity and could inhibit the proliferation and activity of CD8+ T cells. Treatment of G-MDSC with the CpG-STAT3 siRNA successfully induced STAT3 silencing and restored T cell functions. Additional studies that evaluate the effect of CpG-STAT3 on MDSC differentiation are needed to further understand their response to CpG-STAT3. Despite that, the CpG-STAT3 strategy is considered a novel therapeutic approach for targeting immunosuppressive cells in prostate cancer.
Antibody-mediated delivery of RNAi molecules
Antibodies have been successfully used for delivery of anticancer therapeutics including RNAi-molecules to cancer cells. This approach involves conjugating an antibody that binds a certain receptor expressed by cancer cells to a cytotoxic payload through a linker. Several antibody-drug conjugates have been used and approved as cancer therapeutics and more are in various clinical stages (47). While not initially cancer-directed, in 2005 the Lieberman group used a protamine-antibody fusion protein to deliver an siRNA to cells infected with HIV or transfected with the HIV-1 envelope (112). In vitro, HIV replication was inhibited in HIV-infected primary T cells following treatment with an antibody Fab fragment-protamine fusion protein (F105-P) linked to an siRNA targeting the HIV-1 capsid gene gag. To highlight use of the antibody approach for delivery to tumors in vivo, intravenous or intratumoral injection of F105-P-siRNAs targeting c-Myc, Vegf, and Mdm2 in B16 tumors that stably express the HIV envelope specifically suppressed tumor growth. Importantly, there was insignificant induction of IFN-β, Stat-1 or 2′-5′ oligoadenylate synthetase 1 expression following treatment. This body of work also determined that a single-chain antibody directed against ErbB2 (Her2) and fused with protamine specifically delivered siRNAs into ErbB2-expressing cancer cells resulting in silencing of target gene in ErbB2+ cells indicating that this approach could be generalized.
A similar single-chain fragmented antibody (ScFvs) was used for targeting Her2+ human breast cancer cells in vivo (113). Using these cells, the therapeutic potential of Her2-ScFvs-protamine fusion protein (F5-P) complexed to an siRNA targeting PLK1 (F5-P/PLK1-siRNA) was evaluated. Intravenous injection of F5-P/PLK1-siRNA significantly reduced PLK1 expression and inhibited xenograft growth. F5-P/PLK1-siRNA suppressed metastasis and led to prolonged survival of mice bearing Her2+ breast tumors. For future applications and to gain a more comprehensive understanding of the biology, it is important to understand how siRNAs delivered using F5-P/PLK1 are released from the endosomes and what cytosolic concentrations are needed for achieving a response. Nonetheless, an advantage of this approach is that it could be modified for targeting other cancer types by simply changing the fusion protein antibody.
Antibody–siRNA conjugates were developed for targeting transferrin receptor (TfR, or CD71) expressing cells. The transferrin receptor is constitutively internalized by cells which allows the transport of transferrin (Tf) into early endosomes (114). In the acidic pH of endosomes, Tf is dissociated from the TfR which rapidly and repeatedly recycles back to the cell surface (114). A transferrin receptor monoclonal antibody (mAb) was used for delivery of siRNA specifically to brain tumors (115). In this case, TfR–mAb–siRNA conjugates were prepared by conjugating a luciferase-targeting biotin-labeled siRNA to a streptavidin-tagged TfR–mAb. Intravenous injection of TfR–mAb–siRNA targeting luciferase into rat-bearing C6 or RG-2 tumors that stably express the luciferase gene resulted in a 69–81% reduction in luciferase expression in these tumors (115).
Additional studies were conducted to evaluate the use of covalent and noncovalent conjugation between the antibody and siRNA, specifically focusing on endosomal escape. For these studies, a STAT3 siRNA was targeted to Lewis-Y expressing cancer cells using an anti-Lewis-Y monoclonal antibody (Hu3S193) (116). The STAT3 siRNA was covalently linked to the antibody using a cleavable disulfide bond (hu3S193-siRNA) or was noncovalently complexed with an antibody modified with a (D-arginine)9 peptide (9r) using electrostatic interactions (hu3S193-9r(1):siRNA). Activity studies indicated that the covalent conjugate (hu3S193-siRNA) generated a significant reduction in STAT3 expression only when cells were co-treated with an endosomal escape agent such as chloroquine or the arginine peptide (9r). In contrast, the noncovalent conjugate (hu3S193-9r(1):siRNA) alone induced efficient silencing of STAT3 in Lewis-Y-expressing cancer cells, but not in control cells, indicating the importance of the arginine peptide (9r) in mediating siRNA endosomal escape.
Additional antibody-siRNA conjugates were developed to overcome drug resistance, in this case to anti-EGFR antibodies. One mechanism that drives anti-EGFR antibody resistance involves mutations in KRAS, which is downstream of EGFR (117). To overcome KRAS-mediated resistance, an EGFR antibody-KRAS siRNA complex was developed and tested both in vitro and in vivo (117). The EGFR antibody-KRAS siRNA complex was internalized by EGFR-expressing cells, strongly suppressed KRAS expression, and inhibited clonogenic growth of mutant KRAS cells. In vivo, intraperitoneal injection of the EGFR antibody-KRAS siRNA complex significantly inhibited tumor growth in mice-bearing anti-EGFR-resistant cells in comparison to control groups.
To specifically target prostate cancer, an anti-PSMA single chain antibody was engineered to deliver two independent constructs (118). One contained an anti-PSMA single chain antibody fused to a truncated protamine while the other included an endosomal escape peptide HA2 and a furin cleavage site. Both constructs successfully delivered Notch1 siRNA into LNCaP cells, induced efficient knockdown of Notch1, and inhibited LNCaP cell proliferation in vitro and in vivo. However, the inhibition was more robust when the construct containing the HA2 peptide was used. A similar study by Shi et al. successfully delivered a TRIM24 siRNA using a human monoclonal PSMA antibody fused with protamine to target castration-resistant prostate cancer with high PSMA expression resulting in significant suppression of tumor growth (119).
Recently, an antibody-siRNA conjugate has been developed which depends on using dual variable domain (DVD) antibodies that contain an outer variable fragment (Fv) for selective antigen targeting and an inner catalytic Fv which has a uniquely reactive lysine (Lys) for conjugation to a β-lactam-functionalized siRNA (120). Treatment of a multiple myeloma cell line with a SLAMF7, CD138 or BCMA targeting DVD–Antibody conjugated to a β-catenin (CTNNB1) siRNA induced significant knockdown of CTNNB1 expression. This method generates highly homogenous antibody–siRNA conjugates that have a defined structure and are easy to assemble. Despite that, future studies that aim to understand how siRNAs delivered using this approach reach the cytoplasm would be beneficial.
These studies highlight the potential use of antibody-siRNA conjugates as anti-cancer agents. Antibodies have several advantages for delivery of RNAi molecules including clinical relevance, high binding affinity to their antigens, and the ability to deliver an active siRNA. In contrast, slow penetration of solid tumors due to large molecular weight antibodies and potential immune activation could affect the efficacy and safety of the treatment (121). To overcome these issues, several studies as discussed above used Fab fragments or scFv which are smaller and lack the immune activating Fc region. The small size facilitates additional penetration of solid tumors, while removing the Fc region reduces unintended interactions with non-target cells. Additional work to understand, optimize, and enhance the activity of antibody–RNAi conjugates will lend further support to the use of antibody–siRNA conjugates.
High affinity-scaffold proteins
Novel scaffold proteins such as DARPins and Centyrins have been used for delivery of RNAi molecules for targeting extrahepatic diseases. DARPins (designed ankyrin repeat protein) are small non-immunoglobulin proteins that can be selected to bind with high affinity and specificity to virtually any target protein (122). In 2009, an EpCAM-specific DARPin fused to truncated protamine and conjugated to a Bcl-2 siRNA for targeting EpCAM-expressing breast carcinoma cells was generated (123). Cellular uptake studies using FITC-labeled siRNA conjugated to EpCAM-specific DARPin indicated localization of the siRNA in the endosomal compartments, but a diffused cytosolic signal was also detected. Treatment of MCF-7 cells with EpCAM-specific DARPin complexed with the BCL-2 siRNA induced a significant downregulation of BCL2 expression and sensitized the cells to doxorubicin treatment. This indicates that the amount of siRNA released from the endosomes is enough to induce sufficient target gene silencing to influence a combinatorial effect; however, more optimization, including incorporating an endosomal escape agent could result in further enhancement of the therapeutic effect.
Centyrins are small, engineered protein that are based on consensus fibronectin (FN3) domains found in human Tenascin C (124). Centyrins can be engineered to bind any target antigen with high specificity and affinity similar to that of the antibodies (124). For example, an EGFR-binding Centyrin was conjugated to a beta-catenin (CTNNb1) targeting siRNA (EGFR-Cent-CTNNb1) followed by evaluation of its activity in EGFR-expressing cancer cells both in vitro and in vivo (124). Treatment of EGFR-expressing A431 cells with EGFR-Cent-CTNNb1 siRNA conjugates induced a significant downregulation of beta-catenin expression at both mRNA and protein levels. Intravenous injection of EGFR-Cent-CTNNb1 siRNA into mice bearing A431 xenografts resulted in a significant knockdown of beta-catenin expression in the tumor. Similar results were also obtained when the CTNNb1 siRNA was conjugated to Centyrins that bind to PSMA, BCMA, or EpCAM indicating that this approach could be generalized for targeting other cancer types. In addition, when two siRNAs targeting different genes were conjugated to a single EGFR-binding Centyrin, both genes were simultaneously silenced in vitro. The ability to target two genes with the same conjugate provides a way to develop siRNA-based therapeutics that could produce a synergistic antitumor effect or overcome potential resistance mechanisms.
Lipid conjugates for RNAi delivery
The uptake of naked RNAi molecules by cells is limited, in part, by the hydrophilic nature of RNA; thus, conjugating RNAs to a hydrophobic molecule, such as cholesterol, could enhance the uptake of RNAi-molecules by the cells (125). Indeed, cholesterol has been extensively used for delivery of RNAi molecules to various cells and tissues. The uptake of cholesterol-siRNA conjugates by cells occurs rapidly by a selective endocytic process after insertion of the cholesterol conjugates into the plasma membrane (126). Binding of cholesterol with circulating lipoproteins could also facilitate uptake of cholesterol-siRNA conjugates by lipoprotein receptors (126). In 2004, Soutschek et al. conjugated cholesterol to a chemically stabilized siRNA that targets apolipoprotein B (apoB, Chol-apoB-1-siRNA) (127). Intravenous administration of Chol-apoB-siRNA into mice significantly reduced apoB expression in the liver and jejunum as well as total cholesterol levels.
With regard to using cholesterol-siRNA conjugates for treating cancer, cholesterol was conjugated to an siRNA targeting MDR1 (Ch-siMDR) followed by evaluating its activity using KB-8-5 tumor-bearing mice (128). Biodistribution studies using Cy5.5-Labeled Ch-siRNA indicated that conjugation of siRNA to cholesterol enhanced tumor and liver accumulation while reducing retention in the kidney. Intravenous, intraperitoneal, or peritumoral administration of Ch-siMDR into KB-8-5 tumor-bearing mice significantly downregulated the P-glycoprotein level in the tumors. Since siRNAs delivered using this approach might accumulate in other organs in addition to the tumor, genes that are essential for tumor cell growth, while not essential for normal cells, would be preferred targets.
Similarly, cholesterol-siRNA conjugates have been developed for targeting glioblastoma (129). In this case, a fully modified siRNA was conjugated to cholesterol (Chol-hsiRNA) followed by evaluating uptake and activity using primary GBM8 cells. Chol-hsiRNA targeting Cyclophilin B (PPIB) or Huntingtin (HTT) mRNA were rapidly taken up by cells and induced significant target gene silencing in vitro. To evaluate the efficacy of Chol-hsiRNA in vivo, GBM8 cells that stably express firefly luciferase were injected orthotopically into the brains of mice. A single intratumoral injection of Chol-hsiRNAs targeting HTT or firefly luciferase induced 45% reduction in human HTT mRNA and about 90% reduction in firefly luciferase activity seven days post-injection.
In addition to siRNAs, cholesterol, and other lipids such as eicosapentaenoic acid (EPA) and docosanoic acid (DCA) have recently been used to deliver a chemically modified version of the let-7b miRNA for treating non-small cell lung cancer (NSCLC) (130). Uptake of lipid-conjugated let-7b was evaluated using Cy3-labeled let-7b in a NSCLC cell line. Cy3-labeled let-7b conjugated to cholesterol or DCA was significantly taken up by the cells while EPA-let-7b conjugates were not, indicating that the lipid structure might affect conjugate uptake. Significant enrichment of let-7b was detected following treatment of NSCLC cells with different lipid-let-7b conjugates, which was correlated with downregulation of the let-7 target gene HMGA2. The biodistribution of lipid-conjugated let-7b was evaluated in vivo using NSCLC tumor-bearing mice following a single subcutaneous injection. let-7b was enriched in the tumors following treatment with EPA and DCA conjugates. However, the highest level of let-7b was detected in the liver and the spleen. And, while the EPA-hmiR-let-7b treatment downregulated HMGA2 mRNA levels and Ki-67 expression in the tumor, the effect on tumor size was insignificant, which requires further exploration.
Overall, cholesterol and other lipids have been successfully used for delivery of RNAi molecules to various tissues providing an opportunity to modulate gene expression in these tissues, which provides an advantage over naked RNAi molecules. Despite the benefits, the main limitations of lipid or cholesterol-conjugated RNAi molecules include lack of specificity and significant accumulation in the liver, kidney, and spleen. Keeping that in mind, further optimization of the lipid structure could help to minimize non-specific uptake by normal tissue. In addition, careful selection of RNAi-molecules that are only essential for the growth of diseased cells is important for these less-specific delivery approaches. While potentially achievable for siRNA-based therapeutics, this might be difficult in case of miRNAs that have a vast range of targets.
Targeted extracellular vesicles for RNAi delivery
Although synthetic delivery vehicles, such as liposomes, are successfully used for delivery of miRNAs to cancer cells, several limitations stand against routine clinical use, including delivery associated toxicity, non-specific uptake, immunogenicity, and accelerated blood clearance (6,131,132). A great effort has been made to overcome some of these limitations, which was previously reviewed by our group (6) as well as by others (131,132). As an alternative approach, natural extracellular vesicles (EVs) have gained significant scientific attention as potential delivery vehicles. In this section, we discuss recent studies that tested and developed ligand targeted EVs for cancer therapy.
EVs are membrane bound vesicles produced by a variety of cell types, including cancer cells, as a means of intercellular communication. Based on their size and biogenesis, EVs are divided into various subpopulations referred to as exosomes, microvesicles, and apoptotic bodies (133) with exosomes being the most commonly used subpopulation for RNAi delivery. However, due to the inability to selectively purify exosomes, most studies use the more general term, EVs. EVs have emerged as alternative vehicles for delivery of therapeutic miRNAs and siRNAs to diseased cells for many reasons: (i) EVs isolated from normal cells have minimal toxicity and immunogenicity (134,135); (ii) EVs are able to cross natural barriers (136); (iii) compared to liposomes, EVs from certain cell types have enhanced retention in circulation due to CD47 expression, which protects them from phagocytosis (137) and (iv) EVs can be engineered to express a cell-surface ligand that can be used to achieve specific delivery to target cells as discussed below and shown in Figure 3.
Internalization of EVs is mediated by multiple mechanisms, including direct membrane fusion and receptor-mediated endocytosis (138,139) (Figure 3). When ligand decorated EVs are internalized by receptor-mediated endocytosis, they back-fuse with the endosomal membrane and release their content into the cytosol (139). Alternatively, EVs can directly enter the cells through fusion with the outer plasma membrane resulting in the release of their content into the cytosol (139). For instance, folate decorated EVs are mainly taken up by FR expressing cells through membrane fusion (139). After binding to FRs on the cell surface, EVs fuse with the cell membrane and release their content, in this case, siRNAs into the cytosol resulting in target gene silencing (139). Although folate might not be directly involved in the uptake of the EVs in this case, it facilitates the binding of EVs to FR expressing cells.
Peptide-mediated EV delivery was achieved using the GE11 peptide (YHWYGYTPQNVI) that binds selectively to the epidermal growth factor receptor (EGFR) (140). Since EGFR is overexpressed by a variety of tumor cells, including breast cancer cells, GE11-tagged EVs have the potential to specifically deliver tumor suppressive miRNAs or siRNAs to these cancer cells. In this study, HEK293 cells were used as factories to generate the GE11-EVs followed by loading of the EVs with let-7a or a control miRNA. In vitro experiments confirmed the specific uptake of GE11-EVs by EGFR expressing breast cancer cells. In vivo, intravenous injection of let-7a loaded GE11-EVs (1 μg, 1 time weekly for 4 weeks) into mice bearing HCC70 xenografts inhibited tumor growth in comparison to control EVs. Although high EV accumulation was detected in the liver 24 h post-injection, no major tissue damage was observed. Future studies that evaluate the safety and efficacy of this strategy in immunocompetent mice would help advance this technology clinically.
Aptamer-targeted EVs were developed for delivery of miRNAs to breast cancer cells overexpressing the nucleolin receptor (141). In this case, the AS1411 aptamer, which has high affinity for the nucleolin receptor, was displayed on the outer surface of EVs secreted by immature dendritic cells using cholesterol affinity. Afterward, using electroporation, AS1411-modified EVs were loaded with let-7 tagged with a Cy3 fluorophore. In vitro uptake studies indicated efficient uptake of AS1411-EVs-let-7-Cy3 by nucleolin expressing MDA-MB-231 cells resulting in reduced proliferation and migration. Biodistribution was evaluated using nude mice bearing MDA-MB-231 xenografts. Treatment of animals with AS1411-EVs-let-7-Cy3 resulted in a strong fluorescence signal in the tumor tissue in comparison to non-targeting EVs. Both types of EVs accumulated in non-cancerous tissues, including the liver and the brain which might be a concern for generating off-target effects. An additional in vivo study assessed the therapeutic efficacy of AS1411-EVs-let-7. Intravenous injection of AS1411-EVs-let-7 (150 μg/every other day, 12 injections) induced a regression in MDA-MB-231 tumor growth in comparison to mice treated with free let-7 or control EVs. The study showed no signs of tissue damage or immune activation in mice injected with AS1411-EVs-let-7 every other day for 4 days; however, it would be valuable to monitor the safety of this strategy over the course of treatment that led to a therapeutic response.
Another EV targeting strategy was developed for delivery of miRNA and chemotherapeutic drugs using disintegrin and metalloproteinase 15 (A15)-expressing EVs (A15-Exo) to triple-negative breast cancer (142). A15 has high binding affinity to integrin αvβ3, which is overexpressed on the surface of several cancer types including breast, ovarian, glioblastoma, melanoma, and prostate cancer (143,144). This strategy relies on stimulation of THP-1 monocytes with phorbol 12-myristate 13-acetate (PMA) which leads to release of A15-Exo into the cell culture supernatant. After purification from supernatant, A15-Exo were loaded with doxorubicin and cholesterol-modified miR-159 (Co-A15-Exo) followed by evaluating in vivo therapeutic efficacy. Intravenous injection of Co-A15-Exo into MDA-MB-231 tumor-bearing mice resulted in synergistic inhibition of tumor growth in comparison to control groups.
In addition to miRNAs, modified EVs have been used for delivery of siRNAs to Chronic Myeloid Leukemia (CML), capitalizing on upregulation of the interleukin 3 receptor (IL3-R) (145). To achieve selective targeting, HEK293 cells were engineered to produce exosomes that express the exosomal protein Lamp2b, fused to the interleukin 3 fragment (IL3-Lamp2b). Uptake of IL3-Lamp2b exosomes (IL3L Exo) was confirmed in vitro using IL3-R expressing LAMA84 and K562R cell lines. Subsequently, IL3-Lamp2b exosomes were loaded with an siRNA targeting BCR-ABL, a tyrosine kinase which is constitutively active in CML cells (146). In vitro activity assays indicated that IL3L Exo loaded with BCR-ABL siRNA (IL3L Exo BCR-ABL siRNA) inhibited the growth of LAMA84, K562, and Imatinib-resistant K562 cells. Intraperitoneal injection of IL3L Exo BCR-ABL siRNA into NOD/SCID mice bearing Imatinib-resistant K562 xenografts inhibited tumor growth in comparison to mice injected with the negative control.
Another study engineered EVs that displayed folate molecules, PSMA-targeting aptamers, or EGFR-targeting aptamers on their surface for delivery of survivin siRNA to cancer cells overexpressing FR, PSMA, or EGFR, respectively (147). The therapeutic efficacy of PSMA aptamer/EVs, EGFR aptamer/EVs, or folate/EVs loaded with survivin siRNA was evaluated using nude mice bearing prostate cancer, breast cancer, or patient-derived colorectal cancer xenografts, respectively. Significant inhibition of tumor growth was observed in all three cancer models. This work provides an effective strategy for targeting various tumor types by changing the ligand on the EVs surface without affecting EVs composition or integrity.
Overall, these studies indicated that modified EVs can be used to deliver miRNAs/siRNAs either alone or in combination with other drugs for cancer therapy. Despite the therapeutic promise of EVs as delivery vehicles, additional effort is still needed to enhance the therapeutic applicability. Most of these studies loaded the EVs with synthetic miRNA or siRNA through transfecting them into the exosome-secreting cells, through directly transfecting the exosomes, or by electroporation. All these methods have certain limitations and none can be scaled for production (148,149). Transfecting exosome-secreting cells that overexpress miRNA or siRNA may affect the packaging process or the behavior of the EVs. In addition, the loading efficiency could vary greatly based on the RNA sequence (149). Electroporation could be associated with siRNA aggregation or precipitation causing an overestimation of the loaded siRNA (149,150). Direct transfection of EVs with miRNAs has also been used, instead of transfecting the donor cells (151). However, contaminating transfection reagents could remain associated with the EVs resulting in unintended side effects (152). Indeed, poor loading could result in the need to deliver more EVs which may lead to toxicity. To enhance loading, Reshke et al. incorporated an siRNA sequence into the backbone of pre-miR-451, a miRNA that is significantly enriched in EVs of most cell types (150). Significant siRNA enrichment was detected inside the EVs when the siRNA was integrated into the pre-miR-451 backbone but not the pre-miR-16 backbone. In vitro and in vivo uptake and activity studies indicated that this strategy facilitated siRNA delivery to certain cell types and induced efficient gene knockdown at doses lower than lipid nanoparticles or electroporated EVs. Other studies showed efficient loading when cholesterol-modified siRNAs were incubated with the EVs without affecting vesicle integrity (142,148). For example, a study from the Khvorova lab determined that siRNAs that are chemically stabilized, as well as hydrophobically-modified (hsiRNAs), are efficiently loaded into the EVs (148). When these EVs were loaded with an siRNA targeting Huntingtin mRNA, significant silencing was achieved in vitro as well as in vivo. In addition to effective loading, another advantage of this method is the use of chemically stabilized siRNA which can reduce the effective dose required to achieve silencing. Without question, novel, more efficient, and more consistent loading approaches are needed prior to translating EVs into the clinic. Another challenge with using EVs as a delivery vehicle is that secreted EVs are not easy to scale up for production. One strategy that aims to enhance EVs secretions is the immortalization of primary cells (153). For example, mesenchymal stem cells (MSC) can be immortalized by inducing c-Myc overexpression which enhances their expansion and thus EVs secretion (153). Regardless, EVs are considered a promising approach for miRNA and siRNA delivery. However, further understanding of EVs biology such as how different molecules are naturally loaded into the EVs and how EVs are taken up by different cell types will advance EVs as delivery vehicles for RNAi-based therapeutics.
LIMITATIONS AND OPTIMIZATIONS
Ligand-mediated RNAi delivery provides a novel way for targeting various cancer types as discussed above. This approach bypasses many of the limitations of passive targeting, including non-specific delivery and off-target effect on normal cells. In addition, the specific targeting to the diseased cells achieved with ligand-mediated delivery will result in higher accumulation of RNAi molecule in these cells which could enhance effectiveness and potentially reduce dosing. All the methods described also support the ability to deliver various anti-cancer RNAs at the same time resulting in a synergistic effect once potential synergistic RNAs are identified (154).
The major bottleneck limiting the utility of a ligand-targeted approach for RNAi delivery is endosomal entrapment. The internalized RNA must be released into the cytosol or it will be degraded when the late endosomes fuse with the lysosomes. The prevailing hypothesis is that chemically modified RNAi-molecules will be more stable in the acidic compartments and slowly release into the cytosol, similar to what happens in the case of GalNAc-siRNA conjugates. Nonetheless, several strategies have been developed to facilitate more rapid release of RNAi molecules into the cytosol including cell-penetrating or fusogenic peptides such as the EB1 endosomolytic peptide, the influenza-derived fusogenic peptide diINF-7 (34,155), or small molecules such as chloroquine (156). However, some of these techniques are often toxic to cells or stimulate an immune response. Recently, we used the small molecule, nigericin to facilitate endosomal escape of ligand-conjugated miRNAs, which significantly enhanced miRNA activity in the absence of toxicity (73). Regardless, more effort needs to be placed on developing additional novel, safe, and effective endosomal escape agents to further reduce dosing and enhance the activity of RNAi-based therapeutics.
An additional concern includes the inability of the ligand-siRNA conjugate to be released from the receptor, especially if the conjugate does not contain a cleavable linker. Some ligands bind their receptor with so-called ‘excess affinity’ which could lead to continuous binding of the ligand to its receptor. Instead of disengaging from the receptor, the ligand will be recycled back with the receptor to the cell surface reducing cytosolic distribution and subsequent binding and internalization of additional conjugates (114). GalNAc, the successful ligand used for siRNA delivery binds ASGPR with nanomolar affinity (157); thus, ligands with comparable affinity should be considered. However, other ligands might behave differently when bound to their receptor under different endosomal conditions. For example, the level of acid endonucleases in normal cells, such as hepatocytes, could be completely different than that of cancer cells. Further understanding of the endosome environment such as the acidic pH that favors the dissociation of ligand from its receptor will be beneficial to determine when, and in which stage, RNAi molecule will be free in the endosomal lumen for release into the cytosol, which will ultimately enhance the efficacy of ligand-mediated RNAi delivery strategies.
Receptor saturation and rapid clearance following treatment are other issues that greatly influence intracellular RNAi concentration (158). In general, while the binding of high affinity ligands (such as folate) to their receptor occur rapidly after intravenous injection and saturate the receptor in a short amount of time, excess conjugates that did not bind the receptor will be rapidly cleared from the body, perhaps even before the unoccupied receptor recycles back to the surface (47). This will reduce the duration in which the receptor is exposed to the ligand making repetitive dosing a necessity to achieve a therapeutic response.
While there are still multiple hurdles to overcome, significant progress has been made to overcome some of the challenges associated with delivery of RNAi molecules to extrahepatic tissues and will continue. The success of GalNac conjugates is helping to pave the way and the recent use of RNA-based vaccines for protecting individuals from SARS-Cov2 suggests that an exponential increase in RNA therapeutics is on the horizon (159,160). Efforts in achieving specific and efficient delivery, including identifying and developing additional ligands and unique strategies to promote endosomal escape are major areas of focus that need to be tackled in the upcoming years.
CONCLUSION AND FUTURE DIRECTIONS
RNAi-based therapeutics including miRNAs and siRNAs have evolved as ideal therapeutic approaches for the treatment of various diseases, which is not surprising as these molecules can modulate most disease-related genes. However, advancing them as therapeutics for human diseases has been limited by challenges, including non-specific delivery, rapid RNA degradation, and poor cellular uptake (28). Various chemical modifications have been developed that enhanced stability and activity of siRNAs and reduced immune response. These modifications include addition of PS bonds at certain positions of the RNA backbone and replacing the unstable 2′-OH of the ribose sugar by 2′-OMe and 2′-F (29). Despite that, RNAi-based therapeutics for treating oncological diseases have not reached their full potential yet due to lack of safe and specific delivery approaches. Indeed, identifying an efficient delivery system is as important as enhancing the properties of RNAi molecules – both are required for developing RNAi-based therapeutics. GalNAc-siRNA conjugates are an exceptional example that demonstrates how consideration of both features can lead to rapid advances in RNAi-based therapeutics from preclinical to clinical stage. GalNAc binds the ASGPR which is expressed by liver hepatocytes and depends mainly on receptor targeting to achieve selective delivery of the conjugated siRNAs (34). Thus, expanding RNAi-based applications for targeting other tissues requires identifying novel ligands and their receptors. Once identified, RNAi molecules could be directly conjugated to a targeting ligand or packaged into a delivery vehicle that is engineered to display a ligand on its surface. Extensive efforts to identify cancer-targeting ligands have been successful and have resulted in identifying several small molecules, aptamers, antibodies, and peptides that deliver RNAi-molecules specifically to cancer cells as discussed.
Delivery of RNAi molecules using ligand decorated EVs is an emerging area that shows promise in overcoming endosomal entrapment due to the ability of EVs to directly fuse with the cell membrane or back-fuse with endosomal membranes (139). Another advantage of using EVs is that EVs provide a way to deliver RNAi molecules for targeting diseased cells in the central nervous system due to their ability to cross natural barriers (148,161). In addition, EVs are naturally released from cells, and thus, can be obtained from the cells of the patient improving safety. However, it is important to identify optimal EVs secretion and purification conditions, as well as efficient packaging techniques that enable large scale production and reproducible loading conditions prior to clinical applications.
Overall, a great effort has been made to identify various ligand-targeted approaches for delivery of miRNAs and siRNAs specifically to tumor cells and to overcome several delivery challenges. Validating the efficacy and safety of these approaches using immunocompetent and humanized mouse models is required prior to any clinical applications. In addition, developing efficient and non-toxic endosomal escape agents and utilizing these agents to enhance RNAi activity could finally allow us to realize the power of RNAi-based therapeutics for the treatment of human cancers.
ACKNOWLEDGEMENTS
Figures 1–3 and the graphical abstract were created using BioRender.com. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Contributor Information
Ahmed M Abdelaal, Department of Biological Sciences, Purdue University, West Lafayette, IN 47906, USA.
Andrea L Kasinski, Department of Biological Sciences, Purdue University, West Lafayette, IN 47906, USA; Purdue University Center for Cancer Research, Purdue University, West Lafayette, IN 47907, USA.
FUNDING
National Cancer Institute [R01CA226259 (A.L.K.) and R01CA205420 (A.L.K)] (in part); A.M.A. was supported by an Andrew's Fellowship from Purdue University; SIRG Graduate Research Assistantship from the Purdue Center for Cancer Research. Funding for open access charge: The open access publication charge for this paper has been waived by Oxford University Press - NAR Editorial Board members are entitled to one free paper per year in recognition of their work on behalf of the journal.
Conflict of interest statement. None declared.
REFERENCES
- 1. Lin S., Gregory R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer. 2015; 15:321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Croce C.M. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 2009; 10:704–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li W. (Jess), Wang Y., Liu R., Kasinski A.L., Shen H., Slack F.J., Tang D.G. MicroRNA-34a: potent tumor suppressor, cancer stem cell inhibitor, and potential anticancer therapeutic. Front. Cell Dev. Biol. 2021; 9:640587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Peng Y., Croce C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016; 1:15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pal A.S., Bains M., Agredo A., Kasinski A.L. Identification of microRNAs that promote erlotinib resistance in non-small cell lung cancer. Biochem. Pharmacol. 2021; 189:114154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Myoung S., Kasinski A.L. Strategies for safe and targeted delivery of MicroRNA therapeutics. RSC Drug Discovery Series. 2019; 2019-January:386–415. [Google Scholar]
- 7. Yamamura S., Saini S., Majid S., Hirata H., Ueno K., Deng G., Dahiya R. MicroRNA-34a modulates c-Myc transcriptional complexes to suppress malignancy in human prostate cancer cells. PLoS One. 2012; 7:e29722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Östling P., Leivonen S.K., Aakula A., Kohonen P., Mäkelä R., Hagman Z., Edsjö A., Kangaspeska S., Edgren H., Nicorici D. et al. Systematic analysis of microRNAs targeting the androgen receptor in prostate cancer cells. Cancer Res. 2011; 71:1956–1967. [DOI] [PubMed] [Google Scholar]
- 9. Zhang D., Zhou J., Dong M. Dysregulation of microRNA-34a expression in colorectal cancer inhibits the phosphorylation of FAK via VEGF. Dig. Dis. Sci. 2014; 59:958–967. [DOI] [PubMed] [Google Scholar]
- 10. Li L., Yuan L., Luo J., Gao J., Guo J., Xie X. MiR-34a inhibits proliferation and migration of breast cancer through down-regulation of Bcl-2 and SIRT1. Clin. Exp. Med. 2013; 13:109–117. [DOI] [PubMed] [Google Scholar]
- 11. Wang X., Li J., Dong K., Lin F., Long M., Ouyang Y., Wei J., Chen X., Weng Y., He T. et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell. Signal. 2015; 27:443–452. [DOI] [PubMed] [Google Scholar]
- 12. Yan K., Gao J., Yang T., Ma Q., Qiu X., Fan Q., Ma B. MicroRNA-34a inhibits the proliferation and metastasis of osteosarcoma cells both in vitro and in vivo. PLoS One. 2012; 7:e33778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li Y., Guessous F., Zhang Y., DiPierro C., Kefas B., Johnson E., Marcinkiewicz L., Jiang J., Yang Y., Schmittgen T.D. et al. MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res. 2009; 69:7569–7576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kumar M.S., Erkeland S.J., Pester R.E., Chen C.Y., Ebert M.S., Sharp P.A., Jacks T. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:3903–3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee S.-T., Chu K., Oh H.-J., Im W.-S., Lim J.-Y., Kim S.-K., Park C.-K., Jung K.-H., Lee S.K., Kim M. et al. Let-7 microRNA inhibits the proliferation of human glioblastoma cells. J. Neurooncol. 2011; 102:19–24. [DOI] [PubMed] [Google Scholar]
- 16. Liu C., Kelnar K., Liu B., Chen X., Calhoun-Davis T., Li H., Patrawala L., Yan H., Jeter C., Honorio S. et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med. 2011; 17:211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kasinski A.L., Kelnar K., Stahlhut C., Orellana E., Zhao J., Shimer E., Dysart S., Chen X., Bader A.G., Slack F.J. A combinatorial microRNA therapeutics approach to suppressing non-small cell lung cancer. Oncogene. 2015; 34:3547–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Trang P., Medina P.P., Wiggins J.F., Ruffino L., Kelnar K., Omotola M., Homer R., Brown D., Bader A.G., Weidhaas J.B. et al. Regression of murine lung tumors by the let-7 microRNA. Oncogene. 2010; 29:1580–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Esquela-Kerscher A., Trang P., Wiggins J.F., Patrawala L., Cheng A., Ford L., Weidhaas J.B., Brown D., Bader A.G., Slack F.J. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle. 2008; 7:759–764. [DOI] [PubMed] [Google Scholar]
- 20. Dassie J.P., Liu X., Thomas G.S., Whitaker R.M., Thiel K.W., Stockdale K.R., Meyerholz D.K., McCaffrey A.P., McNamara J.O., Giangrande P.H. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat. Biotechnol. 2009; 27:839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gilboa-Geffen A., Hamar P., Le M.T.N., Wheeler L.A., Trifonova R., Petrocca F., Wittrup A., Lieberman J. Gene knockdown by EpCAM aptamer-siRNA chimeras suppresses epithelial breast cancers and their tumor-initiating cells. Mol. Cancer Ther. 2015; 14:2279–2291. [DOI] [PubMed] [Google Scholar]
- 22. Zhang Q., Md Sakib Hossain D., Nechaev S., Kozlowska A., Zhang W., Liu Y., Kowolik C.M., Swiderski P., Rossi J.J., Forman S. et al. TLR9-mediated siRNA delivery for Targeting of normal and malignant human hematopoietic cells in vivo. Blood. 2013; 121:1304–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hossain D.M.S., Dos Santos C., Zhang Q., Kozlowska A., Liu H., Gao C., Moreira D., Swiderski P., Jozwiak A., Kline J. et al. Leukemia cell-targeted STAT3 silencing and TLR9 triggering generate systemic antitumor immunity. Blood. 2014; 123:15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kasinski A.L., Slack F.J. MicroRNAs en route to the clinic: Progress in validating and targeting microRNAs for cancer therapy. Nat. Rev. Cancer. 2011; 11:849–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Adams B.D., Kasinski A.L., Slack F.J. Aberrant regulation and function of MicroRNAs in cancer. Curr. Biol. 2014; 24:R762–R776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Orellana E., Kasinski A. MicroRNAs in Cancer: A historical perspective on the path from discovery to therapy. Cancers (Basel). 2015; 7:1388–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hu B., Zhong L., Weng Y., Peng L., Huang Y., Zhao Y., Liang X.-J. Therapeutic siRNA: state of the art. Signal Transduct. Target. Ther. 2020; 5:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen Y., Gao D.-Y., Huang L. In vivo delivery of miRNAs for cancer therapy: challenges and strategies. Adv. Drug Deliv. Rev. 2015; 81:128–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hassler M.R., Turanov A.A., Alterman J.F., Haraszti R.A., Coles A.H., Osborn M.F., Echeverria D., Nikan M., Salomon W.E., Roux L. et al. Comparison of partially and fully chemically-modified siRNA in conjugate-mediated delivery in vivo. Nucleic Acids Res. 2018; 46:2185–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Khvorova A., Watts J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017; 35:238–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jackson A.L. Position-specific chemical modification of siRNAs reduces ‘off-target’ transcript silencing. RNA. 2006; 12:1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Iribe H., Miyamoto K., Takahashi T., Kobayashi Y., Leo J., Aida M., Ui-Tei K. Chemical modification of the siRNA seed region suppresses off-target effects by steric hindrance to base-pairing with targets. ACS Omega. 2017; 2:2055–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nair J.K., Willoughby J.L.S., Chan A., Charisse K., Alam M.R., Wang Q., Hoekstra M., Kandasamy P., Kel’in A.V., Milstein S. et al. Multivalent N -acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014; 136:16958–16961. [DOI] [PubMed] [Google Scholar]
- 34. Brown C.R., Gupta S., Qin J., Racie T., He G., Lentini S., Malone R., Yu M., Matsuda S., Shulga-Morskaya S. et al. Investigating the pharmacodynamic durability of GalNAc–siRNA conjugates. Nucleic Acids Res. 2020; 48:11827–11844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dowdy S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017; 35:222–229. [DOI] [PubMed] [Google Scholar]
- 36. Debacker A.J., Voutila J., Catley M., Blakey D., Habib N. Delivery of oligonucleotides to the liver with GalNAc: from research to registered therapeutic drug. Mol. Ther. 2020; 28:1759–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Willoughby J.L.S., Chan A., Sehgal A., Butler J.S., Nair J.K., Racie T., Shulga-Morskaya S., Nguyen T., Qian K., Yucius K. et al. Evaluation of GalNAc-siRNA conjugate activity in Pre-clinical animal models with reduced asialoglycoprotein receptor expression. Mol. Ther. 2018; 26:105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lorenzer C., Dirin M., Winkler A.-M., Baumann V., Winkler J. Going beyond the liver: progress and challenges of targeted delivery of siRNA therapeutics. J. Control. Release. 2015; 203:1–15. [DOI] [PubMed] [Google Scholar]
- 39. Janas M.M., Schlegel M.K., Harbison C.E., Yilmaz V.O., Jiang Y., Parmar R., Zlatev I., Castoreno A., Xu H., Shulga-Morskaya S. et al. Selection of GalNAc-conjugated siRNAs with limited off-target-driven rat hepatotoxicity. Nat. Commun. 2018; 9:723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hoy S.M. Patisiran: first global approval. Drugs. 2018; 78:1625–1631. [DOI] [PubMed] [Google Scholar]
- 41. Scott L.J. Givosiran: first approval. Drugs. 2020; 80:335–339. [DOI] [PubMed] [Google Scholar]
- 42. Scott L.J., Keam S.J. Lumasiran: first approval. Drugs. 2021; 81:277–282. [DOI] [PubMed] [Google Scholar]
- 43. Tai W., Li J., Corey E., Gao X. A ribonucleoprotein octamer for targeted siRNA delivery. Nat. Biomed. Eng. 2018; 2:326–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Benešová M., Bauder-Wüst U., Schäfer M., Klika K.D., Mier W., Haberkorn U., Kopka K., Eder M. Linker modification strategies to control the prostate-Specific membrane antigen (PSMA)-targeting and pharmacokinetic properties of DOTA-conjugated PSMA inhibitors. J. Med. Chem. 2016; 59:1761–1775. [DOI] [PubMed] [Google Scholar]
- 45. Kularatne S.A., Zhou Z., Yang J., Post C.B., Low P.S. Design, synthesis, and preclinical evaluation of prostate-specific membrane antigen targeted 99m Tc-radioimaging agents. Mol. Pharm. 2009; 6:790–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Orellana E., Rangasamy L., Tenneti S., Abdelaal A., Low P., Kasinski A. Enhancing microRNA activity through increased endosomal release mediated by nigericin. 2018; bioRxiv doi:11 July 2018, preprint: not peer reviewed 10.1101/367672. [DOI] [PMC free article] [PubMed]
- 47. Srinivasarao M., Galliford C.V., Low P.S. Principles in the design of ligand-targeted cancer therapeutics and imaging agents. Nat. Rev. Drug Discov. 2015; 14:203–219. [DOI] [PubMed] [Google Scholar]
- 48. Chen C., Ke J., Zhou X.E., Yi W., Brunzelle J.S., Li J., Yong E.-L., Xu H.E., Melcher K. Structural basis for molecular recognition of folic acid by folate receptors. Nature. 2013; 500:486–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Leamon C.P., Low P.S. Folate-mediated targeting: from diagnostics to drug and gene delivery. Drug Discov. Today. 2001; 6:44–51. [DOI] [PubMed] [Google Scholar]
- 50. Shen J., Hu Y., Putt K.S., Singhal S., Han H., Visscher D.W., Murphy L.M., Low P.S. Assessment of folate receptor alpha and beta expression in selection of lung and pancreatic cancer patients for receptor targeted therapies. Oncotarget. 2018; 9:4485–4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cresswell G.M., Wang B., Kischuk E.M., Broman M.M., Alfar R.A., Vickman R.E., Dimitrov D.S., Kularatne S.A., Sundaram C.P., Singhal S. et al. Folate receptor beta designates immunosuppressive tumor-associated myeloid cells that can be reprogrammed with folate-targeted drugs. Cancer Res. 2021; 81:671–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kurahara H., Takao S., Kuwahata T., Nagai T., Ding Q., Maeda K., Shinchi H., Mataki Y., Maemura K., Matsuyama T. et al. Clinical significance of folate receptor β–expressing tumor-associated macrophages in pancreatic cancer. Ann. Surg. Oncol. 2012; 19:2264–2271. [DOI] [PubMed] [Google Scholar]
- 53. Shen F., Wu M., Ross J.F., Miller D., Ratnam M. Folate receptor Type .gamma. Is primarily a secretory protein due to lack of an efficient signal for glycosylphosphatidylinositol modification: protein characterization and cell type specificity. Biochemistry. 1995; 34:5660–5665. [DOI] [PubMed] [Google Scholar]
- 54. Yamaguchi T., Hirota K., Nagahama K., Ohkawa K., Takahashi T., Nomura T., Sakaguchi S. Control of immune responses by antigen-specific regulatory T cells expressing the folate receptor. Immunity. 2007; 27:145–159. [DOI] [PubMed] [Google Scholar]
- 55. Tian Y., Wu G., Xing J.-C., Tang J., Zhang Y., Huang Z.-M., Jia Z.-C., Zhao R., Tian Z.-Q., Wang S.-F. et al. A novel splice variant of folate receptor 4 predominantly expressed in regulatory T cells. BMC Immunol. 2012; 13:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Desmoulin S.K., Hou Z., Gangjee A., Matherly L.H. The human proton-coupled folate transporter. Cancer Biol. Ther. 2012; 13:1355–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Alam C., Aufreiter S., Georgiou C.J., Hoque M.T., Finnell R.H., O’Connor D.L., Goldman I.D., Bendayan R. Upregulation of reduced folate carrier by vitamin D enhances brain folate uptake in mice lacking folate receptor alpha. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:17531–17540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cagle P.T., Zhai Q.J., Murphy L., Low P.S. Folate receptor in adenocarcinoma and squamous cell carcinoma of the lung: potential target for folate-linked therapeutic agents. Arch. Pathol. Lab. Med. 2013; 137:241–244. [DOI] [PubMed] [Google Scholar]
- 59. Zhang Z., Wang J., Tacha D.E., Li P., Bremer R.E., Chen H., Wei B., Xiao X., Da J., Skinner K. et al. Folate receptor α associated with triple-negative breast cancer and poor prognosis. Arch. Pathol. Lab. Med. 2014; 138:890–895. [DOI] [PubMed] [Google Scholar]
- 60. Kalli K.R., Oberg A.L., Keeney G.L., Christianson T.J.H., Low P.S., Knutson K.L., Hartmann L.C. Folate receptor alpha as a tumor target in epithelial ovarian cancer. Gynecol. Oncol. 2008; 108:619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shia J., Klimstra D.S., Nitzkorski J.R., Low P.S., Gonen M., Landmann R., Weiser M.R., Franklin W.A., Prendergast F.G., Murphy L. et al. Immunohistochemical expression of folate receptor α in colorectal carcinoma: patterns and biological significance. Hum. Pathol. 2008; 39:498–505. [DOI] [PubMed] [Google Scholar]
- 62. Parker N., Turk M.J., Westrick E., Lewis J.D., Low P.S., Leamon C.P. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal. Biochem. 2005; 338:284–293. [DOI] [PubMed] [Google Scholar]
- 63. Müller C., Schibli R. Folic acid conjugates for nuclear imaging of folate receptor–positive cancer. J. Nucl. Med. 2011; 52:1–4. [DOI] [PubMed] [Google Scholar]
- 64. Leamon C.P., Parker M.A., Vlahov I.R., Xu L.C., Reddy J.A., Vetzel M., Douglas N. Synthesis and biological evaluation of EC20: a new folate-derived, 99mTc-based radiopharmaceutical. Bioconjug. Chem. 2002; 13:1200–1210. [DOI] [PubMed] [Google Scholar]
- 65. Predina J.D., Newton A.D., Connolly C., Dunbar A., Baldassari M., Deshpande C., Cantu E., Stadanlick J., Kularatne S.A., Low P.S. et al. Identification of a folate receptor-targeted near-infrared molecular contrast agent to localize pulmonary adenocarcinomas. Mol. Ther. 2018; 26:390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Leamon C.P., Low P.S. Delivery of macromolecules into living cells: a method that exploits folate receptor endocytosis. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:5572–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Thomas M., Kularatne S.A., Qi L., Kleindl P., Leamon C.P., Hansen M.J., Low P.S. Ligand-Targeted delivery of small interfering RNAs to malignant cells and tissues. Ann. N. Y. Acad. Sci. 2009; 1175:32–39. [DOI] [PubMed] [Google Scholar]
- 68. Rangasamy L., Chelvam V., Kanduluru A.K., Srinivasarao M., Bandara N.A., You F., Orellana E.A., Kasinski A.L., Low P.S. New mechanism for release of endosomal contents: osmotic lysis via nigericin-mediated K+ /H+ exchange. Bioconjug. Chem. 2018; 29:1047–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhang K., Wang Q., Xie Y., Mor G., Sega E., Low P.S., Huang Y. Receptor-mediated delivery of siRNAs by tethered nucleic acid base-paired interactions. RNA. 2008; 14:577–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Salim L., Islam G., Desaulniers J.-P. Targeted delivery and enhanced gene-silencing activity of centrally modified folic acid–siRNA conjugates. Nucleic Acids Res. 2020; 48:75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Orellana E.A., Tenneti S., Rangasamy L., Lyle L.T., Low P.S., Kasinski A.L. FolamiRs: ligand-targeted, vehicle-free delivery of microRNAs for the treatment of cancer. Sci. Transl. Med. 2017; 9:eaam9327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bader A.G. miR-34 – a microRNA replacement therapy is headed to the clinic. Front. Genet. 2012; 3:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Orellana E.A., Abdelaal A.M., Rangasamy L., Tenneti S., Myoung S., Low P.S., Kasinski A.L. Enhancing MicroRNA activity through increased endosomal release mediated by Nigericin. Mol. Ther. - Nucleic Acids. 2019; 16:505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kularatne S.A., Wang K., Santhapuram H.-K.R., Low P.S. Prostate-specific membrane antigen targeted imaging and therapy of prostate cancer using a PSMA inhibitor as a homing ligand. Mol. Pharm. 2009; 6:780–789. [DOI] [PubMed] [Google Scholar]
- 75. Schmittgen T.D., Teske S., Vessella R.L., True L.D., Zakrajsek B.A. Expression of prostate specific membrane antigen and three alternatively spliced variants of PSMA in prostate cancer patients. Int. J. Cancer. 2003; 107:323–329. [DOI] [PubMed] [Google Scholar]
- 76. Bravaccini S., Puccetti M., Bocchini M., Ravaioli S., Celli M., Scarpi E., De Giorgi U., Tumedei M.M., Raulli G., Cardinale L. et al. PSMA expression: a potential ally for the pathologist in prostate cancer diagnosis. Sci. Rep. 2018; 8:4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Liu H., Rajasekaran A.K., Moy P., Xia Y., Kim S., Navarro V., Rahmati R., Bander N.H. Constitutive and antibody-induced internalization of prostate-specific membrane antigen. Cancer Res. 1998; 58:4055–4060. [PubMed] [Google Scholar]
- 78. Tai W., Gao X. Synthetic polymer tag for intracellular delivery of siRNA. Adv. Biosyst. 2018; 2:1800075. [Google Scholar]
- 79. Keefe A.D., Pai S., Ellington A. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010; 9:537–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhou G., Latchoumanin O., Bagdesar M., Hebbard L., Duan W., Liddle C., George J., Qiao L. Aptamer-based therapeutic approaches to target cancer stem cells. Theranostics. 2017; 7:3948–3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Trinh T.Le, Zhu G., Xiao X., Puszyk W., Sefah K., Wu Q., Tan W., Liu C. A synthetic aptamer-drug adduct for targeted liver cancer therapy. PLoS One. 2015; 10:e0136673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bagalkot V., Farokhzad O.C., Langer R., Jon S. An aptamer–doxorubicin physical conjugate as a novel targeted drug-delivery platform. Angew. Chem. Int. Ed. 2006; 45:8149–8152. [DOI] [PubMed] [Google Scholar]
- 83. Esposito C.L., Cerchia L., Catuogno S., De Vita G., Dassie J.P., Santamaria G., Swiderski P., Condorelli G., Giangrande P.H., de Franciscis V. Multifunctional Aptamer-miRNA conjugates for targeted cancer therapy. Mol. Ther. 2014; 22:1151–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Iaboni M., Russo V., Fontanella R., Roscigno G., Fiore D., Donnarumma E., Esposito C.L., Quintavalle C., Giangrande P.H., de Franciscis V. et al. Aptamer-miRNA-212 conjugate sensitizes NSCLC cells to TRAIL. Mol. Ther. - Nucleic Acids. 2016; 5:e289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhang Z., Lee J.C., Lin L., Olivas V., Au V., LaFramboise T., Abdel-Rahman M., Wang X., Levine A.D., Rho J.K. et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012; 44:852–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Russo V., Paciocco A., Affinito A., Roscigno G., Fiore D., Palma F., Galasso M., Volinia S., Fiorelli A., Esposito C.L. et al. Aptamer-miR-34c conjugate affects cell proliferation of Non-Small-Cell lung cancer cells. Mol. Ther. - Nucleic Acids. 2018; 13:334–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Quirico L., Orso F., Esposito C.L., Bertone S., Coppo R., Conti L., Catuogno S., Cavallo F., de Franciscis V., Taverna D. Axl-148b chimeric aptamers inhibit breast cancer and melanoma progression. Int. J. Biol. Sci. 2020; 16:1238–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Tanno T., Zhang P., Lazarski C.A., Liu Y., Zheng P. An aptamer-based targeted delivery of miR-26a protects mice against chemotherapy toxicity while suppressing tumor growth. Blood Adv. 2017; 1:1107–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Catuogno S., Di Martino M.T., Nuzzo S., Esposito C.L., Tassone P., de Franciscis V. An Anti-BCMA RNA aptamer for miRNA intracellular delivery. Mol. Ther. - Nucleic Acids. 2019; 18:981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. McNamara J.O., Andrechek E.R., Wang Y., Viles K.D., Rempel R.E., Gilboa E., Sullenger B.A., Giangrande P.H. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol. 2006; 24:1005–1015. [DOI] [PubMed] [Google Scholar]
- 91. Nuzzo S., Roscigno G., Affinito A., Ingenito F., Quintavalle C., Condorelli G. Potential and challenges of aptamers as specific carriers of therapeutic oligonucleotides for precision medicine in cancer. Cancers (Basel). 2019; 11:1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Iwasaki A., Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004; 5:987–995. [DOI] [PubMed] [Google Scholar]
- 93. Kortylewski M., Moreira D. Myeloid cells as a target for oligonucleotide therapeutics: turning obstacles into opportunities. Cancer Immunol. Immunother. 2017; 66:979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ilvesaro J.M., Merrell M.A., Swain T.M., Davidson J., Zayzafoon M., Harris K.W., Selander K.S. Toll like receptor-9 agonists stimulate prostate cancer invasion in vitro. Prostate. 2007; 67:774–781. [DOI] [PubMed] [Google Scholar]
- 95. Herrmann A., Cherryholmes G., Schroeder A., Phallen J., Alizadeh D., Xin H., Wang T., Lee H., Lahtz C., Swiderski P. et al. TLR9 is critical for glioma stem cell maintenance and targeting. Cancer Res. 2014; 74:5218–5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zhang Q., Hossain D.M.S., Duttagupta P., Moreira D., Zhao X., Won H., Buettner R., Nechaev S., Majka M., Zhang B. et al. Serum-resistant CpG-STAT3 decoy for targeting survival and immune checkpoint signaling in acute myeloid leukemia. Blood. 2016; 127:1687–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zhao X., Zhang Z., Moreira D., Su Y.-L., Won H., Adamus T., Dong Z., Liang Y., Yin H.H., Swiderski P. et al. B cell lymphoma immunotherapy using TLR9-Targeted oligonucleotide STAT3 inhibitors. Mol. Ther. 2018; 26:695–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sirois C.M., Jin T., Miller A.L., Bertheloot D., Nakamura H., Horvath G.L., Mian A., Jiang J., Schrum J., Bossaller L. et al. RAGE is a nucleic acid receptor that promotes inflammatory responses to DNA. J. Exp. Med. 2013; 210:2447–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Gursel M., Gursel I., Mostowski H.S., Klinman D.M. CXCL16 influences the nature and specificity of CpG-induced immune activation. J. Immunol. 2006; 177:1575–1580. [DOI] [PubMed] [Google Scholar]
- 100. Lahoud M.H., Ahmet F., Zhang J.G., Meuter S., Policheni A.N., Kitsoulis S., Lee C.N., O’Keeffe M., Sullivan L.C., Brooks A.G. et al. DEC-205 is a cell surface receptor for CpG oligonucleotides. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:16270–16275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Baumann C.L., Aspalter I.M., Sharif O., Pichlmair A., Blüml S., Grebien F., Bruckner M., Pasierbek P., Aumayr K., Planyavsky M. et al. CD14 is a coreceptor of toll-like receptors 7 and 9. J. Exp. Med. 2010; 207:2689–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zhu P., Liu X., Treml L.S., Cancro M.P., Freedman B.D. Mechanism and regulatory function of CpG signaling via scavenger receptor B1 in primary B cells. J. Biol. Chem. 2009; 284:22878–22887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Nechaev S., Gao C., Moreira D., Swiderski P., Jozwiak A., Kowolik C.M., Zhou J., Armstrong B., Raubitschek A., Rossi J.J. et al. Intracellular processing of immunostimulatory CpG-siRNA: Toll-like receptor 9 facilitates siRNA dicing and endosomal escape. J. Control. Release. 2013; 170:307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Su Y.-L., Wang X., Mann M., Adamus T.P., Wang D., Moreira D.F., Zhang Z., Ouyang C., He X., Zhang B. et al. Myeloid cell–targeted miR-146a mimic inhibits NF-κB–driven inflammation and leukemia progression in vivo. Blood. 2020; 135:167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Boldin M.P., Taganov K.D., Rao D.S., Yang L., Zhao J.L., Kalwani M., Garcia-Flores Y., Luong M., Devrekanli A., Xu J. et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med. 2011; 208:1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zhao J.L., Rao D.S., Boldin M.P., Taganov K.D., O’Connell R.M., Baltimore D. NF- B dysregulation in microRNA-146a-deficient mice drives the development of myeloid malignancies. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:9184–9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Yu H., Kortylewski M., Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007; 7:41–51. [DOI] [PubMed] [Google Scholar]
- 108. Kortylewski M., Swiderski P., Herrmann A., Wang L., Kowolik C., Kujawski M., Lee H., Scuto A., Liu Y., Yang C. et al. In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat. Biotechnol. 2009; 27:925–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Hossain D.M.S., Pal S.K., Moreira D., Duttagupta P., Zhang Q., Won H., Jones J., D’Apuzzo M., Forman S., Kortylewski M. TLR9-Targeted STAT3 silencing abrogates immunosuppressive activity of Myeloid-Derived suppressor cells from prostate cancer patients. Clin. Cancer Res. 2015; 21:3771–3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Bian Z., Shi L., Venkataramani M., Abdelaal A.M., Culpepper C., Kidder K., Liang H., Zen K., Liu Y. Tumor conditions induce bone marrow expansion of granulocytic, but not monocytic, immunosuppressive leukocytes with increased CXCR2 expression in mice. Eur. J. Immunol. 2018; 48:532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Bian Z., Abdelaal A.M., Shi L., Liang H., Xiong L., Kidder K., Venkataramani M., Culpepper C., Zen K., Liu Y. Arginase-1 is neither constitutively expressed in nor required for myeloid-derived suppressor cell-mediated inhibition of T-cell proliferation. Eur. J. Immunol. 2018; 48:1046–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Song E., Zhu P., Lee S.-K., Chowdhury D., Kussman S., Dykxhoorn D.M., Feng Y., Palliser D., Weiner D.B., Shankar P. et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat. Biotechnol. 2005; 23:709–717. [DOI] [PubMed] [Google Scholar]
- 113. Yao Y., Sun T., Huang S., Dou S., Lin L., Chen J., Ruan J., Mao C., Yu F., Zeng M. et al. Targeted delivery of PLK1-siRNA by ScFv suppresses Her2+ breast cancer growth and metastasis. Sci. Transl. Med. 2012; 4:130ra48. [DOI] [PubMed] [Google Scholar]
- 114. Sugo T., Terada M., Oikawa T., Miyata K., Nishimura S., Kenjo E., Ogasawara-Shimizu M., Makita Y., Imaichi S., Murata S. et al. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Control. Release. 2016; 237:1–13. [DOI] [PubMed] [Google Scholar]
- 115. Xia C.-F., Zhang Y., Zhang Y., Boado R.J., Pardridge W.M. Intravenous siRNA of brain cancer with receptor targeting and avidin–biotin technology. Pharm. Res. 2007; 24:2309–2316. [DOI] [PubMed] [Google Scholar]
- 116. Ma Y., Kowolik C.M., Swiderski P.M., Kortylewski M., Yu H., Horne D.A., Jove R., Caballero O.L., Simpson A.J.G., Lee F.-T. et al. Humanized Lewis-Y specific antibody based delivery of STAT3 siRNA. ACS Chem. Biol. 2011; 6:962–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Bäumer S., Bäumer N., Appel N., Terheyden L., Fremerey J., Schelhaas S., Wardelmann E., Buchholz F., Berdel W.E., Müller-Tidow C. Antibody-mediated delivery of Anti– KRAS -siRNA in vivo overcomes therapy resistance in colon cancer. Clin. Cancer Res. 2015; 21:1383–1394. [DOI] [PubMed] [Google Scholar]
- 118. Su Y., Yu L., Liu N., Guo Z., Wang G., Zheng J., Wei M., Wang H., Yang A., Qin W. et al. PSMA specific single chain antibody-mediated targeted knockdown of Notch1 inhibits human prostate cancer cell proliferation and tumor growth. Cancer Lett. 2013; 338:282–291. [DOI] [PubMed] [Google Scholar]
- 119. Shi S.-J., Wang L.-J., Han D.-H., Wu J.-H., Jiao D., Zhang K.-L., Chen J.-W., Li Y., Yang F., Zhang J.-L. et al. Therapeutic effects of human monoclonal PSMA antibody-mediated TRIM24 siRNA delivery in PSMA-positive castration-resistant prostate cancer. Theranostics. 2019; 9:1247–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Nanna A.R., Kel’in A.V., Theile C., Pierson J.M., Voo Z.X., Garg A., Nair J.K., Maier M.A., Fitzgerald K., Rader C. Generation and validation of structurally defined antibody–siRNA conjugates. Nucleic Acids Res. 2020; 48:5281–5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Holliger P., Hudson P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005; 23:1126–1136. [DOI] [PubMed] [Google Scholar]
- 122. Stumpp M.T., Binz H.K., Amstutz P. DARPins: a new generation of protein therapeutics. Drug Discov. Today. 2008; 13:695–701. [DOI] [PubMed] [Google Scholar]
- 123. Winkler J., Martin-Killias P., Plückthun A., Zangemeister-Wittke U. EpCAM-targeted delivery of nanocomplexed siRNA to tumor cells with designed ankyrin repeat proteins. Mol. Cancer Ther. 2009; 8:2674–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Klein D., Goldberg S., Theile C.S., Dambra R., Haskell K., Kuhar E., Lin T., Parmar R., Manoharan M., Richter M. et al. Centyrin ligands for extrahepatic delivery of siRNA. Mol. Ther. 2021; 29:2053–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Zheng Y., Tai W. Insight into the siRNA transmembrane delivery—from cholesterol conjugating to tagging. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020; 12:e1606. [DOI] [PubMed] [Google Scholar]
- 126. Osborn M.F., Khvorova A. Improving siRNA delivery in vivo through lipid conjugation. Nucleic Acid Ther. 2018; 28:128–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Soutschek J., Akinc A., Bramlage B., Charisse K., Constien R., Donoghue M., Elbashir S., Geick A., Hadwiger P., Harborth J. et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004; 432:173–178. [DOI] [PubMed] [Google Scholar]
- 128. Chernikov I.V., Gladkikh D.V., Meschaninova M.I., Ven’yaminova A.G., Zenkova M.A., Vlassov V.V., Chernolovskaya E.L. Cholesterol-containing nuclease-resistant siRNA accumulates in tumors in a carrier-free mode and silences MDR1 gene. Mol. Ther. - Nucleic Acids. 2017; 6:209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Osborn M.F., Coles A.H., Golebiowski D., Echeverria D., Moazami M.P., Watts J.K., Sena-Esteves M., Khvorova A. Efficient gene silencing in brain tumors with hydrophobically modified siRNAs. Mol. Cancer Ther. 2018; 17:1251–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Segal M., Biscans A., Gilles M.-E., Anastasiadou E., De Luca R., Lim J., Khvorova A., Slack F.J. Hydrophobically modified let-7b miRNA enhances biodistribution to NSCLC and downregulates HMGA2 in vivo. Mol. Ther. - Nucleic Acids. 2020; 19:267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Wang H., Jiang Y., Peng H., Chen Y., Zhu P., Huang Y. Recent progress in microRNA delivery for cancer therapy by non-viral synthetic vectors. Adv. Drug Deliv. Rev. 2015; 81:142–160. [DOI] [PubMed] [Google Scholar]
- 132. Xiao Y., Shi K., Qu Y., Chu B., Qian Z. Engineering nanoparticles for targeted delivery of nucleic acid therapeutics in tumor. Mol. Ther. - Methods Clin. Dev. 2019; 12:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Doyle L., Wang M. Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells. 2019; 8:727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Saleh A.F., Lázaro-Ibáñez E., Forsgard M.A.M., Shatnyeva O., Osteikoetxea X., Karlsson F., Heath N., Ingelsten M., Rose J., Harris J. et al. Extracellular vesicles induce minimal hepatotoxicity and immunogenicity. Nanoscale. 2019; 11:6990–7001. [DOI] [PubMed] [Google Scholar]
- 135. Zhu X., Badawi M., Pomeroy S., Sutaria D.S., Xie Z., Baek A., Jiang J., Elgamal O.A., Mo X., Perle K.La et al. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J. Extracell. Vesicles. 2017; 6:1324730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. EL Andaloussi S., Lakhal S., Mäger I., Wood M.J.A. Exosomes for targeted siRNA delivery across biological barriers. Adv. Drug Deliv. Rev. 2013; 65:391–397. [DOI] [PubMed] [Google Scholar]
- 137. Kamerkar S., LeBleu V.S., Sugimoto H., Yang S., Ruivo C.F., Melo S.A., Lee J.J., Kalluri R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017; 546:498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Wiklander O.P.B., Brennan M.Á., Lötvall J., Breakefield X.O., EL Andaloussi S. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019; 11:eaav8521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Zheng Z., Li Z., Xu C., Guo B., Guo P. Folate-displaying exosome mediated cytosolic delivery of siRNA avoiding endosome trapping. J. Control. Release. 2019; 311–312:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Ohno S., Takanashi M., Sudo K., Ueda S., Ishikawa A., Matsuyama N., Fujita K., Mizutani T., Ohgi T., Ochiya T. et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 2013; 21:185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Wang Y., Chen X., Tian B., Liu J., Yang L., Zeng L., Chen T., Hong A., Wang X. Nucleolin-targeted extracellular vesicles as a versatile platform for biologics delivery to breast cancer. Theranostics. 2017; 7:1360–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Gong C., Tian J., Wang Z., Gao Y., Wu X., Ding X., Qiang L., Li G., Han Z., Yuan Y. et al. Functional exosome-mediated co-delivery of doxorubicin and hydrophobically modified microRNA 159 for triple-negative breast cancer therapy. J. Nanobiotechnol. 2019; 17:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Lee H.D., Koo B., Kim Y.H., Jeon O., Kim D. Exosome release of ADAM15 and the functional implications of human macrophage-derived ADAM15 exosomes. FASEB J. 2012; 26:3084–3095. [DOI] [PubMed] [Google Scholar]
- 144. Chen K., Chen X. Integrin targeted delivery of chemotherapeutics. Theranostics. 2011; 1:189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Bellavia D., Raimondo S., Calabrese G., Forte S., Cristaldi M., Patinella A., Memeo L., Manno M., Raccosta S., Diana P. et al. Interleukin 3- receptor targeted exosomes inhibit in vitro and in vivo chronic myelogenous leukemia cell growth. Theranostics. 2017; 7:1333–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Hantschel O. Structure, regulation, signaling, and targeting of Abl kinases in cancer. Genes Cancer. 2012; 3:436–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Pi F., Binzel D.W., Lee T.J., Li Z., Sun M., Rychahou P., Li H., Haque F., Wang S., Croce C.M. et al. Nanoparticle orientation to control RNA loading and ligand display on extracellular vesicles for cancer regression. Nat. Nanotechnol. 2018; 13:82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Didiot M.-C., Hall L.M., Coles A.H., Haraszti R.A., Godinho B.M.D.C., Chase K., Sapp E., Ly S., Alterman J.F., Hassler M.R. et al. Exosome-mediated delivery of hydrophobically modified siRNA for huntingtin mRNA silencing. Mol. Ther. 2016; 24:1836–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Kooijmans S.A.A., Stremersch S., Braeckmans K., de Smedt S.C., Hendrix A., Wood M.J.A., Schiffelers R.M., Raemdonck K., Vader P. Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J. Control. Release. 2013; 172:229–238. [DOI] [PubMed] [Google Scholar]
- 150. Reshke R., Taylor J.A., Savard A., Guo H., Rhym L.H., Kowalski P.S., Trung M.T., Campbell C., Little W., Anderson D.G. et al. Reduction of the therapeutic dose of silencing RNA by packaging it in extracellular vesicles via a pre-microRNA backbone. Nat. Biomed. Eng. 2020; 4:52–68. [DOI] [PubMed] [Google Scholar]
- 151. Zhang D., Lee H., Zhu Z., Minhas J.K., Jin Y. Enrichment of selective miRNAs in exosomes and delivery of exosomal miRNAs in vitro and in vivo. Am. J. Physiol. Cell. Mol. Physiol. 2017; 312:L110–L121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. O’Loughlin A.J., Mäger I., de Jong O.G., Varela M.A., Schiffelers R.M., El Andaloussi S., Wood M.J.A., Vader P. Functional delivery of lipid-conjugated siRNA by extracellular vesicles. Mol. Ther. 2017; 25:1580–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Lai R.C., Yeo R.W.Y., Padmanabhan J., Choo A., de Kleijn D.P.V., Lim S.K. Isolation and characterization of exosome from human embryonic stem cell-derived C-Myc-immortalized mesenchymal stem cells. Methods Mol. Biol. 2016; 1416:477–94. [DOI] [PubMed] [Google Scholar]
- 154. Orellana E.A., Li C., Lisevick A., Kasinski A.L. Identification and validation of microRNAs that synergize with miR-34a – a basis for combinatorial microRNA therapeutics. Cell Cycle. 2019; 18:1798–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Lundberg P., El-Andaloussi S., Sütlü T., Johansson H., Langel Ü. Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. FASEB J. 2007; 21:2664–2671. [DOI] [PubMed] [Google Scholar]
- 156. Du Rietz H., Hedlund H., Wilhelmson S., Nordenfelt P., Wittrup A. Imaging small molecule-induced endosomal escape of siRNA. Nat. Commun. 2020; 11:1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Matsuda S., Keiser K., Nair J.K., Charisse K., Manoharan R.M., Kretschmer P., Peng C.G., Kel’in A.V., Kandasamy P., Willoughby J.L.S. et al. siRNA conjugates carrying sequentially assembled trivalent N-acetylgalactosamine linked through nucleosides elicit robust gene silencing in vivo in hepatocytes. ACS Chem. Biol. 2015; 10:1181–1187. [DOI] [PubMed] [Google Scholar]
- 158. Winkler J. Extrahepatic targeting of oligonucleotides with receptor-binding non-immunoglobulin scaffold proteins. Nucleic Acid Ther. 2018; 28:137–145. [DOI] [PubMed] [Google Scholar]
- 159. Polack F.P., Thomas S.J., Kitchin N., Absalon J., Gurtman A., Lockhart S., Perez J.L., Pérez Marc G., Moreira E.D., Zerbini C. et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N. Engl. J. Med. 2020; 383:2603–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Baden L.R., El Sahly H.M., Essink B., Kotloff K., Frey S., Novak R., Diemert D., Spector S.A., Rouphael N., Creech C.B. et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 2021; 384:403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Alvarez-Erviti L., Seow Y., Yin H., Betts C., Lakhal S., Wood M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011; 29:341–345. [DOI] [PubMed] [Google Scholar]