

Abstract
Nucleotide pyrophosphatase/phosphodiesterase3 (NPP3) catalyzes the hydrolysis of extracellular nucleotides. It is expressed by immune cells and some carcinomas, e.g. of kidney and colon. Together with ecto-5′-nucleotidase (CD73), NPP3 produces immunosuppressive, cancer-promoting adenosine, and has therefore been proposed as a target for cancer therapy. Here we report on the discovery of 4-[(4-methylphthalazin-1-yl)amino]benzenesulfonamide (1) as an inhibitor of human NPP3 identified by compound library screening. Subsequent structure–activity relationship (SAR) studies led to the potent competitive NPP3 inhibitor 2-methyl-5-{4-[(4-sulfamoylphenyl)amino]phthalazin-1-yl}benzenesulfonamide (23, Ki 53.7 nM versus the natural substrate ATP). Docking studies predicted its binding pose and interactions. While 23 displayed high selectivity versus other ecto-nucleotidases, it showed ancillary inhibition of two proposed anti-cancer targets, the carbonic anhydrases CA-II (Ki 74.7 nM) and CA-IX (Ki 20.3 nM). Thus, 23 may act as multi-target anti-cancer drug. SARs for NPP3 were steeper than for CAs leading to the identification of potent dual CA-II/CA-IX (e.g.34) as well as selective CA-IX inhibitors (e.g.31).
The first potent NPP3 inhibitors were discovered, which were found to additionally block cancer-related carbonic anhydrases.
Introduction
Nucleotide pyrophosphatase/phosphodiesterase 3 (NPP3; CD203c, EC 3.1.4.1) is a transmembrane enzyme, which can also be secreted as a soluble enzyme. It belongs to the seven-membered family of nucleotide pyrophosphatases/phosphodiesterases (NPPs).1,2 Several NPPs, namely NPP1, NPP3, NPP4, and NPP5 break down nucleotides, while others (NPP2, NPP6 and NPP7) prefer lipids such as lysophosphatidylcholine or sphingomyelin as substrates.3–12 NPP1 is similar to NPP3 (50% sequence identity), while NPP4 is most closely related to NPP5 (51% sequence identity).3 NPP1 and NPP3 have a single cell membrane-spanning domain with the N-terminus facing the cytosol. Both enzymes are known to hydrolyze a variety of nucleotides including ATP, cAMP, dinucleotide polyphosphates (AP3A and AP4A), NAD+, ADP-ribose and UDP-glucose. NPP4 and NPP5 also have a single membrane-spanning domain, but their N-terminus faces the extracellular milieu.2 NPP4 and NPP5 have been reported to hydrolyze only specific nucleotides, e.g. NPP4 is known to catalyze the hydrolysis of AP3A and AP4A,5,13 while NPP5 was reported to hydrolyze NAD+.12 NPP3 is a zinc-metallohydrolase that cleaves phosphodiester bonds of a variety of substrates including nucleoside triphosphates (e.g. ATP), di-nucleotides and nucleotide sugars yielding the corresponding nucleoside monophosphates (see Fig. 1).2p-Nitrophenyl 5′-thymidine monophosphate (p-Nph-5′-TMP) can be used as a synthetic substrate of NPP3 allowing the colorimetric detection of its hydrolysis product p-nitrophenolate at basic pH value of 9 (intense yellow color, λmax at 400 nm).14–17 This substrate has been previously used for monitoring purified soluble NPP1, NPP1 expressed in cell lines, or non-specified NPPs in cells and organs.14,18–20 This method is well suitable for high-throughput screening of compound libraries to identify novel NPP3 inhibitors, but the compounds' activities have to be subsequently confirmed versus natural substrate.21
Fig. 1. Hydrolysis of various NPP3 substrates: ATP, AP4A, and UDP-glucose are natural substrates of NPP3, p-Nph-5′-TMP is an artificial substrate used for colorimetric enzyme assays. AMP can subsequently be hydrolyzed to adenosine by ecto-5′-nucleotidase (CD73). NPP3 is expressed on immune cells, e.g. activated basophils and mast cells, suppressing immune activation by ATP thereby preventing chronic allergic inflammation.22 Immune cells such as leukocytes and T-lymphocytes can co-express other ATP-hydrolyzing ectonucleotidases such as NPP1 and CD39.23,24 NPP3 is also highly expressed in the proximal region of the intestine and has been implicated in the hydrolysis of dietary nucleotides.2,25.

The enzyme has been reported to possess an immunomodulatory function in the intestine, since it controls plasmacytoid dendritic cells through regulation of intestinal extracellular ATP levels.26 Yano et al. reported an increased expression of NPP3 in human bile duct and colorectal cancers.5,27,28 Moreover, it was found to be expressed in prostate, ovarian and renal carcinomas (www.proteinatlas.org) and on neoplastic mast cells.29 In fact, high NPP3 expression in renal carcinomas was associated with an unfavourable prognosis, and an NPP3–antibody-drug conjugate (AGS16F) has entered clinical trials for the treatment of renal cell carcinoma.30 Along with ecto-5′-nucleotidase (CD73), NPP3 can convert ATP and other adenine nucleotides via AMP to the immunosuppressive, proliferation-, angiogenesis-, and metatastasis-enhancing signaling molecule adenosine.23 These effects of adenosine are mainly mediated by Gs protein-coupled A2A and Gs/Gq protein-coupled A2B adenosine receptors expressed on cancer and immune cells.31,32 Inhibition of NPP3 may thus reduce the formation of extracellular adenosine by diminishing the concentration of extracellular AMP that can be converted to adenosine. At the same time, it prevents the hydrolysis and increases the concentration of ATP, which directly promotes phagocytosis and immunogenicity of immune cells by stimulating P2 nucleotide receptors.33,34 Thus, NPP3 inhibitors have a high potential for the (immuno)therapy of cancers, in particular cancers that show high NPP3 expression.
To further investigate the (patho)physiological roles of NPP3 and its potential as a drug target, potent and selective inhibitors are required. Only few NPP3 inhibitors have been reported to date, and in most cases they additionally inhibit other NPP subtypes or nucleotide-binding proteins. Systematic structure–activity relationship (SAR) analyses of NPP3 inhibitors are still lacking. We previously discovered adenosine 5′-diphosphate-2′,3′-dialdehyde (DialADP, I) to act as a moderately potent inhibitor of human NPP3, showing a Ki value of 9.45 μM vs. p-Nph-5′-TMP as a substrate (Fig. 2).14 Two polysulfonates, Reactive blue 2 (II) and suramin (III), were found to be more potent NPP3 inhibitors, displaying Ki values of 0.710 and 0.040 μM, respectively, tested vs. the natural substrate ATP.15 However, both compounds are highly promiscuous, inhibiting several other ecto-nucleotidases as well as P2 nucleotide receptors.35–37 More recently, Iqbal and collaborators reported on several moderately potent non-nucleotide-derived, neutral, small molecule NPP3 inhibitors (see Fig. 2). These included 1,3,4-oxadiazole-2-thione derivatives (e.g.IV, Ki 1.7 μM),38 and thiooxopyrimidines (e.g.V, IC50 0.57 μM),39 both of which were characterized as non-competitive inhibitors, isoquinolines (e.g.VI, IC50 0.48 μM),40 and thiadiazolopyrimidones (e.g.VII, IC50 0.92 μM),41 which were reported to act as competitive inhibitors, and quinolines (e.g.VIII, IC50 0.36 μM) whose inhibition type has not been studied.42 All of these compounds were tested only against the artificial substrate p-Nph-5′-TMP, which could be problematic (see above), and some of them also inhibit NPP1. Moreover, only limited selectivity testing on other ectonucleotidases has been performed. We previously identified the polyoxometalate [H7P8W48O184]33− (IX) as an inhibitor of human NPP3 with a Ki value of 0.232 μM using ATP as a substrate.43 However, this inorganic cluster compound is 18-fold more potent at inhibiting human NPP1 (Ki = 0.0130 μM). Moreover, it cannot be developed as an orally applicable drug due to its multiple negative charges and high molecular weight.
Fig. 2. Structures of reported NPP3 inhibitors.

The lack of potent NPP3 inhibitors that show selectivity versus other ectonucleotidases prompted us to embark on a high-throughput screening campaign to identify scaffolds suitable for optimization. Various sub-libraries comprising commercial and proprietary compounds were screened using a colorimetric assay employing the artificial substrate p-Nph-5′-TMP. Subsequently, the main hit compound was further characterized versus the natural substrate ATP, optimized, and SARs were analyzed.
The most potent NPP3 inhibitors of the present series were further characterized with regard to their selectivity which led to the discovery of their ancillary carbonic anhydrase (CA) inhibitory activity towards the anti-cancer targets CA-II and CA-IX. Like NPPs, carbonic anhydrases (CA, EC 4.2.1.1) are zinc-metalloenzymes. They catalyze the conversion of CO2 and H2O to HCO3− and H+.44,45 Both CA-II and CA-IX play critical roles in tumor pathophysiology controlling tumor pH value and affecting further processes in the tumor microenvironment that promote cell proliferation, invasion and metastasis.46,47 The expression of CA-II and IX is strongly upregulated in hypoxic tumors including intestinal esophageal and colon cancers, gliomas, and kidney tumours44,47 reducing the pH value in the tumor tissues.
Multi-target drugs are effectively used in clinical practice. In the last decade, polypharmacology has gained increasing importance due to its advantages in the treatment of complex diseases such as cancer.48–50 Interaction of a drug with two or more targets can be highly advantageous and lead to synergistic effects.51 Thus, the optimization of NPP3 inhibitors in the present study, and the serendipitous finding of their ancillary carbonic anhydrase inhibitory activity may lead to potent multi-target drugs for personalized therapy of cancer patients with high expression of these targets, e.g. in kidney or gastrointestinal tumors.
Results and discussion
Screening of compound libraries for identification of human NPP3 inhibitors
A subset of compounds from the PharmaCenter Bonn compound library was selected for screening at NPP3 (https://www.pharmchem1.uni-bonn.de/ak-mueller/bibliothek). These included proprietary purine and pyrimidine derivatives, natural products, our own collection of approved drugs, and commercially available libraries of pharmacological tool compounds. Altogether 4200 compounds were screened in a high-throughput manner for inhibition of NPP3 using a colorimetric assay with p-Nph-5′-TMP as an artificial substrate. In this screening procedure employing test compounds at a concentration of 10 μM, a single hit was identified (defined as showing >70% inhibition) corresponding to a hit rate of only 0.02%. This hit compound (1, 4-[(4-methylphthalazin-1-yl)amino]benzenesulfonamide, Fig. 3), tested at 10 μM concentration, displayed 88% inhibition of human NPP3 activity versus p-Nph-5′-TMP as a substrate. Subsequent determination of concentration–inhibition curves resulted in a Ki value of 0.117 μM in the same assay.
Fig. 3. Hit compound 1 identified as an inhibitor of human NPP3 (Ki 0.117 ± 0.023 μM vs. p-Nph-5′-TMP).
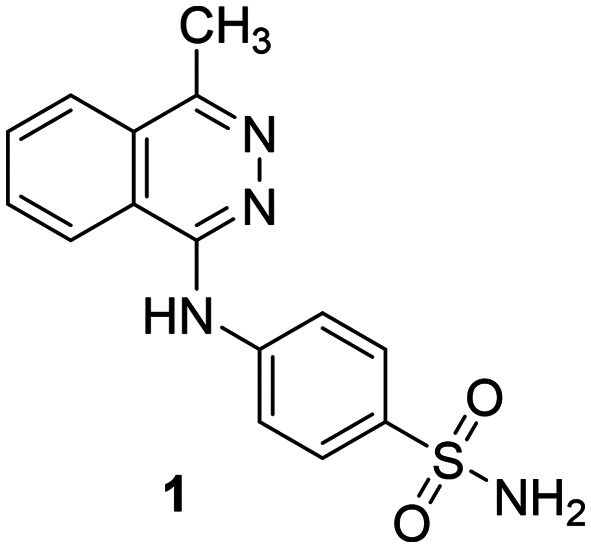
When subsequently tested against the natural substrate ATP, compound 1 displayed approximately 10-fold lower NPP3 inhibitory potency (Ki = 1.11 μM) as compared to the value determined versus the artificial substrate. Such discrepancies have previously been described for NPP1 inhibitors as well. They were attributed to the presence of an allosteric modulatory site for nucleotides (and synthetic analogs) which can affect the conformation of the substrate binding site.21 Therefore, it is important to test the inhibitors using natural substrate. We subsequently optimized this new class of NPP3 inhibitors based on studying SARs employing an assay that utilizes the natural substrate ATP. In a first step, we searched for commercially available derivatives and analogues of 1. Based on the observed SARs we synthesized a series of derivatives, which represent novel compounds not previously described in literature, to complete SARs.
Syntheses
The 4-(phthalazin-1-yl)amino-benzenesulfonamide derivatives (1 and 16–40) were commercially available, while compounds 6a–6g, 11 and 15 were synthesized as described below. The synthesis of target compounds 6a–6g was carried out as outlined in Scheme 1 with the aim to broadly study SARs of substituents on the phenyl ring directly attached to the phthalazine core structure. The compounds were prepared starting from the previously described bromo-substituted phthalazine derivative 3a,53 or the corresponding derivative 3b (see Scheme 1). Compound 3b was obtained by reaction of 1,4-dibromphthalazine (2) with 4-amino-3-methylbenzensulfonamide in the presence of potassium carbonate. Boronate precursors 5a–5c were synthesized by palladium-catalyzed borylation of the appropriate aryl bromide 4 with bis(pinacolato)diboron under inert conditions. The final products were prepared by Suzuki coupling reaction of 4-[(4-bromophthalazin-1-yl)amino]benzenesulfonamide derivative 3a or 3b with the commercially available or synthesized boronate precursors.
Scheme 1. Synthesis of phthalazine derivatives 6a–6g. Reagents and conditions: (a) K2CO3, DMF, 90 °C, overnight, yield: 48%; (b) bis(pinacolate)diboron, KOAc, PdCl2(dppf), dioxane, reflux, overnight; (c) Na2CO3, ethanol/water/toluene, Pd(PPh3)4, 80 °C, 8 h, yield: 13–40%.

Boronate precursors 10 and 14 were obtained as depicted in Scheme 2. Benzoic acid derivative 7 was converted to its acid chloride 8 followed by ammonolysis or hydrazinolysis affording amide 9, or hydrazide 12, respectively. The latter compound was treated with triethyl orthoformate to yield the corresponding oxadiazole derivative 13. Borylation of both building blocks, 9 or 13, respectively, led to the corresponding boronate precursors 10 and 14. The target phthalazine derivatives 11 and 15 were prepared via Suzuki coupling reaction of 3a and boronate precursors 10, or 14, respectively, as described in Scheme 2.
Scheme 2. Syntheses of phthalazine derivatives 11 and 15. Reagents and conditions: (a) (COCl)2, DMF, DCM, rt, overnight; (b) NH3/MeOH-DCM, rt, overnight; (c) bis(pinacolate)diboron, KOAc, PdCl2(dppf), dioxane, reflux, overnight, yield: 10 (45%), 14 (17%); (d) Na2CO3, ethanol/water/toluene, Pd(PPh3)4, 80 °C, 8 h, yield: 11 (44%), 15 (19%); (e) NH2NH2/EtOH, DCM, rt, overnight, yield: 30%; (f) triethylorthoformate, 145 °C, 11 h, yield: 65%.

The purity of the final products was determined by high performance liquid chromatography (HPLC) coupled to electrospray ionization mass spectrometry (ESI-MS) and diode array UV (DAD-UV) spectroscopy, and was found to be at least 95%.
Structure–activity relationships as inhibitors of NPP3
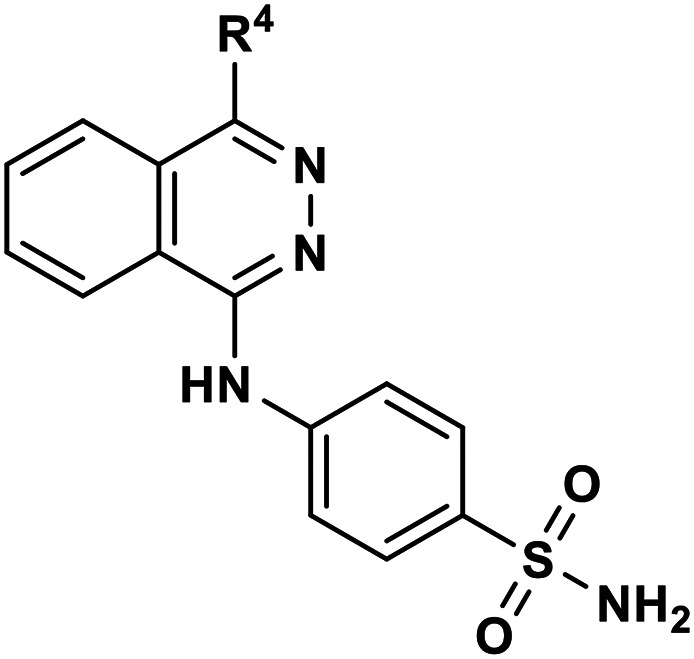
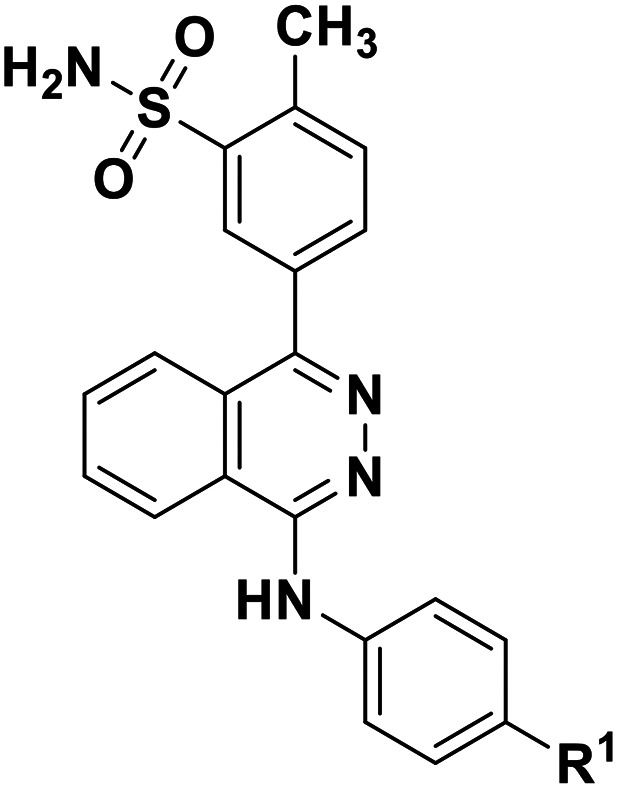
A series of 35 4-(phthalazin-1-yl)aminobenzenesulfonamide derivatives was evaluated at human NPP3 using ATP as a substrate. At first, replacement of the methyl group at the 4-position of the phthalazine scaffold by a phenyl group bearing a variety of substituents was studied (see Table 1). Replacement of methyl (compound 1) by phenyl (compound 16) led to a 10-fold reduction in inhibitory potency. The introduction of a hydroxyl group (17) or a methyl residue (6b) in the p-position of the phenyl ring slightly increased potency by 2-fold, while the introduction of a chloro substituent at that position (in 6a) decreased inhibitory potency by 4-fold. Alkylation of the hydroxyl group in the p-position with methyl, ethyl, or propargyl (compounds 18–20) led to 4–6-fold reduced activities when compared with the parent compound 17 indicating limited space in that position. The introduction of carboxamides in the p-position of the phenyl ring (21, 22) resulted in similarly low potency as observed for the ethers 18–20. However, the introduction of a sulfonamide group in the m-position of the p-methyl-phenyl-substituted derivative 6b resulting in compound 23 led to a boost in potency showing a Ki value of 53.7 nM. This compound was 21-fold more potent in inhibiting human NPP3 activity than the hit compound 1. Removal of the methyl group in the p-position of the phenyl ring (6c) or replacement of the methyl group with a chlorine atom (6d) led to 40-fold reduced activities, when compared to compound 23. Furthermore, replacement of the methyl group with a methoxy (6e) or a hydroxyl group (6f) led to 120-fold decrease in potency. A free amino function of the sulfonamide group in compound 23 was found to be essential for high inhibitory potency. The potency of compound 23 was gradually decreased upon mono- and di-methylation of the amino function (24, 25). Larger substituents like tert-butyl (26), hydroxylethyl (27), hydroxylated tert-butyl (28), pyridylmethyl (29) or furfuryl (30) attached to the amino function significantly decreased inhibitory activity. Integration of the amino function into a cyclic structure as in piperidinyl (31), N-methylpiperazinyl (32) or morpholinyl (33) derivatives also led to significantly reduced inhibitory potencies.
Inhibitory potencies of 4-(phthalazin-1-yl)aminobenzenesulfonamide derivatives with modifications of the 4-substitutent at human NPP3.
| ||
|---|---|---|
| Compd. | R4 | K i ± SEM (nM) |
| II | Reactive blue 2 (ref. 15) | 710 ± 30 |
| 1 | ---CH3 | 1110 ± 40 |
| 16 |
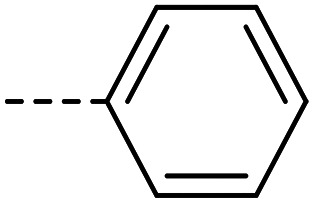
|
10 300 ± 1200 |
| 17 |
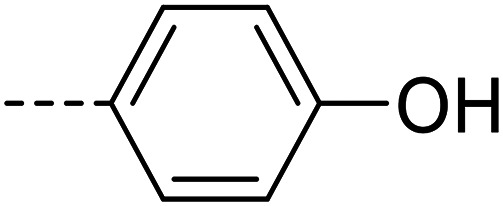
|
5320 ± 120 |
| 6a |
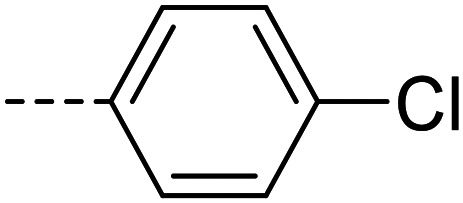
|
20 100 ± 2400 |
| 6b |
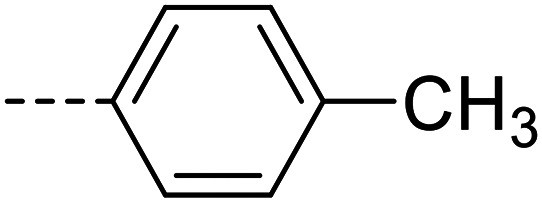
|
5090 ± 240 |
| 18 |
|
20 300 ± 300 |
| 19 |
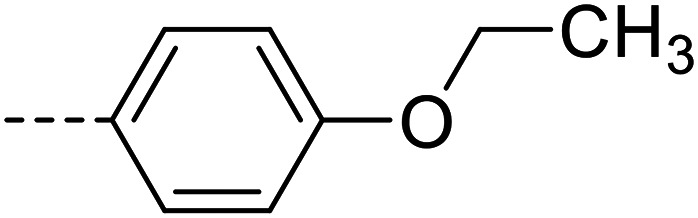
|
24 700 ± 1500 |
| 20 |
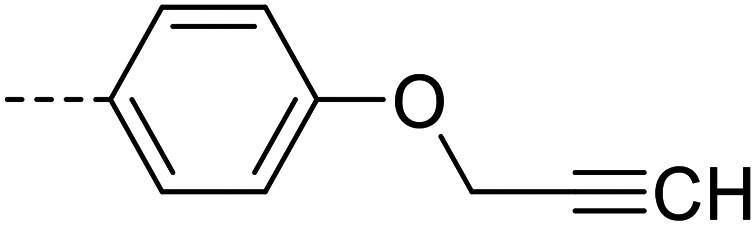
|
30 600 ± 1000 |
| 21 |
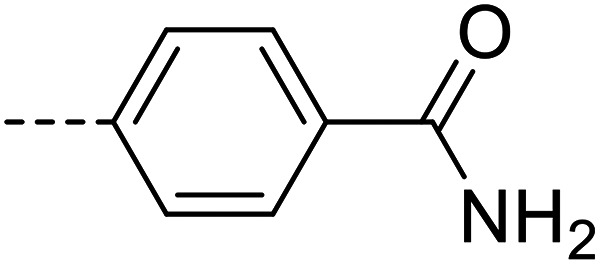
|
25 900 ± 800 |
| 22 |
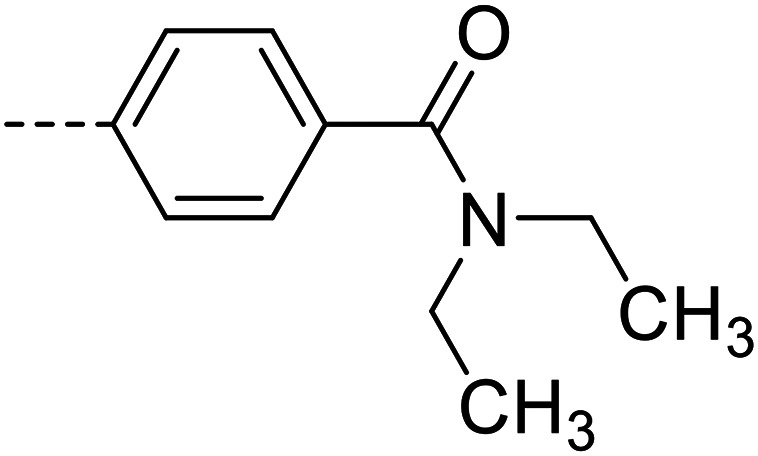
|
25 700 ± 1400 |
| 23 |
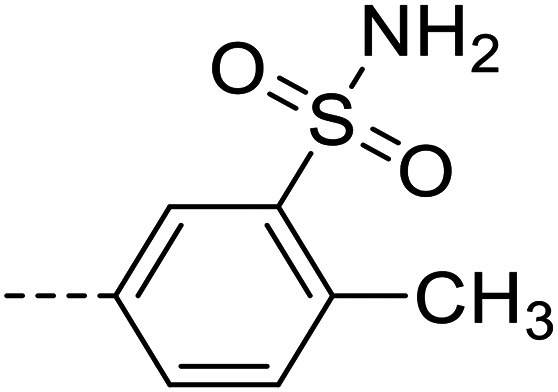
|
53.7 ± 3.1 |
| 6c |
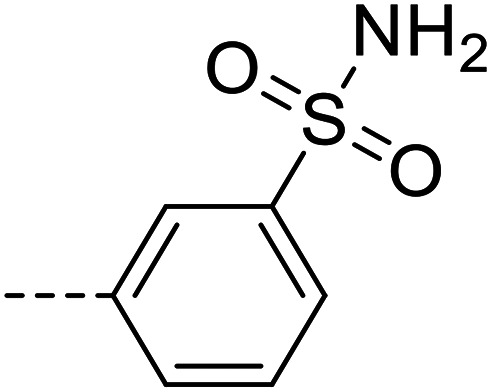
|
2120 ± 130 |
| 6d |
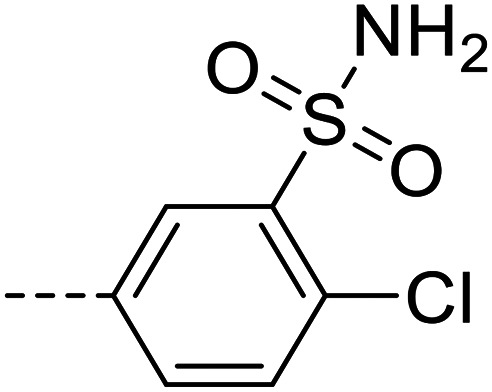
|
3340 ± 650 |
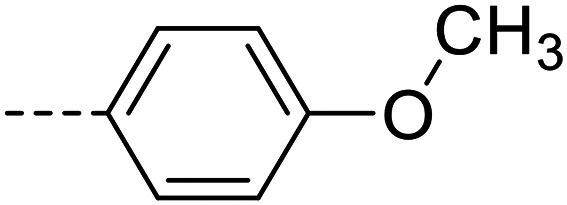
| 6e |
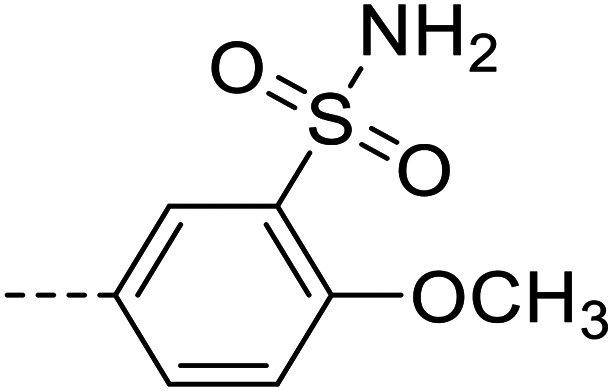
|
15 600 ± 2800 |
| 6f |
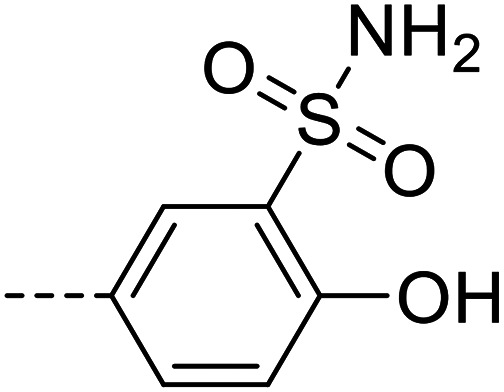
|
25 400 ± 3800 |
| 24 |
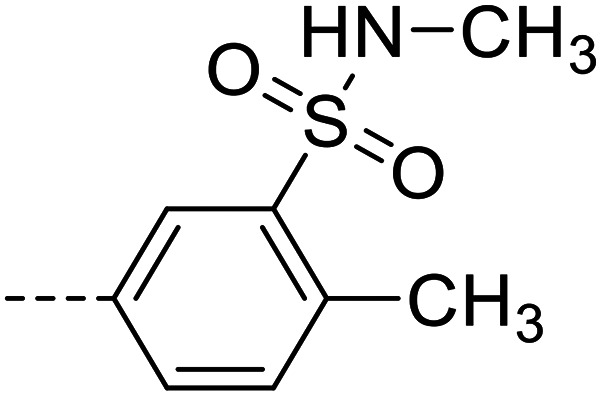
|
4090 ± 140 |
| 25 |
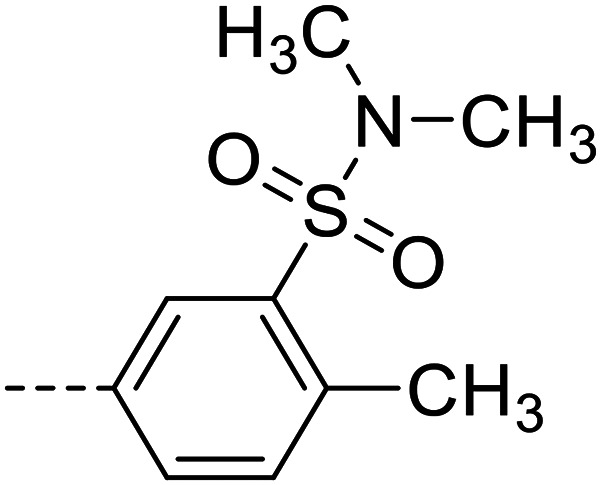
|
30 200 ± 700 |
| 26 |
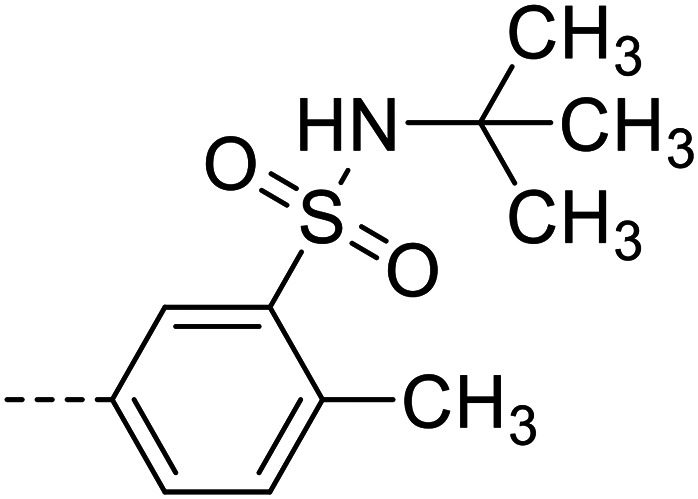
|
30 100 ± 1600 |
| 27 |
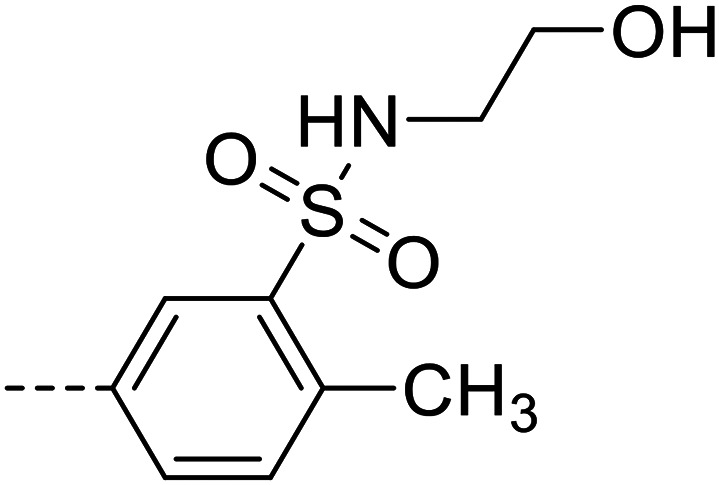
|
25 400 ± 1900 |
| 28 |
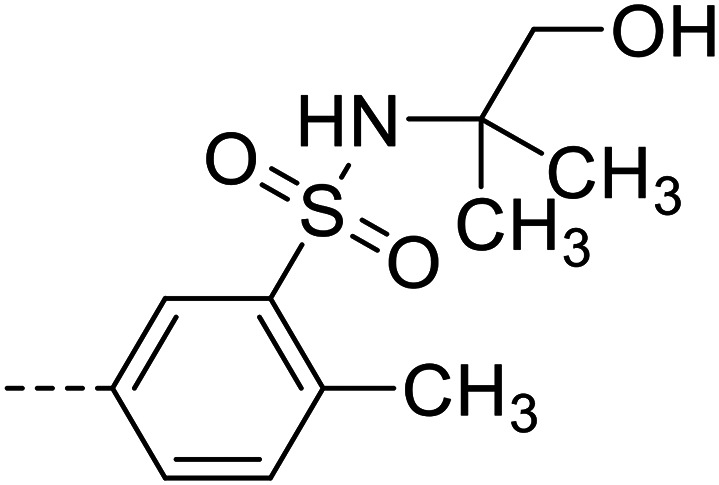
|
23 700 ± 700 |
| 29 |
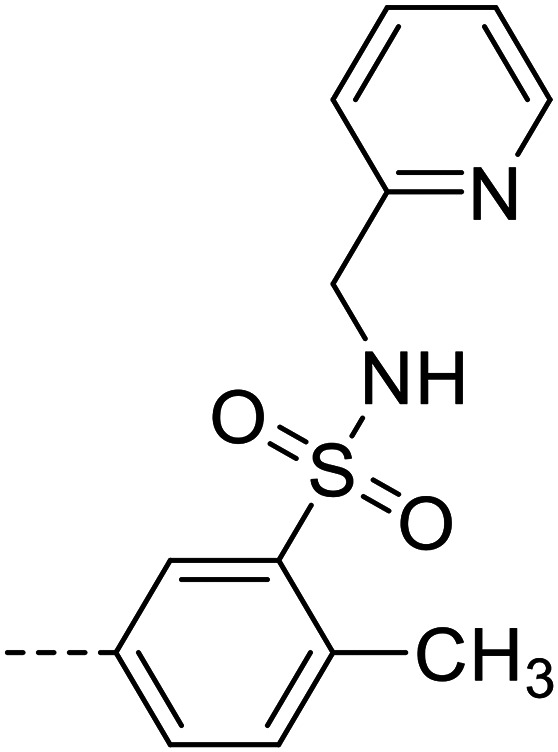
|
5570 ± 450 |
| 30 |
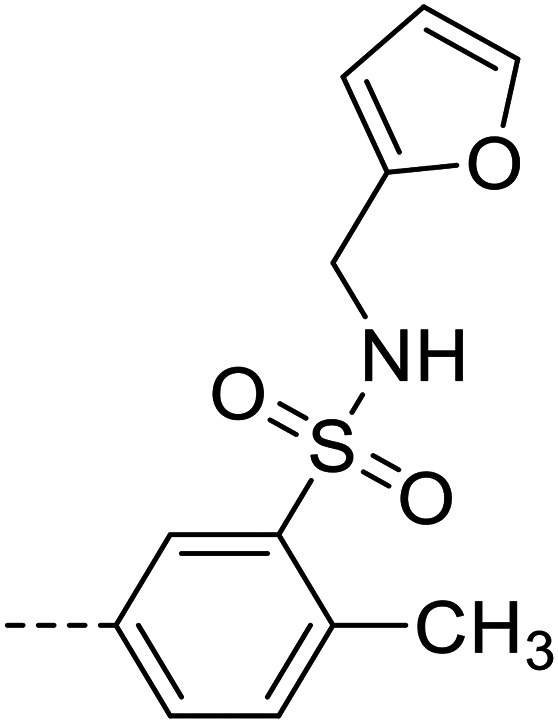
|
8880 ± 120 |
| 31 |
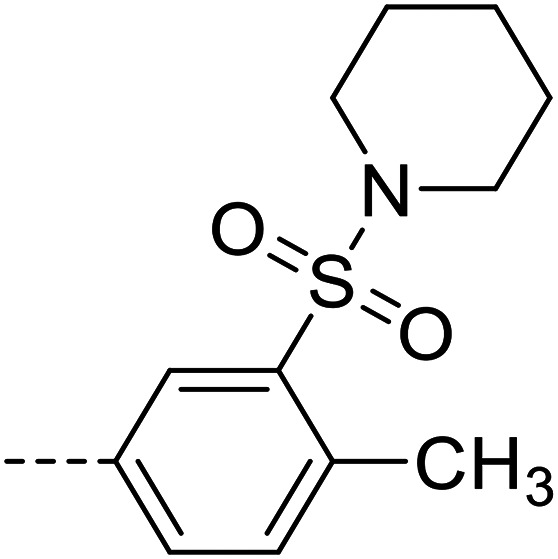
|
50 700 ± 1200 |
| 32 |
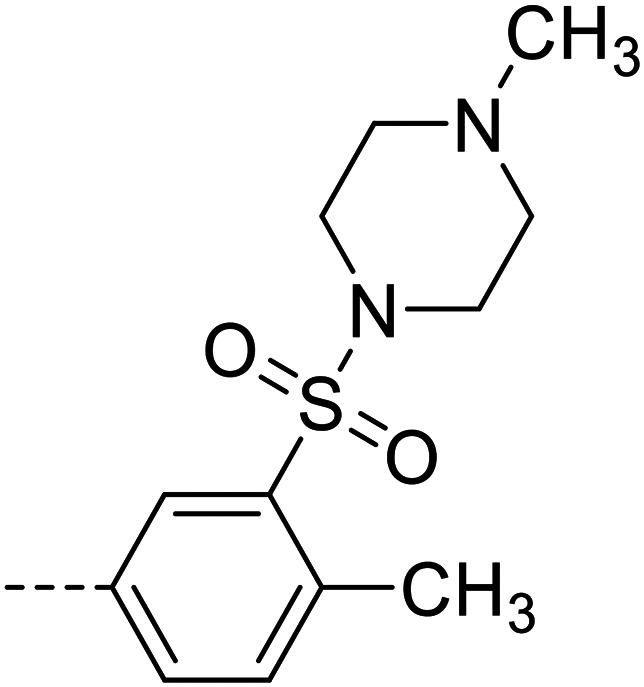
|
49 800 ± 1100 |
| 33 |
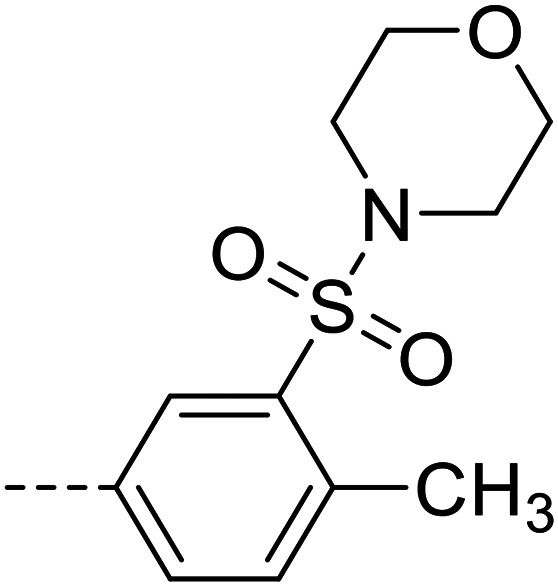
|
45 200 ± 1900 |
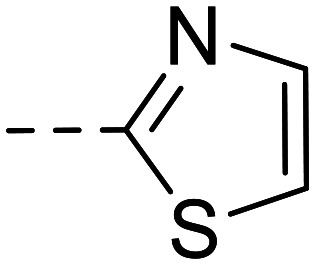
In parallel, modification of the terminal sulfonamide group of hit compound 1 was investigated (Table 2). The introduction of small heterocycles such as 2-thiazolyl (34), 3-(1,2,4)-triazolyl (35) or 5-ethyl-2-(1,3,4)-thiodiazolyl (36) at the sulfonamide group significantly decreased inhibitory potency by 20~30-fold. This implicates that a free amino function on the terminal sulfonamide group of the compounds is important for NPP3 inhibitory potency.
Inhibitory potencies of 4-(phthalazin-1-yl)amino-benzenesulfonamide derivatives at human NPP3.
| Compd. | R1 | R2 | K i ± SEM (nM) | |
|---|---|---|---|---|
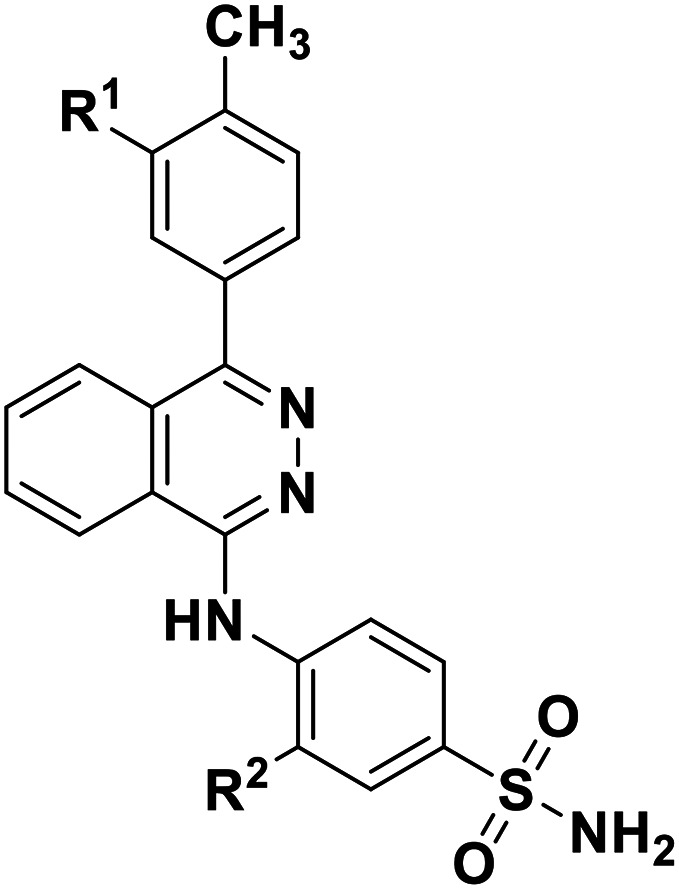
|
6g |
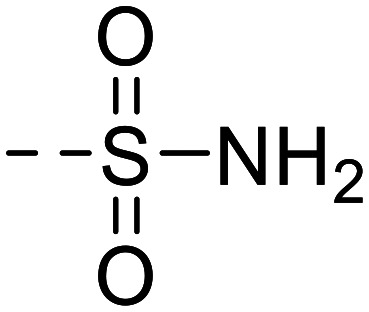
|
--CH3 | 29 900 ± 3400 |
| 11 |
|
--H | 74 200 ± 6800 | |
| 15 |
|
--H | 80 700 ± 9900 | |
|
34 |
|
20 100 ± 800 | |
| 35 |
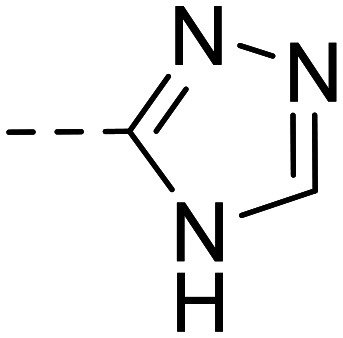
|
19 700 ± 1100 | ||
| 36 |
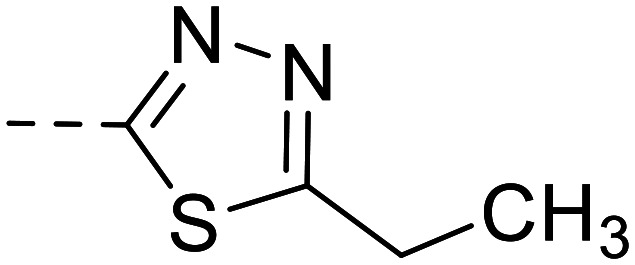
|
30 900 ± 1400 | ||
|
37 | --CH3 | 60 700 ± 3500 | |
| 38 | --Cl | 40 200 ± 2100 | ||
| 39 | ---F | 50 900 ± 3400 | ||
| 40 | --OH | 35 100 ± 1200 |
Subsequently, we modified the so far most potent structure 23, investigating derivatives in which the terminal sulfonamide group was replaced by a methyl (37), chloro (38), fluoro (39) or hydroxyl substituent (40), see the Table 2. This led to dramatically decreased inhibitory activity, indicating that the sulfonamide group in that position is indeed crucial. Next, further modifications were done on the most potent structure 23. An insertion of a methyl group into the o-position of the phenyl ring which had the sulfonamide group in the para-position (6g) led to 1500-fold decreased potency compared with compound 23. Replacement of the sulfonamide group in the m-position of the p-methylphenyl substituent at position 4 of the phthalazine scaffold by a carboxamido function (11) or an oxadiazole ring (15) again led to dramatically reduced inhibitory activities, when compared with 23. This indicates again that the sulfonamide group in the m-position of the p-methylphenyl substituent is essential for high inhibitory potency.
Concentration–inhibition curves for the best NPP3 inhibitors of the present series, hit compound 1 and the lead structure 23 are presented in Fig. 4.
Fig. 4. Concentration–inhibition curves of hit compound 1 (Ki 1110 ± 40 nM, n = 3) and the most potent NPP3 inhibitor of the present series, compound 23 (Ki 53.7 ± 3.1 nM, n = 3) at human NPP3 determined vs. ATP as a substrate.

Structure–activity relationships are summarized in Fig. 5. The key requirements were as follows:
Fig. 5. Summary of the structure–activity relationship studies of 4-(phthalazin-1-yl)amino-benzenesulfonamide derivatives as inhibitors of NPP3. A–D: Modified partial structures.

i) the methyl group in the p-position (partial structure A) seems to be important leading to a significant enhancement. The replacement of this methyl group with a chloro, hydroxyl, methoxy, ethoxy or carbamoyl residue leads to a large decrease in inhibitory potency;
ii) the sulfonamide group in the m-position is important for inhibitory activity at human NPP3 (partial structure B). Substitution of the sulfonamide by others groups or aromatic rings leads to a significant loss of inhibitory potency;
iii) the sulfonamide in the p-position is important for inhibitory activity (partial structure C). As in partial structure B, the amino function of the sulfonamide should be unsubstituted;
iv) methyl substitution in the o-position of partial structure D leads to a complete loss of inhibitory activity.
Mechanism of NPP3 inhibition
The most potent NPP3 inhibitor identified in this study, compound 23, was further investigated to determine its mechanism of inhibition for both the artificial and the natural substrate (p-Nph-5′-TMP and ATP). The Hanes-Woolf plot for both substrates revealed a competitive mechanism of inhibition by showing parallel lines (see the Fig. 6).
Fig. 6. (A) Hanes-Woolf plot of human NPP3 inhibition by compound 23; [S], concentration of substrate p-Nph-5′-TMP in μM; concentration of 23:  , 0 μM;
, 0 μM;  , 0.5 μM;
, 0.5 μM;  , 1.0 μM. (B) Hanes-Woolf plot of human NPP3 inhibition by compound 23; [S], concentration of substrate ATP in μM; concentration of 23:
, 1.0 μM. (B) Hanes-Woolf plot of human NPP3 inhibition by compound 23; [S], concentration of substrate ATP in μM; concentration of 23:  , 0 μM;
, 0 μM;  , 0.15 μM;
, 0.15 μM;  , 0.6 μM.
, 0.6 μM.

Molecular modeling studies
Recently, the complex between human NPP3 and the ATP analog, α,β-methylene-ATP (AMPCPP), which cannot be cleaved by the enzyme and acts as an inhibitor, has been obtained with high resolution of 1.9 Å.54 This crystal structure provided insights into the interactions of AMPCPP with the amino acid residues and the two zinc ions in the substrate binding site on an atomic level. As illustrated in Fig. 7A and D, AMPCPP binds primarily through the α-phosph(on)ate group to the two zinc ions in the binding site interacting with the amino acids of the enzyme and water molecules (not shown) of the enzyme. The nucleobase adenine is bound between the two aromatic residues F206 and Y289 through a strong T-shaped and parallel π-stacking interactions.
Fig. 7. Binding poses of inhibitors AMPCPP, 1 and 23 in the substrate binding site of human NPP3. A. Binding pose of AMPCPP (green) observed in the crystal structure (PBD ID: 6C02). B. Putative binding pose of 1 (magenta), and C. proposed binding mode of 23 (cyan). D, E, and F. Binding poses of AMPCP, 1 and 23 in the substrate binding pocket of human NPP3 shown in surface representation. The important amino acid residues (gray) in the binding pocket are shown as stick models and the zinc ions in the binding site are represented as gray spheres. The oxygen atoms are colored in red, nitrogen atoms in blue, phosphorus atoms in orange, and the interactions are shown as red dotted line.

To rationalize the enzyme-inhibitor interactions of the hit compound 4-[(4-methylphthalazin-1-yl)amino]benzenesulfonamide (1) and the most potent derivative 23, the competitive inhibitors were docked into the AMPCPP binding site of human NPP3. As shown in Fig. 7B and E, docking studies indicated that compound 1 occupies the same binding site as AMPCPP. The sulfonamide group is bound between the two zinc ions and the negatively charged amino acid residues D167 and D325 which are coordinating the zinc ions. Additionally, the sulfonamide group is predicted to interact with the side chain of N226 and the main chain of A204. The phthalazine group is stacked between F206 and Y289, similar to the adenine ring of AMPCPP, forming π-stacking interactions. The amino group (NH2) which connects the phthalazine core and the benzenesulfonamide residue possibly forms a hydrogen bond interaction with the hydroxyl group of Y289. The methyl group at position 4 is predicted to be positioned towards a subpocket formed by the amino acid residues F206, F271, W272, P273, G274 and Y289. Replacing the methyl substituent by a phenyl residue or other aromatic rings such as hydroxybenzene, chlorobenzene, or toluene reduced the inhibitory potency significantly (see Table 1). However, substitution with a p-methyl-m-sulfonamidophenyl residue in 23 led to a large increase in potency (Ki 53.7 nM). Docking of the competitive inhibitor 23 into the substrate binding site of human NPP3 shown in Fig. 7C and F revealed a flipped binding pose in comparison to the predicted binding pose of 1. The m-sulfonamido substitutent on the p-methylphenyl (p-toluyl) residue in 23 is observed to be bound between the two zinc ions interacting with the amino acids coordinating the zinc ions according to our docking studies. In comparison to AMPCPP, the aromatic ring of the methylbenzenesulfonamide residue is likely placed in the same location as the ribose ring of AMPCPP, and the methyl group may occupy a binding cavity where a water molecule is located in the X-ray structure connecting the ribose ring with D325, one of the amino acid residues coordinating the zinc ions (see ESI† Fig. S1). The methyl group of the methylbenzenesulfonamide residue probably directs the orientation of the sulfonamide towards the two zinc ions. Compared to 23, this kind of positioning and interaction with the amino acids in the substrate binding pocket is missing for hit compound 1. However, as visualized in Fig. 7C and F, the flat aromatic phthalazine heterobicycle of 23 is also predicted to be stacked between F205 and Y289 forming a strong hydrophobic π-stacking interaction like in the docked pose of inhibitor 1. The aniline NH function, by which the benzenesulfonamide is connected to the phthalazine core, probably forms a hydrogen bond interaction with Q244, a unique residue found in NPP3 but not in the other NPP subtypes, e.g. NPP1, NPP2 and NPP3 (see sequence alignment provided in ESI† Fig. S2). The sulfonamido group of the benzenesulfonamide residue possibly forms strong interaction with the peptide chain (both keto and amino group) of Y289. This is supported by the determined biological activities of those compounds, in which the sulfonamide group is replaced by longer residues (24–33) or smaller substituents (37–40). Those are much less potent showing the importance of sulfonamide interaction within the binding site.
Selectivity versus different ectonucleotidases
Subsequently, we evaluated the selectivity of the most potent NPP3 inhibitor 23versus other members of the human ecto-nucleotidase family, including NTPDases1–3 (CD39), NPP1 and NPP2, tissue-nonspecific alkaline phosphatase (TNAP), and ecto-5′-nucleotidase (eN, CD73), see Fig. 8. Our data show that 23 does not efficiently block any of the other human ecto-nucleotidases, with selectivities close to or even exceeding 3 orders of magnitude.
Fig. 8. Potency of compound 23 at human ecto-nucleotidases: nucleoside triphosphate diphosphohydrolases1–3 (NTPDases1–3), nucleotide pyrophosphastases/phosphodiesterases 1 and 2 (NPP1; NPP2), tissue-nonspecific alkaline phosphatase (TNAP) and ecto-5′-nucleotidase (eN, CD73). pKi values are given as means ± SEM.

Additional targets: carbonic anhydrases
Despite the high NPP3-selectivity of compound 23versus an array of closely and also more distantly related ectonucleotidases, we next performed a computer search for identifying possible additional targets. This resulted in the identification of carbonic anhydrases (CAs), which are zinc-metallohydrolases like NPP3.53,55 These enzymes were proposed as drug targets for hypoxic solid tumors due to their importance in controlling the pH value of the tumor cells. The enzymes modulate hydrogencarbonate and proton concentrations thereby adjusting the pH value that is optimal for cell survival and proliferation. Many structurally diverse sulfonamides are known to interact with CAs,55 and in fact, some of the compounds identified in the present study as NPP3 inhibitors, have previously been found to inhibit CAs.53 This opens up new perspectives since both CA subtypes have been proposed as novel drug targets for anti-cancer therapy. CA-II (cytosolic) and CA-IX (a transmembrane protein), are known to be unfavorable prognostic markers in renal cancer, just like NPP3. These CAs also play a role in several cancer types for which NPP3 inhibitors may be effective, e.g. gastrointestinal, liver and pancreatic cancer. In fact, a CA-IX inhibitor, namely 4-fluorophenylureidobenzenesulfonamide (SLC-0111), is currently evaluated in clinical trials for the treatment of solid tumors.56
Consequently, we additionally tested hit compound 1 and the majority of its derivatives as inhibitors of human carbonic anhydrases CA-II and CA-IX, the only CA isoenzymes that have potential as anti-cancer drug targets55 (see Fig. 9 and ESI† Tables S1 and S2). Four of these compounds had previously been evaluated by Gao et al. as CA inhibitors.53 They were now re-evaluated, along with the series of new phthalazine derivatives, in the laboratory of Supuran who is a leading medicinal chemist in the field of CAs. The current data obtained in Supuran's laboratory confirmed the CA inhibitory potency of this class of compounds, although the determined values differed to some extent (for comparison see ESI† Tables S1 and S2).
Fig. 9. Potencies (pKi values) of inhibitors at human NPP3, human CA-II and human CA-IX (# indicates a pKi value of <4).

Hit compound 1 as well as the most potent NPP3 inhibitor of the present series, 23, also potently inhibited CA-II and CA-IX. While 1 was about equipotent at all three targets (Ki, NPP3: 1110 nM, CA-II: 948 nM, CA-IX: 1060 nM), 23 showed a very slight preference for CA-IX (Ki, NPP3: 53.7 nM, CA-II: 74.7 nM, CA-IX: 20.3 nM). Most of the other investigated compounds were more potent inhibitors of CA-II and particularly CA-IX. Different SARs were observed at NPP3 as compared to the CAs, and also between the two CA isoenzymes. 4-Phenyl-substitution of the phthalazine core led to potent CA-II and CA-IX inhibitors, especially if an additional para-substituent was present as e.g. in 19 (p-ethoxyphenyl, Ki, NPP3: 24700 nM, CA-II: 285 nM, CA-IX: 19.5 nM). In contrast to NPP3, CAs did not require a sulfonamide residue in the m-position of the 4-phenyl ring for high potency, although it was well tolerated (see e.g.23). The sulfonamide could be substituted, e.g. with methyl (24, Ki NPP3: 4090 nM, CA-II: 72.7 nM, CA-IX: 15.0 nM). CA-IX tolearted larger substituents, and the p-methyl-m-piperidinylsulfonyl derivative 31 was the most selective, potent CA-IX inhibitor of the present series (Ki, NPP3: 49800 nM, CA-II: 941 nM, CA-IX: 27.1 nM; x-fold selectivity). SARs regarding the p-sulfonamidoaniline moiety in the 3-position of the phthalazine core also differed significantly for NPP3 as compared to the CAs. The binding site of the CAs appeared to be much more flexible than that of NPP3. While a sulfonamide function was required for high potency at NPP3, CA-II and CA-IX tolerated its replacement by a 2-thiazolyl group (see e.g.34, Ki NPP3: 20100 nM, CA-II: 73.7 nM, CA-IX: 19.7 nM). In fact, 34 can be envisaged as a new dual CA-II/CA-IX inhibitor which was discovered in the present study (see Fig. 9).
Based on published SARs and molecular modeling studies of CA-IX inhibitors,44,53 it can be assumed that a sulfonamide function is essential for high affinity interaction with the enzyme. Our current results may indicate that the phthalazine derivatives presented in this study may display alternative binding modes depending on the location of the sulfonamide functions. Compounds with only a single sulfonamide present are still highly potent at CAs, but not at NPP3, because they can probably bind in a flipped mode due to the high flexibility of the investigated CAs, especially CA-IX.
Combination therapy aiming at multiple targets is commonly applied in the treatment of cancer to improve efficacy by cooperative or synergistic effects of the drugs, to diminish systemic toxicity due to the dose reduction of each drug, and also to prevent the development of drug resistance because of different mechanisms of drug action.57,58 All three enzymes, NPP3, CA-II and CA-IX, targeted by the developed inhibitors show an overlapping expression profile.30,52 For example, they are overexpressed in many renal cancers, and each of these enzymes represents an unfavorable prognostic marker for renal cancer patients. Thus, multi-target NPP3/CA-II/CA-IX inhibitors represent most promising novel anti-cancer drugs, and a compound like 23 which inhibits all three targets with similar affinity, could lead to high efficacy in cancer therapy, by itself or in combination with other anti-cancer drugs.
Conclusion
In conclusion, we identified 4-(phthalazin-1-yl)aminobenzenesulfonamide derivatives as a new class of potent competitive NPP3 inhibitors. 2-Methyl-5-{4-[(4-sulfamoylphenyl)amino]phthalazin-1-yl}benzenesulfonamide (23, Ki = 53.7 nM determined versus the natural substrate ATP) belongs to the most potent NPP3 inhibitors described to date. In contrast to other NPP3 antagonists, it shows high selectivity versus the other members of the ectonucleotidase family of enzymes. Hit compound 1 and optimized NPP3 inhibitor 23 additionally inhibit a phylogenetically unrelated family of zinc-metallohydrolases, the carbonic anhydrases CA-II and -IX. This is a most fortunate coincidence, partly attributed to the high promiscuity of CAs towards sulfonamides, since all three enzymes can be upregulated in several types of cancers, e.g. renal and gastrointestinal cancers, and their simultaneous inhibition is predicted to result in synergistic effects. Further in vivo studies with this class of multi-target drugs are warranted.
Experimental section
Materials
Adenosine, adenosine 5′-monophosphate (AMP), adenosine 5′-diphosphate (ADP), adenosine 5′-triphosphate (ATP), 4-amino-2,3,-dimethyl-1-phenyl-3-pyrazolin-5-one (4-aminoantipyrine), calcium chloride, choline oxidase, 3-(N-ethyl-3-methylanilino)-2-hydroxypropanesulfonic acid (TOOS), magnesium chloride, p-nitrophenol, p-nitrophenyl phosphate, p-nitrophenyl 5′-thymidine monophosphate (p-Nph-5′-TMP), 1-oleoyl-sn-glycero-3-phosphocholine (LPC (18 : 1)), peroxidase, sodium hydroxide, sodium tetraborate, uridine, uridine 5′-triphosphate (UTP), and zinc chloride were obtained from Sigma (Steinheim, Germany). 2-(N-Cyclohexylamino)ethanesulfonic acid (CHES), 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), sodium dodecyl sulfate (SDS) and tris-(hydroxymethyl)aminomethane (Tris) were from Applichem (Darmstadt, Germany). Disodium hydrogenphosphate was purchased from Carl Roth (Karlsruhe, Germany). Commercially available 4-(phthalazin-1-yl)amino-benzenesulfonamide derivatives (1, and 16–40) were purchased from ChemBridge Corporation (San-Diego, USA), or Vitas-M Laboratory, Ltd. (Apeldoorn, The Netherlands). Compounds 6a-6g, 11 and 15 were synthesized as described below. Human recombinant soluble NTPDase1 and 2, and human recombinant soluble ecto-5′-nucleotidase (eN), expressed in Chinese hamster ovary (CHO) cells, were obtained from R&D Systems GmbH (Wiesbaden, Germany). Human recombinant soluble NTPDase3 and human recombinant soluble tissue non-specific alkaline phosphatase (TNAP), expressed in NS0 cells from murine myeloma, were also obtained from R&D Systems GmbH. Human recombinant soluble NPP1–3, expressed in Sf9 insect cells, were prepared in our laboratory as previously described.43
Inhibitor screening
Commercial and proprietary compound libraries (4200 compounds consisting of purines, pyrimidines, approved drugs and natural products) were screened at a concentration of 10 μM. The assays were performed at 37 °C in a total volume of 100 μl in clear 96-well plates. The reaction mixture contained 1 mM MgCl2, 2 mM CaCl2, 10 mM 2-(N-cyclohexylamino)ethanesulfonic acid (CHES) buffer (pH 9.0), 400 μM p-Nph-5′-TMP, and 10 μM of test compound dissolved in 10% DMSO. The enzyme reactions were initiated by the addition of 2.1 μg of human NPP3, incubated for 30 min at 37 °C, and subsequently terminated by the addition of 20 μl of 1.0 N aqueous NaOH solution. The amounts of p-nitrophenolate liberated were measured at 400 nm using a plate reader (PHERAstar FS, BMG Labtech Germany).
General procedures
Starting materials, reagents and solvents were used as purchased from ABCR, Alfa Aesar, Sigma-Aldrich, Activate Scientific or Fluorochem. The progress of the reactions was monitored by thin-layer chromatography (TLC, Merck, 0,2 mm silica gel 60 F254) followed by analytical LC-MS. Column chromatography was performed on silica gel, 0.060–0.200 mm, pore diameter ca. 6 nm. All synthesized compounds were finally dried in vacuum at 8–12 Pa (0.08–0.12 mbar). 1H and 13C NMR data were collected either on a Bruker Avance 500 MHz NMR spectrometer at 500 MHz (1H) or 126 MHz (13C) or on a Bruker Ascend 600 MHz NMR spectrometer at 600 MHz (1H) or 151 MHz (13C). DMSO-d6 was employed as a solvent at 303 K, unless otherwise noted. Chemical shifts are reported in parts per million relative to the deuterated solvent, that is, DMSO, δ (1H) 2.50 ppm, δ (13C) 39.52 ppm. Coupling constants J are given in Hertz, and spin multiplicities are given as: singlet (s), doublet (d), doublet of doublet (dd), triplet (t), quartet (q), multiplet (m), and broad singlet (br s). The purities of isolated final products were determined by HPLC coupled to a diode array detector (DAD) measuring UV absorption from 200 to 950 nm, and an electrospray ionization (ESI) mass spectrometer (Applied Biosystems API 2000 LCMS/MS, HPLC Agilent 1100) using a Phenomenex Luna 3 μ C18 column (50 mm × 2.00 mm). The compounds were dissolved at a concentration of 1.0 mg mL−1 in acetonitrile containing 2 mM ammonium acetate. Then, 10 μL of the sample were injected into an HPLC column, and elution was performed with a gradient of water/acetonitrile (containing 2 mM ammonium acetate) from 90:10 to 0:100 for 20 min at a flow rate of 300 μL min−1, starting the gradient after 10 min. The purity of the compounds was in all cases ≥95% (for spectra of important final products see ESI†). Compounds 2 and 3a were synthesized as previously described.53
Synthesis of intermediates
Synthesis of 4-[(4-bromophthalazin-1-yl)amino]-3-methylbenzenesulfonamide (3b)
Potassium carbonate (0.42 g, 3.00 mmol) was added to a mixture of 1,4-dibromophthalazine (0.43 g, 1.50 mmol) and 4-amino-3-methylbenzenesulfonamide (0.28 g, 1.50 mmol) in 20 mL DMF. The mixture was heated to 90 °C overnight. Then, the reaction mixture was distilled under reduced pressure to remove most of the DMF. The residue was extracted three times in a separation funnel with water (50 mL each) and dichloromethane (50 mL each), and the collected organic layers were dried over MgSO4 and evaporated under reduced pressure. Purification was performed by flash chromatography on silica gel (3 : 2 petroleum ether 40–60 °C/ethyl acetate), The product was isolated as a white powder (0.28 g, 48% yield). LC-MS (m/z): [M + H]+ calcd. for C15H13BrN4O2S 394.26, found 395.2. Purity by HPLC-UV (254 nm)-ESI-MS: 95.1%.
Synthesis of 5-bromo-2-methylbenzohydrazide (12)
5-Bromo-2-methylbenzoic acid (7, 2.00 g, 9.30 mmol) was suspended in 27 mL of dichloromethane. To the suspension oxalyl chloride (0.90 mL, 10.31 mmol) and 0.03 mL N,N-dimethylformamide were added. The mixture was stirred overnight at room temperature and became a clear solution. The solvent was evaporated under reduced pressure to yield 5-bromo-2-methylbenzoyl chloride (8). A solution of 8 (9.30 mmol) in 8.4 mL of dichloromethane was added dropwise over 30 min to a solution of hydrazine monohydrate (2.10 mL, 42 mmol) in absolute ethanol (10 mL) at 0 °C. The reaction mixture was then allowed to warm up to room temperature, and stirring was maintained overnight. The resulting precipitate was filtered off and thoroughly washed with ethanol/water (1 : 3, 50 mL). The product was isolated as a white powder (0.64 g, 30% yield). 1H NMR (500 MHz, DMSO-d6, 343 K): δ 2.28 (s, 3H, CH3), 4.44 (s, 2H, NH2), 7.21 (d, J = 8.2 Hz, 1H, Ar–H), 7.42 (d, J = 2.2 Hz, 1H, Ar–H), 7.51 (dd, J = 8.2, 2.2 Hz, 1H, Ar–H), 9.48 (s, 1H, NH). 13C NMR (126 MHz, DMSO-d6, 343 K): δ 18.73, 118.05, 129.71, 131.97, 132.55, 135.06, 137.66, 166.76. LC-MS (m/z): [M + H]+ calcd. for C8H9BrN2O 227.99, found 229.1. Purity by HPLC-UV (254 nm)-ESI-MS: 95.3%.
Synthesis of 2-(5-bromo-2methylphenyl)-1,3,4-oxadiazole (13)
A mixture of compound 12 (0.10 g, 0.44 mmol) and triethyl orthoformate (0.93 mL) was heated to 145 °C in a closed vial for 11 h. The resulting mixture was subsequently distilled under reduced pressure and then dried in vacuum (0.2 mm Hg) at 80 °C to afford the desired product (0.07 g, 65% yield). 1H NMR (500 MHz, DMSO-d6, 343 K): δ 1.79 (s, 3H, CH3), 7.42–7.44 (m, 1H, Ar–H), 7.70–7.72 (m, 1H, Ar–H), 8.03 (d, J = 2.2 Hz, 1H, Ar–H), 9.38 (s, 1H, Ar–H). 13C NMR (126 MHz, DMSO-d6, 343 K): δ 20.92, 119.01, 124.68, 131.07, 134.00, 134.20, 137.24, 154.49, 162.71.
General procedure for the synthesis of boronates (5a–c, 10, and 14)
The bromo-substituted precursor 4, 9, or 13 (1 equiv.) was mixed with bis(pinacolate)diboron (1.5 equiv.) and potassium acetate (5 equiv.) in dioxane (10 mL mmol−1 of precursor) under argon. PdCl2(dppf) (40 mg mmol−1 of precursor) was added. After bubbling argon through the solution for 15 min, the reaction mixture was refluxed overnight under argon. The resulting mixture was cooled to rt and filtered through Celite. The filtrate was distilled under reduced pressure, and the remaining residue was purified by column chromatography on silica gel to afford the corresponding boronate (5a–c, 10, or 14).
Synthesis of 2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolane-2-yl)benzamide (10)
The reaction was performed with 5-bromo-2-methylbenzamide 9 (0.15 g, 0.70 mmol). Purification by flash chromatography (99 : 1 dichloromethane/methanol) afforded 10 as a white powder (0.08 g, 45% yield). LC-MS (m/z): [M + H]+ calcd. for C14H20BNO3 261.15, found 262.2. Purity by HPLC-UV (254 nm)-ESI-MS: 97.3%.
Synthesis of 2-[2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolane-2-yl)phenyl]-1,3,4-oxadiazole (14)
The reaction was performed with 2-(5-bromo-2-methylphenyl)-1,3,4-oxadiazole (13, 0.48 g, 2 mmol). Purification was achieved by flash chromatography (99 : 1 dichloromethane/methanol) affording 14 as a white powder (0.10 g, 17% yield). 1H NMR (500 MHz, DMSO-d6, 343 K): δ 1.31 (s, 12H, 4 CH3), 2.66 (s, 3H, CH3), 7.48 (d, J = 7.6 Hz, 1H, Ar–H), 7.76 (dd, J = 7.6, 1.3 Hz, 1H, Ar–H), 8.19 (d, J = 1.3 Hz, 1H, Ar–H), 9.33 (s, 1H, Ar–H). 13C NMR (126 MHz, DMSO-d6, 343 K): δ 21.59, 24.44, 24.62, 24.78, 24.00, 73.47, 82.78, 83.96, 122.18, 137.02, 141.03, 154,04, 163.57.
Synthesis of 4-(phthalazin-1-yl)amino-benzenesulfonamide derivatives
General procedure for the synthesis of the final products (6a–6g, 11 and 15)
The appropriate 4-[(4-bromophthalazin-1-yl)amino]benzenesulfonamide derivative 3a or 3b (0.26 mmol), the appropriate boronate precursor (0.26 mmol) and sodium carbonate (0.056 g, 0.52 mmol) were dissolved in a mixture of ethanol (1.5 mL), water (1.5 mL) and toluene (5 mL) under an argon atmosphere. Then, tetrakis(triphenlyphosphine)palladium (0.02 g) was added and the reaction mixture was stirred under an argon atmosphere for 8 h at 80 °C. The solventwas subsequently distilled off under reduced pressure, and the residue was extracted with ethyl acetate (3 × 50 mL). The combined organic layers were dried over anhydrous MgSO4 and concentrated under reduced pressure. Purification was performed by flash chromatography on silica gel to afford the desired products.
4-((4-(4-Chlorophenyl)phthalazin-1-yl)amino)benzenesulfonamide (6a)
Purification was performed by column chromatography on silica gel (3 : 2 petroleum ether 40–60 °C/ethyl acetate). The product was isolated as a white powder (0.03 g, 32% yield). M.p.: 232–233 °C. 1H NMR (500 MHz, DMSO-d6, 343 K): δ 5.88 (s, 2H, NH2), 6.59–6.61 (m, 2H, Ar–H), 7.66–7.70 (m, 7H, Ar–H), 7.91–7.96 (m, 2H, Ar–H), 8.44 (dd, J = 8.0, 1.2 Hz, 1H), 12.8 (s, 1H, NH). 13C NMR (126 MHz, DMSO-d6, 343 K): δ 112.49, 126.20, 126.46, 126.54, 127.04, 127.86, 128.02, 128.64, 131.31, 132.84, 132.86, 134.34, 134.65, 148.95, 149.17, 152.49. LC-MS (m/z): [M + H]+ calcd. for C20H15ClN4O2S 410.06, found 411.2. Purity by HPLC-UV (254 nm)-ESI-MS: 95.2%.
4-((4-(p-Tolyl)phthalazin-1-yl)amino)benzenesulfonamide (6b)
Purification was performed by column chromatography on silica gel (3 : 2 petroleum ether 40–60 °C/ethyl acetate). The product was isolated as a white powder (0.037 g, 37% yield). M.p.: 273–275 °C. 1H NMR (500 MHz, DMSO-d6, 343 K): δ 2.17 (s, 3H, CH3), 5.97 (s, 2H, NH2), 6.70 (d, J = 8.7 Hz, 2H, Ar–H), 7.46–7.47 (m, 2H, Ar–H), 7.57–7.59 (m, 2H, Ar–H), 7.78–7.81 (m, 3H, Ar–H), 7.97–8.10 (m, 2H, Ar–H), 8.50–8.60 (m, 1H, Ar–H), 12.84 (s, 1H, NH). 13C NMR (126 MHz, DMSO-d6, 343 K): δ 20.87, 112.51, 126.15, 126.45, 126.71, 127.21, 127.97, 128.01, 129.09, 129.30, 131.09, 132.68, 134.55, 139.04, 148.99, 150.21, 152.48. LC-MS (m/z): [M + H]+ calcd. for C21H18N4O2S 390.12, found 391.2. Purity by HPLC-UV (254 nm)-ESI-MS: 98.4%.
3-(4-((4-Sulfamoylphenyl)amino)phthalazin-1-yl)benzenesulfonamide (6c)
Purification was performed by column chromatography on silica gel (98 : 2 dichloromethane/methanol). The product was isolated as a white powder (0.04 g, 34% yield). M.p.: 290–292 °C. 1H NMR (500 MHz, DMSO-d6, 343 K): δ 5.88 (s, 2H, NH2), 6.61 (d, J = 8.76 Hz, 2H, Ar–H), 7.49 (s, 2H, NH2), 7.68–7.70 (m, 3H, Ar–H), 7.77–7.81 (m, 1H, Ar–H), 7.86–7.87 (m, 1H, Ar–H), 7.92–8.10 (m, 4H, Ar–H), 8.47 (dd, J = 1.90, 7.27 Hz, 1H), 12.87 (s, 1H, NH). 13C NMR (126 MHz, DMSO-d6, 343 K): δ 112.50, 126.33, 126.45, 126.55, 126.89, 128.02, 129.40, 132.79, 132.94, 134.64, 134.73, 144.57, 148.96, 152.51. LC-MS (m/z): [M + H]+ calcd. for C20H17N5O4S2 455.07, found 456.1. Purity by HPLC-UV (254 nm)-ESI-MS: 97.1%.
2-Chloro-5-(4-((4-sulfamoylphenyl)amino)phthalazin-1-yl)benzenesulfonamide (6d)
Purification was performed by column chromatography on silica gel (98 : 2 dichloromethane/methanol). The product was isolated as a white powder (0.027 g, 22% yield). M.p.: 250–252 °C. 1H NMR (500 MHz, DMSO-d6, 343 K): δ 5.87 (s, 2H, NH2), 6.60–6.66 (m, 2H, Ar–H), 7.38–7.41 (m, 1H, Ar–H), 7.48 (s, 2H, NH2), 7.68–7.74 (m, 4H, Ar–H), 7.96–8.00 (m, 3H, Ar–H), 8.45–8.47 (d, J = 7.65 Hz, 1H, Ar–H), 12.86 (s, 1H, NH). 13C NMR (126 MHz, DMSO-d6, 343 K): δ 112.51, 119.81, 126.34, 126.57, 126.87, 127.36, 128.03, 129.77, 131.88, 132.29, 132.98, 134.13, 141.52, 148.19, 152.53. LC-MS (m/z): [M + H]+ calcd. for C20H16ClN5O4S2 489.03, found 490.50. Purity by HPLC-UV (254 nm)-ESI-MS: 96.4%.
2-Methoxy-5-(4-((4-sulfamoylphenyl)amino)phthalazin-1-yl)benzenesulfonamide (6e)
Purification was performed by column chromatography on silica gel (97 : 3 dichloromethane/methanol). The product was isolated as a white powder (0.05 g, 40% yield). M.p.: 278–280 °C. 1H NMR (500 MHz, DMSO-d6, 343 K): δ 4.01 (s, 3H, OCH3), 5.87 (s, 2H, NH2), 6.60 (d, J = 8.76 Hz, 2H, Ar–H), 7.22 (s, 2H, NH2), 7.41 (d, J = 8.68 Hz, 1H, Ar–H), 7.68–7.72 (m, 3H, Ar–H), 7.84–7.86 (m, 1H, Ar–H), 7.94–7.99 (m, 3H, Ar–H), 8.45 (d, J = 7.65 Hz, 1H, Ar–H), 12.80 (s, 1H, NH). LC-MS (m/z): [M + H]+ calcd. for C21H19N5O5S2 485.08, found 486.2. Purity by HPLC-UV (254 nm)-ESI-MS: 97.8%.
2-Hydroxy-5-(4-((4-sulfamoylphenyl)amino)phthalazin-1-yl)benzenesulfonamide (6f)
Purification was performed by column chromatography on silica gel (95 : 5 dichloromethane/methanol). The product was isolated as a white powder (0.02 g, 17% yield). M.p.: 240–242 °C. 1H NMR (500 MHz, DMSO-d6, 343 K): δ 5.87 (s, 2H, NH2), 6.66–6.67 (m, 2H, Ar–H), 7.42 (s, 2H, NH2), 7.67–7.69 (m, 3H, Ar–H), 7.98–8.10 (m, 4H, Ar–H), 8.44 (s, 1H, Ar–H), 8.46 (s, 1H, Ar–H), 11.41 (br s, 1H, OH), 12.80 (s, 1H, NH). LC-MS (m/z): [M + H]+ calcd. for C20H17N5O5S2 471.07, found 472.1. Purity by HPLC-UV (254 nm)-ESI-MS: 98.7%.
2-Methyl-5-(4-((2-methyl-4-sulfamoylphenyl)amino)phthalazin-1-yl)benzenesulfonamide (6g)
Purification was performed by column chromatography on silica gel (98 : 2 dichloromethane/methanol). The product was isolated as a white powder (0.016 g, 13% yield). M.p.: 263–265 °C. 1H NMR (500 MHz, DMSO-d6, 343 K): δ 2.08 (s, 3H, CH3), 2.70 (s, 3H, CH3), 5.64 (s, 2H, NH2), 6.66 (d, J = 8.5 Hz, 1H, Ar–H), 7.50 (s, 2H, NH2), 7.57–7.60 (m, 2H, Ar–H), 7.71–7.79 (m, 3H, Ar–H), 7.96–7.97 (m, 2H, Ar–H), 8.09 (s, 1H, Ar–H), 8.44 (s, 1H, Ar–H), 12.81(s, 1H, NH). 13C NMR (126 MHz, DMSO) δ 17.25, 19.69, 112.45, 120.21, 124.83, 125.64, 127.87, 128.31, 129.55, 132.56, 133.11, 134.69, 135.42, 142.45, 148.89, 150.63. LC-MS (m/z): [M + H]+ calcd. for C22H21N5O4S2 483.10, found 484.2. Purity by HPLC-UV (254 nm)-ESI-MS: 95.9%.
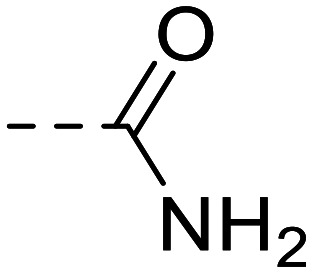
2-Methyl-5-{4-[(4-sulfamoylphenyl)amino]phthalazin-1-yl}benzamide (11)
Purification was performed by column chromatography on silica gel (98 : 2 dichloromethane/methanol). The product was isolated as a white powder (0.05 g, 44% yield). M.p.: 280–282 °C. 1H NMR (500 MHz, DMSO-d6, 343 K): δ 2.47 (s, 3H, CH3), 5.88 (s, 2H, NH2), 6.60 (d, J = 8.7 Hz, 2H, Ar–H), 7.44 (s, 2H, CONH2), 7.57 (d, J = 7.5 Hz, 2H, Ar–H), 7.69 (d, J = 8.7 Hz, 2H, Ar–H), 7.84–7.77 (m, 2H, Ar–H), 8.00–7.91 (m, 2H, Ar–H), 8.45 (dd, J = 8.0, 1.2 Hz, 1H, Ar–H),12.79 (s, 1H, NH). 13C NMR (126 MHz, DMSO-d6, 343 K): δ 19.44, 112.52, 122.32, 126.24, 126.47, 126.78, 127.30, 127.89, 127.92, 128.05, 130.30, 130.83, 131.24, 132.84, 134.69, 136.73, 137.25, 148.98, 149.68, 152.49, 170.33. LC-MS (m/z): [M + H]+ calcd. for C22H19N5O3S 433.12, found 434.3. Purity by HPLC-UV (254 nm)-ESI-MS: 98.2%.
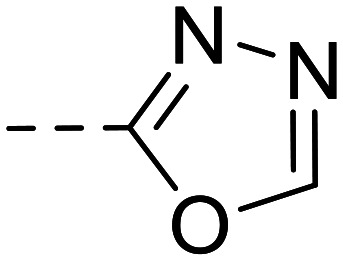
4-({4-[4-Methyl-3-(1,3,4-oxadiazol-2-yl)phenyl]phthalazin-1-yl}amino)benzenesulfonamide (15)
Purification was performed by column chromatography on silica gel (1 : 1 petroleum ether 40–60 °C/ethyl acetate). The product was isolated as a white powder (0.022 g, 19% yield). M.p.: 236–238 °C. 1H NMR (500 MHz, DMSO-d6, 343 K): δ 2.75 (s, 3H, CH3), 5.88 (s, 2H, NH2), 6.61 (d, J = 8.8 Hz, 2H, Ar–H), 7.67 (d, J = 8.0 Hz, 1H, Ar–H), 7.70 (d, J = 8.8 Hz, 2H, Ar–H), 7.81–7.73 (m, 2H, Ar–H), 8.01–7.91 (m, 2H, Ar–H), 8.15 (s, 1H, Ar–H), 8.46 (dd, J = 7.7, 1.5 Hz, 1H, Ar–H), 9.38 (s, 1H, Ar–H), 12.83 (s, 1H, NH). 13C NMR (126 MHz, DMSO-d6, 343 K): δ 21.27, 112.51, 122.83, 126.27, 126.52, 126.54, 127.09, 127.89, 128.03, 128.79, 129.73, 131.39, 131.46, 132.07, 132.34, 132.85, 134.70, 139.08, 149.01, 149.09, 152.51, 154.23, 163.34. LC–MS (m/z): [M + H]+ calcd. for C23H18N6O3S 458.12, found 459.2. Purity by HPLC-UV (254 nm)-ESI-MS: 97.5%.
Concentration–inhibition curves using p-nitrophenyl-5′-TMP as a substrate
The inhibitory effect of 4-(phthalazin-1-yl)aminobenzenesulfonamides on the enzymatic activity of human NPP3 was investigated in reaction buffer containing 1 mM MgCl2, 2 mM CaCl2, 10 mM CHES buffer (pH 9.0), and 400 μM p-Nph-5′-TMP, with inhibitor concentrations ranging from 0.03–100 μM. Incubation and separation conditions were the same as described above for inhibitor screening. Each analysis was performed in duplicates in three separate experiments. The Cheng-Prusoff equation was used to calculate Ki values from IC50 values determined by nonlinear curve fitting using the program Prism 5.0 (GraphPad software, San Diego, CA, USA).59 The Michaelis–Menten constant (Km) for p-Nph-5′-TMP was 432 μM.
Concentration–inhibition curves using capillary electrophoresis assay with ATP as a substrate
The enzyme inhibition assays were performed in 10 mM CHES buffer (pH 9.0) containing 1 mM MgCl2, 2 mM CaCl2, 400 μM ATP, and using a concentration range of 0.03–100 μM of 4-(phthalazin-1-yl)aminobenzenesulfonamide derivatives. The reaction mixture was incubated with 43 μg of human NPP3 at 37 °C for 4 h in a final volume of 100 μl. The reactions were stopped by heating at 90 °C for 3 min and subsequently measured by a capillary electrophoresis (CE) method.60–62 The CE instrumentation and operating conditions were as follows: P/ACE MDQ capillary electrophoresis system (Beckman Instruments, Fullerton, CA, USA) with a DAD detection system, polyacrylamide-coated capillaries of 50 cm effective length × 50 μm (id) obtained from CS Chromatographie GmbH (Langerwehe, Germany), 100 mM phosphate buffer (pH 6.5) as the running buffer, electrokinetic injection (−9 kV, 90 s), and separation voltage of −20 kV. The amounts of AMP produced were quantified by their UV absorption at 260 nm. Data collection and peak area analysis were performed by the 32 Karat software obtained from Beckman Coulter (Fullerton, CA, USA). The Ki values were calculated from the IC50 values with the Cheng–Prusoff equation.59 The Michaelis–Menten constant (Km) for ATP was 18.4 μM.
Determination of inhibition mechanism
The inhibition mechanism of compound 23 was determined using different concentrations of each substrate (from 20 to 1000 μM), and three different concentrations (0, ∼0.5-fold and ∼2-fold of IC50 value) of compound 23. The instrumentation and operation conditions for the experiments were the same as those described above for the determination of the concentration–inhibition curves using p-nitrophenyl-5′-TMP as a substrate and the concentration–inhibition curves determined by capillary electrophoresis assay with ATP as a substrate. Each analysis was performed in three separate experiments. The inhibition type was then determined graphically from the Hanes-Woolf plot.
Molecular modelling studies
The recently published X-ray structure of the human NPP3 (PDB ID: 6C02) in complex with the nucleotide inhibitor α,β-methylene-ATP (AMPCPP) was downloaded from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank.60 The crystal structure was prepared and protonated using the protein preparation tool and Protonate 3D implemented in Molecular Operating Environment (MOE 2018.01).63 The prepared crystal structure of human NPP3 was applied for molecular docking using AutoDock 4.2.64 During the molecular docking simulations the ligand was treated as flexible and the residues of the enzyme were considered rigid. The two selected compounds 1 and 23 were docked into the binding site of the cocrystallized inhibitor AMPCPP. The AutoDockTools were applied to add the atomic partial charges and for computing the three-dimensional energy scoring grid for a box of 60 × 60 × 60 points with a spacing of 0.375 Å.64 Fifty independent docking calculations were performed using the varCPSO-ls algorithm from PSO@Autodock implemented in AutoDock4.2.64,65 The docking calculations were computed by setting the termination criteria as 50 000 evaluation steps. The parameters of the varCPSO-ls algorithm, cognitive coefficient (c1) and social coefficient (c2) were set to 6.05 and the swarm size as 60 individual particles. The default values were applied for the remaining parameters of the algorithm. The docked poses obtained from docking studies for compounds 1 and 23 were explored by visual inspection and the putative binding pose was selected.
Investigation of selectivity versus different human ecto-nucleotidases
The inhibitory effects of compound 23 on human NTPDases was performed in reaction buffer containing 4 mM CaCl2, 4 mM MgCl2, 40 mM HEPES, pH 7.4, 400 μM ATP as substrate, and 10 μM of the inhibitor. The reaction was initiated by adding 20 μL of human NTPDase1 (0.3 μg), human NTPDase2 (0.5 μg) or human NTPDase 3 (1.2 μg), respectively, and then incubated at 37 °C for 30 min. The reaction was subsequently terminated by heating at 99 °C for 5 min. After cooling the reaction samples on ice, they were transferred into CE vials and injected into the CE instrument. The operation conditions for CE analysis were the same as those described above for capillary electrophoresis assay with ATP as substrate. All experiments were performed twice, each in triplicates.
NPP1 inhibition assays were carried out at 37 °C in a final volume of 100 μL. The reaction mixture contained 1 mM MgCl2, 2 mM CaCl2, 10 mM CHES, pH 9.0 and 400 μM ATP as a substrate. Solutions (20 μl) of compound 23 in enzyme assay buffer were added, and the reaction was initiated by the addition of 20 μL human NPP1 (360 ng). The mixture was incubated for 30 min and the reaction was terminated by heating at 90 °C for 3 min. After cooling the reaction samples on ice, they were transferred into CE vials and injected into the CE instrument. The operation conditions for CE analysis were the same as for NTPDase assays.
Inhibitory effects of compound 23 on lysophospholipase D activity of human NPP2 was investigated at 37 °C in a final volume of 50 μL. The reaction mixture contained 5 mM MgCl2, 5 mM CaCl2, 100 mM Tris, pH 9.0, 400 μM 1-oleoyl-sn-glycero-3-phosphocholine (LPC (18 : 1)) as substrate and compound 23 in concentrations of 10–1000 μM. The reaction was initiated by adding 10 μL of human NPP2 (44 μg) and incubation lasted for 60 min at 37 °C. The released choline was quantified colorimetrically at 555 nm after incubation at 37 °C for 10 min with 50 μL of peroxidase reagent (50 mM Tris at pH 9.0, 2 mM 3-(N-ethyl-3-methylanilino)-2-hydroxypropanesulfonic acid (TOOS), 5 U mL−1 peroxidase) and choline oxidase reagent (50 mM Tris at pH 9.0, 2 mM 4-aminoantipyrine, 5 U mL−1 choline oxidase). All experiments were performed twice, each in triplicates.
Ecto-5′-nucleotidase (CD73) assays were conducted at 37 °C in a final volume of 100 μL. The reaction buffer contained 4 mM CaCl2, 4 mM MgCl2, 40 mM HEPES, pH 7.4, 400 μM AMP as a substrate and 10 μM of compound 23. The reaction was initiated by adding 0.11 μg of recombinant human CD73, then the mixture was incubated at 37 °C for 30 min, and the reaction was subsequently terminated by heating at 99 °C for 5 min. After cooling the reaction samples on ice, 50 μl of the reaction mixture were transferred into mini-CE vials containing 50 μl of the internal standard uridine (final concentration 6.25 μM). The CE operation conditions were as follows: P/ACE MDQ capillary electrophoresis system, fused-silica capillary (40 cm [30 cm effective length], × 75.5 μm (id) obtained from Polymicro Technologies, Kehl, Germany), hydrodynamic injection (0.5 psi, 5 s), separation voltage of 15 kV, running buffer (40 mM borax and 100 mM sodium dodecyl sulfate (SDS) at pH 9.0), detection at 260 nm. Between separations, the capillary was washed with 0.1 N aq. NaOH solution for 2 min (30 psi) and subsequently with running buffer for 1 min (30 psi) before each injection. All experiments were performed twice, each in triplicates.
Evaluation of the inhibitory effect of compound 23 on human TNAP was performed in a mixture containing 1 mM CaCl2, 2 mM MgCl2, 10 mM CHES, pH 10.5, 10–1000 μM of compound 23 and 400 μM p-nitrophenyl phosphate as substrate. The enzyme reaction was carried out in a 96-well plate in a total volume of 100 μL. The reaction was started with 0.12 μg of human TNAP, and after 30 min of incubation at 37 °C, the liberated p-nitrophenolate was measured colorimetrically at 400 nm. All experiments were performed twice, each in triplicates.
Selectivity was evaluated versus each ectonucleotidase by calculating and comparing pKi (−log Ki) values from the determined Ki values.
Inhibition of carbonic anhydrase-II and -IX
A stop-flow instrument (Applied Photophysics, Leatherhead, UK) measuring CA-catalyzed CO2 hydration by a spectrophotometric method was applied in order to determine the inhibition constants of the test compounds. Phenol red (0.2 mM) was used as an indicator, and HEPES (20 mM, pH 7.5) was used as a buffer, and the ionic strength was kept constant by the use of 20 mM Na2SO4. CO2 hydration rates were observed for 10–100 s, using CO2 concentrations in a range of 1.7–17 mM. Each inhibitor was tested in triplicate, in serial dilutions starting from 0.01 to 0.1 mM. Inhibitors were preincubated with the enzyme for 15 min. The inhibition constants were calculated using the Cheng–Prusoff equation. The results are means from three determinations.
Abbreviations
- AMPCPP
α,β-Methylene-ATP
- CE
Capillary electrophoresis
- CA
Carbonic anhydrase
- CHES
2-(N-Cyclohexylamino)ethanesulfonic acid
- HEPES
4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid
- LDC (18 : 1)
1-Oleoyl-sn-glycero-3-phosphocholine
- NPP(s)
Ecto-nucleotide pyrophosphatases/phosphodiesterases
- NTPDase(s)
Ecto-nucleoside triphosphate diphosphohydrolases
- p-Nph-5′-TMP
p-Nitrophennyl 5′-thymidine monophosphate
- POM
Polyoxometalate
- PPi
Inorganic pyrophosphate
- SAR(s)
Structure–activity relationship(s)
- SDS
Sodium dodecyl sulfate
- TNAP
Tissue-nonspecific alkaline phosphatase
- TOOS
3-(N-Ethyl-3-methylanilino)-2-hydroxypropanesulfonic acid
- Tris
Tris(hydroxymethyl)aminomethane
Conflicts of interest
There is no conflict of interest to declare.
Supplementary Material
Acknowledgments
We are grateful for support by the Deutsche Forschungsgemeinschaft (SFB1328).
Electronic supplementary information (ESI) available. See DOI: 10.1039/d1md00117e
References
- Zimmermann H. Zebisch M. Sträter N. Purinergic Signalling. 2012;5:437–502. doi: 10.1007/s11302-012-9309-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan C. Jansen S. Bollen M. Trends Biochem. Sci. 2005;30:542–550. doi: 10.1016/j.tibs.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Bollen M. Gijsbers R. Ceulemans H. Stalmans W. Stefan C. Crit. Rev. Biochem. Mol. Biol. 2000;35:393–432. doi: 10.1080/10409230091169249. [DOI] [PubMed] [Google Scholar]
- Nishimasu H. Ishitani R. Aoki J. Nureki O. Trends Pharmacol. Sci. 2012;33:138–145. doi: 10.1016/j.tips.2011.12.004. [DOI] [PubMed] [Google Scholar]
- Goding J. W. Grobben B. Slegers H. Biochim. Biophys. Acta. 2003;1638:1–19. doi: 10.1016/s0925-4439(03)00058-9. [DOI] [PubMed] [Google Scholar]
- Aoki J. Semin. Cell Dev. Biol. 2004;15:477–489. doi: 10.1016/j.semcdb.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Albright R. A. Chang W. C. Robert D. Ornstein D. L. Cao W. Liu L. Redick M. E. Young J. I. De La Cruz E. M. Braddock D. T. Blood. 2012;120:4432–4440. doi: 10.1182/blood-2012-04-425215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albright R. A. Ornstein D. L. Cao W. Chang W. C. Robert D. Tehan M. Hoyer D. Liu L. Tabach P. S. Yang G. De La Cruz E. M. Braddock D. T. J. Biol. Chem. 2014;289:3294–3306. doi: 10.1074/jbc.M113.505867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi M. Okabe T. Okudaira S. Hanaoka K. Fujikawa Y. Terai T. Komatsu T. Kojima H. Aoki J. Nagano T. J. Am. Chem. Soc. 2011;133:12021–12030. doi: 10.1021/ja201028t. [DOI] [PubMed] [Google Scholar]
- Sakagami H. Aoki J. Natori Y. Nishikawa K. Kakehi Y. Natori Y. Arai H. J. Biol. Chem. 2005;280:23084–23093. doi: 10.1074/jbc.M413438200. [DOI] [PubMed] [Google Scholar]
- Duan R. D. Biochim. Biophys. Acta. 2006;1761:281–291. doi: 10.1016/j.bbalip.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Gorelik A. Randriamihaja A. Illes K. Nagar B. FEBS J. 2017;284:3718–3726. doi: 10.1111/febs.14266. [DOI] [PubMed] [Google Scholar]
- Lopez V. Lee S.-Y. Stephan H. Müller C. E. Anal. Biochem. 2020;603:113774. doi: 10.1016/j.ab.2020.113774. [DOI] [PubMed] [Google Scholar]
- Lee S.-Y. Lévesque S. A. Sévigny J. Müller C. E. J. Chromatogr., B. 2012;911:162–169. doi: 10.1016/j.jchromb.2012.10.044. [DOI] [PubMed] [Google Scholar]
- Iqbal J. Lévesque S. A. Sévigny J. Müller C. E. Electrophoresis. 2008;29:3685–3693. doi: 10.1002/elps.200800013. [DOI] [PubMed] [Google Scholar]
- Laketa D. Bjelobaba I. Savic J. Lavrnja I. Stojiljkovic M. Rakic L. Nedeljkovic N. Mol. Cell. Biochem. 2010;339:99–106. doi: 10.1007/s11010-009-0373-1. [DOI] [PubMed] [Google Scholar]
- Henz S. L. Fürstenau C. R. Chiarelli R. A. Sarkis J. J. F. Arch. Oral Biol. 2007;52:916–923. doi: 10.1016/j.archoralbio.2007.03.006. [DOI] [PubMed] [Google Scholar]
- El-Gamal M. I. Ullah S. Zaraei S. O. Jalil S. Zaib S. Zaher D. M. Omar H. A. Anbar H. S. Pelletier J. Sévigny J. Iqbal J. Eur. J. Med. Chem. 2019;181:111560. doi: 10.1016/j.ejmech.2019.07.063. [DOI] [PubMed] [Google Scholar]
- Praetorius H. A. Leipziger J. Purinergic Signalling. 2009;5:433–446. doi: 10.1007/s11302-009-9146-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rücker B. Almeida M. E. Libermann T. A. Zerbini L. F. Wink M. R. Sarkis J. J. Mol. Cell. Biochem. 2007;306:247–254. doi: 10.1007/s11010-007-9576-5. [DOI] [PubMed] [Google Scholar]
- Lee S. Y. Sarkar S. Bhattarai S. Namasivayam V. De Jonghe S. Stephan H. Herdewijn P. El-Tayeb A. Müller C. E. Front. Pharmacol. 2017;8:54. doi: 10.3389/fphar.2017.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai S. H. Kinoshita M. Kusu T. Kayama H. Okumura R. Ikeda K. Shimada Y. Takeda A. Yoshikawa S. Obata-Ninomiya K. Kurashima Y. Sato S. Umemoto E. Kiyono H. Karasuyama H. Takeda K. Immunity. 2015;42:279–293. doi: 10.1016/j.immuni.2015.01.015. [DOI] [PubMed] [Google Scholar]
- Horenstein A. L. Chillemi A. Zaccarello G. Bruzzone S. Quarona V. Zito A. Serra S. Malavasi F. OncoImmunology. 2013;2:e26246. doi: 10.4161/onci.26246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero E. Faini A. C. Malavasi F. Immunol. Lett. 2019;205:51–58. doi: 10.1016/j.imlet.2018.06.008. [DOI] [PubMed] [Google Scholar]
- Stefan C. Jansen S. Bollen M. Purinergic Signalling. 2006;2:361–370. doi: 10.1007/s11302-005-5303-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta Y. Tsai S. Kinoshita M. Fujimoto K. PLoS One. 2017:1–19. doi: 10.1371/journal.pone.0172509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano Y. Hayashi Y. Sano K. Nagano H. Nakaji M. Seo Y. Ninomiya T. Yoon S. Yokozaki H. Kasuga M. Cancer Lett. 2004;207:139–147. doi: 10.1016/j.canlet.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Yano Y. Hayashi Y. Sano K. Shinmaru H. Kuroda Y. Yokozaki H. Yoon S. Kasuga M. Int. J. Mol. Med. 2003;3:763–767. [PubMed] [Google Scholar]
- Zhang Y. Wedeh G. He L. Wittner M. Beghi F. Baral V. Launay J. M. Bibi S. Doñate F. Kouros-Mehr H. Arock M. Louache F. Blood Adv. 2019;3:633–643. doi: 10.1182/bloodadvances.2018026179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J. A. Motzer R. J. Molina A. M. Choueiri T. K. Heath E. I. Redman B. G. Sangha R. S. Ernst D. S. Pili R. Kim S. K. Reyno L. Wiseman A. Trave F. Anand B. Morrison K. Doñate F. Kollmannsberger C. K. Clin. Cancer Res. 2018;24:4399–4406. doi: 10.1158/1078-0432.CCR-18-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastid J. Cottalorda-Regairaz A. Alberici G. Bonnefoy N. Eliaou J.-F. Bensussan A. Oncogene. 2013;32:1743–1751. doi: 10.1038/onc.2012.269. [DOI] [PubMed] [Google Scholar]
- Gessi S. Varani K. Merighi S. Fogli E. Sacchetto V. Benini A. Leung E. Mac-Lennan S. Borea P. A. Purinergic Signalling. 2007;3:109–116. doi: 10.1007/s11302-006-9042-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G. Di Virgilio F. Purinergic Signalling. 2013;9:491–540. doi: 10.1007/s11302-013-9372-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G. Boeynaems J. M. Purinergic Signalling. 2014;10:529–564. doi: 10.1007/s11302-014-9427-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baqi Y. Lee S.-Y. Iqbal J. Ripphausen P. Lehr A. Scheiff A. B. Zimmermann H. Bajorath J. Müller C. E. J. Med. Chem. 2010;53:2076–2086. doi: 10.1021/jm901851t. [DOI] [PubMed] [Google Scholar]
- Müller C. E. Iqbal J. Baqi Y. Zimmermann H. Röllich A. Stephan H. Bioorg. Med. Chem. Lett. 2006;16:5943–5947. doi: 10.1016/j.bmcl.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Malik E. M. Müller C. E. Med. Res. Rev. 2016;36:705–748. doi: 10.1002/med.21391. [DOI] [PubMed] [Google Scholar]
- Raza R. Akhtar T. Hameed S. Lecka J. Iqbal J. Sévigny J. Open Enzyme Inhib. J. 2011;4:17–22. [Google Scholar]
- Andleeb H. Hameed S. Ejaz S. A. Khan I. Zaib S. Lecka J. Sévigny J. Iqbal J. Bioorg. Chem. 2019;88:102893. doi: 10.1016/j.bioorg.2019.03.067. [DOI] [PubMed] [Google Scholar]
- Ausekle E. Ejaz S. Khan S. Ehlers P. Villinger A. Lecka J. Sevigny J. Iqbal J. Langer P. Org. Biomol. Chem. 2016;14:11402–11414. doi: 10.1039/c6ob02236g. [DOI] [PubMed] [Google Scholar]
- Jafari B. Yelibayeva N. Ospanov M. Ejaz S. A. Afzal S. Khan S. U. Abilov Z. A. Turmukhanova M. Z. Kalugin S. N. Safarov S. Lecka J. Sévigny J. Rahman Q. Ehlers P. Iqbal J. Langer P. RSC Adv. 2016;6:107556–107571. [Google Scholar]
- Kuhrt D. Abida S. Afzal S. Khan U. Lecka J. Eur. J. Med. Chem. 2017;138:1–3. doi: 10.1016/j.ejmech.2017.07.017. [DOI] [PubMed] [Google Scholar]
- Lee S.-Y. Fiene A. Li W. Hanck T. Brylev K. A. Fedorov V. E. Lecka J. Haider A. Pietzsch H.-J. Zimmermann H. Sévigny J. Kortz U. Stephan H. Müller C. E. Biochem. Pharmacol. 2015;93:171–181. doi: 10.1016/j.bcp.2014.11.002. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Nat. Rev. Drug Discovery. 2008;7:168–182. doi: 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. J. Enzyme Inhib. Med. Chem. 2017;31:345–360. doi: 10.3109/14756366.2015.1122001. [DOI] [PubMed] [Google Scholar]
- McDonald P. C. Winum J. Supuran C. T. Dedhar S. Oncotarget. 2012;3:84–97. doi: 10.18632/oncotarget.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Kuijk S. Kumar N. Niemans R. Van Gisbergen M. W. Carta F. Vullo D. Pastorekova S. Yaromina A. Supuran C. T. Dubois L. J. Winum J. Lambin P. Eur. J. Med. Chem. 2017;127:691–702. doi: 10.1016/j.ejmech.2016.10.037. [DOI] [PubMed] [Google Scholar]
- Tomaselli D. Lucidi A. Rotili D. Mai A. Med. Res. Rev. 2020;40:190–244. doi: 10.1002/med.21600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antolin A. A. Workman P. Mestres J. Al-Lazikani B. Curr. Pharm. Des. 2016;22:6935–6945. doi: 10.2174/1381612822666160923115828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortuso F. Bagetta D. Maruca A. Talarico C. Bolognesi M. L. Haider N. Borges F. Bryant S. Langer T. Senderowitz H. Alcaro S. Front. Chem. 2018;6:130. doi: 10.3389/fchem.2018.00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognesi M. L. Cavalli A. ChemMedChem. 2016;11:1190–1192. doi: 10.1002/cmdc.201600161. [DOI] [PubMed] [Google Scholar]
- Annan D. A. Maishi N. Soga T. Dawood R. Li C. Kikuchi H. Hojo T. Morimoto M. Kitamura T. Alam M. T. Minowa K. Shinohara N. Nam J. M. Hida Y. Hida K. Cell Commun. Signaling. 2019;17:169. doi: 10.1186/s12964-019-0478-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R. Liao S. Zhang C. Zhu W. Wang L. Huang J. Zhao Z. Li H. Qian X. Xu Y. Eur. J. Med. Chem. 2013;62:597–604. doi: 10.1016/j.ejmech.2013.01.030. [DOI] [PubMed] [Google Scholar]
- Gorelik A. Randriamihaja A. Illes K. Nagar B. FEBS J. 2018;285:2481–2494. doi: 10.1111/febs.14489. [DOI] [PubMed] [Google Scholar]
- Mishra C. B. Tiwari M. Supuran C. T. Med. Res. Rev. 2020;10:1002. doi: 10.1002/med.21713. [DOI] [PubMed] [Google Scholar]
- McDonald P. C. Chia S. Bedard P. L. Chu Q. Lyle M. Tang L. Singh M. Zhang Z. Supuran C. T. Renouf D. J. Dedhar S. A. Am. J. Clin. Oncol. 2020;43:484–490. doi: 10.1097/COC.0000000000000691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vineberg J. G. Zuniga E. S. Kamath A. Chen Y. Seitz J. D. J. Med. Chem. 2014;57:5777–5791. doi: 10.1021/jm500631u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrant W. Patil V. Canzoneri J. C. Oyelere A. K. J. Med. Chem. 2012;55:1465–1477. doi: 10.1021/jm200799p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y.-C. Prusoff W. H. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Iqbal J. Müller C. E. J. Chromatogr. A. 2011;1218:4764–4771. doi: 10.1016/j.chroma.2011.04.085. [DOI] [PubMed] [Google Scholar]
- Iqbal J. Scapozza L. Folkers G. Müller C. E. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2007;846:281–290. doi: 10.1016/j.jchromb.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Iqbal J. Vollmayer P. Braun N. Zimmermann H. Müller C. E. Purinergic Signalling. 2005;1:349–358. doi: 10.1007/s11302-005-8076-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE), 2019.01, Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2019 [Google Scholar]
- Morris G. M. Huey R. Lindstrom W. Sanner M. F. Belew R. K. Goodsell D. S. Olson A. J. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namasivayam V. Gunther R. Chem. Biol. Drug Des. 2007;70:475–484. doi: 10.1111/j.1747-0285.2007.00588.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.