

Abstract
Mitochondrial dysfunction is an underlying pathology in numerous diseases. Delivery of diagnostic and therapeutic cargo directly into mitochondria is a powerful approach to study and treat these diseases. The triphenylphosphonium (TPP+) moiety is the most widely used mitochondriotropic carrier. However, studies have shown that TPP+ is not inert; TPP+ conjugates uncouple mitochondrial oxidative phosphorylation. To date, all efforts toward addressing this problem have focused on modifying lipophilicity of TPP+-linker-cargo conjugates to alter mitochondrial uptake, albeit with limited success. We show that structural modifications to the TPP+ phenyl rings that decrease electron density on the phosphorus atom can abrogate uncoupling activity as compared to the parent TPP+ moiety and prevent dissipation of mitochondrial membrane potential. These alterations of the TPP+ structure do not negatively affect the delivery of cargo to mitochondria. Results here identify the 4-CF3-phenyl TPP+ moiety as an inert mitochondria-targeting carrier to safely target pharmacophores and probes to mitochondria.
Graphical Abstract
INTRODUCTION
Dysfunctional mitochondria are implicated in a plethora of diseases including diabetes and obesity, Alzheimer’s, Parkinson’s, and amyotrophic lateral sclerosis, cardiac ischemia– reperfusion injury and congestive heart failure, cancer, and aging.1–3 One of the pharmacological approaches for the study and treatment of such diseases is the delivery of diagnostic and therapeutic agents directly into mitochondria. Such agents are often targeted to and delivered inside mitochondria by linking them to a mitochondriotropic carrier. Such carriers include diffused lipophilic cations like rhodamine 19 and triphenylphosphonium (TPP+) as well as mitochondria-targeting peptides like the Szeto–Schiller peptides.4–6 The TPP+ moiety is the mostly widely used mitochondria-targeting carrier.7,8
The large ionic radius due to resonance-mediated diffusion of positive charge across three phenyl rings, coupled with a highly lipophilic character, allows the TPP+ moiety to move across biological membranes without the need for transporters8 (Figure 1A). The positively charged TPP+ moiety accumulates inside mitochondria as a result of the −140 to −180 mV membrane potential created across the inner mitochondrial membrane by the proton pumping action of the electron transport chain. There is a 10-fold accumulation of the TPP+ moiety for approximately every 60 mV of membrane potential, resulting in 100- to 500-fold higher accumulation of TPP+ inside mitochondria9 (Figure 1B). This mitochondriotropic property of the TPP+ moiety has been exploited to target a wide variety of cargos to mitochondria including antioxidants (MitoQ, MitoVitE, and MitoTEMPOL)7,10–12 and anticancer agents (MitoMetformin, Mitoporphyrin, and TP187)13–15 as well as probes for detecting ROS (MitoSox and MitoPyl)16,17 and mitochondrial imaging (NPA-TPP)18 with several of them having in vivo efficacy as reported in the pages of this journal.19–24
Figure 1.
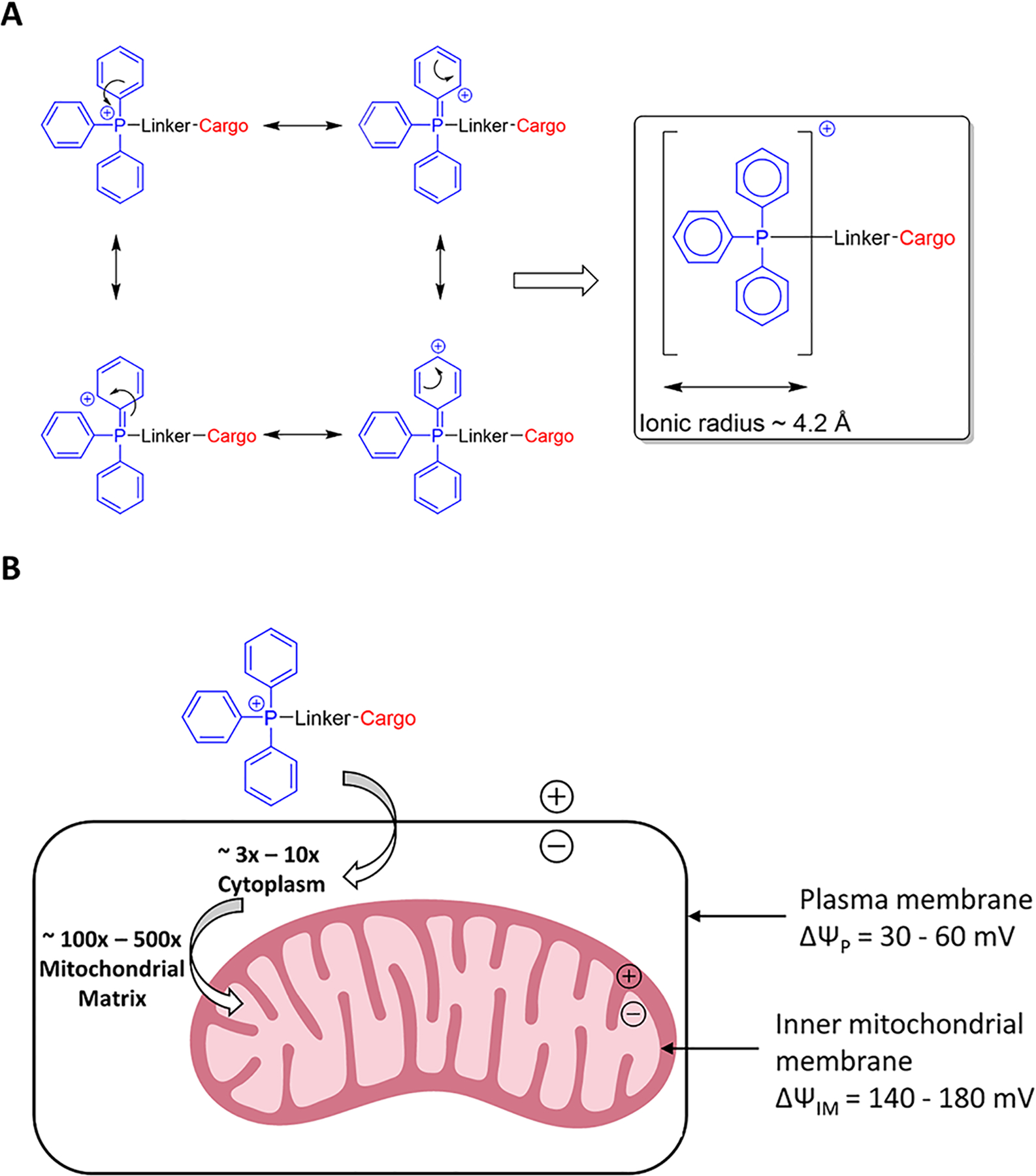
(A) Positive charge on the phosphorus atom of a TPP+ conjugate is diffused through the aryl rings because of the resonance of pi-electrons. (B) Schematic representation of membrane potential-driven uptake of TPP+ conjugates by mitochondria inside intact cells.
The widespread use of TPP+ as the carrier of choice for mitochondrial targeting was based on a belief that the TPP+ moiety was an inert carrier. However, recent studies have established that the TPP+ moiety has detrimental effects on mitochondrial bioenergetics.25–27 TPP+-linker conjugates without any cargo attached increased proton leak and uncoupled mitochondrial oxidative phosphorylation (OXPHOS), thereby decreasing the efficiency of ATP generation. Most structural modifications to TPP+ conjugates have focused on modifying the length of the linker connecting TPP+ to cargo in order to modulate lipophilicity of the entire TPP+ conjugate. However, decreasing lipophilicity and shortening linker length of the conjugates simply lower toxicity by reducing uptake and delivery of attached cargo to the mitochondria.28 Reports describing modification to the phenyl rings of TPP+ conjugates have similarly focused on lipophilicity.29 To date, no structural modification of a TPP+-cargo conjugate has been reported to decrease the toxicity of the TPP+ moiety without a correlative reduction in delivery of cargo to the mitochondria. Moreover, the effect of modification to the TPP+ moiety on mitochondrial toxicity and targeting remains largely unexplored. Toward this end, the current study was undertaken to determine if mitochondrial uptake of TPP+ conjugates could be regulated separately from their potency of OXPHOS uncoupling by systematically characterizing the effects of structural modification on the charge density on the phosphorous atom and the aryl ring lipophilicity. We have identified TPP+ derivatives that dissociate mitochondrial uptake from uncoupling of OXPHOS, affording a new approach to develop improved TPP+ derivatives for targeting cargo to mitochondria.
RESULTS
Electron-Withdrawing and Electron-Donating Substituents on Phenyl Rings of the TPP+ Moiety Alter Hückel Charge and Partition Coefficient.
High lipophilicity and diffusion of cationic charge are necessary for the TPP+ moiety to act as a mitochondrial carrier. Previous research showed that altering lipophilicity of TPP+-cargo conjugates altered mitochondrial accumulation, which in turn directly impacted mitochondrial uncoupling.28,30 However, the effect of altering the electron density on the phosphorus atom of the TPP+ moiety on uncoupling or mitochondrial targeting is unknown. To achieve this goal, we envisioned substitution of the TPP+ phenyl rings with electron-withdrawing and electron-donating groups.
Electron-withdrawing groups at the para- or ortho-position of the phenyl rings of the TPP+ moiety pull electrons away from the phosphorus atom. This increases the charge density on the phosphorus atom (measured as Hückel charge) compared to an unsubstituted TPP+. Conversely, electron-donating substituents at the same positions push electrons toward the phosphorus atom, thereby decreasing the Hückel charge on the phosphorus atom. The effect of a para-substituent on the electron density of the phosphorus atom of TPP+ can be succinctly depicted by an electrostatic potential map (Supplemental Figure S1). Hückel charges on the phosphorus atom for modified triphenylphosphonium decyl conjugates are reported in Supplemental Table S1.
Any modification to the phenyl rings of TPP+ or the linker also alters lipophilicity of the molecule (measured as the partition coefficient, logP). The decyl linker is known to be optimal for mitochondrial uptake of most TPP+ conjugates.31,32 Therefore, TPP+ derivatives were designed as decyl conjugates. Corresponding series with “suboptimal” butyl and benzyl linkers were also designed so that changes in uncoupling potency could be evaluated independent of the linker. Calculated logP (cLogP) values for compounds studied here are reported in Supplemental Table S2.
Based on Hückel charge and cLogP calculations and availability of the modified phosphines, the following substituents were chosen for experimental study: (i) −CF3, strong electron-withdrawing inductive effect; (ii) −F, moderate electron-withdrawing inductive effect; (iii) −Cl, weak electron-withdrawing inductive effect; (iv) −CH3, weak electron-donating inductive effect; and (v) −OCH3, strong electron-donating resonance effect. To prevent steric hindrance only para-(and not ortho-)substituted TPP+ derivatives were synthesized.
The TPP+-linker conjugates were synthesized via an SN2 reaction between selected para-substituted triphenylphosphines (1–6) and excess bromoalkane (1-bromodecane (7) for decyl series (9–14) and 1-bromobutane (8) for butyl series (15–20)) or benzyl bromide (21) for the benzyl series in toluene at varying temperatures for varying duration (Figure 2A). Reactivity of the phosphines varied greatly depending on the nature of the para-substituent. Electron-donating substituents like methoxy (1) and methyl (2) at the para-position of the phosphine phenyl rings resulted in an increased electron density on the phosphorus atom compared to the unsubstituted triphenylphosphine (3). This facilitated the nucleo-philic attack of phosphorus on the carbon alpha to bromine, giving the desired products at lower temperatures and with shorter reaction times. In contrast, phosphines with electron-withdrawing para-substituents like chloro (4), fluoro (5), and trifluoromethyl (6) have decreased electron density on the phosphorus atom compared to 3. As a result, the activation energy for an SN2 attack is higher, necessitating higher temperatures and/or longer reaction times to obtain the desired products. The lower boiling point of 1-bromobutane (8) (101.4 to 102.9 °C) as compared to 1-bromodecane (7) (238 to 244 °C) required the use of higher temperatures as well as periodic replenishment of 1-bromobutane to compensate for losses due to evaporation. Synthesis of butyl tris(4-fluorophenyl)phosphonium bromide (19) and butyl tris(4-trifluoromethylphenyl)phosphonium bromide (20) was particularly challenging due to the low reactivity of the phosphines (5 and 6, respectively), necessitating temperatures higher than the boiling point of 1-bromobutane over an extended period. This is reflected in the poor yields for these particular products. Synthesis of TPP+-benzyl series was much more facile due to much higher reactivity of benzyl bromide (21) as compared to the alkyl bromides. The structure of each first-generation compound in the para-substituted TPP+-linker panel and their assigned acronyms are shown in Supplemental Figure S2.
Figure 2.

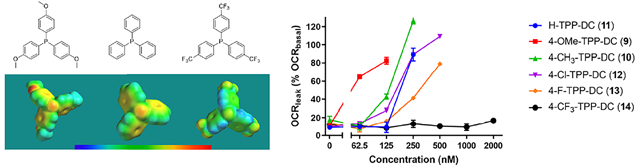
(A) Synthesis of first-generation modified TPP+ compounds of the decyl, butyl, and benzyl series. (B) Bioenergetic profile of C2C12 cells after 20 h of incubation with H-TPP-DC (11) in the cell mito stress test at six different concentrations in a two-fold dilution series between 62.5 nM and 2 μM. Values represent mean ± SE, n = 3. (C) Spare respiratory capacity and proton leak values calculated from data in panel B. (D) Oxygen consumption rate for maximal (solid, black trace) and proton leak (as % basal) (dashed, blue trace) respiration in C2C12 cells post treatment with H-TPP-DC (11). Values represent mean ± SE, n = 3. (E–G) Comparison of the oxygen consumption rate for proton leak (as % basal respiration) in C2C12 cells post treatment with (E) modified TPP+-decyl series analogues (9–14), (F) modified TPP+-butyl series analogues (15–20), and (G) modified TPP+-benzyl series analogues (22–27). Values represent mean ± SE, n = 3.
In response to para-substitution of the phenyl rings of TPP+, Hückel charge on the phosphorus atom decreased in the order of CF3 > F ≈ Cl > H > CH3 > OCH3. Change in linker length or aromaticity did not have an effect on the Hückel charge. For each of the different linker series, the lipophilicity trend in decreasing order was decyl ≫ benzyl > butyl. Within each linker series, TPP+ substitution led to a lipophilicity trend of −CF3 > −Cl > −CH3 > −F > −H > −OCH3.
Modification of Phenyl Rings of TPP+ Conjugates Alters Propensity to Uncouple Mitochondrial Respiration.
Each TPP+ conjugate was evaluated by the Seahorse respirometer “cell mito stress test”. The murine muscle C2C12 cell line was chosen for the study since muscles have a large number of mitochondria and are one of the primary sites for energy consumption. C2C12 myotubes were incubated with each of the TPP+ compounds at six different concentrations for 20 h, after which the cell mito stress test was performed. A representative bioenergetic profile observed after the test with H-TPP-DC (11), a traditional TPP+-linker conjugate, is shown in Figure 2B. Toxicity of traditional TPP+ compounds manifests as a sharp increase in the proton leak accompanied by a decrease in spare respiratory capacity and maximal respiration (Figure 2C). A concise way of depicting these data is shown in Figure 2D. The oxygen consumption rate (OCR) for proton leak as a percent of basal respiration is plotted on the right Y-axis. Under normal conditions, this OCRleak (as % OCRbasal) (dashed, blue trace) is about 10–20% of OCRbasal. For traditional TPP+ compounds, after a certain threshold concentration is reached, a rapid increase in the OCRleak is observed, shown as a spike in the blue trace. This is accompanied by a decrease in maximal respiration (solid, black trace, plotted on left Y-axis). For purpose of data analysis, the lowest concentration that leads to zero spare respiratory capacity (Figure 2C, 500 nM) or the concentration that leads to first decrease in maximal respiration (Figure 2D, 500 nM) was defined as the toxic concentration. To compare the uncoupling potency of different compounds, the OCRleak as percent of the OCRbasal (blue, dashed trace) is plotted up to the toxic concentration. Between two or more compounds, the compound whose trace for the OCRleak (as % OCRbasal) is more to the left is interpreted to be more potent at uncoupling mitochondrial oxidative phosphorylation. (Figure 2E–G). A lead compound for developing a safe mitochondrial cargo delivery agent should show no increase in the OCRleak (as % OCRbasal).
Each decyl conjugate, except for 4-CF3-TPP-DC (14), showed an increase in uncoupling with increasing concentration, resulting in toxicity in the concentration range tested (Figure 2E). The derivatives with electron-donating groups, 4-OMe-TPP-DC (9) and 4-CH3-TPP-DC (10), showed more potent uncoupling than the parent H-TPP-DC (11). Conversely, derivatives with electron-withdrawing substituents, 4-F-TPP-DC (13) and 4-CF3-TPP-DC (14), were less potent uncouplers than H-TPP-DC (11). The 4-Cl-TPP-DC (12) compound also has a weak electron-withdrawing substituent, but unlike the fluoro and trifluoromethyl derivatives, the uncoupling profile of the chloro derivative closely resembled that of the parent TPP+ derivative. The trend in uncoupling potency observed with the decyl series remained constant with the modified TPP+-butyl and TPP+-benzyl series (Figure 2F,G, respectively), thus demonstrating that the effect of TPP+ modification on uncoupling is independent of the linker.
In summary, the Hückel charge on the phosphorus atom of TPP+ had a substantial impact on potency of uncoupling induced by the TPP+ conjugate. A decrease in Hückel charge increased uncoupling potency, while an increase in Hückel charge led to either decreased uncoupling of mitochondrial respiration or complete abolition of mitochondrial uncoupling at concentrations evaluated. As expected, when the same modification of TPP+ was compared across different linker series, the higher the lipophilicity of the linker and conjugate, the more potent the uncoupling of mitochondrial respiration.
Modulation of Hückel Charge on the TPP+ Phosphorus Atom Has a Greater Impact on Uncoupling Potency than Alterations in Lipophilicity.
To delineate the contribution of Hückel charge and lipophilicity to potency of TPP+-mediated uncoupling, analogues of first-generation TPP+ compounds were designed to alter one of these two variables while holding the other constant. Alteration of Hückel charge and lipophilicity, one variable at a time, could be achieved by synthesis of meta-substituted analogues of previous para-substituted TPP+-decyl compounds. When an electron-withdrawing or electron-donating substituent is present at the para-position (or ortho-position) of the phenyl rings of TPP+, the resonance effect increases or decreases the Hückel charge on the phosphorus atom, respectively. In contrast, the resonance effect of a meta-substituent skips the phosphorus atom and has only a minimal effect on its electron density (Supplemental Figure S3). Therefore, comparing two TPP+ conjugates, each with the same substituent at either the para- or meta-position of the phenyl rings of TPP+, should yield differences in Hückel charge but negligible difference in their lipophilicity. To this end, meta-analogues of 4-OMe-TPP-DC (9) and 4-F-TPP-DC (13) were synthesized and evaluated. Calculated Hückel charge for these second-generation compounds and their comparison with the first-generation analogues is given in Supplemental Table S3.
For altering lipophilicity, a decyl tris(naphth-1-yl)-phosphonium compound was designed to compare with HTPP-DC (11). While the goal was to keep Hückel charge unaltered, the electron-withdrawing nature of the fused ring system did increase the Hückel charge when compared to its first-generation analogue 11 (Supplemental Table S3). However, while electron-withdrawing para-substituents on TPP+ phenyl rings showed decreased potency for uncoupling OXPHOS, increasing lipophilicity of the molecule (decyl vs butyl analogues) led to increased uncoupling potency. As such, the naphthyl analogue of H-TPP-DC (11) would decrease uncoupling potency if Hückel charge was the predominant factor affecting the uncoupling potency versus increase it if lipophilicity had a stronger influence. Supplemental Table S4 summarizes the cLogP of all the second-generation TPP+ conjugates and their first-generation analogues. The naphthyl analogue of H-TPP-DC (11) was more lipophilic than the parent. Shifting the para-substituent to the meta-position caused a slight increase in the calculated lipophilicity. This was in accordance with experimentally calculated partition coefficient values of meta- and para-methyl-substituted TPP+.22
Second-generation, meta-substituted TPP+-decyl analogues 30 and 31 were synthesized in a fashion similar to the first-generation, para-substituted compounds (Figure 3A). Attempts to synthesize decyl tri(naphthalen-1-yl)phosphonium (33) using the same reaction conditions were not successful because of two reasons: decreased electron density on the phosphorus atom of tri(naphthalen-1-yl)phosphine (32) deactivates an SN2 attack, and the bigger size of the naphthyl group adds steric hindrance to the SN2 attack. 1-Bromodecane (7) was converted to 1-iododecane in an in situ Finkelstein reaction, which then reacted with the phosphine to give the desired product 32 as an iodide salt (Figure 3A). Structures and acronyms of the second-generation compounds along with the corresponding first-generation analogues are shown in Supplemental Figure S4.
Figure 3.
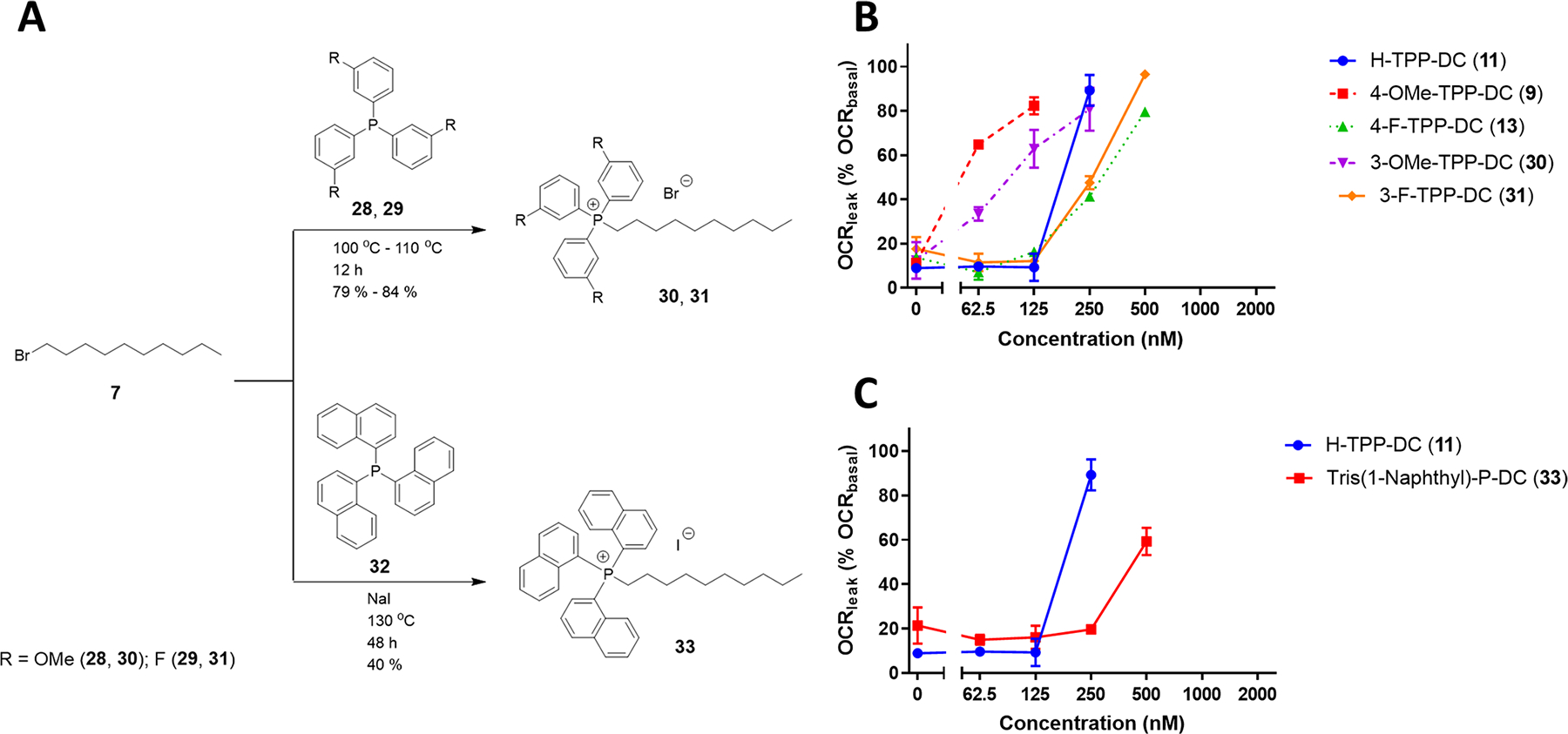
(A) Synthesis of meta-substituted aryl-modified TPP+-decyl derivatives (30 and 31) and decyl tri(naphthalen-1-yl)phosphonium iodide (33). (B, C) Comparison of the oxygen consumption rate for proton leak (as % basal respiration) in C2C12 cells post treatment between (B) second-generation meta-substituted aryl-modified analogues (30 and 31) and cognate first-generation TPP-decyl derivatives (9 and 13) and (C) Tris-(1-naphthyl)-P-DC (33) and cognate H-TPP-DC (11). Values represent mean ± SE, n = 3.
These derivatives were evaluated by the Seahorse cell mito stress test to test their ability to uncouple mitochondrial respiration of C2C12 myotubes at various concentrations. The meta-substituted compound, 3-OMe-TPP-DC (30), was found to be a less potent uncoupler than 4-OMe-TPP-DC (9), while 3-F-TPP-DC (31) was almost identical to 4-F-TPP-DC (13) in uncoupling potency (Figure 3B). Within the meta-modified TPP+ compounds, the uncoupling potency of an electron-withdrawing methoxy derivative was higher than the electron-donating fluoro derivative. The fused ring analogue of H-TPPDC (11), tris(1-naphthyl)-P-DC (33), was found to be a less potent uncoupler than the parent compound (Figure 3C). Taken together, these results indicate that Hückel charge has a more profound effect on the uncoupling potency of TPP+ conjugates than lipophilicity.
Electron-Withdrawing Aromatic Substituents on TPP+ Phenyl Rings Can Abrogate Loss of Mitochondrial Membrane Potential.
To corroborate the respiration data, the effect of TPP+ modification on mitochondrial membrane potential was independently assessed using the potential probe JC-1. Shown in Figure 4A are C2C12 cells stained with JC-1 after 20 h of incubation with either a 125 or 2000 nM dose of H-TPP-DC (11), 4-OMe-TPP-DC (9), and 4-CF3-TPP-DC (14) as indicated. For H-TPP-DC (11) and 4-OMe-TPP-DC (9), the cells maintained membrane potential at the lower dose. However, incubation with 2000 nM dose for both the compounds resulted in the loss of all orange/red fluorescence, consistent with dissipation of mitochondrial membrane potential. Depolarization of mitochondria was also accompanied by a change in the morphology of the myotubes, as seen in Figure 4A. By contrast, after the same period in the presence of both the low and high doses of 4-CF3-TPP-DC (14), cells continued to fluoresce orange/red, consistent with maintenance of mitochondrial membrane potential. No changes in cell density or morphology were observed.
Figure 4.

(A) C2C12 myotubes stained with mitochondrial potential indicator JC-1 after 20 h of incubation with H-TPP-DC (11) (125 nM) (top left); H-TPP-DC (11) (2 μM) (bottom left); 4-OMe-TPP-DC (9) (125 nM) (top middle); 4-OMe-TPP-DC (9) (2 μM) (bottom middle); 4-CF3-TPP-DC (14) (125 nM) (top right); and 4-CF3-TPP-DC (14) (2 μM) (bottom right). (B) Dose-dependent effects of modified TPP+-decyl compounds with electron-donating substitution on the phenyl rings (9, 10, and 30) and electron-withdrawing substitution on the phenyl rings (12, 13, and 14) or decyl Tris(1-Naphthyl)-P-DC (33) on mitochondrial membrane potential in intact C2C12 cells estimated as the ratio of red to green fluorescence of potential sensitive probe JC-1 normalized to the untreated control. Values represent mean ± SE, n = 3, * = p < 0.001 as compared to the untreated control as analyzed by two-way ANOVA and the post-hoc Tukey test using GraphPad prism. (C) Dose-dependent cell viability determined by the MTT assay in C2C12 cells incubated for 20 h with modified TPP+-decyl compounds with electron-donating substitution on the phenyl rings (9, 10, and 30) and electron-withdrawing substitution on the phenyl ring (12–14) or Tris(1-Naphthyl)-P-DC (33). Values represent mean ± SE, n = 3, * = p < 0.001 as compared to the untreated control as analyzed by two-way ANOVA and the post-hoc Dunnett test using GraphPad prism.
The dose-dependent change in membrane potential after treatment with modified TPP+-decyl conjugates is quantified in Figure 4B. Increasing concentration of H-TPP-DC (11) caused a greater decrease in the red to green ratio, consistent with greater depolarization of the mitochondrial membrane potential. A dose-dependent decrease in mitochondrial membrane potential was also observed for compounds with electron-donating substitution on the phenyl ring of TPP+ [4-OMe-TPP-DC (9), 4-CH3-TPP-DC (10), and 3-OMe-TPP-DC (30)]. This dose-dependent change in mitochondrial membrane potential was independent of the position of the methoxy substitution on the TPP+ phenyl ring as both meta-and para-substituted methoxy-TPP+ compounds caused a decrease in the membrane potential to a similar extent at each concentration. At lower concentrations (125 and 250 nM), compounds with electron-donating substituents caused higher membrane depolarization than H-TPP-DC (11) and this is consistent with higher potency of uncoupling mitochondrial membrane potential as observed in the respirometer assays (Figure 2). Among modified TPP+ compounds with electron-withdrawing substitution, 4-Cl-TPP-DC (12) was the most potent uncoupler and also led to the greatest decrease in membrane potential. 4-F-TPP-DC (13) and decyl tri(naphthalen-1-yl)phosphonium iodide (33) showed significant membrane depolarization only at the highest tested concentration of 2000 nM. 4-CF3-TPP-DC (14) did not show any membrane depolarization at any of the tested concentrations. These results further highlight the inertness of the 4-CF3-TPP+ moiety as compared to the parent TPP+, suggesting that it is better suited as a mitochondria-targeting moiety.
Cell Viability Is Directly Proportional to Hückel Charge on the Phosphorus Atom of TPP+ Moiety.
To determine if the changes in potency of uncoupling are directly translated to changes in cell viability, all the synthesized compounds were evaluated in the MTT assay (Figure 4C). Compared to the untreated control, the parent TPP+ compound H-TPP-DC (11) caused a decrease in cell viability with increasing concentration of the compound. The same effect was observed for compounds 4-OMe-TPP-DC (9), 4-CH3-TPP-DC (10), and 3-OMe-TPP-DC (30), with electron-donating substituents on the phenyl ring of TPP+, consistent with results obtained in the JC-1 assay. The decrease in cell viability was independent of the position of the methoxy substitution, and both meta- and para-substituted methoxy-TPP+ compounds were equally toxic at higher concentrations. In contrast, compounds with increased Hückel charge on the phosphorus as compared to the traditional TPP+, either via electron-withdrawing substitution on the phenyl ring or by substitution of the phenyl ring with a fused naphthyl ring, were less toxic than H-TPP-DC (11) (Figure 4C). For 4-F-TPP-DC (13), 4-Cl-TPP-DC (12) and Tris(1-Naphthyl)-P-DC (33) decreased cell viability was observed only at the highest tested concentration of 2000 nM, consistent with decreased membrane potential at that concentration as observed with the JC-1 assay. However, 4-CF3-TPP-DC (14), with the highest Hückel charge did not cause any toxicity up to 2000 nM concentration. These results further reinforce 4-CF3-TPP+ as the lead moiety for developing safer mitochondria-targeting agents.
Modification of the TPP+ Moiety Does Not Prevent Uptake into Isolated Mitochondria.
To determine if modification of the TPP+ moiety was affecting membrane potential-driven uptake into mitochondria, a tetraphenylphosphonium electrode, which selectively detects TPP+ ions, was used to assess uptake of conjugates into isolated mitochondria. As shown in Figure 5, three successive additions of each TPP+-decyl compound tested were made into the chamber to assess the TPP+ electrode response. Addition of mouse muscle mitochondria in the absence of the respiratory substrate resulted in nonspecific uptake of TPP+ compounds, presumably due to lipophilicity of the linker. Following the addition of respiratory substrates, mitochondria began to respire as indicated by an increase in oxygen consumption. This built membrane potential across the inner mitochondrial membrane and drove further uptake of the TPP+ compounds. As seen in Figure 5, neither substitution of the TPP+ phenyl rings nor replacement of phenyl rings with bulkier naphthyl rings prevented mitochondrial uptake. Similar results were obtained with mouse liver mitochondria (Supplemental Figure S5). Importantly, demonstrating the uptake of 4-CF3-TPP-DC (14) into isolated mitochondria implies that its lack of uncoupling in the Seahorse experiment, no dissipation of membrane potential in the JC-1 assay, and lack of toxicity in the MTT assay were not due to the compound being simply excluded from reaching mitochondria. Rather, the increased Hückel charge of the compound seems to abolish effects on respiration associated with the traditional TPP+ moiety (11) while having no effect on mitochondrial uptake.
Figure 5.
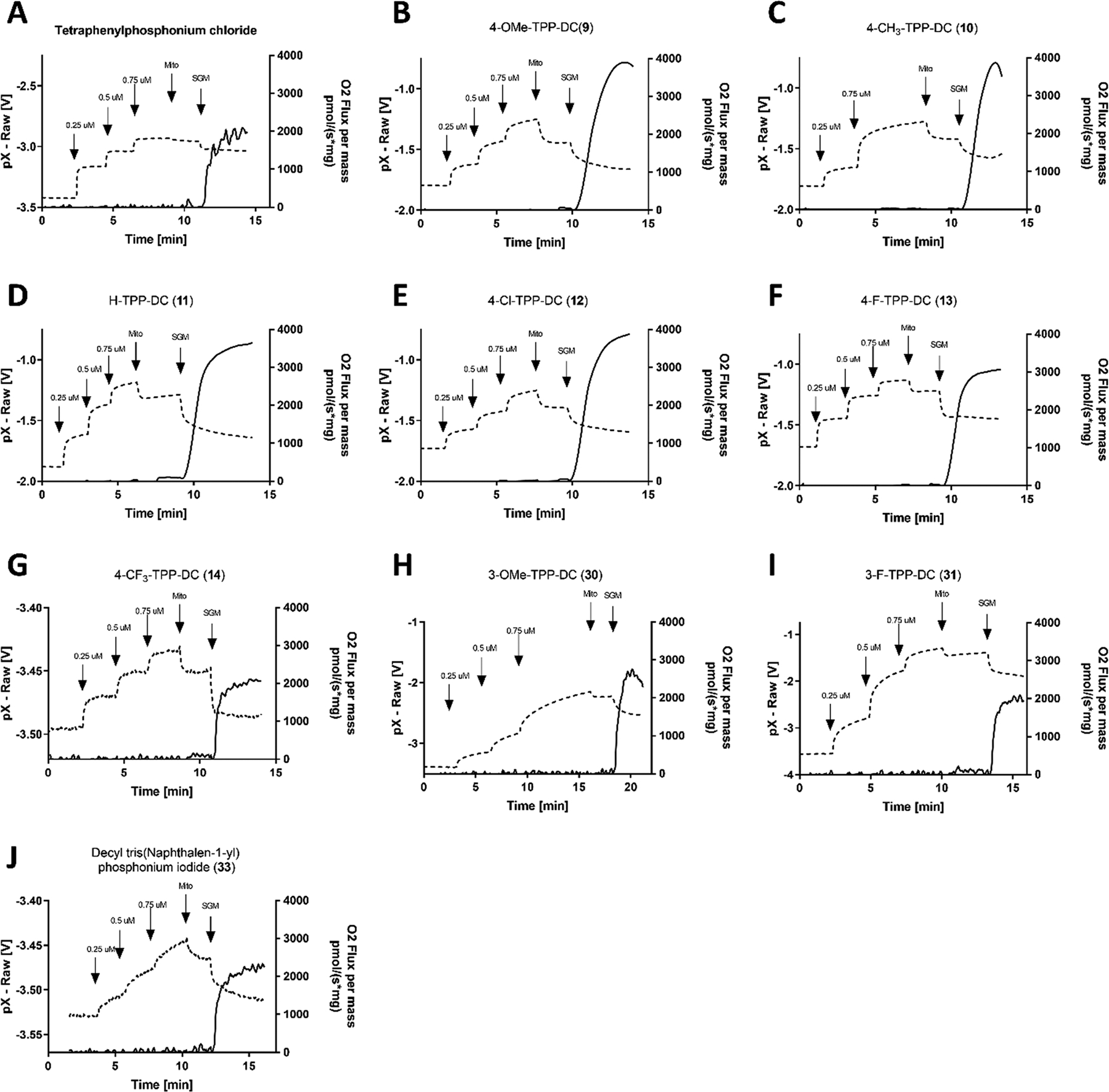
Mitochondrial accumulation of modified TPP+-decyl series assessed by TPP+ ion-selective electrode measurements. Tetraphenylphosphonium electrode response (dashed trace) was calibrated with 3 × 0.25 μM additions of the investigated compound. A greater TPP electrode voltage corresponds to decreased membrane potential when calculated by the Nernst equation. Mouse hind limb mitochondria (mito) (0.05 mg/mL) were added, and finally, mitochondrial substrate combination (5 mM succinate + 5 mM glutamate + 1 mM malate; SGM) was added to the chamber where indicated. The experiment was terminated when steady-state reading was achieved. Corresponding oxygen consumption per unit mass (solid trace) was recorded with a Clark-type oxygen electrode. (A–J) Compounds tested were (A) tetraphenylphosphonium chloride, (B) 4-OMe-TPP-DC (9), (C) 4-CH3-TPP-DC (10), (D) H-TPP-DC (11), (E) 4-Cl-TPP-DC (12), (F) 4-F-TPP-DC (13), (G) 4-CF3-TPP-DC (14), (H) 3-OMe-TPP-DC (30), (I) 4-F-TPP-DC (31), and (J) decyl tri(naphthalen-1-yl)phosphonium iodide (33).
CF3-TPP+ Moiety Delivers Cargo to Mitochondria within Intact Cells.
We next looked to further demonstrate the ability of 4-CF3-TPP+ derivatives to localize inside mitochondria of intact cells. To visualize this, selected parent TPP+ and 4-CF3-TPP+ moieties were conjugated to a fluorescent dye, TAMRA, and colocalization was evaluated with a known fluorescent probe MitoTracker Green FM that selectively accumulates inside mitochondria. Successful use of the combination of MitoTracker and cisplatin-peptide-TAMRA conjugate to assess intracellular mitochondrial localization has been previously reported.33
Synthesis of TPP+-TAMRA conjugates (42 and 43) is shown in Figure 6A. Potassium phthalimide (35) was stirred with excess 1,10-dibromodecane (34) to give 10-bromodecylphthalimide (36). Column chromatography was used to remove the unreacted dibromoalkane. Unsubstituted triphenylphosphine (3) and tris(4-trifluoromethylphenyl)phosphine (6) were then reacted with 10-bromodecylphthalimide (36) to give the intermediates 37 and 38, respectively. Phthalimide deprotection was done by heating with 48% aqueous hydrobromic acid to give compounds with free terminal amine (39 and 40). Finally, both aminoalkyl (39 and 40) intermediates were coupled with 6-TAMRA NHS ester (41) to give the desired products (42 and 43, respectively) after preparative HPLC purification.
Figure 6.

(A) Synthesis of modified TPP-decyl-TAMRA conjugates, H-TPP-DC-TAMRA (42) and 4-CF3-TPP-DC-TAMRA (43). (B) Representative image to show modified TPP+-TAMRA conjugates localizing in mitochondria within intact C2C12 cells. MitoTracker Green FM (top left); 4-CF3-TPP-DC-TAMRA (43) (top right); merged image (bottom left); and merged image showing Hoechst 33342 nuclear stain (bottom right). (C) Mitochondrial accumulation of the 4-CF3-TPP-DC-TAMRA (43) conjugate relative to H-TPP-DC-TAMRA (42) within intact C2C12 cells. Values represent mean ± SE, n ≥ 4.
Representative images revealing colocalization of the TAMRA conjugates with MitoTracker Green FM are shown in Figure 6B. As seen in Figure 6C, 4-CF3-TPP-TAMRA conjugate accumulation inside intact C2C12 myotubes is comparable or better than the TAMRA conjugate with unsubstituted, parent TPP+ at all tested concentrations. The imaging assay proves that modification of the TPP+ moiety by substitution on the phenyl rings does not prevent the modified TPP+ moiety from targeting cargo to the mitochondria within intact cells. In fact, para-substitution of the TPP+ phenyl rings led to a higher mitochondrial accumulation of cargo than that mediated by unmodified TPP+. Combining these data demonstrates that the 4-CF3-TPP+ moiety is a very promising lead for developing inert mitochondria-targeting agents.
DISCUSSION AND CONCLUSIONS
Given the central role of mitochondria in cellular metabolism and the variety of diseases caused due to their dysfunction, mitochondria are prime pharmacologic targets. The TPP+ moiety has been the most widely used mitochondriotropic carrier to deliver both drugs and diagnostic agents to mitochondria. However, TPP+-linker conjugates uncouple mitochondrial OXPHOS, thereby confounding the effect of attached cargo. Here, we have demonstrated that addition of substituents onto the TPP+ phenyl rings is not only an effective way to modulate both charge density and lipophilicity of the phosphonium compounds but also an effective way to dissociate mitochondrial uptake from uncoupling of oxidative phosphorylation.
Substitution of electron-donating groups on the para-position of TPP+ phenyl rings led to a decrease in charge density (Hückel charge) on the phosphorus atom and a more potent uncoupling of OXPHOS in whole cell Seahorse respirometer studies, compared to the unsubstituted TPP+ moiety. Conversely, the presence of electron-withdrawing groups in the same position led to an increase in Hückel charge and a decrease in mitochondrial uncoupling. Substitution of the para-trifluoromethyl group, which is the strongest electron-withdrawing group tested, led to a complete abolition of mitochondrial uncoupling in conjugates tested here. It has been previously reported that lipophilicity of a TPP+ conjugate plays an important role in mitochondrial accumulation21 and extension of mitochondrial uncoupling. Hence, TPP+ compounds with longer, more lipophilic linkers led to better mitochondrial targeting but at the cost of more potent mitochondrial uncoupling. However, we demonstrated here that modulation of Hückel charge can have a greater impact on uncoupling potency, or lack thereof, than modulation of lipophilicity.
While there have been recent reports about various aromatic substitutions on the TPP+ moiety,29,34,35 they have largely focused on one TPP+ modification each. On the contrary, this is the first report of a systematic study of the relationship between the TPP+ structure and its impact on mitochondrial uptake and uncoupling. Most significantly, we identified the 4-CF3-TPP+ moiety as a better alternative to TPP+ for delivery of cargo into mitochondria. The 4-CF3-TPP+ moiety successfully delivers cargo into mitochondria within intact cells without uncoupling mitochondrial respiration or the associated toxicity unlike the traditional TPP+ moiety.
The large ionic radius of the TPP+ moiety, due to resonance-mediated delocalization of the cationic charge on the phosphorus atom, plays an important role in its ability to move across biological membranes (Figure 1A).8 Any aromatic substitution, whether electron withdrawing or donating, will further increase the ionic radius, thereby further facilitating transport across membranes. Why electron-withdrawing para-substitution abrogates uncoupling of mitochondrial OXPHOS, whereas electron-donating para-substitution further intensifies it, remains a matter of further investigation. Theories proposed to explain uncoupling caused by alkyl-TPP+ conjugates include compromise of inner mitochondrial membrane integrity at higher concentration leading to proton leak20 or ionic interactions with endogenous fatty acid anions to stimulate their transport across inner mitochondrial membrane in order to act as protonophores.19 However, these theories consider only the lipophilicity factor of alkyl-TPP+ compounds and cannot explain the absence of uncoupling with 4-CF3-TPP+-alkyl conjugates. In-fact, 4-CF3-TPP+ derivatives are more lipophilic than the unsubstituted TPP+ derivatives studied in these reports. Similarly, 4-OCH3-TPP+ derivatives, which are less lipophilic than unsubstituted TPP+ derivatives, result in more potent uncoupling.
While TPP+ derivatives with electron-donating groups on the aryl rings are unsuitable for mitochondrial cargo delivery due to more potent uncoupling, they may find application as potential anticancer drugs,36,37 where potent uncoupling and/or more selective delivery of toxic agents to the mitochondria in cancer cells is the goal. Further studies toward that end as well as for inert delivery of other functional cargo like antioxidants using 4-CF3-TPP+ are currently underway.
EXPERIMENTAL SECTION
General Synthesis Methods and Equipment.
All reagents and solvents were purchased from Sigma-Aldrich, Alfa Aesar, Fisher Scientific, or Acros Organics and used without further purification. Reaction progress was monitored utilizing thin-layer chromatography (TLC) or analytical high-performance liquid chromatography (HPLC). The TLC plates used were glass-backed 0.25 mm TLC silica gel 60 plates containing fluorescence indicator F264 (EMD Sciences). Analytical HPLC system used for monitoring reaction progress consisted of an LC-10ATvp pump, DGU-14A degasser, FCV-10ALvp mixer, and SPD-10A UV–vis detector operated using Shimadzu (Kyoto, Japan) Client/Server version 7.4 software installed on a Dell OptiPlex GX400 PC, and the column used was Phenomenex Luna C18 100 Å LC column (4.6 mm × 250 mm). The mobile phase was composed of nanopure water with 0.1% trifluoroacetic acid (TFA) (A) and HPLC grade acetonitrile (ACN) with 0.1% TFA (B). For monitoring reaction progress, HPLC analyses were obtained by running a gradient of 10–95% B over 30 min with a flow rate of 1.0 mL/min. For compounds purified by silica gel chromatography, separation was accomplished using silica gel 60 (particle size, 0.040–0.063 mm; 230–400 mesh ASTM) using solvent systems optimized for the specific compound. Removal of organic solvents was accomplished by first drying the organic solvent over Na2SO4, filtering, and then concentrating using a rotary evaporator. Aqueous solutions were concentrated by either rotary evaporation or lyophilization. Compounds requiring HPLC for purification were separated by preparative HPLC employing a Phenomenex Luna 5 μm PFP (2) 100 Å LC column (250 × 21.20 mm) with a Shimadzu HPLC system consisted of an LC-20AP preparative LC pump and SPD-20A UV–vis detector and using LabSolutions software installed on Dell OptiPlex 9020 PC. The gradient used for separation included the mobile phase starting at 40% B and increasing to 95% B linearly over 25 min with a flow rate of 10.0 mL/min. All synthesized compounds were characterized for purity and identity by nuclear magnetic resonance (NMR) and mass spectrometry (MS). 1H and 19F NMR spectra were obtained using a Bruker Ultrashield 300 MHz instrument. The chemical shifts are reported in parts per million and referenced to tetramethylsilane and solvent. All spectra were obtained in CDCl3 or MeOD. Standard abbreviations indicating multiplicity are used as follows: br s = broad, d = doublet, m = multiplet, s = singlet, and t = triplet. Mass spectrometry data were obtained either using (a) Waters ACQUITY TQD mass spectrometer, (b) Shimadzu 2010A 131 LC/MS, or (c) Thermo LCQ Deca mass spectrometer, each utilizing ESI ionization and quadrupole ion trap mass analyzer. Purified compounds were analyzed with an analytical HPLC system mentioned above, to ensure purity greater than 95% prior to use in assays and biological testing. The gradient used, unless otherwise mentioned, was 40–95% ACN (with 0.1% TFA) over 20 min and 95% CAN (with 0.1% TFA) for the next 5 min, with a flow rate of 1.0 mL/min.
Synthesis of Substituted TPP+-Decyl Compounds.
The general synthesis procedure is given here. The temperature and duration of the reaction varied with the triphenylphosphine derivative utilized and are reported for each individual derivative. Para-substituted triphenylphosphine (1 equiv) and 1-bromodecane (7) (10 equiv) were combined neat in a round-bottom flask and set to stir with heating, under a reflux condenser. Reaction progress was monitored using TLC in 5% MeOH in DCM. After the reaction was complete, as determined by the disappearance of the phosphine spot, the reaction mixture was dissolved in DCM and purified by silica gel chromatography using a gradient from 1% MeOH in DCM to 10% MeOH in DCM to give the pure para-substituted TPP+-decyl derivative. The compounds were hygroscopic in nature.
4-OMe-TPP-DC (9): Decyl Tris(4-methoxyphenyl)phosphonium Bromide.
Temperature, 100 °C; duration, 6 h. Product obtained as a white solid crust after purification with silica gel chromatography. Yield, 69% (121.9 mg, 0.213 mmol). 1H NMR (300 MHz, CDCl3): δ 7.60 (dd, J = 11.9, 8.8 Hz, 6H), 7.13 (dd, J = 8.9, 2.4 Hz, 6H), 3.86 (s, 9H), 3.27 (m, 2H), 1.51 (m, 4H), 1.18 (m, 12H), 0.79 (t, J = 6.7 Hz, 3H). MS (ESI) m/z: calculated for (M+), 493.2866; found, 493.2897. HPLC retention time = 18.4 min.
4-CH3-TPP-DC (10): Decyl Tri-4-tolylphosphonium Bromide.
Temperature, 110 °C; duration, 6 h. Product obtained as a white solid crust after purification with silica gel chromatography. Yield, 87% (150.4 mg, 0.286 mmol). 1H NMR (300 MHz, CDCl3): δ 7.49 (dd, J = 12.3, 8.3 Hz, 6H), 7.36 (dd, J = 8.1, 3.1 Hz, 6H), 3.25 (m, 2H), 2.30 (s, 9H), 1.42 (s, 4H), 1.04 (m, 12H), 0.66 (t, J = 6.8 Hz, 3H). MS (ESI) m/z: calculated for (M+), 445.3; found, 445.3. HPLC retention time = 19.8 min.
4-Cl-TPP-DC (12): Decyl Tris(4-chlorophenyl)phosphonium Bromide.
Temperature, 110 °C; duration, 24 h. Product obtained as a white solid crust after purification with silica gel chromatography. Yield, 62% (116.2 mg, 0.198 mmol). 1H NMR (300 MHz, CDCl3): δ 7.84 (dd, J = 12.2, 8.6 Hz, 6H), 7.63 (dd, J = 8.6, 2.6 Hz, 6H), 3.84 (m, 2H), 1.51 (m, 4H), 1.14 (m, 12H), 0.78 (t, J = 6.8 Hz, 3H). MS (ESI) m/z: calculated for (M+), 505.1; found, 505.3. HPLC retention time = 19.0 min.
4-F-TPP-DC (13): Decyl Tris(4-fluorophenyl)phosphonium Bromide.
Temperature, 120 °C; duration, 24 h. Product obtained as a white solid crust after purification with silica gel chromatography. Yield, 83% (141.0 mg, 0.262 mmol). 1H NMR (300 MHz, CDCl3): δ 7.90 (ddd, J = 12.9, 8.1, 5.1 Hz, 6H), 7.38 (m, 6H), 3.78 (m, 2H), 1.53 (m, 4H), 1.14 (m, 12H), 0.78 (t, J = 6.6 Hz, 3H). 19F NMR (282 MHz, CDCl3): δ −99.49 (m, 3F). MS (ESI) m/z: calculated for (M+), 457.23; found, 457.12. HPLC retention time = 19.1 min.
4-CF3-TPP-DC (14): Decyl Tris(4-(trifluoromethyl)phenyl)-phosphonium Bromide.
Temperature, 140 °C; duration, 72 h. Product obtained as a white solid crust after purification with silica gel chromatography. Yield, 63% (93.5 mg, 0.136 mmol). 1H NMR (300 MHz, CDCl3): δ 8.20 (dd, J = 12.2, 8.3 Hz, 5H), 7.97 (dd, J = 8.3, 2.5 Hz, 5H), 4.32 (m, 2H), 1.61 (m, 4H), 1.20 (m, 12H), 0.83 (t, J = 6.8 Hz, 3H). 19F NMR (282 MHz, CDCl3): δ −63.71 (s, 9F). MS (ESI) m/z: calculated for (M+), 607.2171; found, 607.2202. HPLC retention time = 19.1 min.
3-OMe-TPP-DC (30): Decyl Tris(3-methoxyphenyl)phosphonium Bromide.
Temperature, 100 °C; duration, 12 h. Product obtained as a white solid crust after purification with silica gel chromatography. Yield, 84% (135.4 mg, 0.236 mmol). 1H NMR (300 MHz, CDCl3): δ 7.59 (m, 3H), 7.29 (m, 9H), 3.91 (s, 9H), 3.78 (m, 2H), 1.61 (m, 4H), 1.18 (m, 12H), 0.84 (t, J = 6.7 Hz, 3H). MS (ESI) m/z: calculated for (M+), 493.2866; found, 493.2704. HPLC retention time = 17.9 min.
3-F-TPP-DC (31): Decyl Tris(3-fluorophenyl)phosphonium Bromide.
Temperature, 110 °C; duration, 12 h. Product obtained as a white solid crust after purification with silica gel chromatography. Yield, 79% (133.4 mg, 0.248 mmol). 1H NMR (300 MHz, CDCl3): δ 7.95 (dd, J = 12.4, 7.8 Hz, 3H), 7.80 (ddd, J = 7.8, 6.3, 4.0 Hz, 3H), 7.52 (m, 3H), 7.39 (m, 3H), 4.09 (m, 2H), 1.66 (m, 4H), 1.22 (m, 12H), 0.85 (t, J = 6.8 Hz, 3H). 19F NMR (282 MHz, CDCl3): δ−105.98 (m, 3H). MS (ESI) m/z: calculated for (M+), 457.2266; found, 457.2171. HPLC retention time = 16.6 min.
Synthesis of Para-Substituted TPP+-Butyl Compounds.
The general synthesis procedure is given here. The temperature and duration of the reaction varied with the triphenylphosphine derivative utilized and are reported for each individual derivative. Para-substituted triphenylphosphine (1 equiv) was dissolved in toluene (2 mL) in a round-bottom flask. 1-Bromobutane (8) (10 equiv) was added, and the reaction was set to stir at a particular temperature for a particular time, under a reflux condenser. The reaction progress was monitored using TLC in 5% MeOH in DCM. When temperatures higher than the boiling point of 1-bromobutane (100–104 °C) were used for extended periods, 5 equiv of 8 was added every 6 h to approximately replenish the evaporated quantity. After the reaction was complete, as determined by the disappearance of the phosphine spot, toluene was removed in vacuo. The residue was dissolved in DCM and purified by silica gel chromatography using a gradient from 1% MeOH in DCM to 10% MeOH in DCM to give the pure para-substituted TPP+-butyl derivative. Products were hygroscopic.
4-OMe-TPP-BU (15): Butyl Tris(4-methoxyphenyl)phosphonium Bromide.
Temperature, 120 °C; duration, 6 h. Product obtained as a white solid crust after purification with silica gel chromatography. Yield, 73% (104.7 mg, 0.214 mmol). 1H NMR (300 MHz, CDCl3): δ 7.68 (dd, J = 11.9, 8.9 Hz, 6H), 7.16 (dd, J = 9.0, 2.5 Hz, 6H), 3.90 (s, 9H), 3.41 (m, 2H), 1.60 (m, 4H), 0.90 (t, J = 7.0 Hz, 3H). MS (ESI) m/z: calculated for (M+), 409.193; found, 409.170. HPLC retention time = 11.3 min.
4-CH3-TPP-BU (16): Butyl Tri-p-tolylphosphonium Bromide.
Temperature, 120 °C; duration, 6 h. Product obtained as a white solid crust after purification with silica gel chromatography. Yield, 64% (93.2 mg, 0.211 mmol). 1H NMR (300 MHz, CDCl3): δ 7.61 (dd, J = 12.3, 8.1 Hz, 6H), 7.45 (dd, J = 7.8, 2.8 Hz, 6H), 3.50 (m, 2H), 2.44 (s, 9H), 1.60 (m, 4H), 0.86 (t, J = 6.8 Hz, 3H). MS (ESI) m/z: calculated for (M+), 361.2080; found, 361.2045. HPLC retention time = 12.9 min.
H-TPP-BU (17): Butyl Triphenylphosphonium Bromide.
Temperature, 120 °C; duration, 24 h. Product obtained as a white solid crust after purification with silica gel chromatography. Yield, 67% (209.4 mg, 0.524 mmol). 1H NMR (300 MHz, CDCl3): δ 7.74 (m, 15H), 3.77 (dd, J = 15.7, 12.8 Hz, 2H), 1.62 (m, 4H), 0.88 (t, J = 7.1 Hz, 3H). MS (ESI) m/z: calculated for (M+), 319.1610; found, 319.1578. HPLC retention time = 8.7 min.
4-Cl-TPP-BU (18): Butyl Tris(4-chlorophenyl)phosphonium Bromide.
Temperature, 120 °C; duration, 24 h. Product obtained as a white solid crust after purification with silica gel chromatography. Yield, 59% (83.9 mg, 0.170 mmol). 1H NMR (300 MHz, CDCl3): δ 7.86 (dd, J = 12.2, 8.5 Hz, 6H), 7.68 (dd, J = 8.5, 2.7 Hz, 6H), 4.01 (m, 2H), 1.68 (m, 4H), 0.90 (t, J = 7.2 Hz, 3H). MS (ESI) m/z: calculated for (M+), 421.0441; found, 421.0367. HPLC retention time = 12.5 min.
4-F-TPP-BU (19): Butyl Tris(4-fluorophenyl)phosphonium Bromide.
Temperature, 120 °C; duration, 36 h. Product obtained as a white solid crust after purification with silica gel chromatography. Yield, 18% (26.8 mg, 0.059 mmol). 1H NMR (300 MHz, CDCl3): δ 7.95 (ddd, J = 12.1, 8.7, 5.0 Hz, 6H), 7.40 (td, J = 8.4, 2.3 Hz, 6H), 3.98 (m, 2H), 1.69 (m, 2H), 1.55 (m, 2H), 0.90 (t, J = 7.2 Hz, 1H). 19F NMR (282 MHz, CDCl3): δ −99.35 (m, 3F). MS (ESI) m/z: calculated for (M+), 373.1327; found, 373.1294. HPLC retention time = 9.7 min.
4-CF3-TPP-BU (20): Butyl Tris(4-(trifluoromethyl)phenyl)-phosphonium Bromide.
Temperature, 140 °C; duration, 72 h. Product obtained as a white solid crust after purification with silica gel chromatography. Yield, 23% (30.4 mg, 0.504 mmol). 1H NMR (300 MHz, CDCl3): δ 8.19 (dd, J = 12.3, 8.2 Hz, 6H), 7.98 (dd, J = 8.3, 2.6 Hz, 6H), 4.34 (m, 2H), 1.72 (m, 4H), 0.90 (t, J = 7.2 Hz, 3H). 19F NMR (282 MHz, CDCl3): δ −63.70 (s, 9F). MS (ESI) m/z: calculated for (M+), 523.12; found, 523.00. HPLC retention time = 13.7 min.
Synthesis of Para-Substituted TPP+-Benzyl Compounds.
The general synthesis procedure is given here. Para-substituted triphenylphosphine (1 equiv) was dissolved in toluene (2 mL) in a round-bottom flask. Benzyl bromide (21) (10 equiv) was added, and the reaction was set to stir at 80 °C for 24 h. The reaction progress was monitored using TLC in 5% MeOH in DCM. After the reaction was complete, as determined by the disappearance of the phosphine spot, toluene was removed in vacuo. The residue was dissolved in DCM and purified by silica gel chromatography using a gradient from 1% MeOH in DCM to 10% MeOH in DCM to give the pure para-substituted TPP+-benzyl derivative.
4-OMe-TPP-BZ (22): Benzyl Tris(4-methoxyphenyl)phosphonium Bromide.
Product obtained as a white powder after purification with silica gel chromatography. Yield, 75% (113.5 mg, 0.217 mmol). 1H NMR (300 MHz, CDCl3): δ 7.54 (dd, J = 11.9, 8.9 Hz, 6H), 7.08 (m, 11H), 5.01 (d, J = 14.3 Hz, 2H), 3.89 (s, 9H). MS (ESI) m/z: calculated for (M+), 443.177; found, 443.154. HPLC retention time = 10.8 min.
4-CH3-TPP-BZ (23): Benzyl Tri-p-tolylphosphonium Bromide.
Product obtained as a white powder after purification with silica gel chromatography. Yield, 82% (132.5 mg, 0.279 mmol). 1H NMR (300 MHz, CDCl3): δ 7.52 (dd, J = 12.2, 8.3 Hz, 6H), 7.40 (dd, J = 8.2, 3.2 Hz, 6H), 7.13 (m, 5H), 5.15 (d, J = 14.3 Hz, 2H), 2.46 (s, 9H). MS (ESI) m/z: calculated for (M+), 395.1923; found, 395.1892. HPLC retention time = 12.0 min.
H-TPP-BZ (24): Benzyl Triphenylphosphonium Bromide.
Product obtained as a white powder after purification with silica gel chromatography. Yield, 76% (127.3 mg, 0.294 mmol). 1H NMR (300 MHz, CDCl3): δ 7.69 (m, 15H), 7.12 (m, 5H), 5.36 (d, J = 14.4 Hz, 2H). MS (ESI) m/z: calculated for (M+), 353.1454; found, 353.1419. HPLC retention time = 8.5 min.
4-Cl-TPP-BZ (25): Benzyl Tris(4-chlorophenyl)phosphonium Bromide.
Product obtained as a white powder after purification with silica gel chromatography. Yield, 80% (123.6 mg, 0.230 mmol). 1H NMR (300 MHz, MeOD): δ 7.74 (m, 6H), 7.61 (m, 6H), 7.33 (m, 1H), 7.24 (m, 2H), 6.99 (m, 2H), 4.92 (d, J = 15.1 Hz, 2H). MS (ESI) m/z: calculated for (M+), 455.0284; found, 457.0203. HPLC retention time = 12.5 min.
4-F-TPP-BZ (26): Benzyl Tris(4-fluorophenyl)phosphonium Bromide.
Product obtained as a white powder after purification with silica gel chromatography. Yield, 73% (116.9 mg, 0.240 mmol). 1H NMR (300 MHz, CDCl3): δ 7.81 (m, 6H), 7.18 (m, 11H), 5.80 (d, J = 15.2 Hz, 2H). 19F NMR (282 MHz, CDCl3): δ −99.90 (m, 3F). MS (ESI) m/z: calculated for (M+), 407.1171; found, 407.1130. HPLC retention time = 9.6 min.
4-CF3-TPP-BZ (27): Benzyl Tris(4-(trifluoromethyl)phenyl)-phosphonium Bromide.
Product obtained as a white powder after purification with silica gel chromatography. Yield, 67% (93.1 mg, 0.146 mmol). 1H NMR (300 MHz, CDCl3): δ 8.05 (dd, J = 12.3, 8.2 Hz, 6H), 7.68 (dd, J = 8.4, 2.7 Hz, 6H), 7.24 (m, 5H), 6.59 (d, J = 15.8 Hz, 2H). 19F NMR (282 MHz, CDCl3): δ −63.64 (s, 9F). MS (ESI) m/z: calculated for (M+), 557.1075; found, 557.1033. HPLC retention time = 13.7 min.
Miscellaneous.
Tris(1-Naphthyl)-P-DC (33): Decyl tri-(naphthalen-1-yl)phosphonium Iodide.
1-Bromodecane (7) (0.5 mL, 2.424 mmol) and sodium iodide (545.0 mg, 3.636 mmol) were added to a flask and stirred for 15 min. Tris(naphthalen-1-yl)phosphine (32) (100.1 mg, 0.242 mmol) was then added, and the flask was set to heat at 130 °C for 48 h. The reaction mixture was dissolved in DCM and purified by silica gel chromatography using a gradient from 1% MeOH in DCM to 6% MeOH in DCM to give pure Tris(1-Naphthyl)-P-DC (33) as a foamy yellowish solid. Yield, 40% (65.9 mg, 0.097 mmol). 1H NMR (300 MHz, CDCl3): δ 8.33 (d, J = 7.6 Hz, 3H), 8.06 (d, J = 8.0 Hz, 6H), 7.63 (m, 12H), 3.73 (m, 2H), 1.43 (m, 2H), 1.13 (m, 14H), 0.82 (t, J = 7.0 Hz, 3H). MS (ESI) m/z: calculated for (M+), 553.3019; found, 553.3032. HPLC retention time = 20.6 min.
Synthesis of TPP+-TAMRA Conjugates.
Br-DC-Phthalimide (36): 2-(10-Bromodecyl)isoindoline-1,3-dione.
A suspension of potassium phthalimide (35) (1 g, 5.400 mmol) in 15 mL of anhydrous DMF was added to a stirring solution of 1,10-dibromodecane (34) (6.48 g, 21.595 mmol) in anhydrous DMF (20 mL). The mixture was stirred at 90 °C for 4 h under an argon atmosphere. After the reaction was complete, as monitored by the disappearance of the starting material spot on TLC eluted 20% EtOAc in hexanes, the reaction mixture was filtered to remove the white precipitate and the filtrate was evaporated to dryness. The crude product was dissolved in DCM and purified by silica gel chromatography starting elution with hexanes followed by 20% EtOAc in hexanes to give UICK-III-069 as a white powder. Yield, 94% (1.9 g, 5.054 mmol). 1H NMR (300 MHz, CDCl3): δ 7.83 (dt, J = 7.0, 3.5 Hz, 2H), 7.71 (td, J = 5.2, 2.1 Hz, 2H), 3.67 (t, J = 7.0 Hz, 2H), 3.39 (t, J = 6.9 Hz, 2H), 1.83 (m, 2H), 1.66 (m, 2H), 1.33 (m, 12H). The MS (ESI) compound does not ionize well because of its hydrophobic nature.
TPP-DC-Phthalimides (37 and 38).
Either triphenylphosphine (3) (143.1 mg, 0.546 mmol) or tris(4-trifluoromethylphenyl)phosphine (6) (143.36 mg, 0.307 mmol) and Br-DC-Phthalimide (36) (177.9 mg, 0.486 mmol) were dissolved in toluene (3 mL) in a round-bottom flask and set to stir at 120–140 °C for 36–48 h, under a reflux condenser. The reaction progress was monitored using TLC in 5% MeOH in DCM. After the reaction was complete, as determined by the disappearance of the phthalimide spot, the reaction mixture was concentrated in vacuo. The residue was dissolved in DCM and purified by silica gel chromatography using a gradient from 1% MeOH in DCM to 10% MeOH in DCM.
H-TPP-DC-Phthalimide (37): (10-(1,3-Dioxoisoindolin-2-yl)-decyl)triphenylphosphonium Bromide.
Product obtained as a white solid crust. Yield, 45% (135.0 mg, 0.245 mmol). 1H NMR (300 MHz, CDCl3): δ 7.78 (m, 11H), 7.67 (m, 8H), 3.68 (m, 2H), 3.59 (t, J = 7.3 Hz, 3H), 1.57 (m, 4H), 1.17 (m, 12H). MS (ESI) m/z: calculated for (M+), 548.27; found, 547.97.
4-CF3-TPP-DC-Phthalimide (38): (10-(1,3-Dioxoisoindolin-2-yl)-decyl)tris(4-(trifluoromethyl)phenyl)phosphonium Bromide.
Product obtained as a white solid crust. Yield, 44% (101.4 mg, 0.122 mmol). 1H NMR (300 MHz, CDCl3): δ 8.19 (dt, J = 14.6, 7.3 Hz, 6H), 7.96 (dt, J = 10.5, 5.3 Hz, 6H), 7.80 (dd, J = 5.5, 3.0 Hz, 2H), 7.69 (dd, J = 5.5, 3.0 Hz, 2H), 4.30 (m, 2H), 3.61 (m, 2H), 1.61 (m, 2H), 1.20 (m, 14H). 19F NMR (282 MHz, CDCl3): δ −63.69 (s, 9F). MS (ESI) m/z: calculated for (M+), 752.23; found, 752.02.
TPP-DC-NH2 (39 and 40).
TPP-DC-Phthalimides (37 and 38) (0.122–0.245 mmol) were dissolved in 48% aqueous HBr (10 mL) and set to heat at 130 °C for 24 h. The reaction progress was monitored using TLC in 10% MeOH in DCM. The formation of primary amine was detected using ninhydrin stain with subsequent heating. After the reaction was complete, as determined by the disappearance of the phthalimide spot, the reaction mixture was concentrated under vacuum and the resulting residue was purified by silica gel chromatography using 3 to 10% MeOH in DCM.
H-TPP-DC-NH2 (39): (10-Ammoniodecyl)triphenylphosphonium Bromide.
Product obtained as a white solid crust. Yield, 71% (100.5 mg, 0.174 mmol). 1H NMR (300 MHz, MeOD): δ 7.81 (m, 15H), 3.46 (m, 2H), 3.31 (m, 2H, exchangeable), 2.92 (m, 2H), 1.63 (m, 4H), 1.32 (m, 10H). MS (ESI) m/z: calculated for (M+) and (M+ + H+), 418.2658 and 209.6366, respectively; found, 418.2679 and 209.6365, respectively. HPLC retention time = 15.8 min using a linear gradient of 10% ACN (with 0.1% TFA) to 95% ACN (with 0.1% TFA) over 30 min at 1.0 mL/min.
4-CF3-TPP-DC-NH2 (40): (10-Ammoniodecyl)tris(4-(trifluoromethyl)phenyl)phosphonium Bromide.
Product obtained as a white solid crust. Yield, 78% (74.4 mg, 0.095 mmol). 1H NMR (300 MHz, MeOD): δ 8.10 (m, 12H), 3.69 (m, 2H), 3.29 (m, 2H, exchangeable), 2.89 (t, J = 7.6, 2H), 1.65 (m, 6H), 1.29 (m, 10H). 19F NMR (282 MHz, MeOD): δ −65.11 (s, 9F). MS (ESI) m/z: calculated for (M+) and (M+ + H+), 622.2280 and 311.6176, respectively; found, 622.2299 and 311.6166, respectively. HPLC retention time = 20.0 min using a linear gradient of 10% ACN (with 0.1% TFA) to 95% ACN (with 0.1% TFA) over 30 min at 1.0 mL/min.
TPP-DC-TAMRA (42 and 43).
TPP-DC-NH2 (39 and 40) (1.1 equiv) and diisopropylethylamine (1.2 equiv) were dissolved in ACN in a light-protected flask and stirred for 15 min. 6-Carboxytetramethylrhodamine, succinimidyl ester (41) (1 equiv) was added to the flask, and the reaction was stirred for a further 1 h at room temperature. The reaction progress was monitored by analytical HPLC. After the reaction was complete, as determined by the disappearance of the starting material peak, the reaction mixture was diluted with water and purified by preparative HPLC to give the desired modified TPP-DC-TAMRA conjugate (42 and 43).
H-TPP-DC-TAMRA (42): 2-(6-(Dimethylamino)-3-(dimethyliminio)-3H-xanthen-9-yl)-5-((10-(triphenylphosphonio)decyl)-carbamoyl)benzoate Bromide.
Product obtained as a red solid after purification with preparative HPLC. Yield, 80% (25.5 mg, 0.028 mmol). MS (ESI) m/z: calculated for (M+) and (M2+), 830.41 and 415.21, respectively; found, 830.25 and 415.65, respectively. HPLC retention time = 10.1 min.
4-CF3-TPP-DC-TAMRA (43): 2-(6-(Dimethylamino)-3-(dimethyliminio)-3H-xanthen-9-yl)-5-((10-(tris(4-(trifluoromethyl)phenyl)-phosphonio)decyl)carbamoyl)benzoate Bromide.
Product obtained as a red solid after purification with preparative HPLC. Yield, 75% (4.9 mg, 0.004 mmol). MS (ESI) m/z: calculated for (M+) and (M2+), 1034.3703 and 517.1852, respectively; found, 1034.3824 and 517.6877, respectively. HPLC retention time = 12.6 min.
General Cell Culture Conditions.
The maintenance of cell culture, media, preparation of TPP+ compound stocks, and the 6 day procedure for seeding and differentiation of cells in a plate was identical for evaluation in Seahorse respirometer studies of mitochondrial membrane potential, cell viability, and cell uptake as detailed below. C2C12 (ATCC CRL-1772) mouse myoblasts were obtained from ATCC. To form myotubes for the assay, cells were seeded at 5000 cells/well to Seahorse 24-well V7 PS plates or 96-well, black plate with μClear bottom (Greiner Bio-One 655090) plates and grown to confluency in a 37 °C humidified incubator with 10% CO2 over 2 days in a growth medium (DMEM (Thermo Fisher #11965, 4.5 g/L glucose + L-glutamine, no sodium pyruvate) + 10% fetal bovine serum (HyClone, characterized)). The growth medium was changed daily. Wells were then exposed to a differentiation medium (DMEM (Thermo Fisher #11965, 4.5 g/L glucose + L-glutamine, no sodium pyruvate) + 2% equine serum (HyClone)) for the next 3 days, which promotes fusion of myoblasts to form myotubes. The differentiation medium was changed daily. TPP+ compounds were dissolved in 100% ethanol at a concentration of 50 mM. An aliquot was diluted 5-fold with ethanol to make a working stock of 10 mM. All stocks were stored at −20 °C and kept on ice during use. On the third day with the differentiation medium (day before the experiment), 10 mM ethanolic stock of the compounds was diluted with the differentiation medium to prepare a two-fold dilution series from 62.5 to 2000 nM, added to the cell culture plate, and incubated at a 37 °C humidified incubator with 10% CO2 for 20 h. The maximum ethanol concentration in the cell culture medium was 0.004%.
Seahorse Respirometer “Mito Stress Test” Assay.
On the day of the experiment, the overnight medium from the Seahorse plate was replaced with the Seahorse medium (SHM) (bicarbonate-free DMEM (Sigma catalog #D5030), with added 5.55 mM glucose, 2 mM L-glutamine, 1 mM pyruvate, 15 mg/L phenol red, 1.85 g/L NaCl, and 10 mM HEPES, pH 7.45 at 37 °C) and cells were incubated at 37 °C in a humidified incubator with no CO2. After 20 min, the medium in the wells was replaced with the same overnight series of concentrations of the compounds and diluted in the SHM and the plate was incubated in a 37 °C, CO2-free humidified incubator for additional 50 min. Finally, the cells were tested in a “mito stress test” in a Seahorse XF24 by sequential injection of ATP synthase inhibitor oligomycin (final concentration, 2 μM), mitochondrial uncoupler 2,4-dinitrophenol (90 μM), and a combination of rotenone (2 μM, complex I inhibitor) and antimycin A (0.5 μM, complex III inhibitor) to inhibit mitochondrial respiration. Analysis of the data was performed using Wave 2.3 software (Agilent Technologies, Santa Clara, CA) and “XF Mito Stress Test Report” Microsoft Excel macro provided by Seahorse Bioscience.
Determination of Cell Viability.
On the day of the experiment, the medium containing the compounds was removed from the plate and the cells were washed twice with 50 μL of SHM. After that, the SHM and 10 μL of a 5 mg/mL aqueous MTT solution were added to each well and the plate was incubated for 2 h at 37 °C in a CO2-free incubator. Following this, the supernatant was carefully removed and the formazan crystals were dissolved in 200 μL of DMSO. Absorbance was measured at 570 nm using a μQuant microplate spectrophotometer plate reader and Gen5 data analysis software (BioTek Instruments, Inc., Winooski, VT). Percent viability was expressed as a ratio of absorbance of treated well with untreated control well. The experiment was repeated a total of three times with each concentration measured in duplicate.
Determination of Mitochondrial Membrane Potential.
Overnight solution in the 96-well plate was removed, and cells were washed with 50 μL of SHM twice. After this, 0.1 mL of 5 μg/mL solution of JC-1 (Adipogen, San Diego, CA) in the SHM was added and the plate was incubated at 37 °C in a CO2-free incubator for 1 h. The JC-1 solution was removed, and the cells were washed with 50 μL of SHM followed by final addition of 0.1 mL of SHM. The red and green fluorescence was determined at the excitation and emission wavelengths of 544/590 and 485/520, respectively, using a FLUOstar OPTIMA microplate reader and FLUOstar OPTIMA data analysis software (version 2.10) (BMG Labtech GmbH, Ortenberg, Germany). The red to green ratio was calculated as an index of mitochondrial potential, a lower ratio indicative of lower potential. The experiment was repeated a total of three times with each concentration measured in duplicate. An Olympus IX71 inverted compound microscope (Olympus Life Science Solutions, Tokyo, Japan) was used for taking images. SimplePCI version 6.6 software (Hamamatsu Photonics, Japan) was used for image acquisition and processing.
Determination of Cellular Uptake of TPP+-TAMRA Conjugates.
Overnight media containing various concentrations of the TPP+ compounds were aspirated from the plate, and the cells were dosed with 250 nM MitoTracker Green FM (Thermo Fisher Scientific, Waltham, MA) and 1 μg/mL Hoechst 33342 (Thermo Fisher Scientific, Waltham, MA) in 0.1 mL of SHM and incubated for 30 min at 37 °C in a CO2-free atmosphere. Following the incubation, the medium in all wells was replaced with a fresh SHM without the dyes and the plate was read on an Operetta high-content imaging system (PerkinElmer, Inc., Waltham, MA) at 37 °C in a CO2-free atmosphere. The excitation and emission wavelengths used were respectively 552/576 (6-TAMRA), 490/516 (MitoTracker Green FM), 361/497 (Hoechst 33342). Harmony analysis software version 4.1 was used for data analysis. The number of nuclei was measured using the Hoechst 33342 stain as a measure of toxicity. MitoTracker Green FM was used to define the area occupied by mitochondria. TAMRA fluorescence in the area defined by MitoTracker Green FM was measured to express colocalization of TPP-TAMRA conjugates inside the mitochondria within intact cells. The same dilution series of TPP+-TAMRA conjugates was identically dosed in another 96-well, black plate with μClear bottom (Greiner Bio-One 655090) without any cells, and the fluorescence of TAMRA was read with an EnVision plate reader (PerkinElmer, Inc., Waltham, MA) at excitation and emission wavelengths of 552 and 576, respectively. Wallace Envision Manager 1.12 was used for data analysis. The ratio of TAMRA fluorescence in the presence and absence of the cells indicated relative mitochondrial accumulation of the TPP-TAMRA conjugates by eliminating variability in concentration and differences in individual fluorescence intensity of the TPP-TAMRA conjugates. All experiments were performed a minimum of three times with each concentration measured in duplicate.
Mitochondrial Isolation.
Six-week-old, male C57BL/6J mice were ordered from Jackson Laboratory (Bar Harbor, ME). The protocol for harvest of tissue and isolation of mitochondria was approved by our organization’s Institutional Animal Care and Use Committee. All procedures were done on ice or with equipment cooled to 4 °C. Liver and hind limb tissue were harvested and placed in a cold isolation medium (ISO) (0.25 M sucrose, 5 mM HEPES (pH 7.2), and 0.1 mM EDTA) containing 0.1% defatted BSA. Connective tissue, fat, and nerves were removed from the hind limb skeletal muscles, and they were minced with scissors for 4 min in ISO + BSA to aid tissue disruption. Up to 1 g of tissue was homogenized in 14 mL of ISO using a Potter-Elvehjem (Teflon–glass)-type tissue grinder in an ice bucket at approximately 300 rpm for 6 passes. Homogenate was centrifuged at 500g for 10 min, and the suspension was transferred to a Sorvall-type tube (Oak Ridge screw cap). The pellet was discarded. The suspension was then centrifuged at 8500g for 10 min. The resulting suspension was discarded, and the mitochondrial pellet was resuspended with 1 mL of ISO (no BSA) and centrifuged at 8500g for 10 min. This procedure was repeated once more. The final suspension was then discarded, and the pellet was resuspended at ~50% v/v in ISO with BSA to give the crude preparation of either liver or hind limb mitochondria. In addition, mitochondria were purified on a self-generating Percoll (Sigma-Aldrich) gradient using a Beckman XL-80 ultracentrifuge (Beckman Coulter, Inc., Brea, CA) and SW60 swinging bucket rotor as described previously.38 The final washed pellet was resuspended in the BSA-free isolation medium and kept on ice for use in the assay. The protein content was determined by the Bradford method using a Bio-Rad protein assay dye reagent using the manufacturer protocol.
Evaluation of Uptake into Isolated Mitochondria.
Uptake of TPP+ compounds into isolated mitochondria was evaluated using an Oxygraph-2k respirometer with an ion-selective TPP electrode and DatLab 6 data acquisition and analysis software (Oroboros Instruments, Innsbruck, Austria). The TPP and oxygen electrodes were immersed in the respiration medium (105 mM KCl, 10 mM NaCl, 5 mM Na2HPO4, 2 mM MgCl2, 10 mM HEPES (pH 7.2), 1 mM EGTA, and 0.2% BSA), and a stable baseline reading was achieved. TPP+ compounds were added into the chamber in three consecutive 2 μL injections of 250 μM solution, giving final chamber concentrations of 0.25, 0.5, and 0.75 μM. After this, Percoll purified mouse hind limb mitochondria were added to the O2k chamber, giving a final concentration of 0.05 mg/mL. Finally, a combination of mitochondrial substrates (5 mM succinate + 5 mM glutamate + 1 mM malate (SGM)) was added. When a stable reading was achieved, the run was ended. The procedure for evaluation of uptake into liver mitochondria was similar to hind limb mitochondria with the following exceptions. The final liver mitochondria concentrations was 0.1 mg/mL; the final chamber concentrations of TPP+ compounds after each addition were 0.5, 1.0, and 1.5 μM; and the order of addition was the substrate mixture followed by three compound additions and then finally the liver mitochondria.
Computational Calculations of Physicochemical Properties.
To calculate the partition coefficient (cLogP) of the compounds, the structure of the compounds was drawn in ChemBioDraw Ultra 14.0 (PerkinElmer, Waltham, MA) and saved as an SDF file. This file was then opened in MOE 2016.08 (Chemical Computing Group LLC, Montreal, Canada). A 3D wash was performed followed by energy minimization using the MMFF94 force field, and then the “logP (octanol/water)” coefficient was calculated. For measuring the charge density on the phosphorus atom of the TPP+ moiety, structures of the TPP+ compounds were sketched in SYBYL-X (Certara, Princeton, NJ) and all hydrogens were added to the molecule. After this energy minimization was performed using the MMFF94 force field and a gradient of 0.5 kcal/(mol A), Hückel charges were calculated.
Supplementary Material
ACKNOWLEDGMENTS
The work in the laboratory of R.J.K. was supported by the John L. and Carol E. Lach Chair in Drug Delivery Technology and University of Iowa College of Pharmacy research stimulation funds. The work in the laboratory of W.I.S was supported by Merit Review Award 2 I01 BX000285-05 from the U.S. Department of Veterans Affairs, Biomedical Laboratory Research and Development Service, and by the Iowa Fraternal Order of the Eagles. C.A.K. and P.R.C acknowledge the support of predoctoral fellowship from the University of Iowa Center for Biocatalysis and Bioprocessing and of the NIH-sponsored Predoctoral Training Program in Biotechnology (T32 GM008365). C.A.K also acknowledges the support of American Heart Association Postdoctoral Fellowship (#19POST34380212) during the preparation of the manuscript. B.E.G. acknowledges the support of The University of Iowa Summer Research Opportunities Program. Funding for M.W. and the UIHTS Core (R50CA243786-01, P30CA086862, and S10 RR029274-01) are appreciatively acknowledged.
ABBREVIATIONS USED
- TPP+
triphenylphosphonium
- OXPHOS
oxidative phosphorylation
- OCR
oxygen consumption rate
- TFA
trifluoroacetic acid
- ACN
acetonitrile
- MeOH
methanol
- DCM
dichloro-methane
- DMEM
Dulbecco’s modified Eagle’s medium
- SHM
Seahorse medium
- BSA
bovine serum albumi
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01671.
Hückel charge calculations for charge density on the phosphorus atom of various aryl-modified TPP+-decyl conjugates (Tables S1 and S3), logP calculation for all first- and second-generation modified TPP+ compounds (Tables S2 and S4), electrostatic potential maps of various triphenylphosphine moieties (Figure S1), structures for all first- and second-generation modified TPP+ compounds (Figures S2 and S4), resonance of para-and meta-substituted triphenylphosphine and its effect on charge density on the phosphorus atom (Figure S3), accumulation of first-generation TPP+ compounds in mouse liver mitochondria assessed by TPP+ ion-selective electrode measurements (Figure S5), 1H and 19F NMR spectra of synthesized compounds (Figures S6−S39), and HPLC chromatograms of synthesized compounds (Figures S40−S61) (PDF)
Molecular formula string and biochemical data of compounds 9–20, 22–27, 30, 31, 33, 36–40, 42, and 43 (CSV)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.0c01671
The authors declare no competing financial interest.
Contributor Information
Chaitanya A. Kulkarni, Division of Medicinal and Natural Products Chemistry, Department of Pharmaceutical Sciences and Experimental Therapeutics, University of Iowa, Iowa City, Iowa 52242, United States.
Brian D. Fink, Division of Endocrinology and Metabolism, Department of Internal Medicine, University of Iowa and the Iowa City Veterans Affairs Medical Center, Iowa City, Iowa 52246, United States
Bettine E. Gibbs, Division of Medicinal and Natural Products Chemistry, Department of Pharmaceutical Sciences and Experimental Therapeutics, University of Iowa, Iowa City, Iowa 52242, United States.
Pratik R. Chheda, Division of Medicinal and Natural Products Chemistry, Department of Pharmaceutical Sciences and Experimental Therapeutics, University of Iowa, Iowa City, Iowa 52242, United States
Meng Wu, Division of Endocrinology and Metabolism, Department of Internal Medicine, University of Iowa and the Iowa City Veterans Affairs Medical Center, Iowa City, Iowa 52246, United States;; University of Iowa High Throughput Screening (UIHTS) Core, Department of Pharmaceutical Sciences and Experimental Therapeutics, College of Pharmacy, Department of Biochemistry, Carver College of Medicine, University of Iowa, Iowa City, Iowa 52242, United States
William I. Sivitz, Division of Endocrinology and Metabolism, Department of Internal Medicine, University of Iowa and the Iowa City Veterans Affairs Medical Center, Iowa City, Iowa 52246, United States
Robert J. Kerns, Division of Medicinal and Natural Products Chemistry, Department of Pharmaceutical Sciences and Experimental Therapeutics, University of Iowa, Iowa City, Iowa 52242, United States.
REFERENCES
- (1).Lin MT; Beal MF Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [DOI] [PubMed] [Google Scholar]
- (2).Duchen MR; Szabadkai G Roles of mitochondria in human disease. Essays Biochem 2010, 47, 115–137. [DOI] [PubMed] [Google Scholar]
- (3).Nunnari J; Suomalainen A Mitochondria: in sickness and in health. Cell 2012, 148, 1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hoye AT; Davoren JE; Wipf P; Fink MP; Kagan VE Targeting mitochondria. Acc. Chem. Res 2008, 41, 87–97. [DOI] [PubMed] [Google Scholar]
- (5).Jean SR; Ahmed M; Lei EK; Wisnovsky SP; Kelley SO Peptide-mediated delivery of chemical probes and therapeutics to mitochondria. Acc. Chem. Res 2016, 49, 1893–1902. [DOI] [PubMed] [Google Scholar]
- (6).Frantz MC; Wipf P Mitochondria as a target in treatment. Environ. Mol. Mutagen 2010, 51, 462–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Murphy MP; Smith RAJ Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol 2007, 47, 629–656. [DOI] [PubMed] [Google Scholar]
- (8).Ross MF; Kelso GF; Blaikie FH; James AM; Cochemé HM; Filipovska A; Da Ros T; Hurd TR; Smith RAJ; Murphy MP Lipophilic triphenylphosphonium cations as tools in mitochondrial bioenergetics and free radical biology. Biochemistry (Moscow) 2005, 70, 222–230. [DOI] [PubMed] [Google Scholar]
- (9).Murphy MP Targeting lipophilic cations to mitochondria. Biochim. Biophys. Acta, Bioenerg 2008, 1777, 1028–1031. [DOI] [PubMed] [Google Scholar]
- (10).Smith RAJ; Murphy MP Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N. Y. Acad. Sci 2010, 1201, 96–103. [DOI] [PubMed] [Google Scholar]
- (11).Imai Y; Fink BD; Promes JA; Kulkarni CA; Kerns RJ; Sivitz WI Effect of a mitochondrial-targeted coenzyme Q analog on pancreatic β-cell function and energetics in high fat fed obese mice. Pharmacol. Res. Perspect 2018, 6, No. e00393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Fink BD; Guo DF; Kulkarni CA; Rahmouni K; Kerns RJ; Sivitz WI Metabolic effects of a mitochondrial-targeted coenzyme Q analog in high fat fed obese mice. Pharmacol. Res. Perspect 2017, 5, No. e00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Cheng G; Zielonka J; Ouari O; Lopez M; McAllister D; Boyle K; Barrios CS; Weber JJ; Johnson BD; Hardy M; Dwinell MB; Kalyanaraman B Mitochondria-targeted analogues of metformin exhibit enhanced antiproliferative and radiosensitizing effects in pancreatic cancer cells. Cancer Res 2016, 76, 3904–3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lei W; Xie J; Hou Y; Jiang G; Zhang H; Wang P; Wang X; Zhang B Mitochondria-targeting properties and photodynamic activities of porphyrin derivatives bearing cationic pendant. J. Photochem. Photobiol. B 2010, 98, 167–171. [DOI] [PubMed] [Google Scholar]
- (15).Millard M; Pathania D; Shabaik Y; Taheri L; Deng J; Neamati N Preclinical evaluation of novel triphenylphosphonium salts with broad-spectrum activity. PLoS One 2010, 5, No. e13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Robinson KM; Janes MS; Pehar M; Monette JS; Ross MF; Hagen TM; Murphy MP; Beckman JS Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 15038–15043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Dickinson BC; Chang CJ A targetable fluorescent probe for imaging hydrogen peroxide in the mitochondria of living cells. J. Am. Chem. Soc 2008, 130, 9638–9639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Huang S; Han R; Zhuang Q; Du L; Jia H; Liu Y; Liu Y New photostable naphthalimide-based fluorescent probe for mitochondrial imaging and tracking. Biosens. Bioelectron 2015, 71, 313–321. [DOI] [PubMed] [Google Scholar]
- (19).Ye Y; Zhang T; Yuan H; Li D; Lou H; Fan P Mitochondria-targeted lupane triterpenoid derivatives and their selective apoptosis-inducing anticancer mechanisms. J. Med. Chem 2017, 60, 6353–6363. [DOI] [PubMed] [Google Scholar]
- (20).Kuang Y; Sechi M; Nurra S; Ljungman M; Neamati N Design and synthesis of novel reactive oxygen species inducers for the treatment of pancreatic ductal adenocarcinoma. J. Med. Chem 2018, 61, 1576–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Manzano JI; Cueto-Díaz EJ; Olías-Molero AI; Perea A; Herraiz T; Torrado JJ; Alunda JM; Gamarro F; Dardonville C Discovery and pharmacological studies of 4-hydroxyphenyl-derived phosphonium salts active in a mouse model of visceral leishmaniasis. J. Med. Chem 2019, 62, 10664–10675. [DOI] [PubMed] [Google Scholar]
- (22).Jara JA; Castro-Castillo V; Saavedra-Olavarría J; Peredo L; Pavanni M; Jaña F; Letelier ME; Parra E; Becker MI; Morello A; Kemmerling U; Maya JD; Ferreira J Antiproliferative and uncoupling effects of delocalized, lipophilic, cationic gallic acid derivatives on cancer cell lines. Validation in vivo in singenic mice. J. Med. Chem 2014, 57, 2440–2454. [DOI] [PubMed] [Google Scholar]
- (23).Wang J; Yang C-T; Kim Y-S; Sreerama SG; Cao Q; Li Z-B; He Z; Chen X; Liu S 64Cu-Labeled triphenylphosphonium and triphenylarsonium cations as highly tumor-selective imaging agents. J. Med. Chem 2007, 50, 5057–5069. [DOI] [PubMed] [Google Scholar]
- (24).Millard M; Gallagher JD; Olenyuk BZ; Neamati N A selective mitochondrial-targeted chlorambucil with remarkable cytotoxicity in breast and pancreatic cancers. J. Med. Chem 2013, 56, 9170–9179. [DOI] [PubMed] [Google Scholar]
- (25).Reily C; Mitchell T; Chacko BK; Benavides GA; Murphy MP; Darley-Usmar VM Mitochondrially targeted compounds and their impact on cellular bioenergetics. Redox Biol 2013, 1, 86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Khailova LS; Nazarov PA; Sumbatyan NV; Korshunova GA; Rokitskaya TI; Dedukhova VI; Antonenko YN; Skulachev VP Uncoupling and toxic action of alkyltriphenylphosphonium cations on mitochondria and the bacterium Bacillus subtilis as a function of alkyl chain length. Biochemistry (Moscow) 2015, 80, 1589–1597. [DOI] [PubMed] [Google Scholar]
- (27).Trnka J; Elkalaf M; Andél M Lipophilic triphenylphosphonium cations inhibit mitochondrial electron transport chain and induce mitochondrial proton leak. PLoS One 2015, 10, No. e0121837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Asin-Cayuela J; Manas ARB; James AM; Smith RAJ; Murphy MP Fine-tuning the hydrophobicity of a mitochondria-targeted antioxidant. FEBS Lett 2004, 571, 9–16. [DOI] [PubMed] [Google Scholar]
- (29).Hu Z; Sim Y; Kon OL; Ng WH; Ribeiro AJM; Ramos MJ; Fernandes PA; Ganguly R; Xing B; García F; Yeow EKL Unique triphenylphosphonium derivatives for enhanced mitochondrial uptake and photodynamic therapy. Bio-conjugate Chem 2017, 28, 590–599. [DOI] [PubMed] [Google Scholar]
- (30).Li Y; Fawcett JP; Zhang H; Tucker IG Transport and metabolism of some cationic ubiquinone antioxidants (MitoQn) in Caco-2 cell monolayers. Eur. J. Drug Metab. Pharmacokinet 2008, 33, 199–204. [DOI] [PubMed] [Google Scholar]
- (31).James AM; Cochemé HM; Smith RAJ; Murphy MP Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. J. Biol. Chem 2005, 280, 21295–21312. [DOI] [PubMed] [Google Scholar]
- (32).James AM; Sharpley MS; Manas A-RB; Frerman FE; Hirst J; Smith RAJ; Murphy MP Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J. Biol. Chem 2007, 282, 14708–14718. [DOI] [PubMed] [Google Scholar]
- (33).Wisnovsky SP; Wilson JJ; Radford RJ; Pereira MP; Chan MR; Laposa RR; Lippard SJ; Kelley SO Targeting mitochondrial DNA with a platinum-based anticancer agent. Chem. Biol 2013, 20, 1323–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Nikitina EV; Zeldi MI; Pugachev MV; Sapozhnikov SV; Shtyrlin NV; Kuznetsova SV; Evtygin VE; Bogachev MI; Kayumov AR; Shtyrlin YG Antibacterial effects of quaternary bis-phosphonium and ammonium salts of pyridoxine on Staphylococcus aureus cells: A single base hitting two distinct targets? World J. Microbiol. Biotechnol 2016, 32, 5. [DOI] [PubMed] [Google Scholar]
- (35).Chen S; Zhao Z; Zhang Y; Fang W; Lu J; Zhang X Effect of methoxy group position on biological properties of (18)F-labeled benzyl triphenylphosphonium cations. Nucl. Med. Biol 2017, 49, 16–23. [DOI] [PubMed] [Google Scholar]
- (36).Alasadi A; Chen M; Swapna GVT; Tao H; Guo J; Collantes J; Fadhil N; Montelione GT; Jin S Effect of mitochondrial uncouplers niclosamide ethanolamine (NEN) and oxyclozanide on hepatic metastasis of colon cancer. Cell Death Dis 2018, 9, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Pardo-Andreu GL; Nuñez-Figueredo Y; Tudella VG; Cuesta-Rubio O; Rodrigues FP; Pestana CR; Uyemura SA; Leopoldino AM; Alberici LC; Curti C The anti-cancer agent nemorosone is a new potent protonophoric mitochondrial uncoupler. Mitochondrion 2011, 11, 255–263. [DOI] [PubMed] [Google Scholar]
- (38).Fink BD; Herlein JA; Guo DF; Kulkarni C; Weidemann BJ; Yu L; Grobe JL; Rahmouni K; Kerns RJ; Sivitz WI A mitochondrial-targeted coenzyme Q analog prevents weight gain and ameliorates hepatic dysfunction in high-fat-fed mice. J Pharmacol Exp Ther 2014, 351, 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.