

SUMMARY
C9orf72 repeat expansions cause inherited amyotrophic lateral sclerosis/frontotemporal dementia (ALS/FTD) and result in both loss of C9orf72 protein expression and production of potentially toxic RNA and dipeptide repeat proteins. In addition to ALS/FTD, C9orf72 repeat expansions have been reported in a broad array of neurodegenerative syndromes including Alzheimer’s disease. Here we show that C9orf72 deficiency promotes a change in the homeostatic signature in microglia and a transition to an inflammatory state characterized by an enhanced type I IFN signature. Furthermore, C9orf72-depleted microglia trigger age-dependent neuronal defects, in particular enhanced cortical synaptic pruning, leading to altered learning and memory behaviors in mice. Interestingly, C9orf72 deficient microglia promote enhanced synapse loss and neuronal deficits in a mouse model of amyloid accumulation, while paradoxically improving plaque clearance. These findings suggest that altered microglial function due to decreased C9orf72 expression directly contributes to neurodegeneration in repeat expansion carriers independent of gain of function toxicities.
Keywords: Amyotrophic Lateral Sclerosis, Frontotemporal dementia, Alzheimer’s Disease, C9orf72, microglia, neurodegeneration
Graphical Abstract
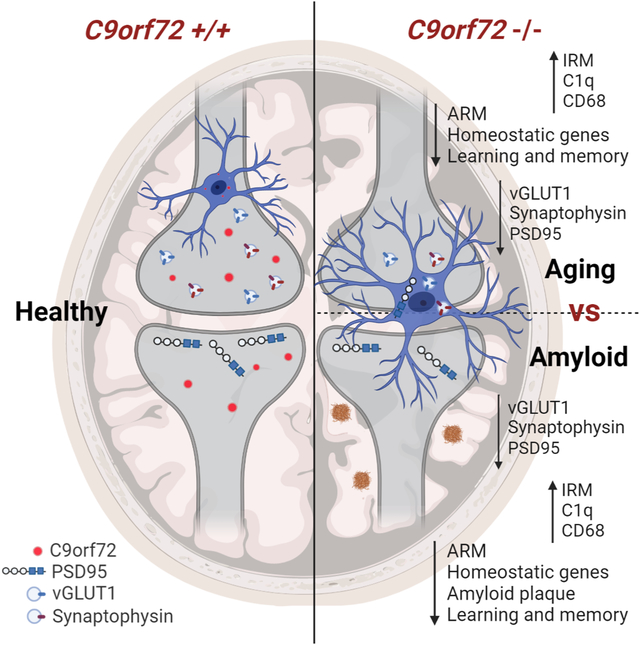
Loss of C9orf72 in microglia changes their transcriptional profile to an enhanced Type 1 IFN signature. Consequently, activated microglia participate in enhanced synaptic pruning, leading to neuronal dysfunction and subsequent deficits in learning and memory in mouse models of ALS/FTD and AD.
INTRODUCTION
Expansion of a noncoding hexanucleotide repeat (GGGGCC) in C9orf72 is the most common genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (Renton et al., 2011, DeJesus-Hernandez et al., 2011). Both diseases are characterized by degeneration of motor neurons of the motor cortex and spinal cord (ALS) and cortical neurons of the frontal and anterior temporal lobes (FTD). It is now widely accepted that ALS and FTD exist as a spectrum disease and share hereditable genetic causes and aggregation pathologies (Gitler and Tsuiji, 2016). The high prevalence of the C9orf72 mutation in both neurodegenerative disorders has led to concentrated efforts in identifying causative mechanisms of neuronal damage and death. Depending on promoter usage, the repeat expansion can be transcribed into RNA species and translated to produce dipeptide repeat (DPR) proteins that accumulate in neurons and cause gain-of-function toxicity (Taylor et al., 2016). However repeat expansions also decrease C9orf72 gene expression at the RNA (DeJesus-Hernandez et al., 2011, Jackson et al., 2020) and protein level (Viode et al., 2018), indicating that haploinsufficiency may contribute to the disease pathogenesis analogous to progranulin (GRN) deficiency related FTD (Baker et al., 2006). In the context of ALS/FTD, recent reports showed that loss of C9orf72 exacerbates autophagic and lysosomal trafficking defects and synergizes with DPR proteins accumulation and toxicity leading to cell death (Abo-Rady et al., 2020, Boivin et al., 2020, Zhu et al., 2020), further implicating a dual-hit pathogenic mechanism.
C9orf72 is widely expressed, including in peripheral myeloid cells and microglia (Rizzu et al., 2016), and loss of C9orf72 in mice leads to enhanced pro-inflammatory responses and lysosomal accumulations in peripheral myeloid cells and microglia (Atanasio et al., 2016, O’Rourke et al., 2016, Sudria-Lopez et al., 2016). In addition, C9orf72−/− myeloid cells showed diminished degradation of the stimulator of interferon gene (STING) protein leading to chronically elevated type I interferon responses in C9orf72 ALS/FTD mouse models and patients (McCauley et al., 2020), and inflammation in these mice is highly dependent on the environment and gut microbiome (Burberry et al., 2020). However, the specific impact of decreased C9orf72 expression on brain microglial function and its potential role in neurodegeneration remains unknown. Here, we show using bulk and single-cell RNAseq (scRNAseq) that loss of C9orf72 directly alters the transcriptomic profile of microglia, intrinsically altering the expression of activated response microglia (ARM) and interferon response microglia (IRM). Furthermore, contrary to prior reports that the knockout mice are neurologically normal, aged C9orf72−/− mice showed aberrant neuronal morphology, enhanced complement-mediated cortical synaptic loss, and learning and memory deficits. Enhanced synapse loss was recapitulated when C9orf72 was selectively absent from microglia in both neuron-microglia co-cultures and in C9orf72fl/fl:Cx3cr1cre+ mice where C9orf72 is selectively depleted in myeloid cells. Finally, microglia lacking C9orf72 drove premature synapse loss and enhanced learning and memory deficits in the 5XFAD model of Alzheimer’s disease (AD), while unexpectedly reducing amyloid accumulation through enhanced plaque clearance. These results reveal a cell autonomous role for C9orf72 in normal microglial function and maintenance of synaptic integrity in the brain.
RESULTS
C9orf72 deficient microglia show altered transcriptional signatures and an enhanced inflammatory state
While some studies using a LacZ reporter mouse suggested microglia do not express C9orf72 (Langseth et al., 2017, Suzuki et al., 2013), we observed C9orf72 protein expression in acutely isolated microglia (Figure S1A,B), in agreement with studies showing C9orf72 mRNA expression in microglia (Zhang et al., 2014). C9orf72 protein expression in microglia is comparable but slightly lower than bone marrow derived macrophages (BMDM) which show a variety of altered inflammatory responses when lacking C9orf72 (O’Rourke et al., 2016) (Figure S1A,B). Presumably the discrepancy between our findings and previous studies is because the LacZ reporter used in those studies spliced only to exon 1a, and not exon 1b (Langseth et al., 2017). We observed in published cell type specific RNA-seq (Zhang et al., 2014) that neurons, oligodendroglia and astrocytes used exon 1a and 1b, while microglia used exclusively exon 1b and therefore would not generate LacZ containing transcripts (Figure S1C,D). This is consistent with the fact that microglia rarely manifest DPR pathology or RNA foci, since these require transcription of exon 1a upstream of the repeat expansion (Saberi et al., 2018, Mackenzie et al., 2013, DeJesus-Hernandez et al., 2017, Mizielinska et al., 2013, Gendron et al., 2013).
To investigate how loss of C9orf72 alters microglial function, we first performed RNAseq on pooled Cd11b+ isolated microglia from young (3-month) or aged (17-month) C9orf72+/+, C9orf72+/− and C9orf72−/− mice. Principal component analyses (PCA) showed all genotypes clustered together at 3 months but C9orf72−/− mice separated from C9orf72+/+ and C9orf72+/− animals at 17 months (Figure 1A). To further characterize dysregulated gene expression in C9orf72 deficient microglia, we analyzed 13 differentially regulated gene modules previously defined via weighted gene correlation network analysis (WGCNA) across multiple mouse models of neurodegeneration (Rexach et al., 2020). This revealed significant upregulation of a single gene expression module at 3 months of age in the C9orf72−/− microglia that was upregulated in both C9orf72+/− and C9orf72−/− microglia at 17 months (Figure S1E). Genes in this module included Trem2/Tyrobp, components of the inflammasome (Il1b, Pycard), and endolysosomal components involved in MHCII antigen presentation (Ctss, CD74) (Figure S1F) suggesting that C9orf72 plays a key role in antigen presentation and inflammation in microglia.
Figure 1: Altered microglia transcriptome profile in C9orf72−/− mice.
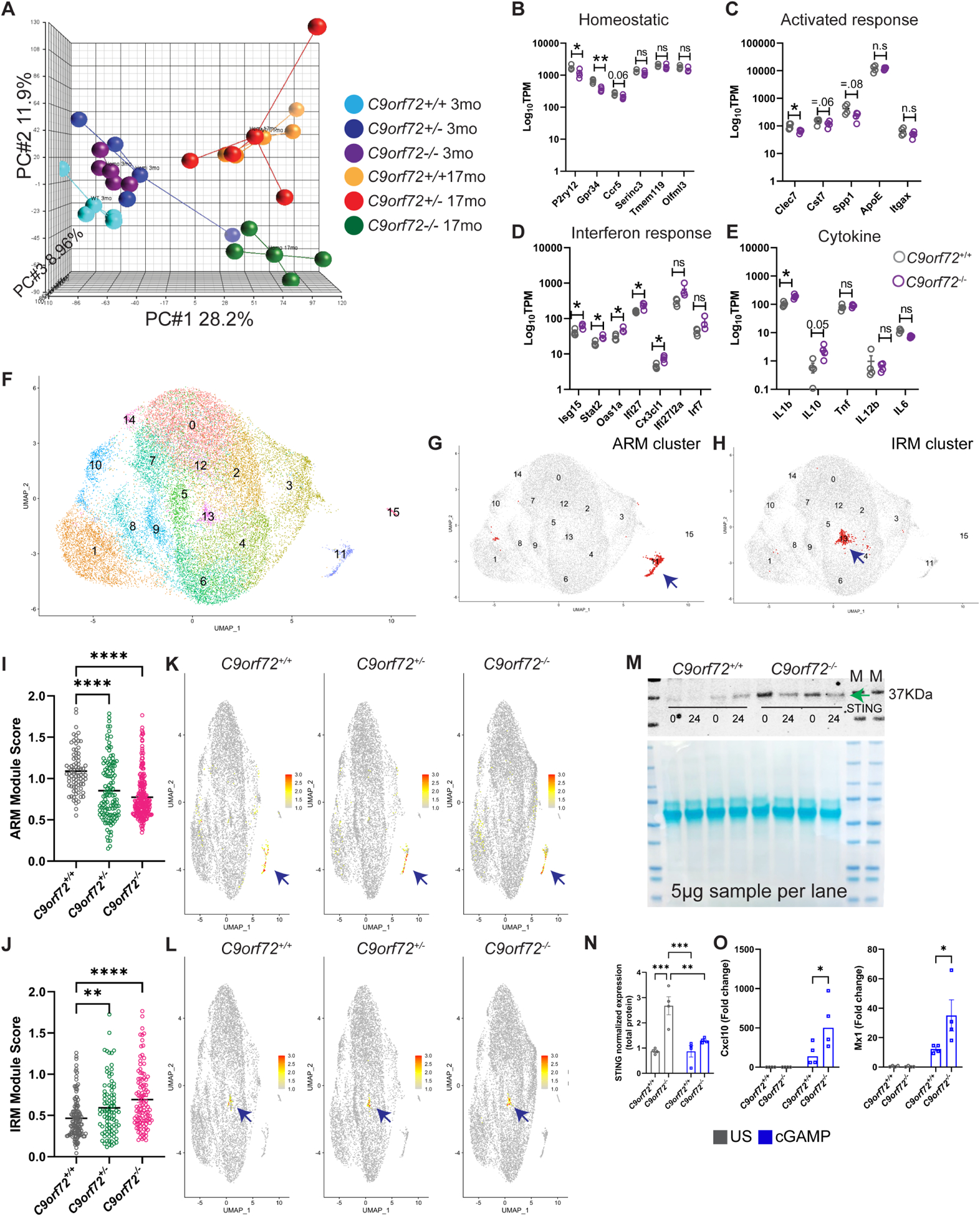
(A) PCA analysis of RNAseq from acutely isolated microglia from 3 and 17-month-old C9orf72+/+, C9orf72+/− and C9orf72−/− mice, n=4 per genotype. (B-E) 17-month TPM graphs of (B) homeostatic, (C) activated response microglia, (D) interferon response microglia, and (E) cytokine genes. (F) scRNAseq analysis showing UMAP of 26,275 microglial cells from all genotypes at 12 months, n=3 per genotype. (G) UMAP plot of ARM cluster. (H) UMAP plot of IRM cluster. (I) ARM module score in C9orf72+/+, C9orf72+/− and C9orf72−/− mice. (J) IRM module score in C9orf72+/+, C9orf72+/− and C9orf72−/− mice. (K) UMAP plot colored for expression for ARM module score separated by genotypes. (L) UMAP plot colored for expression for IRM module score separated by genotypes. (M) Western blot analysis of microglia stimulated with cGAMP (10 μg/ml) for 0 and 24hr showing increased levels of STING in C9orf72−/− vs C9orf72+/+animals (images representative of two experiments with four biological replicates). Arrow indicates band specific for STING. (N) Quantification of western blot. (O) qRT–PCR analysis of CXCL10 and Mx1 production by microglia isolated from C9orf72+/+ and C9orf72−/− mouse brains (n = 4 each) after stimulation with cGAMP (10 μg/ml) for 24hr. Data shown as mean ± SEM. Each dot represents one sample. Two-way ANOVA with Tukey’s multiple comparison test, **p<0.005, ***p<0.0005. For panels B-E, mean ±SEM, Unpaired t test with Welch correction, two-tailed, n=4 per genotype, **p=.0012, *p<0.05. Some genes are shown with exact p-value =.05. ns – not significant. For panels I &J only positively log fold change genes were used for analysis. See also Figure S1 & S2, Table S2.
Recent studies using single cell RNAseq have revealed distinct homeostatic and activation states of microglia in both healthy aging and disease (Friedman et al., 2018, Krasemann et al., 2017, Keren-Shaul et al., 2017, Sala Frigerio et al., 2019). Bulk microglia RNAseq showed that homeostatic microglia markers including P2ry12, Ccr5, and Gpr34 were decreased in aged C9orf72−/− compared to wild-type (WT) microglia, as were markers of ARM (Clec7, Cst7, Spp1) (Figure 1B,C; Figure S1G,H). By contrast, markers of IRM (Isg15, Ifit27, Oas1a, Stat2) (Sala Frigerio et al., 2019) and cytokines (Il1b, Il10) were elevated (Figure 1D,E; Figure S1I). Of note, C9orf72+/− microglia did not show any significant differences in these gene signatures. To determine if these relatively subtle changes in the bulk C9orf72−/− microglia transcriptome were due to an averaged change in all microglial cells, or an alteration in subsets of microglial populations, we performed single-cell RNAseq (scRNAseq) of acutely isolated microglia from cortex of 12-month-old C9orf72+/+, C9orf72+/− and C9orf72−/− mice. A total of 30,872 cells were sequenced (all genotypes) at comparable sequencing depths of ~60,000 reads per cell. On average, 3,430 cells were sequenced per genotype with a median of 2,698 genes identified per cell (Table S1). To identify transcriptionally distinct microglial subpopulations, an initial clustering was performed to identify, curate, and remove contaminating non-microglia cells from further analysis (Figure S2A–D; Table S1).
Clustering analysis of all microglial cells revealed sixteen microglial subpopulations across all genotypes (Figure 1F). Gene expression analyses showed canonical microglial genes (TMEM119, P2ry12, and Trem2) were highly expressed by most of the analyzed cells (Figure S2E). Distinct microglial clusters were identified that were enriched in ARM genes (Cluster 11, Clec7a, Itgax, Apoe, and Cst7), and IRM genes (Cluster 13, Ifit1, Ifit2, Ifi204, and Isg15) (Sala Frigerio et al., 2019) (Figure 1G,H). The proportions of microglia in these clusters were not significantly different between genotypes (Table S2). Unsupervised cluster marker discovery revealed 96 enriched genes in Cluster 11 (ARM) and 86 enriched genes in Cluster 13 (IRM) (Table S2). These two gene lists were then used to assign ARM and IRM module scores to each cell (Table S2, Figure S2F) and compared across genotypes. Module score analyses revealed an overall decrease in the ARM module score in both C9orf72+/− and C9orf72−/− animals (Figure 1I,K, Figure S2G) and an overall increase in the IRM module score (Figure 1J,L, Figure S2H). These data confirm observations from the pooled microglia RNAseq (Figure 1C,D; Figure S1H,I) indicating that C9orf72−/− mice show decreased ARM and increased IRM signature gene expression.
Given the upregulation of IRM signature gene expression in C9orf72−/− microglia, and our recent findings on impaired lysosomal degradation of critical driver of type I interferon response protein STING in C9orf72−/− myeloid cells (McCauley et al., 2020), we examined whether isolated C9orf72−/− microglia also show changes in type I interferon responses and downstream interferon-stimulated genes (ISGs). Similar to peripheral myeloid cells (McCauley et al., 2020), C9orf72−/− microglia showed increased basal STING protein levels, and enhanced production of ISGs CXCL10, and Mx1 (Figure 1M–O) upon stimulation with cyclic GMP–AMP (cGAMP)—an agonist of the cGAMP synthase (cGAS)–STING pathway. Together, these data indicate that loss of C9orf72 in microglia leads to age dependent decreases in expression of activated response microglia genes, and an increased expression of IFN response genes likely driven by enhanced STING activity.
Aged C9orf72−/− mice exhibit lysosomal defects, microgliosis, and enhanced complement mediated pruning of synaptic terminals in motor cortex
C9orf72 is involved in endosomal trafficking, autophagy and lysosomal function (Nassif et al., 2017) and increased lysosomal material is observed in C9orf72 repeat expansion patient microglia (O’Rourke et al., 2016). Therefore, we further examined genes involved in lysosomal function reported in Grn deficient mice (Lui et al., 2016) and found altered expression of a number of genes including Hspe, Cln3, and Lamp2 (Figure S3A). Given this transcriptional dysregulation, we stained C9orf72−/− mouse brains for the microglial marker Iba1 and the lysosomal protein CD68. We observed enhanced accumulation of CD68 positive lysosomes in microglia from aged C9orf72−/− mice compared to WT mice (Figure S3B–D), similar to observations of increased CD68 staining in C9orf72 repeat expansion carrier tissue (Brettschneider et al., 2012).
Prior studies did not observe overt neurodegeneration in mice lacking C9orf72 in all cells (O’Rourke et al., 2016, Burberry et al., 2016, Atanasio et al., 2016, Sudria-Lopez et al., 2016) or selectively in neural progenitors (Koppers et al., 2015). However given the altered inflammatory state and lysosomal accumulations observed in C9orf72−/− microglia, and that microglia are known to regulate complement mediated synapse elimination in both normal development and disease (Stevens et al., 2007, Hong et al., 2016, Schafer et al., 2012), we hypothesized that C9orf72−/− microglia may drive dysfunctional synaptic pruning and thereby cause secondary neuronal dysfunction. We first investigated microglial numbers, and found a small, yet significant increase in the density of Iba1 positive microglia in motor cortex at 12 months in C9orf72−/− animals (Figure S3E,F). Since microglia are the predominant source of C1q in the brain (Fonseca et al., 2017) and increased C1q expression has been observed during aging (Stephan et al., 2013) and in neurodegenerative disorders (Whitelaw, 2018), we immunostained for the complement protein C1q and saw a significant increase in immunoreactivity in the motor cortex of C9orf72−/− animals compared to WT (Figure 2A,B). We also observed a significant decrease in cortical synaptic proteins at 12 months, as assessed by immunostaining and western blotting, with a significant reduction of the synaptic markers synaptophysin and vGLUT1 (Figure 2C–F; Figure S3G,H) and PSD95 (Figure S3G,I). Of note, heterozygous animals did not show significant changes in C1q, synaptophysin, and vGLUT1 levels at 12 months (Figure 2A–F). Increased complement deposition and decreased synaptic markers were accompanied by significantly decreased dendritic arborization and neurite length in C9orf72−/− animals (Figure 2G–I), but not in C9orf72+/− mice (data not shown), indicating that unlike the neuronal specific knockouts which showed no abnormalities (Koppers et al., 2015), the complete knockout of C9orf72 led to marked abnormalities in synaptic content and neuronal morphology.
Figure 2: Synaptic and behavioral defects in aged C9orf72−/− mice.
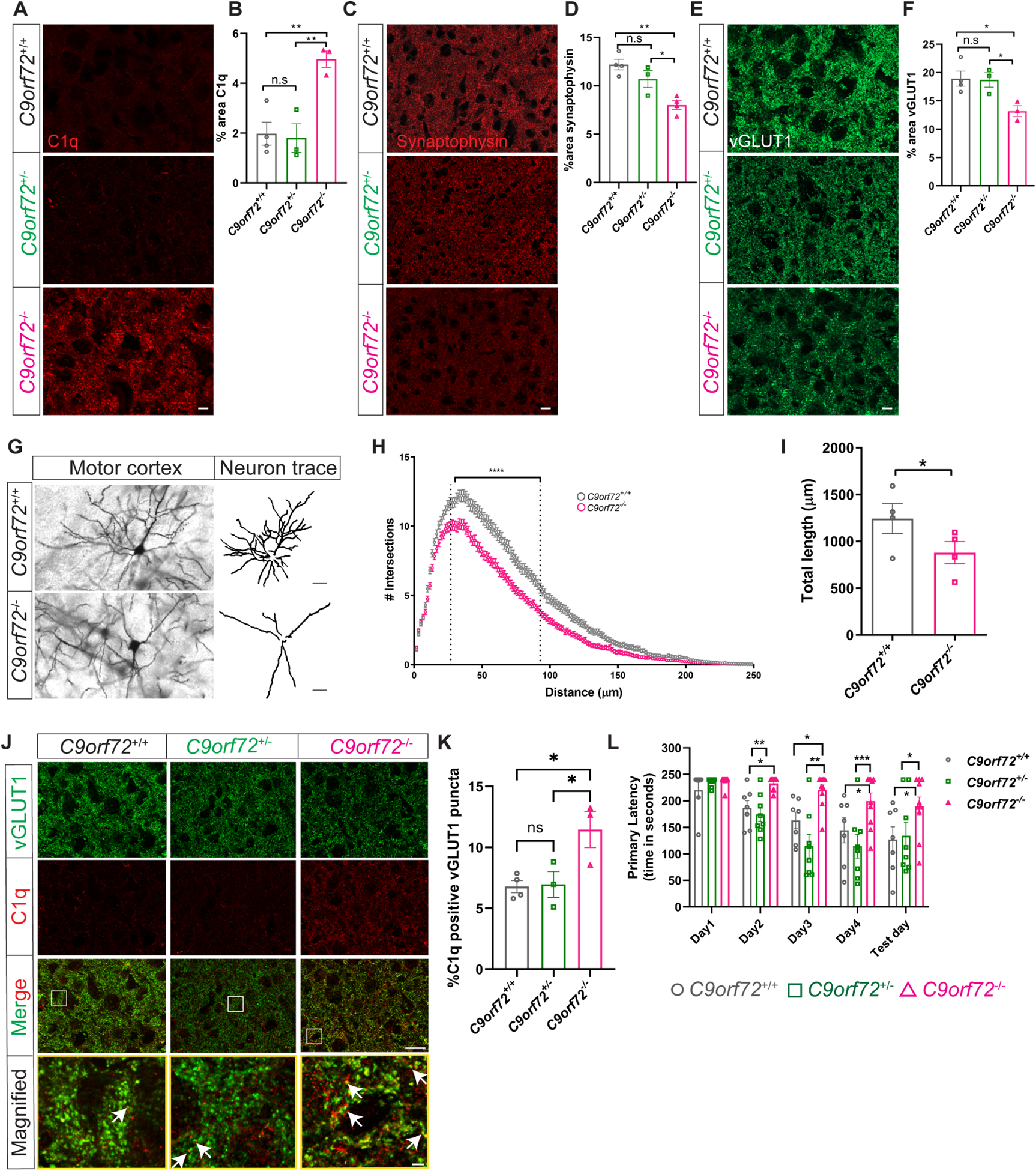
(A) Representative images of C1q immunoreactivity in the motor cortex of C9orf72+/+, C9orf72+/−, and C9orf72−/− mice at 12 months and (B) quantification. mean ±SEM, One-way ANOVA with Tukey’s multiple comparison test, n=3–4 per genotype, **p<0.005. (C) Motor cortex from 12-month-old C9orf72+/+, C9orf72+/−, and C9orf72−/− mice immunostained with synaptophysin and (D) quantification, mean ±SEM, One-way ANOVA with Tukey’s multiple comparison test, n=3–4 per genotype, *p<0.05, **p<0.005. (E) Representative confocal images of vGLUT1 immunoreactivity from 12-month-old C9orf72+/+, C9orf72+/−, and C9orf72−/− mice and (F) quantification. mean ±SEM, One-way ANOVA with Tukey’s multiple comparison test, n=3–4 per genotype, *p<0.05. Scale bars: 10μm. (G) Neuronal morphology tracings from motor cortex of Golgi stained 12-month C9orf72+/+ and C9orf72−/− mice brains; n=4 per genotype. Scale bars: 20μm. (H) Sholl analysis of dendritic arborization, Ordinary Two-way ANOVA, ****p<0.0001. (I) Total length of the neurites in the indicated genotypes, mean ±SEM, Unpaired t-test, two-tailed, n=4 per genotype, *p<0.05. (J) Representative images of vGLUT1 (green) and C1q (red) proteins in the motor cortex of 12-month C9orf72+/+, C9orf72+/−, and C9orf72−/− mice. Rectangular box indicates magnified region. White arrows indicate vGLUT1 positive puncta either co-localized or next to C1q immunoreactive puncta. Scale bars: 10μm, 5μm. (K) Quantification of C1q positive vGLUT1 puncta, mean ±SEM, One-way ANOVA with Tukey’s multiple comparison test, n=3–4 per genotype, *p<0.05. (L) Barnes maze analysis of primary latency from 12-month-old C9orf72+/+ (n=7), C9orf72+/− (n=8), and C9orf72−/− (n=10) mice. Two-way repeated measure ANOVA with Tukey’s multiple comparison test, time x genotype F(8, 88)=2.075, p=0.0467, time effect: F(3.127, 68.79)=16.14, p<0.0001, genotype effect: F(2, 22)=13.30, p=0.0002, *p<0.05, ***p<0.0005, ****p<0.0001. See also Figure S3.
To further determine the relationship between complement deposition and synapse loss, we co-immunolabeled C1q with vGLUT1 in the motor cortex of WT or C9orf72−/− mice. Double immunolabelling revealed a significant increase in co-labeled puncta of C1q with vGLUT1+ in the motor cortex of C9orf72−/− mice (Figure 2J,K), which were rarely observed in WT mice at this age (12 months). We also found significantly enhanced accumulation of vGLUT1 and PSD95 within Iba1+ microglial cells in C9orf72−/− mice compared to WT, indicating they had engulfed synaptic material (Figure S3J–M).
To determine if the synapse loss in aged C9orf72−/− mice leads to behavioral abnormalities in these animals, we used the Barnes maze (BM) to examine spatial learning and memory. C9orf72+/+ and C9orf72+/− mice showed a steady decrease in primary latency over the course of the testing paradigm (Figure 2L) suggesting that spatial learning occurred. In contrast, C9orf72−/− mice showed increased latency in finding the escape tunnel as compared to C9orf72+/+ and C9orf72+/− mice, consistent with an impairment in spatial memory (Figure 2L). Together, these data support that C9orf72−/− microglia contribute to abnormal neuronal dendritic morphology and complement mediated synapse loss in aged mice, resulting in cognitive deficits.
Depletion of C9orf72 in microglia recapitulates synaptic phenotypes observed in C9orf72−/− mice
While C9orf72 is expressed in all cell types in the brain (Langseth et al., 2017) its expression is highest in microglia (O’Rourke et al., 2016). Recent studies have shown that loss of C9orf72 in neurons leads to dendritic morphology defects (Ho et al., 2019) and alterations in synaptic proteins and signaling (Xiao et al., 2019, Fomin et al., 2018). These observations suggest that C9orf72 can manifest cell-autonomous phenotypes in neurons. To further investigate cell vs. non-cell autonomous roles of C9orf72 in different cell types, we designed a co-culture system in which C9orf72+/+ (WT) or C9orf72−/− cortical neurons were cultured in vitro for 14 days (DIV14). At DIV14, C9orf72+/+ or C9orf72−/− microglia were added to WT cortical neuronal cultures and maintained for two additional days before all cultures were fixed for immunostaining and analyses. At DIV14, neither pre-synaptic proteins (synaptophysin, vGLUT1 and Bassoon), nor post-synaptic proteins (Homer and PSD95) showed altered protein expression patterns between WT and C9orf72−/− cortical neurons (Figure 3A–H). These results indicate that under basal conditions in vitro, loss of C9orf72 does not lead to any significant changes in the levels of synaptic proteins in cortical neuronal monocultures. By contrast, quantitative analyses of neuron-microglia co-cultures revealed decreased synaptophysin density in WT neuron/C9orf72−/− microglia compared to WT neuron/WT microglia cultures suggesting increased phagocytic activity of C9orf72−/− microglia (Figure 3I,J). In support of this observation, we found a significant increase of synaptophysin+ puncta inside C9orf72−/− microglia (Figure 3K).
Figure 3. Increased synaptic pruning and phagocytosis by C9orf72−/− microglia.
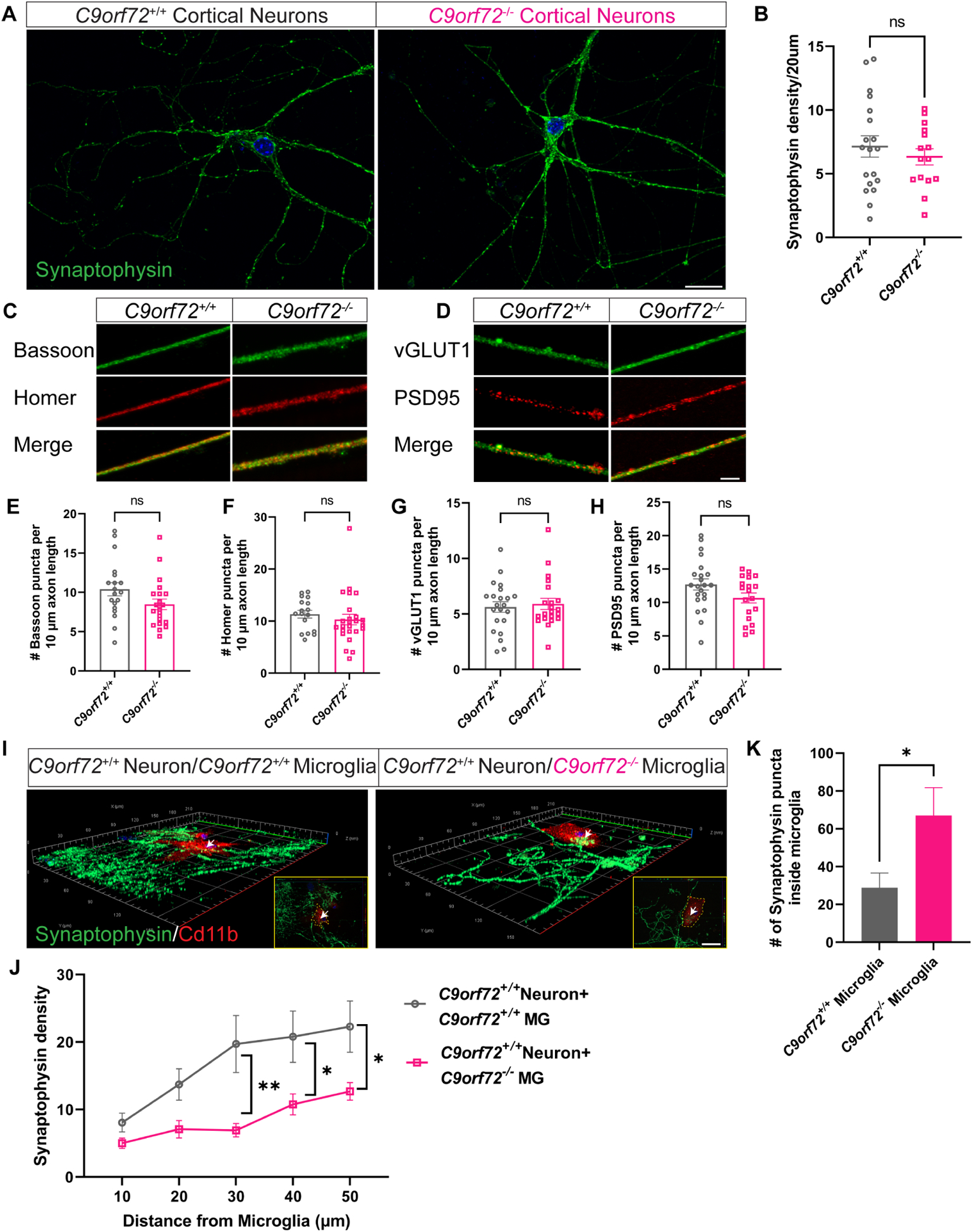
(A) Confocal images showing synaptophysin expression in C9orf72+/+ and C9orf72−/− cortical neuron cultures at 14 days in vitro. Scale bars: 20μm and (B) quantification, mean ±SEM, Unpaired t-test with Welch’s correction. (C) Representative confocal images of presynaptic (Bassoon and vGLUT1) and (D) postsynaptic (Homer and PSD95) markers in neuronal cultures from C9orf72+/+ and C9orf72−/− mice. Scale bars: 10μm. (E-H) Quantification of pre- and post-synaptic marker densities. (I) Representative 3D confocal images of synaptophysin density around C9orf72+/+ and C9orf72−/− Cd11b+ microglia. Inset shows synaptophysin+ puncta inside Cd11b microglia. Scale bars: 20μm. (J) Quantification of synaptophysin density, Two-way ANOVA with Sidak’s multiple comparison test, mean ±SEM, *p<0.05, **p<0.005. (K) Number of synaptophysin puncta inside microglia, mean ±SEM, n=10 cells per genotype, Unpaired t-test, *p<0.05. White arrows indicate synaptophysin puncta inside microglia and is zoomed in white box.
To further evaluate the cell autonomous effects of C9orf72 loss in microglia in vivo, we generated C9orf72fl/fl:Cx3cr1cre+ mice where C9orf72 was selectively depleted in myeloid subpopulations including in microglia. Analyses of motor cortex of 12-month old C9orf72fl/fl:Cx3cr1cre+ mice revealed CD68+ lysosomal accumulations in microglia recapitulating the phenotype observed in C9orf72 complete knockout mice (Figure 4A–C). We also found increased expression of C1q (Figure 4D,E) as well as decreased levels of synaptic proteins synaptophysin and vGLUT1 similar to that in C9orf72−/− total knockouts (Figure 4F–I). Double immunostaining with C1q and vGLUT1 in C9orf72fl/fl:Cx3cr1cre+ mice revealed C1q deposition adjacent to vGLUT1+ puncta supporting that these synapses were tagged for removal (Figure 4J,K). Finally, increased numbers of vGLUT1+ (Figure 4L,M) and PSD95+ (Figure 4N,O) puncta were found within Iba1+ microglia of C9orf72fl/fl:Cx3cr1cre+ animals. Together, these results support that loss of C9orf72 in microglial cells autonomously drives phagocytic activity and induces synapse loss in neurons.
Figure 4: Enhanced synaptic pruning in microglia specific C9orf72 knockout mice.
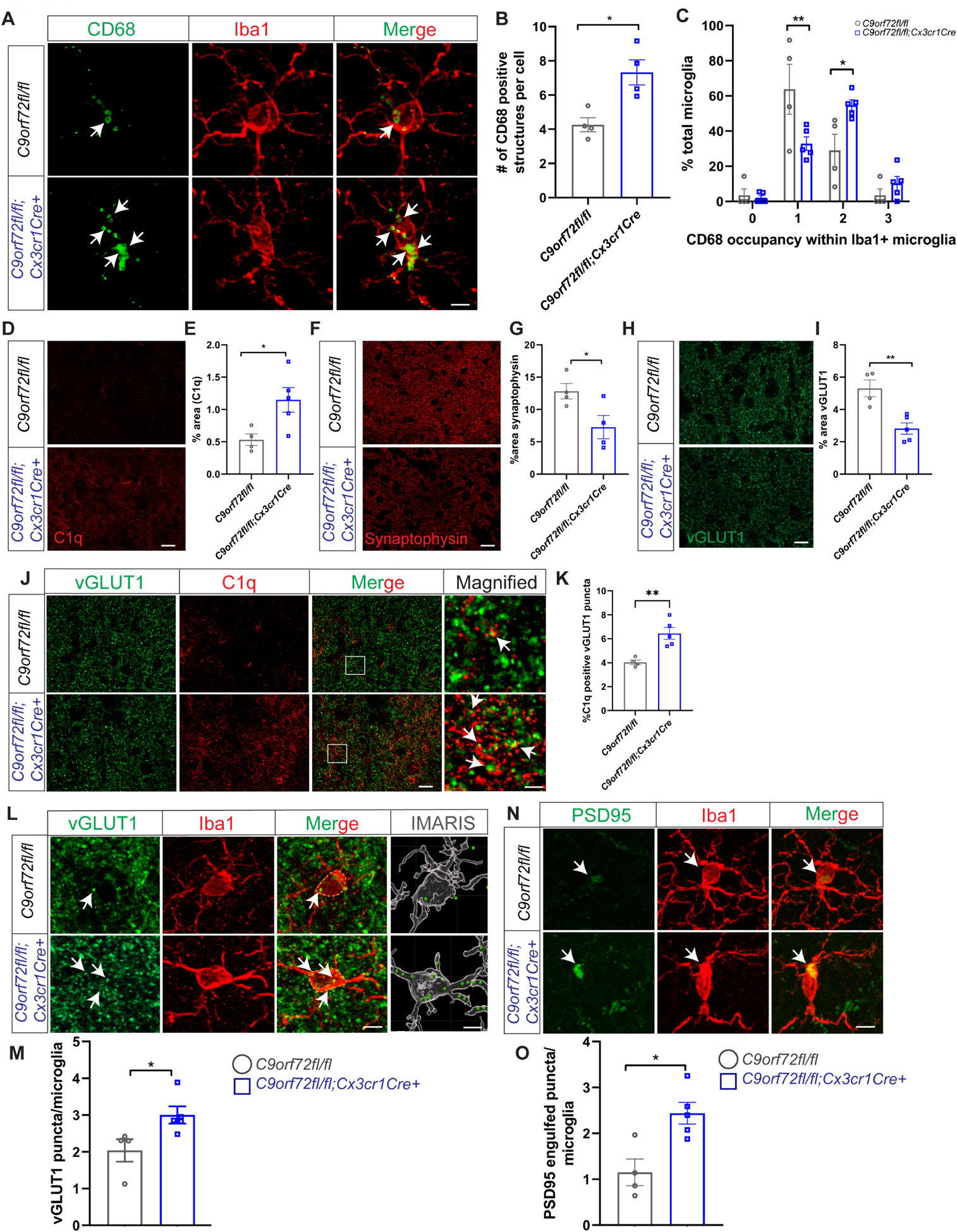
(A) Iba1 and CD68 co-stain in motor cortex of 12-month C9orf72fl/fl and C9orf72-fl/fl:Cx3cr1Cre+ mice showing CD68+ lysosomal accumulations (white arrows). Scale bars: 5μm. (B) & (C) Quantification of cells with CD68+ lysosomal accumulations. For panel B, mean ±SEM, Unpaired t-test with Welch correction, two-tailed, n=4 per genotype, *p<0.05. For panel C, mean ±SEM, Two-way ANOVA with Sidak’s multiple comparison test, n=4–5 per genotype per age. **p < 0.005, *p < 0.05. (D) Representative images of C1q immunoreactivity in the motor cortex of 12-month-old C9orf72fl/fl and C9orf72fl/fl:Cx3cr1Cre+ mice and (E) quantification, mean ±SEM, Unpaired t test with Welch’s correction, two-tailed, n=4–5 per genotype, *p<0.05. (F) Motor cortex from 12-month-old C9orf72fl/fl and C9orf72-fl/fl:Cx3cr1Cre+ mice immunostained with synaptophysin and (G) quantification, mean ±SEM, Unpaired t-test with Welsh correction, two-tailed, n=4–5 per genotype, *p<0.05. Scale bars: 10μm. (H) Representative confocal images of vGLUT1 immunoreactivity in 12-month-old C9orf72fl/fl and C9orf72-fl/fl:Cx3cr1Cre+ mice and (I) quantification, mean ±SEM, Unpaired t test with Welch’s correction, two-tailed, n=4–5 per genotype, **p<0.005. (J) Representative images of vGLUT1 (green) and C1q (red) proteins in the motor cortex of 12-month C9orf72fl/fl and C9orf72-fl/fl:Cx3cr1Cre+ mice. Scale bars: 10μm Rectangular box indicates magnified region. Scale bars: 5μm. White arrows indicate vGLUT1 positive puncta either co-localized or next to C1q immunoreactive puncta and (K) quantification, mean ±SEM, Unpaired t test with Welch’s correction, two-tailed, n=4–5 per genotype, **p<0.005. (L) Confocal and IMARIS reconstruction images showing vGLUT1+ puncta within Iba+ microglia in motor cortex of 12-month C9orf72fl/fl and C9orf72-fl/fl:Cx3cr1Cre+ (white arrows), scale bars 5μm, 1μm and (M) quantification, mean ±SEM, Unpaired t-test with Welsh correction, two-tailed, n=4–5 per genotype, *p< 0.05. (N) Confocal images showing PSD95+ structures within Iba+ microglia in motor cortex of 12-month C9orf72fl/fl and C9orf72-fl/fl:Cx3cr1Cre+ (white arrows) and (O) quantification, mean ±SEM, Unpaired t-test, two-tailed, n=4–5 per genotype, *p< 0.05. Scale bars: 5μm.
C9orf72−/− microglia show enhanced engagement and clearance of amyloid plaques in a mouse model of AD
Although C9orf72 repeat expansions predominantly result in an ALS/FTD phenotype, they have also been associated with a broad range of other neurodegenerative syndromes, ranging from AD, Huntington’s phenocopy, corticobasal syndrome, olivopontocerebellar degeneration, parkinsonism and psychiatric disorders (Majounie et al., 2012, Van Mossevelde et al., 2017, Filikci et al., 2020). While the relationship between C9orf72 expansions and these other degenerative syndromes remains unclear, our findings suggest that decreased expression of C9orf72 in microglia may play a contributory role in other forms of neurodegeneration. For example, microglia are well known to modify Alzheimer’s pathology, as evidenced by microglial expressed genes that alter AD risk, most notably TREM2 which is selectively expressed in myeloid cells (Ulland and Colonna, 2018). To evaluate the effect of C9orf72 deficiency on amyloid pathology, we crossed C9orf72−/− mice to 5XFAD transgenic mice, a model of amyloid deposition previously used to examine the effect of altered microglial function on Aβ accumulation (Figure 5A) (Wang et al., 2015). As this model accumulates Aβ plaques rapidly starting after 3 months, with a large plaque burden by 6 months, we focused analysis at these time points. Staining for Aβ aggregates in matched coronal sections from 5XFAD, 5XFAD/C9orf72+/−, and 5XFAD/C9orf72−/− mice showed minimal plaques with no differences at 3 months (Figure S4A), however at 6 months 5XFAD/C9orf72−/− mice had significantly less Aβ plaque accumulation in cortical and hippocampal regions compared to the other genotypes by HJ3.4 antibody (Figure 5B–D) or ThioS staining (Figure S4D&E). C9orf72 deficiency did not alter soluble Aβ1–40 and Aβ1–42 levels across the genotypes, though there was a trend towards decreased insoluble, guanidine-extracted Aβ1–40 and Aβ1–42 in the cortex of 5XFAD/C9orf72−/− mice in accord with decreased Aβ plaque accumulation (Figure 5E–H). There was also a trend towards decreased guanidine insoluble Aβ42/Aβ40 ratio in 5XFAD/C9orf72−/− mice (Figure S4B–C) suggesting that reduced Aβ42/Aβ40 ratio in 5XFAD/C9orf72−/− mice was due to reduced Aβ42 levels rather than increased abundance of Aβ40 (Figure 5E–H). We did not observe cortical neuronal loss in motor cortex of any of the genotypes at 6 months (Figure S4F,G), consistent with reports that neuronal loss develops after 9-months of age in 5XFAD mice (Eimer and Vassar, 2013). Given the lower plaque accumulation in 5XFAD/C9orf72−/− mice, we hypothesized that the altered functional state of C9orf72−/− microglia enhanced their ability to engage and clear extracellular amyloid plaques. Therefore, we more carefully examined the interaction between microglia and amyloid plaques.
Figure 5: Decreased C9orf72 gene dosage alters amyloid plaque burden and microglial plaque engagement.
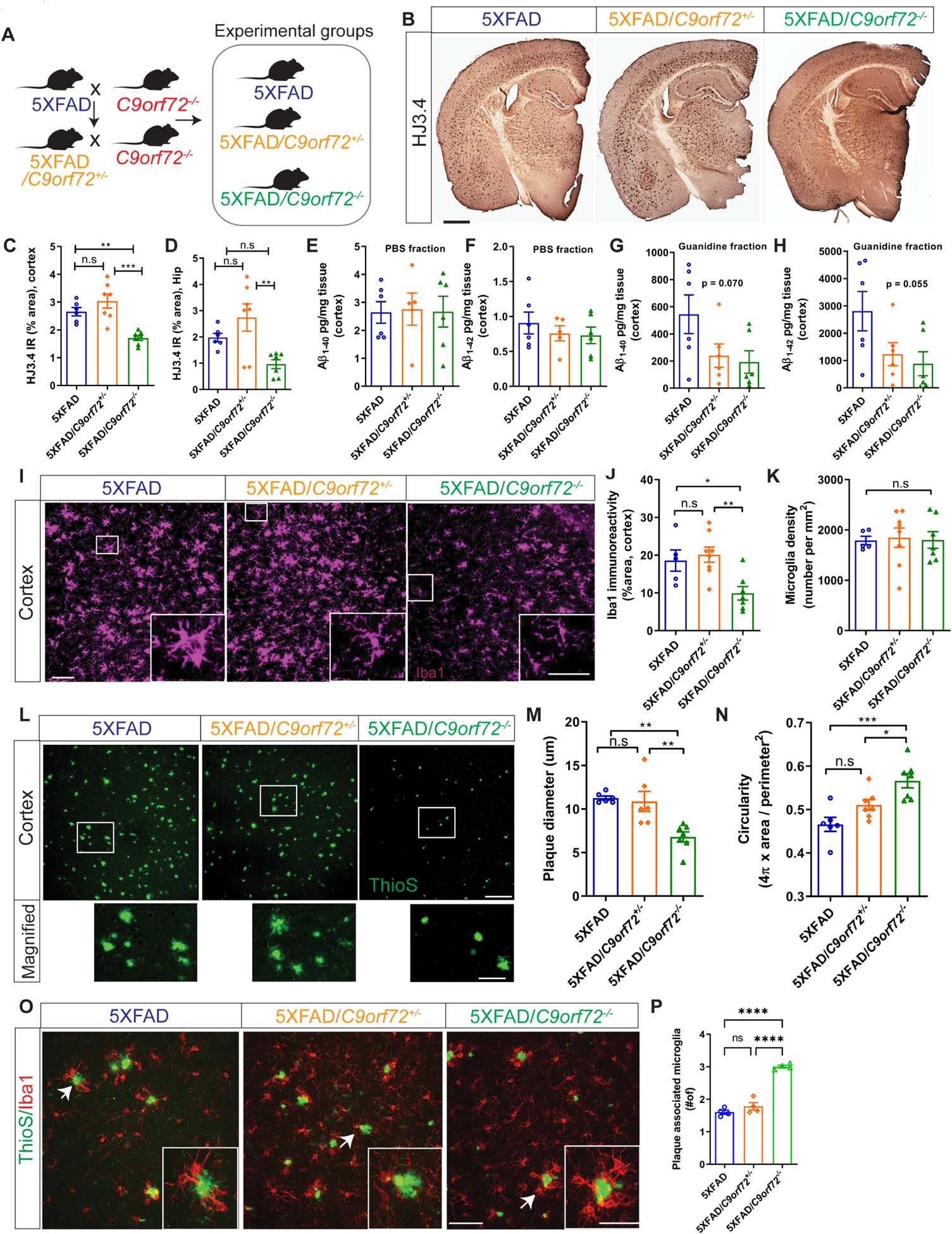
(A) Schematic of breeding strategy for experimental groups. (B) Immunostaining for Aβ with mHJ3.4 antibody in 6-month-old 5XFAD, 5XFAD/C9orf72+/−, and 5XFAD/C9orf72−/− animals. Scale bars: 100μm. (C) & (D) Quantification of Aβ immunoreactivity in the cortex and hippocampus respectively, mean ±SEM, One-way ANOVA with Tukey’s multiple comparison test, **p<0.005, ***p<0.0005. (E-H) Soluble and insoluble Aβ1–40 and Aβ1–42 levels in hippocampus of 2-month-old 5XFAD, 5XFAD/C9orf72+/−, and 5XFAD/C9orf72−/− mice as detected by ELISA. (I) Representative confocal maximum intensity projections images of Iba+ microglia in the cortex of 5XFAD, 5XFAD/C9orf72+/−, and 5XFAD/C9orf72−/− animals at 6 months. Scale bars: 100 μm, inset 25μm. (J) Quantification of Iba+ immunoreactivity and (K) microglial density in cortex, n=5–7 per genotype, mean ±SEM, One-way ANOVA with Tukey’s multiple comparison test, *p<0.05, **p<0.005. (L) Representative images of plaque morphology in 6-month-old 5XFAD, 5XFAD/C9orf72+/−, and 5XFAD/C9orf72−/− mice. Scale bars: 100 μm, inset 25μm. (M) Average plaque size and (N) circularity in indicated genotypes, n=6 mice per group, mean ±SEM, One-way ANOVA with Tukey’s multiple comparison test, *p<0.05, **p<0.005, ***p<0.0005. (O) Images showing interaction between microglia (Iba1+, red) and plaques (ThioS, green) in cortex of 6-month-old 5XFAD, 5XFAD/C9orf72+/−, and 5XFAD/C9orf72−/− mice. Scale bars: 50 μm, inset 20μm. (P) Quantification of plaque-associated microglia (Iba1+). n=4 animals per genotype, mean ±SEM, One-way ANOVA with Tukey’s multiple comparison test, ****p<0.0001. See also Figure S4, Figure S5.
At 6 months, total numbers of Iba1 positive microglia were the same across genotypes, with all “activated” appearing microglia focused around areas of plaque accumulation in 5XFAD and 5XFAD/C9orf72−/− animals (Figure 5I–K). High resolution imaging of ThioS stained amyloid plaques in cortex revealed striking differences in plaque size and morphology in 5XFAD/C9orf72−/− mice. Plaques in 5XFAD/C9orf72−/− mice were smaller, and more compact than in 5XFAD mice (Figure 5L–N). Considering that microglia migrate to sites of plaque deposition and restrict plaque growth (Ulrich et al., 2014), we quantified plaque engagement by C9orf72−/− microglia. We observed more microglia clustering around plaques in 5XFAD/C9orf72−/− mice (average 3.06 microglia/plaque) than those in 5XFAD, or 5XFAD/C9orf72+/− animals (average 1.56 and 1.65 microglia per plaque respectively) (Figure 5O,P).
Given that C9orf72−/− mice have a propensity to generate autoantibodies (Burberry et al., 2016, Atanasio et al., 2016), we further examined whether antibodies to Aβ were present in 5XFAD/C9orf72−/− mice. We observed mouse IgG in association with ThioS+ plaques and Iba1+ microglia in 5XFAD/C9orf72−/− mice but not 5XFAD mice, suggesting that immunoglobulins in the brain parenchyma may contribute to microglial-mediated plaque clearance (Figure S5A–C). We also found increased levels of serum anti-Aβ IgG antibodies in 5XFAD/C9orf72−/− mice, and significantly more anti-Aβ IgG producing B cells in spleen and lymph nodes, however at levels much lower than that observed with direct immunization (Figure S5D–H). In summary, C9orf72−/− microglia are more effectively able to engage and surround amyloid plaques and mitigate their accumulation. This appears to be driven by intrinsically altered microglial function, with a potential contribution from circulating autoantibodies against Aβ.
Enhanced microglia-mediated synaptic pruning in 5XFAD:C9orf72−/− mice
While brain Aβ accumulation is a hallmark of AD, the relationship between plaque accumulation, neuronal dysfunction and clinical phenotype is not completely understood. Animal models with Aβ accumulation have premature synapse loss, which is driven by enhanced complement mediated microglial pruning (Hong et al., 2016). Therefore, we investigated how C9orf72−/− microglia, with their altered functional and inflammatory profile, impacted synaptic integrity in the setting of plaque accumulation in 5XFAD animals. We examined 5XFAD animals at 4 months of age, because at this age plaques have only recently accumulated and no neuronal loss is detected (Eimer and Vassar, 2013). Microglia in 5XFAD/C9orf72−/− animals showed increased accumulation of CD68+ lysosomal material at 4 months (Figure 6A–C), a finding normally only observed in C9orf72−/− microglia after 17 months (see Figure S3B–D, Figure S6A), suggesting they are more phagocytically active in the presence of extracellular amyloid. There was significant reduction in cortical dendritic arborization and total neurite length in 5XFAD/C9orf72−/− animals, which was only minor in 5XFAD animals at this age (Figure 6D–F). We also observed exacerbated reduction of pre-synaptic proteins synaptophysin (Figure S6B,C,F,H), vGLUT1 (Figure S6D,E), and the postsynaptic marker PSD95 in double transgenic 5XFAD/C9orf72−/− animals (Figure S6F,G). To determine if the enhanced synaptic loss was driven by complement mediated pruning, we co-stained for C1q and vGLUT1, and found increased C1q+:vGLUT1+ puncta in the motor cortex in 5XFAD/C9orf72−/− animals compared to 5XFAD alone (Figure 6G–I). We also observed enhanced accumulation of vGLUT1+ and PSD95+ material within Iba1+ microglia of 5XFAD/C9orf72−/− mice, further confirming that microglia are engulfing synaptic proteins (Figure 6,J,K, Figure S6I,J). To determine if there was a behavioral correlate to the enhanced early synapse loss in the 5XFAD/C9orf72−/− double transgenic mice, we performed the Barnes maze test. 5XFAD/C9orf72−/− mice showed increased primary latency compared to all other genotypes including 5XFAD alone, which are normally only minorly impaired at this age, indicating that C9orf72 deficiency exacerbated spatial learning and memory defects in these animals (Figure 6L). Together, these data show that the altered functional state of C9orf72−/− microglia drives enhanced capacity to restrict growth of extracellular amyloid plaques, while at the same time enhancing synapse loss, memory deficits, and neuronal injury in an AD mouse model.
Figure 6: Enhanced microglial lysosome accumulation, neuronal and synaptic defects in 5XFAD:C9orf72−/− mice.
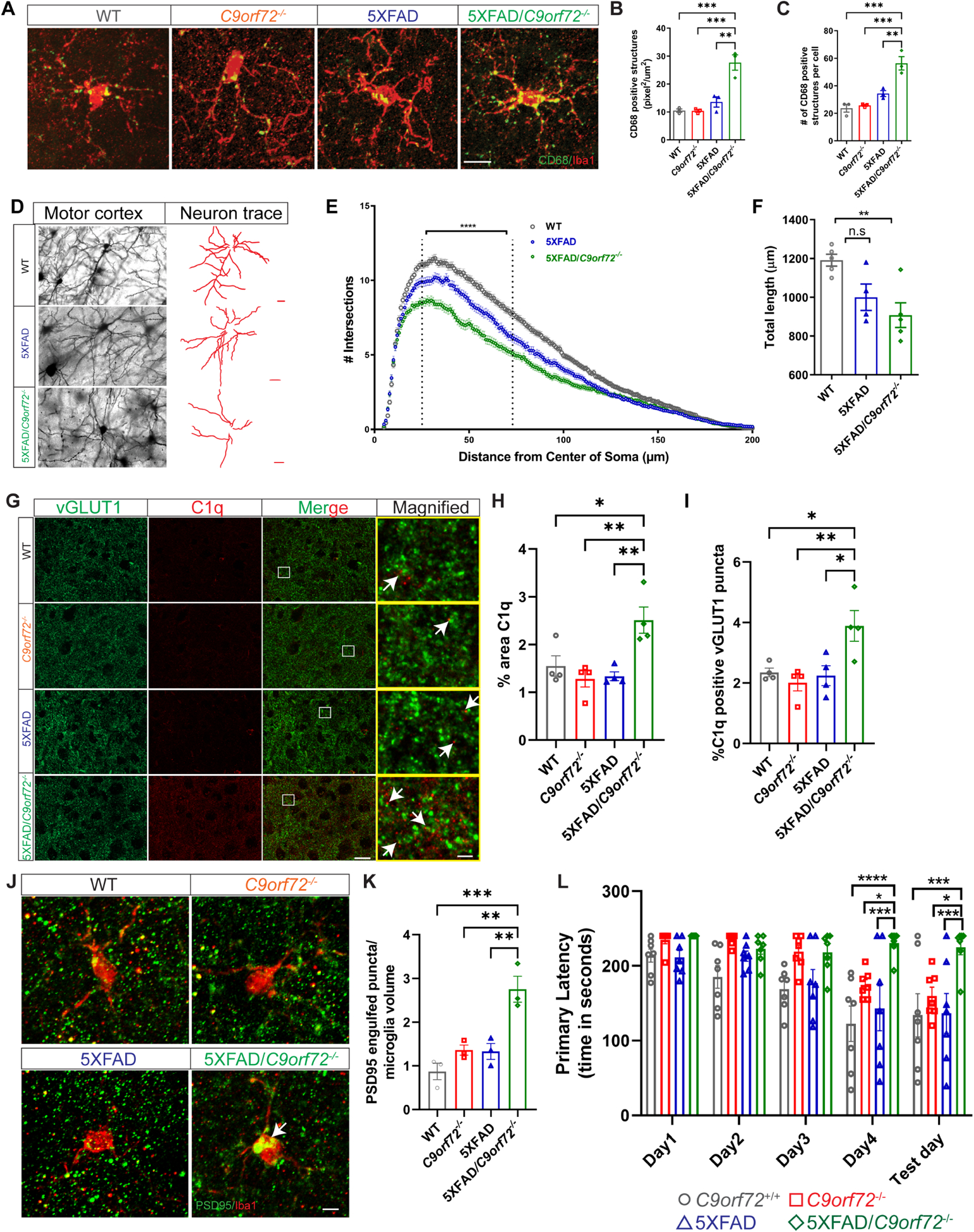
(A) Confocal images of CD68+/Iba1+ microglia from motor cortex of 4-month-old wild-type (WT), C9orf72−/−, 5XFAD, and 5XFAD/C9orf72−/− mice. Scale bars: 25μm. (B) & (C) Quantification of CD68+ total number and volume in different genotypes. Volumes are expressed as pixel2/μm2 per microglia, n=3 mice per genotype, 10–24 microglia per animal per genotype, mean ±SEM, One-way ANOVA with Tukey’s multiple comparison test, **p<0.005, ***p<0.0005. (D) Images of neuronal morphology and neuronal tracings from motor cortex of Golgi stained 4-month-old WT, 5XFAD, and 5XFAD/C9orf72−/− animals. Scale bars: 20μm. (E) Sholl analysis, Two-way ANOVA, ****p<0.001, n=5–6 animals per genotype. (F) Total length of neurites in indicated genotypes, mean ±SEM, Ordinary One-way ANOVA, **p<0.05. (G) Immunostaining of vGLUT1 (green) and C1q (red) in motor cortex of 4-month-old WT, C9orf72−/−, 5XFAD, and 5XFAD/C9orf72−/− animals. Scale bars: 10μm. Rectangular box denotes magnified region. White arrows indicate vGLUT1 positive puncta either co-localized or next to C1q immunoreactive puncta. Scale bars: 5μm. (H) Quantification of total C1q immunoreactivity, n=4 per genotype. mean ±SEM, One-way ANOVA with Tukey’s multiple comparison test, *p<0.05, **p<0.005. (I) Quantification of C1q positive vGLUT1 puncta, mean ±SEM, One-way ANOVA with Tukey’s multiple comparison test, n=4 per genotype, *p<0.05, **p<0.005. (J) Confocal images showing PSD95+ structures within Iba+ microglia (white arrows) in motor cortex of WT, C9orf72−/−, 5XFAD, and 5XFAD/C9orf72−/− mice, scale bars 10μm and (K) quantification, mean ±SEM, n=3 per genotype. One-way ANOVA with Tukey’s multiple comparison test, **p<0.05, ***p<0.0005. (L) Barnes maze analysis of primary latency from 4-month-old WT, C9orf72−/−, 5XFAD, and 5XFAD/C9orf72−/− mice (n=7 per genotype), Two-way repeated measure ANOVA with Tukey’s multiple comparison test, time x genotype, F(12, 96)=1.972, p=0.0351, time effect: F(2.521, 60.51)=16.59, p<0.0001, genotype effect: F(3, 24)=9.832, p=0.0002, *p<0.05, **p<0.005, ***p<0.0005, ****p<0.0001. See also Figure S6.
DISCUSSION
To date it remains unknown whether loss of C9orf72 protein in C9orf72 repeat expansion carriers contributes to neurodegeneration. A recent study found that loss of C9orf72 expression enhanced the toxic gain of function effects of the C9orf72 repeat expansion (Zhu et al., 2020), but the mechanism of this synergistic effect, and which cell types in the brain drive this synergism are still unclear. C9orf72 protein is known to play a role in intracellular trafficking, autophagy, and lysosomal degradation (Farg et al., 2017) and is expressed at high levels in myeloid cells in addition to neurons. Here we found that loss of C9orf72 cell autonomously altered the function of microglia and can drive neuronal injury through enhanced synaptic pruning in aging and in the context of amyloid accumulation.
Numerous scRNAseq studies indicate that microglia are not a uniform population in either normal aging or disease states (Friedman et al., 2018, Keren-Shaul et al., 2017, Hammond et al., 2019, Mathys et al., 2017, Mrdjen et al., 2018, Sala Frigerio et al., 2019). Across all these studies, a common theme is that microglia can be defined as either homeostatic or activated, with a smaller number at any given time that are proliferating. However, the characteristics of subpopulations within the homeostatic and activated categories in healthy and disease states remains an area of active investigation. Our RNAseq data analysis support the notion that compared to WT, aged C9orf72+/− , and C9orf72−/− mice have a reduced ARM and an increased IRM signature (Sala Frigerio et al., 2019, Keren-Shaul et al., 2017). This is interesting in light of a recent report showing that type I IFN drives complement mediated microglial synapse elimination, and that IRM preferentially surrounded Aβ plaques (Roy et al., 2020). Therefore, an increased tendency for C9orf72−/− microglia to adopt an IRM state is consistent with the enhanced complement mediated synaptic engulfment seen in aged C9orf72−/− mice, and the improved plaque clearance observed in the 5XFAD:C9orf72−/− mice. The C9orf72−/− animals also show increased microglial numbers in aged cortex; however these were not of the IRM or ARM identity, and it is likely therefore that there might be other unidentified microglial sub-populations that could be causing the increase.
Autosomal dominant loss of function mutations in the human GRN gene are one of the most common genetic causes of FTD (Baker et al., 2006, Cruts et al., 2006, Ghidoni et al., 2008, Finch et al., 2009). There are many interesting parallels between the effects of loss of C9orf72 and the loss of GRN on microglial function. FTD associated mutations in GRN cause haploinsufficiency, and microglia from Grn-deficient mice similarly show lysosomal dysfunction, an altered inflammatory state and increased synaptic pruning (Lui et al., 2016, Zhang et al., 2020). Additionally, loss of Grn functionally alters the response of microglia to Aβ and the risk of AD (Takahashi et al., 2017, Brouwers et al., 2007, Brouwers et al., 2008). Interestingly both C9orf72 and GRN proteins are involved in lysosomal function, as are a large number of other ALS/FTD risk genes (Almeida and Gao, 2016). This suggests a potential convergence in the pathogenesis of FTD from C9orf72 and GRN mutations, through the alteration of microglial function and response to aging and Aβ pathology.
Although C9orf72 repeat expansions predominantly result in the ALS/FTD phenotype, they have also been observed in a wide array of other neurodegenerative disorders including AD (Harms et al., 2013, Cacace et al., 2013) (Nuytemans et al., 2013, Lindquist et al., 2013, Snowden et al., 2012, Hensman Moss et al., 2014). Though it is possible that the presence of C9orf72 expansions in these other diseases is coincidental, our findings suggest that C9orf72 expansion could directly contribute to their pathogenesis by altering microglial function. In the case of AD it is well established that neuroinflammation and microglia play a key modulatory role (Heneka et al., 2015). Multiple studies using rodent models of Aβ accumulation have shown similar to our observations with C9orf72 that altered levels of Trem2, Grn and even Tardbp expression in microglia can alter their response to Aβ accumulation and contribute to synapse loss and neuronal injury (Wang et al., 2015, Minami et al., 2014, Paolicelli et al., 2017, Lee et al., 2018). Therefore, we hypothesize that an altered state of microglia in C9orf72 carriers could render them susceptible to other environmental or genetic factors, contributing to the wide variety of neurodegenerative phenotypes occasionally observed.
One key point is that while we observed significantly altered gene expression on WGCNA and increased expression of IRM genes in heterozygous C9orf72+/− microglia, other phenotypes such as modification of plaque accumulation and synapse clearance were only observed in homozygous mice. This is relevant as C9orf72 repeat expansion carriers have partial rather than complete loss of protein. While many studies observed phenotypes in the heterozygous mice including altered immune responses in bone marrow macrophages (O’Rourke et al., 2016), development of spontaneous autoimmunity and autoantibody production (Burberry et al., 2016), and enhanced susceptibility to experimental autoimmune encephalitis and resistance to cancer (McCauley et al., 2020), these were generally milder or less penetrant in C9orf72+/− mice compared to C9orf72−/− mice. Interestingly, Grn deficiency also leads to a much milder phenotype in mice than in humans, both in the heterozygous and homozygous states, with phenotypes being generally mild and only being observed at advanced age (Lui et al., 2016, Filiano et al., 2013, Yin et al., 2010, Petkau et al., 2012). Similarly, OPA1 and TBK1 haploinsufficiency are known to cause dominant optic atrophy and ALS/FTD respectively (Marchbank et al., 2002, Freischmidt et al., 2015) but OPA1 and TBK1 heterozygous mice only show mild phenotype which do not recapitulate human disease (Davies et al., 2007, Williams et al., 2011, Brenner et al., 2019). Likewise, knock-in point mutations in genes such as TDP-43 which cause a completely penetrant phenotype in humans, only cause more subtle manifestations in knock-in mice with the exact same genetic lesion (White et al., 2018). Therefore, while our study observed that haploinsufficiency of C9orf72 alters the transcriptome of microglia in aged animals, further studies will be needed to determine whether partial loss of C9orf72 expression in human microglia is sufficient to functionally alter their inflammatory response. Early pathology studies support this idea, as C9orf72 repeat expansion ALS patients were found to have increased CD68 accumulation in motor cortex compared to sporadic ALS, suggesting that the inflammatory response of their microglia was enhanced (Brettschneider et al., 2012). A more recent study also found enhanced type 1 interferon response in brain and peripheral myeloid cells of C9orf72 repeat ALS compared to sporadic ALS patients (McCauley et al., 2020) supporting the idea that patients with C9orf72 ALS/FTD have an altered immune phenotype that is partially driven by microglial dysfunction.
Another interesting finding of our study lies in the identification of a role of C9orf72 in modulating microglia-mediated synaptic engulfment. The complement pathway is known to play an important role in synaptic pruning by microglia during normal postnatal development, aging and potentially in neurodegenerative disorders including AD and FTD (Hong et al., 2016, Schafer et al., 2012, Stevens et al., 2007, Lui et al., 2016). Our data strongly support the idea that microglia-specific loss of C9orf72 is capable of causing synaptic loss and neuronal dysfunction in a non-cell autonomous manner. However, we note that this does not exclude the possibility of additional cell-autonomous defects due to loss of C9orf72 in neurons and effects of other peripheral cells on microglial phenotypes. For example, others previously observed reduced dendritic architecture and neurite length in primary hippocampal cultures from C9orf72−/− animals, indicating a cell-autonomous effect of C9orf72 in neurons at least in culture (Ho et al., 2019, Zhu et al., 2020). Furthermore, recent studies have shown that peripheral type 1 interferon induction can induce unique genetic and phenotypic changes in microglia and can influence amyloid plaque burden in 5XFAD AD mouse model (Aw et al., 2020, Baruch et al., 2015). Further detailed studies deleting C9orf72 specifically from neurons and potentially other cell types (astrocytes, oligodendrocytes) in vivo will be needed to more fully delineate all cell vs. non-cell autonomous effects from loss of C9orf72 function in nervous tissue.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Lead contact: Rita Sattler: rita.sattler@barrowneuro.org
Materials Availability
Further information and requests for resources and reagents may be directed to and will be fulfilled by Dr. Robert H. Baloh: robert.baloh@csmc.edu, robert.baloh@roche.com, Dr. Rita Sattler: rita.sattler@barrowneuro.org, Dr. Deepti.Lall: deepti.lall@cshs.org. This study did not generate new unique reagents.
Data and Code Availability
All datasets generated during and/or analysed during the current study are uploaded and can be downloaded from from the Gene Expression Omnibus website with the following accession numbers: GSE164676 (Superseries), GSE164675 (pooled microglia RNAseq), GSE164674 (Single cell microglia RNAseq). R code for single cell analysis is included as supplementary file 3.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
C9orf72−/− animals were generated as previously described (O’Rourke et al., 2016). 5XFAD mice were purchased from Jackson laboratory (MMRRC Stock No: 34840-JAX) and crossed to C9orf72−/− mice to generate 5XFAD, 5XFAD/C9orf72+/− and 5XFAD/C9orf72−/− mice. C9orf72fl/fl mice were provided by the Pasterkamp lab (Koppers et al., 2015) and were crossed with Cx3cr1Cre (B6J.B6N(Cg)-Cx3cr1tm1.1(cre)Jung/J) purchased from Jackson Laboratories to obtain Cx3cr1Cre;C9orf72fl/fl mice. Mice were sex and age matched for individual experiments. All mice were housed in a 12-hour light: dark cycle and given ad-libitum access to food and water for the duration of the study. All animal experiments were performed under protocols that had been approved by the Institutional Animal Care and Use Committee at Cedars-Sinai Medical Center.
METHOD DETAILS
Preparation of mouse brain samples
For histological analyses, animals from all genotypes were anaesthetized using CO2 and transcardially perfused with ice-cold PBS containing 1 U/ml of heparin. Right brain hemispheres were fixed in 4%PFA for 2 days and cryoprotected in 30% sucrose before freezing and cutting with a cryostat. To analyze Aβ deposition, serial 50 μm free floating sections of the brain were collected from the rostral anterior commissure to caudal hippocampus as landmarks. For all other studies, serial 50 μm free floating sections of the brain were collected and matching sections used for staining and analysis. For biochemical and mRNA expression analysis, cortices and hippocampi of the left-brain hemispheres were carefully dissected, flash frozen in liquid nitrogen and stored at −80°C until further use.
ELISA
Aβ levels were assessed using Sandwich ELISAs as described previously (Ulrich et al., 2014).
Immunohistochemistry
To analyze Aβ deposition, sections were stained with biotinylated anti-Aβ antibody, mHJ3.4 (mouse monoclonal, working dilution 1:500, a generous gift from Dr. David M. Holtzman, Washington University, St. Louis) and developed with DAB using VECTASTAIN Elite ABC Kit per manufacturer’s directions. Stained brain sections were scanned with Leica Aperio Slide scanner. For Nissl staining, matching sections were stained with Cresyl violet (0.1%; Cat #C5042, Sigma-Aldrich, St. Louis, MO, USA) for 30 min, washed in destain solution (70% ethanol, 10% acetic acid) for 1 min, and then dehydrated and cover slipped with DPX mountant. The slides were scanned with Leica Aperio Slide scanner.
Immunofluorescence staining
Immunofluorescence co-staining was performed on free-floating sections with anti- HJ3.4 and anti-Iba1 antibody (rabbit polyclonal, 1:250, Cat #019–19741, Wako chemicals USA, inc, Richmond, VA, USA). The sections were blocked with 2% Normal goat serum/1% Bovine serum albumin/PBS/0.25% Triton X-100/0.02% Sodium azide for 2 hr, followed by incubation with the primary antibodies diluted in the same solution for overnight at 4°C. Next, the sections were incubated for 1 hr with fluorescent secondary antibodies (Molecular Probes, Carlsbad, CA, USA; 1:500 dilution): Alexa Fluor 488 goat anti-mouse IgG (H+L) (Cat #A11029) and Alexa Fluor 555 goat anti-rabbit IgG (H+L) (Cat #A21428). Finally, the processed sections were mounted, briefly air dried, and cover slipped with Prolong Gold Antifade Reagent with DAPI (Invitrogen, Cat # P36931).
Additional immunofluorescence staining was performed with anti vGLUT1 (Guinea pig, 1:500, Synaptic Systems Cat #135304), PSD 95 (Mouse monoclonal (IgG2a) , K28/43, 1: 500, Upstate, Cat #05–494) and anti-synaptophysin (rabbit monoclonal, 1:200, Synaptic Systems Cat #101203) by blocking with 5% Bovine serum albumin/PBS/0.2% Triton X-100 for 1 hr, or co-staining with anti C1q (Rabbit monoclonal, 1:200, Cat. #ab227072), anti-Iba1 and anti-CD68 (rat monoclonal, 1:200, Cat #MCA1957, Bio-Rad Lab. Inc., Hercules, CA, USA). After overnight incubation with the primary antibody/antibodies at 4°C, the sections were incubated for 1 hr with the relevant fluorescent secondary antibodies: Alexa Fluor 488 donkey anti-rat IgG (H+L) (Cat #A21208), Alexa Fluor 555 goat anti-rabbit IgG (H+L), Alexa Fluor 647 donkey anti-mouse IgG (H+L) (Cat #A31571), Alexa Fluor 647 donkey anti-rabbit IgG (H+L) (Cat #A31573).
On additional sets of sections, Thioflavin S (Cat #T1892, Sigma-Aldrich, St. Louis, MO, USA) staining was performed followed by co-staining with anti-glial fibrillary acidic protein (GFAP; mouse monoclonal, 1:400, Cat #MAB360, Millipore, Burlington, MA, USA), or anti-Iba1 and anti-CD68. The brain sections were first permeabilized with TBS/0.25% Triton X-100 for 30 min and then incubated with freshly prepared Thioflavin S (0.025%/50% Ethanol) for 5 min in the dark. The sections were briefly differentiated in 50% Ethanol, blocked with 3% Normal goat serum/TBS/0.25 % Triton X-100 for 1 hr followed by overnight incubation at 4°C with the primary antibody/antibodies diluted in 1% Normal goat serum/TBS/0.25 % Triton X-100. The fluorescent secondary antibodies used were: Alexa Fluor 555 goat anti-mouse IgG (H+L), Alexa Fluor 555 goat anti-rat IgG (H+L), and Alexa Fluor 647 goat anti-rabbit IgG (H+L) (Cat #A21244). Positive and negative controls were run along each of the batches of sections processed.
Western blot
Cortical brain tissues and microglial cell pellets were homogenized using a glass Teflon homogenizer in RIPA lysis and extraction buffer (Thermo Scientific, Cat #89900), supplemented with protease inhibitor cocktail (cOmplete, Roche) and phosphatase inhibitor cocktail (PhosSTOP, Roche). Protein concentration was determined by BCA assay kit (Thermo Fisher). Cell lysates and brain homogenates were separated on protean TGX precast gels (Biorad) and blotted onto nitrocellulose membranes (Biorad). Membranes were blocked for 60 min with Odyssey blocking buffer (PBS, Li-Cor, Cat #927–40000) and incubated overnight at 4°C with anti-PSD95 (Millipore MAB1596, 1: 500), anti-synaptophysin (Abcam ab16659, 1:500), anti-C9orf72 (GeneTex Cat #. GTX634482, 1:1000), anti-STING (Cell Signaling, Cat #. 13647, 1:1000), and anti β tubulin (Sigma-Aldrich T6074, 1:1000) antibodies. After washing membranes were incubated for 60 min with IRDye fluorescent secondary antibodies (Li-Cor). After washing, blots were subsequently analyzed with Li-COR imaging system (Odyssey CLx).
Imaging and quantification
Stained sections were imaged with Olympus BX51 Upright, Zeiss AxioImager Z2 equipped with Apotome structured illumination optics, and Nikon A1R inverted laser confocal microscopes with 20X, 40X or 63X objectives. Depending upon the staining, different step sizes were used to generate z-stacks across the whole tissue thickness. CZI, TIFF and ND2 files were then imported into ImageJ (NIH) to create Z-projection images and quantification of fluorescent signals was performed using ImageJ software with the threshold and analyze particles functions using specific macros (fixed thresholds for signal intensity and voxel) to all the images of the same experiment, in order to produce unbiased signal quantification.
For quantitative analyses of mHJ3.4-biotin staining, scanned images were analyzed with Leica online digital image hub viewer software. Identified objects after thresholding were individually inspected to confirm the object as a plaque. Three brain sections per mouse, each separated by 300mm, were used for quantification. The average of three sections were used to represent the plaque load for each mouse.
For neuronal density analysis, number of neurons in the motor cortex were analyzed by custom drawing a region of interest and counted using the cell counter function of ImageJ and expressed as average densities of nuclei per mm2.
For graphs with percentage area calculations (%area), maximum intensity projection (MaxIP) images were thresholded and the analyze particles function in ImageJ was used to determine positive area. Areas encompassing the motor cortex were chosen (4–5 images per section, 3 sections per animal) for the analyses. Specific macros were applied in an unbiased manner to all the images of the same experiment for quantification.
For plaque morphology analysis, high-resolution images were collected at 40x magnification. Maximum intensity projection of the z-stack for each image was subsequently used for morphological analysis.
For synapses inside microglia, synaptic puncta labelled with vGLUT1 and PSD95 tissues were imaged as described above. Z-stacks were analyzed using the “Co-loc” feature in Fiji ImageJ. Iba1+signals were used to mask microglial outlines, and thresholds were adjusted to overlap with synaptic puncta signal. Data were recorded as number of ROI occupied by the puncta signal.
CD68+ inclusions inside microglia were quantified using previously published protocol (Hong et al., 2016) by investigator blinded to the genotypes of the animal. For microglial levels of CD68, a glycoprotein that localizes to lysosomes and endosomes, CD68 levels were quantified on a scale from 0 to 3. A score of 0 signified no/scarce expression; 1 for punctate expression; and 2 (roughly one-third to two-thirds occupancy) or 3 (greater than two-thirds occupancy) for punctate expression covering an entire cell or aggregated expression.
Endogenous immunoglobulin IgG detection
Free-floating mouse brain cryo-sections were stained with Thioflavin S and then incubated overnight with anti-Iba1 antibody and serum samples (1:100 dilution) collected from 5XFAD and 5XFAD/C9orf72−/− mice. The secondary antibody Alexa Fluor 555 F(ab’)2 fragment of goat anti-mouse IgG (H+L) (1:500; Cat #A21425) was then incubated for 1hr followed by mounting and cover-slipping with Prolong Gold Antifade Reagent with DAPI.
Golgi-immunostaining and dendritic branching analysis
Freshly dissected mouse brains were processed for Golgi staining following the manufacturer’s protocol (FD Neurotechnologies). 150 μm coronal mouse brains sections were generated using a Leica CM3050s cryostat and placed onto gelatin coated slides overnight. Stained sections were imaged using a 40x oil immersion objective of an Axio Observer.Z1 Zeiss microscope at a 1 μm step size. Golgi stained cortical neurons (n=28–30 cells/mouse) were manually processed for filament tracing using the contour graphic tools from the Zeiss Zen 2 Blue Software. Filament tracings were then exported into ImageJ to process for Sholl Analysis (ImageJ Sholl Analysis plugin). The branching complexity of each cortical neuron was determined by the average number of dendrite intersections towards the radial distance from the center of the soma.
Mouse IgG ELISA
To determine endogenous IgG’s against Aβ, sera was collected from 4-month old 5XFAD and 5XFAD/C9orf72−/− mice that received active Aβ immunization. Anti- Aβ titer was measured using a well-validated ELISA as described previously (Davtyan et al., 2010, Davtyan et al., 2014).
B-cell/T-cell reactivity by ELISA and ELISPOT
Aβ producing B and T cells in the spleen and lymph nodes were determined as previously published(Marsh et al., 2016). Briefly spleens, deep cervical lymph nodes (DCLNs) and superficial cervical lymph nodes (SCLNs) were collected and antibody forming B cells specific to Aβ were detected in splenocytes and pooled DCLNs/SCLNs lymphocytes by ELISPOT (Mabtech). Splenocytes or lymphocytes were incubated for 24h in 96-well plates coated with Aβ peptide, and the assay was performed following manufacturer’s protocol (Mabtech). Sera and splenocytes were also collected from mice immunized with Aβ peptide as a positive control (Davtyan et al., 2010, Davtyan et al., 2014).
Microglia isolation
The meninges were removed from the mice brains of different genotypes and dissociated using the Neural Tissue dissociation kit, papain based (Miltenyi Biotech) and GentleMACSdissociator (NTDK Brain setting). The lysate was collected and passed through a 70μm strainer to obtain a single cell suspension. Next, myelin was removed from the cells by incubating with magnetic myelin beads (Myelin Removal Beads II, human, mouse, rat, Miltenyi Biotech) and removed using the AutoMACs. Cells were then incubated with Cd11b+ magnetic beads Cd11b MicroBeads, human and mouse, (Miltenyi Biotech) and sorted using the AutoMACs. After sorting, cells were pelleted and stored at −80°C until further downstream application. For single cell experiment, isolated microglia went directly for downstream processing. For microglia culture, the Cd11b+ cells were counted, plated (200K cells/well) and cultured in Microglia complete media containing DMEM/F-12, GlutaMAX™ with HEPES (Invitrogen), 10% fetal bovine serum, 100μg/ml Penicillin/ Streptomycin, with 10 ng/ml of the following growth factors: Recombinant mouse M-CSF, Recombinant mouse GM-CSF (R&D systems) and 50ng/ml TGF-β1 (Miltenyi Biotech) for 6 days before stimulation. Microglia stimulation: 6 days post culturing, microglia were stimulated with 10ug/ml cGAMP for 24 hrs. Post stimulation, cells were lysed in lysis buffer and RNA isolated using Qiagen Micro RNA isolation kit. RNA quality was determined using Nanodrop and 200ng of RNA was used to make cDNA. Another set of stimulated microglia were used for western blot analysis.
Neuron-Microglia co-culture
For mouse cortical neuron cultures, cortices from E15.5 C9orf72+/+− and C9orf72−/− embryos were dissected in ice-cold dissection buffer (1XHBSS, 30mM glucose, 10mM HEPES, 1mM Sodium pyruvate, 1X Pen/Strep). Cortices from different embryos were pooled and after brief spinning, cortical tissue was dissociated using the Worthington papain cell dissociation system (LK003150) according to manufacturer’s protocol. 300K cells were plated on poly-l-lysine/laminin coated coverslips and cultured for 14 days in vitro. At DIV14, microglia were plated onto the neurons at a 1:3 microglia to neuron ratio. Co-cultures were allowed to continue for 2 days before fixation with 4% PFA. To quantify the synaptic density around microglia in microglia-neuron co-cultures, the presence of microglia and synapses was determined by immunofluorescent staining using antibodies for Cd11b (MCA 711), and synaptophysin (Synaptic systems, SS101203), respectively. For each co-culture coverslip, 10–15 microglia cells were chosen at random for imaging. During capture, microglia cells were centered, and images were acquired using Zeiss AxioImager Z2 equipped with Apotome structured illumination optics at 40X. Synaptophysin density was determined using Scholl analysis at increments of 10μm. Overall synaptophysin density in the neuronal cultures without microglia was also determined to analyze total synaptophysin puncta in different genotypes. Co-culture experiments were repeated twice with new sets of timed pregnancies for C9orf72+/+- and C9orf72−/− animals. At least 3 coverslips from each culture condition were imaged and data averaged for an experiment. Data acquisition and analyses was done by investigator blinded to the experiment.
Mouse behavior (Barnes Maze analyses)
Spatial learning and memory assessment of the animals was determine using Barnes maze paradigm that uses a bright spotlight as mild aversive stimulus. Behavior was performed in animal cohorts at 4 and 12 months of age. Both male and females were used for behavior and animals were singly housed before and during the experiment spanning the whole experimental timeline. The maze (San Diego Instruments, San Diego, CA) is a circular (122 cm in diameter) platform with 20 holes evenly spaced around the perimeter. Distal cues were placed around the room that remained consistent during the whole testing period. On the first day (pretraining), mice were placed on the Barnes maze to roam freely and get habituated to the maze and the surroundings. On the training days (Day1 to Day 4 consecutive days), three trials per day were performed, 4 minutes per trial, with one-hour break between the trials. At the beginning of each session (training and test), mice were placed in the middle of the maze in a cylindrical white start chamber (30.5 cm high, 20.3 cm in diameter). After 30 seconds, chamber was lifted, and the mice were free to explore the maze and find the escape tunnel. If a mouse failed to find the escape tunnel within the 4 min period, it was placed in the tunnel and allowed to stay there for 30 sec prior to removal. Mice were rested on 5th and 6th days. For testing on Day 7, mice were tested using the training paradigm. Following each trial, the maze and escape tunnel were cleaned with 70% ethanol. For each trial primary latency (the latency to locate) and enter (total latency) into the escape tunnel was recorded to assess performance. Behavior was performed by an individual blinded to the genotypes of the animals.
RNA isolation and bulk RNAseq
RNA was isolated using Qiagen RNeasy Micro Kit (Cat #74004) according to manufacturer’s instructions. RNA-seq libraries were generated using 75ng to 180ng total RNA as input for the TruSeq Stranded mRNA Library Prep kit (Illumina) according to the manufacturer’s protocol, and samples were indexed using TruSeq RNA Single Indexes (Illumina). Library preps were analyzed via bioanalyzer (Agilent) and quantified via Qubit fluorometric quantification (Thermo Fisher). Quantified libraries were normalized, pooled, and sequenced on a NextSeq 500 sequencer (Illumina) using the single-end 75 nucleotide setting. Raw sequencing reads were demultiplexed and FASTQ files were uploaded to the Galaxy web platform using the public server at usegalaxy.org (Afgan et al., 2018). FASTQ files were processed using FASTQ Groomer (Galaxy Version 1.1.1) (Blankenberg et al., 2010) and aligned to the mouse genome (mm10) via Tophat (Galaxy Version 2.1.1)(Kim et al., 2013) using default settings. BAM files were downloaded locally and annotated using Partek software v7.17.0918 to generate RPKM values for each gene.
For TPM analyses, raw sequencing reads were demultiplexed, and FASTQ files were used to generate estimated transcript counts against the mouse transcriptome (mm10) via Salmon v0.8.2. TPM values summed to the gene level were generated using the R Bioconductor package DEseq2.
Module Preservation and comparative co-expression network analysis
Co-expression network analyses were performed in R using the WGCNA package (Langfelder and Horvath, 2008). Comparative analysis was used to assess the preservation and trajectory of microglial neurodegeneration modules that were previously defined from transgenic mice expressing human mutant MAPT, including two modules (inflammasome and anti-inflammasome) that were identified in mutant MAPT mice and preserved and up-regulated in microglia isolated from 5XFAD and P2SAPP models of AD pathology (Rexach et al., 2019). To assess the co-expression patterns of these modules in C9orf72 mice, module preservation analysis was applied from the WGCNA package to normalized RPKM counts from the C9orf72 dataset. This measure combines module density and intramodular connectivity metrics to give a composite statistic for which Z<2 suggests lack of module preservation, Z>2 moderate and Z>10 high preservation(Langfelder et al., 2011). For modules with strong associations with disease in both mutant MAPT and C9orf72 datasets, but poor total module preservation in the C9orf72 dataset, the portion of the module that is preserved and up regulated within the C9orf72 dataset (positive kME) was identified. The module eigengene expression trajectories were then measured in C9orf72 microglia compared to WT at 3-months or 17-months of age (unpaired two-sided Wilcox rank test). To identify and visualize protein-protein interactions among module genes, STRING was used (version 10.5; https://string-db.org) with the following settings (organism: Mus musculus; meaning of network edges: confidence; active interaction sources: experiments and databases; minimal required interaction score: medium confidence (0.400), max number of interactors to show: none).
Single cell RNAseq (ScRNAseq)
Microglia were isolated from cortex of mice as previously described in this paper. Both cortices per animal were pooled to get enough microglia for library preparation and sequencing. Three animals per day of mixed genotypes and order each day were processed through the cDNA amplification step. Finally, microglia from all nine animals were taken from cDNA step to the final library preparation step together.
Sequencing library construction using the Chromium platform
Single-cell RNA-Seq libraries were prepared per the Single Cell 3′ v3.1 Reagent Kits User Guide (10x Genomics, Pleasanton, California). Cellular suspensions were loaded on a Chromium Controller instrument (10x Genomics) to generate single-cell Gel Bead-In-EMulsions (GEMs). GEM-RT was performed in a Veriti 96-well thermal cycler (Thermo Fisher Scientific, Waltham, MA). GEMs were then harvested, and the cDNA was amplified and cleaned up with SPRIselect Reagent Kit (Beckman Coulter, Brea, CA). Indexed sequencing libraries were constructed using Chromium Single-Cell 3′ Library Kit for enzymatic fragmentation, end-repair, A-tailing, adapter ligation, ligation cleanup, sample index PCR, and PCR cleanup. The barcoded sequencing libraries were quantified by quantitative PCR using the Collibri Library Quantification Kit (Thermo Fisher Scientific). Libraries were sequenced on a NovaSeq 6000 (Illumina, San Diego, CA) as per the Single Cell 3′ v3.1 Reagent Kits User Guide, with a sequencing depth of ~60,000 reads/cell.
Data Analysis
The demultiplexed raw reads were aligned to the transcriptome using STAR (version 2.5.1) (Dobin et al., 2013) with default parameters, using mouse mm10 transcriptome reference from Ensemble version 84 annotation, containing all protein coding genes, long non-coding RNA genes, and polymorphic pseudogenes. Expression counts for each gene in all samples were collapsed and normalized to unique molecular identifier (UMI) counts using Cell Ranger software version 4.0.0 (10X Genomics). The result is a large digital expression matrix with cell barcodes as rows and gene identities as columns. Filtered digital expression matrices from the CellRanger output (filtered_feature_bc_matrix directory) were loaded into R (version 4.0.3) using the Seurat package (version 3.2.2) in Rstudio (version 1.3.1093), keeping all genes that were expressed in more than one cell. All samples were merged into a single Seurat object (30,872 cells) and filtered by removing all cells with counts, genes, or mitochondrial gene percentage outside of three standard deviations from the mean (30,065 cells). The data were then normalized using SCTransform, regressing out the mitochondrial gene content. Dimensionality reduction was run with Principal Component Analysis (PCA) followed by Uniform Manifold Approximation and Projection (UMAP) using the default 50 principal components (PCs). After visualizing the UMAP plots, the cells appeared to cluster based on the order they were processed, the day they were processed, and the sex of the animal. Integration of the data was performed by using the Harmony R package (version 1.0.0), batch correcting for these covariates (process day, process order, and sex) resulting in a much more uniform clustering between these factors after rerunning UMAP with the Harmony-corrected PCs. Cluster determination was performed using FindNeighbors and FindClusters with a resolution of 0.3 and resulting in an initial discovery of 19 clusters. Cluster markers were determined using FindAllMarkers with the default min.pct = 0.25 and logfc.threshold = 0.4. Investigating these markers, six distinct cell types were discovered (microglia, endothelial cells, astrocytes, granulocytes, neutrophils and lymphocytes). A seventh general cluster was positive for both monocyte and macrophage markers.
In order to investigate microglial state changes between genotypes, all non-microglia cells were removed (26,275 microglia), and the remaining cells were then normalized, centered and scaled (NormalizeData, FindVariableFeatures & ScaleData) followed by Jack Straw Analysis to determine which PCs to use for further analysis. A sharp drop-off of p-values was observed between PC 14 and 15, so 15 PCs were used for microglia analysis. The microglia were then normalized with SCTransform regressed against mitochondrial gene content. Subsequent PCA and UMAP reductions showed similar day, order, and sex bias as the analysis with all the cells, and the microglia were again integrated with Harmony, using the same covariates as before, followed by UMAP. Using FindNeighbors and FindClusters with a resolution of 0.8 discovered 16 clusters. Known ARM genes (Clec7a, Itgax, Apoe, and Cst7) were found to be highly expressed in Cluster 11, and known IRM genes (Ifit1, Ifit2, Ifi204, and Isg15) were found to be highly expressed in Cluster 13. =Using FindAllMarkers (min.pct = 0.25 and logfc.threshold = 0.4), 66 genes were discovered to have a positive average logfc in Cluster 11 (ARM cluster marker genes) and 86 genes were discovered to have a positive average logfc in Cluster 13 (IRM cluster marker genes). These two gene lists were used with the AddModuleScore function to assign ARM and IRM module scores to each cell. Cells in the ARM or IRM clusters were compared across genotypes by ordinary one-way ANOVA (Dunnett’s multiple comparisons test with a single pooled variance) using GraphPad Prism (version 9.0.0 (86) for macOS).
Statistical analysis
For each experiment, the corresponding statistics test is indicated in the figure legend. Number of samples for each group is always shown in the figure and figure legends. Statistical analysis was performed by using GraphPad Prism Version 9.0 (GraphPad Software, La Jolla, CA, USA). Values were presented as mean ± SEM. Statistical significance was determined using Student’s t test and analysis of variance (ANOVA), followed by Tukey’s, Welch correction, or Sidak’s post hoc testing as appropriate.
Supplementary Material
Supplemental table S1 (excel file): Related to Figure 1 and Figure S2: QC data of scRNAseq dataset showing total number of cell sequenced per genotype, average reads per sample, median genes per cell and other cell types identified in analyses.
Supplemental table S2 (excel file): Related to Figure 1: QC data of all microglial clusters identified with scRNAseq and list of genes identified in each microglial cluster.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Aβ antibody, mHJ3.4 | David M. Holtzman | |
| Rabbit polyclonal anti-Iba1 antibody | Wako | Cat #019–19741; RRID:AB_839504 |
| Guinea pig anti vGLUT1 | Synaptic Systems | Cat #135304; RRID:AB_887878 |
| Mouse monoclonal (IgG2a) anti PSD95 | Upstate | Cat #05–494; RRID:AB_2315219 |
| Rabbit monoclonal anti-synaptophysin | Synaptic Systems | Cat #101203; RRID:AB_2810218) |
| Rabbit monoclonal anti C1q | Abcam | Cat. #ab227072; N.A |
| Rat monoclonal anti-CD68 | Bio-Rad Lab | Cat #MCA1957; RRID:AB_322219 |
| Mouse monoclonal anti GFAP | Millipore | Cat #MAB360, RRID:AB_11212597 |
| Mouse monoclonal anti PSD95 | Millipore | Cat #MAB1596, RRID:AB_2092365 |
| Rabbit monoclonal anti-synaptophysin | Abcam | Cat #ab16659; RRID:AB_443419 |
| Mouse monoclonal anti β tubulin | Sigma-Aldrich | Cat # T6074; RRID:AB_477582 |
| mouse monoclonal anti-C9orf72 | GeneTex | Cat #. GTX634482, RRID:AB_2784545 |
| Rabbit polyclonal anti PSD95 | Thermo Fisher Scientific | Catalog # 51–6900; RRID:AB_2533914 |
| Rabbit monoclonal anti STING | Cell Signaling | Catalog # 13647 RRID:AB_ 2732796 |
| Rabbit polyclonal anti-Homer 1 | Synaptic Systems | Cat# 160 002, RRID:AB 2120990 |
| Guinea pig anti Bassoon | Synaptic Systems | Cat# 141 004, RRID:AB_2290619 |
| IRDye® 800CW Goat anti-Rabbit IgG Secondary Antibody | Licor | P/N: 926–32211; RRID:AB_621843 |
| IRDye® 680RD Goat anti-Mouse IgG Secondary Antibody | Licor | P/N: 926–68070; RRID:AB_10956588 |
| Alexa Fluor 488 goat anti-mouse IgG (H+L) | Thermo Fisher Scientific | Cat #A11029; RRID:AB_138404 |
| Alexa Fluor 555 goat anti-rabbit IgG (H+L) | Thermo Fisher Scientific | Cat #A21428; RRID:AB_2535849 |
| Alexa Fluor 488 donkey anti-rat IgG (H+L) | Thermo Fisher Scientific | Cat #A21208; RRID:AB_2535794 |
| Alexa Fluor 647 donkey anti-mouse IgG (H+L) | Thermo Fisher Scientific | Cat #A31571; RRID:AB_162542 |
| Alexa Fluor 647 donkey anti-rabbit IgG (H+L) | Thermo Fisher Scientific | Cat #A31573; RRID:AB_2536183 |
| Alexa Fluor 647 goat anti-rabbit IgG (H+L) | Thermo Fisher Scientific | Cat #A21244; RRID:AB_2535812 |
| Alexa Fluor 555 F(ab’)2 oat anti-mouse IgG (H+L) | Thermo Fisher Scientific | Cat #A21425; RRID:AB_2535846 |
| Prolong Gold Antifade Reagent with DAPI | Thermo Fisher Scientific | Cat # P36931; N.A |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cresyl violet | Sigma-Aldrich | Cat #C5042 |
| cOmplete, Roche protease inhibitor cocktail | Sigma-Aldrich | Cat #5892970001 |
| PhosSTOP phosphatase inhibitor cocktail | Sigma-Aldrich | Cat #4906837001 |
| Critical Commercial Assays | ||
| FD Rapid GolgiStain™ Kit | FD Neuro Technologies, Inc. | Cat #PK401 |
| Mouse IgG ELISpot kit (HRP) | Mabtech | Cat #3825–2H |
| Mouse IFNg ELISPOT Set | BD Biosciences | Cat #551083 |
| Neural Tissue dissociation kit | Miltenyi Biotech | Cat #130–092-628 |
| Myelin Removal Beads II | Miltenyi Biotech | Cat # 30–096-433 |
| CD11b+ magnetic beads | Miltenyi Biotech | Cat #130–093-634 |
| TruSeq Stranded mRNA Library Prep kit | Illumina | Cat #20020594 |
| TruSeq RNA Single Indexes Set A | Illumina | Cat #20020492 |
| TruSeq RNA Single Indexes Set B | Illumina | Cat # 20020493 |
| VECTASTAIN Elite ABC Kit | Vector labs | Cat #PK-6100 |
| Pierce™ BCA Protein Assay Kit | Thermo Fisher Scientific | Cat #23225 |
| Mini-PROTEAN® TGX™ Precast Gels | Bio-Rad | Cat #4561096 |
| Odyssey blocking buffer | Li-Cor | Cat #927–40000 |
| RNeasy Micro Kit | Qiagen | Cat #74004 |
| Single Cell 3′ v3.1 Reagent Kits | 10x Genomics | |
| SPRIselect Reagent Kit | Beckman Coulter | B23319 |
| Chromium Single-Cell 3′ Library Kit | 10x Genomics | PN-120237 |
| Collibri Library Quantification Kit | (Thermo Fisher Scientific | A38524100 |
| Deposited Data | ||
| Bulk RNAseq data Single cell RNAseq data |
This paper | GEO Accession numbers: GSE164676 (superseries) GSE164675 (Bulk RNAseq) GSE164674 (Single Cell RNAseq) |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Mouse: C9orf72 (311004O21Rik) knockout mouse | In house | O’Rourke et.al 2016 |
| Mouse: B6SJL-Tg(APPSwFlLon,PSEN1*M146L*L286V)6799Vas/Mmjax | The Jackson Laboratory | MMRRC Stock No: 34840 |
| Mouse: C9orf72fl/fl | Obtained from R Jeroen Pasterkamp | Koppers et al., 2015 |
| Mouse: (B6J.B6N(Cg)-Cx3cr1tm1.1(cre)Jung/J | The Jackson Laboratory | Stock No: 025524 RRID:IMSR_JAX:025524 |
| Oligonucleotides | ||
| Recombinant DNA | ||
| Software and Algorithms | ||
| Fiji ImageJ | https://doi.org/10.1038/nmeth.2019 | https://imagej.net/Fiji |
| GraphPad Prism | GraphPad Software, La Jolla, CA, USA | Version 9.0.0 https://www.graphpad.com/scientific-software/prism/ |
| WGCNA R package | https://doi.org/10.1186/1471-2105-9-559 |
https://horvath.genetics.ucla.edu/html/ CoexpressionNetwork/Rpackages/ WGCNA |
| STRING | https://doi.org/10.1093/nar/gkw937 | https://string-db.org |
| Galaxy | https://doi.org/10.1093/nar/gky379; https://doi.org/10.1093/nar/gkw343 |
http://usegalaxy.org/ |
| FASTQ Groomer (Galaxy Version 1.1.1) | https://doi.org/10.1093/bioinformatics/btq281 | http://usegalaxy.org/ |
| TopHat (Galaxy Version 2.1.1) | https://doi.org/10.1186/gb-2013-14-4-r36 | http://usegalaxy.org/ |
| Bowtie2 (part of TopHat Galaxy Version 2.1.1) | https://doi.org/10.1038/nmeth.1923 | http://usegalaxy.org/ |
| Samtools (part of TopHat Galaxy Version 2.1.1) | https://doi.org/10.1093/bioinformatics/btp352 | http://usegalaxy.org/ |
| Partek Genomic Suite | Partek® Genomics Suite®, Partek Inc., St. Louis, MO, 2018, version 7.17.0918 | Version 7.17.0918 https://www.partek.com/partek-genomics-suite/ |
| Salmon | https://doi.org/10.1038/nmeth.4197 | Version 0.8.2 https://combine-lab.github.io/salmon/ |
| mm10 (GRCm38) mouse reference genome sequence and transcriptome annotation | http://www.genome.org/cgi/doi/10.1101/gr.213611.116 | hosting webpage: https://support.illumina.com/sequencing/sequencing_software/igenome.html |
| mm10 (continued) | download url used: http://igenomes.illumina.com.s3-website-us-east-1.amazonaws.com/Mus_musculus/UCSC/mm10/Mus_musculus_UCSC_mm10.tar.gz | |
| mm10 (continued) | README.txt file found in the downloaded reference sequence in the following directory ./Mus_musculus/UCSC/mm10/Annotation : “The contents of the annotation directories were downloaded from UCSC on: July 17, 2015. SmallRNA annotation files were downloaded from miRBase release 21.” | |
| DESeq2 R package (Bioconductor version: Release 3.11) | https://doi.org/10.1186/s13059-014-0550-8 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| STAR | https://doi.org/10.1093/bioinformatics/bts635 | Version 2.5.1 |
| Cell Ranger software | 10X Genomics | Version 4.0.0 |
| GO Ontology database | http://doi.org/10.5281/zenodo.4081749 | Release 2020–10-09 |
| MyGeneSet | https://doi.org/10.1038/ni1008-1091 | http://rstats.immgen.org/MyGeneSet_New/index.html |
| R | https://www.R-project.org/ | Version 4.0.3 |
| Rstudio | http://www.rstudio.com | Version 1.3.1093 |
| Seurat R package |
https://doi.org/10.1038/nbt.4096 https://doi.org/10.1016/j.cell.2019.05.031 https://doi.org/10.1186/s13059-019-1874-1 |
Version 3.2.2 |
| Harmony R package | https://doi.org/10.1101/461954 | Version 1.0.0 |
| Tidyverse R package | https://doi.org/10.21105/joss.01686 | Version 1.3.0 |
| Matrix R package | Douglas Bates and Martin Maechler (2019). Matrix: Sparse and Dense Matrix Classes and Methods. | Version 1.2–18 |
| Cowplot R package | Claus O. Wilke (2020). cowplot: Streamlined Plot Theme and Plot Annotations for ‘ggplot2’ | Version 1.1.0 |
| Viridis R package | Simon Garnier (2018). viridis: Default Color Maps from ‘matplotlib’ | Version 0.5.1 |
| Other | ||
| Leica digital image hub | -N-A- | |
| Li-COR imaging system (Odyssey CLx) | -N-A- | |
Highlights.
C9orf72 deficient microglia exhibit altered transcriptional profile and functions
Loss of C9orf72 in microglia leads to non-cell autonomous neuronal dysfunction
Loss of C9orf72 causes defects in learning and memory in ALS/FTD and AD mouse models
ACKNOWLEDGEMENTS
We thank Cedar-Sinai Applied Genomics, Computation & Translational Core for RNAseq, Cedar-Sinai Animal Behavior Core for mice behavior experiments, Dr. Michael Workman and Dr. Ritchie Ho for help with RNAseq analysis and all members of the Baloh and Sattler laboratory for helpful discussions. This work was supported by NIH grant NS097545 (R.H.B), the Robert and Louise Schwab family (R.H.B), the Cedars-Sinai ALS Research Fund (R.H.B.), NIH grants NS090934 (D.M.H), AG047644 (D.M.H), the JPB Foundation (D.M.H), NIH RO1NS085207 (R.S), the Muscular Dystrophy Association (R.S); the ALS Association (R.S); the Robert Packard Center for ALS Research (R.S), the Barrow Neurological Foundation (R.S), Rainwater Charitable Foundation (D.H.G), NIH grant 5R25 NS065723 (J.E.R), NIH grants AG055524 (M.B.J) and AG061895 (H.D).
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. One or more of the authors of this paper received support from a program designed to increase minority representation in science. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
R.H.B is employed by Roche Pharmaceuticals. D.M.H is listed as inventor on a patent licensed by Washington University to C2N Diagnostics on the therapeutic use of anti-tau antibodies. D.M.H co-founded and is on the scientific advisory board of C2N Diagnostics, LLC. C2N Diagnostics, LLC has licensed certain anti-tau antibodies to AbbVie for therapeutic development. D.M.H is on the scientific advisory board of Denali and consults for Genentech, Merck, and Cajal Neurosciences. D.M.H and J.D.U are listed as inventors on a provisional patent from Washington University on TREM2 antibodies. R.S is on the scientific advisory board of Spinogenix Inc.
References
- ABO-RADY M, KALMBACH N, PAL A, SCHLUDI C, JANOSCH A, RICHTER T, FREITAG P, BICKLE M, KAHLERT AK, PETRI S, STEFANOV S, GLASS H, STAEGE S, JUST W, BHATNAGAR R, EDBAUER D, HERMANN A, WEGNER F & STERNECKERT JL 2020. Knocking out C9ORF72 Exacerbates Axonal Trafficking Defects Associated with Hexanucleotide Repeat Expansion and Reduces Levels of Heat Shock Proteins. Stem Cell Reports, 14, 390–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AFGAN E, BAKER D, BATUT B, VAN DEN BEEK M, BOUVIER D, CECH M, CHILTON J, CLEMENTS D, CORAOR N, GRUNING BA, GUERLER A, HILLMAN-JACKSON J, HILTEMANN S, JALILI V, RASCHE H, SORANZO N, GOECKS J, TAYLOR J, NEKRUTENKO A & BLANKENBERG D 2018. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res, 46, W537–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALMEIDA S & GAO FB 2016. Lost & found: C9ORF72 and the autophagy pathway in ALS/FTD. EMBO J, 35, 1251–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ATANASIO A, DECMAN V, WHITE D, RAMOS M, IKIZ B, LEE HC, SIAO CJ, BRYDGES S, LAROSA E, BAI Y, FURY W, BURFEIND P, ZAMFIROVA R, WARSHAW G, ORENGO J, OYEJIDE A, FRALISH M, AUERBACH W, POUEYMIROU W, FREUDENBERG J, GONG G, ZAMBROWICZ B, VALENZUELA D, YANCOPOULOS G, MURPHY A, THURSTON G & LAI KM 2016. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep, 6, 23204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AW E, ZHANG Y & CARROLL M 2020. Microglial responses to peripheral type 1 interferon. J Neuroinflammation, 17, 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAKER M, MACKENZIE IR, PICKERING-BROWN SM, GASS J, RADEMAKERS R, LINDHOLM C, SNOWDEN J, ADAMSON J, SADOVNICK AD, ROLLINSON S, CANNON A, DWOSH E, NEARY D, MELQUIST S, RICHARDSON A, DICKSON D, BERGER Z, ERIKSEN J, ROBINSON T, ZEHR C, DICKEY CA, CROOK R, MCGOWAN E, MANN D, BOEVE B, FELDMAN H & HUTTON M 2006. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature, 442, 916–9. [DOI] [PubMed] [Google Scholar]
- BARUCH K, ROSENZWEIG N, KERTSER A, DECZKOWSKA A, SHARIF AM, SPINRAD A, TSITSOU-KAMPELI A, SAREL A, CAHALON L & SCHWARTZ M 2015. Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer’s disease pathology. Nat Commun, 6, 7967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLANKENBERG D, GORDON A, VON KUSTER G, CORAOR N, TAYLOR J, NEKRUTENKO A & GALAXY T 2010. Manipulation of FASTQ data with Galaxy. Bioinformatics, 26, 1783–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOIVIN M, PFISTER V, GAUCHEROT A, RUFFENACH F, NEGRONI L, SELLIER C & CHARLET-BERGUERAND N 2020. Reduced autophagy upon C9ORF72 loss synergizes with dipeptide repeat protein toxicity in G4C2 repeat expansion disorders. EMBO J, 39, e100574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRENNER D, SIEVERDING K, BRUNO C, LUNINGSCHROR P, BUCK E, MUNGWA S, FISCHER L, BROCKMANN SJ, ULMER J, BLIEDERHAUSER C, PHILIBERT CE, SATOH T, AKIRA S, BOILLEE S, MAYER B, SENDTNER M, LUDOLPH AC, DANZER KM, LOBSIGER CS, FREISCHMIDT A & WEISHAUPT JH 2019. Heterozygous Tbk1 loss has opposing effects in early and late stages of ALS in mice. J Exp Med, 216, 267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRETTSCHNEIDER J, TOLEDO JB, VAN DEERLIN VM, ELMAN L, MCCLUSKEY L, LEE VM & TROJANOWSKI JQ 2012. Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS One, 7, e39216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BROUWERS N, NUYTEMANS K, VAN DER ZEE J, GIJSELINCK I, ENGELBORGHS S, THEUNS J, KUMAR-SINGH S, PICKUT BA, PALS P, DERMAUT B, BOGAERTS V, DE POOTER T, SERNEELS S, VAN DEN BROECK M, CUIJT I, MATTHEIJSSENS M, PEETERS K, SCIOT R, MARTIN JJ, CRAS P, SANTENS P, VANDENBERGHE R, DE DEYN PP, CRUTS M, VAN BROECKHOVEN C & SLEEGERS K 2007. Alzheimer and Parkinson diagnoses in progranulin null mutation carriers in an extended founder family. Arch Neurol, 64, 1436–46. [DOI] [PubMed] [Google Scholar]
- BROUWERS N, SLEEGERS K, ENGELBORGHS S, MAURER-STROH S, GIJSELINCK I, VAN DER ZEE J, PICKUT BA, VAN DEN BROECK M, MATTHEIJSSENS M, PEETERS K, SCHYMKOWITZ J, ROUSSEAU F, MARTIN JJ, CRUTS M, DE DEYN PP & VAN BROECKHOVEN C 2008. Genetic variability in progranulin contributes to risk for clinically diagnosed Alzheimer disease. Neurology, 71, 656–64. [DOI] [PubMed] [Google Scholar]
- BURBERRY A, SUZUKI N, WANG JY, MOCCIA R, MORDES DA, STEWART MH, SUZUKI-UEMATSU S, GHOSH S, SINGH A, MERKLE FT, KOSZKA K, LI QZ, ZON L, ROSSI DJ, TROWBRIDGE JJ, NOTARANGELO LD & EGGAN K 2016. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med, 8, 347ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BURBERRY A, WELLS MF, LIMONE F, COUTO A, SMITH KS, KEANEY J, GILLET G, VAN GASTEL N, WANG JY, PIETILAINEN O, QIAN M, EGGAN P, CANTRELL C, MOK J, KADIU I, SCADDEN DT & EGGAN K 2020. C9orf72 suppresses systemic and neural inflammation induced by gut bacteria. Nature, 582, 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CACACE R, VAN CAUWENBERGHE C, BETTENS K, GIJSELINCK I, VAN DER ZEE J, ENGELBORGHS S, VANDENBULCKE M, VAN DONGEN J, BAUMER V, DILLEN L, MATTHEIJSSENS M, PEETERS K, CRUTS M, VANDENBERGHE R, DE DEYN PP, VAN BROECKHOVEN C & SLEEGERS K 2013. C9orf72 G4C2 repeat expansions in Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging, 34, 1712 e1–7. [DOI] [PubMed] [Google Scholar]
- CRUTS M, GIJSELINCK I, VAN DER ZEE J, ENGELBORGHS S, WILS H, PIRICI D, RADEMAKERS R, VANDENBERGHE R, DERMAUT B, MARTIN JJ, VAN DUIJN C, PEETERS K, SCIOT R, SANTENS P, DE POOTER T, MATTHEIJSSENS M, VAN DEN BROECK M, CUIJT I, VENNEKENS K, DE DEYN PP, KUMAR-SINGH S & VAN BROECKHOVEN C 2006. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature, 442, 920–4. [DOI] [PubMed] [Google Scholar]
- DAVIES VJ, HOLLINS AJ, PIECHOTA MJ, YIP W, DAVIES JR, WHITE KE, NICOLS PP, BOULTON ME & VOTRUBA M 2007. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum Mol Genet, 16, 1307–18. [DOI] [PubMed] [Google Scholar]
- DAVTYAN H, MKRTICHYAN M, MOVSESYAN N, PETRUSHINA I, MAMIKONYAN G, CRIBBS DH, AGADJANYAN MG & GHOCHIKYAN A 2010. DNA prime-protein boost increased the titer, avidity and persistence of anti-Abeta antibodies in wild-type mice. Gene Ther, 17, 261–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVTYAN H, PETRUSHINA I & GHOCHIKYAN A 2014. Immunotherapy for Alzheimer’s disease: DNA- and protein-based epitope vaccines. Methods Mol Biol, 1143, 259–81. [DOI] [PubMed] [Google Scholar]
- DEJESUS-HERNANDEZ M, FINCH NA, WANG X, GENDRON TF, BIENIEK KF, HECKMAN MG, VASILEVICH A, MURRAY ME, ROUSSEAU L, WEESNER R, LUCIDO A, PARSONS M, CHEW J, JOSEPHS KA, PARISI JE, KNOPMAN DS, PETERSEN RC, BOEVE BF, GRAFF-RADFORD NR, DE BOER J, ASMANN YW, PETRUCELLI L, BOYLAN KB, DICKSON DW, VAN BLITTERSWIJK M & RADEMAKERS R 2017. In-depth clinico-pathological examination of RNA foci in a large cohort of C9ORF72 expansion carriers. Acta Neuropathol, 134, 255–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEJESUS-HERNANDEZ M, MACKENZIE IR, BOEVE BF, BOXER AL, BAKER M, RUTHERFORD NJ, NICHOLSON AM, FINCH NA, FLYNN H, ADAMSON J, KOURI N, WOJTAS A, SENGDY P, HSIUNG GY, KARYDAS A, SEELEY WW, JOSEPHS KA, COPPOLA G, GESCHWIND DH, WSZOLEK ZK, FELDMAN H, KNOPMAN DS, PETERSEN RC, MILLER BL, DICKSON DW, BOYLAN KB, GRAFF-RADFORD NR & RADEMAKERS R 2011. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron, 72, 245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EIMER WA & VASSAR R 2013. Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Abeta42 accumulation and Caspase-3 activation. Mol Neurodegener, 8, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FARG MA, SUNDARAMOORTHY V, SULTANA JM, YANG S, ATKINSON RAK, LEVINA V, HALLORAN MA, GLEESON PA, BLAIR IP, SOO KY, KING AE & ATKIN JD 2017. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet, 26, 4093–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FILIANO AJ, MARTENS LH, YOUNG AH, WARMUS BA, ZHOU P, DIAZ-RAMIREZ G, JIAO J, ZHANG Z, HUANG EJ, GAO FB, FARESE RV JR. & ROBERSON ED 2013. Dissociation of frontotemporal dementia-related deficits and neuroinflammation in progranulin haploinsufficient mice. J Neurosci, 33, 5352–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FILIKCI Z, GUSTAFSSON MAK, HENRIKSEN OM, MARNER L & HOGH P 2020. C9ORF72 hexanucleotide repeat expansion with Alzheimer’s disease-like clinical phenotype: A case report with results from neuropsychology, CSF, FDG-PET, and PiB-PET. Clin Case Rep, 8, 3416–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FINCH N, BAKER M, CROOK R, SWANSON K, KUNTZ K, SURTEES R, BISCEGLIO G, ROVELET-LECRUX A, BOEVE B, PETERSEN RC, DICKSON DW, YOUNKIN SG, DERAMECOURT V, CROOK J, GRAFF-RADFORD NR & RADEMAKERS R 2009. Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain, 132, 583–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FOMIN V, RICHARD P, HOQUE M, LI C, GU Z, FISSORE-O’LEARY M, TIAN B, PRIVES C & MANLEY JL 2018. The C9ORF72 Gene, Implicated in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia, Encodes a Protein That Functions in Control of Endothelin and Glutamate Signaling. Mol Cell Biol, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FONSECA MI, CHU SH, HERNANDEZ MX, FANG MJ, MODARRESI L, SELVAN P, MACGREGOR GR & TENNER AJ 2017. Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J Neuroinflammation, 14, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FREISCHMIDT A, WIELAND T, RICHTER B, RUF W, SCHAEFFER V, MULLER K, MARROQUIN N, NORDIN F, HUBERS A, WEYDT P, PINTO S, PRESS R, MILLECAMPS S, MOLKO N, BERNARD E, DESNUELLE C, SORIANI MH, DORST J, GRAF E, NORDSTROM U, FEILER MS, PUTZ S, BOECKERS TM, MEYER T, WINKLER AS, WINKELMAN J, DE CARVALHO M, THAL DR, OTTO M, BRANNSTROM T, VOLK AE, KURSULA P, DANZER KM, LICHTNER P, DIKIC I, MEITINGER T, LUDOLPH AC, STROM TM, ANDERSEN PM & WEISHAUPT JH 2015. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci, 18, 631–6. [DOI] [PubMed] [Google Scholar]
- FRIEDMAN BA, SRINIVASAN K, AYALON G, MEILANDT WJ, LIN H, HUNTLEY MA, CAO Y, LEE SH, HADDICK PCG, NGU H, MODRUSAN Z, LARSON JL, KAMINKER JS, VAN DER BRUG MP & HANSEN DV 2018. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep, 22, 832–847. [DOI] [PubMed] [Google Scholar]
- GENDRON TF, BIENIEK KF, ZHANG YJ, JANSEN-WEST K, ASH PE, CAULFIELD T, DAUGHRITY L, DUNMORE JH, CASTANEDES-CASEY M, CHEW J, COSIO DM, VAN BLITTERSWIJK M, LEE WC, RADEMAKERS R, BOYLAN KB, DICKSON DW & PETRUCELLI L 2013. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol, 126, 829–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GHIDONI R, BENUSSI L, GLIONNA M, FRANZONI M & BINETTI G 2008. Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology, 71, 1235–9. [DOI] [PubMed] [Google Scholar]
- GITLER AD & TSUIJI H 2016. There has been an awakening: Emerging mechanisms of C9orf72 mutations in FTD/ALS. Brain Res, 1647, 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAMMOND TR, DUFORT C, DISSING-OLESEN L, GIERA S, YOUNG A, WYSOKER A, WALKER AJ, GERGITS F, SEGEL M, NEMESH J, MARSH SE, SAUNDERS A, MACOSKO E, GINHOUX F, CHEN J, FRANKLIN RJM, PIAO X, MCCARROLL SA & STEVENS B 2019. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity, 50, 253–271 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARMS M, BENITEZ BA, CAIRNS N, COOPER B, COOPER P, MAYO K, CARRELL D, FABER K, WILLIAMSON J, BIRD T, DIAZ-ARRASTIA R, FOROUD TM, BOEVE BF, GRAFF-RADFORD NR, MAYEUX R, CHAKRAVERTY S, GOATE AM, CRUCHAGA C & CONSORTIUM N-LNFS 2013. C9orf72 hexanucleotide repeat expansions in clinical Alzheimer disease. JAMA Neurol, 70, 736–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HENEKA MT, CARSON MJ, EL KHOURY J, LANDRETH GE, BROSSERON F, FEINSTEIN DL, JACOBS AH, WYSS-CORAY T, VITORICA J, RANSOHOFF RM, HERRUP K, FRAUTSCHY SA, FINSEN B, BROWN GC, VERKHRATSKY A, YAMANAKA K, KOISTINAHO J, LATZ E, HALLE A, PETZOLD GC, TOWN T, MORGAN D, SHINOHARA ML, PERRY VH, HOLMES C, BAZAN NG, BROOKS DJ, HUNOT S, JOSEPH B, DEIGENDESCH N, GARASCHUK O, BODDEKE E, DINARELLO CA, BREITNER JC, COLE GM, GOLENBOCK DT & KUMMER MP 2015. Neuroinflammation in Alzheimer’s disease. Lancet Neurol, 14, 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HENSMAN MOSS DJ, POULTER M, BECK J, HEHIR J, POLKE JM, CAMPBELL T, ADAMSON G, MUDANOHWO E, MCCOLGAN P, HAWORTH A, WILD EJ, SWEENEY MG, HOULDEN H, MEAD S & TABRIZI SJ 2014. C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies. Neurology, 82, 292–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HO WY, TAI YK, CHANG JC, LIANG J, TYAN SH, CHEN S, GUAN JL, ZHOU H, SHEN HM, KOO E & LING SC 2019. The ALS-FTD-linked gene product, C9orf72, regulates neuronal morphogenesis via autophagy. Autophagy, 15, 827–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HONG S, BEJA-GLASSER VF, NFONOYIM BM, FROUIN A, LI S, RAMAKRISHNAN S, MERRY KM, SHI Q, ROSENTHAL A, BARRES BA, LEMERE CA, SELKOE DJ & STEVENS B 2016. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science, 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JACKSON JL, FINCH NA, BAKER MC, KACHERGUS JM, DEJESUS-HERNANDEZ M, PEREIRA K, CHRISTOPHER E, PRUDENCIO M, HECKMAN MG, THOMPSON EA, DICKSON DW, SHAH J, OSKARSSON B, PETRUCELLI L, RADEMAKERS R & VAN BLITTERSWIJK M 2020. Elevated methylation levels, reduced expression levels, and frequent contractions in a clinical cohort of C9orf72 expansion carriers. Mol Neurodegener, 15, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KEREN-SHAUL H, SPINRAD A, WEINER A, MATCOVITCH-NATAN O, DVIR-SZTERNFELD R, ULLAND TK, DAVID E, BARUCH K, LARA-ASTAISO D, TOTH B, ITZKOVITZ S, COLONNA M, SCHWARTZ M & AMIT I 2017. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell, 169, 1276–1290 e17. [DOI] [PubMed] [Google Scholar]
- KIM D, PERTEA G, TRAPNELL C, PIMENTEL H, KELLEY R & SALZBERG SL 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol, 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOPPERS M, BLOKHUIS AM, WESTENENG HJ, TERPSTRA ML, ZUNDEL CA, VIEIRA DE SA R, SCHELLEVIS RD, WAITE AJ, BLAKE DJ, VELDINK JH, VAN DEN BERG LH & PASTERKAMP RJ 2015. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol, 78, 426–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRASEMANN S, MADORE C, CIALIC R, BAUFELD C, CALCAGNO N, EL FATIMY R, BECKERS L, O’LOUGHLIN E, XU Y, FANEK Z, GRECO DJ, SMITH ST, TWEET G, HUMULOCK Z, ZRZAVY T, CONDE-SANROMAN P, GACIAS M, WENG Z, CHEN H, TJON E, MAZAHERI F, HARTMANN K, MADI A, ULRICH JD, GLATZEL M, WORTHMANN A, HEEREN J, BUDNIK B, LEMERE C, IKEZU T, HEPPNER FL, LITVAK V, HOLTZMAN DM, LASSMANN H, WEINER HL, OCHANDO J, HAASS C & BUTOVSKY O 2017. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity, 47, 566–581 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANGFELDER P & HORVATH S 2008. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics, 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANGFELDER P, LUO R, OLDHAM MC & HORVATH S 2011. Is my network module preserved and reproducible? PLoS Comput Biol, 7, e1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANGSETH AJ, KIM J, UGOLINO JE, SHAH Y, HWANG HY, WANG J, BERGLES DE & BROWN SP 2017. Cell-type specific differences in promoter activity of the ALS-linked C9orf72 mouse ortholog. Sci Rep, 7, 5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE CYD, DAGGETT A, GU X, JIANG LL, LANGFELDER P, LI X, WANG N, ZHAO Y, PARK CS, COOPER Y, FERANDO I, MODY I, COPPOLA G, XU H & YANG XW 2018. Elevated TREM2 Gene Dosage Reprograms Microglia Responsivity and Ameliorates Pathological Phenotypes in Alzheimer’s Disease Models. Neuron, 97, 1032–1048 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LINDQUIST SG, DUNO M, BATBAYLI M, PUSCHMANN A, BRAENDGAARD H, MARDOSIENE S, SVENSTRUP K, PINBORG LH, VESTERGAARD K, HJERMIND LE, STOKHOLM J, ANDERSEN BB, JOHANNSEN P & NIELSEN JE 2013. Corticobasal and ataxia syndromes widen the spectrum of C9ORF72 hexanucleotide expansion disease. Clin Genet, 83, 279–83. [DOI] [PubMed] [Google Scholar]
- LUI H, ZHANG J, MAKINSON SR, CAHILL MK, KELLEY KW, HUANG HY, SHANG Y, OLDHAM MC, MARTENS LH, GAO F, COPPOLA G, SLOAN SA, HSIEH CL, KIM CC, BIGIO EH, WEINTRAUB S, MESULAM MM, RADEMAKERS R, MACKENZIE IR, SEELEY WW, KARYDAS A, MILLER BL, BORRONI B, GHIDONI R, FARESE RV JR., PAZ JT, BARRES BA & HUANG EJ 2016. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell, 165, 921–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACKENZIE IR, ARZBERGER T, KREMMER E, TROOST D, LORENZL S, MORI K, WENG SM, HAASS C, KRETZSCHMAR HA, EDBAUER D & NEUMANN M 2013. Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol, 126, 859–79. [DOI] [PubMed] [Google Scholar]
- MAJOUNIE E, ABRAMZON Y, RENTON AE, PERRY R, BASSETT SS, PLETNIKOVA O, TRONCOSO JC, HARDY J, SINGLETON AB & TRAYNOR BJ 2012. Repeat expansion in C9ORF72 in Alzheimer’s disease. N Engl J Med, 366, 283–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARCHBANK NJ, CRAIG JE, LEEK JP, TOOHEY M, CHURCHILL AJ, MARKHAM AF, MACKEY DA, TOOMES C & INGLEHEARN CF 2002. Deletion of the OPA1 gene in a dominant optic atrophy family: evidence that haploinsufficiency is the cause of disease. J Med Genet, 39, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARSH SE, ABUD EM, LAKATOS A, KARIMZADEH A, YEUNG ST, DAVTYAN H, FOTE GM, LAU L, WEINGER JG, LANE TE, INLAY MA, POON WW & BLURTON-JONES M 2016. The adaptive immune system restrains Alzheimer’s disease pathogenesis by modulating microglial function. Proc Natl Acad Sci U S A, 113, E1316–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATHYS H, ADAIKKAN C, GAO F, YOUNG JZ, MANET E, HEMBERG M, DE JAGER PL, RANSOHOFF RM, REGEV A & TSAI LH 2017. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep, 21, 366–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCCAULEY ME, O’ROURKE JG, YANEZ A, MARKMAN JL, HO R, WANG X, CHEN S, LALL D, JIN M, MUHAMMAD A, BELL S, LANDEROS J, VALENCIA V, HARMS M, ARDITI M, JEFFERIES C & BALOH RH 2020. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature, 585, 96–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MINAMI SS, MIN SW, KRABBE G, WANG C, ZHOU Y, ASGAROV R, LI Y, MARTENS LH, ELIA LP, WARD ME, MUCKE L, FARESE RV JR. & GAN L 2014. Progranulin protects against amyloid beta deposition and toxicity in Alzheimer’s disease mouse models. Nat Med, 20, 1157–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIZIELINSKA S, LASHLEY T, NORONA FE, CLAYTON EL, RIDLER CE, FRATTA P & ISAACS AM 2013. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol, 126, 845–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MRDJEN D, PAVLOVIC A, HARTMANN FJ, SCHREINER B, UTZ SG, LEUNG BP, LELIOS I, HEPPNER FL, KIPNIS J, MERKLER D, GRETER M & BECHER B 2018. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity, 48, 599. [DOI] [PubMed] [Google Scholar]
- NASSIF M, WOEHLBIER U & MANQUE PA 2017. The Enigmatic Role of C9ORF72 in Autophagy. Front Neurosci, 11, 442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NUYTEMANS K, BADEMCI G, KOHLI MM, BEECHAM GW, WANG L, YOUNG JI, NAHAB F, MARTIN ER, GILBERT JR, BENATAR M, HAINES JL, SCOTT WK, ZUCHNER S, PERICAK-VANCE MA & VANCE JM 2013. C9ORF72 intermediate repeat copies are a significant risk factor for Parkinson disease. Ann Hum Genet, 77, 351–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’ROURKE JG, BOGDANIK L, YANEZ A, LALL D, WOLF AJ, MUHAMMAD AK, HO R, CARMONA S, VIT JP, ZARROW J, KIM KJ, BELL S, HARMS MB, MILLER TM, DANGLER CA, UNDERHILL DM, GOODRIDGE HS, LUTZ CM & BALOH RH 2016. C9orf72 is required for proper macrophage and microglial function in mice. Science, 351, 1324–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAOLICELLI RC, JAWAID A, HENSTRIDGE CM, VALERI A, MERLINI M, ROBINSON JL, LEE EB, ROSE J, APPEL S, LEE VM, TROJANOWSKI JQ, SPIRES-JONES T, SCHULZ PE & RAJENDRAN L 2017. TDP-43 Depletion in Microglia Promotes Amyloid Clearance but Also Induces Synapse Loss. Neuron, 95, 297–308 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PETKAU TL, NEAL SJ, MILNERWOOD A, MEW A, HILL AM, ORBAN P, GREGG J, LU G, FELDMAN HH, MACKENZIE IR, RAYMOND LA & LEAVITT BR 2012. Synaptic dysfunction in progranulin-deficient mice. Neurobiol Dis, 45, 711–22. [DOI] [PubMed] [Google Scholar]
- RENTON AE, MAJOUNIE E, WAITE A, SIMON-SANCHEZ J, ROLLINSON S, GIBBS JR, SCHYMICK JC, LAAKSOVIRTA H, VAN SWIETEN JC, MYLLYKANGAS L, KALIMO H, PAETAU A, ABRAMZON Y, REMES AM, KAGANOVICH A, SCHOLZ SW, DUCKWORTH J, DING J, HARMER DW, HERNANDEZ DG, JOHNSON JO, MOK K, RYTEN M, TRABZUNI D, GUERREIRO RJ, ORRELL RW, NEAL J, MURRAY A, PEARSON J, JANSEN IE, SONDERVAN D, SEELAAR H, BLAKE D, YOUNG K, HALLIWELL N, CALLISTER JB, TOULSON G, RICHARDSON A, GERHARD A, SNOWDEN J, MANN D, NEARY D, NALLS MA, PEURALINNA T, JANSSON L, ISOVIITA VM, KAIVORINNE AL, HOLTTA-VUORI M, IKONEN E, SULKAVA R, BENATAR M, WUU J, CHIO A, RESTAGNO G, BORGHERO G, SABATELLI M, CONSORTIUM I, HECKERMAN D, ROGAEVA E, ZINMAN L, ROTHSTEIN JD, SENDTNER M, DREPPER C, EICHLER EE, ALKAN C, ABDULLAEV Z, PACK SD, DUTRA A, PAK E, HARDY J, SINGLETON A, WILLIAMS NM, HEUTINK P, PICKERING-BROWN S, MORRIS HR, TIENARI PJ & TRAYNOR BJ 2011. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron, 72, 257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REXACH J, SWARUP V, CHANG T & GESCHWIND D 2019. Dementia risk genes engage gene networks poised to tune the immune response towards chronic inflammatory states. bioRxiv, 597542. [Google Scholar]
- REXACH JE, POLIOUDAKIS D, YIN A, SWARUP V, CHANG TS, NGUYEN T, SARKAR A, CHEN L, HUANG J, LIN LC, SEELEY W, TROJANOWSKI JQ, MALHOTRA D & GESCHWIND DH 2020. Tau Pathology Drives Dementia Risk-Associated Gene Networks toward Chronic Inflammatory States and Immunosuppression. Cell Rep, 33, 108398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIZZU P, BLAUWENDRAAT C, HEETVELD S, LYNES EM, CASTILLO-LIZARDO M, DHINGRA A, PYZ E, HOBERT M, SYNOFZIK M, SIMON-SANCHEZ J, FRANCESCATTO M & HEUTINK P 2016. C9orf72 is differentially expressed in the central nervous system and myeloid cells and consistently reduced in C9orf72, MAPT and GRN mutation carriers. Acta Neuropathol Commun, 4, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROY ER, WANG B, WAN YW, CHIU G, COLE A, YIN Z, PROPSON NE, XU Y, JANKOWSKY JL, LIU Z, LEE VM, TROJANOWSKI JQ, GINSBERG SD, BUTOVSKY O, ZHENG H & CAO W 2020. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest, 130, 1912–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SABERI S, STAUFFER JE, JIANG J, GARCIA SD, TAYLOR AE, SCHULTE D, OHKUBO T, SCHLOFFMAN CL, MALDONADO M, BAUGHN M, RODRIGUEZ MJ, PIZZO D, CLEVELAND D & RAVITS J 2018. Sense-encoded poly-GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with TDP-43 in dendrites of repeat-expanded C9orf72 amyotrophic lateral sclerosis. Acta Neuropathol, 135, 459–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SALA FRIGERIO C, WOLFS L, FATTORELLI N, THRUPP N, VOYTYUK I, SCHMIDT I, MANCUSO R, CHEN WT, WOODBURY ME, SRIVASTAVA G, MOLLER T, HUDRY E, DAS S, SAIDO T, KARRAN E, HYMAN B, PERRY VH, FIERS M & DE STROOPER B 2019. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Abeta Plaques. Cell Rep, 27, 1293–1306 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHAFER DP, LEHRMAN EK, KAUTZMAN AG, KOYAMA R, MARDINLY AR, YAMASAKI R, RANSOHOFF RM, GREENBERG ME, BARRES BA & STEVENS B 2012. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron, 74, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SNOWDEN JS, ROLLINSON S, LAFON C, HARRIS J, THOMPSON J, RICHARDSON AM, JONES M, GERHARD A, NEARY D, MANN DM & PICKERING-BROWN S 2012. Psychosis, C9ORF72 and dementia with Lewy bodies. J Neurol Neurosurg Psychiatry, 83, 1031–2. [DOI] [PubMed] [Google Scholar]
- STEPHAN AH, MADISON DV, MATEOS JM, FRASER DA, LOVELETT EA, COUTELLIER L, KIM L, TSAI HH, HUANG EJ, ROWITCH DH, BERNS DS, TENNER AJ, SHAMLOO M & BARRES BA 2013. A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci, 33, 13460–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEVENS B, ALLEN NJ, VAZQUEZ LE, HOWELL GR, CHRISTOPHERSON KS, NOURI N, MICHEVA KD, MEHALOW AK, HUBERMAN AD, STAFFORD B, SHER A, LITKE AM, LAMBRIS JD, SMITH SJ, JOHN SW & BARRES BA 2007. The classical complement cascade mediates CNS synapse elimination. Cell, 131, 1164–78. [DOI] [PubMed] [Google Scholar]
- SUDRIA-LOPEZ E, KOPPERS M, DE WIT M, VAN DER MEER C, WESTENENG HJ, ZUNDEL CA, YOUSSEF SA, HARKEMA L, DE BRUIN A, VELDINK JH, VAN DEN BERG LH & PASTERKAMP RJ 2016. Full ablation of C9orf72 in mice causes immune system-related pathology and neoplastic events but no motor neuron defects. Acta Neuropathol, 132, 145–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUZUKI N, MAROOF AM, MERKLE FT, KOSZKA K, INTOH A, ARMSTRONG I, MOCCIA R, DAVIS-DUSENBERY BN & EGGAN K 2013. The mouse C9ORF72 ortholog is enriched in neurons known to degenerate in ALS and FTD. Nat Neurosci, 16, 1725–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAKAHASHI H, KLEIN ZA, BHAGAT SM, KAUFMAN AC, KOSTYLEV MA, IKEZU T, STRITTMATTER SM & ALZHEIMER’S DISEASE NEUROIMAGING I 2017. Opposing effects of progranulin deficiency on amyloid and tau pathologies via microglial TYROBP network. Acta Neuropathol, 133, 785–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAYLOR JP, BROWN RH JR. & CLEVELAND DW 2016. Decoding ALS: from genes to mechanism. Nature, 539, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ULLAND TK & COLONNA M 2018. TREM2 - a key player in microglial biology and Alzheimer disease. Nat Rev Neurol, 14, 667–675. [DOI] [PubMed] [Google Scholar]
- ULRICH JD, FINN MB, WANG Y, SHEN A, MAHAN TE, JIANG H, STEWART FR, PICCIO L, COLONNA M & HOLTZMAN DM 2014. Altered microglial response to Abeta plaques in APPPS1–21 mice heterozygous for TREM2. Mol Neurodegener, 9, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN MOSSEVELDE S, VAN DER ZEE J, CRUTS M & VAN BROECKHOVEN C 2017. Relationship between C9orf72 repeat size and clinical phenotype. Curr Opin Genet Dev, 44, 117–124. [DOI] [PubMed] [Google Scholar]
- VIODE A, FOURNIER C, CAMUZAT A, FENAILLE F, NEURO CEBBB, LATOUCHE M, ELAHI F, LE BER I, JUNOT C, LAMARI F, ANQUETIL V & BECHER F 2018. New Antibody-Free Mass Spectrometry-Based Quantification Reveals That C9ORF72 Long Protein Isoform Is Reduced in the Frontal Cortex of Hexanucleotide-Repeat Expansion Carriers. Front Neurosci, 12, 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG Y, CELLA M, MALLINSON K, ULRICH JD, YOUNG KL, ROBINETTE ML, GILFILLAN S, KRISHNAN GM, SUDHAKAR S, ZINSELMEYER BH, HOLTZMAN DM, CIRRITO JR & COLONNA M 2015. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell, 160, 1061–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHITE MA, KIM E, DUFFY A, ADALBERT R, PHILLIPS BU, PETERS OM, STEPHENSON J, YANG S, MASSENZIO F, LIN Z, ANDREWS S, SEGONDS-PICHON A, METTERVILLE J, SAKSIDA LM, MEAD R, RIBCHESTER RR, BARHOMI Y, SERRE T, COLEMAN MP, FALLON JR, BUSSEY TJ, BROWN RH JR. & SREEDHARAN J 2018. TDP-43 gains function due to perturbed autoregulation in a Tardbp knock-in mouse model of ALS-FTD. Nat Neurosci, 21, 552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHITELAW BS 2018. Microglia-mediated synaptic elimination in neuronal development and disease. J Neurophysiol, 119, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILLIAMS PA, MORGAN JE & VOTRUBA M 2011. Mouse models of dominant optic atrophy: what do they tell us about the pathophysiology of visual loss? Vision Res, 51, 229–34. [DOI] [PubMed] [Google Scholar]
- XIAO S, MCKEEVER PM, LAU A & ROBERTSON J 2019. Synaptic localization of C9orf72 regulates post-synaptic glutamate receptor 1 levels. Acta Neuropathol Commun, 7, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YIN F, DUMONT M, BANERJEE R, MA Y, LI H, LIN MT, BEAL MF, NATHAN C, THOMAS B & DING A 2010. Behavioral deficits and progressive neuropathology in progranulin-deficient mice: a mouse model of frontotemporal dementia. FASEB J, 24, 4639–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG J, VELMESHEV D, HASHIMOTO K, HUANG YH, HOFMANN JW, SHI X, CHEN J, LEIDAL AM, DISHART JG, CAHILL MK, KELLEY KW, LIDDELOW SA, SEELEY WW, MILLER BL, WALTHER TC, FARESE RV JR., TAYLOR JP, ULLIAN EM, HUANG B, DEBNATH J, WITTMANN T, KRIEGSTEIN AR & HUANG EJ 2020. Neurotoxic microglia promote TDP-43 proteinopathy in progranulin deficiency. Nature, 588, 459–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG Y, CHEN K, SLOAN SA, BENNETT ML, SCHOLZE AR, O’KEEFFE S, PHATNANI HP, GUARNIERI P, CANEDA C, RUDERISCH N, DENG S, LIDDELOW SA, ZHANG C, DANEMAN R, MANIATIS T, BARRES BA & WU JQ 2014. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci, 34, 11929–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHU Q, JIANG J, GENDRON TF, MCALONIS-DOWNES M, JIANG L, TAYLOR A, DIAZ GARCIA S, GHOSH DASTIDAR S, RODRIGUEZ MJ, KING P, ZHANG Y, LA SPADA AR, XU H, PETRUCELLI L, RAVITS J, DA CRUZ S, LAGIER-TOURENNE C & CLEVELAND DW 2020. Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat Neurosci. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental table S1 (excel file): Related to Figure 1 and Figure S2: QC data of scRNAseq dataset showing total number of cell sequenced per genotype, average reads per sample, median genes per cell and other cell types identified in analyses.
Supplemental table S2 (excel file): Related to Figure 1: QC data of all microglial clusters identified with scRNAseq and list of genes identified in each microglial cluster.
Data Availability Statement
All datasets generated during and/or analysed during the current study are uploaded and can be downloaded from from the Gene Expression Omnibus website with the following accession numbers: GSE164676 (Superseries), GSE164675 (pooled microglia RNAseq), GSE164674 (Single cell microglia RNAseq). R code for single cell analysis is included as supplementary file 3.