

SUMMARY
Myeloid lineage cells use TLRs to recognize and respond to diverse microbial ligands. Although unique transcription factors dictate the outcome of specific TLR signaling, whether lineage-specific differences exist to further modulate the quality of TLR-induced inflammation remains unclear. Comprehensive analysis of global gene transcription in human monocytes, monocyte-derived macrophages, and monocyte-derived dendritic cells stimulated with various TLR ligands identifies multiple lineage-specific, TLR-responsive gene programs. Monocytes are hyperresponsive to TLR7/8 stimulation that correlates with the higher expression of the receptors. While macrophages and monocytes express similar levels of TLR4, macrophages, but not monocytes, upregulate interferon-stimulated genes (ISGs) in response to TLR4 stimulation. We find that TLR4 signaling in macrophages uniquely engages transcription factor IRF1, which facilitates the opening of ISG loci for transcription. This study provides a critical mechanistic basis for lineage-specific TLR responses and uncovers IRF1 as a master regulator for the ISG transcriptional program in human macrophages.
Graphical abstract
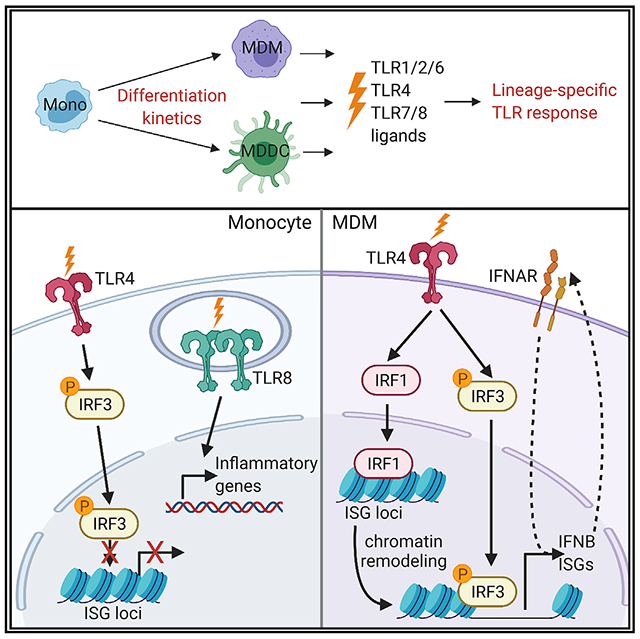
In brief
Song et al. examine the responses of human myeloid populations to bacterial and viral ligands and discover the lineage-specific induction of proinflammatory gene expression. TLR4 signaling in macrophages, but not monocytes, enables nuclear translocation of IRF1, which acts as a pioneer transcription factor to increase the chromatin accessibility of IFNB1 and ISG loci.
INTRODUCTION
The immune system is composed of diverse cell types with defined functions. While T and B lymphocytes have specialized roles in the adaptive immune system, the innate immune system largely relies on mononuclear phagocytes such as monocytes, macrophages, and dendritic cells (DCs) to combat microbial insults (Geissmann et al., 2010; Iwasaki and Medzhitov, 2015; Satpathy et al., 2012). Monocytes originate in the bone marrow from a common myeloid progenitor and are released into the peripheral blood, where they circulate for several days (Geissmann et al., 2010). Although certain populations of macrophages and DCs originate from restricted progenitors, circulating monocytes are also considered to be the natural precursors of macrophages and DCs (Varol et al., 2009). Monocytes can give rise to tissue-resident macrophages and DCs after recruitment to tissues such as the dermis, the intestines, and the lungs (Randolph et al., 1999; Varol et al., 2009). In addition, during infections, monocytes migrate to inflamed tissues and differentiate into macrophages or DCs (Ginhoux and Jung, 2014; Shi and Pamer, 2011). Monocytes, monocyte-derived macrophages (MDMs), and monocyte-derived DCs (MDDCs) mediate fundamental innate immune processes such as the rapid recognition of pathogens, microbial clearance, and inflammatory cytokine production (Geissmann et al., 2010). The differentiation of monocytes to MDMs or MDDCs endows additional lineage-specific functions. Macrophages, for example, increase phagocytic capacity, while DCs upregulate antigen processing machinery to prime adaptive immunity (Geissmann et al., 2010; Satpathy et al., 2012).
Monocytes, macrophages, and DCs express numerous pattern recognition receptors (PRRs), which allows them to recognize and respond to a diverse array of microbial challenges. The Toll-like receptors (TLRs) are one of the best-characterized families of PRRs. TLRs recognize pathogen-associated molecular patterns to activate innate immune responses (Medzhitov et al., 1997; Pasare and Medzhitov, 2004). Following ligand binding, TLRs recruit adaptor proteins such as MyD88 and TRIF, which initiate signal transduction pathways leading to the activation of nuclear factor κB (NF-κB) or interferon regulatory factors (IRFs) to induce the expression of cytokine, chemokine, and type I interferon (IFN) genes (Barbalat et al., 2011; Kawasaki and Kawai, 2014; Lee and Barton, 2014). The specificity of the response downstream of individual TLRs is dictated by the differential use of adaptor molecules and their ability to activate specific transcription factors (TFs) such as activator protein 1 (AP-1), NF-κB, and IRFs (Akira and Takeda, 2004; Barton and Kagan, 2009; Kawasaki and Kawai, 2014; Lim and Staudt, 2013). While selective usage of adapters and unique TFs are known to influence the outcome of TLR signaling (Kawasaki and Kawai, 2014; McGettrick and O’Neill, 2010; Takeuchi and Akira, 2010), several studies suggest that the distinctive chromatin structure of individual genes and their chromatin regulators act in concert with signaling pathways and TFs to generate cell type-specific and stimulus-specific responses (Comoglio et al., 2019; Glass and Natoli, 2016; Ivashkiv, 2013; Medzhitov and Horng, 2009; Nicodeme et al., 2010; Smale, 2012; Smale et al., 2014). Chromatin packaging of genomic DNA creates a barrier against the binding of TFs to nucleosome-associated DNA (Bell et al., 2011). Recent data also demonstrate that during the development of immune cells, lineage-specifying TFs alter chromatin structure to facilitate the binding of stimulus-specific TFs and contribute to lineage-specific responses to a stimulus (Glass and Natoli, 2016; Heinz et al., 2010; Smale et al., 2014). Chromatin accessibility is therefore an important determinant of the quality of inflammatory responses between disparate cell types such as macrophages and fibroblasts (Smale, 2010). Despite extensive study on murine macrophages and DCs, there is limited understanding of whether closely related human innate immune cells, such as macrophages and monocytes, mount disparate transcriptional responses to the same ligand. Furthermore, the extent to which chromatin accessibility dictates lineage-specific inflammatory responses remains unclear. Understanding the molecular basis of cell type-specific inflammatory responses can provide critical insights for targeting specific molecules to modulate inflammation.
In this study, we investigate global changes in gene expression and chromatin accessibility in human monocytes, MDMs, and MDDCs before and after stimulation using several TLR ligands. While we discovered clusters of genes that were induced by all TLRs, TLR4 and TLR7/8 stimulation of monocytes and MDMs revealed insights into the regulation of lineage-specific and TLR-specific transcriptional responses. In particular, we found a surprising dichotomy in the expression of interferon-stimulated genes (ISGs) between monocytes and MDMs when stimulated by TLR4 or TLR7/8 ligands. While both cell types were able to express ISGs, the induction of ISGs was triggered by different TLRs in each cell type. More specifically, TLR7 and TLR8 activation, but not TLR4 activation, led to the induction of ISGs in monocytes. In contrast, TLR4 activation induced ISGs in MDMs, but not monocytes, despite similar signal-induced activation of TF IRF3. We found that TLR4 activation in MDMs enables chromatin opening at ISG loci. Finally, we identified an essential role for IRF1 in regulating chromatin accessibility and expression of ISGs in human macrophages. These results reveal lineage-specific and stimuli-specific heterogeneity within human innate immune responses. These differences underscore the importance of identifying unique regulators of inflammatory gene transcription in different cells of the innate immune system so that they can be specifically targeted to dampen inflammation.
RESULTS
Myeloid lineage-specific responses to TLR signaling
To understand the basis of myeloid lineage-specific responses, we examined transcriptional footprints that define the differentiation trajectory from monocytes into either MDMs or MDDCs. We isolated human monocytes from peripheral blood mononuclear cells (PBMCs) and differentiated them into MDMs or MDDCs using macrophage colony-stimulating factor (M-CSF) or granulocyte-macrophage-CSF (GM-CSF)/interleukin-4 (IL-4), respectively, for 7 days. We performed whole transcriptome sequencing during differentiation at 9 different timepoints (0 h, 4 h, 1 day, 2 days, 3 days, 4 days, 5 days, 6 days, and 7 days) (Figure S1A). To assess the relationships between monocytes, MDMs, MDDCs, and these intermediate populations, we performed principal-component analysis (PCA) of the whole transcriptome. Interestingly, PCA demonstrated that the most substantial gene expression changes occurred in the first 3 days of differentiation, followed by a stable global transcriptome profile from 4 to 7 days (Figure S1B). This result suggests that differentiation factors dictate very early regulation of lineage-specific genes that are maintained to define cell fate.
We posited that these imprinted transcriptomic changes may dictate distinct responses of these 3 related myeloid cell types to PRR stimulation. To this end, we assessed changes in gene expression following the stimulation of monocytes, MDMs, or MDDCs with individual TLR agonists (see Method details). We isolated monocytes from 5 healthy donors and differentiated them into MDMs or MDDCs for 7 days, as described above. Then, we stimulated these 3 populations with TLR2 (Pam3CSK4), TLR4 (lipopolysaccharide [LPS]), TLR7 (R837), and TLR7/8 (R848) agonists for 18 h and analyzed the global gene expression (Figure 1A). We examined the expression of the TLRs in unstimulated monocytes, MDMs, and MDDCs. TLR1, TLR2, TLR4, TLR5, TLR6, and TLR8 were expressed in all 3 cell types from all 5 human donors, with some inter-individual variation in expression (Figure S2A). Interestingly, these myeloid populations also exhibited differential basal expression of other TLRs. For example, TLR3 is expressed (defined by average reads per kilobase of transcript per million mapped reads [RPKM] > 1.5) in differentiated MDMs and MDDCs, but not in monocytes. TLR7 was expressed in monocytes and MDMs, but it was not expressed in MDDCs, while TLR10 was expressed only in MDDCs (Figure S2A).
Figure 1. Monocyte differentiation programming regulates immune responses to TLR agonists.

(A) Schematic of this study.
(B) Clustered heatmap showing expression patterns of differentially expressed genes in primary monocytes, MDMs, and MDDCs from 5 different human donors by stimulation of indicated TLR ligands. Each row represents a gene, and each column represents a donor sample, either unstimulated or stimulated with Pam3CSK4 (1 μg/mL), LPS (10 ng/mL), R837 (10 μg/mL), or R848 (10 μg/mL) for 18 h; n = 5 independent donors.
(C) Summary table showing Ingenuity Pathway Analysis networks or canonical pathways of genes and example of signature genes in each cluster.
(D) qRT-PCR confirmation of expression of signature genes shown in (C). Human monocytes, MDMs, and MDDCs were obtained from 2 additional donors and stimulated with the indicated TLR agonists for 18 h. All plotted values are averages of 2 donors. Error bars indicate SEMs.
We next performed PCA on the global transcriptome of monocytes, MDMs, and MDDCs following stimulation (Figure S2B). The PCA plot demonstrates that the 3 lineages represent distinct profiles at steady state, as shown previously (Figure S1B). Cluster segregation on the PC1 axis reveals that MDMs and monocytes exhibit a more similar direction of transcriptomic change following TLR activation, as compared to MDDCs. In addition, the segregation of PC2 reveals distinct gene expression following LPS stimulation of MDMs compared to other TLR ligands. On the other hand, R837/R848 induced a distinct transcriptional profile in monocytes. Consistent with previous studies, we identified genes regulated in a TLR-specific manner in all 3 cell types (Tables S1 and S2) (Butcher et al., 2018; Cheng et al., 2019; de Marcken et al., 2019; Xue et al., 2014). To further understand the inter-lineage disparities in gene expression following activation of the same TLR, we identified 6 clusters of genes that were uniquely or commonly induced in monocytes, MDMs, and MDDCs by all TLR stimulations (Figure 1B; Table S3). We identified 3 clusters of genes (clusters I, II, III) that were not modulated by TLR stimulation in any lineage (Figure 1B). These clusters consist of lineage-specific regulators that appear to be stably expressed by monocytes, MDMs, or MDDCs. Cluster I contained genes expressed only in monocytes and included genes related to chemokine receptors of homeostatic functions such as CCR2 (Figure 1C). While the expression of these genes does not change in response to TLR stimulation, their expression is lost over the course of differentiation into MDMs or MDDCs. Cluster II comprised genes involved in macrophage phenotype and function, such as CD163 (Figure 1C), and cluster III comprises genes associated with the differentiation of monocytes into DCs and genes associated with antigen presentation, including CD1A, CD1C, HLA-DRA, HLA-DRB1, and HLA-DQA1 (Figure 1C). These data establish the purity of these populations and further indicate that TLR signaling does not promote direct differentiation of monocytes into either macrophages or DCs. Combined with the data shown in Figure S1, we conclude that M-CSF or GM-CSF receptor signaling primarily modulates and imprints the differentiation program that remains unaltered by PRR signaling.
Clusters IV, V, and VI represent lineage-specific responses, irrespective of the type of TLR stimulation (Figures 1B and S2C; Table S4). Cluster IV consists of genes specifically upregulated in monocytes following TLR2, -4, -7, or -7/8 stimulation, such as CD300E and IRAK3 (Figure 1C). Cluster V includes MDM-specific, TLR-responsive genes (Figure 1C), including genes involved in macrophage migration such as MMP9 and genes that are required for endolysosomal trafficking such as VPS37A (Figures 1C and S2C). Cluster VI comprises genes strongly upregulated after TLR stimulation only in MDDCs but not in monocytes or MDMs. Genes related to DC differentiation and co-stimulatory functions such as BATF3, IRF4, and CD86 belong to this cluster (Figures 1C and S2C). We confirmed the expression pattern of signature genes in these clusters by conducting qRT-PCR in cells from 2 additional independent donors (Figure 1D), suggesting that these results are highly conserved across the human population. Overall, these data demonstrate that a common node of the inflammatory response is set in motion following TLR signaling and that this response is independent of the identity of the proximal TLR but dictated by the cell lineage (clusters IV, V, and VI). These genes are different from the lineage-specifying genes (clusters I, II, and II), which remain unchanged. The cellular identity dictates the nature of the inflammatory response, thereby suggesting that the TLR response induced in specific cell types may be imprinted during differentiation.
TLR8 activation leads to the most potent induction of cytokine and chemokine genes in monocytes compared to MDMs and MDDCs
Next, we identified a unique cluster (cluster VII) of genes in monocytes that are highly induced in response to TLR8 stimulation (Figure 2A). This cluster of genes contained cytokines and chemokines such as IL1B, IL6, and CCL3 (Figure 2B). Although these genes are induced in monocytes to some extent following the activation of TLR2, TLR4, and TLR7, the response is of a much higher magnitude when stimulated with TLR7/8 agonist R848, suggesting that TLR8 signaling is a major contributor to the heightened induction of this gene cluster. This selective increased expression of cytokine and chemokine genes by TLR8 signaling was largely lost when monocytes differentiated into MDMs and MDDCs. Although these genes were still upregulated by TLR8 signaling in both MDMs and MDDCs, they were induced at levels comparable to TLR2, TLR4, and TLR7 stimulation (Figure 2A).
Figure 2. Pro-inflammatory cytokine and chemokine genes are highly induced in response to TLR7/8 agonist (R848) in monocytes.

(A) Heatmap showing the expression of genes that are significantly upregulated by R848 stimulation in monocytes.
(B) Summary table showing Ingenuity Pathway Analysis networks or canonical pathways of genes and signature genes in cluster VII.
(C) IL-1β (left) and IL-6 (right) production by monocytes, MDMs, and MDDCs from 5 donors measured by ELISA following TLR stimulation. Monocytes, MDMs, and MDDCs were unstimulated or stimulated with Pam3CSK4 (1 μg/mL), LPS (10 ng/mL), R837 (10 μg/mL), or R848 (10 μg/mL) for 18 h. Data represent the mean values derived from 2 independent experiments with conditions performed in duplicate. Error bars indicate SEMs. Statistics was determined by ordinary 2-way ANOVA. ****p < 0.0001.
TLR7 and TLR8 differ in function, although they are highly structurally related (Gorden et al., 2005). To define the role of TLR7 and TLR8 in the induction of cytokine and chemokine genes in monocytes, we used specific TLR7 (R837), TLR8 (TL8-506), and TLR7/8 (R848) agonists to stimulate monocytes (from 2 independent donors) and examined IL-6 production. TLR8 agonists and TLR7/8 agonists more effectively induced IL-6 production from monocytes than the TLR7 agonist (Figure S3A), suggesting that IL-6 production by monocytes was mainly induced through TLR8. Furthermore, the expression of genes in cluster VII was induced by TLR8 or TLR7/8 agonists, but not TLR7 agonist (Figure S3B). Through the time course RNA sequencing (RNA-seq) analysis, we identified another set of cytokine genes, such as IL1B, CCL3, CCL4, and TNF, which were dramatically induced at an early time point (1 h post-stimulation) by TLR7/8 and TLR8 agonists compared to TLR7 agonist (Figure S3B). R848 remained the only stimuli that induced marked tumor necrosis factor (TNF) secretion in monocytes (Figure S3C). A similar pattern was observed in the secretion of IL-1β and IL-6 (Figure 2C). To investigate the possibility that the different concentrations of the TLR4 or TLR7/8 ligands used in the experiments may account for the observed differences, we titrated the quantities of R848 or LPS to stimulate MDMs or monocytes. We found that even at considerably higher concentrations of LPS treatment, monocytes were unable to induce IL6 expression that is comparable to R848 stimulation, while MDMs responded poorly to both high and low concentrations of R848 when compared to monocytes (Figure S3D). Thus, our analyses revealed a unique sensitivity of monocytes to TLR8 agonists for launching a profound proinflammatory response.
These results prompted us to further delineate the mechanism underlying TLR8 agonist sensitivity in monocytes. Interestingly, the TLR8 gene was expressed 4- to 8-fold higher in monocytes when compared to MDMs and MDDCs from 5 donors (Figure S3E). Concordantly, we found that the expression of TLR8 decreased during differentiation from monocytes to MDMs or MDDCs (Figure S3F). Therefore, these results suggest that the high levels of TLR8 expression in monocytes are associated with the increased levels of cytokines produced following TLR8 stimulation, and that TLR8 expression is downregulated during differentiation from monocytes to MDMs or MDDCs. Whether other molecular mechanisms also contribute to the heightened monocyte response to TLR8 stimulation requires further detailed investigation.
Stimulation of TLR4 in MDMs, but not monocytes, induces persistent expression of ISGs
We next focused on gene cluster VIII, which was strongly induced by TLR4 agonist LPS in MDMs, weakly in MDDCs, but not in monocytes (Figure 3A). This cluster of genes was, however, highly induced by monocytes when they were stimulated using TLR7 or TLR7/8 agonists. The cluster mainly consists of ISGs such as MX1, OAS1, and IFIT3 (Figure 3B). Thus, TLR4 signaling in MDMs, and TLR7 or TLR7/8 signaling in monocytes, is equally potent in initiating the ISG response. Conversely, TLR4 stimulation of monocytes and TLR7/TLR8 stimulation of MDMs are unable to induce ISGs. As shown above, the underlying differences in the levels of TLR8 expressed between monocytes and macrophages may explain their disparate responsiveness to R837 and R848. In contrast, we found no significant difference in the levels of TLR4 expression between monocytes and MDMs from 5 donors (Figure 3C). We confirmed that TLR4 signaling induced ISG expression in MDMs, but not monocytes, from 1 additional donor using qRT-PCR (Figure 3D).
Figure 3. Interferon-stimulated genes are strongly induced by TLR4 agonist (LPS) in MDMs compared to monocytes.

(A) Heatmap showing the expression of genes that are significantly upregulated by LPS stimulation in MDMs and by R837 or R848 in monocytes.
(B) Summary table showing Ingenuity Pathway Analysis networks or canonical pathways of genes and signature genes of cluster VIII.
(C) The expression of TLR4 in monocytes and MDMs from 5 different donors. The expression of the TLR4 gene is shown as normalized gene expression value (RPKM), as determined by RNA-seq. Error bars indicate SEMs. Statistics were determined by Student’s t test; ns, not significant.
(D) The expression of MX1, OAS1, and ISG20 in monocytes and MDMs that were unstimulated or stimulated with LPS (10 ng/mL) for the indicated times. The normalized gene expression value (RPKM), as determined by RNA-seq, of these genes is shown as fold induction compared to the respective unstimulated control.
(E) Heatmap of interferon responsive genes that are highly induced in MDM-like (PMA differentiated) THP-1 cells, compared to monocyte-like (undifferentiated) THP-1 cells, when stimulated with 10 ng/mL LPS for 18 h. Signature genes shown in (B) are marked red.
(F) GSEA plots showing enrichment of interferon responsive genes and interferon β targets in differentiated THP-1 cells compared to undifferentiated THP-1 cells.
(G) The expression of MX1 and OAS1 measured by qRT-PCR in differentiated or undifferentiated THP-1 cells stimulated with LPS (10 ng/mL) for 18 h. The expression of these genes is shown as fold induction compared to the respective unstimulated controls. n = 3, error bars represent SEMs. Statistics were determined by 2-tailed ratio paired t test. **p < 0.01.
The THP-1 acute monocytic leukemia cell line has been widely used as a model to study human monocyte and macrophage functions and signaling pathways (Chanput et al., 2014; Qin, 2012). Following differentiation using phorbol 12-myristate 13-acetate (PMA), THP-1 cells acquire a macrophage-like phenotype, which mimics primary human MDMs in terms of cell morphology, cytokine production, and surface markers (Daigneault et al., 2010; Park et al., 2007). We explored whether PMA-induced, MDM-like THP-1 cells also respond distinctly to TLR4 stimulation. We performed transcriptome profiling of either undifferentiated (monocyte-like) or MDM-like THP-1 cells that were stimulated with LPS for 18 h. We found that 113/277 genes in cluster VIII were also uniquely expressed in MDM-like, compared to monocyte-like, THP-1 cells (Figure 3E). Among these genes, we found ISGs to be highly induced in MDM-like THP-1 cells, which is consistent with our observation in primary MDMs (Figure 3E). Gene set enrichment analysis (GSEA) also identified significant enrichment of IFN responsive genes and IFN-β targets in LPS-stimulated MDM-like THP-1 cells (Figure 3F). Consistent with our previous finding in primary monocyte and MDMs, TLR4 stimulation led to higher expression of MX1 and OAS1 in MDM-like THP-1 cells (Figure 3G), supporting the idea that myeloid differentiation imparts changes that elicit distinct responses to TLR activation.
Chromatin accessibility coincides with the induction of IFNB1 and ISGs by TLR agonists in monocytes and MDMs
Our results point to the possibility that distinct downstream events engaged by TLR4 in monocytes and MDMs, rather than differences in TLR expression, were responsible for the observed differences in ISG induction. TLR4 stimulation leads to the phosphorylation and translocation of IRF3 to the nucleus, where IRF3 interacts with its binding site and induces the expression of IFNB1 and primary response ISGs (Honda et al., 2006; Tong et al., 2016). Subsequently, IFN-β can also acts in cis and trans by signaling through IFN-α/βR-ISG factor 3 (ISGF3) complex, thereby amplifying secondary ISG expression (Honda et al., 2006; Tong et al., 2016). We found that LPS induces IFNB1 expression at 1 h post-stimulation in MDMs but not in monocytes (Figure S4A). In comparison, R848 induced IFNB1 as well as various subtypes of IFNA genes in monocytes, but IFNA genes were not upregulated by LPS stimulation in MDMs (Figure S4A). We examined the activation of IRF3 or NF-κB in LPS-stimulated monocytes and MDMs. We did not find any major differences in IRF3 phosphorylation between monocytes and MDMs downstream of TLR4 activation (Figure 4A, left). At some time points, a higher phosphorylation level of IRF3 was observed in MDMs. Nevertheless, TLR4 stimulation induced a substantial increase in IRF3 phosphorylation with the same kinetics in both monocytes and MDMs, suggesting that the stark differences in ISGs induction cannot be explained by differential activation of IRF3 (Figures 4A and S4B). Similarly, we did not find differences in the magnitude and kinetics of NF-κB activation, as measured by IκB phosphorylation, between LPS-stimulated monocytes and MDMs (Figure S4C). However, we observed that LPS strongly increased IFNB1, RSAD2, and IFITM3 transcripts in MDMs as early as 1 h post-stimulation, which was completely absent in monocytes (Figures 4B, 4D, and S4D). These data suggest that, despite activation, phospho-IRF3 in monocytes is unable to induce the transcription of IFNB1 and ISGs.
Figure 4. Chromatin accessibility precedes the induction of ISGs by LPS in MDMs.

(A) Total IRF3 (t-IRF3) and phosphorylated IRF3 (p-IRF3) measured by western blot in monocytes and MDMs stimulated with LPS (10 ng/mL) for 0, 5, 15, 30, 60, and 120 min. Data are representative of 2 independent donors. Normalized band intensity from 3 independent donors is shown at right. Error bars indicate SEMs. Statistics were determined by 2-way ANOVA with Sidak’s correction. **p < 0.01; ***p < 0.001.
(B–E) Normalized gene expression value (RPKM), as determined by RNA-seq, of IFNB1 (B) and RSAD2 (D), with corresponding IGV browser tracks showing chromatin accessibility (C and E) at the 2 gene loci in monocytes and MDMs stimulated with LPS (10 ng/mL) for 0, 1, 6, or 18 h, respectively. The red shading area in (C) and (E) indicates changes in ATAC-seq peaks.
(F and G) Heatmaps showing chromatin accessibility at the ISG promoter region (F), determined by ATAC-seq read abundance on all datasets around the peak center (±2.5 kb/2.0 kb) of promoters for ISG genes, and the expression of these ISGs (G), measured by RNA-seq, in monocytes and MDMs stimulated with LPS (10 ng/mL) for 0, 1, 6, or 18 h.
(H) ATAC-seq peak enrichment within promoters of ISGs in MDMs and monocytes. The genomic coordinates of promoter regions of expressed ISGs (±1 kb to the transcription start site [TSS]) were intersected with various categories of ATAC-seq peaks (e.g., the set of peaks that are stronger in MDMs compared to monocytes 1 h post-stimulation). The y axis indicates the proportion of expressed ISG promoters that contain a monocyte- or MDM-specific ATAC-seq peak at the given time point. p values were calculated using an “N-1” chi-square proportions test comparing the 2 values at each time point.
Since histone modifications and chromatin structure can influence the binding of TFs to regulatory elements that control transcription, we hypothesized that differences in signal-induced chromatin accessibility between monocytes and MDMs may be dictating the ability of IRF3 to access the relevant regions of chromatin. To identify such differences, we performed assay for transposase-accessible chromatin using sequencing (ATAC-seq) paired with RNA-seq analysis of monocytes and MDMs following 1, 6, and 18 h of TLR4 stimulation. Chromatin accessibility of the ISG loci, such as the 5′, 3′ region and proximal gene body of IFNB1, and the promoters of RSAD2 and IFITM3, increased in MDMs 1 h post-LPS stimulation, which correlated with changes in their mRNA expression (Figures 4B-4E and S4D). In contrast, chromatin opening and the corresponding changes in gene expression in these regions were not observed in monocytes (Figures 4B-4E and S4D). We found that LPS stimulation broadly opens the chromatin of ISG promoter loci as early as the 1-h time point in MDMs (Figure 4F), thereby enabling transcription from these regions as reflected by ISG induction at 6 and 18 h (Figure 4G). In monocytes, there was no dynamic or early change in the chromatin structure following TLR4 stimulation (Figure 4F), and there was no subsequent induction of ISGs at 6 and 18 h post-stimulation (Figure 4G). The percentage of ISG loci with open chromatin (ATAC-seq peaks) are also significantly higher in MDMs than in monocytes at all time points, indicating a clear correlation between chromatin opening and ISG transcription in MDMs (Figure 4H). Monocytes are functionally capable of opening ISG loci and inducing their transcription following TLR7 and TLR8 stimulation or following exposure to exogenous IFNβ (Figures 3A, S4E, and S4F). The lack of ISG transcription downstream of TLR4 signaling is therefore unlikely to be the result of lineage-defining factors or defective IFN-α/β receptor (IFNAR) signaling, but is rather due to the chromatin remodeling that TLR4 signaling fails to induce in monocytes. Although IFNB1 produced following 1 h post-stimulation could activate the IFN-α/βR-ISGF3 pathway to induce ISG transcription (Figure 4B), it does not explain the marked differences in the opening of ISG promoters at 1 h following TLR4 stimulation (Figure 4F). Open chromatin at ISG promoters as early as 1 h therefore implies that early chromatin remodeling at ISG loci contribute directly to the competent ISG transcription. The inability of IRF3 downstream of TLR4 to drive the transcription of ISGs because of the lack of accessibility likely explains the primary defect in monocytes (Collins et al., 2004; Grandvaux et al., 2002; Yoneyama et al., 1998). These observations are consistent with previous findings that ISGs can be induced in the absence of IFNAR signaling (Collins et al., 2004; Wathelet et al., 1992, 1998; Weaver et al., 1998; Yoneyama et al., 1998). However, it is highly likely that the induction of early IFNB1 in MDMs plays a role in amplifying the ISG response at later time points. Overall, these data suggest that differences in chromatin accessibility, rather than the robustness of early signal transduction, account for the differences in the expression of ISGs upon TLR4 stimulation in myeloid cells.
Enrichment of IRF binding motifs in open chromatin and increase in nuclear translocation of IRF1 in MDMs upon TLR4 stimulation
We hypothesized that TLR4 signaling in MDMs leads to chromatin remodeling by a modifier, a process that is lacking in monocytes. To identify this modifier, we performed a global analysis of the enrichment of TF binding site motifs in accessible chromatin regions. Specifically, we used HOMER to calculate the enrichment of each TF motif in open chromatin (ATAC-seq peaks) specific to MDMs, compared to monocytes, at various times post-stimulation (Heinz et al., 2010). These analyses revealed that the open loci in MDMs had enrichment for the binding sites of IRFs (Figure 5A), which play essential roles in type I IFN and ISG induction (Honda et al., 2006; Mogensen, 2019; Negishi et al., 2018; Platanitis and Decker, 2018). It has been reported that promoters of inducible inflammatory genes in unstimulated macrophages commonly remain in an open or poised chromatin state (Foster et al., 2007; Hargreaves et al., 2009; Ivashkiv, 2013; Ramirez-Carrozzi et al., 2009). Despite that, we observed highly enriched IRF binding motifs in both enhancers and promoters of MDMs following stimulation (Figure S5A). In contrast, signal transducer and activator of transcription (STAT) binding motifs are relatively lowly enriched in ISG promoters and enhancers, and showed no significant bias between MDM and monocyte (Figures 5A and S5A). Furthermore, IRF motifs that are predicted by HOMER to be bound by IRF1 were significantly enriched in MDMs compared to monocytes in response to TLR4 stimulation (Figure 5B). We next examined the expression of the IRF1 protein and found it to be substantially increased in MDMs immediately following TLR4 stimulation, while it remained stable and at lower levels in LPS-stimulated monocytes (Figure 5C). At 6 and 18 h post-stimulation, we also observed a significant difference in IRF1 expression between monocytes and MDMs (Figures 5D and S5B). While this could be due partially to feedback of IFN-β, the elevation of IRF1 may further sustain the opening of ISG loci. A similar, 2-fold higher expression of IRF1 was also observed in differentiated THP-1 cells stimulated with LPS (Figure S5C). Importantly, IRF1 in MDMs, but not monocytes, entered the nucleus rapidly after TLR4 stimulation (Figure 5C), suggesting that TLR4 signaling in MDMs enables IRF1 transcription and facilitates its translocation into the nucleus. These results suggest that activated IRF1 is a critical factor for ISG chromatin accessibility in TLR4-activated MDMs.
Figure 5. Enrichment of IRF binding motifs and increase of IRF1 translocation to the nucleus in MDMs upon TLR4 stimulation.

(A) Comparison of genome-wide TF motif enrichment in ATAC-seq peaks specific to monocytes or MDMs. Cells were stimulated for indicated time points with 10 ng/mL LPS. Chromatin accessibility was assessed using ATAC-seq. For each time point, the DNA sequences within peaks unique to monocytes or MDMs were assessed for the enrichment of TF binding site motifs using HOMER and motifs contained in the Cis-BP database (see Method details). The significance (−log10 p value) of the motif enrichment in monocytes (y axis) and MDMs (x axis) is shown as a dot. The dots are colored based on the family of TFs. See Figure S5A for the same analysis performed separately in promoter and enhancer regions.
(B) IRF1 binding motifs in genome-wide open chromatin regions and the enrichment p values in monocytes and MDMs stimulated with LPS (10 ng/mL) for 1, 6, and 18 h.
(C) Western blot analysis of IRF1 abundance in total cell lysate and nuclear extracts from monocytes and MDMs stimulated with LPS (10 ng/mL) for 0, 5, 15, 30, 60, and 120 min. β-Actin and histone H3 were used for the loading control of total cell lysate and nuclear fraction, respectively.
(D) Normalized gene expression value (RPKM), as determined by RNA-seq, of IRF1 in unstimulated and LPS-stimulated (18 h) monocytes and MDMs. Error bars indicate SEMs. Statistics were determined by 2-way ANOVA with Tukey’s correction. ns, not significant; **p < 0.01.
IRF1 deficiency leads to impaired chromatin accessibility and expression of ISGs
To further investigate the role of IRF1 in regulating chromatin accessibility in human macrophages, we used the THP-1 differentiation model used previously (Figure 3). We generated clonal cultures of IRF1-deficient (knockout [KO]) and IRF-sufficient control (Ctrl) THP-1 cells by CRISPR/Cas9 editing (Figure S5D). We then differentiated Ctrl and IRF1 KO THP-1 clones with PMA to mimic human MDMs and stimulated them using LPS to examine the transcription of ISGs at 1 and 6 h. IRF1 deficiency completely abrogated the ability of differentiated THP-1 cells to induce ISGs, such as MX1 and OAS1, following LPS stimulation (Figure 6A). Notably, IRF1 deficiency did not impair the expressions of ISGs such as IFIT1 and OAS1 upon TLR7/8 stimulation (Figure S5E), suggesting that different TLRs use unique chromatin modifiers to induce the expression of a shared set of genes. Similar to what we observed in primary MDMs, we did not find any significant differences in TLR4-induced IRF3 phosphorylation between PMA-differentiated Ctrl and IRF1 KO THP-1 clones (Figure 6B). We next investigated the impact of IRF1 deficiency on chromatin accessibility at the ISG loci. We performed a combined RNA-seq and ATAC-seq experiment to examine ISG expression and chromatin accessibility in differentiated Ctrl and IRF1 KO clones following LPS stimulation. We found a decreased global expression of all ISGs in IRF1 KO clones when compared to Ctrl clones (Figure 6C, left). Furthermore, the impaired ISG expression directly correlated with a lack of chromatin accessibility in KO cells (Figure 6C, right). While chromatin of ISG loci in Ctrl THP-1 cells became accessible at 1 and 6 h, these regions remained inaccessible in IRF1 KO THP1 cells at all time points. Surprisingly, the deletion of IRF1 also reduced the basal accessibility of ISG loci in resting unstimulated cells (Figure 6C, right). To determine whether IRF1 is required for the accessibility of the conserved IFN-sensitive response element (ISRE), we used HOMER to assess the status of ISRE motifs that are occupied by IRF or by the ISGF3 complex. ISRE enrichment in open chromatin regions in IRF1 KO was significantly less compared to the Ctrl clone as early as 1 h (Figure 6D). Overall, these results suggest that IRF1 is an essential factor contributing to chromatin remodeling that facilitates ISG induction in TLR4-stimulated human macrophages.
Figure 6. IRF1 deficiency impairs expression and chromatin accessibility of ISGs in LPS-stimulated, macrophage-like THP-1 cells.

(A) The expression of MX1 and OAS1 measured by quantitative RT-PCR in PMA-differentiated control (Ctrl) or IRF1 KO THP-1 clones, following stimulation with LPS (10 ng/mL) for 0, 1, or 6 h. The expression of these genes is shown as fold induction compared to the respective unstimulated (0 h) control. Data represent 3 distinct Ctrl and IRF1 KO clones. Error bars represent SEMs. Statistics were determined by repeated-measure 2-way ANOVA with Sidak correction. **p < 0.01.
(B) Total IRF3 (t-IRF3) and p-IRF3 were detected by western blot in PMA-differentiated control (Ctrl) or IRF1 KO THP-1 clones following stimulation with LPS (10 ng/mL) for 0, 5, 15, 30, 60, and 120 min. Data are representative of 2 independent experiments.
(C) Heatmaps showing the expression (left, determined by RNA-seq) of ISGs and chromatin accessibility (right, determined by ATAC-seq) at ISG promoter loci (±2.5 kb/2.0 kb around the peak center) in PMA-differentiated control (Ctrl) or IRF1 KO THP-1 clones. Cells were stimulated with LPS (10 ng/mL) for 0, 1, or 6 h. Data depict experiments with 1 Ctrl or 2 different IRF1 KO THP-1 clones (RNA-seq) or with 1 Ctrl or 2 IRF1 KO THP-1 clone (ATAC-seq).
(D) PMA-differentiated control and IRF1 KO THP-1 clones were stimulated with 10 ng/mL LPS, and chromatin accessibility was assessed using ATAC-seq. For each time point, the DNA sequences within peaks unique to IRF1 KO and Ctrl were assessed for the enrichment of ISRE motifs (see method details). The significance (−log10 p value) of each ISRE motif enrichment in IRF1 KO (y axis) and Ctrl (x axis) is shown.
DISCUSSION
A critical feature of the innate immune system is the ability of specific stimuli to elicit distinct transcriptional responses in different cell types (Medzhitov and Horng, 2009; Monticelli and Natoli, 2017; Natoli et al., 2011; Smale, 2010). The basis for TLR-specific transcriptional responses was mostly attributed to the use of distinct signaling adaptor molecules, while the underlying mechanism for the lineage-specific response is usually rooted in differential receptor expression (Kawasaki and Kawai, 2014; Takeuchi and Akira, 2010). However, we found that differential TLR4 expression could not account for the ability of LPS to induce IFNB1 and ISGs in MDMs but not monocytes. Our data revealed that the differential induction of IFNB1 and ISGs strongly correlated with the differential chromatin accessibility between monocytes and MDMs. This result is consistent with emerging concepts that chromatin dynamically regulates gene expression by altering its architecture by repositioning, assembling, and restructuring nucleosomes (Amit et al., 2011; Smale and Fisher, 2002; Smale et al., 2014; Struhl and Segal, 2013; Winter and Amit, 2014). A recent study characterized chromatin accessibility, enhancer activity, and TF motif enrichments across myeloid lineages, including monocytes, macrophages, and DCs (Yoshida et al., 2019). Another study showed marked differences in chromatin landscape and regulatory TFs between monocyte-derived DCs and plasmacytoid DCs (pDCs), which demarcate transcriptional response to all TLR stimulations between these 2 cell types (Bornstein et al., 2014). However, 2 important questions remain unanswered. First, pDCs have been shown to originate from various lymphoid and myeloid progenitors, and their chromatin landscape is distant from the cluster of other myeloid cells (Reizis et al., 2011; Yoshida et al., 2019). Thus, it is still not clear whether chromatin accessibility of more closely related myeloid populations regulate their inflammatory responses. Second, it remains unknown whether the chromatin landscape regulates stimuli-specific responses, thereby dictating myeloid cell sensitivity to different TLR ligands. In this study, we further highlighted the differences in regulatory motif accessibility that emerged following specific classes of TLR stimulation in monocytes and their direct descendants, MDMs and MDDCs. Our findings show that the most distinctively enriched motifs in the open chromatin regions of MDMs upon TLR4 activation are the IRF family, which play critical roles in the induction of type I IFN and downstream mechanisms (Honda et al., 2006; Mogensen, 2019; Negishi et al., 2018; Platanitis and Decker, 2018). We demonstrated a critical role for IRF1 in enabling ISG expression downstream of TLR4 signaling in human macrophages and macrophage-like cells. Our findings reveal that the differential inflammatory output of related, yet functionally distinct myeloid cells is centered on the regulation of chromatin accessibility.
Activation of the human IFNB1 gene has been studied in great detail in the past. Many of these studies, which were performed in HeLa cells following viral infections, established the formation of an enhanceosome through the cooperative binding of several key TFs, including NF-κB, IRF3/7, and ATF-2/c-Jun (Thanos and Maniatis, 1995). Examination of the IFNB1 locus has also led to a general understanding of selective gene activation. Signaling by plasma membrane TLRs, which activate NF-κB and ATF2/c-Jun but not IRF3, does not lead to the transcription of IFNB1 or the secondary ISGs (Doyle et al., 2002). The activation of IRF3 has been proposed to be a central mechanism by which TLR3 and TLR4, but not TLR2, induce IFNB1 and the antiviral gene program. IRF3 has been implicated in promoting nucleosome remodeling at the loci of both primary and secondary response genes in LPS-stimulated macrophages (Ramirez-Carrozzi et al., 2009). Removal of the nucleosome barrier allowed NF-κB and ATF2-c-Jun to stimulate IFNB1 promoter activity in the absence of IRF3. Thus, chromatin remodeling at the IFNB1 and ISG loci has been proposed as a critical regulator of transcription. Our studies suggest that IRF1 may position higher in the hierarchy of TFs than IRF3 in initiating IFNB1 and ISG transcription. We note here that studies reporting a role for IRF3 in promoting nucleosome remodeling were performed in murine macrophages. Whether the absence of IRF3 affected the expression of IRF1 or whether these 2 TFs have differential roles in regulating IFNB1 and ISG expression between murine and human cells remains to be examined.
It has been previously observed that the overexpression of IRF1 results in the induction of type I IFN genes and/or ISGs, and that the deficiency of IRF1 leads to impaired expression of type I IFN and/or ISGs upon TLR stimulation in monkey kidney (COS) cells, mouse embryonic fibroblasts, human hepatoma (Huh-7) cells, and human bronchial epithelial (BEAS-2B) cells (Fujita et al., 1989; Matsuyama et al., 1993; Panda et al., 2019; Schoggins et al., 2011). Furthermore, 2 recent studies have provided insight into the critical role played by IRF1 in chromatin remodeling to control gene expression (Karwacz et al., 2017; Panda et al., 2019). IRF1 deficiency altered the chromatin landscape during type 1 regulatory T (Tr1) cell differentiation, suggesting that IRF1 increases chromatin accessibility and facilitates the binding of additional Tr1 TFs (Karwacz et al., 2017). In addition, it was reported that IRF1 modulates H3K4me1 at promoter and enhancer regions to maintain the expression of IRF1-dependent genes (Panda et al., 2019). Here, we show that the enrichment of IRF1 binding motifs and translocation of IRF1 into the nucleus increased in MDMs compared to monocytes upon TLR4 stimulation. In addition, we revealed that IRF1 deficiency impairs the expression and chromatin accessibility of IFNB1 and ISGs following TLR4 stimulation. Mechanistically, IRF1 appears to be critical for preparing the chromatin landscape at ISG loci upon TLR4 stimulation. A recent study examined the ability of different TFs to bind to mitotic chromosomes, a predictor of pioneer activity, and found IRF1 to be highly enriched in the mitotic chromosome-bound fraction (Raccaud et al., 2019). Thus, our findings, along with evidence from previous studies, strongly suggest that IRF1 may act as a pioneer factor. Alternatively, as a critical non-pioneer factor (Mayran et al., 2019), IRF1 may be associating with other pioneer factors to open ISG chromatin following TLR4 stimulation in macrophages.
Another intriguing finding of this study is the differential responses of human monocytes and macrophages to TLR7/8 ligands. Our experiments revealed that monocytes, but not MDMs, transcribe IFNB1 and ISGs in response to TLR7/8 stimulation. The ISG loci are available after 1 h of TLR7/8 stimulation in monocytes. This observation indicates that TFs and chromatin modifiers downstream of TLR7/8 signaling are sufficient to open up these chromatin regions. In contrast, to trigger ISG transcription, TLR4 signaling seems to additionally require nuclear IRF1 activity, which is lacking in monocytes but proficient in MDMs. As a result, monocytes cannot sufficiently respond to TLR4 in terms of IFNB1/ISGs induction. The exact mechanisms by which TLR7/8 signaling regulates chromatin accessibility of ISGs in monocytes remains a future area of investigation.
Using both primary human myeloid cells and THP-1 cell lines, we have shown that following their differentiation into macrophages, monocytes lose the ability to respond to TLR7/8 ligands and gain the ability to respond to TLR4 stimulation to induce transcription of ISGs. These data demonstrate that chromatin accessibility at IFNB1 and ISG loci is not solely determined by cell identity, but rather by lineage-specific TLR signaling. We do not currently understand the biological need for such gain and loss of TLR sensitivity between monocytes and macrophages. We speculate that this may be the reflection of the anatomical location of these cell types. Since monocytes are primarily circulating cells, there may be a greater need for them to produce type I IFNs to drive the ISG response to systemic viral infections. However, it could be critical for macrophages to produce IFN-β and the protein products of ISGs in the tissues to clear localized bacterial infections. Further studies that examine tissue resident macrophage responses may reveal the molecular and biological bases of these dichotomies.
Apart from IFNB1 and ISGs, monocytes from human donors also produce very high levels of inflammatory cytokines, including IL-1β, IL-6, IL-12, and TNF, in response to TLR7/8 ligation. It is now well documented that a “cytokine storm” is a hallmark of severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) infections, leading to considerable morbidity and mortality (Mehta et al., 2020; Ye et al., 2020; Zhang et al., 2020a). In light of these findings, we hypothesize that circulating monocytes that are inherently hyperresponsive to TLR7/8 ligands could be responding to single-stranded RNA (ssRNA) from SARS-CoV-2 to drive this cytokine storm in viremic patients. Recent clinical studies observed significant increases in IL-6-producing CD14+ monocytes in the peripheral blood of severe cases of coronavirus disease 2019 (COVID-19) patients (Gomez Rial et al., 2020; Zhang et al., 2020b). Our study suggests that one way to mitigate this systemic condition would be to dampen TLR7/8 signaling and/or regulate the exit of monocytes from the bone marrow. However, aberrant IRF1 activation is found in lupus patient peripheral blood monocytes associates with global interferon signatures and disease severity (Liu et al., 2017; Zhang et al., 2015). In addition, TNFR signaling in mouse macrophages has been shown to induce IFN-β in an IRF1-dependent manner, and IFN-β in turns sustains the expression of inflammatory genes in synergy with other TNF-inducing ligands such as TLRs (Yarilina et al., 2008). TNF receptor (TNFR) signaling in endothelial cells also drives IRF1-dependent IFN-β production that leads to the accumulation of monocytes and macrophages in inflamed renal tissues (Venkatesh et al., 2013). Thus, suppressing IRF1 activity may serve to prevent the tissue-specific inflammation and systemic inflammation that underlies autoimmune etiology. Targeting IRF1 could be a therapeutic approach in dampening IFN production in these types of autoimmune and autoinflammatory diseases.
There remain some limitations in the present study that leave questions to be tackled in future endeavors. First, we have profiled the transcriptomes based on synthetic TLR7/8 ligands R848 and R837, although we predict a corollary dichotomy in response to natural TLR7/8 ligands such as ssRNA and RNA viral infections. Future studies using these natural ligands and infectious agents may reveal physiological meaning and need for lineage-specific TLR responses. Second, although multiple lines of evidence in this study have pointed out that IRF1 can directly regulate the chromatin accessibility of ISGs in the primary phase of the TLR ligand response, the relative contribution of this direct regulation of primary ISG chromatin accessibility, as compared to secondary augmentation by IFNβ-IFNAR that likely amplifies the magnitude of ISG transcription remains to be determined. Third, we have used THP-1 human monocytic cell lines to genetically dissect the role of IRF1 by CRISPR/Cas9-mediated knockout, due to the technical difficulty of creating IRF1-deficient primary human myeloid cells. A recent technological advance may be harnessed in the near future to consolidate the role of IRF1 in primary MDMs (Freund et al., 2020).
In summary, our study provides sophisticated insights into the transcriptional programs that are generated within closely related cell types of the human innate immune system. We reveal the dynamic chromatin remodeling in the transcriptional response of human monocytes and macrophages that could be instrumental to understanding inflammatory cytokine production in response to various microbial insults. Finally, our results suggest that by regulating chromatin accessibility, IRF1 plays an essential role in the regulation of IFNB1 and ISG expression by TLR4 stimulation in human macrophages. Our findings highlight the importance of understanding the molecular basis of lineage-specific responses in the human innate immune system to combat inflammation in both autoimmunity and microbial infections.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Chandrashekhar Pasare (chandrashekhar.pasare@cchmc.org). All unique/stable reagents generated in this study are available from the lead contact with a completed Material Transfer Agreement.
Materials availability
This study did not generate new unique reagents.
Data and code availability
MATLAB code used for RNA-seq analysis is described in RNA-seq analysis and is available on request. Illustrations were created with Biorender (https://biorender.com). RNA-seq and ATAC-seq data for this study have been deposited at NCBI’s Gene Expression Omnibus (GEO). The acession number is GEO: GSE147314.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human PBMC donors
Peripheral blood was obtained from donors of masked identity (unknown gender or age) via Carter Blood Care (Dallas, TX). IRB-approved protocols (UT Southwestern Medical Center) were used to collect, transport, and store all the biological samples.
Primary cell cultures and differentiation
Human peripheral blood mononuclear cells (PBMCs) were enriched by density gradient centrifugation of peripheral blood from healthy human donors through a Ficoll-Paque PLUS (GE Healthcare) gradient. Monocytes were isolated from PBMCs by negative selection using the EasySep Human Monocyte Isolation Kit (STEMCELL Technologies) according to the manufacturer’s instructions. Monocytes were immediately stimulated with Pam3CSK4 (1 μg/ml), LPS (10ng/ml), R837 (10 μg/ml), TLR8-506 (0.5 μg/ml) or R848 (10 μg/ml, InvivoGen) after isolation from PBMCs for the indicated times. Human monocyte-derived macrophages (MDMs) or Human monocyte-derived dendritic cells (MDDCs) were generated in RPMI-1640 medium with 10% FBS, 2mM L-glutamine, 10mM HEPES, 1mM sodium pyruvate, 100U/ml penicillin, and 100 μg/mL streptomycin and recombinant human M-CSF (50ng/mL) or recombinant human GM-CSF (100ng/mL) and recombinant human IL-4 (50ng/mL), respectively. The culture medium which contained fresh recombinant human M-CSF or recombinant human GM-CSF and recombinant human IL-4 was replaced every 2 days. After 7 days, MDMs were collected by Accutase (Innovative Cell Technologies) and MDDCs were harvested by pipetting. MDMs were allowed to adhere overnight and stimulated with TLR ligands for the indicated times. MDDCs were immediately stimulated with TLR ligands for the indicated times.
Cell lines
THP-1 cells were cultured in T-75 flasks between 2-8 × 105 cells/mL in growth medium containing RPMI-1640, 10% FBS, 2mM L-glutamine, 10mM HEPES, 1mM sodium pyruvate, 100U/ml penicillin, and 100μg/mL streptomycin. THP-1 monocytes were differentiated into macrophages by a 48-hr incubation with 50ng/ml phorbol 12-myristate 13-acetate (PMA) followed by 48 hr without PMA. THP-1 or PMA-differentiated THP-1 cells were stimulated with LPS (10ng/ml) or R848 (10 μg/ml, InvivoGen) for the indicated times.
METHOD DETAILS
ELISA
The concentrations of IL-1β, TNFα, and IL-6 were quantified using ELISA kits (R&D Systems) according to the manufacturers’ instructions.
Western blotting
Cells were lysed using CelLytic M Cell Lysis Reagent (Sigma-Aldrich) supplemented with protease/phosphatase inhibitor cocktail (Cell Signaling). 10ug of proteins were separated by SDS-PAGE and blotted onto nitrocellulose membranes. The membranes were stained with primary antibodies against IRF1 (Cell Signaling), IκBα, phospho-IκBα, IRF3, or phospho-IRF3 (Abcam), followed by HRP-conjugated secondary antibodies (Cell Signaling). Staining was revealed with SuperSignal West Pico Chemiluminescent Substrate (Pierce). Protein densitometric analysis was performed with ImageJ software on scanned blots. Band Intensity was measured using ImageJ.
RNA sequencing library preparation
Monocytes, MDMs or MDDCs were stimulated with Pam3CSK4 (1 μg/ml), LPS (10ng/ml), R837 (10 μg/ml), TLR8-506 (0.5 μg/ml) and R848 (10 μg/ml, InvivoGen) for the indicated times. RNA was extracted using TRIzol reagent (Life Technologies) and RNeasy Mini Kit (QIAGEN) according to the manufacturer’s protocol. Quantity and quality of RNA samples were measured by an Agilent Bioanalyzer 2100 (Agilent Technologies). RNA-seq libraries were prepared with the Illumina TruSeq RNA Sample Preparation kit (Illumina) according to the manufacturer’s protocol. Libraries were validated on an Agilent Bioanalyzer 2100. Indexed libraries were equimolarly pooled and sequenced on a SE50 (single-end 50 base pair) Illumina HiSeq2500 lane, which yielded an average of about 30x106 reads/sample.
CLC Genomics Workbench 7 was used for bioinformatics and statistical analysis of the sequencing data. This approach used by CLC Genomics Workbench is based on method developed by Mortazavi et al. (2008). Human Genome GRCh37 was used as reference sequence.
RNA-sequencing analysis
Methods for data normalization and analysis are based on the use of “internal standards” that characterize some aspects of the system’s behavior, such as technical variability, as presented elsewhere (Dozmorov and Centola, 2003; Dozmorov and Lefkovits, 2009; Dozmorov et al., 2011). Created initially for the analysis of microarray data they were slightly modified to the needs of RNA-seq data analysis.
Differential gene expression analysis includes the following steps:
Construction of the ‘reference group’ by identifying a group of genes expressed above background with inherently low variability as determined by an F-test. The ‘reference group’ presents an internal standard of equal expression. As such, the ‘reference group’ is used to assess the inherent variability resulting from technical factors alone (technological variation). By creating an estimate of the technological variation we are able to select a group of biologically stable genes.
Selection of replicates using the commonly accepted significance threshold of p < 0.05 with a Student t test. This selection maintains the commonly accepted sensitivity level; however, a significant proportion of genes identified as differentially expressed at this threshold will represent false positive determinations.
An Associative t test in which the replicated residuals for each gene from the experimental group are compared with the entire set of residuals from the reference group defined above. The Ho hypothesis is checked to determine whether the levels of gene expression in the experimental group presented as replicated residuals (deviations from the averaged control group profile) is associated with a highly representative (several hundred members) normally distributed set of residuals of gene expression values in the reference group. The significance threshold is then corrected to render the appearance of false positive determinations improbable: in current case p < 0.0001. Only genes that pass both tests are presented in the final selections. Additional restrictions were applied to the minimal gene expression level (RPKM > 2) and fold of changes (> 1.5).
HV-gene analysis. Even when working with samples from a homogeneous group, we are able to observe a portion of genes that have high variability among individuals, which can be explained by experimental error or evidence about some non-synchronized dynamic events in an otherwise homogeneous group. We developed a method for selection of these groups of genes of statistically significant higher variability and named them the ‘hypervariable genes (HV genes)’ based on analysis of residuals of normalized expression using an F-criterion (Dozmorov et al., 2004, 2011). To reveal among them groups of genes demonstrating synchronized variations of expression there can be used any clustering procedure, usually applied to analysis of the genes dynamical behavior. We applied clustering procedure based on the correlation analysis of HV-gene profiles, created and used for analysis of gene expression variability in numerous experimental and clinical data (Dozmorov et al., 2003; Hasan et al., 2013; Hu et al., 2015; Jarvis et al., 2004). In brief, the selected genes were clustered with procedure based on the correlation as a measure of the profile similarity. The threshold for selection of the cluster – correlation coefficient – was chosen with use simulation technique. The natural expression data were substituted with random data having for each gene over all samples the same Average and SD values. In our case we had significant differences in the content of the clusters for normal and simulated data at the threshold CC > 0.7.
All analytical procedures, including two-step normalization, associative analysis of differentially expressed genes, HV-gene selection and clustering are implemented in MATLAB (Mathworks, MA). Functional analysis of identified genes was performed with Ingenuity Pathway Analysis (IPA; Ingenuity® Systems, Redwood City, CA, https://wwwqiagenbioinformatics.com).
Quantitative real-time PCR (RT-qPCR)
500 ng of RNA was reverse transcribed using the Superscript ® III First-Strand Synthesis System (Invitrogen) according to manufacturer’s protocol. RT-qPCR was performed using PowerUp SYBR® Green Master Mix (applied biosystems) on the QuantStudio 7 Flex Real-Time PCR System (applied biosystems) according to the manufacturer’s directions. Primers were as follows: GAPDH, 5′-tctctgccccctctgctg-3′ (forward) and 5′-agtccttccacgataccaaa-3′ (reverse); CCR2, 5′-gaaacgagaagaagaggcatag-3′ (forward) and 5′-ccccaacgaggcataga-3′ (reverse); HLA-DRB1, 5′-ttcctgtggcagcctaagag-3′(forward) and 5′-aaccccgtagttgtgtctgc-3′ (reverse); CD300E, 5′-agagaaggtggagaggaatgg-3′ (forward) and 5′-aggaagatgggaggtgtgg-3′ (reverse); MMP9, 5′-cgtcttccccttcactttcc-3′ (forward) and 5′-ccccacttcttgtcgctgt-3′ (reverse); BATF3, 5′-aggaaggtccgaaggagaga-3′ (forward) and 5′-gaggcactggcacaaagttc-3′ (reverse); MX1, 5′-ctgggattttggggcttt-3′ (forward) and 5′-gggatgtggctggagatg-3′ (reverse); and OAS1, 5′-tcagaaataccccagccaaa-3′ (forward) and 5′-gagccaccctttaccacctt-3′ (reverse). GAPDH was used as an internal control. The comparative CT (ΔΔCT) method was used for data analysis.
Assay for transposase-accessible chromatin using sequencing (ATAC-seq)
ATAC-seq was performed as previously described (Buenrostro et al., 2013). Briefly, 50,000 monocytes or MDMs were centrifuged 500 g for 5 min at 4°C. After washing with cold PBS, cell pellets were resuspended in 50 μL of lysis buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL CA-630) and nuclei were pelleted by centrifugation for 10 min at 500 g, 4°C. Nuclei were resuspended in the transposase reaction mix (25 μL 2 × TD buffer (Nextera DNA Sample Preparation Kit), 2.5 μL Illumina Tn5 transposase and 22.5 μL nuclease-free water). The transposition reaction was incubated at 37°C for 30 min. After tagmented DNA was purified using a MinElute Purification Kit (QIAGEN), PCR amplification was performed by combining tagmented DNA with NEBNext 2x PCR Mater Mix and Custom Nextera PCR Primers. An initial amplification was performed using the following PCR conditions: 72°C for 5 min; 98°C for 30 s; and 5 cycles of 98 °C for 10 s, 63 °C for 30 s, and 72 °C for 1 min. 5 μL of the PCR reaction was subjected to an additional 20 cycles of SYBR green based qPCR to determine the additional number of cycles needed for the remaining 45 μL reaction. The libraries were purified using AMPure XP beads (Beckman Coulter). Library quality and quantity were assessed on a BioAnalyzer High Sensitivity DNA chip (Agilent Technologies). Libraries were pooled at equimolar ratios and sequenced on Illumina NextSeq 550.
We used TrimGalore version 0.4.1 (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) on the raw reads to remove reads shorter than 35bp or with phred quality scores less than 20 and then aligned trimmed reads to the human reference genome (GRCh37) using default parameters in BWA version 0.7.12 (Li and Durbin, 2009). The aligned reads were subsequently filtered for quality and uniquely mappable reads were retained for further analysis using Samtools version 1.3 (Li et al., 2009) and Sambamba version 0.6.6 (Tarasov et al., 2015). Library complexity was measured using BEDTools version 2.26.0 (Quinlan and Hall, 2010) and meets ENCODE data quality standards (Landt et al., 2012). Peaks were called using MACS version 2.1.0 (Feng et al., 2012) with a p value cutoff of 1x10−2.
ATAC-seq density heatmap and profile
For heatmaps and profiles of ATAC-seq intensities, we used deepTools version 2.5.0 (Ramírez et al., 2016) to generate read abundance from all ChIP-seq datasets around the peak center (±2.5kb/2.0kb), using ‘computeMatrix’. These matrices were then used to create heatmaps and profiles, using the deepTools commands ‘plotHeatmap’ or ‘plotProfile’ respectively.
ATAC-seq differential accessibility analysis and peak comparison
We used The MAnorm software package (Shao et al., 2012) was used to identify regions of differential chromatin accessibility in macrophages and monocytes. A fold change cutoff of greater than or equal to 1.5 and a p value cutoff of less than or equal to 0.05 were used to identify significant differential peaks for each pairwise comparison between untreated and treatment conditions. The macrophage and monocyte specific peaks identified by MAnorm were further partitioned into those located within promoters and those that are not (‘enhancers’) using HOMER annotation tool (Heinz et al., 2010). The enrichment of macrophage and monocyte specific peaks within the promoters of expressed ISG genes was performed using the RELI tool (Harley et al., 2018).
Transcription factor (TF) binding site motif enrichment analysis
To identify specific TFs that might bind the regions identified in our ATAC-seq experiments, we performed HOMER (Heinz et al., 2010) TF binding site motif enrichment analysis. Specifically, we used HOMER to calculate the enrichment of each motif in ATAC-seq peaks specific to monocytes compared to MDMs at various times after LPS stimulations. HOMER was modified to use the large library of human position weight matrix (PWM) binding site models contained in build 2.0 of the CisBP database (Weirauch et al., 2014) and a log base 2 log likelihood scoring system.
Lentiviral transduction
VSV-G pseudotyped, self-inactivating lentivirus was prepared by transfecting the 6 well plate of 1x106 293T cells with 1.25 μg of pVSV-G, 1.875 μg of psPAX-2, and 2.5 μg of the pRRL lentiCRISPR vector for 6 hr and then aspirating and replacing the media with fresh media. The medium was collected 60 hr after transfection and filtered through 0.45 μm-pore-size filters. 2x105 THP-1 cells were transduced with lentiviral supernatant. 48 hr post-transduction, the cells were placed under selection with either 5 μg/mL puromycin (Thermo Fisher) or 10 μg/mL blasticidin (Thermo Fisher) for 5 days.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed in Prism (Graphpad) using methods as indicated in the figure legends. Data are presented as mean ± SEM. Significance was considered at *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns = not significant. Computational methods for RNA-sequencing and ATAC-sequencing analysis can be found in detail in the respective method sections.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Recombinant Anti-IRF3 (phospho S386) antibody | abcam | Cat# ab76493; RRID:AB_1523836 |
| IRF-3 (D6I4C) XP® Rabbit mAb | Cell Signaling Technology | Cat# 11904S; RRID:AB_2722521 |
| IRF-1 (D5E4) XP® Rabbit mAb | Cell Signaling Technology | Cat# 8478S; RRID:AB_10949108 |
| IκBα (L35A5) Mouse mAb | Cell Signaling Technology | Cat# 4814S; RRID:AB_390781 |
| Phospho-IκBα (Ser32) (14D4) Rabbit mAb | Cell Signaling Technology | Cat# 2859S; RRID:AB_561111 |
| Histone H3 (D1H2) XP® Rabbit mAb | Cell Signaling Technology | Cat# 4499S; RRID:AB_10544537 |
| β-Actin (8H10D10) Mouse mAb | Cell Signaling Technology | Cat# 3700S; RRID:AB_2242334 |
| Biological samples | ||
| Buffy coats | Carter BloodCare | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Human M-CSF Protein | R&D systems | Cat# 216-MC-025 |
| Recombinant Human GM-CSF Protein | R&D systems | Cat# 215-GM-050 |
| Recombinant Human IL-4 Protein | R&D systems | Cat# 204-IL-050 |
| Pam3CSK4 | InvivoGen | Cat# tlrl-pms |
| LPS | InvivoGen | Cat# tlrl-3pelps |
| Imiquimod (R837) | InvivoGen | Cat# tlrl-imqs |
| R848 (Resiquimod) | InvivoGen | Cat# tlrl-r848 |
| TL8-506 | InvivoGen | Cat# tlrl-tl8506 |
| RPMI 1640 | HyClone | Cat# SH30027.01 |
| Penicillin-Streptomycin | HyClone | Cat# SV30010 |
| Sodium Pyruvate | HyClone | Cat# SH30239.01 |
| HEPES | HyClone | Cat# SH30237.01 |
| L-Glutamine | Sigma-Aldrich | Cat# G7513-100ml |
| FBS | HyClone | Cat# SH30070.03 |
| Ficoll | GE Healthcare | Cat# 17-1440-03 |
| TRIzol Reagent | Thermo Fisher Scientific | Cat# 15596018 |
| IGEPAL CA-630 | Sigma-Aldrich | I8896-50ML |
| Critical commercial assays | ||
| EasySep Human Monocyte Isolation Kit | STEMCELL technologies | Cat# 19359 |
| SuperScript III First-Strand Synthesis System | Thermo Fisher Scientific | Cat# 18080051 |
| RNeasy Mini Kit | QIAGEN | Cat# 74106 |
| Human IL-1 beta/IL-1F2 DuoSet ELISA | R&D systems | Cat# DY201-05 |
| Human IL-6 DuoSet ELISA | R&D systems | Cat# DY206-05 |
| Human TNF-alpha DuoSet ELISA | R&D systems | Cat# DY210 |
| Illumina TruSeq RNA Sample Preparation kit | Illumina | Cat# FC-122-1001 |
| MinElute PCR Purification Kit | QIAGEN | Cat# 28004 |
| Nextera DNA Library Preparation Kit | Illumina | Cat# FC-121-1031 |
| Deposited data | ||
| RNA-seq and ATAC-seq data | This Paper | GEO: GSE147314 |
| Experimental models: cell lines | ||
| THP-1 | ATCC | ATCC Cat# TIB-202, RRID:CVCL_0006 |
| Oligonucleotides | ||
| Primers for RT-qPCR | See method details | N/A |
| Recombinant DNA | ||
| psPAX2 | Provided by Dr. Neal Alto | N/A |
| pVSV-G | Provided by Dr. Neal Alto | N/A |
| IRF1 gRNA lentiCRISPRv2 (gRNA sequence: GCTCAGCTGTGCGAGTGTAC) | Provided by Dr. Daniel Stetson | N/A |
| Software and algorithms | ||
| GraphPad Prism | GraphPad Software, Inc. | N/A |
| CLC Genomics Workbench 7 | QIAGEN | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-clc-genomics-workbench/ |
| MATLAB | Mathworks | https://www.mathworks.com/products/matlab.html |
| Ingenuity Pathway Analysis | QIAGEN | https://wwwqiagenbioinformatics.com |
| BEDTools version 2.26.0 | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| MACS version 2.1.0 | Feng et al., 2012 | https://github.com/macs3-project/MACS |
| MAnorm software package | Shao et al., 2012 | https://pypi.org/project/MAnorm/1.0.1/ |
| HOMER | Heinz et al., 2010 | http://homer.ucsd.edu/homer/download.html |
| TrimGalore version 0.4.1 | the Babraham Institute | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
Highlights.
Human myeloid cells show lineage-specific transcriptional responses to TLR activation
Monocytes display an exaggerated pro-inflammatory profile in response to TLR8 ligand
TLR4 engagement elicits robust ISG response in macrophages but not in monocytes
IRF1 governs chromatin accessibility at ISG loci in TLR4-stimulated macrophages
ACKNOWLEDGMENTS
We thank the members of the Genomics and Microarray Core at UT Southwestern Medical Center for their help with the RNA sequencing experiments and analysis. Daniel Stetson, PhD, and Michael Gale, Jr., PhD, at the University of Washington kindly provided lentiviral CRISPR constructs targeting IRF1. Neal Alto, PhD, kindly provided the lentiviral packaging constructs. C.P. is supported by grants from the National Institutes of Health (AI113125, GM120196, and AI123176). E.K.W. is supported by the Edwin L. Cox Distinguished Chair in Immunology and Genetics Bioinformatics Core Facility (BICF) at UT Southwestern Medical Center, and V.M. is supported by the Cancer Prevention and Research Institute of Texas (RP150596). M.T.W. is supported by the National Institutes of Health (R01 HG010730, R01 NS099068, R01 AR073228, and R01 GM055479) and a Cincinnati Children’s Hospital Research Foundation Endowed Scholar Award.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.108891.
REFERENCES
- Akira S, and Takeda K (2004). Toll-like receptor signalling. Nat. Rev. Immunol 4, 499–511. [DOI] [PubMed] [Google Scholar]
- Amit I, Regev A, and Hacohen N (2011). Strategies to discover regulatory circuits of the mammalian immune system. Nat. Rev. Immunol 11, 873–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbalat R, Ewald SE, Mouchess ML, and Barton GM (2011). Nucleic acid recognition by the innate immune system. Annu. Rev. Immunol 29, 185–214. [DOI] [PubMed] [Google Scholar]
- Barton GM, and Kagan JC (2009). A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nat. Rev. Immunol 9, 535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell O, Tiwari VK, Thomä NH, and Schübeler D (2011). Determinants and dynamics of genome accessibility. Nat. Rev. Genet 12, 554–564. [DOI] [PubMed] [Google Scholar]
- Bornstein C, Winter D, Barnett-Itzhaki Z, David E, Kadri S, Garber M, and Amit I (2014). A negative feedback loop of transcription factors specifies alternative dendritic cell chromatin States. Mol. Cell 56, 749–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher SK, O’Carroll CE, Wells CA, and Carmody RJ (2018). Toll-Like Receptors Drive Specific Patterns of Tolerance and Training on Restimulation of Macrophages. Front. Immunol 9, 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanput W, Mes JJ, and Wichers HJ (2014). THP-1 cell line: an in vitro cell model for immune modulation approach. Int. Immunopharmacol 23, 37–45. [DOI] [PubMed] [Google Scholar]
- Cheng Q, Behzadi F, Sen S, Ohta S, Spreafico R, Teles R, Modlin RL, and Hoffmann A (2019). Sequential conditioning-stimulation reveals distinct gene- and stimulus-specific effects of Type I and II IFN on human macrophage functions. Sci. Rep 9, 5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SE, Noyce RS, and Mossman KL (2004). Innate cellular response to virus particle entry requires IRF3 but not virus replication. J. Virol 78, 1706–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comoglio F, Simonatto M, Polletti S, Liu X, Smale ST, Barozzi I, and Natoli G (2019). Dissection of acute stimulus-inducible nucleosome remodeling in mammalian cells. Genes Dev. 33, 1159–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigneault M, Preston JA, Marriott HM, Whyte MK, and Dockrell DH (2010). The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS ONE 5, e8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Marcken M, Dhaliwal K, Danielsen AC, Gautron AS, and Dominguez-Villar M (2019). TLR7 and TLR8 activate distinct pathways in monocytes during RNA virus infection. Sci. Signal 12, eaaw1347. [DOI] [PubMed] [Google Scholar]
- Doyle S, Vaidya S, O’Connell R, Dadgostar H, Dempsey P, Wu T, Rao G, Sun R, Haberland M, Modlin R, and Cheng G (2002). IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity 17, 251–263. [DOI] [PubMed] [Google Scholar]
- Dozmorov I, and Centola M (2003). An associative analysis of gene expression array data. Bioinformatics 19, 204–211. [DOI] [PubMed] [Google Scholar]
- Dozmorov I, and Lefkovits I (2009). Internal standard-based analysis of microarray data. Part 1: analysis of differential gene expressions. Nucleic Acids Res. 37, 6323–6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dozmorov I, Saban MR, Knowlton N, Centola M, and Saban R (2003). Connective molecular pathways of experimental bladder inflammation. Physiol. Genomics 15, 209–222. [DOI] [PubMed] [Google Scholar]
- Dozmorov I, Knowlton N, Tang Y, Shields A, Pathipvanich P, Jarvis JN, and Centola M (2004). Hypervariable genes–experimental error or hidden dynamics. Nucleic Acids Res. 32, e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dozmorov IM, Jarvis J, Saban R, Benbrook DM, Wakeland E, Aksentijevich I, Ryan J, Chiorazzi N, Guthridge JM, Drewe E, et al. (2011). Internal standard-based analysis of microarray data2–analysis of functional associations between HVE-genes. Nucleic Acids Res. 39, 7881–7899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Liu T, Qin B, Zhang Y, and Liu XS (2012). Identifying ChIP-seq enrichment using MACS. Nat. Protoc 7, 1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster SL, Hargreaves DC, and Medzhitov R (2007). Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 447, 972–978. [DOI] [PubMed] [Google Scholar]
- Freund EC, Lock JY, Oh J, Maculins T, Delamarre L, Bohlen CJ, Haley B, and Murthy A (2020). Efficient gene knockout in primary human and murine myeloid cells by non-viral delivery of CRISPR-Cas9. J. Exp. Med 217, e20191692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita T, Kimura Y, Miyamoto M, Barsoumian EL, and Taniguchi T (1989). Induction of endogenous IFN-alpha and IFN-beta genes by a regulatory transcription factor, IRF-1. Nature 337, 270–272. [DOI] [PubMed] [Google Scholar]
- Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, and Ley K (2010). Development of monocytes, macrophages, and dendritic cells. Science 327, 656–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, and Jung S (2014). Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat. Rev. Immunol 14, 392–404. [DOI] [PubMed] [Google Scholar]
- Glass CK, and Natoli G (2016). Molecular control of activation and priming in macrophages. Nat. Immunol 17, 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez Rial J, Curras Tuala MJ, Rivero Calle I, Gomez Carballa A, Cebey Lopez M, Rodriguez Tenreiro C, Dacosta Urbieta A, Rivero Velasco C, Rodriguez Nunez N, Trastoy Pena R, et al. (2020). Increased serum levels of sCD14 and sCD163 indicate a preponderant role for monocytes in COVID-19 immunopathology. Front Immunol. 11, 560381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorden KB, Gorski KS, Gibson SJ, Kedl RM, Kieper WC, Qiu X, Tomai MA, Alkan SS, and Vasilakos JP (2005). Synthetic TLR agonists reveal functional differences between human TLR7 and TLR8. J. Immunol 174, 1259–1268. [DOI] [PubMed] [Google Scholar]
- Grandvaux N, Servant MJ, tenOever B, Sen GC, Balachandran S, Barber GN, Lin R, and Hiscott J (2002). Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J. Virol 76, 5532–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves DC, Horng T, and Medzhitov R (2009). Control of inducible gene expression by signal-dependent transcriptional elongation. Cell 138, 129–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley JB, Chen X, Pujato M, Miller D, Maddox A, Forney C, Magnusen AF, Lynch A, Chetal K, Yukawa M, et al. (2018). Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity. Nat. Genet 50, 699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan M, Koch J, Rakheja D, Pattnaik AK, Brugarolas J, Dozmorov I, Levine B, Wakeland EK, Lee-Kirsch MA, and Yan N (2013). Trex1 regulates lysosomal biogenesis and interferon-independent activation of antiviral genes. Nat. Immunol 14, 61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Takaoka A, and Taniguchi T (2006). Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25, 349–360. [DOI] [PubMed] [Google Scholar]
- Hu W, Jain A, Gao Y, Dozmorov IM, Mandraju R, Wakeland EK, and Pasare C (2015). Differential outcome of TRIF-mediated signaling in TLR4 and TLR3 induced DC maturation. Proc. Natl. Acad. Sci. USA 112, 13994–13999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivashkiv LB (2013). Epigenetic regulation of macrophage polarization and function. Trends Immunol. 34, 216–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A, and Medzhitov R (2015). Control of adaptive immunity by the innate immune system. Nat. Immunol 16, 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis JN, Dozmorov I, Jiang K, Frank MB, Szodoray P, Alex P, and Centola M (2004). Novel approaches to gene expression analysis of active polyarticular juvenile rheumatoid arthritis. Arthritis Res. Ther 6, R15–R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karwacz K, Miraldi ER, Pokrovskii M, Madi A, Yosef N, Wortman I, Chen X, Watters A, Carriero N, Awasthi A, et al. (2017). Critical role of IRF1 and BATF in forming chromatin landscape during type 1 regulatory cell differentiation. Nat. Immunol 18, 412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki T, and Kawai T (2014). Toll-like receptor signaling pathways. Front. Immunol 5, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. (2012). ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 22, 1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BL, and Barton GM (2014). Trafficking of endosomal Toll-like receptors. Trends Cell Biol. 24, 360–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KH, and Staudt LM (2013). Toll-like receptor signaling. Cold Spring Harb. Perspect. Biol 5, a011247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Berthier CC, and Kahlenberg JM (2017). Enhanced Inflammasome Activity in Systemic Lupus Erythematosus Is Mediated via Type I Interferon-Induced Up-Regulation of Interferon Regulatory Factor 1. Arthritis Rheumatol. 69, 1840–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama T, Kimura T, Kitagawa M, Pfeffer K, Kawakami T, Watanabe N, Kündig TM, Amakawa R, Kishihara K, Wakeham A, et al. (1993). Targeted disruption of IRF-1 or IRF-2 results in abnormal type I IFN gene induction and aberrant lymphocyte development. Cell 75, 83–97. [PubMed] [Google Scholar]
- Mayran A, Sochodolsky K, Khetchoumian K, Harris J, Gauthier Y, Bemmo A, Balsalobre A, and Drouin J (2019). Pioneer and nonpioneer factor cooperation drives lineage specific chromatin opening. Nat. Commun 10, 3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGettrick AF, and O’Neill LA (2010). Localisation and trafficking of Toll-like receptors: an important mode of regulation. Curr. Opin. Immunol 22, 20–27. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, and Horng T (2009). Transcriptional control of the inflammatory response. Nat. Rev. Immunol 9, 692–703. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Preston-Hurlburt P, and Janeway CA Jr. (1997). A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388, 394–397. [DOI] [PubMed] [Google Scholar]
- Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, and Manson JJ; HLH Across Speciality Collaboration, UK (2020). COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395, 1033–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen TH (2019). IRF and STAT Transcription Factors - From Basic Biology to Roles in Infection, Protective Immunity, and Primary Immunodeficiencies. Front. Immunol 9, 3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monticelli S, and Natoli G (2017). Transcriptional determination and functional specificity of myeloid cells: making sense of diversity. Nat. Rev. Immunol 17, 595–607. [DOI] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, and Wold B (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. [DOI] [PubMed] [Google Scholar]
- Natoli G, Ghisletti S, and Barozzi I (2011). The genomic landscapes of inflammation. Genes Dev. 25, 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negishi H, Taniguchi T, and Yanai H (2018). The Interferon (IFN) Class of Cytokines and the IFN Regulatory Factor (IRF) Transcription Factor Family. Cold Spring Harb. Perspect. Biol 10, a028423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, et al. (2010). Suppression of inflammation by a synthetic histone mimic. Nature 468, 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda D, Gjinaj E, Bachu M, Squire E, Novatt H, Ozato K, and Rabin RL (2019). IRF1 Maintains Optimal Constitutive Expression of Antiviral Genes and Regulates the Early Antiviral Response. Front. Immunol 10, 1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EK, Jung HS, Yang HI, Yoo MC, Kim C, and Kim KS (2007). Optimized THP-1 differentiation is required for the detection of responses to weak stimuli. Inflamm. Res 56, 45–50. [DOI] [PubMed] [Google Scholar]
- Pasare C, and Medzhitov R (2004). Toll-like receptors: linking innate and adaptive immunity. Microbes Infect. 6, 1382–1387. [DOI] [PubMed] [Google Scholar]
- Platanitis E, and Decker T (2018). Regulatory Networks Involving STATs, IRFs, and NFκB in Inflammation. Front. Immunol 9, 2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z (2012). The use of THP-1 cells as a model for mimicking the function and regulation of monocytes and macrophages in the vasculature. Atherosclerosis 221, 2–11. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raccaud M, Friman ET, Alber AB, Agarwal H, Deluz C, Kuhn T, Gebhardt JCM, and Suter DM (2019). Mitotic chromosome binding predicts transcription factor properties in interphase. Nat. Commun 10, 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44 (W1), W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Carrozzi VR, Braas D, Bhatt DM, Cheng CS, Hong C, Doty KR, Black JC, Hoffmann A, Carey M, and Smale ST (2009). A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell 138, 114–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolph GJ, Inaba K, Robbiani DF, Steinman RM, and Muller WA (1999). Differentiation of phagocytic monocytes into lymph node dendritic cells in vivo. Immunity 11, 753–761. [DOI] [PubMed] [Google Scholar]
- Reizis B, Bunin A, Ghosh HS, Lewis KL, and Sisirak V (2011). Plasmacytoid dendritic cells: recent progress and open questions. Annu. Rev. Immunol 29, 163–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, Wu X, Albring JC, and Murphy KM (2012). Re(de)fining the dendritic cell lineage. Nat. Immunol 13, 1145–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, and Rice CM (2011). A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z, Zhang Y, Yuan GC, Orkin SH, and Waxman DJ (2012). MAnorm: a robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol. 13, R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, and Pamer EG (2011). Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol 11, 762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smale ST (2010). Selective transcription in response to an inflammatory stimulus. Cell 140, 833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smale ST (2012). Transcriptional regulation in the innate immune system. Curr. Opin. Immunol 24, 51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smale ST, and Fisher AG (2002). Chromatin structure and gene regulation in the immune system. Annu. Rev. Immunol 20, 427–462. [DOI] [PubMed] [Google Scholar]
- Smale ST, Tarakhovsky A, and Natoli G (2014). Chromatin contributions to the regulation of innate immunity. Annu. Rev. Immunol 32, 489–511. [DOI] [PubMed] [Google Scholar]
- Struhl K, and Segal E (2013). Determinants of nucleosome positioning. Nat. Struct. Mol. Biol 20, 267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, and Akira S (2010). Pattern recognition receptors and inflammation. Cell 140, 805–820. [DOI] [PubMed] [Google Scholar]
- Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, and Prins P (2015). Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanos D, and Maniatis T (1995). Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell 83, 1091–1100. [DOI] [PubMed] [Google Scholar]
- Tong AJ, Liu X, Thomas BJ, Lissner MM, Baker MR, Senagolage MD, Allred AL, Barish GD, and Smale ST (2016). A Stringent Systems Approach Uncovers Gene-Specific Mechanisms Regulating Inflammation. Cell 165, 165–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varol C, Yona S, and Jung S (2009). Origins and tissue-context-dependent fates of blood monocytes. Immunol. Cell Biol 87, 30–38. [DOI] [PubMed] [Google Scholar]
- Venkatesh D, Ernandez T, Rosetti F, Batal I, Cullere X, Luscinskas FW, Zhang Y, Stavrakis G, García-Carderña G, Horwitz BH, and Mayadas TN (2013). Endothelial TNF receptor 2 induces IRF1 transcription factor-dependent interferon-β autocrine signaling to promote monocyte recruitment. Immunity 38, 1025–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wathelet MG, Berr PM, and Huez GA (1992). Regulation of gene expression by cytokines and virus in human cells lacking the type-I interferon locus. Eur. J. Biochem 206, 901–910. [DOI] [PubMed] [Google Scholar]
- Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM, and Maniatis T (1998). Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Mol. Cell 1, 507–518. [DOI] [PubMed] [Google Scholar]
- Weaver BK, Kumar KP, and Reich NC (1998). Interferon regulatory factor 3 and CREB-binding protein/p300 are subunits of double-stranded RNA-activated transcription factor DRAF1. Mol. Cell. Biol 18, 1359–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, et al. (2014). Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter DR, and Amit I (2014). The role of chromatin dynamics in immune cell development. Immunol. Rev 261, 9–22. [DOI] [PubMed] [Google Scholar]
- Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, et al. (2014). Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40, 274–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarilina A, Park-Min K-H, Antoniv T, Hu X, and Ivashkiv LB (2008). TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat. Immunol 9, 378–387. [DOI] [PubMed] [Google Scholar]
- Ye Q, Wang B, and Mao J (2020). The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect 80, 607–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, and Fujita T (1998). Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 17, 1087–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Lareau CA, Ramirez RN, Rose SA, Maier B, Wroblewska A, Desland F, Chudnovskiy A, Mortha A, Dominguez C, et al. ; Immunological Genome Project (2019). The cis-Regulatory Atlas of the Mouse Immune System. Cell 176, 897–912.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Shi L, Song L, Ephrem E, Petri M, and Sullivan KE (2015). Interferon regulatory factor 1 marks activated genes and can induce target gene expression in systemic lupus erythematosus. Arthritis Rheumatol. 67, 785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Wu Z, Li JW, Zhao H, and Wang GQ (2020a). Cytokine release syndrome in severe COVID-19: interleukin-6 receptor antagonist tocilizumab may be the key to reduce mortality. Int. J. Antimicrob. Agents 55, 105954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Guo R, Lei L, Liu H, Wang Y, Wang Y, Dai T, Zhang T, Lai Y, Wang J, et al. (2020b). COVID-19 infection induces readily detectable morphological and inflammation-related phenotypic changes in peripheral blood monocytes, the severity of which correlate with patient outcome. medRxiv, 2020.2003.2024.20042655. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
MATLAB code used for RNA-seq analysis is described in RNA-seq analysis and is available on request. Illustrations were created with Biorender (https://biorender.com). RNA-seq and ATAC-seq data for this study have been deposited at NCBI’s Gene Expression Omnibus (GEO). The acession number is GEO: GSE147314.