

Abstract
Both the development of the mammalian innate immune system and the antagonistic strategies acquired by alphaherpesviruses to dismantle it have been shaped by co-evolving virus-host interactions over millions of years. Here, we review mechanisms employed by mammalian cells to detect pathogen molecules, such as viral glycoproteins and nucleic acids, and induce innate immune signaling upon infection with alphaherpesviruses. We further explore strategies acquired by these viruses to bypass immune detection and activation, thereby supporting virus replication and spread. Finally, we discuss the contributions of advanced “omics” and microscopy methods to these discoveries in immune signaling and highlight emerging technologies that can help to further our understanding of the dynamic interplay between host innate immune responses and virus immune evasion.
Introduction
Mammalian innate immunity has been shaped by ancestral and continuous challenges from viruses for millions of years. Phylogenetic analyses have predicted that the alphaherpesvirinae subfamily of herpesviruses arose approximately 180 to 220 million years ago, and that mammalian herpesviruses have been cospeciating with hosts as far back as placental mammals have existed (McGeoch and Cook, 1994; McGeoch et al., 1995). The capacity of mammalian cells to induce immune responses against human alphaherpesviruses, including herpes simplex virus types 1 and 2 (HSV-1, HSV-2) and Varicella-Zoster virus (VZV), is a critical arm of host defense. HSV initiates primary infection via local inoculation of mucocutaneous tissue or oral mucosa, causing localized lesions, while VZV infects upper respiratory mucosa that results in varicella (Arvin, 1996; Karasneh and Shukla, 2011). Upon mucosal cell entry, VZV interacts locally with immune cells, which traffic to the skin and clinically manifest as vesicular skin lesions. Exemplifying their success as pathogens, a defining attribute of alphaherpesviruses is their ability to latently persist in sensory neurons of the nervous system. Thus, not only adaptive immune responses, but also innate immune responses within differentiated cells of the skin and epithelium, are critical for controlling both primary and reactivated alphaherpesvirus infections.
The last decade has provided significant advances in the identification and characterization of host restriction factors that drive cellular innate immunity during infection. Indispensable components that initiate immune signal transduction are specialized pattern recognition receptors (PRR) that survey intracellular spaces for pathogen-associated molecular patterns (PAMP). Alphaherpesvirus glycoproteins and nucleic acids are two PAMP classes that elicit immune signaling from the cell surface or intracellularly, respectively. Viral double-stranded DNA (dsDNA) or dsRNA intermediates are recognized by PRRs in multiple subcellular compartments, including endosomes, the cytosol, and the nucleus. These recognition events signal through adaptor molecules in a PRR-dependent manner and lead to the transcription of antiviral interferons (IFN) and other cytokines. IFN expression further amplifies the transcription of additional IFN molecules and interferon-stimulated genes (ISG) in both the host and neighboring cells (Platanias, 2005). The activated ISGs represent several hundred genes that regulate a diverse set of immune modulatory functions, prime cells for pathogen detection, arrest the cell cycle, or inhibit stages of the viral life cycle (Schneider et al., 2014).
Concurrently, the establishment of a productive viral infection relies on the ability of the virus to evade these IFN- and cytokine-stimulatory programs. Indeed, despite the sophisticated innate immune arsenal, human alphaherpesviruses maintain a close relationship with humans throughout the lifetime of the host via latent persistence in the central nervous system. To accomplish this, alphapherpesviruses have undergone successive cycles of adaptation with mammalian cells, acquiring effective mechanisms that use virus-host interactions to dismantle the host defense repertoire.
Here, we review the current understanding of the host innate immune response to alphaherpesvirus infection. Specifically, we discuss the central host PRRs that detect HSV and VZV components, and the mechanisms underlying immune signaling cascades. We further explore the strategies evolved by these viruses to inhibit innate immune responses by either limiting the production or the downstream signal transduction of ISGs. In closing, we examine how technological advances in omics and microscopy have contributed to these findings and highlight the value of emerging technologies that promise to help further uncover the complex relationship between alphaherpesviruses and mammalian cells.
Innate immunity against alphaherpesviruses
Thus far, we understand that the mammalian innate immune system has established capabilities to detect two classes of alphaherpesvirus PAMPs - glycoproteins embedded in the lipid envelopes of virion particles and intracellular virus-derived nucleic acids (dsDNA and RNA). Both PAMP classes present challenges to host cells because, unlike bacterial and fungal pathogens that contain pathogen-specific biochemical signatures in their cell walls, viruses lack conserved physical characteristics across virus types (Kawai and Akira, 2006; Medzhitov, 2007; Barbalat et al., 2009). Sensing viral glycoproteins may appear inefficient when accounting for both the absence of consistent protein homology across viruses and the potential of viruses to evade immune detection via mutations that maintain protein function(s). Despite this, several PRRs have been identified that demonstrate viral glycoprotein recognition and innate immune activation. Similarly, considering the essentiality of the viral DNA and RNA intermediates for viral gene expression and replication, the ability of the immune system to target pathogenic nucleic acids seems strategically effective. However, the likeness of viral and host DNA molecules led to the initial thinking that viral DNA can only be sensed in subcellular compartments devoid of human DNA.
Indeed, PRRs that sense alphaherpesvirus DNA have been identified in endolysosomes and the cytoplasm. One conundrum of this DNA sensing mechanism is that the virus DNA is protected by the capsid following virus entry into the cell and its trafficking towards the nucleus. A possible answer to this puzzle was given by the observation that the integrity of the capsid can be compromised during infection by proteasomal degradation in certain cell types, such as macrophages, thereby resulting in viral genome leakage and sensing of viral DNA within the cytoplasm (Horan et al., 2013a). Additionally, in recent years it was further recognized that mammalian cells have the ability to sense viral DNA in the nuclei of infected cells. In this section, we review the broad classes of innate immune sensing of alphaherpesviruses by the aforementioned PAMPs and place this knowledge in the context of the subcellular locations for PAMP detection.
Cell surface glycoprotein detection
At the plasma membrane, mammalian cells are equipped with Toll-like receptors (TLR), transmembrane PRRs that have been shown to recognize a variety of microbial membrane components, including lipids, proteins (e.g., lipoproteins, glycoproteins, flagellin), polysaccharides, and other glycans (Lester and Li, 2014). Specific to alphaherpesviruses, TLR detection of viral glycoproteins has been shown for HSV-1, VZV, and certain HSV-2 strains (Kurt-Jones et al., 2004; Wang et al., 2005; Sato et al., 2006). TLR2 is known to form heterodimers in vivo with either TLR1 or TLR6, enabling extracellular binding of ligands (Figure 1, step 1a) (Lien et al., 1999; Hajjar et al., 2001). Ligand-bound TLR2 heterodimers initiate immune signal transduction via signaling domains that project into the cytosol and recruit the adaptor protein MyD88. MyD88 activates the cytoplasmic-to-nuclear translocation of several transcription factors, including NF-κB, IRF3, and IRF7. In non-immune stimulated cells, NF-κB remains in an inhibited state via association with IκB. To initiate NF-κB translocation, MyD88 facilitates the activation of the IκB kinase (IKK) complex, which mediates the phosphorylation and subsequent proteasomal degradation of the NF-κB inhibitor, IκB. Via its death domain, MyD88 further recruits a set of interleukin (IL)-1 receptor-associated kinases (IRAK)-1, IRAK-2, and IRAK-4, IRF7, and the E3 ubiquitin ligase TRAF6 (Takeda et al., 2003). The recruited complex both phosphorylates IRF7, thereby activating its translocation to the nucleus, as well as activates the TANK-binding kinase 1 (TBK-1), which subsequently also phosphorylates IRF3. These transcription factors are responsible for inducing the expression of type I IFNs (IFNα, IFNβ) and pro-inflammatory cytokines (IL-6, IL-8, IL-12) (West et al., 2012) (Figure 1).
Figure 1. Mechanisms of innate immune signaling during alphaherpesvirus infection.
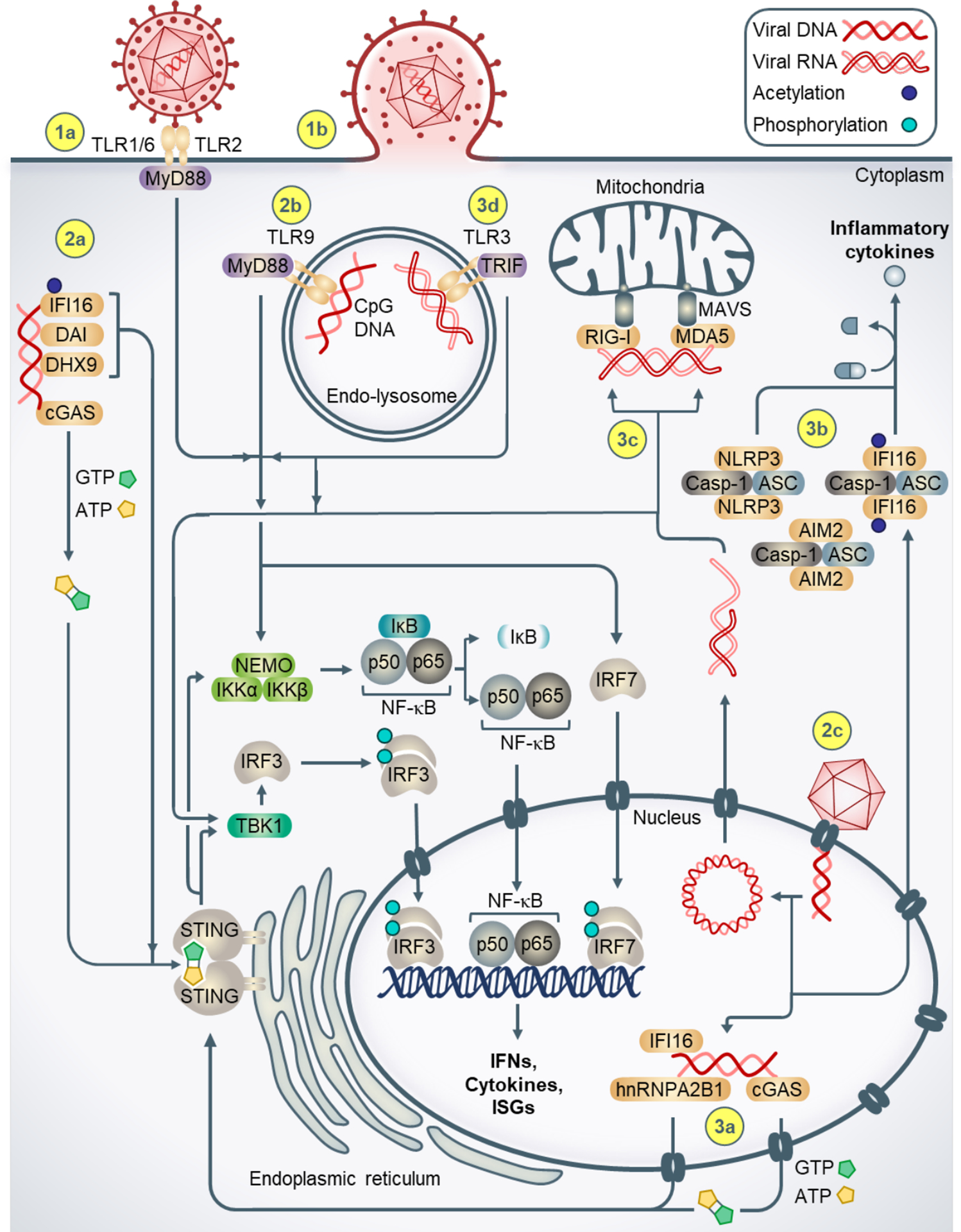
(1a) TLR2 forms heterodimers with TLR1/6, binds extracellular viral glycoproteins, and intracellularly recruits the adaptor protein MyD88. MyD88 activates the transcription factors NF-κB, IRF3, IRF7, which upregulate type I IFNs, cytokines, and ISGs. (1b) Virion fusion with the plasma membrane facilitates entry of the viral nucleocapsid and tegument proteins. (2a) Cytosolic DNA sensors bind viral DNA genomes and signal to STING at the ER. Activated cGAS generates cGAMP, which binds STING. Activated STING induces immune signaling via TBK-1 and the IKK complex, resulting in cytoplasmic to nuclear translocation of IRF3 and NF-κB and induction of type I IFNs, cytokines, and ISGs. (2b) Endosomal detection of CpG-rich viral DNA occurs via TLR9, which also recruits MyD88 to induce immune signaling. (2c) Viral nucleocapsids docking at the nuclear pore eject dsDNA genomes into the nucleus, (3a) facilitating DNA sensing by nuclear IFI16, hnRNPA2B1, and cGAS. (3b) Viral DNA-bound IFI16, NLRP3, and AIM2 inflammasomes in the cytoplasm generate pro-inflammatory cytokines. (3c) RNA intermediates generated during viral replication are sensed by RIG-I and MDA5 in the cytoplasm. (3d) Endosomal RNA is sensed by TLR3, which recruits the adaptor TRIF to activate downstream transcription factors.
TLR sensing of alphaherpesviral glycoprotein was first demonstrated during HSV-1 infection (Kurt-Jones et al., 2004). In HEK 293 cells transfected with TLR2, IL-6 expression increased significantly upon HSV-1 infection in a NF-κB-dependent manner, whereas infected TLR2−/− mice demonstrated attenuated IL-6 expression compared to wild-type (WT) or TLR4−/− mice. Surprisingly, the authors additionally observed that TLR2 activity may not necessarily confer a global protective state in vivo. Specifically, infected TLR2−/− mice exhibited lower severity of viral encephalitis, suggesting that TLR2 expression contributes to lethal HSV-1-induced encephalitis. Supporting a mechanism of TLR2 heterodimerization in immune signaling upon alphaherpesviral infection, TLR2 and TLR6, and to a lesser extent TLR2 and TLR1, were shown to contribute to cytokine induction in HEK 293T cells infected with clinical and laboratory isolates of TLR2-activating HSV-1 (Sato et al., 2006).
It was later recognized that virions from WT or ultraviolet (UV)-inactivated HSV-2 similarly induced cytokines (IL-6 and IL12p40) and IFNα/β in mouse-derived dendritic cells via a TLR2-dependent mechanism (Sorensen et al., 2008). This finding solidified that TLR2 immune signaling did not require active virus replication or gene expression. Yet until more recently, the alphaherpesviral components recognized by TLR2 remained unknown. HSV-1 glycoproteins gH/gL and gB, which are conserved across all human herpesviruses and are essential components for virus attachment and entry into permissive cells (Reske et al., 2007), were identified as independent TLR2 ligands (Leoni et al., 2012; Cai et al., 2013). While gH/gL was found sufficient for activating TLR2-mediated immunity via NF-κB, the activation by gB remains less certain and warrants future in vivo investigation. Thus far the reports have been divided, as Leoni et al. did not observe expression of NF-κB-responsive genes in HEK 293T cells transfected with TLR2 and a NF-κB luciferase reporter plasmid. In opposition, Cai et al. observed an effect when using a similar transfection method and cells. Additionally, gB was found to co-immunoprecipitate not only with TLR2, but also with TLR1 and TLR6 (Cai et al., 2013).
Similar to sensing HSV-1 and HSV-2, TLR2 also recognizes VZV virion components and induces immune signaling. VZV infection of human monocyte-derived macrophages induced IL-6 expression in a TLR2- and NF-κB-dependent manner (Wang et al., 2005). Wang et al. determined that CD14, an accessory molecule and common co-receptor of TLR2, was also required to elicit cytokine expression. It was subsequently determined by Cai, et al. that TLR2 sensing during HSV-1 infection did not necessitate CD14, indicating differences between immune activation by alphaherpesviruses (Cai et al., 2013). Further emphasizing the divergence of TLR2 immune responses between HSV and VZV infections, no NF-κB activation was observed in mouse cell lines infected with VZV (Wang et al., 2005). However, human cells expressing murine TLR2 were capable of activating NF-κB, suggesting that mouse cells either lack TLR2-specific adaptor molecule(s) upstream of NF-κB, or VZV has evolved a species-specific strategy to subvert immune signaling in mouse cells.
Endolysosomal DNA sensing
Aside from cell surface TLRs that sense alphaherpesviral glycoproteins, other PRRs function intracellularly to sense viral nucleic acids once virion particles have entered into cells. The first PRR discovered to detect herpesviral DNA was TLR9, which is localized to endolysosomes, and contains ligand-binding motifs that face the endolysosomal lumen (Figure 1, step 2b) (Lund et al., 2003; Roers et al., 2016). Based on experiments using cells derived from TLR9−/− mice, as well as treatments with UV-inactivated virus, inhibitory CpG oligonucleotides, and inhibitors of endosome acidification, TLR9 was proposed to upregulate IFNα expression during HSV-2 infection in a MyD88-dependent fashion. This suggested that TLR9 senses CpG-rich herpesviral DNA from virion particles that have entered mammalian cells via endocytosis and traversed the cytoplasm through the endocytic pathway. Consistent with these findings, in comparison to beta- and gamma-herpesviruses and adenoviruses, alphaherpesviruses (HSV-1, HSV-2, VZV, and bovine herpesvirus-1) were found to contain the highest number of CpG hexamer motifs, when normalized to the respective genome sizes (Lundberg et al., 2003). It was later clarified that TLR9 induced IFN expression by recruiting MyD88 and subsequently activating NF-κB and IRF7 (Kawai and Akira, 2011). In plasmacytoid dendritic cells infected with VZV, introduction of inhibitory CpG oligodeoxynucleotides suppressed IFNα levels, indicating that TLR9 contributed to VZV-induced IFNα expression (Yu et al., 2011). Yet, Yu et al. also showed that cells treated with UV-inactivated VZV induced lower IFNα levels than cells treated with WT VZV, further suggesting that additional TLR9-independent innate immune signaling exists during VZV infection. Indeed, the concurrent characterization of nucleic acid sensing pathways localized outside of the endolysosomal compartment have started to build a view of the complex immune response mechanisms against alphaherpesviruses.
Cytosolic DNA sensing
Over the past decade, seminal discoveries have been made in clarifying the immune signaling pathways underlying cytosolic detection of alphaherpesviral DNA genomes. We now understand that several DNA sensors exist in the cytosol (Figure 1, step 2a), including cyclic GMP-AMP synthase (cGAS, also known as C6ORF150 and Mab-21 Domain Containing 1 (MB21D1)), interferon-inducible protein 16 (IFI16), DEAD-box helicase 41 (DDX41), RNA polymerase III (RNAP III), absent in melanoma 2 (AIM2), DHX9, DHX36, leucine-rich repeat flightless-interacting protein 1 (LRRFIP1), DNA-dependent activator of IFN-regulatory factors (DAI), and DNA-dependent protein kinase (DNA-PK). As some of these sensors have since been identified to function redundantly, emphasis will be placed on describing recently characterized sensors and pathways that strongly induce innate immune responses during alphaherpesviral infection.
Historically, the central immune signaling axis that is activated by viral DNA detection was characterized prior to the discovery of the DNA sensors. This axis is comprised of the endoplasmic reticulum adapter protein STING (also known as ERIS, MITA, and TMEM173), TBK-1, and IRF3 (Figure 1). Initially, several human and murine cDNA screening studies identified STING as a potent inducer of IFNs and ISGs (Ishikawa and Barber, 2008; Zhong et al., 2008; Sun et al., 2009). STING was subsequently implicated in immune signaling when STING−/− mice were shown to exhibit greater sensitivity to HSV-1 infection (Ishikawa et al., 2009; Parker et al., 2015). It has since been demonstrated that, upon its activation, STING dimerizes and traffics from the ER to both an intermediate ER-Golgi compartment and the Golgi apparatus (Saitoh et al., 2009; Ishikawa et al., 2009; Dobbs et al., 2015). Activated STING thereafter recruits TBK-1, facilitating TBK-1 phosphorylation and subsequent phosphorylation and mobilization of IRF3 to the nucleus, where it binds to consensus interferon-stimulated response elements (Pomerantz and Baltimore, 1999; Tanaka and Chen, 2012). Additionally, STING activates IKK, relieving NF-κB inhibition and facilitating its nuclear translocation for transcriptional activity (Ishikawa and Barber, 2008).
Characterization of the STING-TBK-1-IRF3 immune signaling axis fueled efforts to identify the cytosolic DNA sensor(s) that activate this pathway during viral infection. Specific to sensing alphaherpesvirus DNA, not long after the elucidation of this axis, IFI16 was shown to bind to cytosolic viral DNA, associate with STING, and contribute to IFN production in monocyte-derived THP-1 macrophages (Unterholzner et al., 2010). Upon HSV-1 infection, knockdown of IFI16 or the mouse ortholog p204 in macrophage cells resulted in loss of IRF3 and NF-κB activation, as well as the accompanying IFN and ISG induction (Unterholzner et al., 2010). The localization of IFI16 to the cytoplasm or nucleus was found to be impacted by acetylation within a multi-partite nuclear localization signal (NLS) between its oligomerizing pyrin and DNA-binding HIN200 domains, whereupon acetylation retained cytoplasmic IFI16 by inhibiting its nuclear import (Li et al., 2012). As further evidence of IFI16 immune activity during infection, in THP-1 macrophages, STING and IFI16 were observed to co-localize in the cytoplasm during HSV-1 infection (Horan et al., 2013b). In myeloid dendritic cells, DDX41 was characterized as another DNA sensor that required STING to promote IFN expression upon HSV-1 infection (Zhang et al., 2011). Horan, et al. proceeded to show that knockdown of DDX41 additionally repressed IFNβ levels upon HSV-1 infection in THP-1 cells (Horan et al., 2013b). Despite these findings, it remains unclear if and how cytosolic IFI16 and DDX41 directly activate STING during infection, and whether these DNA sensors function cooperatively.
Soon after, cGAS was discovered as another DNA sensor and the uncovered signaling mechanism provided a compelling explanation for how STING can be activated. cGAS was found to generate a cyclic dinucleotide that binds directly to STING, thereby robustly activating IFN and ISG expression via the STING-TBK-1-IRF3 pathway (Figure 1, step 2a) (Sun et al., 2013; Wu et al., 2013). Specifically, upon binding to viral DNA, cGAS catalyzes the production of the cyclic second messenger 2’,3’ cGAMP from intracellular pools of ATP and GTP. This molecule, containing unique mixed 2’–5’ and 3’–5’ phosphodiester bonds, binds and activates STING to induce the production of type I IFNs. The discovery of cGAS and 2’3’ cGAMP cemented a fundamental function for cyclic dinucleotides (CDN) as second messengers in anti-pathogen innate immune signaling of mammalian cells. This was at first unexpected, as prior to this finding, CDNs were believed to function predominantly in prokaryotes, which possess exclusive 3’–5’ linked CDNs (Danilchanka and Mekalanos, 2013).
Following these studies, several critical observations were made that solidified a role for cGAS in sensing HSV-1 via the STING-TBK-1-IRF3 pathway. 2’,3’ cGAMP was detected by mass spectrometry in HSV-1-infected human fibroblasts (Sun et al., 2013). cGAS−/− mice and lung fibroblasts from these mice exhibited attenuated IFN responses to HSV-1 infection (Li et al., 2013b). Lung fibroblasts containing a functionally-inactive form of STING, STINGgt/gt, were similarly incapable of mounting a type I IFN response to HSV-1. Although cGAS has demonstrated immune signaling capacity during HSV-1 infection, its possible role during VZV infection has yet to be fully clarified. Recently, it was shown that knockdown of STING in human dermal fibroblasts and immortalized HaCaT keratinocytes depressed levels of phosphorylated IRF3 and IFNβ upon VZV infection (Kim et al., 2017). IFNλ levels were also attenuated in VZV-infected cells lacking STING, suggesting that STING promotes an antiviral state in VZV-infected cells via the expression of both Type I and III IFNs (Kim et al., 2017). Considering the apparent parallels in the virus replication cycles and structure between HSV and VZV, assessing the possibility of shared cGAS sensing amongst alphaherpesviruses warrants future investigation.
A DNA sensor that was found to detect VZV DNA was the DNA-dependent RNAP III. RNAP III was shown to bind cytosolic AT-rich DNA, converting it into 5’-triphosphate RNA, which served as a ligand for the type I IFN-inducing RNA sensor, retinoic acid-induced gene I (RIG-I) (Chiu et al., 2009). Several children exhibiting severe VZV infections were found to have heterozygous missense mutations in the RNAP III subunits, POLR3A and POLR3C (Ogunjimi et al., 2017). During VZV infection, leukocytes from these patients were defective in producing IFN, which was restored upon transduction with respective WT alleles of the subunits. Consistent with these findings, mutations in POLR3A and POLR3E were identified in adults with reactivated VZV in the central nervous system (Carter-Timofte et al., 2018). Cells from these patients similarly exhibited impaired IFN expression when challenged with poly(dA:dT) and higher viral gene expression. Considering that the VZV genome contains several spans of AT-rich regions only otherwise partially found in HSV (Paludan et al., 2011; Khalil et al., 2015), these findings support the notion that RNAP III senses VZV genomes via its AT-rich regions. Indeed, another report showed that cGAS-deficient lung fibroblasts derived from mice did not have impaired IFN responses to transfection with AT-rich DNA in the form of poly (dA:dT) (Li et al., 2013b). The role of RNAP III in sensing HSV-1 remains unclear and warrants future research, as several reports uncover conflicting results in mouse and human cells lines (Chiu et al., 2009; Choi et al., 2009; Melchjorsen et al., 2010; Unterholzner et al., 2010).
In addition to the induction of type I IFNs, DNA sensors can also mediate the production of pro-inflammatory cytokines. Within the cytosol and the nucleus, this is accomplished in the presence of pathogen-derived DNA by the formation of distinct multimeric protein assemblies known as inflammasomes (Gram et al., 2012). These structures were evident during alphaherpesvirus infection via the PRRs NLRP3, AIM2, and IFI16 (Figure 1, step 3b), although the roles of IFI16 and AIM2 remain contentious. Upon recognizing viral PAMPs, these sensors recruit the adaptor protein ASC, which cleaves procaspase-1 into its mature and active form, caspase-1. Caspase-1 subsequently promotes the maturation of cytokines IL-18 and IL-1β. NLRP3 was found to generate cytosolic inflammasomes during VZV infection in THP-1 cells and during HSV-1 infection in primary human fibroblasts (Nour et al., 2011; Johnson et al., 2013). Johnson, et al. additionally identified a role for IFI16 inflammasomes during HSV-1 infection, in which IFI16-DNA puncta were first seen in the nucleus and later translocated with ASC to the cytosol (Johnson et al., 2013). These observations of nuclear IFI16 puncta during infection were consistent with a growing field of research on DNA sensing within the nucleus.
Nuclear DNA sensing
While the actions of most DNA sensors have been predominantly identified and studied in the cytosol and endolysosomes, more recent evidence has provided an additional focus on sensing pathogen-derived DNA in the host nucleus. This is particularly relevant to alphaherpesviruses, which deposit their DNA genomes into the nucleus for replication (Figure 1, step 2c). Although IFI16 was initially characterized as a cytoplasmic DNA sensor (Unterholzner et al., 2010), several observations pointed to an additional nuclear function. IFI16 was reported to localize predominantly to the nucleus in several cell types, including epithelial cells, endothelial cells, primary fibroblast cells, and immune cells (Choubey and Lengyel, 1992; Aglipay et al., 2003; Cristea et al., 2010; Unterholzner et al., 2010; Kerur et al., 2011; Li et al., 2012). Additionally, the ability of IFI16 to bind to the viral genome in the nucleus was reported even prior to the knowledge of its viral DNA sensing function (Cristea et al., 2010). Yet, this was observed during infection with the betaherpesvirus human cytomegalovirus (HCMV) and was connected to a transcriptional regulatory function. IFI16 was subsequently demonstrated to sense nuclear viral DNA during infection with the gamma herpesvirus Kaposi’s sarcoma-associated herpesvirus (KSHV) (Kerur et al., 2011). Kerur, et al. reported that nuclear IFI16 formed inflammasomes upon co-localization with KSHV DNA.
Next, the finding of the IFI16 multi-partite NLS and its acetylation enabled interrogation of localization-dependent IFI16 DNA sensing, this time upon infection with the alphaherpesvirus HSV-1 (Li et al., 2012). Li, et al. showed that NLS mutants of IFI16 that could not localize to the nucleus were significantly impaired in their ability to bind nuclear HSV-1 DNA and induce IFN response upon infection. Orzalli, et al. subsequently demonstrated the necessity of IFI16 and STING for effective IFN and ISG expression in primary human fibroblasts during infection with a mutant HSV-1 strain known to promote strong cytokine expression relative to WT HSV-1, d109 (Orzalli et al., 2012). Prevention of HSV-1 genome release during d109 HSV-1 infection via proteasome inhibitor treatment with tolylsulfonylphenylalanyl chloromethyl ketone, impaired STING- and IFI16-dependent innate immune response.
Subsequently, IFI16 was shown to form nuclear aggregates via its pyrin domain (Li et al., 2013a). Consistent with this phenotype, Morrone, et al. established that IFI16 forms filaments and cooperatively assembles onto DNA (Morrone et al., 2014). More recently, during HSV-1 infection in primary fibroblasts, IFI16 was shown to form these oligomeric puncta at the nuclear periphery upon viral nucleocapsid docking (Cuchet-Lourenco et al., 2013; Everett, 2016; Diner et al., 2016; Lum et al., 2019). Characterization of IFI16 pyrin oligomerization via predicted solvent-exposed charged residues further suggested that IFI16 rapidly binds to incoming viral DNA at nuclear pores in an oligomerization-dependent manner (Lum et al., 2019). In these studies, CRISPR-mediated knockout of IFI16 or expression of oligomerization-incapable forms of IFI16 significantly impaired both cytokine expression and suppression of viral genes during infection.
Another member of the protein family containing pyrin and HIN200 domains, the interferon-inducible protein X (IFIX), was also demonstrated to have the capacity to bind to dsDNA in a sequence-independent manner and to induce IFN response (Diner et al., 2015a). Similar to IFI16, IFIX was reported to be enriched at the nuclear periphery during early HSV-1 infection and to further promote nucleus-derived immune signaling in fibroblasts (Crow and Cristea, 2017). Altogether, these findings suggested a role for nuclear DNA sensors in directly signaling to the STING-TBK-1-IRF3 pathway for cytokine induction. In support of this possibility, Dunphy et al. demonstrated the capacity of nuclear IFI16 to translocate from the nucleus to STING-containing membranous fractions upon DNA damage (Dunphy et al., 2018). Most recently, the heterogeneous nuclear ribonucleoprotein A2B1 (hnRNPA2B1) was identified as a nuclear-to-cytoplasmic translocating DNA sensor against HSV-1 (Wang et al., 2019). Upon HSV-1 infection in murine macrophages and human THP-1 cells, nuclear hnRNPA2B1 was found to associate with HSV-1 DNA, engaging its homodimerization, demethylation, and translocation from the nucleus to the cytoplasm, where it associated with STING and TBK-1.
Several of these observations of STING activation by DNA sensors during HSV-1 infection incited efforts to explore the possibility of synergistic, cooperative, or sequential activities among the sensors (e.g., nuclear and cytosolic IFI16 pools signaling to STING without known IFI16-specific ligands; the identification of a cGAS-specific ligand that directly activates STING; characterization of nuclear-to-cytoplasmic translocating hnRNPA2B1 that binds STING and TBK-1). Yet, the precise mechanisms governing many of these observations are still being uncovered. For instance, how do IFI16 immune signals reach the ER- and Golgi apparatus-associated STING, and are there pools of nuclear cGAS that impact the antiviral state of the cell during alphaherpesvirus infection? Indeed, initial explorations into the localization of endogenous cGAS using antibodies and immunoblotting suggested that a subset of cGAS resides within the nucleus of primary human fibroblasts and normal oral keratinocytes (Orzalli et al., 2015). cGAS was also found to interact with nuclear IFI16, promoting IFI16 protein-level stability during HSV-1 infection. Such an interaction implicates cGAS in maintaining the presence of IFI16-derived immune signals, or sequentially coordinating with IFI16 to activate STING upon infection. Several studies have since identified roles for nuclear cGAS during viral infection and DNA damage (Lahaye et al., 2018; Liu et al., 2018). In further support of DNA sensor-mediated maintenance of immune responses, hnRNPA2B1 was found to promote N6-methyladenosine modification and nuclear-to-cytoplasmic translocation of IFI16, cGAS, and STING mRNAs (Wang et al., 2019). This served to maintain and amplify STING-activated immune responses. Taken together, these findings further highlight how the regulation of nuclear DNA sensing during viral infection is only just beginning to be clarified.
In addition to directly regulating immune signaling pathways, nuclear innate immune proteins can also modulate alphaherpesviral transcription. For example, IFI16 was found to epigenetically promote nucleosome loading and addition of heterochromatin-associated markers onto HSV-1 genomes (Orzalli et al., 2013). These observations suggest that IFI16 functions may be in-part associated with those of a set of sub-nuclear multi-protein complexes, Nuclear Domain 10 (ND10) bodies (also known as PML nuclear bodies). Components of ND10 bodies are IFN-inducible and are known to rapidly associate with viral DNA genomes being extruded into the nucleus from viral capsids docked at the nuclear pore. This association functions to suppress viral gene expression (Everett et al., 2006; Cuchet et al., 2011). In primary human fibroblasts, stable knockdowns of ND10 complex components, including PML, hDaxx, ATRX, or Sp100, increased titers of an HSV-1 strain lacking ICP0, which is an immediate-early viral protein that degrades several ND10 body components (Chelbi-Alix and de The, 1999). Consistent with the concerted action of innate immune factors, interactions between the DNA sensors IFI16 and IFIX with ND10 body components and mediators of chromatin assembly and transcription have been identified (Diner et al., 2015a; Diner et al., 2015b; Diner et al., 2016; Lum et al., 2019).
RNA sensing
During the replication of DNA viruses, RNA intermediates have long been known to accumulate within the cytoplasm (Jacquemont and Roizman, 1975; Weber et al., 2006). This occurrence is thought to be facilitated in-part by symmetrical, overlapping transcription that generates RNA-RNA annealing. As mammalian cells are armed with an ensemble of RNA sensing components, these pathways have been found to contribute to the induction of an innate immune response in cells during alphaherpesvirus infection. Upon HSV-1 and HSV-2 infections, dsRNA intermediates have been shown to serve as agonists of the endolysosome-localized TLR3 sensor and adaptor protein, TIR-domain-containing adapter-inducing interferon-β (TRIF) (Figure 1, step 3d) (Zhang et al., 2007; Peri et al., 2008; Davey et al., 2010; Willmann et al., 2010). Notably, two children with HSV-1 encephalitis were found to be homozygous for a dominant-negative form of TLR3, which could not be identified in over 3,000 chromosomes examined in healthy control individuals (Zhang et al., 2007). Yet how viral RNA is localized to endolysosomes is still unclear. Most recently, TLR3-mediated IFN expression was shown to additionally rely upon coordination with the receptor, TRAF3 (Sato et al., 2018).
While initially induced by AT-rich DNA sensing by RNAP III, 5’ triphosphate RNA generated by RNAP III is sensed by RIG-I, which signals to the adaptor protein, mitochondrial antiviral signaling protein (MAVS), to activate TBK-1 and IKK for inducing IFNs (Chiu et al., 2009; Choi et al., 2009). An additional RIG-I-independent mechanism for sensing RNA during HSV-1 infection has since been characterized via melanoma differentiation-associated protein 5 (MDA5) and MAVS (Melchjorsen et al., 2010) (Figure 1, step 3c). In primary macrophages, knockdown of MDA5 resulted in reduced levels of IFNβ and IFNλ1 upon HSV-1 infection, whereas knockdown of RIG-I had no effect. The authors further observed an increase in TNFα levels during infection that was independent of either RIG-I or MDA5, indicating the potential for alternative pathways, such as via TLR3, in promoting a cytokine-rich antiviral state in the cell.
Alphaherpesvirus innate immune evasion strategies
The ability of alphaherpesviruses to rapidly engage host factors and subvert innate immune responses is a fundamental determinant of the outcome of infection. Highlighting the multifunctionality of viral proteins, several have known capabilities to inhibit immune response via diverse mechanisms, from avoiding PRR detection of PAMPs to preventing downstream cytokine and effector ISG expression (Table 1, Figure 2). This functional versatility enables alphaherpesviruses to effectively constrain host defenses by simultaneously impeding multiple steps of immune response pathways. To ensure a productive viral infection, the implementation of these immune evasion strategies has to match the rapidity of host immune response and, hence, to take place early in infection. This is illustrated by the fact that a considerable number of known immune modulatory viral proteins are either already present as high-copy number components within virion particles that invade a permissive cell (i.e., tegument proteins), or are expressed quickly during productive infection (i.e., immediate-early proteins). In this section, we explore the array of mechanisms employed by alphaherpesviruses to circumvent host immune activation, including the degradation of immune signaling components, as well as their inactivation via binding, post-translational modifications, and sequestration.
Table 1.
Alphapherpesvirus innate immune evasion strategies.
| Immune evasion strategy | Virus | Viral Protein | Functional consequence | Reference(s) |
|---|---|---|---|---|
| Degradation | HSV-1 | vhs | Degradation of ZAP, viperin, IFI16, cGAS mRNA | (Shen et al., 2014; Su et al., 2015; Orzalli et al., 2016) (Su and Zheng, 2017) |
| ICP0 | Degradation of MyD88, Mal | (van Lint et al., 2010) | ||
| HSV-1, HSV-2 | ICP0 | Degradation of IFI16, ND10 components, DNA-PKcs, NF-κB p50 | (Parkinson et al., 1999; Boutell et al., 2003; Orzalli et al., 2012; Cuchet-Lourenco et al., 2013; Zhang et al., 2013; Diner et al., 2015b) | |
| BHV-1 | ICP0 | Degradation of IRF3 | (Saira et al., 2007) | |
| VZV | ORF61p | Degradation of IRF3 | (Zhu et al., 2011) | |
| PRV | pUL50 | Degradation of IFNAR1 | (Zhang et al., 2017) | |
| PTM | HSV-1 | pUL37 | Deamidation of cGAS | (Zhang et al., 2018) |
| pUL36 | Deubiquitination of TRAF3, IKKα | (Wang et al., 2013a) (Ye et al., 2017) | ||
| pUS3 | Atypical IRF3, NF-κB p65 phosphorylation | (Wang et al., 2013b; Wang et al., 2014) | ||
| Hyperphosphorylation, deacetylation prevention of HDAC1/2 | (Poon et al., 2006; Walters et al., 2010; Shapira et al., 2016) | |||
| VZV | ORF47p | Atypical IRF3 phosphorylation | (Vandevenne et al., 2011) | |
| ORF66p | Hyperphosphorylation of HDAC1/2 | (Walters et al., 2009) | ||
| Molecular sink, sequestration | FHV-1 | pUS3 | Kinase-independent prevention of IRF3 dimerization | (Tian et al., 2018) |
| HSV-1 | pUS11 | Binding of PKR, RIG-I, and MDA5 | (Peters et al., 2002) (Xing et al., 2012) | |
| VP22 | Binding of cGAS, prevention of AIM2 inflammasome formation | (Huang et al., 2018) (Maruzuru et al., 2018) | ||
| pUL46 | Binding of STING, TBK-1 | (Deschamps and Kalamvoki, 2017) | ||
| Molecular sink, sequestration | HSV-1 | ICP27 | Binding of TBK-1 | Christensen et al., 2016 |
| γ134.5 | Binding of TBK-1 | Verpooten et al., 2009) | ||
| ICP0 | Sequestration of IRF3, CBP/p300 in subnuclear puncta | (Melroe et al., 2007) | ||
| VP24 | Binding of IRF3 | (Zhang et al., 2016) | ||
| VP16 | Binding of IRF3, CBP, NF-κB p65 | (Xing et al., 2013) | ||
| pUL36 | Binding of IFNAR2 | (Yuan et al., 2018) | ||
| Unknown | pUL13 | Attenuation of CXCL9 expression | (Koyanagi et al., 2017) | |
| Unknown | pUS3 | Inhibition of TLR3 expression | (Peri et al., 2008) | |
| Unknown | VZV | IE62 | Prevention of IRF3 phosphorylation, independent of binding TBK-1, IRF3 | (Sen et al., 2010) |
| Unknown | PRV | gE, gI | Attenuate IFNα expression | (Lamote et al., 2017) |
Figure 2. Mechanisms of alphaherpesvirus-mediated innate immune evasion.
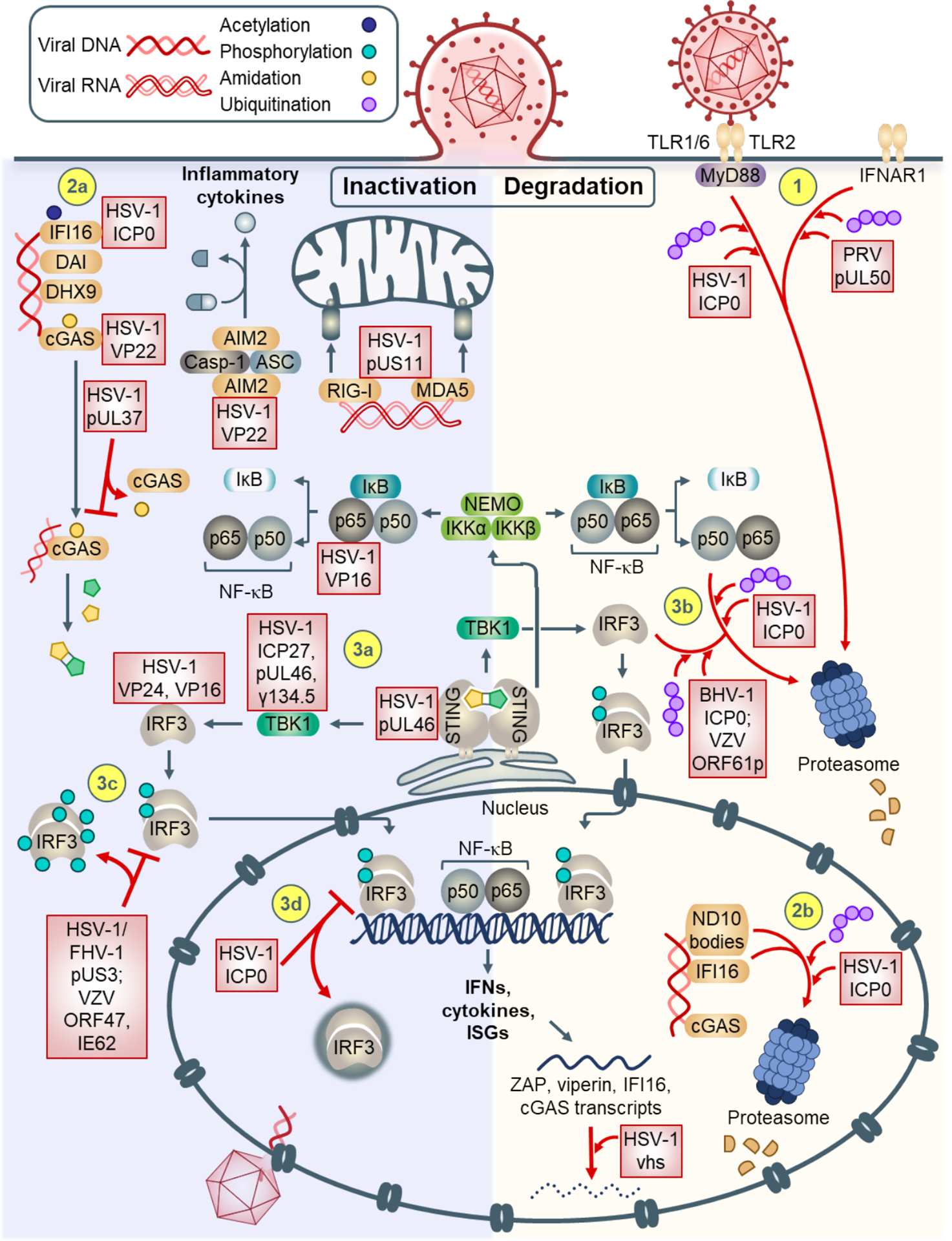
Signal transduction can be inhibited via inactivation (left), or degradation (right) of immune response components. (1) At the plasma membrane, IFNAR1 and TLR2-recruited MyD88 can be targeted for proteasomal degradation. (2a) Cytosolic DNA sensors IFI16 and cGAS are inhibited by direct binding. (2b) In the nucleus, ND10 body components and IFI16 are targeted for proteasomal degradation via HSV-1 ICP0. Upon activation of the STING-TBK-1-IRF3 pathway, (3a) STING, TBK-1, IRF3, and NF-κB p65 are inhibited by alphaherpesvirus proteins via binding. (3b) Additionally, IRF3 and NF-κB p50 can be targeted for degradation. (3c) IRF3 can also be atypically phosphorylated, preventing proper dimerization and nuclear translocation. (3d) HSV-1 ICP0 can also sequester IRF3 into nuclear puncta, preventing its transcriptional activity. (4) Degradation can occur at the RNA level (e.g., ISG transcripts ZAP, viperin, IFI16, cGAS). (5) Sensors AIM2, RIG-I, and MDA5 can be inhibited by binding of HSV-1 proteins.
Degradation of innate immune factor mRNA and protein
Perhaps one of the most understood and straight-forward mechanisms of virus immune evasion is the ability of alphaherpesviruses to target host innate immune factors for degradation at either the transcriptional or protein level (Figure 2). The major transcriptional strategy employed by HSV-1 is host shutoff via global mRNA destabilization. Once virion particles enter the cell, the abundant HSV-1 tegument protein, virion host shutoff (vhs), functions as an mRNA-specific endonuclease that rapidly degrades host and viral mRNA in the cytoplasm prior to de novo viral gene expression (Elgadi et al., 1999). This allows the virus to co-opt cellular transcriptional and translational machinery, as well as engage in the temporal cascade of viral gene expression, which relies in-part on the prior degradation of host and viral mRNA as signaling cues (Read and Frenkel, 1983; Oroskar and Read, 1987, 1989) (Table 1). Notably included as degradation targets are ISGs, such as ZAP and viperin. During WT HSV-1 infection, ZAP and viperin mRNA levels were attenuated relative to those in uninfected cells. In cells infected with HSV-1 strains lacking vhs, both ZAP and viperin mRNA destabilization was prevented (Shen et al., 2014; Su et al., 2015). In line with this function, vhs was more recently demonstrated to additionally target cGAS mRNA for degradation (Su and Zheng, 2017). Interestingly, vhs was found to specifically target adenosine and uridine (AU)-rich elements in the 3’ untranslated portions of mRNA, and was shown to be recruited to AU-rich elements via the cellular protein, tristetraprolin (von Roretz et al., 2011; Shu et al., 2015; Rivas et al., 2016). These findings are consistent with the observation that mRNA encoded by many immune response genes are enriched in AU-rich elements (Schott and Stoecklin, 2010; Schwerk and Sayan, 2015), suggesting a sequence-specific mechanism for vhs-dependent destabilization of immune-related mRNA targets.
Alphaherpesvirus-induced degradation at the protein level relies on the exploitation of the cellular proteasome pathway. To maintain a balanced intracellular state, the proteasome pathway is essential for mediating the homeostatic turnover of proteins. Relevant to active innate immune signal transduction, cells require the proteasomal degradation of IκB to release the inhibition of NF-κB and promote its translocation into the nucleus (Karin and Ben-Neriah, 2000). Upon recognition and binding of proteins covalently modified with poly-ubiquitin chains, the proteasomal 26S unfolds and catabolizes proteins into oligopeptides (Lecker et al., 2006).
A prime example of virus-mediated degradation of cellular immune proteins involves the HSV-1 immediate-early protein ICP0. ICP0 contains an E3 ubiquitin ligase activity via a RING finger domain and promotes the proteasome-dependent degradation of several cellular and viral proteins via ubiquitination. ICP0 is known to target a growing number of host defense receptors for degradation, including a glycoprotein sensor, DNA sensor, chromatin modulators, and transcription factors. During infection, van Lint et al. determined that TLR2-mediated immune signaling, via IL-6 production and NF-κB-directed transcription, was suppressed due to the ICP0 E3 ubiquitin ligase activity on the TLR2 adaptor proteins, MyD88 and Mal (van Lint et al., 2010).
Critical to nuclear sensing of HSV-1, ICP0 was found to promote the degradation of IFI16 in the nucleus, at least in-part via its E3 ubiquitin ligase function (Orzalli et al., 2012; Cuchet-Lourenco et al., 2013; Diner et al., 2015b). Specifically, ICP0 was found to target the oligomerizing pyrin domain of IFI16 (Diner et al., 2016). Cells infected with an HSV-1 strain containing an E3 ligase-null form of ICP0 (ICP0-RF HSV-1) showed a rescue in IFI16 protein levels. Yet, these initial reports conducted in HFFs and HEK 293Ts remain divided over whether ICP0 is sufficient to induce IFI16 degradation, or whether additional viral or hijacked cellular cofactors are also involved in this process. Consistent with this notion, the HSV-1-mediated degradation of the DNA sensor IFIX was not relieved upon infection with ICP0-RF HSV-1 (Crow and Cristea, 2017). Furthermore, the mode of IFI16 degradation by HSV-1 was observed to be cell type-dependent, as HSV-1 infection resulted in reduced IFI16 protein levels in HFFs, normal oral keratinocytes, and HeLa cells, but not in U2OS cells. In HeLa cells, the HSV-1 tegument protein vhs additionally facilitated IFI16 mRNA turnover, consistent with its known roles in engaging ribonuclease-induced mRNA destabilization (Orzalli et al., 2016). Noteworthy, ICP0 was shown to be necessary and sufficient to target several other host defense proteins for degradation, including ND10 body components (i.e., PML, ATRX, hDaxx, Sp100, SUMO1), the DNA sensor DNA-PKcs, and the NF-κB subunit p50 (Parkinson et al., 1999; Boutell et al., 2003; Cuchet-Lourenco et al., 2013; Zhang et al., 2013). Highlighting an interesting conservation of this ICP0 immune-inhibiting function across strains and species, ICP0 encoded by bovine herpesvirus 1 was shown to target IRF3 for degradation in HEK 293s (Saira et al., 2007), yet the capacity of HSV-1 ICP0 to accomplish this has yet to be observed.
Given the uncontested prominence of the STING-TBK-1-IRF3 pathway in promoting an antiviral state in the cell, one might expect several more examples of alphaherpesviral proteins that target these immune components for degradation. Indeed, the VZV immediate-early protein ORF61p co-immunoprecipitated with IRF3, leading to IRF3 ubiquitination and proteasome-dependent degradation. IRF3 degradation was subsequently rescued by treatment with the proteasome inhibitor, MG132 (Zhu et al., 2011). Furthermore, the porcine alphaherpesvirus pseudorabies virus (PRV) encodes pUL50, which abrogates IFNα expression by degrading interferon α/β receptor subunit 1 (IFNAR1) via lysosomes (Zhang et al., 2017).
Inactivation by virus-induced post-translational modifications
In light of the dynamic and temporal role of protein post-translation modifications (PTM) in signal transduction, it is not surprising that alphaherpesviruses encode proteins that catalyze both the addition and removal of PTMs from cellular immune signaling components. At the PRR level, the HSV-1 inner tegument protein pUL37 was shown to deamidate human and mouse cGAS (Zhang et al., 2018). A critical asparagine residue, N210, was identified in the human and mouse cGAS activation loop, which impinges upon the conformational rearrangement of the cGAMP-producing catalytic site (Zhang et al., 2018). In both human THP-1 macrophages and murine L929 fibroblasts, this site was deamidated by pUL37 during HSV-1 infection, suppressing cGAS capacity to generate 2,3’-cGAMP and induce cytokines. pUL37 deamidation did not affect cGAS abilities to bind DNA and dimerize. Infection with a pUL37 deamidation-deficient HSV-1 strain in human macrophages and in BL6 mice resulted in higher cGAS- and STING-dependent cytokine expression relative to infection with WT HSV-1. Unexpectedly, pUL37 had no effect on non-human primate cGAS, suggesting that HSV-1 has evolved to exploit the natural species sequence variation of cGAS. Consistent with this finding, the authors identified that the N210 residue was not conserved in non-human primate species. Further supporting a crucial role for deamidation in subverting host innate immunity during HSV-1 infection, pUL37 was also shown to deamidate the RNA sensor, RIG-I, thereby attenuating IFN expression (Zhao et al., 2016). As pUL37 is evolutionarily conserved across all three herpesvirus subfamilies (alpha, beta, and gamma), and both cGAS and RIG-I have been shown to promote IFN immune signaling upon infection with these herpesviruses, it is possible that pUL37 has a similar PRR deamidation function as these other types of herpesvirus infection.
Downstream of PRR activation, virus-induced deubiquitination and phosphorylation, via both cellular- and virus-encoded enzymes, have been shown to inactivate components of the TLR and STING-TBK-1-IRF3 signaling pathways. Sun et al. showed that the cellular protein USP13 catalyzed the deconjugation of K27-linked polyubiquitin chains from STING in mouse embryonic fibroblasts, thereby preventing TBK-1 activation and expression of antiviral cytokines during HSV-1 infection (Sun et al., 2017). This finding suggests that HSV-1 may hijack USP13 to inactivate STING-dependent IFN and ISG expression.
For those immune-evasive enzymes encoded by viruses, the capacity to recognize and modify several host receptors confers an efficient and adaptive advantage against the host defense. This broad host substrate specificity has been identified for several alphaherpesviral proteins. The HSV-1 ubiquitin-specific protease, pUL36, was demonstrated to suppress IFN and cytokine production by modifying both the receptor TRAF3 and NF-KB inhibitor, IKKα. Wang, et al. showed that these pUL36 functions relied upon the activity of the deubiquitinase (DUB) motif that was previously identified by Schlieker, et al. (Schlieker et al., 2005; Wang et al., 2013a). Via a series of ectopic expression tests in HEK 293Ts and HSV-1 infection in C57BL/6 mice, the authors observed that pUL36-mediated deubiquitination of TRAF3 prevented the formation of a TRAF3-TBK-1 signaling complex, which interfered with IRF3 dimerization. It was later shown that pUL36 was further capable of cleaving K48-linked polyubiquitin chains from IKKα in HEK 293Ts and HFFs, protecting it from proteasomal degradation, and thus maintaining NF-κB in an inactive state (Ye et al., 2017).
Indicating the potential for conserved immune-evasion activity of viral enzymes, the HSV-1 kinase pUS3 and the predicted VZV homolog ORF47p, are known to atypically phosphorylate several cellular factors that promote IFN and ISG induction. Canonically, activated TBK-1 phosphorylates IRF3 at Ser396. However, VZV ORF47p expression in cells was found to interact with IRF3, preventing Ser396 phosphorylation and subsequent transcription of target genes (Vandevenne et al., 2011). HSV-1 pUS3 was observed to hyperphosphorylate IRF3 at Ser175, inhibiting its homodimerization, and the NF-κB subunit p65 at Ser75, preventing its translocation into the nucleus (Wang et al., 2013b; Wang et al., 2014). To identify the specific residues phosphorylated by pUS3, the authors conducted either IRF3 or p65 immunoaffinity isolations in cells co-expressing epitope-tagged US3 and either IRF3 or p65, and analyzed the eluates by mass spectrometry. Consistent with the immune-activating role of Ser396 phosphorylation, this modification was not detected upon pUS3 expression, further indicating that atypical modification of IRF3 impedes the ability of TBK-1 to modify and activate it. An astute demonstration of virus strain-specific divergence in pUS3 inhibition of IRF3 was given in feline kidney cells infected with the alphaherpesvirus, feline herpesvirus 1 (FHV-1) (Tian et al., 2018). FHV-1 pUS3 similarly prevented IRF3 homodimerization and subsequent IFNβ expression yet accomplished this via a kinase-independent mechanism by competitively binding the IRF association domain. As this study identified several other FHV-1 ORFs that inhibit IFN expression (i.e., UL1, UL4, UL3.5, UL11, UL27, UL45, UL48, and UL55), future explorations may reveal additional layers of virus-host interactions and cooperative or distinct immune suppression mechanisms.
Host factor inactivation and sequestration
To obstruct signal transduction, alphaherpesviruses encode proteins that effectively act as molecular sinks for host innate immune factors by associating with and saturating these receptors. At the level of PAMP detection, the dsRNA sensor protein kinase R (PKR), was found to be antagonized by the HSV-1 tegument protein, pUS11. In uninfected cells, PKR binds RNA, facilitating its autophosphorylation (Romano et al., 1998). Subsequently, PKR activates the translation initiation factor eIF-2α, which engages in translational inhibition and thus blocks viral protein synthesis. As strong expression of pUS11 was previously found to rescue premature translational stasis during infection of cells with a mutant HSV-1 strain (Mohr and Gluzman, 1996; Mulvey et al., 1999), Peters, et al. hypothesized that pUS11 could relieve the translational repression imparted by PKR (Peters et al., 2002). Indeed, in vitro affinity purification assays revealed that the HSV-1 tegument protein pUS11 was capable of binding directly to PKR, preventing the function of a PKR activator, PACT, and thus inhibiting PKR-mediated translational shutdown in cells.
In addition to PKR inhibition, the HSV-1 tegument protein VP22 was found to bind to the DNA sensors, cGAS and AIM2, suppressing their immune activities (Huang et al., 2018; Maruzuru et al., 2018). With estimates of well over 1,000 copies per virion particle (Heine et al., 1974; Leslie et al., 1996), it may not be surprising that VP22 is simultaneously capable of binding and nearly saturating endogenous levels of these sensors. In HFFs, knocking down cGAS promoted the replication of an HSV-1 strain lacking VP22, but not WT HSV-1, further supporting the cGAS-specific antagonism of this viral protein. The identified VP22-AIM2 association prevented AIM2 oligomerization, formation of inflammasome complexes, and expression of the pro-inflammatory IL-1β and IL-18 in human macrophages (Maruzuru et al., 2018). In a similar fashion, the HSV-1 tegument protein pUS11 was found to bind RIG-I and MDA5. These associations prevented their oligomeric aggregation with the MAVS adaptor for inducing downstream cytokine expression in HEK 293Ts (Xing et al., 2012).
Inactivation of the STING-TBK-1-IRF3 pathway via physical and stable virus-host protein interaction has been demonstrated for each pathway component during HSV-1 infection. Deschamps and Kalamvoki established that the abundant tegument protein pUL46 binds STING to prevent IFN expression in immortalized HEL cells (Deschamps and Kalamvoki, 2017). When treated with 2’,3’-cGAMP, cells exogenously expressing pUL46 were incapable of rescuing IFN production. This finding suggested that the pUL46-STING association prevents 2’3’-cGAMP and/or IFI16 binding to STING, STING dimerization, or STING recruitment and activation of TBK-1. The authors also found that pUL46 associated with TBK-1, indicating that pUL46 may physically inhibit the STING-TBK-1 complex. Additional examples of HSV-1 inhibition of TBK-1 are provided by the associations with the immediate-early protein ICP27 and leaky-late protein γ134.5 (Verpooten et al., 2009; Christensen et al., 2016). As expected, these associations prevented IRF3 phosphorylation and activation in each of the studies.
At the level of antagonizing transcription factors downstream of STING activation, the HSV-1 immediate-early protein ICP0 has been shown to re-localize activated IRF3 and its transcriptional co-activating binding partners, CBP and p300, into subnuclear puncta (Melroe et al., 2007). This sequestration strategy prevents IRF3 from docking at ISRE regions to promote the expression of target host genes. Subsequently, Zhang, et al. demonstrated that HSV-1 encodes the serine protease, VP24, which directly bound IRF3, inhibiting its activation by TBK-1, its phosphorylation, and homodimerization (Zhang et al., 2016). In a similar manner, the HSV-1 tegument protein VP16 was found to bind IRF3 and its coactivator CBP, preventing the formation of active IRF3-CBP transcriptional complexes (Xing et al., 2013). In support of this finding, the authors did not see an effect of VP16 on IRF3 homodimerization or nuclear translocation. VP16 was additionally observed to inhibit the activation of NF-κB via a VP16-p65 subunit association.
Furthermore, as IFNs are secreted from infected cells to signal to neighboring cells, it would stand to reason that alphaherpesviruses have evolved strategies to prevent this immune activity. Indeed, aside from its deubiquitinase capacity, HSV-1 pUL36 was shown to competitively bind the IFN-α/β receptor, IFNAR2. This association prevented binding of the kinase-signal transducer and activator of transcription, JAK1 (Yuan et al., 2018).
Contributions of advances in omic and microscopy techniques
Concurrent improvements in technologies and methodology have facilitated many of the above-discussed biological discoveries in immune response and virus immune subversion strategies. In this section, we highlight how transcriptomics, proteomics, and microscopy approaches have contributed to our understanding of innate immunity during alphaherpesvirus infection.
Gene profiling reveals immune modulating factors
The application of transcriptomic approaches to exploring virus-host dynamics has promoted the identification of cellular immune factors and associated alphaherpesviral antagonists. In these studies, genes were assessed for their capacity to modulate IFN and ISG expression. Prior to characterizing the DNA sensing activities of cGAS, Schoggins, et al. used an ISG screening approach to discover that cGAS was one of the genes that possessed broad IFN-based antiviral function against several viruses, including yellow fever virus, chikungunya virus, Venezuelan equine encephalitis virus, and Sindbis virus (Schoggins et al., 2011). Cells were treated with a lentiviral vector bicistronically bearing one of several hundred ISGs and a red fluorescent protein (RFP) tag. Upon infection with different viruses expressing a distinct green fluorescent protein (GFP) tag, FACS was used to quantify virus replication in RFP-positive cells. Similarly, the ability of FHV-1 pUS3 to antagonize two downstream effector proteins of cGAS, STING and IRF3, was identified from screens of cellular and viral genes, respectively, which affected IFN and ISG levels (Ishikawa and Barber, 2008; Tian et al., 2018). Furthermore, the HSV-1 pUL37 was initially considered as a candidate for suppressing cGAS-mediated IFNβ based on RNA sequencing of host defense genes in samples infected with HSV-1 strains containing mutant pUL37 (Zhang et al., 2018).
In addition to the use of transcriptomic strategies, concurrent explorations into protein interactions uncovered the DNA sensors and associated immune-modulatory protein complexes. These protein interaction studies complemented the findings from gene profiling and enabled investigations of associations between immune proteins with either DNA, other proteins, or small molecules.
Protein interaction studies identify DNA sensors and immune complexes
Over the past decade, the use of immunoaffinity purification coupled to mass spectrometry (IP-MS) has contributed to several landmark discoveries, including the identification of several viral DNA sensors (i.e., AIM2, DHX9, DHX36, IFI16, DNA-PKcs, IFIX, and cGAS) and alphaherpesviral antagonists. The first of these proteins to have been identified and characterized as a DNA sensor was AIM2 in 2009 (Burckstummer et al., 2009). In this study, the authors indeed sought to discover a cytoplasmic DNA sensor that mediated inflammasome formation. In three different mouse cell lines, the authors incubated cell extracts with biotinylated 45-base pair interferon-stimulatory DNA (ISD) immobilized to streptavidin resin and subjected eluates to mass spectrometry analysis. As an orthogonal approach, a microarray analysis of genes induced by IFNβ was conducted in parallel. From these assays, AIM2 was uncovered as one of three DNA-binding proteins belonging to the interferon-inducible PYHIN protein family that was additionally transcriptionally regulated by IFNβ. Further experimentation demonstrated that AIM2 was cytoplasmic and capable of forming inflammasomes in the presence of DNA. As would be expected based on the DNA sensors that are known today, one of the other PYHIN proteins identified, but not further explored in the study, was the mouse homolog of IFI16, IFI204. In fact, not long after this initial study, IFI16 was similarly identified as a bona fide cytoplasmic DNA sensor (Unterholzner et al., 2010). Using biotinylated oligomers of 70 base pairs derived from Vaccinia virus, IP-MS was conducted from the cytosolic extracts of THP-1 cells.
The versatility of IP-MS to identify both proteins and small molecules facilitated the identifications of cGAS and cGAMP, respectively. cGAS was discovered as a robust DNA sensor using a modified mass spectrometry approach on lysates from cells pre-stimulated to produce cGAMP (Wu et al., 2013). Via a series of three separate biochemical fractionation strategies conducted in parallel, in which IP-MS using biotinylated ISD was conducted following one of the strategies, cGAS was found to co-purify with cGAMP. cGAMP was also characterized by IP-MS, upon its co-isolation with Flag-tagged STING (Sun et al., 2013). Sun, et al. subsequently employed a sensitive targeted mass spectrometry approach, selected reaction monitoring, to quantify cGAMP levels and demonstrate that fractions containing cGAMP were those associated with activated IRF3.
The application of IP-MS to studying DNA sensors has since further expanded to exploring their protein-protein interactions. An interaction network for all PYHIN proteins (AIM2, IFI16, IFIX and MNDA) was generated using cells inducibly expressing each GFP-tagged PYHIN protein (Diner et al., 2015a). The study revealed shared associations involved in functions such as chromatin remodeling and antiviral response, as well as IFIX interactions with PML and other ND10 components. From this study, IFIX was also classified as the second nuclear DNA sensor and was shown to restrict HSV-1 replication. IFI16 interactions from this study and from additional IP-MS studies using an HSV-1 strain that lacks the ability to degrade IFI16 highlighted associations important for immune signaling, such as with ND10 body components (Diner et al., 2015b). An MS-based approach was also used to confirm the interaction of IFI16 with cGAS in multiple cell types (Orzalli et al., 2015). Domain-specific IP-MS analyses were later performed to assign different IFI16 interactions to its pyrin or HIN domains (Orzalli et al., 2015; Diner et al., 2015b; Diner et al., 2016). More recently, a broader cGAS protein interaction network during HSV-1 infection in primary fibroblasts was also characterized using IP-MS analyses (Lum et al., 2018). In support of previous findings that HSV-1 pUL37 and pUL49 directly antagonize cGAS-mediated production of 2’,3’-cGAMP, this study also identified these viral protein associations with cGAS, along with additional HSV-1 proteins, providing a future resource to study hypothesis-driven cGAS regulation and antagonism by HSV-1. Additionally, a cGAS interaction was also identified the 2’-5’-oligoadenylate synthase-like protein OASL, a protein associated with RNA sensing. Further investigation in this study showed that OASL suppresses cGAS-mediated IFN response, suggesting its role as a regulatory negative feedback loop for controlling cytokine induction.
Proteome alterations associated with immune response
Knowledge of PRRs, downstream effector pathways, and their protein interactions were also placed in a broader proteome context during infection to provide a clearer understanding of immune regulatory changes throughout the virus replication cycle. Kulej, et al. applied an epigenomics-proteomics approach to generate a resource of global protein abundance changes in both cellular and viral proteins, as well as phosphorylation sites and chromatin-bound histone acetylations during multiple time points of HSV-1 infection in primary human fibroblasts (Kulej et al., 2017). At different time points across HSV-1 infection, fibroblast lysates were sequentially digested with LysC and trypsin. Phosphorylated peptides were enriched using a titanium dioxide-based chromatographic resin, and then subjected to MS analysis. To identify histone acetylations, histones were purified from the lysates of infected cells, and assessed by MS.
Temporal proteome alterations during HSV-1 infection were further explored in the context of cGAS-mediated immune signaling. Lum, et al. placed their identified cGAS interactions in the context of the respective endogenous protein abundances during HSV-1 infection (Lum et al., 2018). This was accomplished via whole proteome analysis of primary human fibroblasts using isobaric tag-based quantitative mass spectrometry. Targeted MS using parallel reaction monitoring further validated several protein abundance changes throughout infection, and enabled the assessment of proteins with low endogenous abundances. Providing an additional layer of insight into immune regulation, proteome changes were compared between cells infected with wild type or mutant HSV-1 strains that induced varying degrees of cytokines and apoptosis.
Discovery of PTMs
The contribution of proteomics has extended beyond the identification of DNA sensors and protein-protein interactions. As highlighted in the above sections, PTMs (i.e., phosphorylation, acetylation, deamidation, and ubiquitination) are indispensable for the temporal and spatial modulation of both host immune signal transduction and effective circumvention of this defense by alphaherpesviruses. In addition to investigating global PTM mapping of specific modifications in cells, as mentioned above in the study by Kulej, et al, IP-MS has also been used to identify multiple PTMs on immune signaling proteins. For example, Li, et al. used a combinatorial IP-MS strategy to identify PTMs on endogenous IFI16 in primary fibroblasts and CEM T cells by digesting immunoisolated samples separately with the endoproteinases, trypsin and GluC (Li et al., 2012). This strategy facilitated the detection of multiple acetylation and phosphorylation sites on IFI16, including the aforementioned acetylation within its NLS, which the authors showed to serve as a toggle for nuclear-cytoplasmic localizations. Downstream of PRR function, two studies identified the capacity of HSV-1 pUS3 to hyperphosphorylate both IRF3 and NF-κB p65 via immunoisolations of epitope-tagged pUS3 and trypsin-based MS analyses (Wang et al., 2013b; Wang et al., 2014).
More recently, this approach was employed to identify both cellular and viral proteins that regulate the PTM state of cGAS, and thus regulate IFNβ induction, during HSV-1 infection. Seo, et al. first conducted IP-MS in cells expressing Flag-tagged cGAS and identified that the cellular protein kinase Akt promoted the addition of a phosphorylation on Ser305 of cGAS (Seo et al., 2015). This modification inhibited cGAS-mediated IFN expression and HSV-1 replication during infection upon exogenous Akt expression, suggesting that Akt either functions as a regulator of cGAS activity, or is co-opted by HSV-1. Recently, IP-MS was used to identify cGAS deamidation by HSV-1 pUL37 (Zhang et al., 2018), emphasizing how this approach is amenable for exploring both anti-viral and pro-viral pathways.
Microscopy in determining the localization dynamics of DNA sensors
The implementation of advanced microscopy approaches has facilitated important insights into the dynamic localization of immune and viral proteins during infection. As an example, microscopy-based investigations into how and when IFI16 binds nuclear HSV-1 DNA have provided an elegant understanding of its mechanism of action with great temporal and spatial resolution. First via confocal microscopy, nuclear IFI16 was observed to re-localize from a diffuse state to distinct puncta asymmetrically at the nuclear periphery, coincident with viral genome-associated proteins (Cuchet-Lourenco et al., 2013). Imaging via in vitro single-molecule fluorescence, as well as via time-lapse total internal reflection fluorescence microscopy and Förster resonance energy transfer, fluorescently-labeled IFI16 was observed to track along biotinylated, euchromatinized DNA and assemble into oligomeric structures (Stratmann et al., 2015). This clustering was estimated to be comprised of eight to ten protomers. Subsequently, Diner, et al. conducted live cell imaging in primary fibroblasts infected with HSV-1 (Diner et al., 2016). Upon stably expressing fluorescently-tagged IFI16 in cells, IFI16 was observed to dynamically enrich first to incoming HSV-1 genomes within minutes of nucleocapsids docking at the nuclear periphery, and subsequently re-localize to diffuse nucleoplasmic puncta within hours after infection, and then result in loss of signal. These secondary localizations were consistent with previous indirect fluorescence microscopy reports of IFI16 localizing to nuclear puncta in an ICP0-dependent manner prior to its degradation during infection (Orzalli et al., 2012; Cuchet-Lourenco et al., 2013). Thus, microscopy techniques enhanced the understanding of IFI16-mediated signal transduction and antagonism by ICP0.
Concluding remarks and future trends
As we have seen, many alphaherpesvirus-sensing PRRs converge upon shared immune signaling pathways. With the ongoing characterization of PRRs in the cytosol and nucleus, our knowledge of how host defense components act in a synergistic fashion, in distinct immune signaling pathways, operate sequentially, or function redundantly is expected to grow in the coming years. We have also discussed several viral proteins with broad host substrate specificity, providing viruses with multiple opportunities to dismantle host immune signaling. Given the multifunctional capacity of many viral proteins, this ability to both evade immune induction and promote virus replication may explain why the host innate immune response contains such a complex network of PRRs and signaling cascades. As an example, while HSV-1 US3 is known to inactivate IRF3 and NF-κB via atypical phosphorylation, it also relieves the transcriptional repression of viral genes by hyperphosphorylating and preventing deacetylation of histone deacetylases, HDAC1 and HDAC2 (Poon et al., 2006; Walters et al., 2010; Shapira et al., 2016). Similarly, the homolog in VZV, ORF66p, was also shown to hyperphosphorylate HDAC1 and HDAC2 (Walters et al., 2009). In this way, it is perhaps not surprising that we continue to uncover additional functions for proteins involved in both cellular innate immunity and virus immune evasion.
Thus far, many intriguing examples of alphaherpesvirus-mediated IFN suppression have additionally been identified without defined mechanisms. For instance, in the central nervous system of mice, the HSV-1 kinase pUL13 was shown to subvert CD8+ T cell infiltration by attenuating expression of the chemokine attractant CXCL9 (Koyanagi et al., 2017). The VZV immediate-early protein IE62 was found to prevent IFN-inducing IRF3 phosphorylation at serines 396, 398, 402, yet was not found to bind to either TBK-1 or IRF3 (Sen et al., 2010). In plasmacytoid dendritic cells infected with PRV, the glycoproteins gE and gI were observed to antagonize IFNα expression (Lamote et al., 2017). As more sensitive technology emerges, these techniques may be leveraged to provide more mechanistic insight into existing and as-of-yet discovered virus immune evasion strategies.
Altogether, the implementation of developing omic technology has significantly enriched our understanding of innate immunity and alphaherpesvirus immune evasion. Ongoing improvements in mass spectrometry instrumentation with unprecedented resolution and sensitivity provide a powerful tool to be leveraged for future investigations. The additional incorporation of biochemical and molecular tools (i.e., microscopy, chemical crosslinking, alpha screens) is expected to provide greater mechanistic insight and depth. We envisage that these emerging technologies will continue to facilitate exciting opportunities for future research in 1) identifying virus-host protein interactions; 2) uncovering dynamic PTMs on viral and cellular proteins across infection time points and subcellular locations; 3) quantifying global changes in protein abundance; 4) integrating information among multi-omics data (i.e., genomic, transcriptomic, lipidomic) for a systems-level view of alphaherpesvirus replication and host response under different biological contexts (i.e., alphaherpesvirus-induced disease states, variations in clinical isolates, mutant virus strains). These areas of research will be critical in the coming years to address more sophisticated questions regarding the modulation of innate immune signaling during alphaherpesvirus infection.
References
- Aglipay JA, Lee SW, Okada S, Fujiuchi N, Ohtsuka T, Kwak JC, Wang Y, Johnstone RW, Deng CX, Qin J, et al. (2003). A member of the Pyrin family, IFI16, is a novel BRCA1-associated protein involved in the p53-mediated apoptosis pathway. Oncogene 22, 8931–8938. 10.1038/sj.onc.1207057 [DOI] [PubMed] [Google Scholar]
- Arvin AM (1996). Varicella-zoster virus. Clin Microbiol Rev 9, 361–+. 10.1128/Cmr.9.3.361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbalat R, Lau L, Locksley RM, and Barton GM (2009). Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat Immunol 10, 1200–1207. 10.1038/ni.1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutell C, Orr A, and Everett RD (2003). PML residue lysine 160 is required for the degradation of PML induced by herpes simplex virus type 1 regulatory protein ICP0. Journal of Virology 77, 8686–8694. 10.1128/Jvi.77.16.8686-8694.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burckstummer T, Baumann C, Bluml S, Dixit E, Durnberger G, Jahn H, Planyavsky M, Bilban M, Colinge J, Bennett KL, et al. (2009). An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nature Immunology 10, 266–272. 10.1038/ni.1702 [DOI] [PubMed] [Google Scholar]
- Cai M, Li M, Wang K, Wang S, Lu Q, Yan J, Mossman KL, Lin R, and Zheng C (2013). The herpes simplex virus 1-encoded envelope glycoprotein B activates NF-kappaB through the Toll-like receptor 2 and MyD88/TRAF6-dependent signaling pathway. PLoS One 8, e54586. 10.1371/journal.pone.0054586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter-Timofte ME, Hansen AF, Christiansen M, Paludan SR, and Mogensen TH (2018). Mutations in RNA Polymerase III genes and defective DNA sensing in adults with varicella-zoster virus CNS infection. Genes and Immunity 10.1038/s41435-018-0027-y [DOI] [PubMed] [Google Scholar]
- Chelbi-Alix MK, and de The H (1999). Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 18, 935–941. 10.1038/sj.onc.1202366 [DOI] [PubMed] [Google Scholar]
- Chiu YH, MacMillan JB, and Chen ZJJ (2009). RNA Polymerase III Detects Cytosolic DNA and Induces Type I Interferons through the RIG-I Pathway. Cell 138, 576–591. 10.1016/j.cell.2009.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi MK, Wang Z, Ban T, Yanai H, Lu Y, Koshiba R, Nakaima Y, Hangai S, Savitsky D, Nakasato M, et al. (2009). A selective contribution of the RIG-I-like receptor pathway to type I interferon responses activated by cytosolic DNA. Proc Natl Acad Sci U S A 106, 17870–17875. 10.1073/pnas.0909545106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choubey D, and Lengyel P (1992). Interferon Action - Nucleolar and Nucleoplasmic Localization of the Interferon-Inducible 72-Kd Protein That Is Encoded by the Ifi-204 Gene from the Gene-200 Cluster. J Cell Biol 116, 1333–1341. 10.1083/jcb.116.6.1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen MH, Jensen SB, Miettinen JJ, Luecke S, Prabakaran T, Reinert LS, Mettenleiter T, Chen ZJ, Knipe DM, Sandri-Goldin RM, et al. (2016). HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. Embo J 35, 1385–1399. 10.15252/embj.201593458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristea IM, Moorman NJ, Terhune SS, Cuevas CD, O’Keefe ES, Rout MP, Chait BT, and Shenk T (2010). Human cytomegalovirus pUL83 stimulates activity of the viral immediate-early promoter through its interaction with the cellular IFI16 protein. J Virol 84, 7803–7814. 10.1128/JVI.00139-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow MS, and Cristea IM (2017). Human Antiviral Protein IFIX Suppresses Viral Gene Expression during Herpes Simplex Virus 1 (HSV-1) Infection and Is Counteracted by Virus-induced Proteasomal Degradation. Mol Cell Proteomics 16, S200–S214. 10.1074/mcp.M116.064741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuchet-Lourenco D, Anderson G, Sloan E, Orr A, and Everett RD (2013). The Viral Ubiquitin Ligase ICP0 Is neither Sufficient nor Necessary for Degradation of the Cellular DNA Sensor IFI16 during Herpes Simplex Virus 1 Infection. Journal of Virology 87, 13422–13432. 10.1128/Jvi.02474-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuchet D, Sykes A, Nicolas A, Orr A, Murray J, Sirma H, Heeren J, Bartelt A, and Everett RD (2011). PML isoforms I and II participate in PML-dependent restriction of HSV-1 replication. J Cell Sci 124, 280–291. 10.1242/jcs.075390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilchanka O, and Mekalanos JJ (2013). Cyclic dinucleotides and the innate immune response. Cell 154, 962–970. 10.1016/j.cell.2013.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey GM, Wojtasiak M, Proietto AI, Carbone FR, Heath WR, and Bedoui S (2010). Cutting Edge: Priming of CD8 T Cell Immunity to Herpes Simplex Virus Type 1 Requires Cognate TLR3 Expression In Vivo. Journal of Immunology 184, 2243–2246. 10.4049/jimmunol.0903013 [DOI] [PubMed] [Google Scholar]
- Deschamps T, and Kalamvoki M (2017). Evasion of the STING DNA-Sensing Pathway by VP11/12 of Herpes Simplex Virus 1. J Virol 91 10.1128/JVI.00535-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diner BA, Li T, Greco TM, Crow MS, Fuesler JA, Wang J, and Cristea IM (2015a). The functional interactome of PYHIN immune regulators reveals IFIX is a sensor of viral DNA. Mol Syst Biol 11, 787. 10.15252/msb.20145808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diner BA, Lum KK, Javitt A, and Cristea IM (2015b). Interactions of the Antiviral Factor Interferon Gamma-Inducible Protein 16 (IFI16) Mediate Immune Signaling and Herpes Simplex Virus-1 Immunosuppression. Mol Cell Proteomics 14, 2341–2356. 10.1074/mcp.M114.047068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diner BA, Lum KK, Toettcher JE, and Cristea IM (2016). Viral DNA Sensors IFI16 and Cyclic GMP-AMP Synthase Possess Distinct Functions in Regulating Viral Gene Expression, Immune Defenses, and Apoptotic Responses during Herpesvirus Infection. MBio 7. 10.1128/mBio.01553-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbs N, Burnaevskiy N, Chen DD, Gonugunta VK, Alto NM, and Yan N (2015). STING Activation by Translocation from the ER Is Associated with Infection and Autoinflammatory Disease. Cell Host Microbe 18, 157–168. 10.1016/j.chom.2015.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jonsson KL, Jakobsen MR, Nevels MM, Bowie AG, and Unterholzner L (2018). Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-kappa B Signaling after Nuclear DNA Damage. Mol Cell 71, 745–+. 10.1016/j.molcel.2018.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgadi MM, Hayes CE, and Smiley JR (1999). The herpes simplex virus vhs protein induces endoribonucleolytic cleavage of target RNAs in cell extracts. Journal of Virology 73, 7153–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD (2016). Dynamic Response of IFI16 and Promyelocytic Leukemia Nuclear Body Components to Herpes Simplex Virus 1 Infection. J Virol 90, 167–179. 10.1128/JVI.02249-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Rechter S, Papior P, Tavalai N, Stamminger T, and Orr A (2006). PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J Virol 80, 7995–8005. 10.1128/JVI.00734-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gram AM, Frenkel J, and Ressing ME (2012). Inflammasomes and viruses: cellular defence versus viral offence. J Gen Virol 93, 2063–2075. 10.1099/vir.0.042978-0 [DOI] [PubMed] [Google Scholar]
- Hajjar AM, O’Mahony DS, Ozinsky A, Underhill DM, Aderem A, Klebanoff SJ, and Wilson CB (2001). Cutting edge: Functional interactions between toll-like receptor (TLR) 2 and TLR1 or TLR6 in response to phenol-soluble modulin. Journal of Immunology 166, 15–19. 10.4049/jimmunol.166.1.15 [DOI] [PubMed] [Google Scholar]
- Heine JW, Honess RW, Cassai E, and Roizman B (1974). Proteins specified by herpes simplex virus. XII. The virion polypeptides of type 1 strains. J Virol 14, 640–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horan KA, Hansen K, Jakobsen MR, Holm CK, Soby S, Unterholzner L, Thompson M, West JA, Iversen MB, Rasmussen SB, et al. (2013a). Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J Immunol 190, 2311–2319. 10.4049/jimmunol.1202749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horan KA, Hansen K, Jakobsen MR, Holm CK, Soby S, Unterholzner L, Thompson M, West JA, Iversen MB, Rasmussen SB, et al. (2013b). Proteasomal Degradation of Herpes Simplex Virus Capsids in Macrophages Releases DNA to the Cytosol for Recognition by DNA Sensors. J Immunol 190, 2311–2319. 10.4049/jimmunol.1202749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, You HJ, Su CH, Li YX, Chen SH, and Zheng CF (2018). Herpes Simplex Virus 1 Tegument Protein VP22 Abrogates cGAS/STING-Mediated Antiviral Innate Immunity. Journal of Virology 92. 10.1128/JVI.00841-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, and Barber GN (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–U674. 10.1038/nature07317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, and Barber GN (2009). STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–U740. 10.1038/nature08476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont B, and Roizman B (1975). Ribonucleic-Acid Synthesis in Cells Infected with Herpes-Simplex Virus - Characterization of Viral High Molecular-Weight Nuclear-Rna. Journal of General Virology 29, 155–165. 10.1099/0022-1317-29-2-155 [DOI] [PubMed] [Google Scholar]
- Johnson KE, Chikoti L, and Chandran B (2013). Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J Virol 87, 5005–5018. 10.1128/JVI.00082-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasneh GA, and Shukla D (2011). Herpes simplex virus infects most cell types in vitro: clues to its success. J Virol 8. 10.1186/1743-422x-8-481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, and Ben-Neriah Y (2000). Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol 18, 621–663. 10.1146/annurev.immunol.18.1.621 [DOI] [PubMed] [Google Scholar]
- Kawai T, and Akira S (2006). Innate immune recognition of viral infection. Nat Immunol 7, 131–137. 10.1038/ni1303 [DOI] [PubMed] [Google Scholar]
- Kawai T, and Akira S (2011). Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity 34, 637–650. 10.1016/j.immuni.2011.05.006 [DOI] [PubMed] [Google Scholar]
- Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, and Chandran B (2011). IFI16 Acts as a Nuclear Pathogen Sensor to Induce the Inflammasome in Response to Kaposi Sarcoma-Associated Herpesvirus Infection. Cell Host Microbe 9, 363–375. 10.1016/j.chom.2011.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil MI, Sommer MH, Hay J, Ruyechan WT, and Arvin AM (2015). Varicella-zoster virus (VZV) origin of DNA replication oriS influences origin-dependent DNA replication and flanking gene transcription. Virology 481, 179–186. 10.1016/j.virol.2015.02.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JA, Park SK, Seo SW, Lee CH, and Shin OS (2017). STING Is Involved in Antiviral Immune Response against VZV Infection via the Induction of Type I and III IFN Pathways. J Invest Dermatol 137, 2101–2109. 10.1016/j.jid.2017.03.041 [DOI] [PubMed] [Google Scholar]
- Koyanagi N, Imai T, Shindo K, Sato A, Fujii W, Ichinohe T, Takemura N, Kakuta S, Uematsu S, Kiyono H, et al. (2017). Herpes simplex virus-1 evasion of CD8(+) T cell accumulation contributes to viral encephalitis. J Clin Invest 127, 3784–3795. 10.1172/Jci92931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulej K, Avgousti DC, Sidoli S, Herrmann C, Della Fera AN, Kim ET, Garcia BA, and Weitzman MD (2017). Time-resolved Global and Chromatin Proteomics during Herpes Simplex Virus Type 1 (HSV-1) Infection. Mol Cell Proteomics 16, S92–S107. 10.1074/mcp.M116.065987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, and Finberg RW (2004). Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci U S A 101, 1315–1320. 10.1073/pnas.0308057100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahaye X, Gentili M, Silvin A, Conrad C, Picard L, Jouve M, Zueva E, Maurin M, Nadalin F, Knott GJ, et al. (2018). NONO Detects the Nuclear HIV Capsid to Promote cGAS-Mediated Innate Immune Activation. Cell 175, 488–501 e422. 10.1016/j.cell.2018.08.062 [DOI] [PubMed] [Google Scholar]
- Lamote JAS, Kestens M, Van Waesberghe C, Delva J, De Pelsmaeker S, Devriendt B, and Favoreel HW (2017). The Pseudorabies Virus Glycoprotein gE/gI Complex Suppresses Type I Interferon Production by Plasmacytoid Dendritic Cells. Journal of Virology 91. 10.1128/JVI.02276-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecker SH, Goldberg AL, and Mitch WE (2006). Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol 17, 1807–1819. 10.1681/ASN.2006010083 [DOI] [PubMed] [Google Scholar]
- Leoni V, Gianni T, Salvioli S, and Campadelli-Fiume G (2012). Herpes simplex virus glycoproteins gH/gL and gB bind Toll-like receptor 2, and soluble gH/gL is sufficient to activate NF-kappaB. J Virol 86, 6555–6562. 10.1128/JVI.00295-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie J, Rixon FJ, and McLauchlan J (1996). Overexpression of the herpes simplex virus type 1 tegument protein VP22 increases its incorporation into virus particles. Virology 220, 60–68. 10.1006/viro.1996.0286 [DOI] [PubMed] [Google Scholar]
- Lester SN, and Li K (2014). Toll-like receptors in antiviral innate immunity. J Mol Biol 426, 1246–1264. 10.1016/j.jmb.2013.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Chen J, and Cristea IM (2013a). Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe 14, 591–599. 10.1016/j.chom.2013.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Diner BA, Chen J, and Cristea IM (2012). Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc Natl Acad Sci U S A 109, 10558–10563. 10.1073/pnas.1203447109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XD, Wu J, Gao D, Wang H, Sun L, and Chen ZJ (2013b). Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341, 1390–1394. 10.1126/science.1244040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien E, Sellati TJ, Yoshimura A, Flo TH, Rawadi G, Finberg RW, Carroll JD, Espevik T, Ingalls RR, Radolf JD, et al. (1999). Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J Biol Chem 274, 33419–33425. 10.1074/jbc.274.47.33419 [DOI] [PubMed] [Google Scholar]
- Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, Jiang Y, Fei Y, Zhu C, Tan R, et al. (2018). Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 563, 131–136. 10.1038/s41586-018-0629-6 [DOI] [PubMed] [Google Scholar]
- Lum KK, Howard TR, Pan C, and Cristea IM (2019). Charge-Mediated Pyrin Oligomerization Nucleates Antiviral IFI16 Sensing of Herpesvirus DNA. mBio 10. 10.1128/mBio.01428-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum KK, Song B, Federspiel JD, Diner BA, Howard T, and Cristea IM (2018). Interactome and Proteome Dynamics Uncover Immune Modulatory Associations of the Pathogen Sensing Factor cGAS. Cell Syst 7, 627–642 e626. 10.1016/j.cels.2018.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund J, Sato A, Akira S, Medzhitov R, and Iwasaki A (2003). Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med 198, 513–520. 10.1084/jem.20030162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg P, Welander P, Hang X, and Cantin E (2003). Herpes simplex virus type 1 DNA is immunostimulatory in vitro and in vivo. Journal of Virology 77, 11158–11169. 10.1128/Jvi.77.20.11158-11169.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruzuru Y, Ichinohe T, Sato R, Miyake K, Okano T, Suzuki T, Koshiba T, Koyanagi N, Tsuda S, Watanabe M, et al. (2018). Herpes Simplex Virus 1 VP22 Inhibits AIM2-Dependent Inflammasome Activation to Enable Efficient Viral Replication. Cell Host Microbe 23, 254–265 e257. 10.1016/j.chom.2017.12.014 [DOI] [PubMed] [Google Scholar]
- McGeoch DJ, and Cook S (1994). Molecular phylogeny of the alphaherpesvirinae subfamily and a proposed evolutionary timescale. J Mol Biol 238, 9–22. 10.1006/jmbi.1994.1264 [DOI] [PubMed] [Google Scholar]
- McGeoch DJ, Cook S, Dolan A, Jamieson FE, and Telford EA (1995). Molecular phylogeny and evolutionary timescale for the family of mammalian herpesviruses. J Mol Biol 247, 443–458. 10.1006/jmbi.1995.0152 [DOI] [PubMed] [Google Scholar]
- Medzhitov R (2007). Recognition of microorganisms and activation of the immune response. Nature 449, 819–826. 10.1038/nature06246 [DOI] [PubMed] [Google Scholar]
- Melchjorsen J, Rintahaka J, Soby S, Horan KA, Poltajainen A, Ostergaard L, Paludan SR, and Matikainen S (2010). Early Innate Recognition of Herpes Simplex Virus in Human Primary Macrophages Is Mediated via the MDA5/MAVS-Dependent and MDA5/MAVS/RNA Polymerase III-Independent Pathways. Journal of Virology 84, 11350–11358. 10.1128/Jvi.01106-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melroe GT, Silva L, Schaffer PA, and Knipe DM (2007). Recruitment of activated IRF-3 and CBP/p300 to herpes simplex virus ICP0 nuclear foci: Potential role in blocking IFN-beta induction. Virology 360, 305–321. 10.1016/j.virol.2006.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr I, and Gluzman Y (1996). A herpesvirus genetic element which affects translation in the absence of the viral GADD34 function. Embo J 15, 4759–4766. 10.1002/j.1460-2075.1996.tb00853.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrone SR, Wang T, Constantoulakis LM, Hooy RM, Delannoy MJ, and Sohn J (2014). Cooperative assembly of IFI16 filaments on dsDNA provides insights into host defense strategy. Proc Natl Acad Sci U S A 111, E62–71. 10.1073/pnas.1313577111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvey M, Poppers J, Ladd A, and Mohr I (1999). A herpesvirus ribosome-associated, RNA-binding protein confers a growth advantage upon mutants deficient in a GADD34-related function. Journal of Virology 73, 3375–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nour AM, Reichelt M, Ku CC, Ho MY, Heineman TC, and Arvin AM (2011). Varicella-zoster virus infection triggers formation of an interleukin-1beta (IL-1beta)-processing inflammasome complex. J Biol Chem 286, 17921–17933. 10.1074/jbc.M110.210575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogunjimi B, Zhang SY, Sorensen KB, Skipper KA, Carter-Timofte M, Kerner G, Luecke S, Prabakaran T, Cai Y, Meester J, et al. (2017). Inborn errors in RNA polymerase III underlie severe varicella zoster virus infections. J Clin Invest 127, 3543–3556. 10.1172/Jci92280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oroskar AA, and Read GS (1987). A Mutant of Herpes-Simplex Virus Type-1 Exhibits Increased Stability of Immediate-Early (Alpha) Messenger-Rnas. Journal of Virology 61, 604–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oroskar AA, and Read GS (1989). Control of Messenger-Rna Stability by the Virion Host Shutoff Function of Herpes-Simplex Virus. Journal of Virology 63, 1897–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzalli MH, Broekema NM, Diner BA, Hancks DC, Elde NC, Cristea IM, and Knipe DM (2015). cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. P Natl Acad Sci USA 112, E1773–E1781. 10.1073/pnas.1424637112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzalli MH, Broekema NM, and Knipe DM (2016). Relative Contributions of Herpes Simplex Virus 1 ICP0 and vhs to Loss of Cellular IFI16 Vary in Different Human Cell Types. J Virol 90, 8351–8359. 10.1128/JVI.00939-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzalli MH, Conwell SE, Berrios C, DeCaprio JA, and Knipe DM (2013). Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc Natl Acad Sci U S A 110, E4492–4501. 10.1073/pnas.1316194110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzalli MH, DeLuca NA, and Knipe DM (2012). Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci U S A 109, E3008–3017. 10.1073/pnas.1211302109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paludan SR, Bowie AG, Horan KA, and Fitzgerald KA (2011). Recognition of herpesviruses by the innate immune system. Nature Reviews Immunology 11, 143–154. 10.1038/nri2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker ZM, Murphy AA, and Leib DA (2015). Role of the DNA Sensor STING in Protection from Lethal Infection following Corneal and Intracerebral Challenge with Herpes Simplex Virus 1. Journal of Virology 89, 11080–11091. 10.1128/Jvi.00954-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson J, Lees-Miller SP, and Everett RD (1999). Herpes simplex virus type 1 immediate-early protein Vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. Journal of Virology 73, 650–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peri P, Mattila RK, Kantola H, Broberg E, Karttunen HS, Waris M, Vuorinen T, and Hukkanen V (2008). Herpes Simplex Virus Type 1 Us3 Gene Deletion Influences Toll-like Receptor Responses in Cultured Monocytic Cells. Virol J 5. 10.1186/1743-422x-5-140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters GA, Khoo D, Mohr I, and Sen GC (2002). Inhibition of PACT-mediated activation of PKR by the herpes simplex virus type 1 Us11 protein. J Virol 76, 11054–11064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platanias LC (2005). Mechanisms of type-I-and type-II-interferon-mediated signalling. Nat Rev Immunol 5, 375–386. 10.1038/nri1604 [DOI] [PubMed] [Google Scholar]
- Pomerantz JL, and Baltimore D (1999). NF-kappa B activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. Embo J 18, 6694–6704. 10.1093/emboj/18.23.6694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon AP, Gu H, and Roizman B (2006). ICP0 and the US3 protein kinase of herpes simplex virus 1 independently block histone deacetylation to enable gene expression. Proc Natl Acad Sci U S A 103, 9993–9998. 10.1073/pnas.0604142103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read GS, and Frenkel N (1983). Herpes-Simplex Virus Mutants Defective in the Virion-Associated Shutoff of Host Polypeptide-Synthesis and Exhibiting Abnormal Synthesis of Alpha-(Immediate Early) Viral Polypeptides. Journal of Virology 46, 498–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reske A, Pollara G, Krummenacher C, Chain BM, and Katz DR (2007). Understanding HSV-1 entry glycoproteins. Rev Med Virol 17, 205–215. 10.1002/rmv.531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas HG, Schmaling SK, and Gaglia MM (2016). Shutoff of Host Gene Expression in Influenza A Virus and Herpesviruses: Similar Mechanisms and Common Themes. Viruses 8, 102. 10.3390/v8040102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roers A, Hiller B, and Hornung V (2016). Recognition of Endogenous Nucleic Acids by the Innate Immune System. Immunity 44, 739–754. 10.1016/j.immuni.2016.04.002 [DOI] [PubMed] [Google Scholar]
- Romano PR, Garcia-Barrio MT, Zhang XL, Wang QZ, Taylor DR, Zhang F, Herring C, Mathews MB, Qin J, and Hinnebusch AG (1998). Autophosphorylation in the activation loop is required for full kinase activity in vivo of human and yeast eukaryotic initiation factor 2 alpha kinases PKR and GCN2. Mol Cell Biol 18, 2282–2297. 10.1128/Mcb.18.4.2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saira K, Zhou Y, and Jones C (2007). The infected cell protein 0 encoded by bovine herpesvirus 1 (bICP0) induces degradation of interferon response factor 3 and, consequently, inhibits beta interferon promoter activity. J Virol 81, 3077–3086. 10.1128/JVI.02064-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, Matsunaga K, Kageyama S, Omori H, Noda T, et al. (2009). Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. P Natl Acad Sci USA 106, 20842–20846. 10.1073/pnas.0911267106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato A, Linehan MM, and Iwasaki A (2006). Dual recognition of herpes simplex viruses by TLR2 and TLR9 in dendritic cells. Proc Natl Acad Sci U S A 103, 17343–17348. 10.1073/pnas.0605102103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato R, Kato A, Chimura T, Saitoh SI, Shibata T, Murakami Y, Fukui R, Liu KW, Zhang Y, Arii J, et al. (2018). Combating herpesvirus encephalitis by potentiating a TLR3-mTORC2 axis. Nature Immunology 19, 1071–+. 10.1038/s41590-018-0203-2 [DOI] [PubMed] [Google Scholar]
- Schlieker C, Korbel GA, Kattenhorn LM, and Ploegh HL (2005). A deubiquitinating activity is conserved in the large tegument protein of the herpesviridae. J Virol 79, 15582–15585. 10.1128/JVI.79.24.15582-15585.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, and Rice CM (2014). Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32, 513–545. 10.1146/annurev-immunol-032713-120231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, and Rice CM (2011). A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485. 10.1038/nature09907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott J, and Stoecklin G (2010). Networks controlling mRNA decay in the immune system. Wires Rna 1, 432–456. 10.1002/wrna.13 [DOI] [PubMed] [Google Scholar]
- Schwerk J, and Sayan R (2015). Translating the Untranslated Region. Journal of Immunology 195, 2963–2971. 10.4049/jimmunol.1500756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N, Sommer M, Che XB, White K, Ruyechan WT, and Arvin AM (2010). Varicella-Zoster Virus Immediate-Early Protein 62 Blocks Interferon Regulatory Factor 3 (IRF3) Phosphorylation at Key Serine Residues: a Novel Mechanism of IRF3 Inhibition among Herpesviruses. Journal of Virology 84, 9240–9253. 10.1128/Jvi.01147-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo GJ, Yang A, Tan B, Kim S, Liang Q, Choi Y, Yuan W, Feng P, Park HS, and Jung JU (2015). Akt Kinase-Mediated Checkpoint of cGAS DNA Sensing Pathway. Cell Rep 13, 440–449. 10.1016/j.celrep.2015.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira L, Ralph M, Tomer E, Cohen S, and Kobiler O (2016). Histone Deacetylase Inhibitors Reduce the Number of Herpes Simplex Virus-1 Genomes Initiating Expression in Individual Cells. Front Microbiol 7, 1970. 10.3389/fmicb.2016.01970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen G, Wang K, Wang S, Cai M, Li ML, and Zheng C (2014). Herpes simplex virus 1 counteracts viperin via its virion host shutoff protein UL41. J Virol 88, 12163–12166. 10.1128/JVI.01380-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu M, Taddeo B, and Roizman B (2015). Tristetraprolin Recruits the Herpes Simplex Virion Host Shutoff RNase to AU-Rich Elements in Stress Response mRNAs To Enable Their Cleavage. J Virol 89, 5643–5650. 10.1128/JVI.00091-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen LN, Reinert LS, Malmgaard L, Bartholdy C, Thomsen AR, and Paludan SR (2008). TLR2 and TLR9 synergistically control herpes simplex virus infection in the brain. J Immunol 181, 8604–8612. [DOI] [PubMed] [Google Scholar]
- Stratmann SA, Morrone SR, van Oijen AM, and Sohn J (2015). The innate immune sensor IFI16 recognizes foreign DNA in the nucleus by scanning along the duplex. Elife 4. 10.7554/eLife.11721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C, Zhang J, and Zheng C (2015). Herpes simplex virus 1 UL41 protein abrogates the antiviral activity of hZAP by degrading its mRNA. Virol J 12, 203. 10.1186/s12985-015-0433-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C, and Zheng C (2017). Herpes Simplex Virus 1 Abrogates the cGAS/STING-Mediated Cytosolic DNA-Sensing Pathway via Its Virion Host Shutoff Protein, UL41. J Virol 91. 10.1128/JVI.02414-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Zhang Q, Jing YY, Zhang M, Wang HY, Cai Z, Liuyu T, Zhang ZD, Xiong TC, Wu Y, et al. (2017). USP13 negatively regulates antiviral responses by deubiquitinating STING. Nat Commun 8, 15534. 10.1038/ncomms15534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun LJ, Wu JX, Du FH, Chen X, and Chen ZJJ (2013). Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 339, 786–791. 10.1126/science.1232458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun WX, Li Y, Chen L, Chen HH, You FP, Zhou X, Zhou Y, Zhai ZH, Chen DY, and Jiang ZF (2009). ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. P Natl Acad Sci USA 106, 8653–8658. 10.1073/pnas.0900850106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Kaisho T, and Akira S (2003). Toll-like receptors. Annu Rev Immunol 21, 335–376. 10.1146/annurev.immunol.21.120601.141126 [DOI] [PubMed] [Google Scholar]
- Tanaka Y, and Chen ZJJ (2012). STING Specifies IRF3 Phosphorylation by TBK1 in the Cytosolic DNA Signaling Pathway. Sci Signal 5. 10.1126/scisignal.2002521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Liu YX, Liu XX, Sun X, Zhang JK, and Qu LD (2018). Feline Herpesvirus 1 US3 Blocks the Type I Interferon Signal Pathway by Targeting Interferon Regulatory Factor 3 Dimerization in a Kinase-Independent Manner. Journal of Virology 92. 10.1128/JVI.00047-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin TC, Latz E, Xiao TS, et al. (2010). IFI16 is an innate immune sensor for intracellular DNA. Nature Immunology 11, 997–U942. 10.1038/ni.1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Lint AL, Murawski MR, Goodbody RE, Severa M, Fitzgerald KA, Finberg RW, Knipe DM, and Kurt-Jones EA (2010). Herpes Simplex Virus Immediate-Early ICP0 Protein Inhibits Toll-Like Receptor 2-Dependent Inflammatory Responses and NF-kappa B Signaling. Journal of Virology 84, 10802–10811. 10.1128/Jvi.00063-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandevenne P, Lebrun M, El Mjiyad N, Ote I, Di Valentin E, Habraken Y, Dortu E, Piette J, and Sadzot-Delvaux C (2011). The Varicella-Zoster Virus ORF47 Kinase Interferes with Host Innate Immune Response by Inhibiting the Activation of IRF3. Plos One 6. 10.1371/journal.pone.0016870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verpooten D, Ma YJ, Hou SW, Yan ZP, and He B (2009). Control of TANK-binding Kinase 1-mediated Signaling by the gamma(1)34.5 Protein of Herpes Simplex Virus 1. J Biol Chem 284, 1097–1105. 10.1074/jbc.M805905200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Roretz C, Di Marco S, Mazroui R, and Gallouzi IE (2011). Turnover of AU-rich-containing mRNAs during stress: a matter of survival. Wiley Interdiscip Rev RNA 2, 336–347. 10.1002/wrna.55 [DOI] [PubMed] [Google Scholar]
- Walters MS, Erazo A, Kinchington PR, and Silverstein S (2009). Histone Deacetylases 1 and 2 Are Phosphorylated at Novel Sites during Varicella-Zoster Virus Infection. Journal of Virology 83, 11502–11513. 10.1128/Jvi.01318-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters MS, Kinchington PR, Banfield BW, and Silverstein S (2010). Hyperphosphorylation of histone deacetylase 2 by alphaherpesvirus US3 kinases. J Virol 84, 9666–9676. 10.1128/JVI.00981-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JP, Kurt-Jones EA, Shin OS, Manchak MD, Levin MJ, and Finberg RW (2005). Varicella-zoster virus activates inflammatory cytokines in human monocytes and macrophages via Toll-like receptor 2. J Virol 79, 12658–12666. 10.1128/JVI.79.20.12658-12666.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KZ, Ni LW, Wang S, and Zheng CF (2014). Herpes Simplex Virus 1 Protein Kinase US3 Hyperphosphorylates p65/RelA and Dampens NF-kappa B Activation. Journal of Virology 88, 7941–7951. 10.1128/Jvi.03394-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wen M, and Cao X (2019). Nuclear hnRNPA2B1 initiates and amplifies the innate immune response to DNA viruses. Science 365. 10.1126/science.aav0758 [DOI] [PubMed] [Google Scholar]
- Wang S, Wang KZ, Li J, and Zheng CF (2013a). Herpes Simplex Virus 1 Ubiquitin-Specific Protease UL36 Inhibits Beta Interferon Production by Deubiquitinating TRAF3. Journal of Virology 87, 11851–11860. 10.1128/Jvi.01211-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Wang KZ, Lin RT, and Zheng CF (2013b). Herpes Simplex Virus 1 Serine/Threonine Kinase US3 Hyperphosphorylates IRF3 and Inhibits Beta Interferon Production. Journal of Virology 87, 12814–12827. 10.1128/Jvi.02355-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber F, Wagner V, Rasmussen SB, Hartmann R, and Paludan SR (2006). Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol 80, 5059–5064. 10.1128/JVI.80.10.5059-5064.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JA, Gregory SM, and Damania B (2012). Toll-like receptor sensing of human herpesvirus infection. Front Cell Infect Microbiol 2, 122. 10.3389/fcimb.2012.00122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmann O, Ahmad-Nejad P, Neumaier M, Hennerici MG, and Fatar M (2010). Toll-Like Receptor 3 Immune Deficiency May Be Causative for HSV-2-Associated Mollaret Meningitis. Eur Neurol 63, 249–251. 10.1159/000287585 [DOI] [PubMed] [Google Scholar]
- Wu JX, Sun LJ, Chen X, Du FH, Shi HP, Chen C, and Chen ZJJ (2013). Cyclic GMP-AMP Is an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Science 339, 826–830. 10.1126/science.1229963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing JJ, Ni LW, Wang S, Wang KZ, Lin RT, and Zheng CF (2013). Herpes Simplex Virus 1-Encoded Tegument Protein VP16 Abrogates the Production of Beta Interferon (IFN) by Inhibiting NF-kappa B Activation and Blocking IFN Regulatory Factor 3 To Recruit Its Coactivator CBP. Journal of Virology 87, 9788–9801. 10.1128/Jvi.01440-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing JJ, Wang S, Lin RT, Mossman KL, and Zheng CF (2012). Herpes Simplex Virus 1 Tegument Protein US11 Downmodulates the RLR Signaling Pathway via Direct Interaction with RIG-I and MDA-5. Journal of Virology 86, 3528–3540. 10.1128/Jvi.06713-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye RJ, Su CH, Xu HY, and Zheng CF (2017). Herpes Simplex Virus 1 Ubiquitin-Specific Protease UL36 Abrogates NF-kappa B Activation in DNA Sensing Signal Pathway. Journal of Virology 91. 10.1128/JVI.02417-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu HR, Huang HC, Kuo HC, Sheen JM, Ou CY, Hsu TY, and Yang KD (2011). IFN-alpha production by human mononuclear cells infected with varicella-zoster virus through TLR9-dependent and -independent pathways. Cell Mol Immunol 8, 181–188. 10.1038/cmi.2010.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, You J, You H, and Zheng CF (2018). Herpes Simplex Virus 1 UL36USP Antagonizes Type I Interferon-Mediated Antiviral Innate Immunity. Journal of Virology 92. 10.1128/JVI.01161-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Su C, and Zheng C (2016). Herpes Simplex Virus 1 Serine Protease VP24 Blocks the DNA-Sensing Signal Pathway by Abrogating Activation of Interferon Regulatory Factor 3. J Virol 90, 5824–5829. 10.1128/JVI.00186-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wang K, Wang S, and Zheng C (2013). Herpes simplex virus 1 E3 ubiquitin ligase ICP0 protein inhibits tumor necrosis factor alpha-induced NF-kappaB activation by interacting with p65/RelA and p50/NF-kappaB1. J Virol 87, 12935–12948. 10.1128/JVI.01952-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhao J, Xu S, Li J, He S, Zeng Y, Xie L, Xie N, Liu T, Lee K, et al. (2018). Species-Specific Deamidation of cGAS by Herpes Simplex Virus UL37 Protein Facilitates Viral Replication. Cell Host Microbe 24, 234–248 e235. 10.1016/j.chom.2018.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Xu A, Qin C, Zhang Q, Chen S, Lang Y, Wang M, Li C, Feng W, Zhang R, et al. (2017). Pseudorabies Virus dUTPase UL50 Induces Lysosomal Degradation of Type I Interferon Receptor 1 and Antagonizes the Alpha Interferon Response. J Virol 91. 10.1128/JVI.01148-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, et al. (2007). TLR3 deficiency in patients with herpes simplex encephalitis. Science 317, 1522–1527. 10.1126/science.1139522 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Yuan B, Bao M, Lu N, Kim T, and Liu YJ (2011). The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol 12, 959–965. 10.1038/ni.2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Zeng Y, Xu S, Chen J, Shen G, Yu C, Knipe D, Yuan W, Peng J, Xu W, et al. (2016). A Viral Deamidase Targets the Helicase Domain of RIG-I to Block RNA-Induced Activation. Cell Host Microbe 20, 770–784. 10.1016/j.chom.2016.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao FC, Lei CQ, He X, Zhang L, Tien P, et al. (2008). The Adaptor Protein MITA Links Virus-Sensing Receptors to IRF3 Transcription Factor Activation. Immunity 29, 538–550. 10.1016/j.immuni.2008.09.003 [DOI] [PubMed] [Google Scholar]
- Zhu H, Zheng C, Xing J, Wang S, Li S, Lin R, and Mossman KL (2011). Varicella-zoster virus immediate-early protein ORF61 abrogates the IRF3-mediated innate immune response through degradation of activated IRF3. J Virol 85, 11079–11089. 10.1128/JVI.05098-11 [DOI] [PMC free article] [PubMed] [Google Scholar]