

Abstract
The FDA recently approved eight targeted therapies for acute myeloid leukemia (AML), including the BCL-2 inhibitor venetoclax. Maximizing efficacy of these treatments requires refining patient selection. To this end, we analyzed two recent AML studies profiling the gene expression and ex vivo drug response of primary patient samples. We find that ex vivo samples often exhibit a general sensitivity to (any) drug exposure, independent of drug target. We observe that this “general response across drugs” (GRD) is associated with FLT3-ITD mutations, clinical response to standard induction chemotherapy, and overall survival. Further, incorporating GRD into expression-based regression models trained on one of the studies improved their performance in predicting ex vivo response in the second study, thus signifying its relevance to precision oncology efforts. We find that venetoclax response is independent of GRD but instead show that it is linked to expression of monocyte-associated genes by developing and applying a multi-source Bayesian regression approach. The method shares information across studies to robustly identify biomarkers of drug response and is broadly applicable in integrative analyses.
Subject terms: High-throughput screening, Acute myeloid leukaemia, Cancer genomics, Gene expression analysis, Predictive markers
Introduction
Acute myeloid leukemia (AML) is genetically, epigenetically, and transcriptionally heterogeneous. Nevertheless, patient treatment has been uniform and for decades standard first-line treatment has been “7 + 3” induction chemotherapy with cytarabine and an anthracycline. In addition, a significant portion of AML patients are not considered fit enough to tolerate induction chemotherapy and are instead typically treated with low-dose cytarabine (LDAC) and hypomethylating agents (HMAs), e.g., azacitidine or decitabine, that extend survival but are rarely curative. Together, outcomes for adult AML patients (aged ≥20 years) remain very poor, with the current 5-year relative survival rate at 24%1. The US Food and Drug Administration (FDA) has recently shifted this therapeutic landscape by approving eight drugs (enasidenib, gemtuzumab ozogamicin, glasdegib, ivosidenib, midostaurin, gilteritinib, venetoclax, and a liposome-encapsulated combination of daunorubicin and cytarabine) for newly diagnosed or relapse refractory patients, alone or in combination with LDAC or HMAs2,3. Maximizing clinical benefit from these treatments will require biomarkers for optimized patient selection and rational drug combination strategies.
We used data from two independent studies that comprehensively profiled AML patient cohorts to address this need. In both studies, ex vivo functional drug testing was performed on freshly isolated mononuclear cells from AML patients and cell viability was assessed following drug exposure across a concentration range. The multi-center Beat AML initiative, led by Oregon Health & Science University (OHSU), profiled 562 patient samples, including 411 with RNA sequencing (RNA-seq), 531 with exome sequencing, and 363 across a panel of 122 small-molecule inhibitors4. The AML Individualized Systems Medicine program at the Institute for Molecular Medicine Finland (FIMM) profiled 37 patients using RNA-seq, exome sequencing, and across a panel of 470 inhibitors5. We performed a comparative analysis, training regression models in one study to predict ex vivo drug response in the other. In both studies, we observed a tendency of a patient-derived sample to respond consistently across all drugs. This “general response across drugs” (GRD) correlated with complete response to standard induction therapy and with patient leukemia-free survival. Including GRD in the models significantly improved their performance.
Several drugs did not conform to this trend, including the BCL-2 inhibitor venetoclax6, which was recently approved in combination with HMAs or LDAC for the treatment of AML in newly diagnosed elderly patients or in those unfit for intensive chemotherapy. Venetoclax in combination with azacitidine or decitabine showed a favorable overall response rate [complete remission (CR) + CR with incomplete count recovery (CRi)] of 67% in a phase 1b study of 145 patients aged ≥65 years with newly diagnosed AML7, whereas an overall response rate of 28% has been observed for a similar patient population treated with azacitidine alone8. Nevertheless, a large minority of patients do not respond to venetoclax, while the majority who do ultimately relapse7.
As such, recent efforts have attempted to refine patient selection for venetoclax treatment. An ex vivo study9 suggested that patients with monocytic AML have reduced sensitivity to venetoclax. An in vivo study10 additionally demonstrated that intra-patient heterogeneity arising from monocytic subclones contributes to venetoclax resistance. Here we show that the degree of patient monocyticity accounts for the majority of inter-patient variation in resistance. We quantify this effect through a robust signature comprised of monocyte-associated genes identified via a Bayesian multi-source regression (BMSR) method. BMSR nominates drug biomarkers by performing a joint multi-source analysis across the OHSU and FIMM datasets. The method is broadly applicable in integrating multiple expression datasets to overcome technical variation, biological heterogeneity, and small sample size that contribute to the low reproducibility of biomarker studies11.
We extend BMSR to additionally perform simultaneous multi-task analysis across multiple drugs to identify their common biomarkers. Applying it to three mitogen-activated protein kinase kinase (MEK) inhibitors (trametinib, PD184352, and selumetinib) reveals that their response is correlated with the monocytic venetoclax resistance signature. This, coupled with the observation that venetoclax treatment selects for pre-existing monocytic subclones10, provides independent rationale for combination therapies targeting the BCL-2 and MEK pathways12.
Results
GRD is associated with improved patient outcome
The 122 OHSU drug panel and the 470 FIMM drug panel shared 87 drugs (71 of which are kinase inhibitors). We used these common drugs to assess the consistency of drug response across the two studies. We first quantified response as area under the dose–response curve (AUC; Supplementary Figs. 1 and 2, Supplementary Table 1, and Fig. 1). Responses were positively correlated across drugs in each dataset [OHSU: 84% of Pearson correlation rs are positive, mean r = 0.22, 95% confidence interval (CI) = −0.22 to 0.59; FIMM: 85% positive r, mean r = 0.33, 95% CI = −0.29 to 0.83; Supplementary Fig. 3]. We measured the cross-study correlation of the intra-study drug–drug correlations, i.e., the “correlation of correlations,” a general measure of interstudy consistency (r = 0.35, p < 10−10; Supplementary Fig. 3). As expected, correlations were higher when restricted to drugs with a common target, including for 5 cyclin-dependent kinase inhibitors (r = 0.76), 5 epidermal growth factor receptor inhibitors (r = 0.51), 7 vascular endothelial growth factor receptor inhibitors (r = 0.61), 6 fms-like tyrosine kinase 3 (FLT3) inhibitors (r = 0.84), and 4 mammalian target of rapamycin/phosphoinositide 3-kinase inhibitors (r = 0.95). We also calculated each drug’s mean response across patients and found these to be highly concordant between the datasets (r = 0.67; p = 2.04 × 10−12; Supplementary Fig. 4). Finally, we assessed the consistency of each individual drug’s response across the two datasets. To do so, we represented each drug in each dataset by a vector of its correlations to all other drugs. For each drug, we then calculated the correlation between these dataset- and drug-specific correlation vectors (Supplementary Fig. 5). This cross-dataset drug correlation was positively associated with the drug’s range of response [i.e., interquartile range (IQR) of unnormalized AUCs] in both the OHSU (r = 0.45; p = 1.02 × 10−5) and FIMM (r = 0.47; p = 4.55 × 10−6) datasets. We found no evidence that drug correlation was associated with its class (analysis of variance p = 0.37).
Fig. 1. A patient’s ex vivo responses are similar across common drugs.

AUCs calculated across patient-derived ex vivo samples (columns) and 87 drugs (rows) common to OHSU (a; n = 338) and FIMM (b; n = 37) datasets. Red values correspond to higher AUC or more sensitive samples, blue are less sensitive, black are filtered, and gray are missing. Standardized AUCs (i.e., with mean zero and standard deviation one across patients) displayed in heatmap. Raw AUCs displayed in top and side panels. General response across drugs (GRD) is mean of raw AUCs for an individual patient over drugs; mean response across patients (MRP) is mean of raw AUCs for an individual drug over patients. Samples ordered by GRD in each dataset. Drugs ordered by MRP in OHSU dataset. One sample displayed per patient, with sample assayed across the highest number of drugs displayed in cases with multiple samples per patient. Class drug class, Diff/epi differentiation/epigenetic, HSP heat shock protein.
Each patient sample tended to respond uniformly across all drugs in the panel—e.g., many responded relatively poorly to all drugs (blue; leftmost samples, Fig. 1), while a smaller number responded relatively strongly to most drugs (red; rightmost samples, Fig. 1). We describe this phenomenon as a sample’s GRD, i.e., its mean AUC across all drugs. Notably, this trend held also across the full drug panels of each dataset (OHSU: 122 drugs; FIMM: 470 drugs; Supplementary Fig. 6), which are less biased toward tyrosine kinase inhibitors (TKIs) than the shared set of drugs. More specifically, we found that GRD computed across all drugs in a dataset was strongly correlated (r > 0.9) with that computed from the common set of drugs, the set of drugs that excludes class III TKIs (of which FLT3 inhibitors are members), and the set of drugs that excludes all TKIs (Supplementary Fig. 7). Consistently, we found that GRD calculated from a random selection of drugs was highly concordant with GRD calculated from all drugs in the respective dataset for both the OHSU (mean r across 100 bootstrap samples = 0.97; 95% CI = 0.95–0.98) and FIMM (mean r = 0.99; 95% CI = 0.98–1.00) datasets. An observation similar to GRD, General Levels of Drug Sensitivity (GLDS), has previously been reported across cell lines representing various cancer types13.
GRD was higher in samples from patients who achieved a CR or a CRi to standard induction chemotherapy relative to those refractory to induction (two-sided Wilcoxon rank-sum test p = 0.02; Fig. 2a; GRD computed across common drugs). Consistently, CR/CRi patients were enriched among those with high GRD (enrichment p = 7.5 × 10−3; Supplementary Fig. 8). Notably, patients in the top quartile of GRD showed improved overall survival relative to those in the bottom quartile [Cox proportional hazard ratio (HR) = 0.96, log-rank p = 0.01; Fig. 2b and Supplementary Fig. 9]. These findings held when GRD was instead computed across all drugs (two-sided Wilcoxon rank-sum test p = 8.2 × 10−4; HR = 0.95; log-rank p = 0.01; Supplementary Fig. 10).
Fig. 2. Ex vivo general response across drugs is associated with clinical response and improved patient outcome.

a GRD in patients who achieve complete remission (CR) or complete remission with incomplete hematologic recovery (CRi) to standard induction chemotherapy (n = 118) versus those refractory to induction (n = 50) in OHSU dataset. *Wilcoxon rank-sum test p < 0.05. b Kaplan–Meier survival curves of patients in OHSU dataset with GRD above the upper quartile (red; “responders”; n = 42) and of those with GRD below the lower quartile (blue; “non-responders”; n = 42). Data are right censored at 610 days. HR: Cox proportional hazard ratio. a, b GRD is computed across drugs common to OHSU and FIMM datasets.
Despite these associations, refractory patients were not statistically enriched in extreme GRD values (enrichment p = 0.15; Supplementary Fig. 8). To understand this heterogeneity in response, we examined clinical features, genes, and pathways that differentiated refractory from CR/CRi patients among those with low (bottom quartile) GRD. Increased age was associated with refractory response among GRD-low patients (two-sided Wilcoxon rank-sum test p = 9.0 × 10−3), though the trend was not significant after correcting for testing of the multiple clinical variables [Benjamini–Hochberg (BH)-adjusted p = 0.26; Supplementary Table 2]. One hundred and twenty-two genes were differentially expressed (false discovery rate (FDR) < 20%; Supplementary Tables 3 and 4), with lymphocyte costimulation, T cell receptor signaling, antigen binding, and antigen receptor-mediated signaling (all significant at an FDR < 20%) upregulated in refractory patients and among the strongest enrichments (Supplementary Table 5). Conversely, in examining GRD-high patients, we found that higher creatinine levels trended with CR/CRi (two-sided Wilcoxon rank-sum test p = 0.02; BH-adjusted p = 0.62; Supplementary Table 6). Forty-six differentially expressed genes (FDR < 20%; Supplementary Table 7) were most strongly enriched within extracellular pathways upregulated in CR/CRi patients (Supplementary Table 8).
GRD is associated with FLT3-ITD
Presence of internal tandem duplication in FLT3 (FLT3-ITD) was positively associated with GRD in the OHSU dataset (BH-adjusted two-sided Wilcoxon rank-sum test p = 6.86 × 10−8; Supplementary Table 9). This positive association remained significant and independent of NPM1 mutation status, ethnicity, age, and sex in a multivariate analysis [R2 = 0.26; F-statistic p = 6.97 × 10−10; two-sided t test p = 4.95 × 10−8; Supplementary Fig. 11 and Supplementary Table 10). We validated the FLT3-ITD/GRD association in an independent AML dataset profiling expression and drug response of ex vivo samples published by Tavor and colleagues14 (one-sided Wilcoxon rank-sum test p < 0.01; Supplementary Fig. 12). We also observed a consistent trend in the FIMM dataset (one-sided Wilcoxon rank-sum test p = 0.08). Significantly, we found that FLT3-ITD status remained associated with GRD even when the latter was computed from a subset of drugs that excluded FLT3 inhibitors (Supplementary Fig. 13).
To determine whether GRD could be modeled using gene expression data, we trained an expression-based ridge regression model of GRD using the OHSU dataset (Supplementary Figs. 14–19; Supplementary Table 3; see “Methods”). The model was validated in the FIMM dataset, demonstrating good predictive performance (r = 0.67; p = 5.6 × 10−6; Fig. 3a). We hypothesized that robust biomarkers of GRD should be consistent between the OHSU and FIMM datasets (e.g., having large positive model coefficients in both datasets relative to other genes). To test this, we trained a GRD model on the FIMM dataset and compared the model coefficients associated with each gene between the OHSU- and FIMM-trained models. Unexpectedly, this did not reveal candidate biomarkers with outlying coefficients in both datasets (Fig. 3b). Nevertheless, we did confirm that the ABCB1 gene [i.e., Multidrug Resistance Protein 1 (MDR1)], encoding a drug efflux pump and previously observed to be associated with GLDS13, was negatively correlated with GRD in both datasets (Fig. 3b). Further, the ABC transporter family was among the gene sets and pathways having the strongest negative association with GRD (Supplementary Tables 11–15).
Fig. 3. Integrative analysis reveals growth-, apoptosis-, and drug efflux-associated biomarkers of general response across drugs.

a, c Observed (x axis) versus model-predicted (y axis) GRD. a Expression-based ridge regression model trained on OHSU samples (n = 292) and tested on FIMM samples (n = 37). c Expression-based Bayesian multi-source regression (BMSR) model trained using fivefold cross-validation on combined OHSU and FIMM datasets (n = 263) and tested on held-out fold yielding median performance across the fivefolds (n = 66). b, d Coefficients of genes (n = 2132) in OHSU (x axis) or FIMM (y axis) datasets following b training of ridge regression model independently on both datasets or d training of BMSR model simultaneously on entirety of both datasets (n = 329). GRD is computed across drugs common to OHSU and FIMM datasets. r: Pearson correlation; dashed line: identity line; blue line: linear regression fit; gray shading: 95% confidence interval. Labeled genes were those having extremal (Stouffer’s p < 0.01) combined coefficients across both datasets, as well as ABCB1 (i.e., MDR1).
BMSR identifies biomarkers jointly across studies
We reasoned that our inability to detect consistent biomarkers through independent analysis of the two datasets resulted from the highly correlated expression of subsets of genes (e.g., within a pathway), thereby dampening the effect of any single gene and hindering the identification of genes significantly impacting response. This is further complicated by study site-specific technical artifacts, biological variation, and small sample sizes, all of which compound noise. These factors could be ameliorated by performing regularization across multiple datasets simultaneously, which would shrink the coefficients of all but one or a few of the highly correlated genes toward zero.
To do so, we developed an integrative BMSR method that detects consistent and robust biomarkers through joint analysis of the two datasets. BMSR assumes that the biomarker expression/drug response relationship is similar across multiple datasets but allows for relatively small differences due to technical noise, biological variation, or clinical heterogeneity of different patient populations. It achieves this by modeling the contribution to the response by gene g in dataset d (i.e., the regression coefficient ) as arising from a mean contribution (i.e., βg) for that gene that is shared across datasets (see “Methods”). BMSR effectively performs regularization on the shared βg rather than the dataset-specific coefficient through a prior distribution, which acts as a penalty term in a non-Bayesian/frequentist formulation. For two genes g1 and g2 that are correlated with response, this approach encourages (but does not guarantee) that the shared coefficient of one of them be shrunk to zero by exploiting correlations in the data rather than explicit biological annotations (e.g., pathways).
Modeling GRD with the BMSR approach (Fig. 3c) revealed candidate biomarkers (Fig. 3d and Supplementary Fig. 20) that were distinctly separated from the bulk of non-contributing genes (i.e., those with coefficients near zero in both datasets). Candidate biomarkers included genes involved in cell proliferation [IL7R15 and NIBAN216], cell cycle [MAPK1217], cell growth [BCL218 and S100A8/S100A919], apoptosis [BCL220, NIBAN221, S100A8/S100A922], and drug response [BCL223]). Notably, these genes were near the periphery of the ridge coefficient distribution (Fig. 3b), thus demonstrating the consistency of the two methods. As expected, candidate GRD biomarkers were also consistently correlated with response to individual drugs (Supplementary Fig. 21).
Drug response is robustly predicted by gene expression
Responses could be significantly predicted (p < 0.01) using expression-based ridge regression for 31 of the 87 drugs (Fig. 4 and Supplementary Table 16; median r = 0.33; 25th–75th percentile = 0.04–0.51; all significant correlations were positive; see “Methods”). Significantly predicted drugs included the heat shock protein inhibitor tanespimycin, the immunomodulatory agent lenalidomide, the bromodomain inhibitor JQ1, two apoptotic modulators (nutlin-3 and venetoclax), and 26 kinase inhibitors. Prediction performance was correlated with the spread of response (i.e., IQR of unnormalized AUCs) in the FIMM validation dataset (Supplementary Fig. 22; r = 0.36; p = 6.19 × 10−4). These results reinforce the consistency of the two ex vivo studies demonstrated by the drug–drug correlations above.
Fig. 4. Expression-based predictions of drug response indicate concordance of independent ex vivo datasets and may be improved by incorporating general response across drugs.
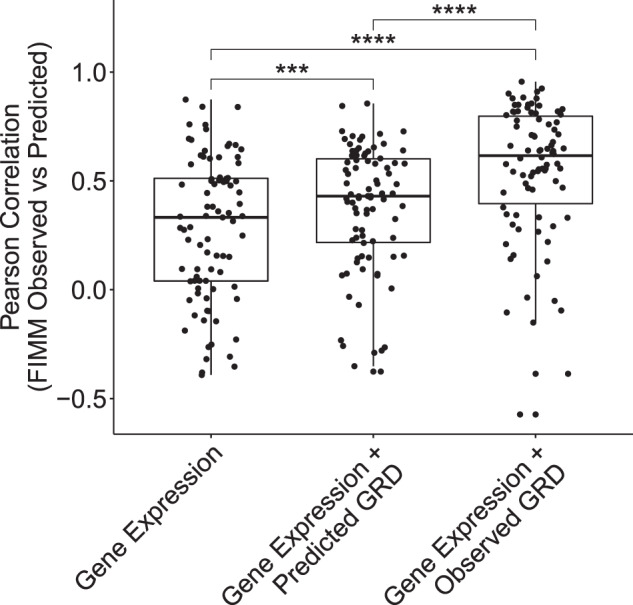
Performance (Pearson correlation between observed and model-predicted drug response; y axis) of ridge regression models trained on OHSU data and tested on FIMM data using genes as predictors (Gene Expression), genes and GRD predicted by applying ridge regression to gene expression (Gene Expression + Predicted GRD), or genes and GRD calculated from drug response data (Gene Expression + Observed GRD). Each point corresponds to a drug (n = 87). Drug d is excluded from observed and predicted GRD in modeling d’s response. ****One-sided paired Wilcoxon signed rank p < 0.0001; ***p < 0.001.
Using predicted GRD as a model variable in addition to gene expression improved response-modeling performance (median r = 0.43; 25th–75th percentile = 0.17–0.60) relative to modeling based on gene expression variables alone (one-sided paired Wilcoxon signed rank p = 3.45 × 10−4; Fig. 4). Using observed, rather than predicted, GRD as a variable in addition to gene expression further improved performance (median r = 0.63; 25th–75th percentile = 0.39–0.75) relative to using gene expression alone (one-sided paired Wilcoxon signed rank p = 4.71 × 10−11; Fig. 4).
These results were largely independent of whether GRD was computed across common drugs or all drugs in a dataset (correlation r between common drug and all drug-based models of observed versus predicted drug response correlations ≥0.96; Supplementary Figs. 23 and 24). This was true despite decreased performance in predicting GRD across all drugs (r = 0.52; Supplementary Fig. 25) relative to across common drugs (r = 0.67; Fig. 3).
A previous study proposed a filtering strategy to combat experimental and technical noise anticipated in large-scale drug screens24. To ensure that our findings were robust to such noise, we developed and applied a related outlier-removal approach (Supplementary Figs. 26–31; see “Methods”). Since prediction performance was not significantly different after removing outliers (Supplementary Fig. 32; two-sided Wilcoxon rank-sum test p = 0.89; Supplementary Tables 17 and 18), subsequent analysis does not exclude outliers.
Monocytic signature predicts venetoclax resistance
Having determined potential biomarkers of GRD above, we next asked whether responses of individual drugs were driven solely by GRD. To isolate drug-specific responses from general effects, we compared prediction performance of models using both gene expression and GRD as variables to that of models using only GRD (i.e., effectively drug–GRD correlations). Drugs showing the greatest specificity were the MEK1/2 inhibitors trametinib, PD184352, and selumetinib and the BCL-2 inhibitor venetoclax (Supplementary Fig. 33). Of these, gene-based prediction performance was highest for venetoclax (r = 0.84; p = 7.50 × 10−8; Supplementary Fig. 34A).
BMSR analysis revealed that venetoclax response had a positive association with the gene BCL2 encoding the drug target, as well as strong negative correlations with CD14 and SLC15A3 (Fig. 5 and Supplementary Figs. 34 and 35). SLC15A3 (Solute Carrier Family 15 Member 3) is highly expressed in monocytes at the protein25 and mRNA26,27 levels, while CD14 encodes a canonical (classical) monocyte cell surface marker28. As such, BMSR analysis indicated that monocyte-associated genes are correlated with venetoclax resistance. We confirmed that genes having expression correlated with venetoclax resistance are enriched for a monocyte signature (p = 2.0 × 10−4; Supplementary Fig. 36 and Supplementary Table 19). Both results are consistent with findings from Kuusanmäki and colleagues that the myeloid differentiation stage of AML cells impacts venetoclax response, with monocytic cells exhibiting resistance to BCL-2 inhibition9.
Fig. 5. Integrative analysis reveals monocyte-associated biomarkers predictive of venetoclax resistance.

a Observed (x axis) versus BMSR-predicted (y axis) venetoclax response. Expression-based Bayesian regression model trained using fivefold cross-validation on combined OHSU and FIMM datasets (n = 159) and tested on held-out fold yielding median performance across the fivefolds (n = 37). b Coefficients of genes (n = 2132) in OHSU (x axis) or FIMM (y axis) datasets following training of Bayesian regression modeling simultaneously on both datasets (n = 196). r: Pearson correlation; dashed line: identity line; blue line: linear regression fit; gray shading: 95% confidence interval. Labeled genes were those having extremal (Stouffer’s p < 0.01) combined coefficients across both datasets.
As with GRD above, these candidate biomarkers contribute consistently (i.e., with the same sign) to the ridge regression model (Supplementary Fig. 34), but BMSR prioritizes a smaller set of features with large coefficients. As a comparison, we also applied minimum redundancy maximum relevance (mRMR), a method that performs feature selection within a single dataset to identify parsimonious feature sets29,30. mRMR identified a subset of the BMSR-prioritized, monocyte-associated genes across both datasets, but the individual genes selected differed. In particular, across 200 random (bootstrap) samples of the datasets, mRMR selected SLC15A3 across all random samples in the OHSU dataset but only once in the FIMM dataset and, conversely, selected PSAP across all random samples in the FIMM dataset but only once in the OHSU dataset. mRMR identified all BMSR-prioritized, monocyte-associated genes across one or more random samples; however, no gene was selected more than twice across both datasets (Supplementary Table 20). Collectively, these results demonstrate the biological consistency of the BMSR, ridge regression, and mRMR analyses, while highlighting BMSR’s motivating contribution of identifying a small set of features concordantly across datasets.
Intriguingly, genes associated with venetoclax resistance were also enriched for T and/or B cell-mediated pathways (Supplementary Tables 21–25). Further, we observed that venetoclax response was positively associated with blast percentage of both peripheral blood (r = 0.37; two-sided Wilcoxon rank-sum test p = 4.6 × 10−4) and bone marrow (r = 0.43; p = 5.9 × 10−5; Supplementary Fig. 37). Nevertheless, several lines of evidence suggest that our results are unlikely to be compromised by impure leukemic samples and/or lymphocyte contamination. First, predicted levels of lymphocytes are lower and less variable than those of monocytes in both datasets (Supplementary Fig. 38). Second, genes associated with venetoclax resistance are more enriched for markers of monocytes and other cell types within the monocytic lineage (i.e., macrophages) than for markers of lymphocytes and are also strongly enriched in myeloid dendritic cells (Supplementary Table 19). Finally, we controlled for potential confounding effects by including lymphocyte levels as covariates in our ridge regression models. Our findings were consistent with the original ridge regression models: prediction results across drugs were highly correlated between the two models (r > 0.99; Supplementary Fig. 39) and genes contributing most to the lymphocyte-controlled model continued to show strong enrichments for markers of the monocytic lineage (Supplementary Table 26).
Because technical or biological variation may contribute noise to individual genes, we next sought to develop a robust signature of venetoclax resistance that would mitigate these fluctuations. To do so, we focused attention on monocyte-associated genes prioritized by BMSR (see “Methods”), which, in addition to CD14 and SLC15A3, included BCL3, LILRB1, LRP1, MAFB, PSAP, and SLC7A7 (Fig. 5b). We compressed the expression of these genes into a single enrichment-based signature of venetoclax resistance (see “Methods”).
We sought to validate this signature (and its constituent genes) across several independent conditions, drugs, and/or ex vivo functional and transcriptomic profiles of AML (Fig. 6), including a study by Lee and colleagues of drug sensitivity that profiled the BCL-2/BCL-XL inhibitor navitoclax31, the ex vivo study by Tavor and colleagues that profiled both venetoclax and navitoclax14, and a second FIMM dataset that profiled venetoclax and navitoclax in a stroma-derived conditioned medium (CM) that differed from the mononuclear cell medium (MCM) of the above FIMM dataset32. The signature was inversely correlated with venetoclax response (i.e., correlated with resistance) in the Tavor dataset (r = −0.44; p = 3.2 × 10−3). It also trended with resistance in the FIMM CM dataset (r = −0.29; p = 0.17), with a lack of significance consistent with the specific dampening of venetoclax response in CM relative to MCM32. Despite this, the signature smoothed out the weak anti-correlations of several genes (e.g., CD14 and LILRB1) as intended. The monocytic signature was strongly anti-correlated with navitoclax response in three datasets: FIMM (CM) (r = −0.60; p = 2.22 × 10−4), FIMM (MCM) (r = −0.75; p = 1.06 × 10−7), and Lee (r = −0.78; p = 2.83 × 10−7). All genes in the signature, with the exception of BCL3, also validated against navitoclax in these three datasets, though their correlation was often not as strong as that of the signature itself. The signature trended with navitoclax resistance in the Tavor dataset (r = −0.25; p = 0.11), with BCL3 again having the weakest association.
Fig. 6. A monocyte expression signature robustly predicts resistance to BCL-2 inhibitors.

Pearson correlation of response of the indicated drug (venetoclax or navitoclax; top) versus expression of the indicated gene or venetoclax monocyte signature (Signature) across FIMM (MCM), OHSU, FIMM (CM), Lee, or Tavor datasets. Dataset/drug combinations are indicated as “Training” if they were used to derive the signature and biomarkers and “Validation” otherwise. ****p < 0.0001; ***p < 0.001; **p < 0.01; *p < 0.05.
As a further validation of the robustness of the signature and of the generality of our Bayesian approach, we used BMSR to jointly analyze the Tavor and FIMM (CM) datasets. Following the approach above, we defined a signature from the monocyte-associated genes prioritized by the analysis. This revised signature was highly correlated with the original FIMM/OHSU-derived signature in the FIMM, FIMM (CM), OHSU, and Tavor datasets (r ≥ 0.75; Supplementary Fig. 40). Finally, to demonstrate its applicability to more than two datasets, we similarly applied BMSR to three datasets, FIMM, OHSU, and Tavor, and again observed that the resulting signature was highly correlated with the original FIMM/OHSU-derived signature (r ≥ 0.95; Supplementary Fig. 41).
Several biomarkers of venetoclax response in AML have been proposed at the protein [BCL-XL, MCL-1, and BCL-233] and mRNA [BCL2, BCL2/MCL1 ratio, BCL2A1, CD11b, CD14, CD68, CD86, CLEC7A (CD369), HOX gene family members, MCL1, S100A8, and S100A99,34–37] levels. Of these, the monocytic signature, BCL2A1, CD68, CD86, and CLEC7A were robust predictors across both BCL-2-inhibitors (venetoclax or navitoclax), different cell culture conditions, and datasets, with none performing best across all conditions (Supplementary Fig. 42). Strikingly, all five were strongly correlated with one another (Supplementary Fig. 43) and are highly expressed in monocytes27,38. These findings validate our own monocytic signature and previously proposed, monocyte-associated biomarkers as predictors of resistance to BCL-2 inhibition in AML.
Monocytic signature predicts MEK inhibitor response
Similar to venetoclax, gene expression data improved the prediction performance of the three MEK inhibitors, trametinib, PD184352, and selumetinib, beyond that provided by GRD alone (Supplementary Fig. 33). We therefore next sought to determine biomarkers of their response. As the response of the MEK inhibitors are strongly correlated with one another (r > 0.48; Supplementary Fig. 44), we developed a Bayesian multi-source multi-task regression (BMSMTR) approach that simultaneously analyzes multiple datasets (i.e., multi-source, as above) and also multiple drugs (i.e., multi-task; see “Methods”). Joint analysis of trametinib, PD184352, and selumetinib with BMSMTR identified monocytic genes as candidate biomarkers, including LRP1 and CD300E (Supplementary Fig. 45), that are positively associated with MEK inhibitor response. Hence, we hypothesized that the monocytic signature defined above to be correlated with venetoclax resistance would be correlated with MEK inhibitor response. Indeed, we observed a positive correlation between the response of each of the three MEK inhibitors and the monocytic signature in both the OHSU and FIMM datasets, where BCL-2 inhibitor response is inversely correlated with MEK inhibitor response (Supplementary Fig. 44). The association did not hold in the FIMM CM dataset, where BCL-2 inhibitor response is not significantly (inversely) (Supplementary Fig. 44) correlated with MEK inhibitor response and where our hypothesis would not be expected to hold.
Discussion
We demonstrated consistency between two large-scale AML studies that profiled ex vivo drug sensitivity and gene expression. In both datasets, we observed that patient-derived samples exhibited a GRD, i.e., a sample often responded uniformly across drugs independent of target or mechanism of action—relatively strongly to all drugs or relatively weakly to all drugs. GRD was further associated with clinical endpoints. Finally, we developed a BMSR method for biomarker discovery and applied it to reveal a robust monocytic signature of BCL-2 inhibition in AML.
We demonstrated that the two studies were consistent: first, drug–drug correlations were conserved across studies (Supplementary Fig. 3); second, the mean response of each drug across patients relative to that of the other drugs was also conserved across studies (Supplementary Fig. 4); and, finally, regression models trained on gene expression data in one study predicted response in the second, independent study for 31 of the 87 drugs (Fig. 4). Similar efforts comparing39 in vitro drug screens40,41 reported discordance in drug responses across the datasets. This finding spurred considerable activity in the research community24,42–47, which ultimately resolved certain discrepancies between the Cancer Cell Line Encyclopedia (CCLE) and the Genomics of Drug Sensitivity in Cancer (GDSC) studies, in part, by harmonizing curve fitting methods applied across datasets and by quantifying drug response via a modified AUC44,48. We also employed AUC since it both intuitively and empirically (Supplementary Figs. 1 and 2) provides a more robust summary of a multi-parameter curve fit than does a single parameter such as EC50 or IC50. A retrospective re-analysis of these in vitro studies applied quality control filters to exclude curves that grossly violated the assumptions that sensitivities range between 0 and 100% and that they increase monotonically with drug concentration24. We developed and applied related quality control measures (Supplementary Figs. 26–31) and found they had little impact on prediction performance of most drugs (Supplementary Fig. 32). Nevertheless, even low-frequency technical noise is expected to be observed in large-scale studies, and hence, we concur with Safikhani and colleagues24 that its impact should be carefully investigated, as we have done here.
Geeleher and colleagues13 reported a phenomenon similar to GRD—GLDS—across cell lines spanning cancer types40,41,49,50. They showed that conditioning on GLDS eliminated spurious biomarker predictions while identifying evidence-supported biomarkers that otherwise went undetected. We similarly showed that including GRD in prediction models improved overall accuracy (Fig. 4), thus demonstrating that the general trend detected in vitro is active in patient-derived AML samples as well. Thus, precision oncology studies should account for both target-specific and target-agnostic effects when correlating drug response with biomarkers.
Our findings directly relate this trend to clinical endpoints: increased GRD is associated with complete response to induction therapy and to improved overall survival (Fig. 2). This generalizes the previous observation that ex vivo response to individual drugs may be correlated with AML remission status31. Further, we found that patients with FLT3-ITD mutations have higher GRD. This result held even when GRD was calculated from subsets of drugs that excluded FLT3 inhibitors. Relatedly, Tavor and colleagues found that patient-derived samples with high sensitivity across TKIs and several other drugs were enriched in FLT3-ITD mutations relative to resistant samples14. Collectively, these results suggest that FLT3-ITD mutations may confer a generalized drug sensitivity. One such mechanism for doing so may be through its association with reduced levels of the ABCB1 drug efflux pump51. Indeed, our expression-based analysis revealed that ABCB1 expression was inversely correlated with GRD. Nevertheless, prior results relating ABCB1 expression to ex vivo response to agents used in induction therapy have been inconsistent51–53. Additionally, the modest correlation of candidate biomarkers with GRD (Supplementary Fig. 20), particularly relative to the consistent correlation observed for venetoclax biomarkers (Supplementary Fig. 35), suggests that mechanisms underlying GRD may involve a complex interplay of multiple genes, possibly conditioned on FLT3 status.
We developed BMSR to mitigate factors that limit reproducibility in prioritizing biomarkers in high-dimensional gene feature spaces, including small sample sizes, correlated expression of functionally related genes, technical variation across datasets, and heterogeneity of patient populations54–57. It does so by performing integrated analysis11,58 across multiple heterogeneous datasets to increase cumulative sample size, both of which reduce the likelihood of overfitting. BMSR shares information across the datasets: gene expression values (or, more formally, the gene expression coefficients) in each dataset are modeled as arising from a shared prior distribution. BMSR differs from meta-analysis methods that first analyze datasets independently before combining effect sizes or p values across datasets. By regularizing the prior’s hyperparameters, BMSR effectively selects features simultaneously across the two datasets to prioritize a sparse set of biomarkers. This is manifested in a small number of genes having coefficients that are well separated from the majority of gene coefficients in both datasets, in contrast to independent ridge regression analysis across the two datasets in which coefficients are evenly distributed with no clear separation indicating candidate biomarkers (Fig. 3 and Supplementary Fig. 34).
We demonstrated BMSR’s generalizability by jointly analyzing different pairs of datasets [FIMM and OHSU; FIMM (CM) and Tavor] and by analyzing a dataset trio (FIMM, OHSU, and Tavor; Supplementary Figs. 40 and 41). As such, we anticipate BMSR will have broad applicability in integrative biomarker studies. However, such analyses are often plagued by the effort required to harmonize multiple datasets. To address this issue, we collaborated with the developers of ORCESTRA in the Haibe-Kains laboratory to make several of the datasets (the Tavor and OHSU datasets) more broadly and easily accessible59. ORCESTRA is a cloud-based platform that provides automated processing of pharmacogenomic profiles and packages them into a fully documented and DOI-indexed “PharmacoSet” (PSet). These PSets are compatible with PharmacoGx, an open-source computational framework that facilitates integrative studies of multiple pharmacogenomic datasets through routines for standardized access and analysis60. To provide a template for others in applying BMSR for integrative analysis, we included a demo in our BMSR GitHub repository (https://github.com/suleimank/bmsr) that downloads the Tavor (10.5281/zenodo.4585705) and OHSU (10.5281/zenodo.4582786) datasets from ORCESTRA and uses the functions of PharmacoGx to predict biomarkers of venetoclax response. Through our ongoing collaboration, we will also make these datasets available through PharmacoDB (https://pharmacodb.ca), a web application that similarly assembles pharmacogenomic datasets into a single database for cross-study analyses61. The online, interactive capabilities of PharmacoDB will make these resources accessible beyond computational biologists and those with programming skills.
BMSR revealed that venetoclax resistance is correlated with expression of monocyte-associated genes (Fig. 5). We combined these genes into a single signature that is robust to the variations of its individual genes62 across datasets (Fig. 6). Additionally, we showed that the venetoclax-derived signature strongly predicts resistance to the BCL-2/BCL-XL inhibitor navitoclax. Resistance to both venetoclax and navitoclax was significantly correlated with the signature in standard culture conditions (MCM), though the trends in a stroma-derived CM reached significance only for navitoclax and not for venetoclax. This may have resulted from the reduced sensitivity of AML cells to venetoclax observed in CM relative to MCM, mediated by a switch from BCL-2- to BCL-XL-dependent cell survival that has a less pronounced effect on navitoclax32.
Our findings extend and support recent reports demonstrating that monocytic cells are resistant to BCL-2 inhibition, whereas myeloid progenitors exhibit sensitivity. Kuusanmäki and colleagues showed that differentiated cells from AML samples expressing monocytic markers are less sensitive to venetoclax than immature blasts9. They further showed that sensitivity to venetoclax decreased along the differentiation spectrum, from less differentiated AML samples [French-American-British subtype M1] to more differentiated samples with significant monocytic differentiation (M5 subtype). They concluded by associating increased expression of monocyte markers CD14, CD11b, CD86, and CD68 with decreased ex vivo responsiveness to venetoclax. Among other identified biomarkers, Zhang and colleagues also implicated high expression of CD14, as well as the monocyte-associated CLEC7A (CD369), with reduced sensitivity to venetoclax36. We provide independent validation of these biomarkers, confirming that leukemic cells expressing high levels of CD68, CD86, and CLEC7A, in particular, are more resistant to BCL-2 inhibition via both venetoclax and navitoclax (Supplementary Fig. 42). Further, we identified a mostly non-overlapping, but strongly correlated (Supplementary Fig. 43), set of genes highly expressed in monocytes: BCL3, CD14, LILRB1, LRP1, MAFB, PSAP, SLC15A3, and SLC7A7. We combined these genes into a per-sample monocytic score using a straightforward approach that did not require additional training or parameter turning. This signature was competitive with CD68, CD86, and CLEC7A across BCL-2 inhibitors, culture conditions, and datasets. Since neither the signature nor the genes nominated by these two studies outperformed the others across all conditions, it may be beneficial to combine them for increased robustness, much as the signature itself smoothed out variation in its constituent genes. Such an approach should be validated in the future using independent data.
These results are likely to be of clinical relevance, as Pei and colleagues demonstrated that phenotypically primitive AML is sensitive to venetoclax in vivo, whereas monocytic AML is more resistant10. Further, they showed that venetoclax (in combination with azacitidine) selects for monocytic subclones present at diagnosis. This resistance is associated with a loss of BCL-2 expression and a concomitant shift to MCL1 for survival that is inherent in monocytic differentiation. MCL1 is itself stabilized by extracellular signal-regulated kinase (ERK)63,64. As such, targeting ERK via a MEK inhibitor has been shown to sensitize cells to the BCL-2/BCL-XL inhibitor ABT-737 in vitro and in xenograft models65. Targeting MCL1 has also been shown to forestall acquired resistance to venetoclax in vitro66, while leukemic blasts resistant to venetoclax are sensitive to MEK inhibition via trametinib ex vivo9. Finally, combination of venetoclax with the MEK inhibitor cobimetinib exhibited synergy in vitro, inhibited growth ex vivo, and reduced leukemia burden in xenografts12. Our work has implications for the rational selection of patient groups for combination therapies targeting these two pathways. Since the monocytic signature is correlated with BCL-2 inhibitor resistance and MEK inhibitor response, it may provide a means of prospectively identifying patients for combination treatment.
Methods
Drug–response curve fitting and filtering
We fit 3- (LL3) and 4-parameter log-logistic (LL4) curves to the dose–response data using PharmacoGx60 and drc67 in R68, respectively. We excluded non-AML patients or those exhibiting gross dissimilarities across replicates from analysis. We excluded any drug–sample pair having a concentration range outside the most common (dataset-specific) concentration range for that corresponding drug. We further excluded a drug–sample pair if it did not include all concentration points and only analyzed one sample per drug–patient pair (see Supplementary Methods). Additionally, we assessed the impact of an outlier-removal strategy that excluded drug–sample pairs: (1) whose fits were not monotonically increasing, (2) that had large differences between fits that did (LL3) and did not (LL4) constrain the curve to asymptote to zero response at low drug concentration, or (3) had a replicate screen (technical in OHSU and biological in FIMM) to which it strongly differed (Supplementary Figs. 26–31). However, we found that this outlier-removal strategy had little impact on prediction performance (Supplementary Fig. 32) and hence did not apply it in the analyses.
Bayesian regression analysis
BMSR models the gene coefficients in each dataset as a sample from a shared, underlying data generating distribution and by regularizing the mean of that distribution to provide parsimony. Formally, it performs joint regression across all datasets constrained to a set of NG common genes as
| 1 |
where is the response vector for a particular drug across the Nd patient samples in dataset d ∈ {FIMM, OHSU}, is the corresponding expression matrix over NG genes, is the gene regression coefficient vector, I is the NG × NG identity matrix, and the standard deviation σ(d) has a non-informative noise prior
with IG(α, β) the Inverse Gamma distribution.
BMSR models coefficient vectors β(d) using the joint hierarchical prior with the shared mean coefficient vector . It regularizes the scalar components βg using the Finnish horse-shoe prior69
where C+(μ, σ) is the half-Cauchy distribution with location μ and scale σ, the scalar λg induces localized gene-wise regularization, and the scalar τ is the global regularization parameter that induces the number of active genes (p0) a priori. Collectively, this formulation encourages the dataset-specific coefficients to either have large magnitude in both datasets (i.e., representing genes whose expression makes a large contribution to the response) or small magnitude in both datasets (i.e., genes with little or no contribution) and to have the same direction (i.e., sign) in both datasets.
BMSMTR simultaneously analyzes multiple datasets (i.e., multi-source, as in BMSR) and also multiple drugs (i.e., multi-task) in a set of drugs . It does so by generalizing Eq. (1) according to
with the response vector for dataset d and drug distributed about a mean that is a product of a factor X(d)β(d) common to drugs in and a factor w(i) ~ N(0.5, 0.5) specific to drug i.
The BMSR and BMSMTR models were implemented in STAN70, a platform for statistical modeling that provides efficient automatic procedures for Bayesian statistical inference. We performed inference using MCMC sampling with 500 samples of the posterior and a burn-in of 500. BMSR and BMSMTR are available at https://github.com/suleimank/bmsr.
Statistical analysis
Statistical analyses were performed in R68. Wilcoxon rank-sum test was performed using wilcox.test. Fisher’s exact test was performed with fisher.test. Linear regression was performed with lm. Forest plot for multivariate linear regression was generated using the forestmodel package.
Gene expression analysis
Ridge regression was performed using glmnet71. Genes used as variables in (ridge, BMSR, and BMSMTR) regression models were filtered to exclude those with low expression (Supplementary Figs. 14–17) and low variance (Supplementary Figs. 18–19). Gene set enrichment was performed using fgsea72, including relative to the set of monocyte marker genes defined in CIBERSORT73. Genes differentially expressed in monocytes relative to other cell populations were determined by applying limma74 to the expression dataset GSE2475926. Immune cell fractions were computed with CIBERSORT using non-log expression data and parameters QN=TRUE, absolute=TRUE, and abs_method=“no.sumto1”. Expression of the genes BCL3, CD14, LILRB1, LRP1, MAFB, PSAP, SLC15A3, and SLC7A7 was compressed into a single enrichment score using GSVA75. This enrichment score is the monocytic signature. Additional details are provided in Supplementary Methods.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
We are grateful to Sisira Nair, Petr Smirnov, and Benjamin Haibe-Kains for creating PSets for the Tavor and OHSU datasets and for making them available on ORCESTRA. B.J.D. and C.E.T. were partially supported by NIH grant 1R01CA214428-01A1. B.S.W., M.J.M., M.A., S.P., K.W., and J.G. were partially supported by a grant from St. Baldrick’s Foundation Genius Award (ID 319134). T.A. was partially supported by research grants from the Academy of Finland (grants 279163, 292611, 310507, 313267) and the Cancer Society of Finland. S.A.K. was supported by a postdoctoral grant from the Academy of Finland (grant 296516).
Author contributions
T.A., K.W. and J.G. conceived and supervised the project. B.S.W., S.A.K., M.J.M., M.A., T.A., K.W. and J.G. designed computational experiments. B.S.W., S.A.K., M.J.M. M.A. and S.P. analyzed the data. S.A.K. and M.A. developed software methodology (BMSR and/or BMSMTR). D.M., H.K., B.J.D., C.A.H., O.K., S.E.K., K.P., C.E.T. and J.W.T. contributed data. B.S.W., S.A.K., D.M., H.K., C.A.H., C.E.T., J.W.T., T.A., K.W. and J.G. interpreted results. B.S.W., S.A.K., T.A., K.W. and J.G. wrote the paper. All authors read, edited, and approved the final manuscript.
Data availability
We re-analyzed four datasets in this study. The OHSU/Beat AML dataset is available on the Synapse data-sharing platform (https://www.synapse.org/#!Synapse:syn2942337/wiki/390658), at dbGaP (study ID 30641 and accession ID phs001657.v1.p1), as supplementary data within the original manuscript4, and via ORCESTRA (10.5281/zenodo.4582786). Additionally, the OHSU dataset can be interactively browsed using Vizome (www.vizome.org). Our re-analysis of the OHSU/Beat AML drug response data are included in Supplementary Tables 1 and 17. The FIMM (MCM) and FIMM (CM) RNA-seq data are available for re-analysis upon request at a secure and GDPR-compliant data analysis data lake environment. Only aggregate data can be downloaded from the data lake. The Lee dataset is available as Supplementary Data within the original manuscript31. The Tavor dataset is available via ORCESTRA (10.5281/zenodo.4585705)14.
Code availability
The BMSR and BMSMTR code is available on GitHub (https://github.com/suleimank/bmsr).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Brian S. White, Suleiman A. Khan.
These authors jointly supervised this work: Tero Aittokallio, Krister Wennerberg, Justin Guinney.
Supplementary information
The online version contains supplementary material available at 10.1038/s41698-021-00209-9.
References
- 1.American Cancer Society. Cancer facts & figures. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2019/cancer-facts-and-figures-2019.pdf (2019).
- 2.Bohl, S. R., Bullinger, L. & Rucker, F. G. New targeted agents in acute myeloid leukemia: new hope on the rise. Int. J. Mol. Sci.20, 1983 (2019). [DOI] [PMC free article] [PubMed]
- 3.Short, N. et al. Advances in the treatment of acute myeloid leukemia: new drugs and new challenges. Cancer Discov. 10, 506–525 (2020). [DOI] [PubMed]
- 4.Tyner JW, et al. Functional genomic landscape of acute myeloid leukaemia. Nature. 2018;562:526–531. doi: 10.1038/s41586-018-0623-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malani D, et al. Identification and clinical exploration of individualized targeted therapeutic approaches in acute myeloid leukemia patients by integrating drug response and deep molecular profiles. Blood. 2017;130:854–854. doi: 10.1182/blood.V130.Suppl_1.854.854. [DOI] [Google Scholar]
- 6.Sharma P, Pollyea DA. Shutting down acute myeloid leukemia and myelodysplastic syndrome with BCL-2 family protein inhibition. Curr. Hematol. Malig. Rep. 2018;13:256–264. doi: 10.1007/s11899-018-0464-8. [DOI] [PubMed] [Google Scholar]
- 7.DiNardo, C. et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood133, 7–17 (2019). [DOI] [PMC free article] [PubMed]
- 8.Dombret H, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood. 2015;126:291–299. doi: 10.1182/blood-2015-01-621664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuusanmaki, H. et al. Phenotype-based drug screening reveals association between venetoclax response and differentiation stage in acute myeloid leukemia. Haematologica105, 708–720 (2020). [DOI] [PMC free article] [PubMed]
- 10.Pei S, et al. Monocytic subclones confer resistance to venetoclax-based therapy in patients with acute myeloid leukemia. Cancer Discov. 2020;10:536–551. doi: 10.1158/2159-8290.CD-19-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamid, J. S. et al. Data integration in genetics and genomics: methods and challenges. Hum. Genomics Proteomics2009, 869093 (2009). [DOI] [PMC free article] [PubMed]
- 12.Han L, et al. Concomitant targeting of bcl2 with venetoclax and mapk signaling with cobimetinib in acute myeloid leukemia models. Haematologica. 2020;105:697–707. doi: 10.3324/haematol.2018.205534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geeleher P, Cox NJ, Huang RS. Cancer biomarker discovery is improved by accounting for variability in general levels of drug sensitivity in pre-clinical models. Genome Biol. 2016;17:190. doi: 10.1186/s13059-016-1050-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tavor S, et al. Dasatinib response in acute myeloid leukemia is correlated with FLT3/ITD, PTPN11 mutations and a unique gene expression signature. Haematologica. 2020;105:2795–2804. doi: 10.3324/haematol.2019.240705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reche, P. A. et al. Human thymic stromal lymphopoietin preferentially stimulates myeloid cells. J. Immunol. 167, 336–343 (2001). [DOI] [PubMed]
- 16.Zhang X, et al. Maternally expressed gene 3, an imprinted noncoding RNA gene, is associated with meningioma pathogenesis and progression. Cancer Res. 2010;70:2350–2358. doi: 10.1158/0008-5472.CAN-09-3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, et al. Involvement of the MKK6-p38gamma cascade in gamma-radiation-induced cell cycle arrest. Mol. Cell. Biol. 2000;20:4543–4552. doi: 10.1128/MCB.20.13.4543-4552.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lam M, et al. Evidence that BCL-2 represses apoptosis by regulating endoplasmic reticulum-associated Ca2+ fluxes. Proc. Natl Acad. Sci. USA. 1994;91:6569–6573. doi: 10.1073/pnas.91.14.6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vogl T, Gharibyan AL, Morozova-Roche LA. Pro-inflammatory S100A8 and S100A9 proteins: self-assembly into multifunctional native and amyloid complexes. Int. J. Mol. Sci. 2012;13:2893–2917. doi: 10.3390/ijms13032893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol. Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen S, Evans HG, Evans DR. FAM129B/MINERVA, a novel adherens junction-associated protein, suppresses apoptosis in HeLa cells. J. Biol. Chem. 2011;286:10201–10209. doi: 10.1074/jbc.M110.175273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghavami S, et al. S100A8/A9 induces autophagy and apoptosis via ROS-mediated cross-talk between mitochondria and lysosomes that involves BNIP3. Cell Res. 2010;20:314–331. doi: 10.1038/cr.2009.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joshi AD, et al. ATM, CTLA4, MNDA, and HEM1 in high versus low CD38 expressing B-cell chronic lymphocytic leukemia. Clin. Cancer Res. 2007;13:5295–5304. doi: 10.1158/1078-0432.CCR-07-0283. [DOI] [PubMed] [Google Scholar]
- 24.Safikhani Z, et al. Revisiting inconsistency in large pharmacogenomic studies. F1000Res. 2016;5:2333. doi: 10.12688/f1000research.9611.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fishilevich, S. et al. Genic insights from integrated human proteomics in GeneCards. Database2016, baw030 (2016). [DOI] [PMC free article] [PubMed]
- 26.Novershtern N, et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011;144:296–309. doi: 10.1016/j.cell.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bagger FO, Kinalis S, Rapin N. BloodSpot: a database of healthy and malignant haematopoiesis updated with purified and single cell mRNA sequencing profiles. Nucleic Acids Res. 2019;47:D881–D885. doi: 10.1093/nar/gky1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ziegler-Heitbrock L, et al. Nomenclature of monocytes and dendritic cells in blood. Blood. 2010;116:74–80. doi: 10.1182/blood-2010-02-258558. [DOI] [PubMed] [Google Scholar]
- 29.Ding C, Peng H. Minimum redundancy feature selection from microarray gene expression data. J. Bioinform. Comput. Biol. 2005;3:185–205. doi: 10.1142/S0219720005001004. [DOI] [PubMed] [Google Scholar]
- 30.De Jay N, et al. mRMRe: an R package for parallelized mRMR ensemble feature selection. Bioinformatics. 2013;29:2365–2368. doi: 10.1093/bioinformatics/btt383. [DOI] [PubMed] [Google Scholar]
- 31.Lee SI, et al. A machine learning approach to integrate big data for precision medicine in acute myeloid leukemia. Nat. Commun. 2018;9:42. doi: 10.1038/s41467-017-02465-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karjalainen R, et al. JAK1/2 and BCL2 inhibitors synergize to counteract bone marrow stromal cell-induced protection of AML. Blood. 2017;130:789–802. doi: 10.1182/blood-2016-02-699363. [DOI] [PubMed] [Google Scholar]
- 33.Pan R, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014;4:362–375. doi: 10.1158/2159-8290.CD-13-0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Niu X, et al. Acute myeloid leukemia cells harboring MLL fusion genes or with the acute promyelocytic leukemia phenotype are sensitive to the Bcl-2-selective inhibitor ABT-199. Leukemia. 2014;28:1557–1560. doi: 10.1038/leu.2014.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kontro M, et al. HOX gene expression predicts response to BCL-2 inhibition in acute myeloid leukemia. Leukemia. 2017;31:301–309. doi: 10.1038/leu.2016.222. [DOI] [PubMed] [Google Scholar]
- 36.Zhang H, et al. Integrated analysis of patient samples identifies biomarkers for venetoclax efficacy and combination strategies in acute myeloid leukemia. Nat. Cancer. 2020;8:826–839. doi: 10.1038/s43018-020-0103-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karjalainen R, et al. Elevated expression of S100A8 and S100A9 correlates with resistance to the BCL-2 inhibitor venetoclax in AML. Leukemia. 2019;33:2548–2553. doi: 10.1038/s41375-019-0504-y. [DOI] [PubMed] [Google Scholar]
- 38.Rapin N, et al. Comparing cancer vs normal gene expression profiles identifies new disease entities and common transcriptional programs in AML patients. Blood. 2014;123:894–904. doi: 10.1182/blood-2013-02-485771. [DOI] [PubMed] [Google Scholar]
- 39.Haibe-Kains B, et al. Inconsistency in large pharmacogenomic studies. Nature. 2013;504:389–393. doi: 10.1038/nature12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garnett MJ, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barretina J, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geeleher P, Gamazon ER, Seoighe C, Cox NJ, Huang RS. Consistency in large pharmacogenomic studies. Nature. 2016;540:E1–E2. doi: 10.1038/nature19838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Safikhani, Z. et al. Safikhani et al. reply. Nature540, E2–E4 (2016). [DOI] [PubMed]
- 44.Mpindi, J. P. et al. Consistency in drug response profiling. Nature540, E5–E6 (2016). [DOI] [PubMed]
- 45.Safikhani, Z. et al. Safikhani et al. reply. Nature540, E6–E8 (2016). [DOI] [PubMed]
- 46.Bouhaddou, M. et al. Drug response consistency in CCLE and CGP. Nature540, E9–E10 (2016). [DOI] [PMC free article] [PubMed]
- 47.Safikhani Z, et al. Safikhani et al. reply. Nature. 2016;540:E11–E12. doi: 10.1038/nature20581. [DOI] [PubMed] [Google Scholar]
- 48.Yadav B, et al. Quantitative scoring of differential drug sensitivity for individually optimized anticancer therapies. Sci. Rep. 2014;4:5193. doi: 10.1038/srep05193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seashore-Ludlow B, et al. Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov. 2015;5:1210–1223. doi: 10.1158/2159-8290.CD-15-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rees MG, et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 2016;12:109–116. doi: 10.1038/nchembio.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boyer, T. et al. Clinical significance of ABCB1 in acute myeloid leukemia: a comprehensive study. Cancers11, 1323 (2019). [DOI] [PMC free article] [PubMed]
- 52.Guerci A, et al. Predictive value for treatment outcome in acute myeloid leukemia of cellular daunorubicin accumulation and P-glycoprotein expression simultaneously determined by flow cytometry. Blood. 1995;85:2147–2153. doi: 10.1182/blood.V85.8.2147.bloodjournal8582147. [DOI] [PubMed] [Google Scholar]
- 53.Walter RB, et al. CD33 expression and P-glycoprotein-mediated drug efflux inversely correlate and predict clinical outcome in patients with acute myeloid leukemia treated with gemtuzumab ozogamicin monotherapy. Blood. 2007;109:4168–4170. doi: 10.1182/blood-2006-09-047399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.He Z, Yu W. Stable feature selection for biomarker discovery. Comput. Biol. Chem. 2010;34:215–225. doi: 10.1016/j.compbiolchem.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 55.Buness A, Ruschhaupt M, Kuner R, Tresch A. Classification across gene expression microarray studies. BMC Bioinformatics. 2009;10:453. doi: 10.1186/1471-2105-10-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Di Camillo B, et al. Effect of size and heterogeneity of samples on biomarker discovery: synthetic and real data assessment. PLoS ONE. 2012;7:e32200. doi: 10.1371/journal.pone.0032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Michiels S, Koscielny S, Hill C. Prediction of cancer outcome with microarrays: a multiple random validation strategy. Lancet. 2005;365:488–492. doi: 10.1016/S0140-6736(05)17866-0. [DOI] [PubMed] [Google Scholar]
- 58.Ma S, Huang J, Wei F, Xie Y, Fang K. Integrative analysis of multiple cancer prognosis studies with gene expression measurements. Stat. Med. 2011;30:3361–3371. doi: 10.1002/sim.4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mammoliti, A. et al. Orchestrating and sharing large multimodal data for transparent and reproducible research. Preprint at bioRxiv10.1101/2020.09.18.303842v2 (2021). [DOI] [PMC free article] [PubMed]
- 60.Smirnov P, et al. PharmacoGx: an R package for analysis of large pharmacogenomic datasets. Bioinformatics. 2016;32:1244–1246. doi: 10.1093/bioinformatics/btv723. [DOI] [PubMed] [Google Scholar]
- 61.Smirnov P, et al. Pharmacodb: an integrative database for mining in vitro anticancer drug screening studies. Nucleic Acids Res. 2018;46:D994–D1002. doi: 10.1093/nar/gkx911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Loscalzo, S., Yu, L. & Ding, C. Consensus group stable feature selection. In Proc. 15th ACM SIGKDD International Conference of Knowledge Discovery and Data Mining 567–576 (ACM Press, 2009).
- 63.Townsend KJ, et al. Regulation of mcl1 through a serum response factor/elk-1-mediated mechanism links expression of a viability-promoting member of the bcl2 family to the induction of hematopoietic cell differentiation. J. Biol. Chem. 1999;274:1801–1813. doi: 10.1074/jbc.274.3.1801. [DOI] [PubMed] [Google Scholar]
- 64.Domina AM, Vrana JA, Gregory MA, Hann SR, Craig RW. MCL1 is phosphorylated in the pest region and stabilized upon ERK activation in viable cells, and at additional sites with cytotoxic okadaic acid or taxol. Oncogene. 2004;23:5301–5315. doi: 10.1038/sj.onc.1207692. [DOI] [PubMed] [Google Scholar]
- 65.Konopleva M, et al. Mek inhibition enhances ABT-737-induced leukemia cell apoptosis via prevention of ERK-activated MCL-1 induction and modulation of MCL-1/BIM complex. Leukemia. 2012;26:778–787. doi: 10.1038/leu.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 66.Lin KH, et al. Targeting MCL-1/BCL-XL forestalls the acquisition of resistance to ABT-199 in acute myeloid leukemia. Sci. Rep. 2016;6:27696. doi: 10.1038/srep27696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ritz C, Baty F, Streibig JC, Gerhard D. Dose-response analysis using R. PLoS ONE. 2015;10:e0146021. doi: 10.1371/journal.pone.0146021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2014).
- 69.Piironen, J. & Vehtari, A. Sparsity information and regularization in the horseshoe and other shrinkage priors. Electron. J. Stat. 11, 5018–5051 (2017).
- 70.Carpenter, B. et al. Stan: a probabilistic programming language. J. Stat. Softw. 76, 1–32 (2017). [DOI] [PMC free article] [PubMed]
- 71.Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 2010;33:1–22. doi: 10.18637/jss.v033.i01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Korotkevich, G. et al. Fast gene set enrichment analysis. Preprint at bioRxiv. https://www.biorxiv.org/content/early/2021/02/01/060012 (2021).
- 73.Newman AM, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We re-analyzed four datasets in this study. The OHSU/Beat AML dataset is available on the Synapse data-sharing platform (https://www.synapse.org/#!Synapse:syn2942337/wiki/390658), at dbGaP (study ID 30641 and accession ID phs001657.v1.p1), as supplementary data within the original manuscript4, and via ORCESTRA (10.5281/zenodo.4582786). Additionally, the OHSU dataset can be interactively browsed using Vizome (www.vizome.org). Our re-analysis of the OHSU/Beat AML drug response data are included in Supplementary Tables 1 and 17. The FIMM (MCM) and FIMM (CM) RNA-seq data are available for re-analysis upon request at a secure and GDPR-compliant data analysis data lake environment. Only aggregate data can be downloaded from the data lake. The Lee dataset is available as Supplementary Data within the original manuscript31. The Tavor dataset is available via ORCESTRA (10.5281/zenodo.4585705)14.
The BMSR and BMSMTR code is available on GitHub (https://github.com/suleimank/bmsr).