

SUMMARY
Background
Accurate non-invasive prediction of long-term hepatocellular carcinoma (HCC) risk in advanced liver fibrosis is urgently needed for cost-effective HCC screening; however, this currently remains an unmet need.
Methods
A serum-protein-based prognostic liver secretome signature (PLSec) was bioinformatically derived from previously validated hepatic transcriptome signatures and optimized in 79 patients with advanced liver fibrosis. We independently validated PLSec for HCC risk in 331 cirrhosis patients with mixed etiologies (validation set 1 [V1]) and thereafter developed a score with clinical prognostic variables. The score was then validated in two independent cohorts: validation set 2 (V2): 164 patients with advanced liver fibrosis due to hepatitis C virus (HCV) infection cured after direct-acting antiviral therapy; validation set 3 (V3): 146 patients with advanced liver fibrosis with successfully-treated HCC and cured HCV infection.
Findings
An 8-protein blood-based PLSec recapitulated transcriptome-based hepatic HCC risk status. In V1, PLSec was significantly associated with incident HCC risk (adjusted hazard ratio [aHR], 2.35; 95% confidence interval [CI], 1.30–4.23). A composite score with serum alpha-fetoprotein (PLSec-AFP) was defined in V1, and validated in V2 (adjusted odds ratio, 3.80 [95%CI, 1.66–8.66]) and V3 (aHR, 3.08 [95%CI, 1.78–5.31]; c-index, 0.74). PLSec-AFP outperformed AFP alone (Brier score, 0.165 vs. 0.186 in V2; 0.196 vs. 0.206 in V3, respectively).
Conclusions
The blood-based PLSec-AFP can accurately stratify patients with advanced liver fibrosis for long-term HCC risk and thereby guide risk-based tailored HCC screening.
Graphical Abstract
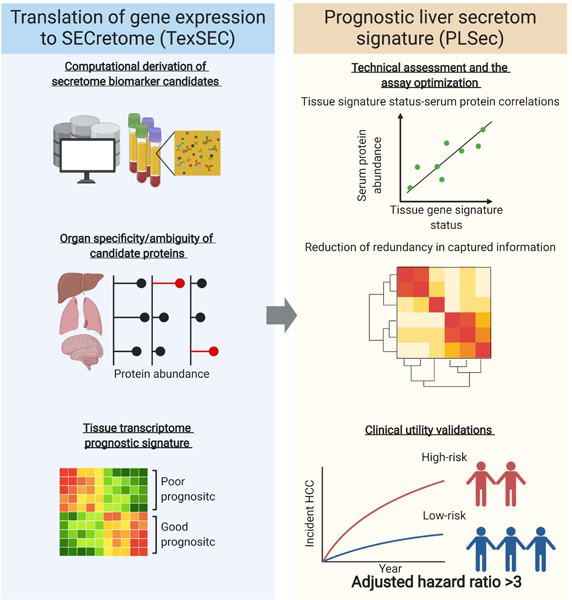
eTOC blurb
Fujiwara et al. developed a computational pipeline to translate tissue transcriptome to secretome signature, named TexSEC. TexSEC identified an 8-protein serum-based secretome signature predictive of liver cancer risk in patients with advanced liver fibrosis, which was validated in three independent cohorts with various clinical scenarios.
INTRODUCTION
Cirrhosis, the terminal stage of progressive liver fibrosis from viral and metabolic etiologies, affects 1–2% of the global population and leads to 1.32 million deaths annually with a 15% increase over the past decade.1,2 Reducing cirrhosis-related death was recently identified as a high priority in the Healthy People 2030 Initiative (health.gov/healthypeople). Hepatocellular carcinoma (HCC) is a major life-limiting complication of cirrhosis and represents the fastest-rising cause of cancer-related death in the U.S. and the fourth most common cancer mortality worldwide.3 Therapeutic clearance of hepatitis viruses does not eliminate HCC risk when advanced fibrosis is present, and there is no treatment for an emerging metabolic HCC etiology, non-alcoholic fatty liver disease (NAFLD).1
Given the strong association between early detection and improved survival, professional society guidelines recommend semi-annual HCC screening in all patients with advanced liver fibrosis and cirrhosis.4 However, with this “one-size-fits-all” strategy, the large at-risk patient population overburdens limited medical resources, as evidenced by the low utilization of HCC screening (<25%).5 As a consequence, the majority of HCCs are diagnosed at late stages, not amenable to curative treatment, which accounts for its overall poor prognosis (5-year survival <15%). Thus, precise prediction of future HCC risk could enable more effective HCC screening by identifying a subset of cirrhosis patients at higher HCC risk and allocating limited resources to high-risk patients.6
We previously identified and validated a hepatic-transcriptome-based Prognostic Liver Signature (PLS) that predicts long-term HCC risk in patients with cirrhosis across major liver disease etiologies and guides discovery of novel HCC chemoprevention strategies.7–13 Despite the confirmed prognostic capability of the signature, the need for liver biopsy limits its widespread use in clinical practice. To address this important and long-standing challenge, we aimed to develop a blood-based surrogate of PLS, Prognostic Liver Secretome signature (PLSec), using our integrative bioinformatics pipeline to translate tissue transcriptome signatures into secretome markers, and externally validate its clinical utility in three independent patient cohorts that represent the major clinical scenarios of HCC risk prediction.
RESULTS
Computational pipeline to translate a gene signature to secretome signature
Transcriptome profiling of diseased organ tissue has been widely used as the first step to reliably identify pathogenic and prognostic molecular dysregulation. However, the need for invasive tissue biopsy generally limits its clinical applicability. On the other hand, direct biomarker discovery in circulation obscures the source organ that releases the biomolecules. To overcome the challenge and enable less-invasive monitoring of organ-specific biological dysregulation, we developed a computational pipeline to systematically translate a tissue-transcriptome-based molecular signature into a list of proteins inferred to be released into circulation from an organ of interest, named Translation of gene expression to SECretome (TexSEC) (www.texsec-app.org). Briefly, this pipeline consists of two parts: (i) assembled proteome databases with an algorithm to infer organ specificity of proteins in circulation and (ii) a list of genes (i.e., gene signature) specific to a disease context. For (i), we integrated proteome databases that complementarily cover bioinformatically-predicted secretable proteins as well as empirically detected proteins in human body fluids. For (ii), we used our previously defined tissue-based PLS as well as proteins in associated molecular pathways (Data S1). By taking intersection of (i) and (ii), we derived prognostic liver secretome biomarker candidates. See STAR Methods and Supplementary Methods for the details.
Derivation of PLSec as a blood-based long-term HCC risk biomarker
Our computational pipeline identified 43 candidate serum proteins, for which validated antibodies are available for quantitative multiplex assessment (Figure 1). This preliminary panel included proteins that were previously reported as potential HCC risk biomarkers, e.g., interleukin 6 (IL-6), osteopontin, and midkine,14–16 supporting the validity of our unbiased secretome biomarker derivation pipeline. Based on association with the prognostic tissue transcriptome and the least information redundancy among the probes in the optimization set, we ultimately selected 6 high-risk-associated serum proteins, including vascular cell adhesion molecule 1 (VCAM-1), insulin-like growth factor-binding protein 7 (IGFBP-7), gp130, matrilysin, IL-6, and C-C motif chemokine ligand 21 (CCL-21), and 2 low-risk-associated serum proteins, including angiogenin and protein S. We observed high within-plate reproducibility (r2 = 0.9997, p = 1.1×10-11), inter-plate/batch reproducibility (r2 = 0.971, p = 7.7×10-6) of technical replicates, and sensitivity for positive control proteins (99.9% ± 2.5%), supporting the assay reliability as a clinical test. The normalized protein abundance measurements were converted into an aggregated score, named PLSec, and a cut-off of ≥4 was defined to identify patients with a high-risk prediction in the optimization set (See STAR Methods and Supplementary Methods for details). PLSec recapitulated 93% of dysregulated molecular pathways and all involved cell types associated with the original tissue transcriptome signatures (Data S1), indicating that PLSec conveys equivalent biological information as the PLS.
Figure 1. Study design.
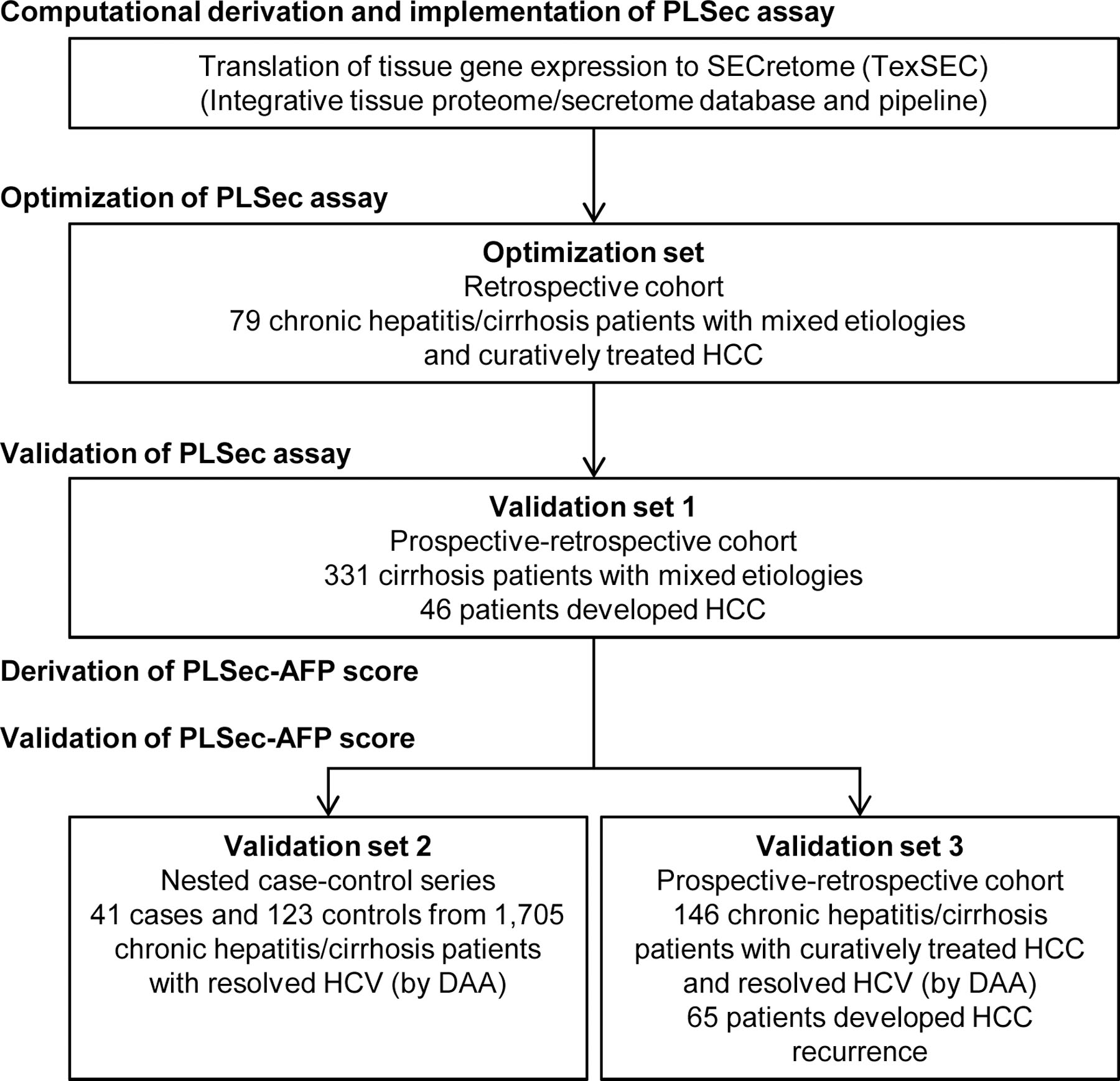
See also Supplementary Methods, and Data S1.
PLSec, Prognostic Liver Secretome signature; HCC, hepatocellular carcinoma; AFP, alpha-fetoprotein; HCV, hepatitis C virus; DAA, direct-acting antiviral.
Validation of PLSec assay in validation set 1: Incident HCC risk in patients with cirrhosis
Among 331 cirrhosis patients from various etiologies, PLSec was significantly associated with incident HCC (adjusted hazard ratio [aHR], 2.35; 95% confidence interval [CI], 1.30–4.23, p=0.004) (Figure 2A, 2B; Figure S1A). The association with HCC risk remained similar when death and liver transplantation were considered as competing risks (adjusted sub-distribution HR, 1.91; 95% CI, 1.07–3.41). Annual HCC incidence rates in low-risk (n=208) and high-risk (n=123) patients were 1.5 and 3.6 per 100 person-years, respectively. HCC incidence at 5 and 10 years were 6.2% and 15.7% among low-risk patients, compared to 19.5% and 23.9% among high-risk patients, respectively.
Figure 2. Clinical utility validation 1: cirrhosis with mixed HCC etiologies (prospective-retrospective cohort).
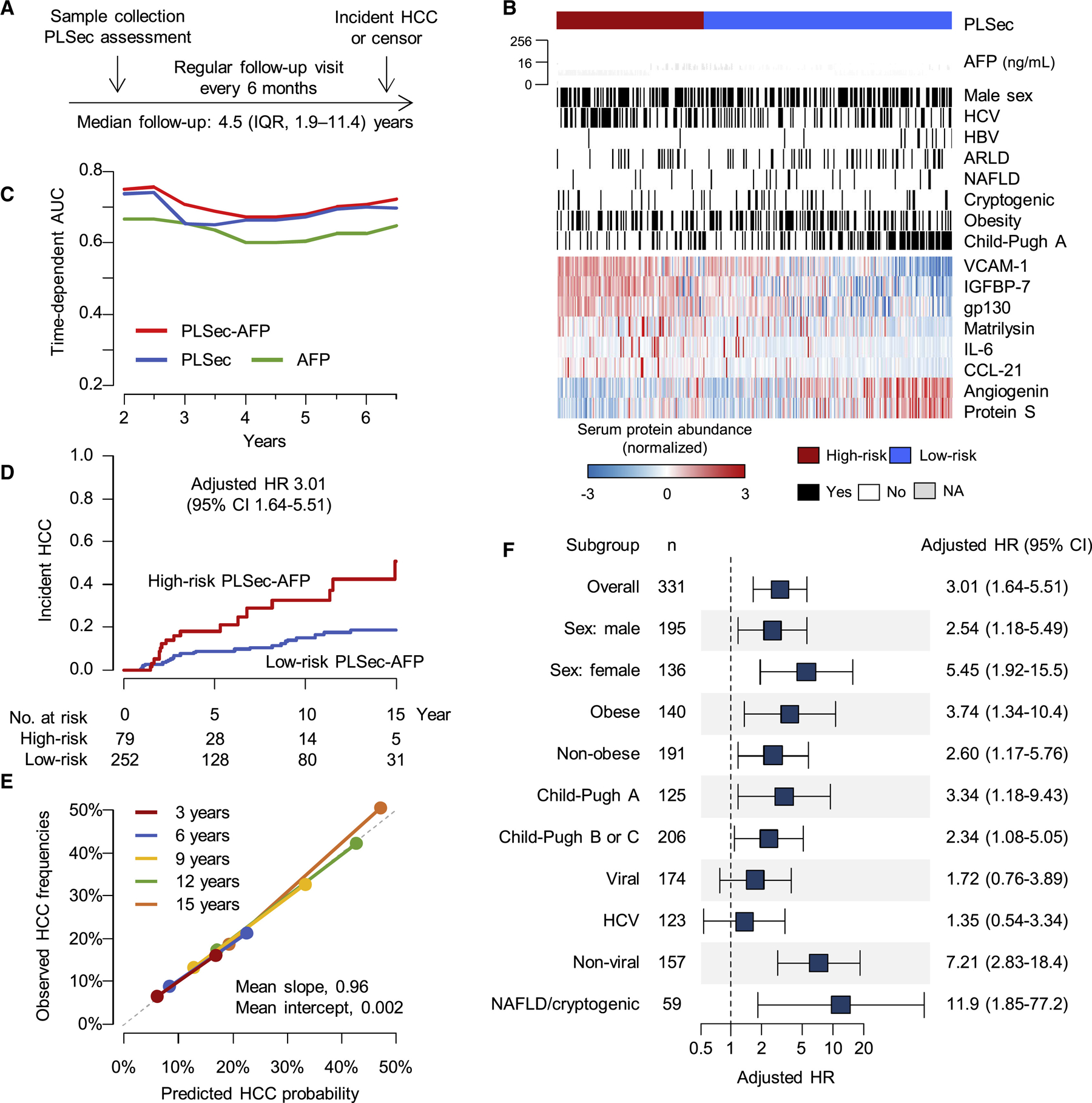
(A) Study design. (B) Patten of the PLSec protein abundance and associated clinical variables. (C) Time-dependent AUC of PLSec-AFP score, PLSec alone, and AFP alone. (D) Association of PLSec-AFP with incident HCC. (E) Calibration plot of PLSec-AFP at various time points. The grey dash line indicates ideal calibration. (F) Association of PLSec-AFP with incident HCC in various subgroups.
See also Figure S1 and Table S1–3.
PLSec, prognostic liver secretome signature; HCC, hepatocellular carcinoma; IQR, interquartile range; AFP, alpha-fetoprotein; HCV, hepatitis C virus; HBV, hepatitis B virus; ARLD, alcohol-related liver disease; NAFLD, non-alcoholic fatty liver disease; AUC, area under the receiver operating characteristic curve; HR, hazard ratio; CI, confidence interval.
Derivation of PLSec-AFP score in validation set 1
Among available clinical variables, AFP was also associated with HCC risk (aHR, 1.38; 95% CI, 1.14–1.68) independent of PLSec (Table S1), consistent with evidence that AFP can be a future HCC risk marker (reflecting chronic hepatocyte injury and proliferation without HCC),17 especially after HCV cure1, as well as early detection marker.4 If elevated AFP is attributable to presence of malignant cells which are still clinically undetectable, it is expected that the very early subclinical HCC nodule grows and becomes detectable in a few years given the anticipated tumor doubling time around 5 months.18 In this cohort, proportion of HCC incidence is consistent throughout the 17 years of follow up irrespective of AFP levels, suggesting that AFP elevation in this cohort is more likely attributable to carcinogenesis-permissive hepatic tissue microenvironment (so-called “field effect”) rather than undetectable subclinical tumor (Figure S1B). High-risk PLSec was associated with HCC risk even in patients with low AFP (<5ng/mL) (n = 199, aHR, 3.49, 95% CI, 1.40–8.72). The aHRs of high AFP (≥5ng/mL) for viral and non-viral etiologies were 1.52 (95% CI, 0.64–3.61) and 3.50 (95% CI, 1.34–9.12), respectively. By incorporating AFP, we developed an integrated PLSec-AFP score (see STAR Methods, Figure S1C; Table S2), which was significantly associated with HCC risk (aHR, 2.71, 95% CI;1.69–4.33, p<0.001) and demonstrated better predictive performance than either variable alone (c-indices for PLSec-AFP, PLSec, and AFP were 0.73, 0.69, and 0.66, respectively) (Figure 2C; Table 2).
Table 2.
Prognostic association and model performance of PLSec-AFP.
| Prognostic association | Overall performance | Discrimination | Goodness of fit | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Variable | Adjusted HR or OR* | (95% CI) | Brier score** | (95% CI)*** | c-index | (95% CI)*** | AIC | (95% CI)*** | BIC | (95% CI)*** |
| Validation set 1 (n=331): Cirrhosis with mixed etiology (prospective–retrospective cohort) | ||||||||||
| PLSec-AFP | 2.71 | (1.69–4.33) | 0.116 | (0.091–0.139) | 0.73 | (0.59–0.84) | 448 | (332–575) | 450 | (333–577) |
| PLSec | 1.28 | (1.08–1.52) | 0.120 | (0.094–0.142) | 0.69 | (0.56–0.80) | 457 | (338–581) | 459 | (340–583) |
| AFP | 1.39 | (1.16–1.66) | 0.119 | (0.092–0.142) | 0.66 | (0.45–0.80) | 459 | (340–584) | 461 | (342–586) |
| Validation set 2 (n=41:123): Resolved HCV hepatitis/cirrhosis (nested case-control series) | ||||||||||
| PLSec-AFP | 3.80 | (1.66–8.66) | 0.165 | (0.131–0.184) | - | - | 104 | (97–133) | 106 | (99–135) |
| AFP | 1.69 | (0.74–3.86) | 0.186 | (0.176–0.187) | - | - | 114 | (123–137) | 116 | (125–139) |
| Validation set 3 (n=146): Resolved HCV hepatitis/cirrhosis after HCC therapies (prospective–retrospective cohort) | ||||||||||
| PLSec-AFP | 3.08 | (1.78–5.31) | 0.196 | (0.165–0.214) | 0.74 | (0.64–0.84) | 590 | (479–690) | 592 | (481–693) |
| AFP | 1.75 | (1.03–2.98) | 0.206 | (0.183–0.222) | 0.64 | (0.52–0.77) | 602 | (496–702) | 605 | (498–705) |
HRs were adjusted for age (as continuous), sex, obesity, diabetes, and active hazardous alcohol drinking. ORs were adjusted for obesity, diabetes, and active hazardous alcohol drinking with conditioning on the pairs of cases and the matched controls.
For cohort studies, integrated Brier scores were demonstrated.
95% confidence intervals were estimated by 1,000-time bootstrapping of the samples.
PLSec, prognostic liver secretome signature; AFP, alpha-fetoprotein; HR, hazard ratio; OR, odds ratio; CI, confidence interval; AIC, Akaike information criterion; BIC, Bayesian information criterion; HCV, hepatitis C virus; HCC, hepatocellular carcinoma.
Subsequently, we defined a cut-off of 1.66 to classify low- vs. high-risk. Annual HCC incidence rates in low-risk (n=252) and high-risk (n=79) patients were 1.5 and 4.8 per 100 person-years, respectively. HCC incidence rates at 5 and 10 years were 8.8% and 15.2% among low-risk patients, respectively, compared to 18.1% and 32.7% among high-risk patients (aHR, 3.01, 95% CI;1.64–5.51, p<0.001, Figure 2D). PLSec-AFP was well-calibrated over time (Figure 2E) and showed robust prognostic association after adjustment for other clinical variables (Table S3). Subgroup analyses suggested enhanced magnitude of association in patients with early-stage, i.e., compensated liver disease (Child-Pugh class A) as well as NAFLD or cryptogenic etiology (often associated with history of NAFLD19), a patient population in greatest need of HCC risk stratification (Figure 2F). We observed modest prognostic association in patients with active HCV infection, a vanishing population with widespread direct-acting antivirals (DAA) use. These data collectively supported successful independent validation of PLSec and warranted further validation of PLSec-AFP.
Validation set 2: Incident HCC risk after HCV cure
In contrast to the decrease of patients with active HCV infection, HCV-cured patients are sharply increasing. In this nested case-control series of patients after sustained virologic response (SVR) achievement (Table 1; Figure 3A), high-risk PLSec-AFP was significantly associated with future HCC occurrence (adjusted odds ratio [aOR], 3.80; 95% CI, 1.66–8.66, p=0.002) (Figure 3B; Table S3 and S4). The association remained significant in the subset of patients with cirrhosis (aOR, 3.12; 95% CI, 1.27–7.65). Overall sensitivity and specificity of PLSec-AFP for long-term HCC risk were 56% and 72%, respectively. PLSec-AFP showed stable sensitivity and specificity and consistently improved prognostic association compared to AFP alone (time-dependent AUC is approximately 0.70 over time) (Figure 3C; Figure S1D). Additionally, PLSec-AFP showed better model performance and fitness compared to AFP alone (Brier score, 0.165 vs. 0.186; Akaike information criterion [AIC], 104 vs. 114; Bayesian information criterion [BIC], 106 vs. 116 for PLSec-AFP and AFP, respectively) (Table 2). Previous clinical studies reported the annual HCC incidence rate is 1% to 2% in cirrhosis patients after achieving SVR with DAA therapy, and 2% to 3.5% in cirrhosis patients with a high FIB-4 index, a clinical indicator of liver fibrosis.20–22 Based on results in the validation set 2, high-risk PLSec-AFP was estimated to identify a subgroup of patients with approximately 3-fold higher annual HCC incidence rate, up to 7% of the group (Figure 3D).
Table 1.
Clinical demographic of validation sets
| Variable | Validation set 1 (n=331): Cirrhosis with mixed etiology (prospective–retrospective cohort) | Validation set 2 (n=41:123): Cured HCV hepatitis/cirrhosis (nested case-control series)* | Validation set 3 (n=146): Cured HCV hepatitis/cirrhosis after HCC therapies (prospective– retrospective cohort) |
|---|---|---|---|
| Age (y) | 52 (47 – 57) | 72 (62 – 77) : 72 (64 – 76) | 73 (66 – 78) |
| Male sex | 195 (59%) | 23 (56%) : 69 (56%) | 66 (45%) |
| Cirrhosis | 331 (100%) | 30 (73%) : 92 (75%) | 117 (80%) |
| Etiology: HCV/HBV/ARLD/NAFLD/cryptogenic/others | 123/13/60/20/39/76 (37%/4%/18%/6%/12%/23%) | 164/0/0/0/0/0** (100%/0%/0%/0%/0%/0%) | 146/0/0/0/0/0** (100%/0%/0%/0%/0%/0%) |
| Race/ethnicity: white/black/Hispanic/Asian/others | 311/9/8/1/2 (94%/3%/2%/0.3%/1%) | 0/0/0/164/0 (0%/0%/0%/100%/0%) | 0/0/0/146/0 (0%/0%/0%/100%/0%) |
| besity | 140 (42%) | 10 (24%) : 30 (25%) | 20 (14%) |
| Diabetes | 76 (23%) | 5 (12%) : 18 (15%) | 25 (18%) |
| Active hazardous alcohol drink | 34 (11%) | 5 (14%):7 (6%) | 12 (9%) |
| Albumin (g/dL) | 3.4 (2.9 – 3.8) | 3.5 (3.4 – 3.7) : 3.5 (3.3 – 3.8) | 3.6 (3.2 – 3.7) |
| Total bilirubin (mg/dL) | 1.2 (0.8 – 1.9) | 0.9 (0.8 – 1.2) : 0.9 (0.8 – 1.3) | 0.9 (0.7 – 1.1) |
| ALT (IU/L) | 49 (34 – 79) | 20 (17 – 28) : 20 (15 – 28) | 18 (14 – 24) |
| Platelet count (×103/uL) | 95 (67 – 136) | 116 (83 – 152) : 128 (97 – 162) | 115 (91 – 163) |
| AFP (ng/mL) | 3.9 (2.3 – 7.9) | 7 (4–12) : 6 (4–8) | 6 (4 – 10) |
| HCV genotype 1 | n.a. | 36 (88%) : 103 (84%) | 134 (92%) |
| DAA regimen: sofosbuvir-based | - | 11 (27%) : 48 (39%) | 30 (21%) |
| HCC AJCC stage I | - | - | 101 (69%) |
| HCC therapy: resection/ablation/TACE/SRBT | - | - | 43/100/3/2 (29%/68%/2%/1%) |
| Child-Pugh class (A/B/C) | 122/179/25 (37%/55%/8%) | 35/6/0 (85%/15%/0%): 109/14/0 (87%/13%/0%) | 132/14/0 (90%/10%/0%) |
| Follow-up time (y) | 4.5 (1.9 – 11.4) | 1.1 (0.5 – 2.1) : 4.3 (4.0 – 4.6) | 2.9 (0.9–4.1) |
Categorical variables are shown as n (%). Continuous variables are shown as median (IQR).
Case:control.
All patients achieved sustained virologic response with direct-acting antiviral therapy.
HCV, hepatitis C virus; HBV, hepatitis B virus; ARLD, alcohol-related liver disease; NAFLD, non-alcoholic fatty liver disease; ALT, alanine transaminase; AFP, alpha-fetoprotein; DAA, direct acting antiviral; HCC, hepatocellular carcinoma; AJCC, American Joint Committee of Cancer; TACE, transarterial chemoembolization; SRBT, stereotactic body radiation therapy; IQR, interquartile range; HCC, hepatocellular carcinoma.
Figure 3. Clinical utility validation 2: HCC risk after HCV cure by DAA (nested case-control series).
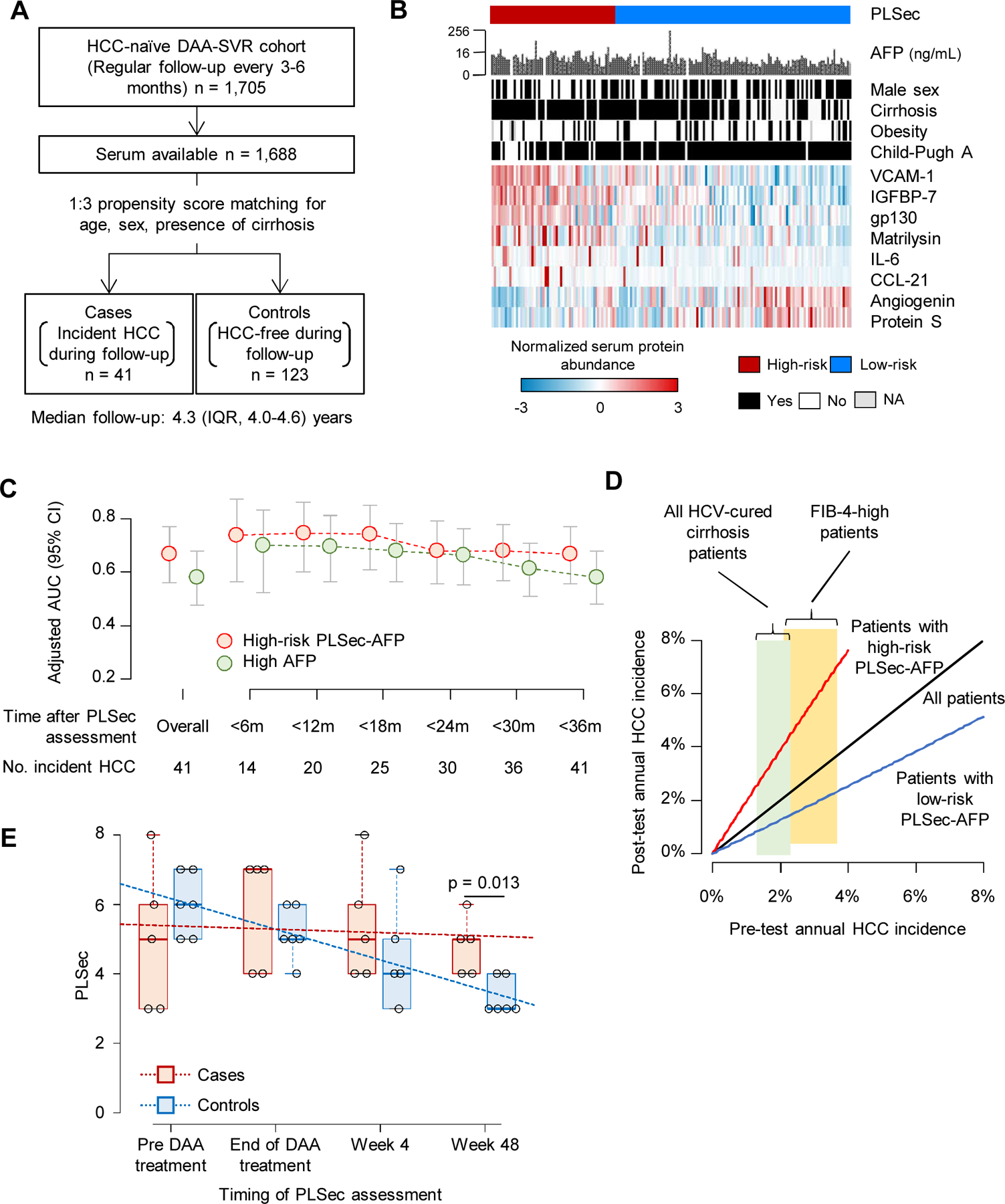
(A) Study design. (B) Patten of the PLSec protein abundance and associated clinical variables. (C) Adjusted AUC of PLSec-AFP score (≥ 1.66) and AFP (≥ 5.5 ng/mL) over time. (D) Pre- and post-test annual HCC incidence rate estimated based on the performance of PLSec-AFP in validation set 2. Widths of light green and yellow boxes indicate ranges of reported annual HCC incidence in all HCV-cured cirrhosis patients and their subset with high FIB-4 index, respectively. (E) Change in PLSec over the course of DAA-based anti-HCV treatment and post-treatment follow-up. Trend of change in PLSec over time was tested by Jonckheere-Terpstra test (p=0.43 for the cases; p <0.001 for the controls). PLSec values at week 48 were lower in the controls compared to the cases (Wilcoxon rank-sum test, p=0.013).
See also Figure S1 and Table S2, 3.
HCC, hepatocellular carcinoma; DAA, direct-acting antivirals; SVR, sustained virologic response; IQR, interquartile range; PLSec, prognostic liver secretome signature; AFP, alpha-fetoprotein; AUC, area under receiver operating characteristic curve; CI, confidence interval; HCV, hepatitis C virus.
Among analyzed patients, time-series PLSec assessment was performed in 11 patients (5 cases and 6 controls) (Figure 3E). PLSec significantly declined after treatment in the controls, whereas it remained stably elevated among the cases, suggesting that the kinetic change of molecular HCC risk status measured by PLSec may also be used to monitor prognostic efficacy of anti-HCV or chemoprevention therapies.
Validation set 3: de novo HCC recurrence after complete HCC treatment response and HCV cure
DAA therapy is increasingly considered in conjunction with curative HCC treatment because of an observed survival benefit.23 However, these patients remain at risk of de novo HCC recurrence (i.e., newly initiated HCC in remnant diseased liver clonally unrelated to the initially treated tumor) 20,24 and therefore need HCC risk prediction. We evaluated PLSec-AFP in a cohort of 146 patients with history of treated HCC, with confirmed complete response, and SVR after DAA therapy (Table 1; Figure 4A, 4B). At time of PLSec-AFP assessment, patients were recurrence-free for a median of 1.5 years, indicating that observed HCC incidences during the follow-up were more likely de novo recurrence. High-risk PLSec-AFP showed a significant association with recurrence (aHR, 3.08; 95% CI, 1.78–5.31; p<0.001) (Table 2). The association remained significant in the subset of patients with cirrhosis (aHR, 3.44; 95% CI, 1.86–6.36), whereas presence of cirrhosis was not associated with HCC recurrence (aHR, 0.88, 95% CI, 0.46–1.68). Cumulative incidences of recurrent HCC at 1 and 3 years were 18.5% and 30.8% among 104 low-risk patients, and 38.2% and 69.7% among 42 high-risk patients, respectively (Figure 4C). Prognostic association and model fitness for PLSec-AFP were superior to AFP alone (integrated Brier score, 0.196 vs. 0.206; c-index, 0.74 vs. 0.64; AIC, 590 vs. 602; BIC, 592 vs. 605 for PLSec-AFP and AFP, respectively) (Table 2; Figure S1E). Time-dependent AUC showed stably superior prognostic performance of PLSec-AFP over time (Figure 4D). Both high-risk PLSec-AFP and high AFP were comparably well calibrated (Figure 4E; Figure S1F). Interestingly, when DAA therapy was initiated >1 year after HCC cure, high-risk PLSec-AFP showed an enhanced association with recurrent HCC (aHR, 7.32; 95% CI, 2.86–18.8) (Figure S1G).
Figure 4. Clinical utility validation 3: HCC risk after complete response to HCC therapies and HCV cure by DAA (prospective-retrospective cohort).
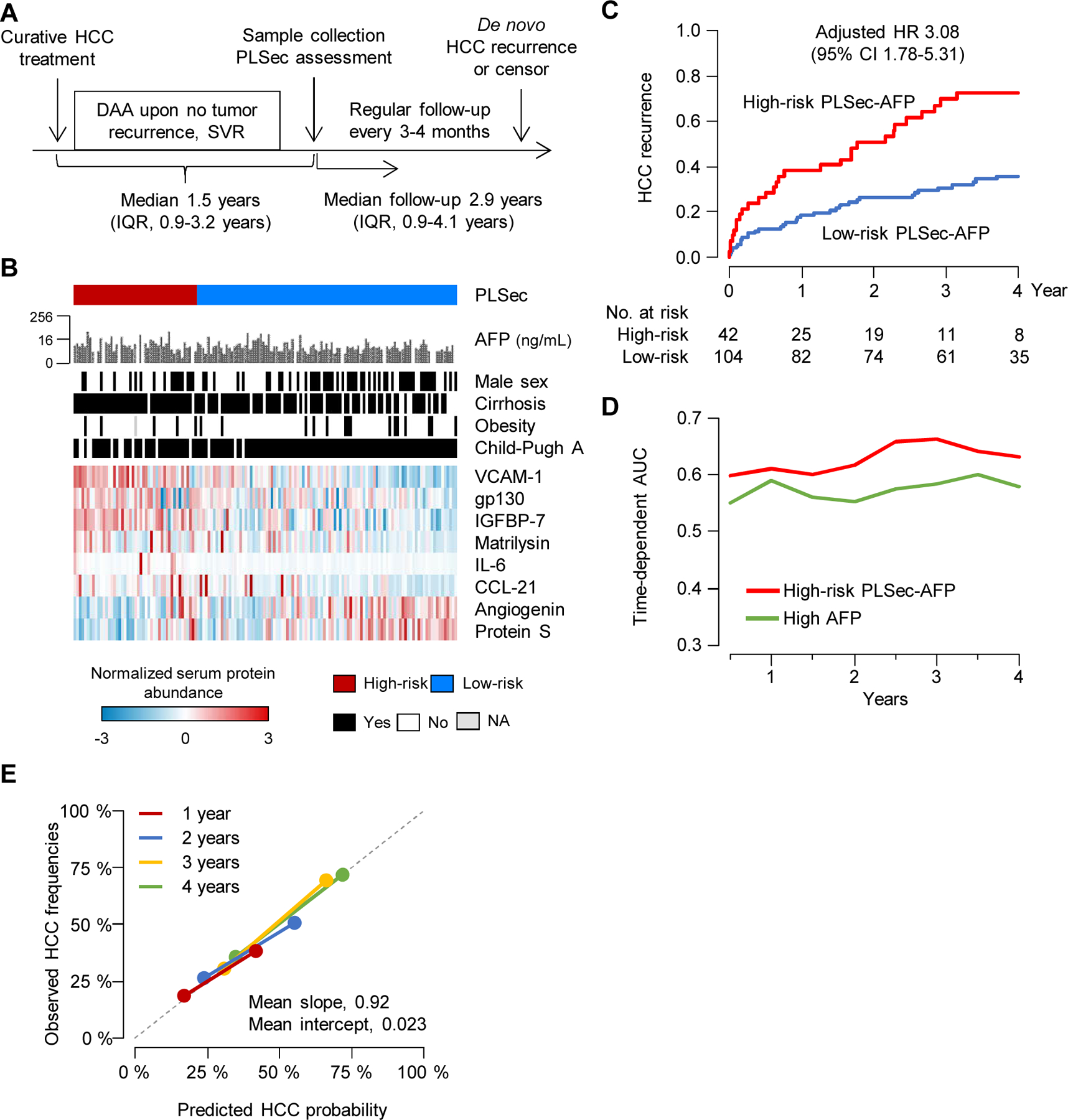
(A) Study design (B) Patten of the PLSec protein abundance and associated clinical variables. (C) Association of PLSec-AFP with HCC recurrence. (D) Time-dependent AUC of high-risk PLSec-AFP and high AFP (≥ 5.5 ng/mL). (E) Calibration plots of high-risk PLSec-AFP at various time points. The grey dash line indicates ideal calibration.
See also Figure S1 and Table S3, 4.
HCC, hepatocellular carcinoma; DAA, direct-acting antivirals; PLSec, prognostic liver secretome signature; IQR, interquartile range; AFP, alpha-fetoprotein; HR, hazard ratio; CI, confidence interval; AUC; area under receiver operating characteristic curve.
DISCUSSION
The rapid increase in the number of patients with NAFLD and cured HCV25 has created a pressing need for stratifying the vast patient population based on long-term HCC risk because their HCC incidence is relatively low. Long-term HCC risk biomarkers may identify those who benefit most from close monitoring for disease progression and emerging chemopreventive interventions.1,26,27 Our previous simulation-based study showed that HCC-risk-biomarker-based stratified HCC screening prolongs overall survival with minimal increase in net medical care costs when the biomarker identifies two-fold or higher HCC risk,6 which was achieved by our PLSec-AFP in this study. Furthermore, PLSec-AFP also predicts recurrence after curative HCC treatment. Such upfront risk stratification will enable individual-risk-based personalized HCC screening by guiding allocation of the limited medical resources for the semi-annual HCC screening (that accommodate only <25% of the guideline-recommended target patient population for the HCC screening5) to a subset of patients with elevated HCC risk. Moreover, the PLSec-based high-risk patients will be the rational target for HCC screening with new high-performance modalities (e.g., circulating cell-free methylated DNA associated with HCC occurrence within 8 months28) to improve early tumor detection and prolong patient survival. In fact, our previous Markov model-based simulation study demonstrated that such individual-risk-based personalized HCC screening strategies are substantially more cost-effective compared to the current “one-size-fits-all” HCC screening.6
Identification of low-risk patients is also important to avoid cancer screening with low likelihood of benefit given potential physical, psychological, and financial harms.29 PLSec-AFP is a continuous score with linear correlation with time to incident HCC, and therefore a cut-off to define such distinctly low-risk patients should be explored in future clinical studies. Indeed, among patients within the lowest quintile of PLSec-AFP in the validation set 1, annual HCC incidence rate was only 0.6% - well below the cost-effectiveness threshold from prior decision analyses.6,30
There is also increased interest in HCC chemoprevention using generic agents such as aspirin and statins1. PLSec-AFP can refine assessment of risk-benefit ratio for these drugs, which can cause adverse events such as bleeding and hepatotoxicity, according to individual HCC risk. HCC chemoprevention clinical trials can also be made time- and cost-efficient by identifying and enrolling only high-risk patients using PLSec-AFP. Moreover, given that PLS can be therapeutically modulated,10,13 PLS/PLSec could also be considered as surrogate endpoints for HCC chemoprevention trials to estimate long-term prognostic benefit of the experimental therapies. This concept has been sought in our ongoing and planned trials with PLS/PLSec as companion biomarkers (NCT02273362, NCT04172779).
Several proteins in the PLSec have been acknowledged for their potential roles in hepatocarcinogenesis, and may serve as rational chemoprevention targets to be monitored by PLSec. For example, IL-6 has been known as an HCC driver, which is detectable in serum.31 Soluble gp130 is a natural inhibitor of IL-6 signaling, but paradoxically associated with progressive liver disease in patients.32 Its association with elevated HCC risk may indicate counteracting response to activated IL-6 pathway. Soluble VCAM-1 is a chemotactic factor in the liver and associated with severity of chronic liver disease.33 CCL-21 is a member of the molecular signature of the ectopic lymphoid structure, which serves as micro-niches for hepatocarcinogenesis.34
In summary, our PLSec-based score shows the utility of individual-prognostic-risk-based management of patients with advanced liver fibrosis for cost-effective care and transformative improvement of patient prognosis. This has a significant clinical and medical economic impact given the high prevalence of liver fibrosis and HCC that affect 1.5 billion people and account for 3.5% of all deaths globally.2,25 It also promises to introduce a precision-medicine approach to chronic liver disease management. Furthermore, our generic pipeline of translating prognostic tissue transcriptome signatures into surrogate circulating markers can facilitate non-invasive prognostic molecular biomarker discovery in other diseases.
Limitations of Study
Despite our promising results, we acknowledge several limitations. First, besides cured HCV, dedicated evaluation in patients with other major HCC etiologies, NAFLD and HBV infection, should be pursued in future studies. Second, previous studies have reported association of advanced fibrosis with post-SVR HCC risk.21,22 It is of interest to assess its joint prognostic performance with PLSec in future studies. Third, prospective evaluation of the PLSec-AFP-based risk-stratified HCC screening is still needed. Fourth, the time-series PLSec assessment for its dynamic change over time warrants future evaluation in larger cohorts to further understanding of natural history of chronic liver disease and response to therapeutic intervention. Finally, we leveraged the PRoBE design with archived specimens,35 therefore other potential risk stratification variables/biomarkers were not available for comparison.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Yujin Hoshida (Yujin.Hoshida@UTSouthwestern.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Serum protein abundance data are available from Mendeley Data at http://dx.doi.org/10.17632/5r7c48xkbw.1. The R codes for Gene Set Enrichment Index based on single-sample-based signature enrichment analysis (eseach algorithm)10 and TexSEC are available from the corresponding author upon reasonable request. The research team will provide an email address for communication once the information sharing is approved. The proposal should include detailed aims, statistical plan, and other information/materials to guarantee the rationality of requirement and the security of the data. The related patient data will be shared after review and approval of the submitted proposal and any related requested materials. Of note, data with patient names and other identifiers cannot be shared.
EXPERIMENTAL MODEL AND SUJECT DETAILS
Analysis of archived de-identified samples and clinical information was approved as an exempt study (category 4) by the institutional review board (approval numbers: STU 062018–058, STU 082017–013).
Optimization set for PLSec
PLSec was optimized based on recapitulation of prognostic hepatic transcriptome and the least redundant information among the protein probes defined by the least absolute shrinkage and selection operator algorithm36 in a cohort of 79 chronic hepatitis/cirrhosis patients from our previous study7 (optimization set) (please refer to Supplementary Methods). The serum samples were collected approximately 3 months (median 92 [IQR 75–107] days) after HCC resection to minimize the influence of surgical procedure and proteins released from HCC tumor. Late recurrence was defined as HCC tumor recurrence 2 years after the surgical resection of the primary HCC, which was shown to be clonally independent in our previous study.7 The follow-up time was defined as the interval between the date of blood collection and the primary endpoints (late recurrence) or the last observation date without the clinical events. For the time-to-HCC recurrence analyses, death without HCC recurrence was handled as a censoring.
Patients for validation of PLSec and a construction of PLSec-AFP
The optimized PLSec was first validated in an independent cohort of cirrhosis patients (validation set 1, cohort study) for its association with HCC risk. Subsequently, we developed a composite score with other clinical variables associated with HCC risk, which was further validated in two independent cohorts (validation set 2 [nested case-control series] and 3 [cohort study]) (Figure 1; Table 1), utilizing a prospective specimen collection, retrospective blinded evaluation (PRoBE) design.35 HCC was diagnosed based on histological or imaging-based examinations according to practice guidelines.4,37
Validation set 1 (prospective–retrospective cohort):
A total of 331 cirrhosis patients with mixed etiologies were consecutively and prospectively enrolled at University of Michigan between January 2004 and September 2006, and regularly followed up using ultrasonography with AFP every 6 months for incident HCC for a median of 4.5 years (IQR, 1.9–11.4 years). During the clinical follow-up, 46 patients developed HCC. Time to HCC development was defined as the interval between the dates of PLSec assessment and HCC diagnosis or the last follow-up or death as a censored observation. HCC was diagnosed based on histological or imaging-based (contrast-enhanced multiphase CT and/or MRI) examinations according to the American Association of the Study of Liver Disease (AASLD) practice guideline.4 Liver disease etiology was defined for each patient following the AASLD guidelines (www.aasld.org/publications/practice-guidelines). Diagnosis of cirrhosis was based on liver histology or clinical-, laboratory-, and/or imaging-based evidence. Obesity was defined as body mass index (BMI) ≥ 30 kg/m2 according to WHO criteria.38 Diabetes was based on medical history or a 75-gram oral glucose tolerance test. Active hazardous alcohol drinking was defined as alcohol consumption of ≥ 48 grams per day for men and ≥ 24 grams per day for women.39
Validation sets for PLSec-AFP
Validation set 2 (nested case-control series):
A total of 1,705 patients were consecutively treated with DAA for chronic hepatitis C and achieved SVR, defined as no HCV RNA detection at 24 weeks after DAA treatment, at Toranomon Hospital, Tokyo between September 2014 and October 2017.40 Serum samples collected at 4 weeks after DAA treatment completion from 1,688 patients were available for the PLSec assessment. All patients were regularly screened for incident HCC with ultrasonography along with AFP and des-gamma-carboxy prothrombin every 3–6 months after completion of DAA treatment. Time to HCC development was defined as the interval between the dates of PLSec assessment and HCC diagnosis or the last follow-up or death as a censored observation. During a median follow-up of 4.3 years (IQR, 4.0–4.6 years), 41 patients developed HCC and were designated as cases. From the rest of the patients, 123 patients were selected as controls (HCC-free for at least 3.7 years) using the propensity score matching for age at DAA initiation, sex, and presence of cirrhosis using MatchIt R package (1:3 matching) (Table 1). HCC diagnosis was based on the Japan Society for Hepatology practice guidelines.37 Obesity was defined as BMI ≥ 25 kg/m2 according to the Asian-Pacific criteria.41 Active hazardous alcohol drinking was defined by alcohol consumption at the PLSec assessment of ≥ 40 grams per day for men and ≥ 20 grams per day for women.42 High AFP was defined by ≥ 5.5 ng/mL.43,44
Validation set 3 (prospective–retrospective cohort):
A total of 146 patients were consecutively treated with DAA and achieved SVR after confirming complete response to HCC treatment at Toranomon Hospital between November 2014 and January 2018.45 The patients were diagnosed for early-stage HCC (American Joint Committee of Cancer, T1/2 tumor without extrahepatic lesion) and received HCC treatment (surgical resection, thermal ablation, transarterial chemoembolization, or stereotactic body radiation therapy). The absence of residual tumor was histologically (as no microscopic tumor cells at/near surgical resection margin) and/or radiologically (as no enhanced lesion with contrast-enhanced multiphase CT and/or MRI) confirmed before initiating DAA therapy. Patients who had HCC recurrence during DAA treatment were excluded. Blood samples were collected at 4 weeks after DAA treatment completion and used for the PLSec assessment. All patients were regularly followed up for HCC recurrence with multiphase CT and/or MRI every 3–4 months. Time to HCC development was defined as the interval between the dates of PLSec assessment and HCC diagnosis or the last follow-up or death as a censored observation. During a median follow-up of 2.9 years (IQR, 0.9–4.1 years), 65 patients developed HCC recurrence. At the date of PLSec assessment, the patients were already recurrence-free for a median of 1.5 years (IQR, 0.9–3.2 years) since the previous HCC treatment, therefore the observed recurrences are assumed to be dominantly de novo HCC recurrence. HCC diagnosis as well as the determination of cirrhosis, obesity, diabetes, active hazardous alcohol drinking, and high AFP was similarly performed as in the validation set 2.
METHOD DETAILS
Computational derivation of Prognostic Liver Secretome signature (PLSec) – overview
Transcriptome profiling of diseased organ tissue has been widely used as the first step to reliably identify pathogenic and prognostic molecular dysregulation. However, as a clinical biomarker, the need for invasive tissue biopsy generally limits its clinical applicability especially in the setting of risk assessment for the future emergence of adverse outcomes in asymptomatic individuals. On the other hand, direct biomarker discovery in circulation obscures the source organ that releases the biomolecules. To overcome the challenge and enable less-invasive monitoring of organ-specific biological dysregulation, we developed a computational pipeline to systematically translate a tissue-transcriptome-based molecular signature into a list of proteins inferred to be released into circulation from an organ of interest. A previous proof-of-concept study demonstrated the feasibility of this approach by integrating proteome databases in ovarian cancer.46 We have streamlined the strategy by integrating (i) assembled proteome databases with an algorithm to infer organ specificity of proteins in circulation (liver secretome biomarker candidates), and (ii) a list of genes (i.e., gene signature) specific to a disease context, e.g., hepatocellular carcinoma (HCC) risk prediction in cirrhosis (prognostic liver proteome biomarker candidates) to identify a list of circulating proteins for prognostic prediction (prognostic liver secretome biomarker candidates) (please refer to Supplementary Methods) as detailed in the following sections.
Derivation of liver secretome biomarker candidates
We developed a versatile pipeline to infer a list of proteins that are likely secreted into circulation from an organ of interest, called Translation of tissue gene expression to secretome (TexSEC) (www.texsec-app.org). The pipeline consists of two components: (i) computational derivation of secretome biomarker candidates, and (ii) organ specificity/ambiguity assessment. First, the secretome biomarker candidates were derived as follows (please refer to Supplementary Methods). Circulating proteins include actively secreted proteins with functional roles outside their source cells/organs (at high abundance for housekeeping purposes, e.g., albumin, or with occasional secretion when needed, e.g., cytokines) and leaked proteins from injured cells. To broadly survey these types of proteins as candidate circulating biomarkers, we integrated proteome databases that complementarily cover bioinformatically-predicted secretable proteins as well as empirically detected proteins in human body fluids. The following three algorithms to predict extracellular secretion based on signal peptide sequences were applied to a total of 20,431 non-redundant proteins encoded by 20,103 genes from UniProtKB (www.uniprot.org)47. SignalP-5.048 is an algorithm that predicts secretory signal peptides transported by the Sec translocon and cleaved by Signal Peptidase I using a deep neural network (www.cbs.dtu.dk/services/SignalP-5.0/) and identified 3,528 proteins. DeepSig v.1.049 focuses on signal peptide sequences located at N-terminus in the membrane and other proteins (deepsig.biocomp.unibo.it) and identified 3,133 proteins. TOPCONS250 is a topology-based method combining five algorithms, OCTOPUS, Philius, PolyPhobius, SCAMPI, and SPOCTOPUS (topcons.cbr.su.se) and identified 3,772 proteins. The intersection of the three prediction methods, including 2,875 proteins, was further considered as secretome biomarker candidates.
As complementary wet-lab-based experimental evidence of detection in human body fluids, we integrated the following three proteome databases. A list of mass-spectrometry-based human plasma proteins was retrieved from Human Plasma Protein Project PeptideAltas database51 (www.peptideatlas.org/hupo/c-hppp), including 3,485 “evidence level 1” proteins. Plasma Protein Database52 is a literature-based collection of human plasma and serum proteins (www.plasmaproteomedatabase.org), in which 3,742 proteins reported in two or more studies were included. Protein Abundance Across Organisms (PAXdb) v.4.153 is a database of human protein abundance measured in 15 organs, plasma, and urine (pax-db.org), from which 4,312 proteins were retrieved. A total of 3,274 proteins detected in at least two of the three databases were included in subsequent analysis. Finally, the union of the two types of proteins, i.e., computationally-inferred and experimentally-detected proteins, were regarded as a pool of secretome biomarker candidates that includes 5,134 proteins encoded by 5,044 genes.
Organ specificity/ambiguity of the candidate proteins was estimated as follows (please refer to Supplementary Methods). Proteins released from non-target organs especially at high baseline levels will obscure pathogenic change in abundance of proteins released from the target organ of interest (i.e., liver in this study).54,55 To maximize the chance to identify candidate biomarker proteins to monitor the target organ, we used the PAXdb, a comprehensive database of human proteins quantified in 15 organs (liver, brain, colon, rectum, esophagus, female gonad, gallbladder, heart, kidney, lung, pancreas, prostate, skin, testis, and uterus), plasma, and urine from healthy individuals, in which quantitative protein abundance data are available for 4,491 out of the 5,134 proteins.53 Among the proteins, 211 proteins (5%) in plasma + urine are substantially more abundant compared to the sum of the 15 organs (at least 5-fold higher), suggesting that these proteins are released from other organ(s) not covered in the database or immediately released from source organ(s) among the 15 organs. These proteins were regarded as released from “unknown origin” as a pseudo organ (e.g., leptin secreted from adipose tissues), and the rest of the proteins were assessed for their association with the 15 organs. The proteins were classified into the following three categories based on their relative abundance across the organs: organ-specific, i.e., > 5-fold higher in one organ compared to the rest; organ-group-specific, > 5-fold higher in two to five organ (median) compared to the rest; non-specific. We retained proteins excessively secreted into urine and absent or scarce in plasma, defined by urine-to-plasma ratio > 10-fold, regardless of organ specificity because such proteins may still serve as liver-related biomarkers in case pathogenic secretion from liver exceeds the excretion into urine and becomes detectable in plasma (such as a prostate-specific protein, growth/differentiation factor 15 [GDF-15], associated with chronic liver disease and HCC when detected in blood56,57). These parameters are modifiable for an organ of user’s interest in the TexSEC web application. We kept 643 secretome biomarker candidates unavailable in the PAXdb for the organ specificity/ambiguity assessment and finally selected 4,125 proteins as liver secretome biomarker candidates to be merged with the list of proteins specifically related to the prognostic tissue transcriptome gene signatures described in the next section.
Derivation of prognostic liver proteome biomarker candidates
As the source of tissue transcriptome signatures to be translated into blood secretome panel, we used our previously defined 186-gene prognostic liver signature (PLS) and 132-gene late recurrence signature (LRS).7 In addition to proteins encoded by the signature member genes, we considered proteins in molecular pathways associated with the tissue transcriptome signature as follows. To systematically identify relevant pathways in an unbiased manner across the major liver disease etiologies, we surveyed 1,316 gene sets of well-defined molecular pathways from Molecular Signature Database (MSigDB, v6.2)58 using gene set enrichment analysis59 in the training data sets, including four independent cohorts of 523 chronic liver disease patients affected with hepatitis B virus (HBV), hepatitis C virus (HCV) (including resolved HCV), alcohol-related liver disease (ARLD), and non-alcoholic fatty liver disease (NAFLD) (please refer to Supplementary Methods). Enrichment of each pathway gene set in each cohort was assessed on rank-ordered genes by correlation with that of the prognostic gene signature in each patient. The enrichment of each pathway gene set across the cohorts was synthesized using Fisher’s inverse chi-square statistic,60 and 466 pathways showed association in the same direction with statistical significance (random permutation test-based false discovery rate <0.25) (Data S1). For each of the associated pathways, proteins encoded by leading-edge genes59 contributing to the enrichment as well as their putative upstream signals were added to the list of candidate proteins. With proteins encoded by the tissue transcriptome signature genes themselves, we identified 1,631 proteins (1,020 high- and 611 low-risk-associated proteins) as prognostic liver proteome biomarker candidates, which were validated for their association with tissue PLS/LRS status in the validation data sets of 16 cohorts of 1,034 chronic liver disease patients (please refer to Supplementary Methods).
Identification of prognostic liver secretome biomarker candidates
Finally, we took the intersection of the two lists of proteins, i.e., liver secretome biomarker candidates and prognostic liver proteome biomarker candidates, and derived prognostic liver secretome biomarker candidates, including 697 proteins (431 high- and 266 low-risk-associated proteins) proteins, for subsequent assay implementation.
Implementation and optimization of PLSec assay
Among the 697 prognostic liver secretome biomarker candidates, validated antibodies are available for multiplex assay for 43 proteins (41 high- and 2 low-risk-associated proteins), which were implemented in an FDA-approved multiplex clinical diagnostic technology, xMAP platform (Luminex), as a preliminary version of the Prognostic Liver Secretome signature (PLSec) assay, and run on the Bio-Plex 200 systems (Bio-Rad) at UT Southwestern BioCenter according to the manufacturer’s protocol. The abundance of each protein was measured as median fluorescent intensity (MFI) corrected for background signals from negative control probes and normalized to built-in dilution series of positive control probes as the standards in each 96-well assay plate, with which plate-to-plate adjustment of MFI values was performed. Please see Supplementary Methods.
We first assessed the correlation between serum protein abundance and tissue gene signature status (not expression level of genes that encode the proteins, but collective induction or suppression of each of high- or low-risk-associated signature genes quantified by gene-set-enrichment-based statistic as a measure of the molecular status of the liver as detailed below) in the optimization set. Among the 43 assayed proteins, the abundance of 27 proteins (63%) was significantly correlated with modulation of each of the high- or low-risk-associated genes in the hepatic transcriptome signatures (i.e., PLS and LRS) measured by Gene Set Enrichment Index based on single-sample-based signature enrichment analysis (eseach algorithm)10 (Spearman correlation test, false discovery rate < 0.25). Next, we evaluated correlation across the 27 proteins in a correlation matrix, which revealed that there are several groups of proteins sharing a highly similar pattern of abundance across the patients (i.e. redundancy in captured information) in the optimization set (please refer to Supplementary Methods). Because a larger number of assay probes generally makes the development of clinical diagnostic assay more complex and costly due to increased burden of developing and validating the assay probes, we attempted to perform dimensionality reduction to shave the redundant probes without sacrificing the prognostic association by using the least absolute shrinkage and selection operator (LASSO) algorithm.36,61 We analyzed high- and low-risk-associated proteins separately to resolve the redundancy within each direction of the prognostic association. We repeated the feature selection based on 10-fold cross-validation scheme 1,000 times and chose the most frequently selected 8 features, i.e., 6 high-risk-associated proteins (vascular cell adhesion molecule 1 [VCAM-1], insulin-like growth factor-binding protein 7 [IGFBP-7], gp130, matrilysin, interleukin-6 [IL-6], and C-C motif chemokine ligand 21 [CCL-21]), and 2 low-risk-associated proteins (angiogenin and protein S). We finally termed the 8-protein secretome signature as PLSec. We also evaluated the technical validity of the assay. We observed high within-plate reproducibility (r2 = 0.9997, p = 1.1×10-11), inter-plate/batch reproducibility (r2 = 0.971, p = 7.7×10-6) of technical replicates, and sensitivity of positive control proteins (99.9% ± 2.5%), supporting reliable protein quantification with the assay platform as a clinical diagnostic test (please refer to Supplementary Methods).
To use the PLSec assay to assist clinical decision making according to the predicted prognosis, we converted the multi-analyte measurements into a single value as follows. In general, antibody-based protein quantification is sensitive to change in experimental conditions such as lot of antibody reagents, and it makes assay calibration more challenging, especially in the clinical setting. To minimize the influence of the potential variation in the measurements and ensure the robust prognostic performance of the assay in a clinic, we converted the continuous protein abundance values (i.e., normalized MFI) into high or low abundance by top quartile cut-off in the optimization set, and calculated a semi-quantitative score as follows:
The cut-off value for the PLSec of 4 was chosen to maximize prognostic association for late recurrence based on log-rank test p-value in the optimization set. Assessment of area under receiver operating characteristic curve (AUC) to the risk prediction by the tissue-based PLS showed that the selection of the 27 proteins out of the 43 all assayed proteins improved performance of the panel as expected, and the reduction to 8 proteins with LASSO maintained the panel’s performance. More importantly, significant prognostic associations for late recurrence were maintained. Please refer to Supplementary Methods.
Lastly, we assessed whether the finally optimized 8-protein PLSec panel recapitulated the full biological information associated with the original tissue-transcriptome-based gene signatures with regard to involved molecular pathways and cell types present in the liver. For the derivation of prognostic liver proteome biomarker candidates, we identified 466 molecular pathway gene sets associated with the tissue transcriptome signatures. We asked how many of these gene sets are dysregulated in association with the PLSec-based outcome prediction as we observed for the tissue transcriptome signatures. In the genome-wide transcriptome dataset of the optimization set, genes were rank-ordered by Spearman’s rank correlation (rho) with the PLSec score, and enrichment of the gene sets was assessed by Gene Set Enrichment Analysis (GSEA)59. We confirmed that 435 gene sets (93%) were enriched as observed for the tissue transcriptome signatures (Data S1). Besides, in single-cell transcriptome data of three human cirrhotic livers (NCBI Gene Expression Omnibus, GSE136103)62, induction of the 466 pathways across the cell types present in each liver was assessed by the eseach algorithm, which depicted involvement of diverse cell types, covering parenchymal, stromal, and infiltrating cell types (please refer to Supplementary Methods). We could also confirm that all of the involved cell types were recapitulated by the 8-protein PLSec panel. Thus, we could technically validate and optimize the 8-protein PLSec assay, maintaining association with prognosis and relevant biological information for the original tissue-based transcriptome signatures. These results collectively support that the PLSec assay is ready for subsequent clinical utility validation.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
All statistical analyses were performed using the R statistical language (www.r-project.org). For time-to-event analyses (validation sets 1 and 3), prognostic associations of the clinical variables and PLSec were assessed using Kaplan-Meier curves and uni/multivariable Cox regression modeling. Annual incidence rates were calculated per 100 person-years, and cumulative incidences at certain time points were estimated by Kaplan-Meier method. Proportional-hazards assumption was confirmed by using cox.zph function in R survival package (Table S2). Sample size to detect hazard ratio of 3 as statistically significant is 232 under assumption that 25% of the patients show high-risk score and 15% of the patients develop HCC at statistical power of 0.8 and alpha error of 0.05. The case-control series (validation set 2) was analyzed by multivariable conditional logistic regression. To develop a composite prognostic score combining PLSec and clinical variables in the validation set 1, PLSec and AFP were chosen based on multivariable Cox regression p-value less than 0.05 (Table S1).
It is clinically known that AFP can increase at low level in association with non-malignant conditions such as chronic hepatic inflammation accompanied with hepatocyte regeneration.63 Even with the clinically used cut-off of > 20 ng/mL, HCC is present only in up to 60% of the patients.4 In this cohort, vast majority of the patients (93%) showed AFP levels even below the cut-off. If a high AFP is indicative of already existing HCC in the cohort, the tumor nodule should be clinically diagnosed in 2–3 years given the tumor volume doubling time is 4.7 months according to a recent meta-analysis.64 This is not the case in majority of our cohort as shown by consistent incidence rates over time (Figure S1B) and proportional hazard of incident HCC irrespective of AFP levels (Table S2). Collectively, we assume that AFP in this cohort reflected more likely tumor initiating microenvironment in liver (so-called “field effect”) rather than an existing but undetected tumor.
Both PLSec and AFP are linearly correlated with time to HCC development according to non-linearity test on log relative risk plot (Figure S1C) and are independently associated with time to HCC development (p = 0.548 for their interaction term in multivariable Cox regression). The composite PLSec-AFP score was derived by using regression coefficients from multivariable Cox regression as follows: PLSec-AFP = 0.175×PLSec + 0.325×log2(1+AFP). The risk-predictive performance of the PLSec-AFP score was assessed and compared to that of AFP alone using integrated Brier score65 and c-indix66 calculated by pec R package,67 and time-dependent AUCs in validation set 1 and 3, and Brier score and covariate-adjusted AUC68 in the validation set 2 (Table 2). Fitness of the models was assessed by Akaike information criterion (AIC) and Bayesian information criterion (BIC). The confidence intervals were estimated by bootstrapping of the samples (n=1,000). A predefined subgroup analysis was performed in patients with cirrhosis in the validation set 2 and 3.
The cut-off of 1.66 was defined to determine high-risk patients based on maximally selected rank statistics using maxstat R package.69 Hazard ratios in validation set 1 and 3 were primarily adjusted for known clinical variables that influence liver disease prognosis, i.e., age (as continuous), sex, obesity, diabetes, and active hazardous alcohol drinking as defined above. The odds ratios in validation set 2 were adjusted for obesity, diabetes, and active hazardous alcohol drinking with conditioning on the pairs of cases and the matched controls. Besides, the hazard ratios and odds ratios were also adjusted for liver function reserve (Child-Pugh class A vs. the rest), a liver fibrosis indicator, FIB-4 index (≥3.25 vs. <3.25),21 and all variables used in each model as sensitivity analyses in all validation cohorts (Table S3). Missing data on the covariates used in multivariable adjustments were imputed using classification and regression trees.70,71 For each incomplete case, we identified five sets of imputed values. The incomplete case’s missing value was replaced with median of the five values for continuous variables and the most frequent value for categorical variables. Proportion of missing values in each variable was ≤10%.72
In a matched case-control study, the receiver operating characteristic curve is substantially biased when the data does not acknowledge covariate matching.68 Therefore, we evaluated the performance of high-risk PLSec-AFP in validation set 2 using covariate-adjusted AUCs, including variables used in the matching, i.e., age, sex, and presence of cirrhosis, with semiparametric Bayesian inference, calculated by AROC R package (Figure 3C). Besides, covariate-adjusted AUC was used to compare a performance of PLSec-AFP to that of AFP alone in validation set 2.
In the validation set 2, the effect of PLSec-AFP on HCC risk discrimination was estimated according to Bayes’ theorem as follows:
,where x (%) denotes pre-test annual HCC risk, based on sensitivity (%) and specificity (%) for incident HCC within 3 years after PLSec-APF assessment based on their confirmed stability over time (Figure S1D).
Supplementary Material
KEY RESOURCES
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Serum from patients who underwent curative HCC resection (optimization set) | Toranomon Hospital | N/A |
| Serum from patients with cirrhosis (validation set 1) | University of Michigan | N/A |
| Serum from patients with or without HCC after hepatitis C virus cure (validation set 2) | Toranomon Hospital | N/A |
| Serum from patients with HCV-related liver fibrosis consecutively treated with DAA and achieving SVR after curative HCC treatment (validation set 3) | Toranomon Hospital | N/A |
| Critical Commercial Assays | ||
| Human Magnetic Luminex Assay | R&D | Cat No. LXSAHM |
| Bio-Plex 200 systems | BioRad | 171000201 |
| Deposited Data | ||
| Serum protein abundance data | This manuscript | http://dx.doi.org/10.17632/5r7c48xkbw.1 |
| Human liver biopsy specimens from HCV, early-stage liver cirrhosis, microarray | Hoshida et al. | GEO Accession GSE15654 |
| Human non-tumor liver tissues from patients with HBV-related HCC undergoing surgical resection, microarray | Roessler et al. | GEO Accession GSE14520 |
| Human liver biopsy specimens from NASH, microarray | Moylan et al. | GEO Accession GSE49541 |
| Human liver biopsy specimens from alcohol-related cirrhosis, microarray | Trepo et al. | GEO Accession GSE94417 |
| Human non-tumor liver tissues from patients with HCV-related HCC undergoing surgical resection, microarray | Tsuchiya et al. | GEO Accession GSE17856 |
| Human liver specimens from patients with HCV infection, microarray | Archer et al. | GEO Accession GSE17967 |
| Human liver biopsy specimens after HCV cure with DAA, microarray | Meissner et al. | GEO Accession GSE51699 |
| Human liver biopsy specimens after hepatitis virus cure with direct-acting antivirals, microarray | Meissner et al. | GEO Accession GSE70779 |
| Human non-tumor liver tissues from patients with HCC undergoing surgical resection, microarray | Kim et al. | GEO Accession GSE39791 |
| Human non-tumor liver tissues from patients with HBV-related HCC undergoing surgical resection, microarray | Halgand et al. | GEO Accession GSE47197 |
| Human non-tumor liver tissues from patients with HCC undergoing surgical resection, microarray | Chaisaingmongkol et al. | GEO Accession GSE76297 |
| Human non-tumor liver tissues from patients with HCC undergoing surgical resection, microarray | Grinchuk et al. | GEO Accession GSE76427 |
| Human non-tumor liver tissues from patients with HBV-related HCC undergoing surgical resection, microarray | Zhou et al. | GEO Accession GSE83148 |
| Human non-tumor liver tissues from patients with HCC undergoing surgical resection, microarray | Wang et al. | GEO Accession GSE84044 |
| Human non-tumor liver tissues from patients with HBV-related HCC undergoing surgical resection, microarray | Hui et al. | GEO Accession GSE121248 |
| Human liver biopsy specimens of different phases from control to NASH, microarray | Ahrens et al. | GEO Accession GSE48452 |
| Human liver biopsy specimens from adolescents undergoing bariatric surgery, microarray | Xanthakos et al. | GEO Accession GSE66676 |
| Human liver biopsy specimens of different phases from control to NAFLD, microarray | Arendt et al. | GEO Accession GSE89632 |
| Human liver biopsy specimens from obese patients, microarray | Francque et al. | GEO Accession GSE83452 |
| Human non-tumor liver tissues from patients with HCC undergoing surgical resection, microarray | Makowska et al. | GEO Accession GSE64041 |
| Software and Algorithms | ||
| R (ver. 3.6.1) | CRAN | https://cran.r-project.org/ |
| MSigDB v6.2 | Broad Institute | https://www.gsea-msigdb.org/gsea/msigdb/ |
| TexSEC (Translation of tissue gene expression to secretome) | This manuscript | www.texsec-app.org |
| eseach | Nakagawa et al. | http://www.gparc.org/ |
INCLUSION AND DIVERSITY STATEMENT.
We worked to ensure gender balance in the recruitment of human subjects. We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Highlights.
Pipeline to translate tissue mRNA to secretome signature (TexSEC) is developed
TexSEC identified a secretome signature (PLSec) predictive of liver cancer risk
PLSec together with AFP (PLSec-AFP) predicts liver cancer risk in cirrhosis
PLSec-AFP predicts liver cancer risk even after curing chronic hepatitis C
Context and Significance.
Accurate non-invasive prediction of long-term hepatocellular carcinoma (HCC) risk in advanced liver fibrosis is urgently needed for early tumor detection to improve the poor HCC prognosis. Hepatic transcriptome signatures have been validated for their HCC risk-predictive capability, but the need for liver biopsy limits their applicability in clinic. Fujiwara et al. developed a computational pipeline to translate tissue transcriptome into secretome signature, TexSEC, which identified an 8-protein blood-based prognostic liver secretome signature (PLSec). PLSec predicts long-term HCC risk in patients with advanced liver fibrosis, and a composite score with alpha-fetoprotein (PLSec-AFP) predicts HCC risk even after curing chronic hepatitis C. The PLSec-AFP will enable individual-risk-based personalized HCC screening to optimize allocation of limited medical resources and maximize likelihood of early HCC detection.
ACKNOWLEDGEMENT
This work was supported by Uehara Memorial Foundation, U.S. NIH (DK099558, CA233794, CA226052, R01CA222900, U01CA230694, U01CA230669, R01CA237659), European Commission (ERC-2014-AdG-671231), Cancer Prevention and Research Institute of Texas (RR180016), LABEX HepSYS (ANR-10-LABX-0028_HEPSYS), ARC Foundation (IHU201901299), IUF and Inserm Plan Cancer (HCCMICTAR), MGH Research Scholars Program.
Funding
NIH (DK099558, CA233794, CA222900, CA230694, CA230669, CA237659, CA226052), European Commission (ERC-2014-AdG-671231), CPRIT (RR180016), LABEX (ANR-10-LABX-0028-HEPSYS), ARC (IHU201901299), Inserm (HCCMICTAR).
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
Y.H. serves as an advisory board member for Helio Health and founding shareholder for Alentis Therapeutics, and received a research funding from Morphic Therapeutics. T.F.B. serves as advisor and is a founding shareholder or Alentis Therapeutics. R.T received a lecture fee from Bayer, Chugai, Eisai, Takeda and Wako/Fujifilm. N.P. has served as a consultant for Bristol Myers-Squibb, Exact Sciences, Eli Lilly, and Freenome. A.G.S. has served on advisory boards of Genentech, Eisai, Bayer, Exelixis, Wako/Fujifilm and has received research funding from Bayer, Target Pharmasolutions, Exact Sciences, and Glycotest.
SUPPLEMENTARY EXCEL TABLE TITLE AND LEGENDS
Data S1. PLS-/LRS-associated gene sets and their shared leading edge genes, Related to STAR Methods.
REFERENCES
- 1.Fujiwara N, Friedman SL, Goossens N, Hoshida Y. Risk factors and prevention of hepatocellular carcinoma in the era of precision medicine. J Hepatol 2018;68:526–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collaborators GBDCoD. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018;392:1736–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 4.Marrero JA, Kulik LM, Sirlin CB, et al. Diagnosis, Staging, and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 2018;68:723–50. [DOI] [PubMed] [Google Scholar]
- 5.Wolf E, Rich NE, Marrero JA, Parikh ND, Singal AG. Use of Hepatocellular Carcinoma Surveillance in Patients With Cirrhosis: A Systematic Review and Meta-Analysis. Hepatology 2020. [DOI] [PMC free article] [PubMed]
- 6.Goossens N, Singal AG, King LY, et al. Cost-Effectiveness of Risk Score-Stratified Hepatocellular Carcinoma Screening in Patients with Cirrhosis. Clin Transl Gastroenterol 2017;8:e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoshida Y, Villanueva A, Kobayashi M, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med 2008;359:1995–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoshida Y, Villanueva A, Sangiovanni A, et al. Prognostic gene expression signature for patients with hepatitis C-related early-stage cirrhosis. Gastroenterology 2013;144:1024–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.King LY, Canasto-Chibuque C, Johnson KB, et al. A genomic and clinical prognostic index for hepatitis C-related early-stage cirrhosis that predicts clinical deterioration. Gut 2015;64:1296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakagawa S, Wei L, Song WM, et al. Molecular Liver Cancer Prevention in Cirrhosis by Organ Transcriptome Analysis and Lysophosphatidic Acid Pathway Inhibition. Cancer Cell 2016;30:879–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goossens N, Hoshida Y, Song WM, et al. Nonalcoholic Steatohepatitis Is Associated With Increased Mortality in Obese Patients Undergoing Bariatric Surgery. Clin Gastroenterol Hepatol 2016;14:1619–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ono A, Goossens N, Finn RS, et al. Persisting risk of hepatocellular carcinoma after hepatitis C virus cure monitored by a liver transcriptome signature. Hepatology 2017;66:1344–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamdane N, Juhling F, Crouchet E, et al. HCV-Induced Epigenetic Changes Associated With Liver Cancer Risk Persist After Sustained Virologic Response. Gastroenterology 2019;156:2313–29e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakagawa H, Fujiwara N, Tateishi R, et al. Impact of serum levels of interleukin-6 and adiponectin on all-cause, liver-related, and liver-unrelated mortality in chronic hepatitis C patients. J Gastroenterol Hepatol 2015;30:379–88. [DOI] [PubMed] [Google Scholar]
- 15.Sun T, Li P, Sun D, Bu Q, Li G. Prognostic value of osteopontin in patients with hepatocellular carcinoma: A systematic review and meta-analysis. Medicine (Baltimore) 2018;97:e12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang BH, Li B, Kong LX, Yan LN, Yang JY. Diagnostic accuracy of midkine on hepatocellular carcinoma: A meta-analysis. PLoS One 2019;14:e0223514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hughes DM, Berhane S, Emily de Groot CA, et al. Serum Levels of alpha-Fetoprotein Increased More Than 10 Years Before Detection of Hepatocellular Carcinoma. Clin Gastroenterol Hepatol 2020. [DOI] [PMC free article] [PubMed]
- 18.Rich NE, John BV, Parikh ND, et al. Hepatocellular Carcinoma Demonstrates Heterogeneous Growth Patterns in a Multicenter Cohort of Patients With Cirrhosis. Hepatology 2020;72:1654–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Castello B, Aguilera V, Blazquez MT, et al. Post-transplantation outcome in non-alcoholic steatohepatitis cirrhosis: Comparison with alcoholic cirrhosis. Ann Hepatol 2019;18:855–61. [DOI] [PubMed] [Google Scholar]
- 20.Baumert TF, Juhling F, Ono A, Hoshida Y. Hepatitis C-related hepatocellular carcinoma in the era of new generation antivirals. BMC Med 2017;15:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanwal F, Kramer J, Asch SM, Chayanupatkul M, Cao Y, El-Serag HB. Risk of Hepatocellular Cancer in HCV Patients Treated With Direct-Acting Antiviral Agents. Gastroenterology 2017;153:996–1005e1. [DOI] [PubMed] [Google Scholar]
- 22.Ioannou GN, Beste LA, Green PK, et al. Increased Risk for Hepatocellular Carcinoma Persists Up to 10 Years After HCV Eradication in Patients with Baseline Cirrhosis or High FIB-4 Scores. Gastroenterology 2019. [DOI] [PMC free article] [PubMed]
- 23.Singal AG, Rich NE, Mehta N, et al. Direct-Acting Antiviral Therapy for Hepatitis C Virus Infection Is Associated With Increased Survival in Patients With a History of Hepatocellular Carcinoma. Gastroenterology 2019;157:1253–63e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manthravadi S, Paleti S, Pandya P. Impact of sustained viral response postcurative therapy of hepatitis C-related hepatocellular carcinoma: a systematic review and meta-analysis. Int J Cancer 2017;140:1042–9. [DOI] [PubMed] [Google Scholar]
- 25.Moon AM, Singal AG, Tapper EB. Contemporary Epidemiology of Chronic Liver Disease and Cirrhosis. Clin Gastroenterol Hepatol 2019. [DOI] [PMC free article] [PubMed]
- 26.Simon TG, Duberg AS, Aleman S, et al. Lipophilic Statins and Risk for Hepatocellular Carcinoma and Death in Patients With Chronic Viral Hepatitis: Results From a Nationwide Swedish Population. Ann Intern Med 2019. [DOI] [PMC free article] [PubMed]
- 27.Simon TG, Duberg AS, Aleman S, Chung RT, Chan AT, Ludvigsson JF. Association of Aspirin with Hepatocellular Carcinoma and Liver-Related Mortality. N Engl J Med 2020;382:1018–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qu C, Wang Y, Wang P, et al. Detection of early-stage hepatocellular carcinoma in asymptomatic HBsAg-seropositive individuals by liquid biopsy. Proc Natl Acad Sci U S A 2019;116:6308–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Atiq O, Tiro J, Yopp AC, et al. An assessment of benefits and harms of hepatocellular carcinoma surveillance in patients with cirrhosis. Hepatology 2017;65:1196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parikh ND, Singal AG, Hutton DW, Tapper EB. Cost-Effectiveness of Hepatocellular Carcinoma Surveillance: An Assessment of Benefits and Harms. Am J Gastroenterol 2020. [DOI] [PMC free article] [PubMed]
- 31.Schmidt-Arras D, Rose-John S. IL-6 pathway in the liver: From physiopathology to therapy. J Hepatol 2016;64:1403–15. [DOI] [PubMed] [Google Scholar]
- 32.Migita K, Abiru S, Maeda Y, et al. Serum levels of interleukin-6 and its soluble receptors in patients with hepatitis C virus infection. Hum Immunol 2006;67:27–32. [DOI] [PubMed] [Google Scholar]
- 33.Diaz-Sanchez A, Matilla A, Nunez O, et al. Serum level of soluble vascular cell adhesion molecule in patients with hepatocellular carcinoma and its association with severity of liver disease. Ann Hepatol 2013;12:236–47. [PubMed] [Google Scholar]
- 34.Finkin S, Yuan D, Stein I, et al. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat Immunol 2015;16:1235–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fujiwara N, Liu PH, Athuluri-Divakar SK, Zhu S, Hoshida Y. Risk Factors of Hepatocellular Carcinoma for Precision Personalized Care. In: Hoshida Y, ed. Hepatocellular Carcinoma: Translational Precision Medicine Approaches Cham (CH)2019:3–25. [PubMed] [Google Scholar]
- 36.Friedman J, Hastie T, Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- 37.Kokudo N, Takemura N, Hasegawa K, et al. Clinical practice guidelines for hepatocellular carcinoma: The Japan Society of Hepatology 2017 (4th JSH-HCC guidelines) 2019 update. Hepatol Res 2019;49:1109–13. [DOI] [PubMed] [Google Scholar]
- 38.WHO. Physical status: the use and interpretation of anthropometry. Report of a WHO Expert Committee. World Health Organ Tech Rep Ser 1995;854:1–452. [PubMed] [Google Scholar]
- 39.Elmadhun NT, Sellke FW. Is there a link between alcohol consumption and metabolic syndrome? Clinical Lipidology 2013;8:5–8. [Google Scholar]
- 40.Ogata F, Kobayashi M, Akuta N, et al. Outcome of All-Oral Direct-Acting Antiviral Regimens on the Rate of Development of Hepatocellular Carcinoma in Patients with Hepatitis C Virus Genotype 1-Related Chronic Liver Disease. Oncology 2017;93:92–8. [DOI] [PubMed] [Google Scholar]
- 41.Pan WH, Yeh WT. How to define obesity? Evidence-based multiple action points for public awareness, screening, and treatment: an extension of Asian-Pacific recommendations. Asia Pac J Clin Nutr 2008;17:370–4. [PubMed] [Google Scholar]
- 42.Osaki Y, Kinjo A, Higuchi S, et al. Prevalence and Trends in Alcohol Dependence and Alcohol Use Disorders in Japanese Adults; Results from Periodical Nationwide Surveys. Alcohol Alcohol 2016;51:465–73. [DOI] [PubMed] [Google Scholar]
- 43.Carrat F, Fontaine H, Dorival C, et al. Clinical outcomes in patients with chronic hepatitis C after direct-acting antiviral treatment: a prospective cohort study. Lancet 2019;393:1453–64. [DOI] [PubMed] [Google Scholar]
- 44.Nagata H, Nakagawa M, Asahina Y, et al. Effect of interferon-based and -free therapy on early occurrence and recurrence of hepatocellular carcinoma in chronic hepatitis C. J Hepatol 2017;67:933–9. [DOI] [PubMed] [Google Scholar]
- 45.Ikeda K, Kawamura Y, Kobayashi M, et al. Direct-Acting Antivirals Decreased Tumor Recurrence After Initial Treatment of Hepatitis C Virus-Related Hepatocellular Carcinoma. Dig Dis Sci 2017;62:2932–42. [DOI] [PubMed] [Google Scholar]
- 46.Vathipadiekal V, Wang V, Wei W, et al. Creation of a Human Secretome: A Novel Composite Library of Human Secreted Proteins: Validation Using Ovarian Cancer Gene Expression Data and a Virtual Secretome Array. Clin Cancer Res 2015;21:4960–9. [DOI] [PubMed] [Google Scholar]
- 47.UniProt C. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 2019;47:D506–D15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Almagro Armenteros JJ, Tsirigos KD, Sonderby CK, et al. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol 2019;37:420–3. [DOI] [PubMed] [Google Scholar]
- 49.Savojardo C, Martelli PL, Fariselli P, Casadio R. DeepSig: deep learning improves signal peptide detection in proteins. Bioinformatics 2018;34:1690–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsirigos KD, Peters C, Shu N, Kall L, Elofsson A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res 2015;43:W401–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schwenk JM, Omenn GS, Sun Z, et al. The Human Plasma Proteome Draft of 2017: Building on the Human Plasma PeptideAtlas from Mass Spectrometry and Complementary Assays. J Proteome Res 2017;16:4299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nanjappa V, Thomas JK, Marimuthu A, et al. Plasma Proteome Database as a resource for proteomics research: 2014 update. Nucleic Acids Res 2014;42:D959–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang M, Herrmann CJ, Simonovic M, Szklarczyk D, von Mering C. Version 4.0 of PaxDb: Protein abundance data, integrated across model organisms, tissues, and cell-lines. Proteomics 2015;15:3163–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Diamandis EP. Cancer biomarkers: can we turn recent failures into success? J Natl Cancer Inst 2010;102:1462–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Uhlen M, Fagerberg L, Hallstrom BM, et al. Proteomics. Tissue-based map of the human proteome. Science 2015;347:1260419. [DOI] [PubMed] [Google Scholar]
- 56.Liu X, Chi X, Gong Q, et al. Association of serum level of growth differentiation factor 15 with liver cirrhosis and hepatocellular carcinoma. PLoS One 2015;10:e0127518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koo BK, Um SH, Seo DS, et al. Growth differentiation factor 15 predicts advanced fibrosis in biopsy-proven non-alcoholic fatty liver disease. Liver Int 2018;38:695–705. [DOI] [PubMed] [Google Scholar]
- 58.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fisher RA. Statistical Methods for Research Workers London: Oliver and Boyd; 1932. [Google Scholar]
- 61.Shi H, Liu S, Chen J, Li X, Ma Q, Yu B. Predicting drug-target interactions using Lasso with random forest based on evolutionary information and chemical structure. Genomics 2019;111:1839–52. [DOI] [PubMed] [Google Scholar]
- 62.Ramachandran P, Dobie R, Wilson-Kanamori JR, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019;575:512–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Galle PR, Foerster F, Kudo M, et al. Biology and significance of alpha-fetoprotein in hepatocellular carcinoma. Liver Int 2019;39:2214–29. [DOI] [PubMed] [Google Scholar]
- 64.Nathani P, Gopal P, Rich N, et al. Hepatocellular carcinoma tumour volume doubling time: a systematic review and meta-analysis. Gut 2021;70:401–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Graf E, Schmoor C, Sauerbrei W, Schumacher M. Assessment and comparison of prognostic classification schemes for survival data. Stat Med 1999;18:2529–45. [DOI] [PubMed] [Google Scholar]
- 66.Harrell FE Jr., Lee KL, Mark DB. Multivariable prognostic models: issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Stat Med 1996;15:361–87. [DOI] [PubMed] [Google Scholar]
- 67.Mogensen UB, Ishwaran H, Gerds TA. Evaluating Random Forests for Survival Analysis using Prediction Error Curves. J Stat Softw 2012;50:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pepe MS, Fan J, Seymour CW. Estimating the receiver operating characteristic curve in studies that match controls to cases on covariates. Acad Radiol 2013;20:863–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lausen B, Schumacher M. Maximally Selected Rank Statistics. Biometrics 1992;48:73–85. [Google Scholar]
- 70.Doove L, van Buurenca S, Dusseldorp E. Recursive partitioning for missing data imputation in the presence of interaction effects. Computational Statistics and Data Analysis 2014;72:92–104. [Google Scholar]
- 71.van Buurenca S, Groothuis-Oudshoorn K. Mice: multivariate imputation by chained equations in R. Journal of Statistical Software 2011;45:1–67. [Google Scholar]
- 72.Dong Y, Peng CY. Principled missing data methods for researchers. Springerplus 2013;2:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Serum protein abundance data are available from Mendeley Data at http://dx.doi.org/10.17632/5r7c48xkbw.1. The R codes for Gene Set Enrichment Index based on single-sample-based signature enrichment analysis (eseach algorithm)10 and TexSEC are available from the corresponding author upon reasonable request. The research team will provide an email address for communication once the information sharing is approved. The proposal should include detailed aims, statistical plan, and other information/materials to guarantee the rationality of requirement and the security of the data. The related patient data will be shared after review and approval of the submitted proposal and any related requested materials. Of note, data with patient names and other identifiers cannot be shared.