

Abstract
PREMISE
Hybrids contain divergent alleles that can confound phylogenetic analyses but can provide insights into reticulated evolution when identified and phased. We developed a workflow to detect hybrids in target capture data sets and phase reads into parental lineages using a similarity and phylogenetic framework.
METHODS
We used Angiosperms353 target capture data for Nepenthes, including known hybrids to test the novel workflow. Reference mapping was used to assess heterozygous sites across the data set and to detect hybrid accessions and paralogous genes. Hybrid samples were phased by mapping reads to multiple references and sorting reads according to similarity. Phased accessions were included in the phylogenetic framework.
RESULTS
All known Nepenthes hybrids and nine additional samples had high levels of heterozygous sites, had reads associated with multiple divergent clades, and were phased into accessions resembling divergent haplotypes. Phylogenetic analysis including phased accessions increased clade support and confirmed parental lineages of hybrids.
DISCUSSION
HybPhaser provides a novel approach to detect and phase hybrids in target capture data sets, which can provide insights into reticulations by revealing origins of hybrids and reduce conflicting signal, leading to more robust phylogenetic analyses.
Keywords: alleles, Angiosperm353, HybPiper, introgression, Nepenthes, paralogs, polyploidy, reticulation
Reticulation events caused by hybridization are common and important sources of novelty in angiosperm evolution (Wood et al., 2009; Palfalvi et al., 2020). The detection, investigation, and representation of hybridization remains a challenge in phylogenomics (Kellogg, 2016; Mallet et al., 2016; Spooner et al., 2020). The combination of divergent genomes in hybrids (herein used for any organism that contains divergent genomes due to a hybridization event, e.g., many polyploids) introduces conflicting phylogenetic signal and can lead to topologically incorrect or poorly resolved phylogenetic trees (McDade, 1992; Soltis et al., 2008). However, the advancement of target capture data and universal probe kits such as Angiosperms353 (Johnson et al., 2018) provides an opportunity to gain insight into historical reticulations in angiosperm evolution and reduce phylogenetic conflict, if ortholog (or homeolog in polyploids) gene variants can be identified and separated (phased).
Previously, inclusion of phased gene variants in phylogenetic studies has been used to confirm hybrid status of organisms (Sang and Zhang, 1999), determine the origin of polyploids (Popp and Oxelman, 2001), reveal parental lineages (Triplett et al., 2012; Estep et al., 2014), enable reconstruction of past reticulations (Estep et al., 2014; Brassac and Blattner, 2015), and date ancient hybridization events (Marcussen et al., 2015). Using Sanger sequencing, single‐gene studies generated sequences for each variant separately using cloning (Sang and Zhang, 1999; Popp and Oxelman, 2001), and multi‐gene studies linked these gene variants using their phylogenetic association in single‐gene phylogenies (Triplett et al., 2012; Estep et al., 2014; Marcussen et al., 2015). However, linking phased gene variants was limited by the resolution of single‐locus phylogenies to detect parental clades and the low numbers of nuclear genes available using Sanger sequencing to generate robust data sets. Universal target capture sequencing bait kits such as Angiosperms353 (Johnson et al., 2018) have revolutionized the study of angiosperm evolution by enabling the recovery of sequence reads from all variants of hundreds of nuclear genes across broad phylogenetic groups. Two challenges remain: assembling reads into phased sequences from each gene variant and linking genes according to their origin to investigate phased accessions.
De novo assembly, as implemented in common pipelines such as PHYLUCE (Faircloth, 2016), HybPiper (Johnson et al., 2016), and SECAPR (Andermann et al., 2018), can only recover complete phased gene variants when all polymorphic sites are frequent enough to connect all sequence reads across the whole gene locus (Fig. 1A). If this is not the case, sequences from different gene variants (including paralogs) can be unwittingly combined to generate chimeric contigs (Kates et al., 2018). Some approaches utilize paired‐end reads to increase the connectivity of polymorphisms across reads (Andermann et al., 2019); however, phased blocks do not always span across all gene loci (Kates et al., 2018). Even if all gene variants are perfectly phased, the problem of linking phased sequences across gene loci and unconnected exons to generate phased haplotypes persists.
FIGURE 1.
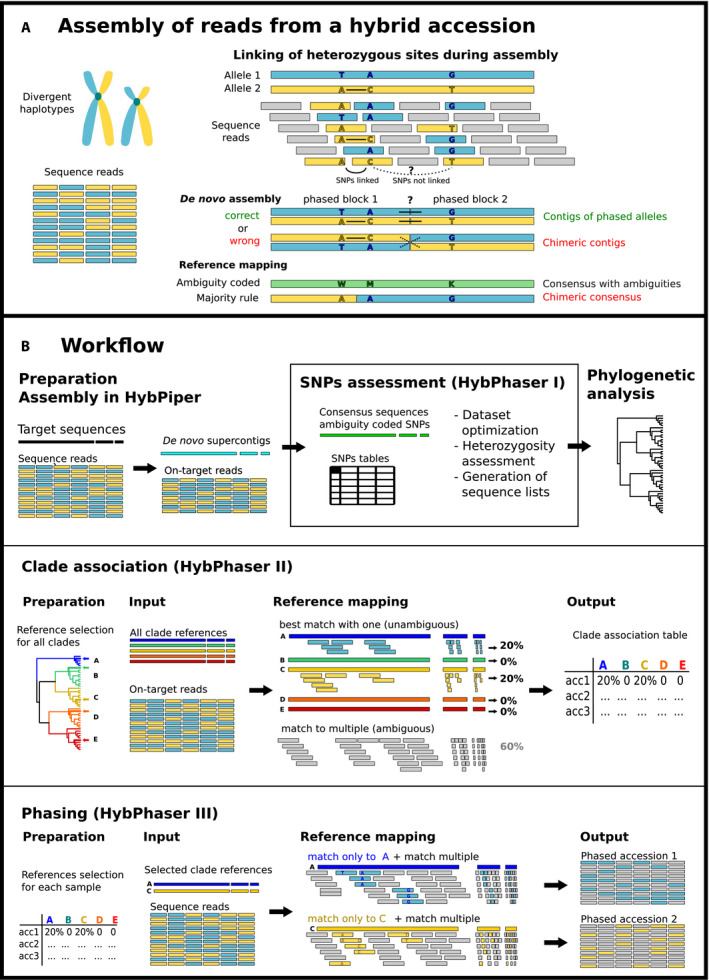
(A) Illustration of the linkage of heterozygous sites in the assembly of hybrid accessions that contain reads from two divergent haplotypes (blue = TAG, yellow = ACT). The two SNPs on the left can be linked (continuous line), but they cannot be linked to the third SNP on the right (dotted line). Reference mapping can result in consensus sequence coding SNPs as ambiguities (WMK) or represent the most common nucleotide (AAG) generating a chimeric sequence. De novo assembly can either connect phased blocks correctly into phased alleles (TAG/ATC) or generate chimeric sequences (ACG/TAT). (B) Illustration of major parts and concepts of the workflow: (1) assembly in HybPiper; (2) SNPs assessment in HybPhaser; (3) phylogenetic analysis; (4) clade association; and (5) phasing.
An alternative method for sequence assembly is reference mapping, where reads are mapped to a single reference (as implemented in pipelines such as HybPhyloMaker [Fér and Schmickl, 2018]). Reads from all gene variants are mapped, and the presence of heterozygous sites generated by multiple haplotypes can be handled in two ways (Fig. 1A): (1) the nucleotide is called using a majority rule (as in HybPhyloMaker) or (2) an International Union of Pure and Applied Chemistry (IUPAC) ambiguity code is used to accommodate divergent nucleotides and prevent the creation of chimeric sequences (Uribe‐Convers et al., 2016; Kates et al., 2018). However, phasing of hybrid accessions is not possible.
Here we present HybPhaser, a bioinformatic workflow that can phase hybrid accessions by simultaneously mapping sequence reads to references from the parental clades and sorting reads accordingly to generate accessions that approximate phased haplotypes. This approach performs the phasing before the assembly and thus avoids difficulties with linking of heterozygous sites and gene loci but requires a phylogenetic framework and method to select suitable references.
HybPhaser is built as an extension to the de novo assembly of HybPiper and consists of three parts (Fig. 1B, Table 1): (1) assessment of heterozygous sites in assembled sequences to detect putative hybrid accessions; (2) creation of a read‐to‐clade association framework; and (3) the phasing of read files based on the clade association framework. It extends the HybPiper pipeline further by enabling the modulation of paralog detection based on the heterozygosity of the data set, facilitating the optimization of the data set by cleaning samples and loci with poor recovery, and enabling the collation of consensus sequences (with ambiguity codes) as well as de novo contigs into sequence lists for subsequent analyses.
TABLE 1.
HybPhaser workflow overview containing all steps with a short description, the script/software used, required input, and generated output for each step.
| Part/Step | Description | Script/software | Input | Output |
|---|---|---|---|---|
| 1. Assessment of SNPs | ||||
| Consensus sequence generation | Reads are mapped to the de novo–assembled contigs to generate consensus sequences with SNPs where reads differ. | Bash 1 (BWA, bcftools) | de novo contigs (HybPiper), reads mapped to each locus (HybPiper) | Consensus sequences |
| Consensus sequence assessment | Proportion of SNPs and length of consensus sequences for each locus are collected. | R1a | Consensus sequences | Tables with SNPs/locus and sequence length |
| Data set optimization | Missing data can be reduced and putative paralogs removed. | R1b | Tables with SNPs/locus and sequence length | Lists with samples and loci to be removed |
| Assessment of heterozygosity and allele divergence | Summary tables are generated. | R1c | Tables with SNPs/locus and sequence length (cleaned) | Summary table and graphs for the assessment of heterozygosity and the detection of hybrids |
| Sequence lists generation | Sequences from HybPiper and HybPhaser folders are collated into sequence lists. | R1d | Contigs and consensus sequences and list with samples/loci to be removed | Sequence lists for loci or samples with contigs or consensus sequences, raw or cleaned (optimized) |
| 2. Clade association | ||||
| Phylogenetic analysis | Alignments and phylogenetic analysis | e.g., MAFFT*, IQ‐TREE* | Sequence lists | Phylogenetic tree |
| Selection of clade references | Taxa that represent major clades are selected by the user. | Information from phylogeny and summary table | Table (csv) with names of clade references | |
| Extraction of mapped reads | Generation of read files that contain only reads that mapped on the target sequences | Bash 2 (samtools) | Bam file from HybPiper | Read files (mapped only) |
| BBSplit script preparation/execution | Generate and run BBSplit script to match reads (mapped only) to clade references | R2a | Table (csv) with names of clade references | BBSplit stats files with proportions of reads mapped to each reference |
| Collation of BBSplit results | Generation of summary table for clade association | R2b | BBSplit stats files, summary table | Clade association summary table |
| 3. Phasing | ||||
| Selection of accessions for phasing | Clade association summary table | Table (csv) with names of accessions to phase with respective references | ||
| BBSplit phasing script preparation and execution | Generate and run BBSplit script to map and phase read files | R3a | Table (csv) with names of accessions to phase with respective references, sequence read files | Read files of phased accessions, BBSplit stats files |
| Collation of BBSplit phasing results | Generation of summary table for phasing | R3b | BBSplit stats files | Summary table for phasing stats |
| 4. Data set merging | ||||
| Assembly of phased accessions | Phased accessions are assembled using HybPiper and HybPhaser (part 1) | HybPiper, HybPhaser | Read files of phased accessions, target sequence list | Sequence lists of phased accessions |
| Merging of data sets | Sequences of phased accessions are merged with sequences of non‐phased accessions | R4 | Sequence lists of phased and non‐phased accessions | Merged sequence lists of phased and non‐phased accessions |
| Phylogenetic analysis | Alignments and phylogenetic analysis | e.g., MAFFT*, IQ‐TREE* | Merged sequence lists | Phylogenetic tree including phased and non‐phased accessions |
Software marked with an asterisk are not part of the workflow.
Herein we detail the HybPhaser (version 1.0) workflow and demonstrate its utility for detecting hybrids and identifying parental lineages using Angiosperms353 data from the carnivorous plant genus Nepenthes L. (Nepenthaceae).
METHODS
Preparation of the data set and assembly
Study group
Nepenthes is a paleotropical genus of ca. 160 species known to hybridize readily in horticulture and nature (Clarke et al., 2018). Molecular phylogenetic studies of the genus based on Sanger‐sequenced loci have resulted in poorly resolved and supported trees due to limited resolution of the markers, incongruence between plastid and nuclear phylogenies, and the possible inclusion of ITS paralogs (Meimberg and Heubl, 2006; Alamsyah and Ito, 2013). Recently, phylogenomic approaches including genome skimming (Nauheimer et al., 2019) and Angiosperms353 target capture (Murphy et al., 2020) have provided great advances in resolving the Nepenthes phylogeny but were unable to clarify past reticulations evident in the data sets. In a study that sampled almost all recognized species in the genus, Murphy et al. (2020) included two known hybrids and inferred one putative hybrid accession from conflict between supermatrix and gene tree analyses. Given the availability of suitable data with good species coverage and presence of known natural and cultivated hybrids, Nepenthes is a suitable model group for demonstrating hybrid detection and phasing using HybPhaser (version 1.0) and the Angiosperms353 probe set.
Input data
We generated Angiosperms353 target capture data for 125 samples of 68 Nepenthes species, including 15 samples of 12 different horticultural hybrids. In addition, we downloaded the Murphy et al. (2020) Nepenthes raw read files from the National Center for Biotechnology Information (NCBI) Sequence Read Archive (BioProject PRJEB35235, 185 accessions including one known and one suspected hybrid) and added them to the data set to increase species coverage across the genus. In total, 310 accessions representing 157 Nepenthes taxa including 17 accessions of 14 known hybrid taxa were analyzed (Appendix S1).
DNA extraction, library enrichment, and sequencing
Total genomic DNA was isolated from leaf material stored in silica gel for 125 Nepenthes samples. DNA was extracted using the cetyltrimethylammonium bromide (CTAB) method (Doyle and Doyle, 1987), and DNA extracts were further processed at the Australian Genomic Research Facility (AGRF; Melbourne, Australia) for library preparation (using the NEBNext Ultra II DNA Kit protocol [New England BioLabs, Ipswich, Massachusetts, USA] per the manufacturer’s instructions) and sequencing. Target sequence capture was carried out using the ‘Angiosperms353 v1’ universal probe set (catalog no. 308196; Arbor Biosciences, Ann Arbor, Michigan, USA) following Johnson et al. (2018). Sequencing was performed on an Illumina HiSeq 2500 sequencer (Illumina, San Diego, California, USA) producing 150‐bp paired‐end reads.
Read trimming
The sequence reads were trimmed using Trimmomatic (v.0.39; Bolger et al., 2014) to remove sequencing adapters and poor‐quality bases (illuminaclip 2:30:10, leading 20, trailing 20, sliding window 4:20). Final reads of less than 30 nucleotides were excluded.
HybPiper sequence assembly
De novo sequence assembly was performed with HybPiper (version 1.3.1) using the Angiosperms353 target file (https://github.com/mossmatters/Angiosperms353) in order to efficiently generate gene sequences consisting of concatenated exons and extract reads matching each gene. HybPiper performs reference mapping to pre‐select reads matching each gene for an efficient de novo assembly of relevant reads only. If genes consist of multiple exons, assembled contigs are mapped onto the target gene and concatenated to generate a complete gene sequence. We used the nucleotide format and BWA (Li and Durbin, 2009) for mapping, but the workflow can also handle amino acid sequences and BLASTX mapping. The script ‘intronerate.py’ was run to retrieve gene sequences that included intron regions. Summary statistics were generated using the script ‘hybpiper_stats.py’ (Appendix S2).
HybPhaser part I: SNPs assessment
Reference mapping of reads from divergent gene variants leads to heterozygous sites or single‐nucleotide polymorphisms (SNPs) where gene variants diverge (Fig. 1A). Assessing the distribution of SNPs across samples and loci can provide insights into the occurrence of hybrid accessions and paralogous genes, both of which are expected to have considerable divergence between gene versions. HybPhaser generates consensus sequences through reference mapping and codes SNPs as IUPAC ambiguity characters, which can be recorded and quantified using custom scripts in R (R Core Team, 2014) and assessed in generated graphs and tables.
Generation of consensus sequences
HybPhaser relies on the output of HybPiper assembly for remapping and consensus sequence generation using the Bash script ‘Generate_consensus_sequences.sh’. For each sample and gene, the HybPiper de novo contig was used as reference to which the pre‐selected on‐target reads were mapped using BWA, generating a BAM file. Consensus sequence files containing ambiguity codes at SNP sites were generated using bcftools (v1.9, http://samtools.github.io/bcftools/bcftools.html). To prevent sequencing artifacts being coded as SNPs, variants are only called as SNPs if the read depth is ≥10, the alternative nucleotide count is ≥4, and the proportion of alternative nucleotides is ≥0.15. The script ‘Rscript_1a_count_snps_in_consensus_seqs.R’ was then used to collate information on the proportions of SNPs and length of sequences from all samples and loci (Appendix S3).
Reducing proportions of missing data
Missing data can have negative consequences for downstream analyses (e.g., for gene tree summary analyses; Mirarab et al., 2014), and low sequence recovery can indicate poor assembly quality. The HybPhaser script ‘R1b_optimize_dataset.R’ can be used to explore loci and sample quality, and to exclude loci with poor sequence recovery. Thresholds for minimum proportions of loci recovered per sample and samples recovered per locus, as well as minimum proportions of target sequence length, can be adjusted in the configuration script ‘Configure_1_SNPs_assessment.R’. An initial run without data set optimization resulted in phylogenies with poorly placed low‐quality samples. To improve the quality of the data set while keeping important samples, we set thresholds to remove all loci that had sequences recovered for less than 20% of samples or less than 25% of the target sequence length recovered and all samples that had less than 20% of loci or less than 45% of the target sequence length recovered.
Removal of putative paralogous genes
Genes that had been subject to gene or genome duplications can be retained as paralogs, leading to the sequencing of multiple gene variants that are potentially assembled into chimeric gene sequences (Morales‐Briones et al., 2018) or to high proportions of SNPs in reference mapping (Andermann et al., 2020).
The assessment of heterozygosity in HybPhaser provides a method to detect putative paralogous genes because they are expected to have higher values compared to non‐paralogous genes. HybPhaser flags genes as putative paralogs that have an unusually high proportion of SNPs compared to other genes, first as averaged across all samples to detect paralogs shared across many samples, and second in each sample individually to detect paralogs that might not be shared with other samples. HybPhaser generates a graph and boxplot to visualize SNP distribution, and manually sets a threshold for removing these putative paralogs for all samples. Alternatively, one can choose to automatically remove samples and loci with SNP frequencies that are statistical outliers (here defined as more than 1.5× the interquartile range above the third quartile). Here we used the statistical outlier method to flag loci with a frequency of SNPs greater than 1.5× the interquartile range as paralogs (Appendix S4). This was done across the whole Nepenthes data set to identify and flag loci that are paralogous in all Nepenthes samples, as well as within each sample, so that paralogous loci unique to each sample could also be identified.
Assessment of locus heterozygosity and allele divergence
Hybrids inherit divergent alleles from their parental species; therefore, hybrid samples are expected to have a high proportion of loci that contain SNPs (here called locus heterozygosity or LH) and a high proportion of SNPs across all loci (here called allele divergence or AD), which should correlate to the divergence of the parental lineages. Using the script ‘Rscript_1c_summary_table.R’, we generated tables and figures that summarized the LH and AD of each sample to identify putative hybrid Nepenthes samples (Fig. 2A, Appendix S5). Finally, the script ‘Rscript_1d_generate_sequence_lists.R’ was used to collate sequence lists for downstream analyses. The script collates all loci for each sample or all samples for each loci, either HybPiper de novo contigs or HybPhaser consensus sequences, and either with data set optimization (paralogs and poor‐quality data removed) or without.
FIGURE 2.
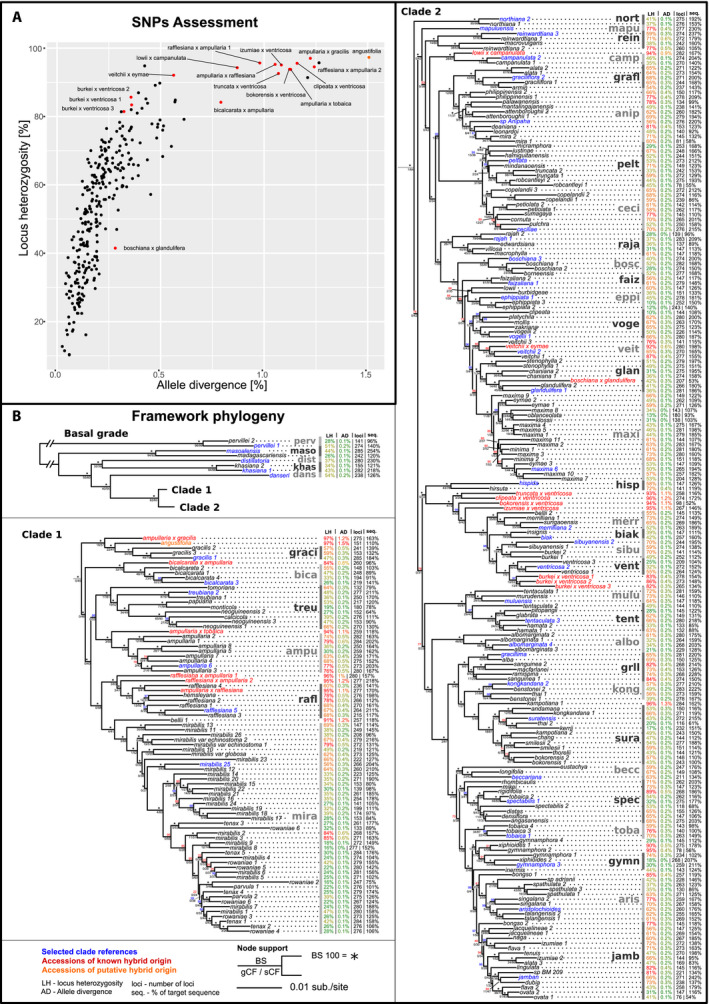
(A) Scatterplot displaying the locus heterozygosity and allele divergence of samples. Known hybrids (red dots) and putative hybrid (orange dot) are labeled. (B) Phylogenetic tree of the consensus supermatrix displayed in three parts: the basal grade with two diverging clades, clade 1 below, and clade 2 on the right. Summary statistics for each accession are given: locus heterozygosity (LH), allele divergence (AD), number of loci (loci), and proportion of target sequence recovered (seq.). Clades selected for clade association are shown in gray with bars. Clade references are displayed in blue, known hybrids in red, and the putative hybrid in orange. Node support is shown above the node in bootstrap (BS) (*=BS100), and below the node in gene and site concordance factors (gCF/sCF).
Framework phylogeny
The sequence lists produced by HybPhaser can be used for alignment and further phylogenetic analyses. Here, we first inferred phylogenetic relationships in Nepenthes based on unphased accessions to act as a framework for phasing later in the HybPhaser workflow. To do this, we used the consensus sequences for each locus after reducing missing data and removing putative paralogs. We preferred to use consensus sequences over contigs, as they mask heterozygous sites with ambiguity codes and therefore reduce conflicting phylogenetic signal from any hybrids. We aligned the consensus sequences using MAFFT (version 7.467; Katoh and Standley, 2013) and removed columns with more than 50% gaps using TrimAl (version 1.4.rev22; Capella‐Gutierrez et al., 2009). Phylogenetic analyses were performed using maximum likelihood in the IQ‐TREE2 package (version 2.0.5; Minh et al., 2020b). A supermatrix phylogeny was created by concatenating all locus alignments and using the ‘edge proportional’ partition model with single loci as partitions. Phylogenetic clade support was estimated using ultra‐fast bootstrap with 1000 replicates (Hoang et al., 2018) as well as gene and site concordance factors (gCF, sCF; Minh et al., 2020a), which indicate the concordance of single gene trees and of 1000 randomly chosen parsimony informative sites with the phylogeny of the concatenated data set. To determine gCF, single‐gene phylogenies were generated using IQ‐TREE2 applying the best‐fit model for each locus selected by ModelFinder (Kalyaanamoorthy et al., 2017). In addition to the supermatrix phylogeny, we summarized the gene trees using ASTRAL‐III (version 5.6.3; Zhang et al., 2018) to generate a phylogeny that is consistent under the multi‐species coalescence model (Degnan and Rosenberg, 2009). While this model is only consistent when incomplete lineage sorting is the only source for gene tree discordance, the comparison with the supermatrix analysis can provide valuable insights and corroborate supported clades. The resulting phylogenies were rooted using Nepenthes pervillei Blume, the sister species to all other Nepenthes species (Murphy et al., 2020).
HybPhaser part II: Clade association
To select suitable references for phasing, the association of sequence reads with divergent clades must be established. Hybrid accessions contain reads from divergent parental clades, which can be detected by mapping sequence reads to multiple references simultaneously. If the references are chosen carefully, reads from hybrid accessions will associate with parental clades. In HybPhaser part II, the software BBSplit (BBMap v38.47; B. Bushnell, https://sourceforge.net/projects/bbmap/) is used to map reads simultaneously to multiple references and record the proportion of reads matching to references from clades across the phylogeny (Fig. 1B). The reads of samples with non‐hybrid origin are expected to only map to one clade reference, whereas the reads of hybrid samples are expected to map to two clades, reflecting the divergent origins of their alleles. Reads of higher‐level polyploids may map to more than two references. This establishes the read‐to‐clade association required for the phasing in part III.
Reference selection
The selection of clade references from the framework phylogeny is critical to the efficacy of the clade association step and should be carefully considered. Clade references should be evenly distributed across the phylogeny and have low LH as well as low AD but high coverage of target sequences. References that are too closely related will result in many ambiguous matches, while references that are too distantly related can lead to reads not matching either reference. The optimal distance between clade references depends on the divergence between samples and the AD of putative hybrids, as well as factors like the allele diversity and the extent of incomplete lineage sorting or introgression in the study group. Determining a suitable number of clade references might require multiple iterations of clade association. Here, we selected a relatively high number of clade references (44) from the framework phylogeny (Fig. 2A). This enables the capture of putative hybrids with low AD (e.g., >0.4 in N. burkei × ventricosa) but also increases the chance for multiple clade associations of non‐hybrids.
Clade association
Sequence read files varied in the proportion of reads that matched to the target sequences. To only take into account on‐target reads, we extracted those reads from the BAM file generated by HybPiper using the Bash script ‘extract_mapped_reads.sh’. The software BBSplit (BBMap v38.47) was then used to map each on‐target read file simultaneously to all 44 selected clade references, recording the proportions of reads that mapped to either reference unambiguously. The script ‘Rscript_2a_prepare_BBSplit_script.R’ was used to generate a BBSplit commands file with the selected clade reference names and abbreviations. The mapping results were summarized using the script ‘Rscript_2b_collate_BBSplit_results.R’ (Appendix S6).
HybPhaser part III: Phasing
Phasing of sequence reads
Once hybrid samples have been detected and their parental clades identified, the putative hybrid accessions can be phased by simultaneously mapping the sequence reads to the relevant references and separating the reads accordingly into new read files that represent phased haplotype accessions. In this step, BBSplit is used to distribute reads that match unambiguously to one clade reference into the respective read file and then write read files that match similarly well onto multiple references into all read files for that sample (Fig. 1B). Therefore, the resulting phased accessions differ only in the reads that contain sites in which the references diverge. Ideally, hybrids containing one haplotype from each parent will be phased into two accessions representing both haplotypes. Similarly, polyploids containing multiple haplotypes can be phased into multiple accessions, in which case the proportions of reads can indicate the proportions of haplotypes.
For phasing, we considered samples with multiple clade associations as well as a high LH (>80%). Although there are samples with multiple clade associations that have low LH, they are unlikely to be hybrids that can be unambiguously phased. However, we make one exception, the known hybrid Nepenthes boschiana Korth. × glandulifera Chi. C. Lee, which had low LH (41.6%) likely due to low sequence recovery impacting variant calling. In total, we selected 26 samples for phasing, which included all 17 accessions of known hybrids and nine additional samples that had not previously been identified as hybrids.
The R script ‘Rscript_3a_prepare_phasing_script.R’ was used to generate a Bash script to run all 26 BBSplit commands that mapped the original sequence read files to the selected references for each chosen sample. Finally, the script ‘Rscript_3b_collate_phasing_stats.R’ was used to collate phasing statistics and generate a table with the proportions of reads mapped to either reference, which can give insights into the proportions of haplotypes and ploidy levels (Appendix S7).
Processing of phased accessions in HybPiper and HybPhaser part I
The newly generated read files of the phased accessions were passed through the HybPiper and HybPhaser part I pipelines using the R scripts ‘Configure_1_SNPs_assessment.R’ and ‘Rscript_1a_count_snps_in_consensus_sequences.R’ to ‘Rscript_1d_generate_sequence_lists.R’ with the same settings as for the non‐phased samples. For the data set optimization step, we removed the same 24 loci flagged as putative paralogs for the non‐phased accessions and performed the paralog removal step for each sample, individually flagging and removing statistical outliers using the scripts (Appendix S8).
Combined data set of phased and non‐phased accessions
In order to analyze the phased accessions in the context of the whole genus, we generated a data set that combined the phased accessions with all non‐phased accessions using the R scripts ‘Configure_4_Combining_sequence_lists.R’ and ‘Rscript_4a_combine_phased_with_normal_sequence_lists.R’. Alignments and phylogenetic analyses were then performed with the same methodology as was used for the framework phylogeny using the consensus sequences.
HybPhaser is compatible with Linux and consists of a collection of Bash and R scripts (Table 1) that are protected under the terms of a free GNU General Public License; the scripts, along with the manual, are available online on GitHub (https://github.com/LarsNauheimer/HybPhaser). Sequence data generated for this study are available at the NCBI Sequence Read Archive (PRJNA706038).
RESULTS
HybPiper sequence assembly
The HybPiper assembly was performed successfully for 307 of the 310 samples (Appendix S2). Paralog warnings were issued for 38 samples and between one and five genes per sample (average 0.16 paralogous genes per sample).
Reducing proportions of missing data
A total of 20 samples and 41 loci were excluded from the data set (Appendix S4) due to poor sequence recovery. Ten of the excluded samples had less than 20% of loci recovered, and all 20 had a total sequence length of less than 45% of the target sequence length recovered. Thirty‐nine loci had sequences recovered for less than 20% of the samples, and five loci had on average across all samples less than 50% of the target sequence length of the gene recovered. The remaining data set comprised 290 samples and 312 loci.
Removal of paralogous genes
Twenty‐four loci were flagged as putative paralogous genes across all samples, with an average proportion of SNPs of 0.0062 or greater (Appendix S4). On average, 7.5 loci per sample were flagged as putative paralogs by comparing the SNPs between all genes in each sample individually.
Assessment of locus heterozygosity and allele divergence
The sampled known hybrids displayed considerably higher values of LH and AD compared to other samples, although the distinction between hybrids and non‐hybrids was not clear cut. Across all samples, LH varied gradually between 10.3% and 97.4% and AD varied between 0.03% and 1.51%. Known and suspected hybrids had an average of 89.4% LH and 0.86% AD, while all other samples had an average of 52.3% LH and 0.21% AD (Fig. 2A, Appendix S5). All included hybrids except one had an LH of more than 80%. The hybrid accession Nepenthes boschiana × glandulifera, which had poor sequence recovery (50.5% of target sequence length before optimization), had very low values of LH (41.6%) and AD (0.28%) compared to the other hybrids.
Framework phylogeny alignment generation
The supermatrix data set consisted of 285 genes and 290 taxa, with an aligned sequence length of 423,105 bp and 30.4% missing data. Overall, clades in the framework phylogeny were well supported, and largely corresponded to the clades retrieved by Murphy et al. (2020) (Fig. 2B). Many clades obtained maximum bootstrap support, although the gCF values were often low. Some of the backbone nodes received very low support. Most hybrid accessions fell within the clade containing one of the parental taxa where the hybrid was often found in a basally diverging position within the clade (e.g., N. ampullaria Jack × gracilis Korth.; Fig. 2B). The gene tree summary phylogeny (Appendix S9) is largely congruent with the supermatrix phylogeny, with the only exception being the position of the ‘albo’ clade, which groups with moderate support in a different clade in each phylogeny. Hybrid accessions group either basal in one of the parental clades or between parental clades.
Clade association
Interpretation of clade association must be done for each sample individually, taking into consideration the phylogenetic distance between that sample and the clade references, the phylogenetic distance between clade references, and the sequence length of the clade reference sequences. Most samples had a single clade association with a higher proportion of reads matching unambiguously to one clade reference compared to the others (Fig. 3, Appendix S6). In contrast, all known hybrids showed associations with multiple, often divergent clades. In most cases, the association with both clades is strong (e.g., N. ampullaria × gracilis), whereas in others the association with one clade is weak (e.g., N. truncata Macfarl. × ventricosa Blanco) or very weak (e.g., N. clipeata Danser × ventricosa). Even N. boschiana × glandulifera, which had low values of LH and AD, showed an association to both parental clades. Nine non‐hybrid accessions with high LH (>80%) showed associations with two clades (e.g., N. kampotiana‐1 Lecomte, N. bellii‐1 K. Kondo, N. xiphioides‐1 B. R. Salmon & Maulder). Samples with lower LH values generally showed either a single clade association or multiple associations of closely related references.
FIGURE 3.
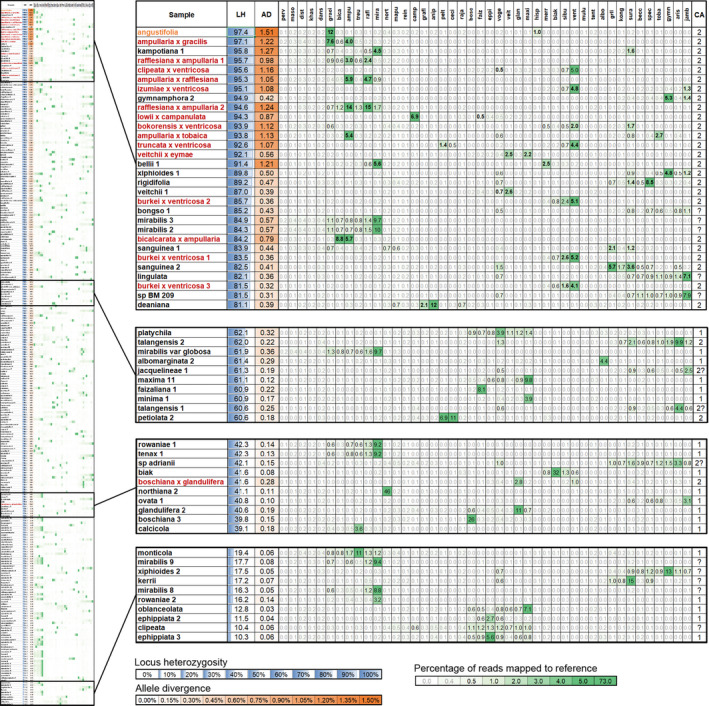
Clade association table and heatmap displaying the percentage of reads matching to each of the 44 clade references. The complete table is on the left, with extracts of example rows shown on the right. The table includes locus heterozygosity (LH) and allele divergence (AD), percentages of reads matching to each reference, and the number of clade associations (CA). Known hybrids are in red, and the putative hybrid is in orange.
The highest proportions of matching reads were found in phylogenetically distinct, basally diverging lineages (e.g., >60% in N. khasiana Hook. f.), while multiple clade associations were often found in taxa with low LH and AD when they were not closely related to one or were in between two clade references (e.g., N. petiolata Danser, N. clipeata). Few samples had reads associated with multiple divergent clades but had medium to low values for LH and AD (e.g., N. talangensis Nerz & Wistuba or N. sp ‘adrianii’).
Phasing of sequence reads
In mapping onto the selected references, an average of 4.9% (1.1–13.9%) of original sequence reads mapped unambiguously to a single reference. Thirteen phased accessions had approximately equal proportions of unambiguous mapped reads, six accessions had a proportion of approximately 3 : 2, nine accessions had a proportion of approximately 2 : 1, and four accessions had a proportion of roughly 1 : 3 (Appendix S7).
Processing of phased accessions
Most phased accessions had considerably lower LH and AD compared to the non‐phased accessions before phasing (Appendix S8). Both phased accessions of N. boschiana × glandulifera had very low sequence recovery (35.5% and 10.5%) and were removed from the data set. The paralog detection for each sample individually resulted in flagging of on average 6.8 genes per sample as putative paralogs that were removed from the data set.
Combined data set of phased and non‐phased accessions
The combined data set consisted of 285 genes and 315 taxa, with an aligned sequence length of 400,685 bp and 32.6% missing data. The topology of the supermatrix phylogeny based on the combined data set including phased accessions is congruent with the framework phylogeny in all nodes, but displays stronger support (Fig. 4). The support for the backbone nodes and for the clades that contained hybrids increased compared to the framework phylogeny with non‐phased accessions due to the reduction of conflicting phylogenetic signal. This was especially prominent regarding gene concordance factors showing that phasing hybrids reduced conflicting information and increased concordance in the data set.
FIGURE 4.
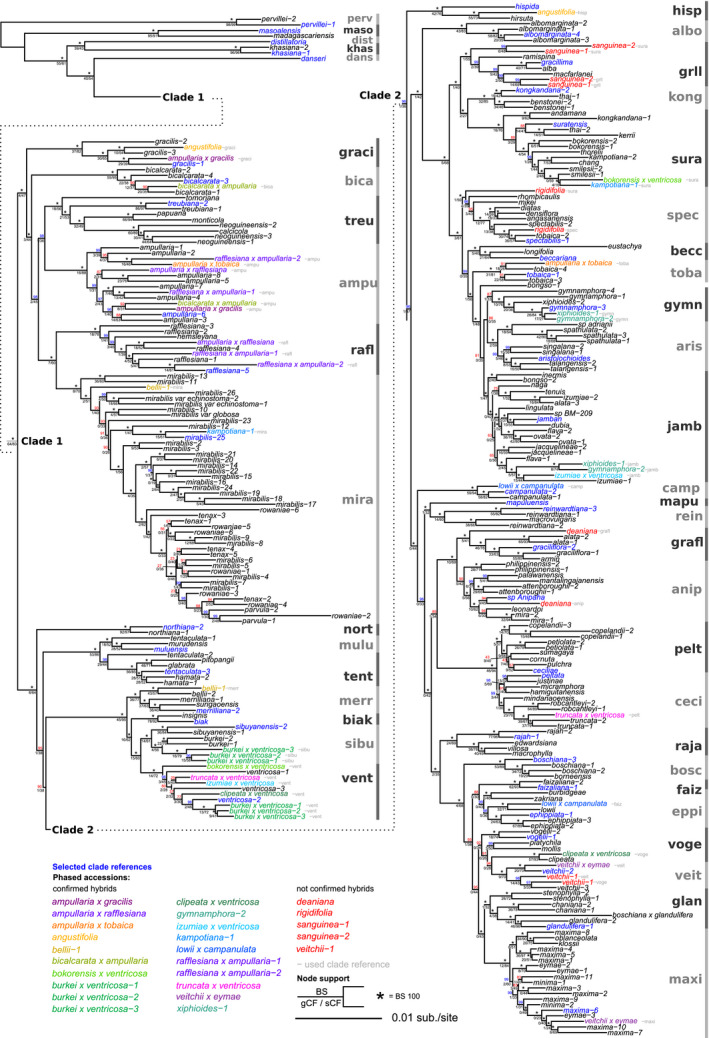
Phylogenetic tree of the consensus supermatrix including phased haplotype accessions. Phased accessions are displayed in different colors. Clade references are displayed in blue. Node support is shown above the node in bootstrap (BS) (*=BS100) and below the node in gene and site concordance factors (gCF/sCF).
The phased haplotype accessions from all 16 known hybrids grouped in the clade of their respective parental species, mostly as sister to the parental species, e.g., N. ampullaria × gracilis grouped with N. gracilis‐1 in the graci clade and N. ampullaria × gracilis grouped with N. ampullaria‐6 and N. ampullaria‐3 in the ampu clade. The phased accessions of the putative hybrid N. angustifolia grouped in divergent clades with N. gracilis‐3 and N. hirsuta Hook. f. Of the nine accessions previously considered to be non‐hybrids but with high LH and multiple clade associations, five had phased accessions grouping in divergent clades; three of these groupings were with putative parent taxa: N. kampotiana‐1 with N. mirabilis‐25 and N. smilesii‐1 Hemsl., N. bellii‐1 with N. mirabilis‐12 and N. bellii‐2, and both N. gymnamphora‐2 Reinw. ex Nees as well as N. xiphioides‐1 with N. gymnamphora‐3, and N. izumiae‐1. The phased accessions of N. deaniana Macfarl. grouped more ambiguously with one clade containing N. sp‐Anipaha and N. leonardoi and another clade of N. alata‐1,2 and N. graciflora‐1,2. The phased haplotypes of four more accessions, N. rigidifolia, N. sanguinea‐1,2, and N. veitchii‐1 did not separate into divergent clades. The summary gene tree phylogeny (Appendix S10) is largely concordant with the supermatrix tree (Fig. 4), but with considerably lower support for many nodes. Supported topological differences are found in the positions of the phased accessions within the graci clade, as well as N. boschiana × glandulifera, which had low sequence recovery, and the phased accessions of N. veitchii‐1, which had not been phased successfully.
DISCUSSION
Read phasing with HybPhaser
With the Nepenthes data set, we have shown that phasing reads prior to sequence assembly through HybPhaser is an effective way to detect and characterize haplotypes in hybrid accessions. Previous approaches perform phasing during or after the sequence assembly (Kates et al., 2018; Andermann et al., 2019); however, linking reads using de novo assembly requires frequent heterozygous sites to connect reads across the loci (Fig. 1A), which can be difficult to achieve even with read‐backed phasing methods using paired‐end reads assembly (Kates et al., 2018). Linking phased genes to generate a concatenated multi‐gene data set can be performed by assigning genes to clades based on single‐locus phylogenies, which requires sufficient phylogenetic signal in each locus for successful clade assignment (Triplett et al., 2012; Estep et al., 2014; Marcussen et al., 2015). In HybPhaser, sequence reads are phased by mapping to multiple reference sequences representing parental clades prior to their assembly, thereby avoiding these issues.
Although this pre‐assembly phasing approach of HybPhaser is advantageous, it must be emphasized that the phased haplotypes only represent an approximation of the parental haplotypes. BBSplit, as used in the HybPhaser workflow, assigns reads to a single phased accession that matches unambiguously to its reference, while all other reads are assigned to all phased accessions. Therefore, the differences between the references determine the phasing. Theoretically, haplotypes of hybrids will phase perfectly (100% unambiguous matches) if the references are the actual parents of the hybrid sample and no random mutations or cross‐over events occurred. However, if the selected references diverge from the actual parental lineages, are heterozygous, or differ in the sequence length or locus coverage, haplotypes will not be phased perfectly and will only approximate the parental haplotypes.
Furthermore, the efficacy of the HybPhaser phasing approach is sensitive to the selection of appropriate reference sequences. First, successful phasing will be dependent on the phylogenetic divergence between references; when references are closely related, more reads will be ambiguous and the number of successfully matched reads will be low. Second, the divergence between the clade reference and the parental lineage of the hybrid has an influence on clade association; with increasing divergence from the clade reference, fewer reads will match unambiguously. We therefore recommend that selected references represent samples that are evenly distributed across the phylogeny, have high loci coverage, and contain little allele divergence. In Nepenthes, we chose 44 clade references based on a phylogenetic framework and the assessment of heterozygosity across the data set. This included relatively closely related references in order to resolve known hybrids with closely related parents (N. burkei × ventricosa and N. veitchii × eymae), as well as putative hybrids with similarly high LH but low AD (e.g., N. sanguinea, N. gymnamphora‐2, or N. xiphioides‐1). Choosing suitable reference sequences was crucial for successfully phasing Nepenthes hybrids with parents of differing relatedness using HybPhaser.
Hybrid detection with HybPhaser
In this study, we have shown the utility of HybPhaser not only in characterizing the parentage of known hybrids, but also in detecting previously unknown, putative hybrids. Almost all known hybrid accessions of Nepenthes had high LH (>80%) and an AD that reflected the divergence of the parental lineages (most had >0.6%). This contrasts with most samples, which had LH between 20% and 70% and an AD of 0.1% and 0.3%, reflecting the occurrence of allelic variation in species. Several samples had an AD between 0.3% and 0.6% or low to medium LH and multiple clade associations; both of these might indicate the occurrence of introgression through backcrossing of hybrids into one parental population. Evolution is dynamic, and a variety of processes (e.g., introgression, polyploidization and diploidization, gene duplications and losses, as well as incomplete lineage sorting) or a combination of these can impact LH and AD values as well as the clade association analysis (Mallet et al., 2016; Soltis et al., 2016). In addition, low coverage of sequence reads can lead to low LH and AD values due to the method of variance calling. Here, the hybrid N. boschiana × glandulifera had by far the lowest LH (41.6%) and AD (0.42%) of all included hybrids. This might be an underestimate due to low sequence coverage, which is indicated by the low sequence recovery of the original accession (50.5% of the target sequence length) and the very low sequence recovery of the phased accessions (10.6% and 35.5%). Although N. bokorensis Mey × ventricosa and N. gymnamphora‐2 had similarly low sequence recovery in the original mapping, they had normal values of LH and AD and a sequence recovery of phased accessions similar to the normal accessions.
The assessment of heterozygous sites and thus the conflict between the gene variants or divergent haplotypes in the data set provides a simple way to gain insights into the reticulation of samples. Nepenthes is known for including a variety of horticultural and natural hybrids (Clarke et al., 2018). We found a gradient of LH and AD across the genus, with many samples having intermediate values between known hybrids and most other samples, which indicates introgression and the presence of unknown hybrid accessions. Fourteen samples had LH (>80%) and AD (>0.3%) similar to the closely related hybrids, of which nine had reads associated with multiple clades and were phased. After phasing and inclusion of phased accession into the phylogenetic analyses, four samples had phased accessions clearly grouping in divergent clades with specific taxa confirming their hybrid status and revealing parental lineages: N. bellii‐1 (bellii × mirabilis), N. kampotiana‐1 (smilesii × mirabilis), N. gymnamphora‐2 (gymnamphora × izumiae), and N. xiphioides‐1 (gymnamphora × izumiae). Furthermore, the AD of phased accessions combined from each of the samples was lower than the AD of the non‐phased accessions. Four samples were not confirmed as hybrids based on the phasing, because the phased accessions did not group in divergent clades (N. rigidifolia, N. sanguinea‐1,2, and N. veitchii‐1). One sample was more ambiguous; N. deaniana had phased accessions grouping in divergent clades but not clearly associated with other samples, thus reducing the phylogenetic support of the affected clades. Furthermore, the AD of the phased accessions combined (0.36% + 0.21%) was much higher than the AD of the non‐phased accessions (0.39%). Therefore, it is unlikely that N. deaniana is a hybrid between species of these clades.
Paralog detection with HybPhaser
Our results suggest that the paralog detection of HybPhaser greatly improves on existing methods. In approaches such as the paralog investigator of HybPiper, a paralog warning is only issued when multiple de novo contigs are recovered with comparable size, i.e., when multiple contigs have at least 75% of the whole target sequence length (Johnson et al., 2016). To successfully detect a paralogous gene, heterozygous sites must occur frequently enough to connect reads of all versions across most of the locus and, because the target length is used as a reference, the gene must not consist of multiple exons shorter than 75% of the gene. The paralog detection provided in HybPhaser is based on the idea that paralogous genes have higher rates of heterozygous sites than normal genes, and therefore can detect paralogs independent of exon size or distribution of heterozygous sites across the gene.
In Nepenthes, HybPiper issued warnings for only 0.16 of genes per sample, while the approach taken by HybPhaser flagged 31.5 genes per sample (ca. 10% of the genes). This large difference is due not only to the methodology, but also to the chosen conservative approach to remove more rather than fewer genes. First, flagging genes that have unusually high proportions of SNPs across all samples (here 24 genes) will lead to removing those genes for samples without multiple variants. This might be desired, however, as gene loss can lead to leaving only one version active, which is not guaranteed to be the ortholog. Second, this method flags genes that have unusually high rates of SNPs as a result of reasons other than paralog variants (e.g., sequencing or assembly artifacts, indels, or non‐paralogous but similar sequences from other genes). HybPhaser provides graphs and tables to assess the distribution of SNPs in each locus either averaged across all samples or for each sample individually. It further provides methods to remove putative paralogous loci from the data set. The assessment of individual BAM files can give more insights into the cause of the dubious loci.
Reconciling phylogenetic conflict with HybPhaser
In this study, we have demonstrated how HybPhaser can be used to handle phylogenetic conflict in target capture data sets and to more accurately estimate phylogenies using supermatrix and gene tree summary approaches. Hybrids introduce phylogenetic conflict in the data set, leading to poorly resolved clades or incorrect topology (McDade, 1992; Soltis et al., 2008). The effect will differ depending on the method of sequence generation and handling heterozygous sites. Hybrids might group together with one parent, basal to one parent, or in between multiple parental clades. Here, we used ambiguity codes in the non‐phased data set, and most hybrids were in a basal position in the clade of one parent with a shorter total branch length. Phasing of hybrid accessions reduced the conflicting signal and improved the clade support of affected clades, especially in gene and site concordance factors. To further decrease the conflicting signal in the data set, one might consider removing all samples with high LH or AD values. This can be especially useful as a means to increase clade support for the framework phylogeny for clade association.
Concluding remarks
HybPhaser provides a novel workflow to detect and phase hybrid accessions in target capture data sets. In this study, we have used HybPhaser to untangle the reticulate evolutionary history of Nepenthes and demonstrated its utility for phasing reads into parental haplotypes, revealing and characterizing hybrids, detecting putative paralogous genes, and resolving phylogenetic conflict.
AUTHOR CONTRIBUTIONS
L.N. developed the workflow, performed analyses, and wrote the manuscript. N.W. contributed to data curation and analysis. C.C. provided sample collections and plant identifications. E.J. tested the workflow and contributed to manuscript preparation. D.C. and K.N. contributed to project administration, supervision, and manuscript preparation. C.C., D.C., and K.N. acquired funding for the project. All authors read and approved the final manuscript.
Supporting information
APPENDIX S1. List of accessions included in this study with DNA or SRA number, short name used in this paper, voucher information, geographic origin, and whether the sample was used in Murphy et al. (2020) or was generated for this study.
APPENDIX S2. Statistics retrieved from HybPiper script ‘hybpiper_stats.py’ for the normal contigs (not those including intron regions).
APPENDIX S3. Tables with recovered sequence length and proportion of SNPs for each sample and locus, before and after data set optimization.
APPENDIX S4. Graphs and tables generated in the data set optimization step.
APPENDIX S5. Summary table from heterozygosity assessment.
APPENDIX S6. Table from clade association analysis.
APPENDIX S7. Input table and summary statistics from phasing analysis.
APPENDIX S8. Graphs and tables from data set optimization and heterozygosity assessment of phased accessions.
APPENDIX S9. Gene tree summary phylogeny from normal data set (framework phylogeny).
APPENDIX S10. Gene tree summary phylogeny from data set including phased accessions.
ACKNOWLEDGMENTS
This work was supported by the Australian Biological Resource Study (Department of the Environment, Australian Government; ABRS grant RFL214‐62) and the Australian and Pacific Science Foundation (APSF 16/4). We are thankful to Melissa Harrison (Australian Tropical Herbarium) for support in the laboratory and Todd McLay and Chris Jackson (Royal Botanic Gardens Victoria, Genomics for Australian Plants Consortium) for feedback on the HybPhaser workflow.
Nauheimer, L. , Weigner N., Joyce E., Crayn D., Clarke C., and Nargar K.. 2021. HybPhaser: A workflow for the detection and phasing of hybrids in target capture data sets. Applications in Plant Sciences 9(7): e11441.
DATA AVAILABILITY STATEMENT
All workflow scripts are available through the HybPhaser repository on GitHub (https://github.com/LarsNauheimer/HybPhaser; Nauheimer, 2021). Sequence data are available through the National Center for Biotechnology Information (NCBI) (BioProjects: P RJNA706038 [this study] and PRJEB35235 [Murphy et al., 2020]).
LITERATURE CITED
- Alamsyah, F. , and Ito M.. 2013. Phylogenetic analysis of Nepenthaceae, based on internal transcribed spacer nuclear ribosomal DNA sequences. Acta Phytotaxonomica et Geobotanica 64: 113–126. [Google Scholar]
- Andermann, T. , Cano Á., Zizka A., Bacon C., and Antonelli A.. 2018. SECAPR—A bioinformatics pipeline for the rapid and user‐friendly processing of targeted enriched Illumina sequences, from raw reads to alignments. PeerJ 6: e5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andermann, T. , Fernandes A. M., Olsson U., Töpel M., Pfeil B., Oxelman B., Aleixo A., et al. 2019. Allele phasing greatly improves the phylogenetic utility of ultraconserved elements. Systematic Biology 68: 32–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andermann, T. , Torres Jiménez M. F., Matos‐Maraví P., Batista R., Blanco‐Pastor J. L., Gustafsson A. L. S., Kistler L., et al. 2020. A guide to carrying out a phylogenomic target sequence capture project. Frontiers in Genetics 10: 10.3389/fgene.2019.01407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse M., and Usadel B.. 2014. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brassac, J. , and Blattner F. R.. 2015. Species‐level phylogeny and polyploid relationships in Hordeum (Poaceae) inferred by next‐generation sequencing and in silico cloning of multiple nuclear loci. Systematic Biology 64: 792–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella‐Gutierrez, S. , Silla‐Martinez J. M., and Gabaldon T.. 2009. TrimAl: A tool for automated alignment trimming in large‐scale phylogenetic analyses. Bioinformatics 25: 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, C. , Schlauer J., Moran J., and Robinson A.. 2018. Systematics and evolution of Nepenthes . In Ellison A. M. and Adamec L. [eds.], Carnivorous plants: Physiology, ecology, and evolution. Oxford University Press, Oxford, United Kingdom. [Google Scholar]
- Degnan, J. H. , and Rosenberg N. A.. 2009. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends in Ecology & Evolution 24: 332–340. [DOI] [PubMed] [Google Scholar]
- Doyle, J. J. , and Doyle J. L.. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin 19: 11–15. [Google Scholar]
- Estep, M. C. , McKain M. R., Diaz D. V., Zhong J., Hodge J. G., Hodkinson T. R., Layton D. J., et al. 2014. Allopolyploidy, diversification, and the Miocene grassland expansion. Proceedings of the National Academy of Sciences, USA 111: 15149–15154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faircloth, B. C. 2016. PHYLUCE is a software package for the analysis of conserved genomic loci. Bioinformatics 32: 786–788. [DOI] [PubMed] [Google Scholar]
- Fér, T. , and Schmickl R. E.. 2018. HybPhyloMaker: Target enrichment data analysis from raw reads to species trees. Evolutionary Bioinformatics 14: 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang, D. T. , Chernomor O., A. von Haeseler , Minh B. Q., and Vinh L. S.. 2018. UFBoot2: Improving the ultrafast bootstrap approximation. Molecular Biology and Evolution 35: 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, M. G. , Gardner E. M., Liu Y., Medina R., Goffinet B., Shaw A. J., Zerega N. J. C., and Wickett N. J.. 2016. HybPiper: Extracting coding sequence and introns for phylogenetics from high‐throughput sequencing reads using target enrichment. Applications in Plant Sciences 4: 1600016. 10.3732/apps.1600016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, M. G. , Pokorny L., Dodsworth S., Botigué L. R., Cowan R. S., Devault A., Eiserhardt W. L., et al. 2018. A universal probe set for targeted sequencing of 353 nuclear genes from any flowering plant designed using k‐medoids clustering. Systematic Biology 68: 594–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyaanamoorthy, S. , Minh B. Q., Wong T. K. F., A. von Haeseler , and Jermiin L. S.. 2017. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nature Methods 14: 587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kates, H. R. , Johnson M. G., Gardner E. M., Zerega N. J. C., and Wickett N. J.. 2018. Allele phasing has minimal impact on phylogenetic reconstruction from targeted nuclear gene sequences in a case study of Artocarpus . American Journal of Botany 105: 404–416. [DOI] [PubMed] [Google Scholar]
- Katoh, K. , and Standley D. M.. 2013. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molecular Biology and Evolution 30: 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg, E. A. 2016. Has the connection between polyploidy and diversification actually been tested? Current Opinion in Plant Biology 30: 25–32. [DOI] [PubMed] [Google Scholar]
- Li, H. , and Durbin R.. 2009. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet, J. , Besansky N., and Hahn M. W.. 2016. How reticulated are species? BioEssays 38: 140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcussen, T. , Heier L., Brysting A. K., Oxelman B., and Jakobsen K. S.. 2015. From gene trees to a dated allopolyploid network: Insights from the angiosperm genus Viola (Violaceae). Systematic Biology 64: 84–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDade, L. A. 1992. Hybrids and phylogenetic systematics II. The impact of hybrids on cladistic analysis. Evolution 46: 1329–1346. [DOI] [PubMed] [Google Scholar]
- Meimberg, H. , and Heubl G.. 2006. Introduction of a nuclear marker for phylogenetic analysis of Nepenthaceae. Plant Biology 8: 831–840. [DOI] [PubMed] [Google Scholar]
- Minh, B. Q. , Hahn M. W., and Lanfear R.. 2020a. New methods to calculate concordance factors for phylogenomic datasets. Molecular Biology and Evolution 37: 2727–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh, B. Q. , Schmidt H. A., Chernomor O., Schrempf D., Woodhams M. D., A. von Haeseler , and Lanfear R.. 2020b. IQ‐TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Molecular Biology and Evolution 37: 1530–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirarab, S. , Reaz R., Bayzid Md. S, Zimmermann T., Swenson M. S., and Warnow T.. 2014. ASTRAL: Genome‐scale coalescent‐based species tree estimation. Bioinformatics 30: i541–i548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales‐Briones, D. F. , Liston A., and Tank D. C.. 2018. Phylogenomic analyses reveal a deep history of hybridization and polyploidy in the Neotropical genus Lachemilla (Rosaceae). New Phytologist 218: 1668–1684. [DOI] [PubMed] [Google Scholar]
- Murphy, B. , Forest F., Barraclough T., Rosindell J., Bellot S., Cowan R., Golos M., et al. 2020. A phylogenomic analysis of Nepenthes (Nepenthaceae). Molecular Phylogenetics and Evolution 144: 106668. [DOI] [PubMed] [Google Scholar]
- Nauheimer, L. 2021. Data from: HybPhaser: A workflow for the detection and phasing of hybrids in target capture data sets [posted 4 May 2021]. Available at Zenodo repository: 10.5281/zenodo.4735820 [accessed 23 June 2021]. [DOI] [PMC free article] [PubMed]
- Nauheimer, L. , Cui L., Clarke C., Crayn D. M., Bourke G., and Nargar K.. 2019. Genome skimming provides well‐resolved plastid and nuclear phylogenies, showing patterns of deep reticulate evolution in the tropical carnivorous plant genus Nepenthes (Caryophyllales). Australian Systematic Botany 32: 243–254. [Google Scholar]
- Palfalvi, G. , Hackl T., Terhoeven N., Shibata T. F., Nishiyama T., Ankenbrand M., Becker D., et al. 2020. Genomes of the Venus flytrap and close relatives unveil the roots of plant carnivory. Current Biology 30: 2312–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp, M. , and Oxelman B.. 2001. Inferring the history of the polyploid Silene aegaea (Caryophyllaceae) using plastid and homoeologous nuclear DNA sequences. Molecular Phylogenetics and Evolution 20: 474–481. [DOI] [PubMed] [Google Scholar]
- R Core Team . 2014. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. Website http://www.R‐project.org/ [accessed 11 June 2021]. [Google Scholar]
- Sang, T. , and Zhang D.. 1999. Reconstructing hybrid speciation using sequences of low copy nuclear genes: Hybrid origins of five Paeonia species based on Adh gene phylogenies. Systematic Botany 24: 148–163. [Google Scholar]
- Soltis, D. E. , Mavrodiev E. V., Doyle J. J., Rauscher J., and Soltis P. S.. 2008. ITS and ETS sequence data and phylogeny reconstruction in allopolyploids and hybrids. Systematic Botany 33: 7–20. [Google Scholar]
- Soltis, D. E. , Visger C. J., Marchant D. B., and Soltis P. S.. 2016. Polyploidy: Pitfalls and paths to a paradigm. American Journal of Botany 103: 1146–1166. [DOI] [PubMed] [Google Scholar]
- Spooner, D. M. , Ruess H., Ellison S., Senalik D., and Simon P.. 2020. What is truth: Consensus and discordance in next‐generation phylogenetic analyses of Daucus . Journal of Systematics and Evolution 58(6): 1059–1070. [Google Scholar]
- Triplett, J. K. , Wang Y., Zhong J., and Kellogg E. A.. 2012. Five nuclear loci resolve the polyploid history of switchgrass (Panicum virgatum L.) and relatives. PLoS ONE 7: e38702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uribe‐Convers, S. , Settles M. L., and Tank D. C.. 2016. A phylogenomic approach based on PCR target enrichment and high throughput sequencing: Resolving the diversity within the South American species of Bartsia L. (Orobanchaceae). PLoS ONE 11: e0148203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood, T. E. , Takebayashi N., Barker M. S., Mayrose I., Greenspoon P. B., and Rieseberg L. H.. 2009. The frequency of polyploid speciation in vascular plants. Proceedings of the National Academy of Sciences, USA 106: 13875–13879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, C. , Rabiee M., Sayyari E., and Mirarab S.. 2018. ASTRAL‐III: Polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics 19: 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
APPENDIX S1. List of accessions included in this study with DNA or SRA number, short name used in this paper, voucher information, geographic origin, and whether the sample was used in Murphy et al. (2020) or was generated for this study.
APPENDIX S2. Statistics retrieved from HybPiper script ‘hybpiper_stats.py’ for the normal contigs (not those including intron regions).
APPENDIX S3. Tables with recovered sequence length and proportion of SNPs for each sample and locus, before and after data set optimization.
APPENDIX S4. Graphs and tables generated in the data set optimization step.
APPENDIX S5. Summary table from heterozygosity assessment.
APPENDIX S6. Table from clade association analysis.
APPENDIX S7. Input table and summary statistics from phasing analysis.
APPENDIX S8. Graphs and tables from data set optimization and heterozygosity assessment of phased accessions.
APPENDIX S9. Gene tree summary phylogeny from normal data set (framework phylogeny).
APPENDIX S10. Gene tree summary phylogeny from data set including phased accessions.
Data Availability Statement
All workflow scripts are available through the HybPhaser repository on GitHub (https://github.com/LarsNauheimer/HybPhaser; Nauheimer, 2021). Sequence data are available through the National Center for Biotechnology Information (NCBI) (BioProjects: P RJNA706038 [this study] and PRJEB35235 [Murphy et al., 2020]).