

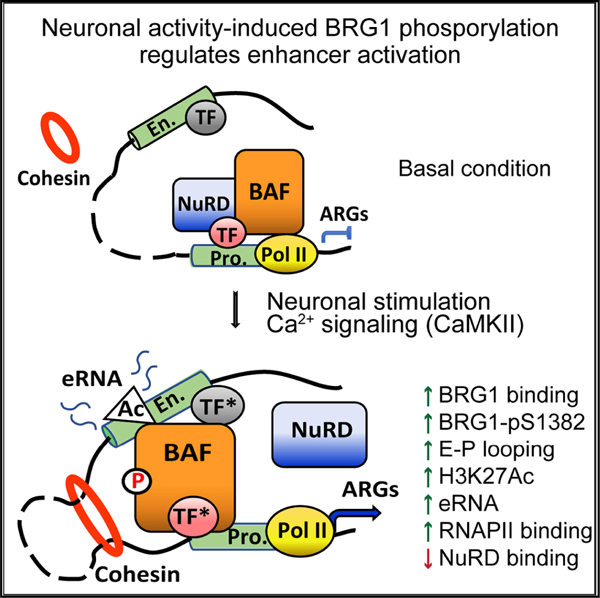
SUMMARY
Neuronal activity-induced enhancers drive gene activation. We demonstrate that BRG1, the core subunit of SWI/SNF-like BAF ATP-dependent chromatin remodeling complexes, regulates neuronal activity-induced enhancers. Upon stimulation, BRG1 is recruited to enhancers in an H3K27Ac-dependent manner. BRG1 regulates enhancer basal activities and inducibility by affecting cohesin binding, enhancer-promoter looping, RNA polymerase II recruitment, and enhancer RNA expression. We identify a serine phosphorylation site in BRG1 that is induced by neuronal stimulations and is sensitive to CaMKII inhibition. BRG1 phosphorylation affects its interaction with several transcription co-factors, including the NuRD repressor complex and cohesin, possibly modulating BRG1-mediated transcription outcomes. Using mice with knockin mutations, we show that non-phosphorylatable BRG1 fails to efficiently induce activity-dependent genes, whereas phosphomimic BRG1 increases enhancer activity and inducibility. These mutant mice display anxiety-like phenotypes and altered responses to stress. Therefore, we reveal a mechanism connecting neuronal signaling to enhancer activities through BRG1 phosphorylation.
Graphical abstract
In brief
Kim et al. find a critical function of the BRG1 chromatin remodeler in regulating basal activities and inducibilities of neuronal activity-induced enhancers. Neuronal activity-induced BRG1 phosphorylation regulates its interaction with other transcription co-factors and target gene activation. This study provides insights to BRG1 function in neural development and plasticity.
INTRODUCTION
Activity-regulated gene (ARG) expression plays a central role in short-term neural responses, as well as in long-term memory formation, homeostasis, and adaptation (West and Greenberg, 2011; Yap and Greenberg, 2018). Altered ARG expression may lead to behavior defects in stress responses, learning and memory, addiction, and psychiatric disorders (Gallo et al., 2018; Manning et al., 2017). ARGs include immediate early genes (IEGs) that are induced within minutes and late response genes (LRGs) that are induced over hours. ARG expression is directed through activity-induced enhancers by specific neuronal stimuli (Kim et al., 2010; Nord and West, 2020; Tyssowski et al., 2018). Activated distal enhancers marked by H3K27Ac interact with promoters to form enhancer-promoter (E-P) loops (Kim and Shiekhattar, 2015). Cohesin-mediated E-P looping facilitates RNA polymerase II (RNA Pol II) recruitment and enhancer RNA (eRNA) expression, which drives mRNA transcription (Kagey et al., 2010; Kim and Shiekhattar, 2015). Thousands of neuronal activity-induced enhancers marked by increased H3K27Ac have been identified in cultured primary cortical neurons in response to KCl-mediated depolarization (Malik et al., 2014). Many chromatin regulators, such as histone acetyl transferase (HAT) CBP and histone deacetylases (HDACs), methyl DNA recognizing protein MECP2, and chromatin remodeling regulators BRG1/BRM-Associated Factor (BAF) and Nucleosome Remodeling Deacetylase (NuRD) complexes, have been shown to play important roles in ARG regulation (Chen et al., 2003, 2019; Qiu and Ghosh, 2008; Yang et al., 2016; Zhang et al., 2015). Importantly, it remains largely unclear how these factors functionally interact with each other to regulate enhancer activity and modulate transcription outcomes.
Mammalian SWItch/Sucrose Non-Fermentable (SWI/SNF)-like ATP-dependent chromatin remodeling BAF complexes, which contain core ATPase subunits BRG1/SMARCA4 or BRM/SMARCA2 and 10–12 tightly associated subunits, use energy derived from ATP hydrolysis to modulate chromatin structures and regulate transcription (Son and Crabtree, 2014). Mutations in BAF subunits are the genetic causes of Coffin-Siris syndrome (Santen et al., 2012; Tsurusaki et al., 2012; Van Houdt et al., 2012), which results in severe neural developmental defects. In addition, de novo functional mutations in genes encoding BAF subunits are observed in patients with autism spectrum disorders, amyotrophic lateral sclerosis, and schizophrenia (De Rubeis et al., 2014; Halgren et al., 2012; Helsmoortel et al., 2014; Neale et al., 2012). Moreover, BRG1 is a key node in the autism spectrum disorder gene network (De Rubeis et al., 2014). Animal studies demonstrated important functions of BAF subunits in neural development and plasticity (Sokpor et al., 2017; Son and Crabtree, 2014; Wu, 2012). BRG1 and BAF complexes preferentially bind to enhancers during development and cancer progression (Alexander et al., 2015; Alver et al., 2017; Yu et al., 2013). Mechanistic analyses are complicated because BAF complexes can function as activators or as repressors. Previously, we and others have shown that neuronal BAF complexes play important but context-dependent roles in ARG regulation. It has been shown that BAF subunits could repress ARG basal expression or either limit or promote ARG induction in different contexts (Qiu and Ghosh, 2008; Wenderski et al., 2020; Wu et al., 2007; Zhang et al., 2015). However, it remains unclear how BAF complexes execute specific functions in basal and activated neuronal conditions.
Neuronal activities, which trigger Ca2+ influx, initiate multiple signaling pathways into the nucleus to regulate transcription. Intermediates, such as cyclic AMP/protein kinase A (cAMP/PKA), RAS/mitogen-activated protein kinase (MAPK), Ca2+/calmodulin-dependent protein kinase (CaMK), and calcineurin, can phosphorylate or dephosphorylate a number of transcription factors and co-factors that serve as activity switches to regulate ARG expression (Ebert and Greenberg, 2013; Wong and Ghosh, 2002; Yap and Greenberg, 2018). Phosphorylation of transcription regulators influences a dynamic protein interaction network that allows neurons to produce rapid and diverse responses to enable adaptation to the changing environment. Despite the identification of phosphorylation sites in BAF subunits in various conditions (Kimura et al., 2014; Kwon et al., 2015; Padilla-Benavides et al., 2020; Wang et al., 2013), it is not clear whether BAF subunits undergo activity-induced protein modifications nor how post-translational modifications regulate BAF activities. Here, we examined how BRG1 coordinates co-factor activities in ARG regulation. We demonstrated a critical role of BRG1 in regulating enhancer activities and identified a phosphorylation event that fine-tunes BRG1 function in response to neuronal signaling. Our study provides significant insights into the function of chromatin remodeling complexes in neural development and plasticity.
RESULTS
BRG1 regulates neuronal ARG activation
Using BAF53b-Cre, we specifically deleted Brg1 in developing neurons, resulting in the conditional Brg1 knockout mice (Brg1cko). Previously, we showed that in cultured Brg1cko cortical neurons, Brg1 deletion specifically affected the expression of a number of MEF2-regulated ARGs, such as Bdnf and Nr4a1, after KCl-mediated depolarization (Zhang et al., 2015). RNA sequencing (RNA-seq) results showed that 1,253 genes were significantly activated as measured at 6 h after KCl treatment in wild-type (WT) neurons (p < 0.05, fold change > 1.3; Table S1) and thus were likely ARGs. A comparison between WT and Brg1cko neurons in KCl-treated conditions showed that 76 ARGs were significantly impaired in induction in Brg1cko neurons (p < 0.05) (Figures 1A and S1A). Interestingly, most of these genes (72/76) were not significantly changed at basal conditions, suggesting that BRG1 is specifically required for the activation of these ARGs in response to neuron depolarizations. Many of the BRG1-activated ARGs were LRGs, possibly because of the 6-h time point analyzed. A time-course expression study of prototypical ARGs after KCl-induced depolarization showed that Brg1 deletion led to the impaired expression of not only LRGs but also of IEGs, such as c-Fos and Arc (Figures 1B and S1B). Although many ARGs were affected by Brg1 deletion, several genes, such as Gadd45b and Junb, were not (Figures 1A and S1B) (Zhang etal., 2015), indicating functional specificities of BRG1 and the overall normal Ca2+ signaling in depolarized neurons. In addition to BRG1-activated ARGs, a group of ARGs displayed significantly increased basal and/or KCl-induced expression in Brg1cko neurons (Figure S1A), suggesting that BRG1 could also function as a repressor for a subset of ARGs at basal and/or depolarized conditions. In this study, we focused on the BRG1 function in the activation of ARGs in response to neuronal activation.
Figure 1. BRG1 regulates ARG induction and binds to H3K27Ac marked enhancers in response to neuronal activities.
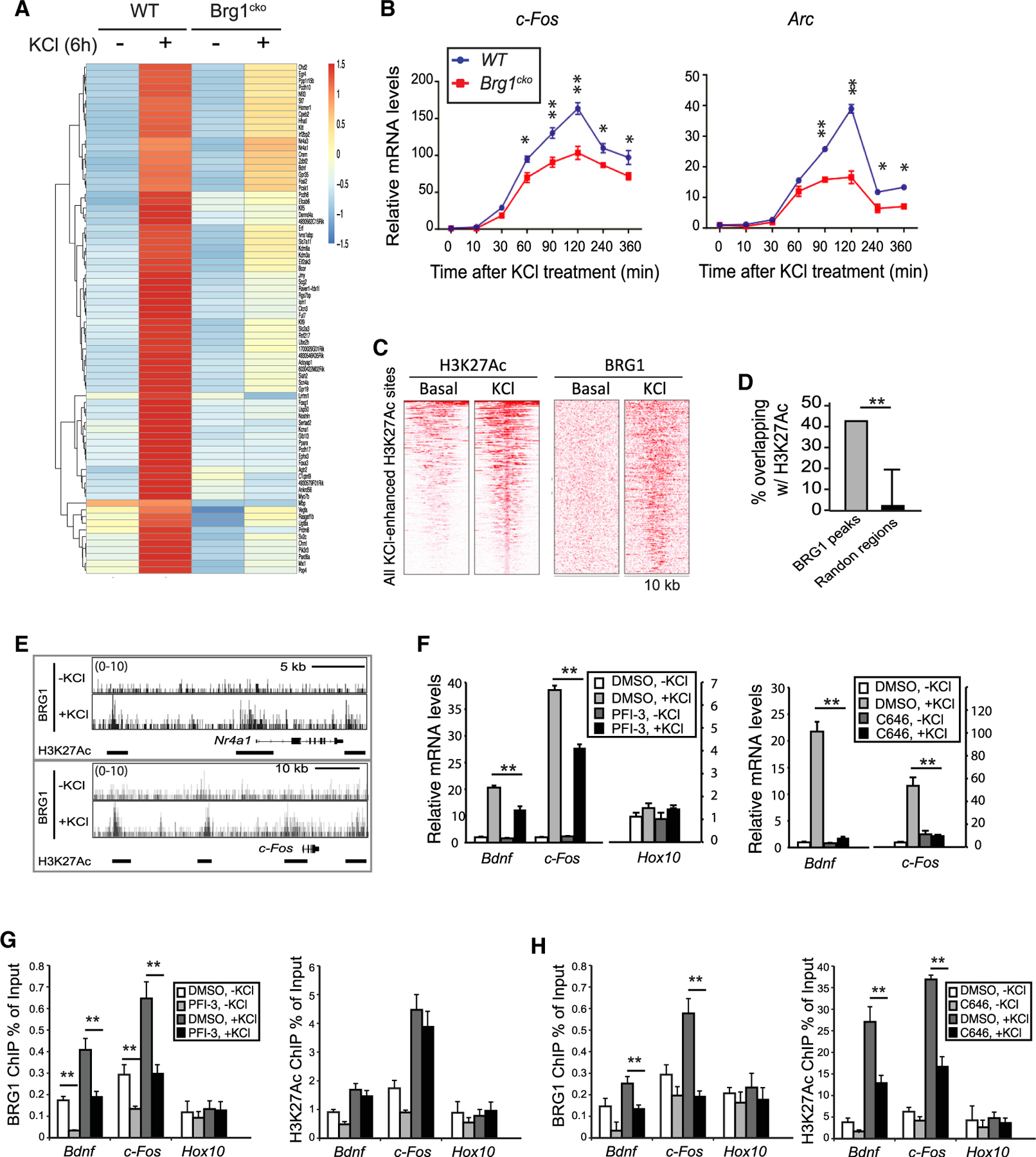
(A) Heatmap showing RNA-seq signals of ARGs that were differentially expressed in wild-type and Brg1cko neurons at 6 h after KCl treatment.
(B) qRT-PCR analyses of c-Fos and Arc mRNA in cultured Brg1cko and control neurons after KCl treatment.
(C) Heatmap of H3K27Ac and BRG1 ChIP-seq signals in 10-kb regions surrounding all H3K27Ac peaks that showed increased signals upon depolarization in cultured cortical neurons (1 h).
(D) Percent overlap between BRG1 peaks and randomly selected regions with H3K27Ac peaks in depolarized neurons.
(E) BRG1 ChIP-seq signals in resting and depolarized neurons around activity-induced genes Nr4a1 and c-Fos. H3K27Ac peaks are shown as bars below the BRG1 ChIP-seq plot.
(F) qRT-PCR analyses of cultured cortical neurons (6 h, ± KCl) treated with PFI-3 or C646.
(G and H) ChIP-qPCR assays of BRG1 and H3K27Ac levels in cultured cortical neurons treated with or without PFI-3 (G) or C646 (H) (0.5 h, ± KCl).
Student’s t test (n = 3), *p < 0.05, **p < 0.01. See also Figures S1 and S2 and Table S1.
Activity-induced BRG1 binding to enhancers requires H3K27Ac
To determine where BRG1 binds on chromatin and how binding changes in response to neuronal activities, we performed BRG1 chromatin immunoprecipitation sequencing (ChIP-seq) in resting and KCl-depolarized cortical neuron cultures. We found that the induction of neuronal activity significantly increased BRG1 binding: peak numbers increased from 708 to 20,962. Previous genome-wide studies revealed that dynamic H3K27Ac marks were also increased by neuron depolarization in a similar experimental setting (Malik et al., 2014). An intersection between BRG1 and H3K27Ac peaks showed that almost all activity-induced H3K27Ac sites also had increased BRG1 binding upon depolarization (Figure 1C). A distribution analysis showed a significant enrichment of BRG1 sites that overlapped with H3K27Ac peaks in depolarized neurons compared with random sites (Figure 1D). Detailed examination of the regulatory regions of ARGs, such as c-Fos and Nr4a1, showed extensive co-occupancies by BRG1 and H3K27Ac in enhancer peaks in depolarized neurons (Figure 1E). Thus, despite the limited numbers of BRG1-activated ARGs identified at one time point (Figure 1A), BRG1 binds extensively to activity-induced enhancers in response to neuronal depolarization.
BAF complexes could be recruited to specific chromatin sites through interactions with transcription factors or through interactions with histones (Wu et al., 2009). Transcription factors such as MEF2, SP1, and AP-1 have been shown to be important for BRG1 recruitment to ARG regulatory regions (Qiu and Ghosh, 2008; Vierbuchen et al., 2017; Zhang et al., 2015). Because the bromodomain of BRG1 binds to H3K27Ac with low affinity (Filippakopoulos et al., 2012), BAF complexes could also be recruited to enhancers through interactions with H3K27Ac (Malik et al., 2014). A time-course study of H3K27Ac and BRG1 binding to representative ARGs showed that the increase of H3K27Ac at the promoters of Bdnf and c-Fos could be detected at 5 min after depolarization. BRG1 binding to these promoters has similar kinetics as H3K27Ac (Figure S2). To determine whether the BRG1 bromodomain is important for BAF recruitment to active enhancers, we treated cultured neurons before KCl treatment with the BRG1 bromodomain inhibitor PFI-3 (Gerstenberger et al., 2016) or with C646, an inhibitor of p300/CBP HATs (Bowers et al., 2010). Both PFI-3 and C646 impaired activity-induced BRG1 target gene expression (Figure 1F). Importantly, ChIP-qPCR showed that PFI-3 impaired BRG1 binding to enhancers but did not significantly affect activity-induced local H3K27Ac levels shortly after depolarization (Figure 1G). In contrast, C646 treatment not only impaired the induced H3K27Ac levels but also decreased activity-induced BRG1 binding to the enhancers (Figure 1H). These data suggest that in early stages of ARG induction, the increased H3K27Ac helps recruit BRG1 to activity-induced enhancers through an interaction with the BRG1 bromodomain.
BRG1 regulates activity-induced E-P looping and enhancer activities
Consistent with a function of BRG1 in ARG activation, we observed a reduction in RNA pol II recruitment to the promoters of representative ARGs upon BRG1 deletion (Figure 2A). Because BRG1 binds to enhancers, we examined the levels of the active enhancer marker H3K27Ac in WT and Brg1cko neurons. Brg1 deletion reduced H3K27Ac levels at several well-characterized activity-induced enhancers (Figure 2B), including at c-Fos enhancer 2 (e2), and at the Arc enhancer (Schaukowitch et al., 2014). Although the initial rapid increase of H3K27Ac is required for BRG1 recruitment, BRG1 is also required for later maximum induction of H3K27Ac. Thus, chromatin regulators, including both BAF complexes and p300/CBP H3K27 HATs, work cooperatively to activate ARGs. Because eRNA expression levels correlate with enhancer activities, we examined the levels of eRNAs previously reported to influence c-Fos and Arc expression (Schaukowitch et al., 2014). We observed defects in the activity-induced expression of these eRNAs upon Brg1 deletion (Figure 2C).
Figure 2. BRG1 regulates activity-induced E-P looping and enhancer activities.
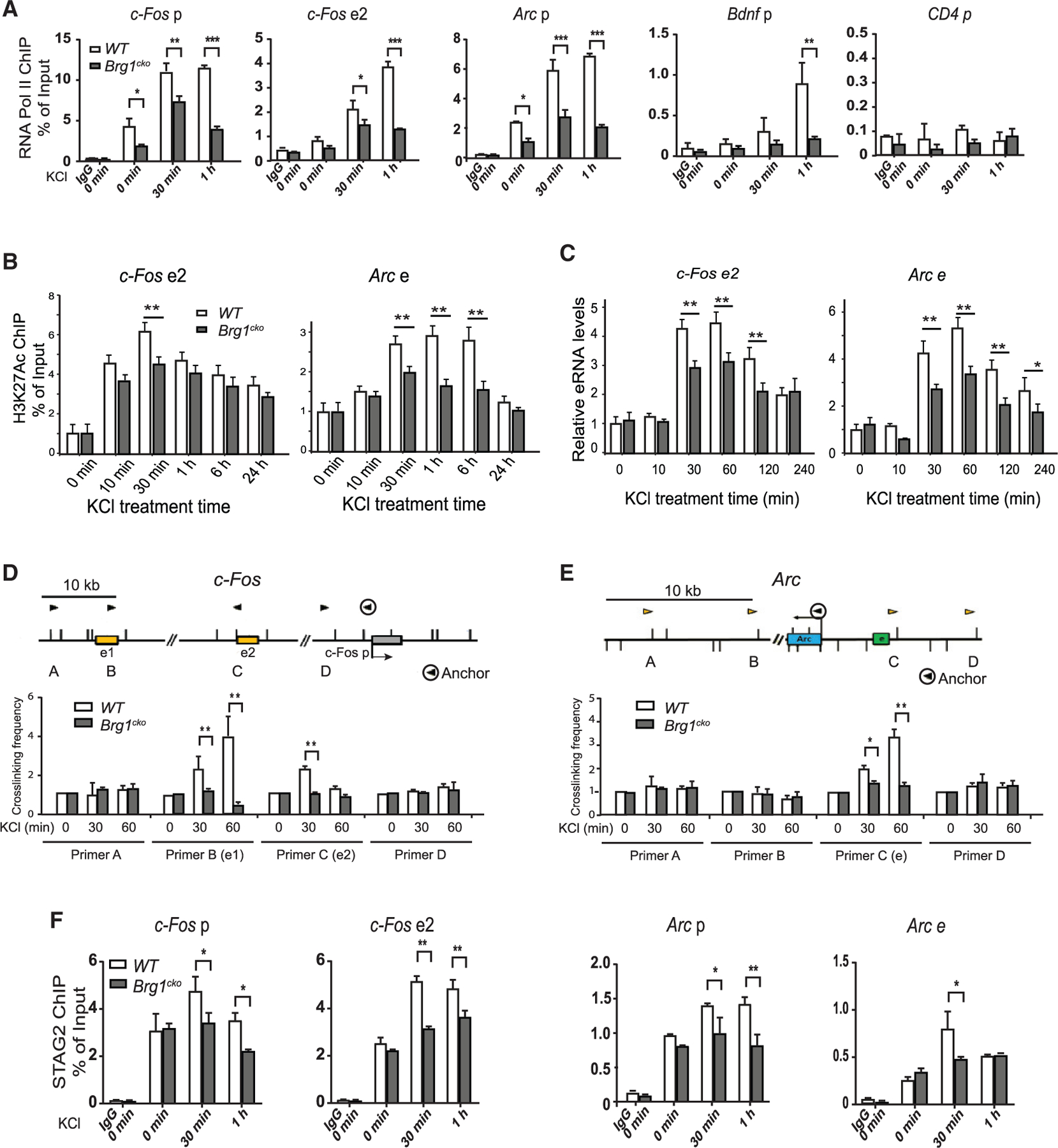
(A) ChIP-qPCR assay of RNA Pol II binding to the promoters and enhancers of ARGs in wild-type and Brg1cko neurons after KCl stimulation. The CD4 gene was shown as a negative control.
(B) ChIP-qPCR analyses of H3K27Ac at c-Fos e2 and Arc enhancer (e) in wild-type and Brg1cko neurons after KCl stimulation.
(C) qRT-PCR analyses of eRNA levels of c-Fos e2 and Arc e in Brg1cko and control neurons after KCl treatment.
(D and E) E-P crosslinking frequency in 3C experiments of c-Fos (D) and Arc (E) in basal or depolarized wild-type and Brg1cko neurons. The genomic structures and the PCR primer locations (triangles) are shown above the results. The anchor primer was paired with other primers to measure the interactions.
(F) ChIP-qPCR analyses of STAG2 levels at enhancers and promoters of c-Fos and Arc in wild-type and Brg1cko neurons after KCl stimulation.
Student’s t test (n = 3), *p < 0.05, **p < 0.01. See also Figure S3.
To determine the direct function of BRG1 at enhancers, we examined an early critical step of enhancer activation: E-P looping. Using the chromatin conformation capture (3C) assay, we examined the function of BRG1 in E-P looping in the c-Fos and Arc gene-regulatory regions. We confirmed the previously reported activity-induced interactions of c-Fos enhancers (e1 and e2) with its promoter (Joo et al., 2016) in WT neurons. Brg1 deletion significantly impaired c-Fos E-P looping (Figure 2D). Similarly, the activity-induced interaction between the enhancer and promoter of Arc (Schaukowitch et al., 2014) was also impaired by Brg1 deletion (Figure 2E). Cohesin complexes are required for E-P interaction. By performing cohesin subunit STAG2 ChIP-qPCR, we observed decreased cohesin binding to activity-induced enhancers and promoters in Brg1cko neurons (Figure 2F). Therefore, BRG1 plays a critical role in modulating enhancer activities by regulating cohesin binding and E-P interactions.
It has been shown that neuronal activity induced a global accessibility change in adult brain in vivo (Su et al., 2017). Previous reports also suggested that BRG1 functions together with the transcription factor AP-1 in enhancer generation during development (Vierbuchen et al., 2017). In cultured neurons, we performed ATAC-seq to identify open chromatin regions and enhancer accessibilities. We identified 9,070 and 5,751 open chromatin sites in basal and depolarized conditions, respectively (Figure S3A). These sites largely overlapped with H3K27Ac marks identified in neurons (Malik et al., 2014), suggesting that they are active enhancers. Interestingly, neither KCl treatment nor Brg1 deletion significantly changed ATAC-seq signals (Figure S3B). It is possible that, as reported, cultured neurons have a significantly different chromatin accessibility state from neurons in vivo (Sinnamon et al., 2019). Thus, in neuron cultures, the deletion of Brg1 impairs enhancer activities but does not significantly change enhancer selection.
BRG1 phosphorylation at S1382 is induced by neuronal activities
To determine how BRG1 and BAF complexes respond to neuronal activities, we affinity-purified endogenous BAF complexes and interactomes from nuclear extracts of resting and depolarized cortical neuron cultures using an anti-BRG1/BRM antibody (Figure 3A). The mass spectroscopy showed that the purifications yielded 272 proteins in the basal condition and 305 proteins in depolarized neurons. These proteins include BRG1 and most known BAF subunits and interacting proteins (Table S2). Based on the silver staining of the purified complexes and the mass spectroscopy results, there were no obvious subunit differences under the two conditions (Figure 3B; Table S2). Comparison of phosphorylated peptides under basal and depolarized conditions (Table S3) revealed that the peptides corresponding to the phosphorylation of BRG1 at S1382 (BRG1-pS1382) were detected only in the depolarized condition and not in the basal condition (Figure 3C). BRG1-pS1382 was previously demonstrated by proteomic analyses in various human and murine tissues, including developing brains (Dephoure et al., 2008; Liao et al., 2008; Mertins et al., 2014). BRG1-pS1382 is dynamic with levels affected by factors, including the cell cycle, signaling stimulation, and the cell’s neural differentiation state. Serine-1382 is conserved in all mammalian BRG1 and in Drosophila BRM, but not in the yeast SWI2/SNF2 protein (Figure 3D). Interestingly, the site in mammalian BRM protein is an alanine despite the close resemblance between BRG1 and BRM in this region (Figure 3D). Therefore, the phosphorylation of BRG1 at S1382 could result from a mechanism conserved in Drosophila and could be indicative of a function of BRG1 that is distinct from that of BRM.
Figure 3. Neuronal activity-induced BRG1 phosphorylation at S1382.
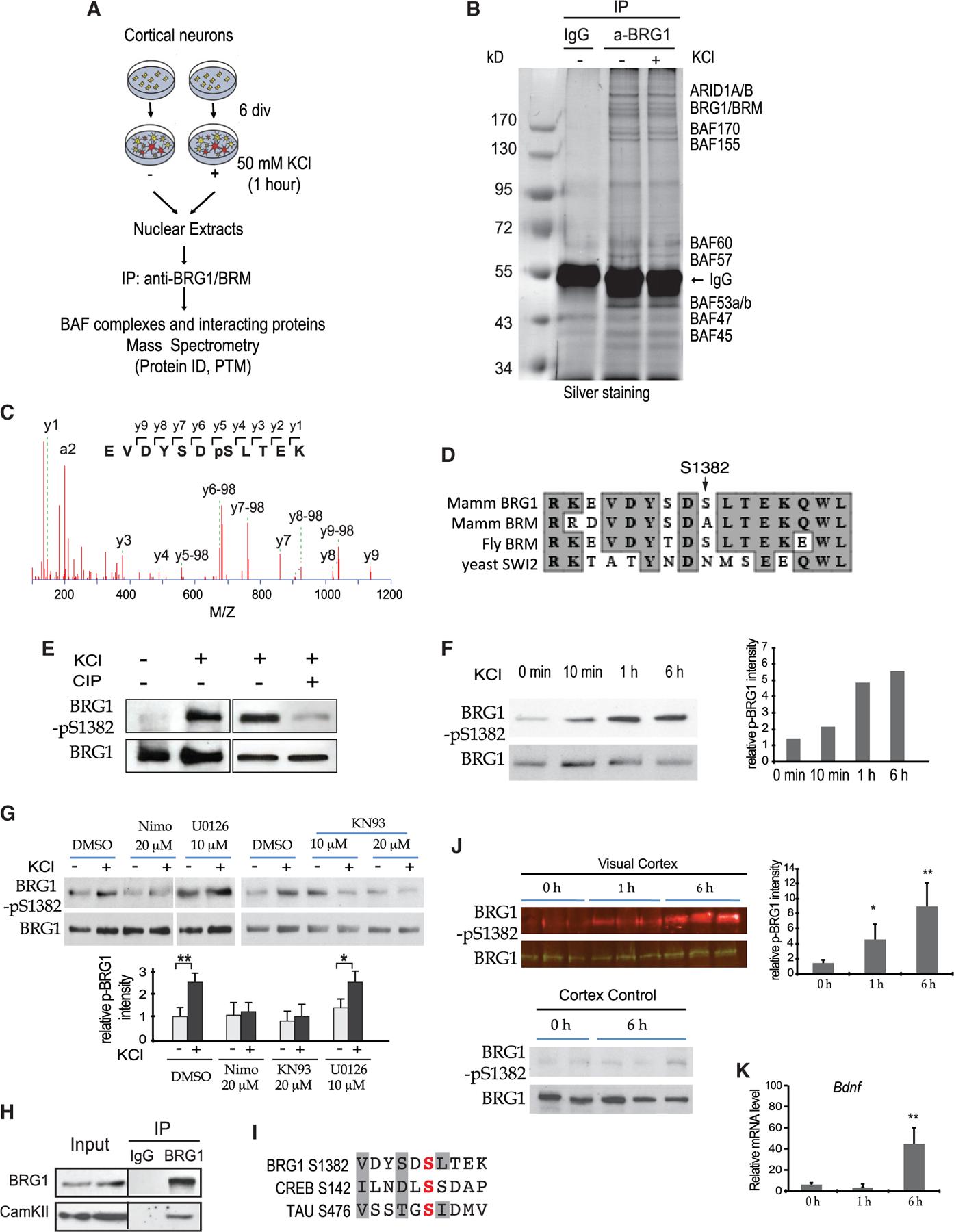
(A) A schematic of the purification and proteomic analyses of BAF complexes and interacting proteins from cortical neurons under basal and depolarized conditions.
(B) Photograph of a silver-stained gel of affinity-purified neuronal BAF complexes.
(C) Tandem mass spectrometry (MS/MS) fragment ion spectrum with peak assignments for the BRG1 peptide containing phosphorylated S1382.
(D) Comparison of protein sequences around mammalian BRG1 S1382 among BRG1 homologs.
(E) Western blot for KCl-induced BRG1 phosphorylation in cultured neurons and its sensitivity to CIP.
(F) BRG1-pS1382 in cortical neurons analyzed by western blot after KCl treatment. Quantifications are shown on the right.
(G) Western blot analysis of BRG1-pS1382 in cortical neurons treated with and without indicated inhibitors (1 h, ± KCl). Quantifications are shown at the bottom (n = 3).
(H) Western blot analyses of CaMKII in samples immunoprecipitated (IPed) from nuclear extracts from P5 cortices using antibodies against BRG1.
(I) Comparison of protein sequences around BRG1 S1382 and phosphorylation sites (in red) in CREB and TAU. The conserved residues important for CaMKII are shaded.
(J) Western blot analysis of light-induced BRG1-pS1382 in mouse visual cortex. Quantifications are shown on the right (n = 3).
(K) qRT-PCR analysis of Bdnf mRNA in visual cortices after light exposure (n = 3).
Student’s t test, *p < 0.05, **p < 0.01. See also Figure S4 and Tables S2 and S3.
We generated an antibody that specifically recognizes BRG1-pS1382 in western blot analyses. Using this antibody, we confirmed the increased BRG1-pS1382 in response to neuron depolarization in culture, which is sensitive to treatment with calf intestinal phosphatase (CIP) (Figure 3E). Time-course studies showed that BRG1-pS1382 was increased at 10 min after KCl stimulation, reached the highest level at the 1-h time point, and remained high at 6 h after stimulation (Figure 3F). Using this antibody, we also demonstrated the enrichment of BRG1-pS1382 in M phase HeLa cells (Figure S4A), in agreement with a previous proteomic study (Dephoure et al., 2008). The antibody also recognized Drosophila BRM in its phosphorylated state in S2 cells (Figure S4B).
Neuronal cell depolarization results in Ca2+ influx through L-type Ca2+ channels, which activates CaMKs and/or MAPK/ERK kinase/extracellular-signal-regulated kinase (MEK/ERK) pathways (Deisseroth et al., 2003). We therefore tested the sensitivity of BRG1 phosphorylation to the inhibitors of these pathways. Depolarization-induced BRG1-pS1382 was sensitive to nimodipine, an L-type Ca2+ channel inhibitor, and to KN93, a CaMKII inhibitor, but not to U0126, a MEK/ERK inhibitor (Figure 3G). We identified CaMKIIb as a BRG1 interacting protein from our proteomic analyses (Table S2). Endogenous CaMKII was co-immunoprecipitated (coIPed) by the anti-BRG1 antibody from P5 cortices (Figure 3H). The peptide sequence surrounding BRG1 S1382 shares similarities with the phosphorylation sites in several CaMKII substrates, such as CREB and Tau (White et al., 1998) (Figure 3I). These results suggest that CaMKII is likely one kinase responsible for neuronal activity-induced BRG1 phosphorylation.
To determine whether BRG1 is phosphorylated in response to physiological stimulation in vivo, we examined BRG1-pS1382 levels in murine visual cortices after light stimulation. At post-natal day (P) 21 (P21), mice were put in the dark for 3 days before being exposed to light. At different time points following light stimulation, visual cortices and the anterior part of the cortex as a control were examined for BRG1 phosphorylation by western blot and for gene activation by qRT-PCR. BRG1-pS1382 levels in the visual cortex were significantly increased after light exposure (Figure 3J), and the expression of ARGs, such as Bdnf, was upregulated (Figure 3K). Thus, BRG1-pS1382 is induced by neuronal activation in vivo, possibly through Ca2+ signaling and CaMKII.
BRG1 phosphorylation regulates neuronal ARG activation
To determine whether BRG1 phosphorylation influences ARG expression, we performed rescue experiments with a phosphomimic and a non-phosphorylatable BRG1 mutant at S1382 (BRG1-SE and BRG1-SA, respectively). In KCl-treated Brg1cko neurons, c-Fos expression was induced by exogenous BRG1 and BRG1-SE. BRG1-SA failed to rescue impaired c-Fos expression (Figure 4A). Expressing BRG1 or mutant proteins in WT neurons had little effect on c-Fos expression (Figure 4A). In the Brg1cko neurons that expressed BRG1-SA, BRM (which lacks the serine site), or BRG1-KR (which is an ATPase-inactive mutant) (Khavari et al., 1993), activity-induced expression of ARGs, such as c-Fos and Arc, at 1 h after KCl treatment was lower than that in cells expressing WT BRG1 or BRG1-SE (Figures 4B and 4C). These results indicate that both BRG1 phosphorylation and its ATPase activity are important for its function in activating ARGs in neurons. This function is also BRG1 specific and cannot be compensated by BRM. Interestingly, 1 h after KCl stimulation, in Brg1cko neurons that expressed BRG1-SE, LRGs such as Bdnf were upregulated; in contrast, expression of WT BRG1 did not result in this upregulation (Figure 4C). This observation suggests that constitutive phosphorylation of BRG1 S1382 potentiates Bdnf enhancers for activation. Thus, a precise regulation of BRG1 phosphorylation is required to fine-tune ARG induction kinetics and inducibilities in response to neuronal signals.
Figure 4. BRG1 S1382 phosphorylation regulates ARG activation.
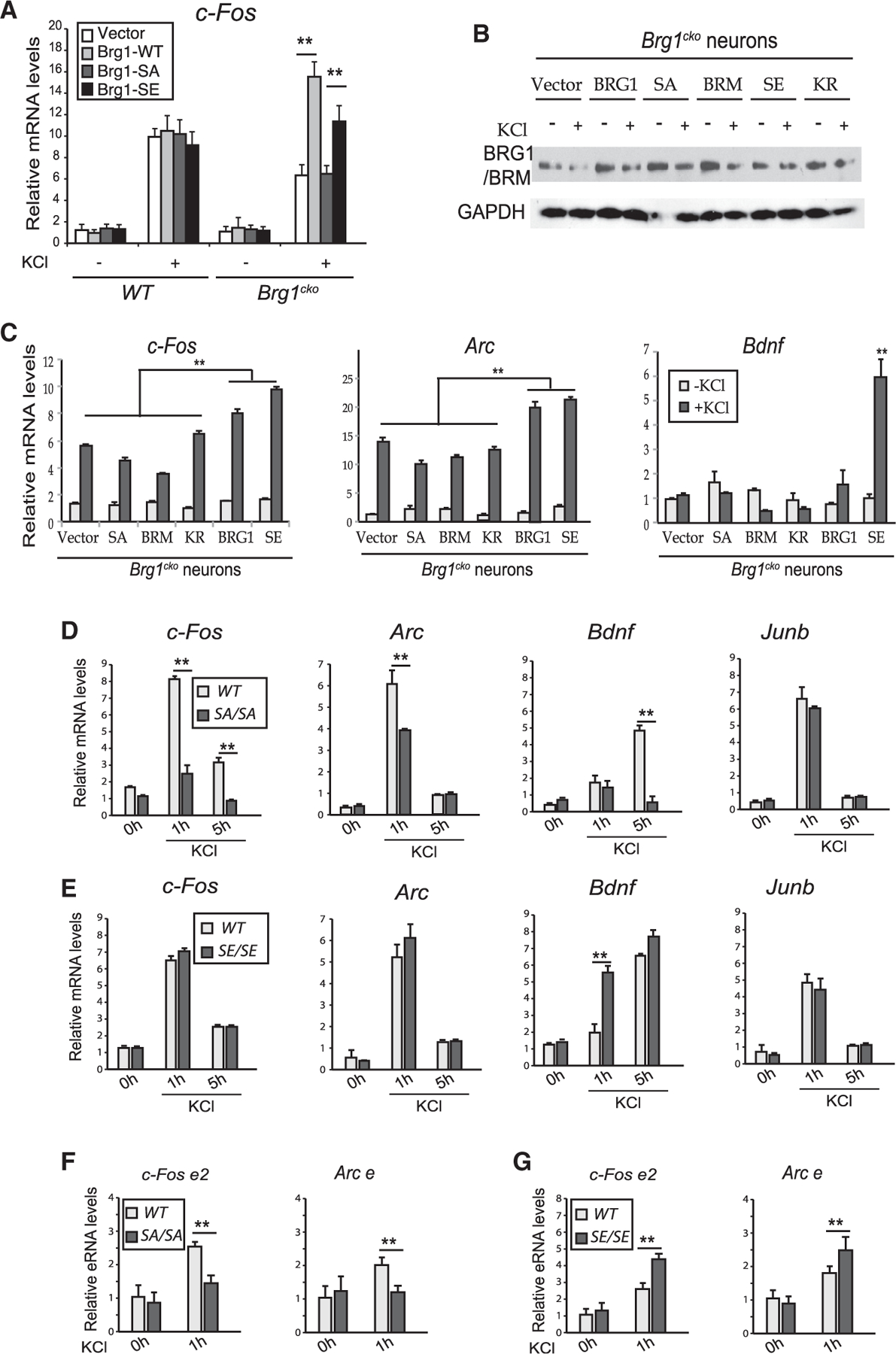
(A) qRT-PCR analysis of c-Fos mRNA in wild-type and Brg1cko neurons expressing BRG1 mutant proteins (1 h, ± KCl).
(B) Western blot using the J1 antibody showing the expression of the exogenous wild-type and mutant BRG1 and BRM proteins in Brg1cko neurons in experiments performed in (C).
(C) qRT-PCR analyses of c-Fos, Arc, and Bdnf in Brg1cko neurons expressing wild-type BRG1 or mutant proteins (1 h, ± KCl). ANOVA post hoc t tests between each condition (n = 3). **p < 0.01.
(D and E) qRT-PCR analyses of ARG mRNA levels in WT and SA neurons (D) or WT and SE neurons (E) after KCl treatment.
(F and G) qRT-PCR analyses of eRNA levels of c-Fos e2 and Arc e in WT and SA neurons (F) or WT and SE neurons (G) after KCl treatment.
Student’s t test (n = 3), *p < 0.05, **p < 0.01. See also Figure S5.
Generating Brg1-S1382A and Brg1-S1382E knockin mice
To further understand the function of BRG1 phosphorylation, we generated knockin mice that harbor the point mutations present in either BRG1-SA or BRG1-SE using the CRISPR-Cas9 method (Figure S5A). The homozygous Brg1-S1382A (SA) and Brg1-S1382E (SE) mice express similar levels of BRG1 and BRM and are grossly normal and fertile. Cortical neurons cultured from SA mice had impaired activity-induced expression of ARGs compared with WT neurons at 1 and 5 h after KCl treatment (Figures 4D and S5B). Junb, which is not a BRG1-dependent IEG, was expressed at the same level in SA and in WT neurons (Figures 4D and S5B). When we compared WT and SE cortical neurons, SE neurons displayed similar or more sensitive induction of these genes in response to depolarization (Figures 4E and S5C). Consistently, KCl-induced ARG enhancer activities, as measured by eRNA expression at c-Fos e2 and the Arc enhancer, were impaired in SA neurons but increased in SE neurons compared with WT neurons (Figures 4F and 4G). To further understand the function of BRG1-pS1382 in ARG induction, we performed an RNA-seq analysis of SA and SE cortical neurons compared with their corresponding WT littermate controls at basal and KCl depolarized conditions (1 h). Within the 732 genes induced by KCl (p < 0.05, fold change > 2 in any genotype), we examined the induced gene levels in SA or SE neurons compared with their WT controls (Figure S5D). A subset of genes (n = 273) displayed reduced expression in SA neurons but increased expression in SE neurons (Figure S5D). These include IEGs such as c-Fos, Fosl2, and Npas4. A comparison of these 273 genes with the 76 BRG1-regulated ARGs identified at the 6-h time points (Figure 1A) revealed 13 common genes, including Bdnf, Nr4a1, Homer1, and Nr4a3 (Figure S5E). These results confirmed that BRG1 phosphorylation is required for the maximum induction of a significant subset of ARGs in response to neuron depolarization, whereas constitutive phosphorylation sensitizes ARGs to induction.
BRG1 phosphorylation regulates BAF interactions with transcription co-factors
To understand how BRG1 phosphorylation regulates neuronal gene activation, we examined the biochemical functions of BRG1-SA and BRG1-SE proteins. In SW13 cells where BRG1 and BRM are both absent (Liu et al., 2001), we expressed WT and mutant BRG1 proteins using lentiviral vectors. BRG1-SA, BRG1-SE, and BRG1-KR mutants were localized in the nucleus alongside the BRG1-WT (Figure 5A). In BRG1, S1382 is in the histone-interacting Snf2 ATP Coupling (SnAC) domain downstream of the ATPase domain (Sen et al., 2013). We affinity-purified BAF complexes containing WT and mutant BRG1 and compared their ATPase activities. Unlike the complexes containing ATPase-inactive BRG1-KR, complexes containing BRG1-SA and BRG1-SE had ATPase activities similar to that of the WT BAF complex (Figure 5B). Therefore, BRG1 phosphorylation at S1382 does not affect its ATPase activity.
Figure 5. BRG1 phosphorylation affects its interaction with other co-factors without affecting BAF complex formation or ATPase activities.
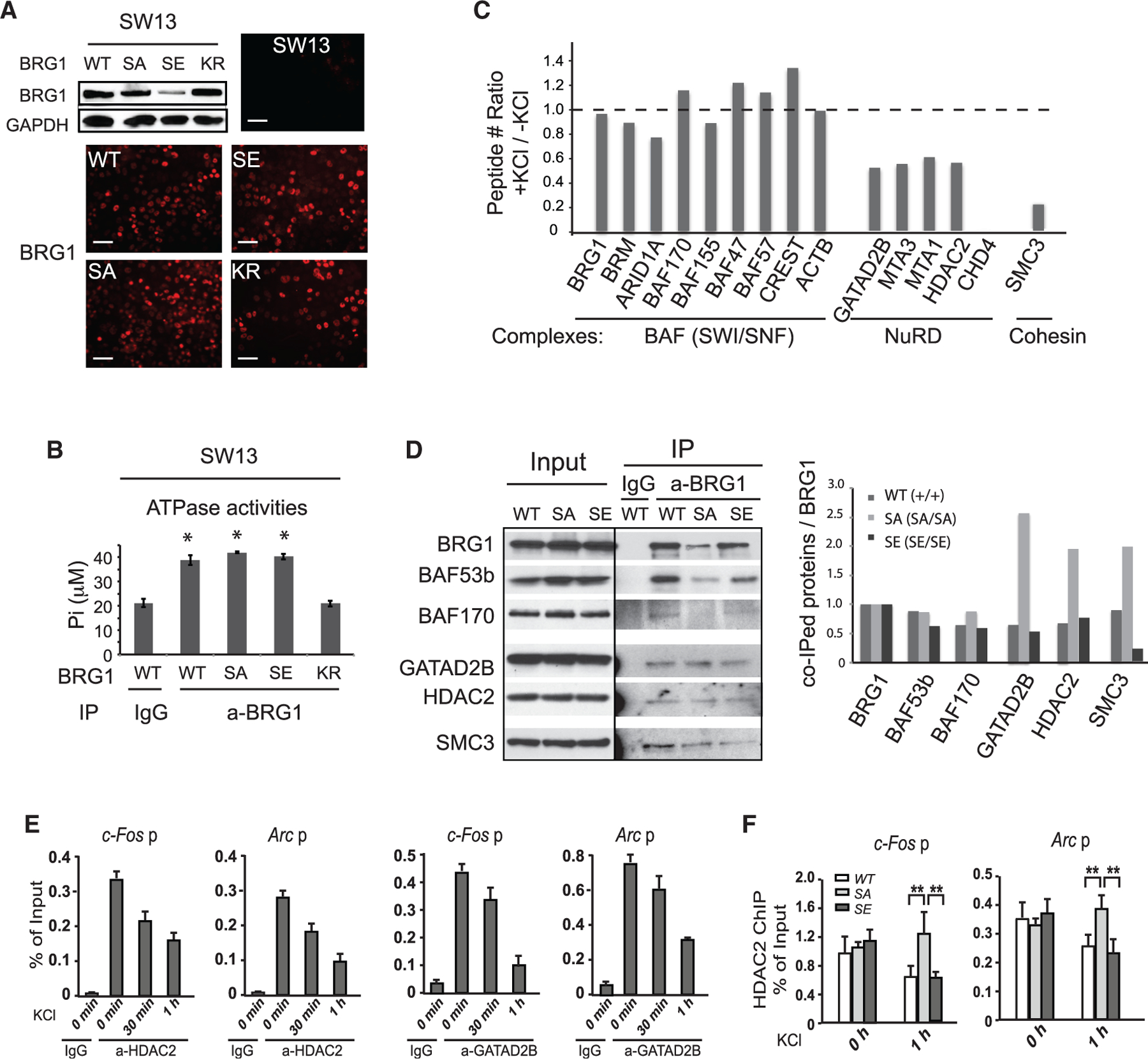
(A) Expression and localization of exogenous BRG1 and BRG1 mutant proteins in SW13 cells as detected by western blot and immunostaining. Scale bars, 50 µm.
(B) ATPase activity assay of BAF complexes purified from SW13 cells expressing BRG1 or mutant proteins. ANOVA post hoc t test (n = 3), *p < 0.05.
(C) Ratios of peptide numbers identified from mass spectrometry for representative BAF subunits, NuRD subunits, and cohesin subunits from proteomic analyses of neurons between depolarized and basal conditions.
(D) Western blot analyses of samples IPed from nuclear extracts from WT and SA or SE. P5 cortices using antibodies against BRG1. Quantifications of co-purified BRG1 interacting proteins relative to IPed BRG1 are shown on the right.
(E) ChIP-qPCR analyses of HDAC2 and GATAD2B levels at promoters of c-Fos and Arc in cultured cortical neurons after KCl stimulation.
(F) ChIP-qPCR analysis of HDAC2 levels at promoters of c-Fos and Arc in WT, SA, and SE neurons after KCl stimulation.
Student’s t test (n = 3), **p < 0.01. See also Figure S6.
Recent cryoelectron microscopy study of BAF complexes showed that the region of S1382 is likely located at the complex surface (He et al., 2020; Mashtalir et al., 2020). To determine whether BRG1 phosphorylation affects its ability to bind to other proteins, we examined the BRG1-interacting proteins identified from our proteomic analyses and compared the abundances of peptides in basal and depolarized conditions (Table S2). Although all BAF subunits and most known interacting proteins were detected with similar coverage percentages and peptide counts in both conditions (ratios between 0.7 and 1.4; Figure 5C; Table S2), four subunits of the NuRD co-repressor complex, GA-TAD2B, MTA1, MTA3, and HDAC2 (Xue et al., 1998), had relatively high peptide numbers under basal conditions and were all reduced to 70% or less in the depolarized condition (Figure 5C). For the NuRD core ATPase subunit CHD4, eight peptides were recovered from resting neurons, but none from the depolarized neurons (Table S2; Figure 5C). Similarly, four peptides corresponding to cohesin subunit SMC3 were obtained in basal conditions, but only one was detected in the depolarized condition (Table S2; Figure 5C). We performed BRG1 immunoprecipitation from nuclear extracts of P5 WT, SA, and SE cortices when the cortex consists mostly of neurons. We observed that although similar ratios of BAF subunits were pulled down from all three genotypes relative to BRG1 levels, more NuRD subunits and SMC3 proteins were co-purified with BRG1-SA than BRG1-WT or with BRG1-SE (Figures 5D and S6). These results support the hypothesis that the BRG1 binding affinities for transcriptional co-factors are affected by neuronal activity-induced BRG1 phosphorylation. Importantly, ChIP-qPCR analyses of NuRD subunits GATAD2B and HDAC2 in cultured cortical neurons showed reduced binding of both to ARG promoters upon KCl treatment (Figure 5E). The KCl-induced HDAC2 departure from c-Fos and Arc promoters was impaired in SA neurons (Figure 5F). These results suggest that neuronal activity-induced BRG1 phosphorylation facilitates the release of NuRD repressor complex from ARGs, which may contribute to the activation of ARGs in depolarized neurons.
BRG1-pS1382 modulates basal activities of the c-Fos enhancer in vivo
Inducible enhancers are precisely regulated to remain repressed but poised for induction under basal conditions. The basal activities of inducible enhancers may also affect gene induction kinetics in response to signal stimulation. To understand how BRG1 phosphorylation influences basal enhancer activities in vivo, we examined levels of H3K27Ac and key transcription regulators at the promoter and the enhancers of c-Fos in P5 WT, SA, and SE cortices. Differences in phosphorylation of BRG1 at S1382 did not affect the amount of BRG1 bound to the c-Fos promoter or to c-Fos e2 as indicated by BRG1 ChIP-qPCR (Figure 6A). Interestingly, H3K27Ac ChIP showed that there was a slight but significant increase of H3K27Ac at c-Fos e2 in the cortical samples from SE mice compared with those from WT and SA mice (Figure 6B), suggesting an increased basal activity of c-Fos e2 in SE brains. Consistently, in the P5 cortex, the cohesin subunit STAG2 was also observed at higher levels at the c-Fos promoter and c-Fos e2 in SE than in SA cortices (Figure 6C). We next performed 3C experiments to examine the interaction between c-Fos e2 and the c-Fos promoter. A significantly higher interaction rate was observed in SE cortices than in SA and WT samples (Figure 6D). Collectively, these results indicate that there is a higher basal c-Fos enhancer activity in SE mutant neurons. Consequently, there was a slight but significant increase of the basal expression of c-Fos in SE cortices than in WT or SA cortices as shown by qRT-PCR (Figure 6E). Thus, BRG1 phosphorylation could increase c-Fos enhancer basal activities and expression noise.
Figure 6. BRG1 phosphorylation regulates c-Fos enhancer basal activities in vivo.
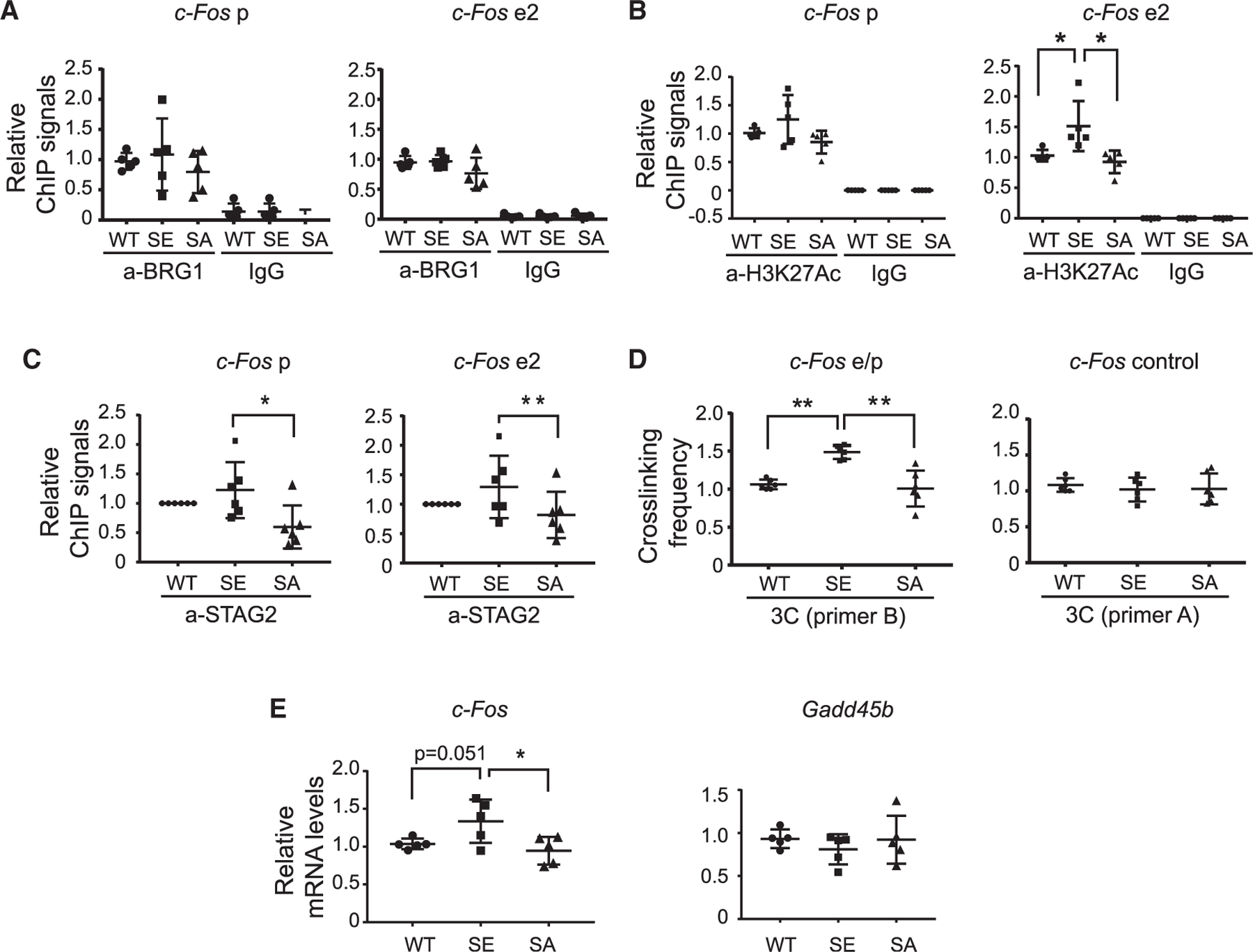
(A–C) ChIP-qPCR analyses of BRG1 (A), H3K27Ac (B), and STAG2 (C) at c-Fos promoter and enhancer regions in P5 SA and SE cortices relative to signals in WT tissue. In STAG2 CHIP-qPCR, six sets of ChIP assays were performed. Each included one WT, one SA, and one SE sample. All WT samples were normalized to 1, and the averages of the relative ChIP signals of the SA and SE samples were compared.
(D) Crosslinking frequency between the c-Fos enhancer and promoter and control regions in P5 SA and SE cortices measured in a 3C experiment relative to signals in WT tissue.
(E) qRT-PCR analyses of c-Fos and control Gadd45b mRNAs in P5 WT and SA and SE cortices.
ANOVA post hoc t test (n = 5), *p < 0.05, **p < 0.01.
BRG1 phosphorylation regulates brain ARG induction in response to stress, and mutants display anxiety-like behaviors
ARG expression affects many aspects of neuronal function and behaviors, especially in response to stress (Gallo et al., 2018). To determine whether BRG1-pS1382 is important for in vivo neurological functions, we performed behavioral tests of the age-matched WT, SA, and SE mice, all of which are from the C57BL/6J background. In the light-dark box test, both SA and SE mice tended to stay for less time in the lit region and more time in the dark region than did WT mice (Figure 7A). Although both SA and SE mice displayed normal activities in general, SE mice tended to be more active in the dark and less active in the light box than were WT mice (Figure 7A). In the elevated maze test, both SA and SE mice spent less time in open arms than did WT mice (Figure 7B). Further, SA mice entered the open arms less frequently and spent significantly more time in the closed arm than did WT and SE mice (Figure 7B). These results indicate that alterations in BRG1 phosphorylation cause anxiety-like behaviors.
Figure 7. Alterations of BRG1 phosphorylation states cause anxiety-like behaviors and changes in c-FOS expression in response to stress.
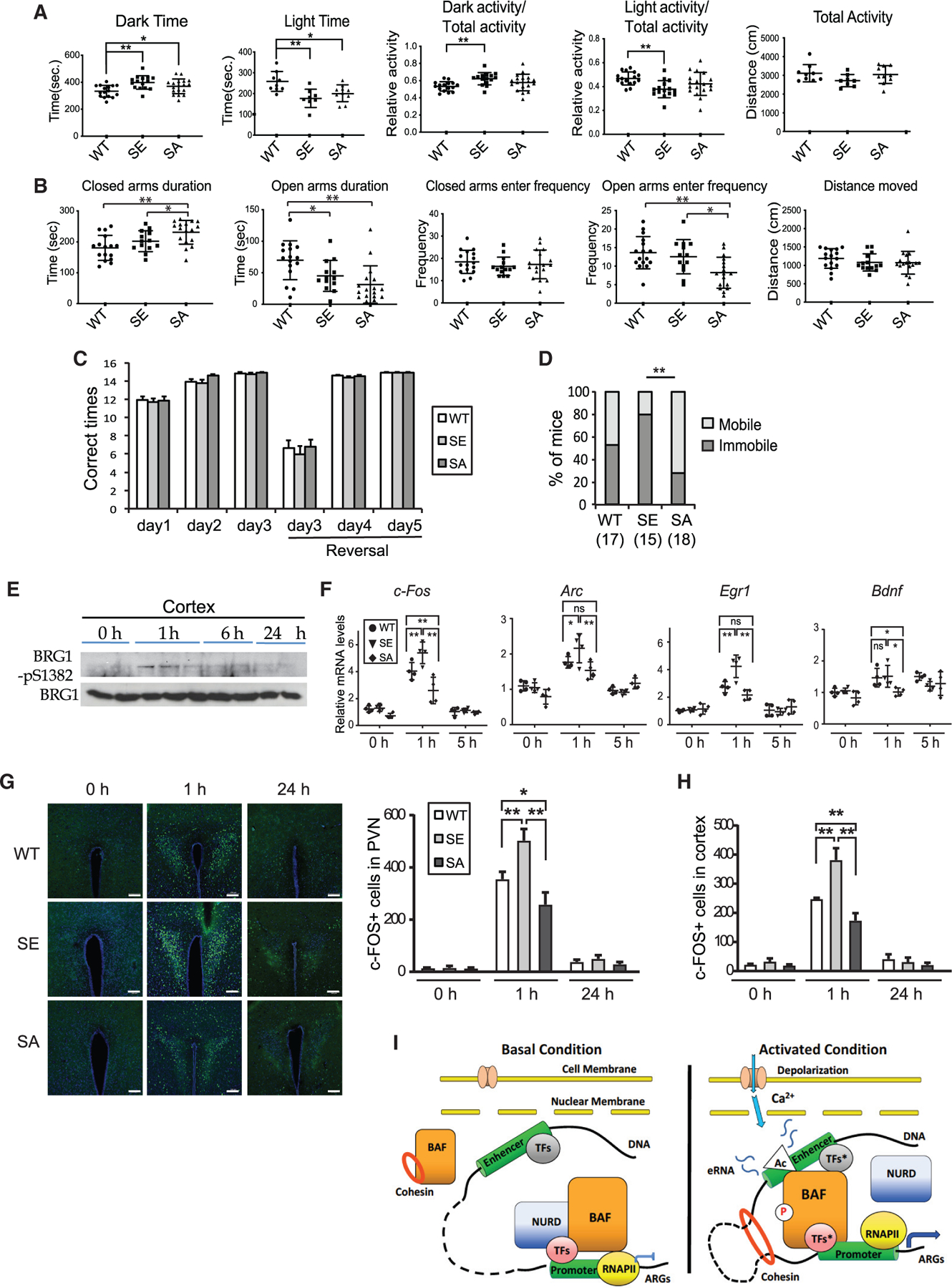
(A) Time spent in the dark or light box in the light-dark box test, relative distances traveled in the dark, or light box and total distance traveled for WT (n = 17), SA (n = 15), and SE (n = 18) mice.
(B) Time spent in open and closed arms of the elevated maze and entering frequency to open or closed arms for WT (n = 17), SA (n = 15), and SE (n = 18) mice.
(C) Number of times out of 15 trials that WT (n = 17), SA (n = 15), and SE (n = 18) mice found the platform in each session of the Y maze reversal learning swimming test.
(D) The percentage of mice that were immobile for longer than 30 s once put in water in any trials for WT (n = 17), SA (n = 15), and SE (n = 18) groups. Two-proportion Z test, **p < 0.01.
(E) Western blot analysis of BRG1-pS1382 in P60 cortices after six swimming trials.
(F) qRT-PCR measurement of ARG expression in the cortices of WT, SA, and SE mice after six swimming trials (n = 5–7).
(G) Representative images of c-FOS-stained paraventricular nucleus (PVN) regions of brain sections of mice before and after swim test. Scale bars, 200 µm. Quantification of c-FOS+ cells in PVN is shown below (n = 3).
(H) Quantification of c-FOS+ cells in the somatosensory cortex before and after swim test (n = 3). ANOVA post hoc t test (A, B, and F–H), *p < 0.05, **p < 0.01.
(I) Model of BRG1 phosphorylation regulating neuronal activity-induced enhancer activation. In resting neurons, the basal binding of unphosphorylated BRG1, together with the repressive NuRD complex, prevents the activation of enhancers of a group of ARGs. There is a low level of cohesin at inactive enhancers and no E-P looping. Upon neuronal stimulation, rapidly increased H3K27Ac and activated transcription factors (TFs*) at enhancers recruit more BRG1/BAF to enhancers. Active Ca2+ signaling also induces BRG1 S1382 phosphorylation, which leads to the local dissociation of the NuRD complex, as well as an increase of cohesin-mediated E-P lopping. Together, these events lead to further increase of H3K27Ac and RNA pol II recruitment, expression of eRNAs, and ultimately ARG mRNA expression.
In a Y maze reversal learning swimming test, both SA and SE mice learned to find the correct targets in the initial and reversal learning phases with frequencies similar to that of WT mice (Figure 7C). In the first several trials on the first day of swimming training, all mice started swimming shortly after being put in water. After several rounds of swimming over a time period of about 1–2 h, some mice became immobile for longer than 30 s in subsequent trials, possibly as an adaptation to swimming stress. The number of mice that displayed immobile behaviors was significantly different among the different genotypes. SE mice showed the highest percentage with this phenotype, whereas SA mice showed the lowest percentage (Figure 7D). We examined somatosensory cortices of the WT mice and observed increased BRG1-pS1382 at 1 h after swimming stress, which then decreased gradually (Figure 7E). In the cortex, using qRT-PCR, we observed a higher induction of IEGs, including c-Fos, Arc, and Egr1, in SE neurons upon swimming stress than in WT and SA neurons, and an impaired induction of IEGs and Bdnf in SA neurons compared with in WT and/or SE neurons (Figure 7F). Using immunostaining, we examined c-FOS protein expression in various regions important for stress response at 1 h after the swimming test. We observed consistently higher c-FOS expression in SE brains in regions such as the hypothalamus paraventricular nucleus (PVN) and somatosensory cortex than in the same regions in WT brains, whereas SA brains expressed less c-FOS than their WT counterparts (Figures 7G and 7H). Thus, the elevated basal c-Fos enhancer activities in SE brains likely resulted in a stronger induction of c-Fos expression, whereas c-Fos induction was impaired in SA brains without BRG1 phosphorylation. Thus, BRG1 phosphorylation potentiates ARG enhancers for induction during neuronal responses to stress and stimulation.
DISCUSSION
In this study, we uncovered molecular mechanisms underlying the function of BRG1 in enhancer activation in response to neuronal activities (Figure 7I). Neuronal depolarization-triggered Ca2+ signaling likely activates BRG1 function in ARG induction in two aspects: increasing the recruitment of BRG1 to inducible enhancers and inducing S1382 phosphorylation. We demonstrated that nonphosphorylated BRG1 helps maintain enhancers in an inactive state under basal conditions, and that BRG1 phosphorylation in response to neuronal activities facilitates the activation of these signaling-inducible enhancers. BRG1 phosphorylation does not affect BRG1 binding to DNA but does affect its interactions with important transcription co-factors, which could contribute to the transcription outcomes of ARGs. In addition, we observed defects in stress-induced ARG expression and anxiety-like behaviors in BRG1 phospho-mutant mice. Thus, we revealed another level of regulation of BRG1 responsible for fine-tuning signal-induced enhancer activation and gene expression, which could be important for BRG1 function in neural development and plasticity.
We demonstrated that an interaction between the BRG1 bromodomain and H3K27Ac contributes to the activity-induced recruitment of BRG1 to enhancers. A specific and precise targeting of BAF complexes to enhancers is likely dependent on the coordinated work of BAF interacting transcription factors, co-factors, and chromatin features. At enhancers, BAF complexes have context-dependent functions and can act as activators or as repressors depending on the subunit composition, chromatin environment, and interacting transcription co-factors (Hodges et al., 2016; Kadoch, 2019; Wu, 2012; Wu et al., 2009). In our study, we took advantage of the relatively synchronized ARG induction system to evaluate BRG1 function in enhancer activation. We showed that although BRG1 binds extensively to activity-induced enhancers, only a subset of ARGs were affected by BRG1 deletion or phosphorylation mutations. It is possible that multiple pathways contribute to the activation of ARGs, and that BRG1 is one of the modulators fine-tuning the enhancer activities. Alternatively, BRG1 may employ multiple mechanisms regulating target genes, as we measured a combined effect on gene expression resulting from a BRG1 mutation. A previous study showed that the inhibition of BRG1 in cultured cortical neurons resulted in the induction of c-Fos at a lower concentration of KCl (Qiu and Ghosh, 2008). A recent study also showed that the deletion of a neuron-specific BAF subunit BAF53b/ACTL6B led to the derepressed expression of several ARGs at the basal condition (Wenderski et al., 2020). Our observation that phosphomimic BRG1 in vivo failed to keep the c-Fos enhancer inactive is consistent with these previous reports (Figure 6). In cultured neurons, BRG1 can function as an activator or a repressor for ARGs (Figure S1A). The repression of a subset of ARGs by BRG1 occurs at both basal and induced conditions, and thus likely through alternative neuronal activity-independent repression mechanisms.
In this study, we discovered that BRG1 plays a central role in coordinating the interactions of transcription co-factors at enhancers to maintain precise activities under basal and activated conditions. Brg1 deletion in activated neurons reduced cohesin binding, E-P looping, H3K27Ac levels, RNA pol II recruitment, and eRNA expression levels at inducible enhancers (Figure 2). H3K27Ac is an important indicator of enhancer activities. BRG1 recruitment relies on the rapid increase of H3K27Ac, whereas BRG1 deletion led to decreased H3K27Ac (Figures 1 and 2). BRG1 may regulate enhancer H3K27Ac levels either directly or indirectly. BAF complexes could directly recruit more p300 to increase local H3K27Ac levels as previously reported (Alver et al., 2017). In this study, we showed that BRG1 phosphorylation facilitates the departure of the NuRD complex, which could increase H3K27 levels by reducing local HDAC levels. Alternatively, BRG1 may affect H3K27Ac levels through eRNA expression, because it has been shown that eRNA stimulates p300/CBP HAT activities (Bose et al., 2017).
We identified a dynamic BRG1 phosphorylation site at S1382 that affects BRG1 interactions with several transcription co-factors. We showed that the interaction of BRG1 with the NuRD complex was reduced by BRG1-pS1382 (Figures 5C and 5D). The NuRD complex is an important co-repressor that has been shown to bind to enhancers and to antagonize BAF complexes to fine-tune enhancer activities (Bornelö v et al., 2018; Bracken et al., 2019; Gao et al., 2009). In cerebellar granule neurons, NuRD complexes repress ARG expression after activation (Yamada et al., 2014; Yang et al., 2016). We showed that NuRD subunits were present at ARG regulatory regions and decreased upon neuron depolarization in a BRG1-pS1382-dependent manner (Figures 5E and 5F). Therefore, phosphorylation-mediated dynamic interactions of BRG1 with NuRD complexes could underlie the dual functions of BRG1 in repressing basal levels and activating signaling-induced ARGs (Figure 7I). Another important enhancer regulator that displayed differential interaction with BRG1 upon phosphorylation is cohesin subunit SMC3. Interestingly, although Brg1 deletion led to decreased activity-induced cohesin binding to enhancers, the interaction of BRG1 with cohesin was reduced by neuronal activation and by BRG1-pS1382 (Figures 5C and 5D). Although the possibility that cohesin plays a repressor role at enhancers at basal conditions could not be excluded, BRG1 more likely regulates cohesin recruitment to enhancers through an indirect mechanism. One possibility is that the dynamic and low-affinity BRG1-cohesin interaction may be important for keeping a pool of dynamic cohesin. Cohesin dynamics have been shown to be important for their function in mediating E-P interactions (Kueng et al., 2006; Liu et al., 2021). The reduced interactions with BRG1 upon depolarization may increase cohesin dynamics and change cohesin binding to E-P regions. Future studies will be needed to investigate how BRG1 phosphorylation affects the activities of cohesin and other enhancer regulators.
BRG1 has been shown to be phosphorylated during mitosis, which correlates with its inactivation and dissociation from condensed chromatin (Muchardt et al., 1996; Sif et al., 1998). Recent studies showed that Casein kinase 2 phosphorylates BRG1 during mitosis at multiple sites (Padilla-Benavides et al., 2017, 2020). During myogenesis, phosphorylation of BRG1 by PKCβ1 and dephosphorylation by calcineurin regulate gene expression (Nasipak et al., 2015). In Drosophila, phosphorylation of BRM by cyclin-dependent kinases is necessary for cell proliferation and the development of wing epithelium (Roesley et al., 2018). In addition, BRG1 has been shown to be phosphorylated upon DNA-damage signaling by the kinase ATM, resulting in the binding of the ATPase to γ-H2AX-containing nucleosomes (Kwon et al., 2015). Besides BRG1, BAF60 (Simone et al., 2004), PHF10/BAF45a, and BAF155 could also be phosphorylated in different cell types (Brechalov et al., 2016; Foster et al., 2006). We found that BRG1-pS1382 in postmitotic neurons could be induced by multiple types of stimulation. We further showed that BRG1-pS1382 is regulated by CaMKII. In addition to CaMKII in neurons, it is possible that other kinases and phosphatases could regulate BRG1 phosphorylation to modulate enhancer activities in response to various signals and cell-cycle progression during normal development and in cancers.
Finally, using knockin mice, we demonstrated that phosphomimic and non-phosphorylatable BRG1 mutations both led to abnormal neuronal responses to stress, changes in ARG expression, and anxiety-like behaviors, indicating the importance of BRG1 phosphorylation for normal neuronal development and functions in vivo. Brg1 phospho-mutant mice were grossly normal, indicating that BRG1 phosphorylation is not required for overall brain development. The behavioral defects could be caused by alterations in ARG expression in neurons directly or by altered neural circuits or abnormal neuronal activities that result from impaired ARG expression during development. We observed that both SA and SE mice displayed anxiety-like behaviors (Figures 7A and 7B). These behaviors could be caused by altered ARG expression and/or developmental adaptations in the phospho-mutant mice. On the other hand, the differential swimming-induced ARG expression in the brain of phospho-mutant mice corelates with BRG1 phosphorylation states and their stress-response behaviors (Figures 7D–7H), suggesting a direct function of BRG1-pS1382 in ARG activation in response to stress. It would be important to further characterize the Brg1 phospho-mutant mice to determine the short-term and long-term effects of BRG1 phosphorylation and ARG expression changes.
In summary, this study not only demonstrated that BRG1 functions to coordinate the chromatin regulation of enhancer activities but also identified a signaling pathway that regulates ARG expression through BRG1 phosphorylation. These functions are important for normal neural responses to stress. Both Brg1 mutations and altered BRG1 phosphorylation may contribute to human neurological and psychiatric diseases.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| J1 (BRG1/BRM) | Khavari et al., 1993 | N/A |
| H3K27Ac | Abcam | Cat# ab4729, RRID: AB_2118291 |
| RNA pol II | Abcam | Cat# ab817, RRID:AB_306327 |
| GATAD2B | Novus | Cat# NB100–60646, RRID:AB_922172 |
| HDAC2 | Thermo Fisher Scientific | Cat# PA1–861, RRID:AB_2118520 |
| STAG2 | Gift from Dr. Hongtao Yu, UTSW | N/A |
| BRG1(G7) | Santa Cruz Biotechnology | Cat# sc-17796, RRID:AB_626762 |
| SMC3 | Santa Cruz Biotechnology | Cat# sc-376352, RRID:AB_10989741 |
| BAF53b | Wu et al., 2007 | N/A |
| BAF170 | Wang et al., 1996 | N/A |
| GAPDH | Sigma-Aldrich | Cat# G9545, RRID:AB_796208 |
| H3S10P | Cell Signaling Technology | Cat# 9701, RRID:AB_331535 |
| Rabbit BRG1-S1382 phospho-specific antibody | Generated against peptide antigen VDYSD(pS)LTEKQ by Ab-Mart (Shanghai) | N/A |
| c-FOS | Cell Signaling Technology | Cat# 2250, RRID:AB_2247211 |
| Chemicals, peptides, and recombinant proteins | ||
| PolyJet | Signagen | Cat# SL100688 |
| Poly-L-ornithine | Sigma Aldrich | Cat# P4957 |
| Fibronectin | Sigma Aldrich | Cat# F1141 |
| Neurobasal medium | GIBCO | Cat# 21103049 |
| B27 plus supplement | GIBCO | Cat# A3582801 |
| TTX | Tocris | Cat# 1078 |
| APV | Tocris | Cat# 0106 |
| PFI-3 | Tocris | Cat# 5072 |
| C646 | Tocris | Cat# 4200 |
| Nimodipine | Tocris | Cat# 0600 |
| KN93 | Tocris | Cat# 1278 |
| U0126 | Tocris | Cat# 1144 |
| TRIzol reagent | ThermoFisher Scientific | Cat# 15596018 |
| iScript cDNA synthesis kit | Bio-Rad | Cat# 1708891 |
| PFA | ELECTRON MICROSCOPY SCIENCES | Cat# 15710 |
| DSG | Pierce | Cat# A35392 |
| Protein A-coated magnetic Dynabeads | Invitrogen | Cat# 10002D |
| 2X Sample Buffer | Bio-Rad | Cat# 1610737 |
| FastFlow Sepharose Protein A beads | GE Healthcare | Cat# GE17–5280-01 |
| SacI | New England Biolabs | Cat# R0156S |
| BgIII | New England Biolabs | Cat# R0144S |
| NcoI | New England Biolabs | Cat# R0193S |
| T4 DNA ligase | New England Biolabs | Cat# M0202S |
| Critical commercial assays | ||
| Illumina RNA-Seq Preparation Kit | Illumina | Cat# RS-122–2001 |
| High-sensitivity colorimetric ATPase assay kit | Innova Biosciences | Cat# 601–0120 |
| Silver staining kit | Invitrogen | Cat# LC6070 |
| Nextra kit | Illumina | Cat# FC-121–1030 |
| Deposited data | ||
| Genomic Data (combined) | NCBI | GEO: GSE174585 |
| ATAC-seq | NCBI | GEO: GSE174579 |
| BRG1 CHIP-seq | NCBI | GEO: GSE174581 |
| RNA-seq (wt, Brg1cko) | NCBI | GEO: GSE174583 |
| RNA-seq (WT, SA, SE) | NCBI | GEO: GSE174584 |
| Experimental models: Cell lines | ||
| Human: HEK293T cells | ATCC | Cat# CRL-1573 |
| Human: SW13 cells | ATCC | Cat# CCL-105 |
| Mouse: Primary Cortical Neuron | Wu et al., 2007; Zhang et al., 2015 | N/A |
| Experimental models: Organisms/Strains | ||
| Mouse: Brg1F/F BAF53b-Cre (Brg1cko), | Zhang et al., 2015 | N/A |
| Mouse: Brg1-SA (SA), Brg1-SE (SE) | This paper | N/A |
| Oligonucleotides | ||
| See Table S4 | N/A | |
| Recombinant DNA | ||
| pSin4-EF2-IRES-Puro-BRG1 | Zhan et al., 2011 | N/A |
| pSin4-EF2-IRES-Puro-BRM | Zhan et al., 2011 | N/A |
| pSin4-EF2-IRES-Puro-BRG1-SA | This paper | N/A |
| pSin4-EF2-IRES-Puro-BRG1-SE | This paper | N/A |
| pSin4-EF2-IRES-Puro-BRG1-KR | Zhan et al., 2011 | N/A |
| Software and algorithms | ||
| Kallisto program | Bray et al., 2016 | https://pachterlab.github.io/kallisto/ |
| EdgeR program | McCarthy et al., 2012 | https://bioconductor.org/ |
| Mascot search engine (version 2.3) | Perkins et al., 1999 | https://www.matrixscience.com/ |
| Bowtie 2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| MACS2 | Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| BWA aligner | Li and Durbin, 2009 | https://github.com/lh3/bwa |
| SICER | Zang et al., 2009 | https://zanglab.github.io/SICER2/#introduction |
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jiang Wu (jiang9.wu@utsouthwestern.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
The accession number for the next generation sequencing raw data reported in this paper is NCBI Gene Expression Omnibus: GSE174585. No new code was generated in this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Brg1F/F BAF53b-Cre (Brg1cko) and control embryos (Zhan et al., 2015; Zhang et al., 2015) were generated from crosses between Brg1F/+ BAF53b-Cre and Brg1F/F mice and were maintained on a mixed genetic background. Brg1-SA (SA) and Brg1-SE (SE) mice were generated by injecting Cas9 recombinant protein, gRNA targeting the 5′-ccaCCGCAAGGAGGTAGACTACA-3′ site upstream of the codon encoding S1382 in BRG1, and repair single-stranded DNA oligonucleotides into C57BL/6J pronuclei. Progenies were screened by sequencing the target sites (Figure S5A) and backcrossed to wild-type C57BL/6J mice to obtain heterozygous mice and subsequent homozygotes. All mice are housed at UT Southwestern Medical Center Animal Facility. All procedures were performed in accordance with the IACUC-approved protocols. Cortical neurons were isolated from E16.5 embryos for primary cultures. Mice at P5 were used for gene expression in the cortex. Light stimulation experiments were performed on mice at P21. Behavior tests were performed on 2–6 months old mice. In all animal experiments, both males and females were used, and there were no significant differences found between genders.
Cell lines
HEK293T cells and SW13 cells were obtained from ATCC and cultivated by a standard method in DMEM (Dulbecco Modified Eagle Medium) and L-15 medium, respectively. The media were supplemented with 10% fetal bovine serum (Sigma), 2 mM glutamine, and 1× antibiotic-antimycotic (Thermo Fisher Scientific). Primary cortical neurons were cultured from E16.5 embryos as previously described (Wu et al., 2007; Zhang et al., 2015). Dissociated cortical neurons were plated on poly-L-ornithine- and fibronectin-coated wells. Culture media contained neurobasal plus with B27 plus supplement (GIBCO). The cells were kept at 37°C in a humidified atmosphere containing 5% CO2.
METHOD DETAILS
Plasmid construction, virus preparation, and transfection or infection
The constructs for expression of BRG1, BRM, BRG1-SA, BRG1-SE, and BRG1-KR were generated by inserting the coding regions of these genes into pSin4-EF2-IRES-Puro lentiviral vectors (Zhan et al., 2011). Lentiviruses were propagated in HEK293T cells according to a previously described procedure (Zhan et al., 2011), followed by ultracentrifugation for concentration. Attached cultured SW13 and primary neurons were infected at an MOI of 5 for 24–48 h. PolyJet (Signagen) was used for plasmid transfection of cultured cells.
Cortical neuron culture treatments
Cortical cultures were infected with lentiviruses on 4 days in vitro (div) and analyzed at 6 div. For depolarization, the cultures were pretreated with TTX (1 µM) and APV (100 µM) to inhibit spontaneous activation on 5 div and 50 mM KCl was added to the cultures for 1 to 6 h on 6 div as reported (Flavell et al., 2006). For inhibitor treatment, PFI-3 (final concentration 20 µM), C646 (25 µM), or DMSO (as a control) was added 4 h before KCl treatment on 6 div. Nimodipine, KN93, and U0126 treatments were performed with indicated concentrations on 5 div before KCl treatment on 6 div.
RT-PCR and q-PCR
RNAs from cells or ground tissues were extracted with TRIZOL (Invitrogen). cDNAs were synthesized by reverse transcription using iScript (Bio-Rad), followed by PCR or quantitative PCR analysis. A Bio-Rad real-time PCR system (C1000 Thermal Cycler) was used for quantitative PCR. Levels of RNAs of interest were normalized to GAPDH mRNA. Bar graphs shown are representative of experiments performed in triplicate unless otherwise indicated. The experiments were repeated at least three times. Dot graphs represent results from individual samples. Standard errors were calculated according to a previously described method (Zhan et al., 2011). The sequences of all the primers are listed in Table S4.
RNA-seq
Cultured cortical neurons were treated with KCl as described above (50 mM, 1 h or 6h). Total RNAs were extracted, and RNA-seq libraries were prepared using the Illumina RNA-Seq Preparation Kit and sequenced on a HiSeq 2500 sequencer at the University of Texas Southwestern Sequencing Core Facility. The expression levels of genes were quantified by a Kallisto program (Bray et al., 2016), and the differentially expressed genes were detected by an EdgeR program (McCarthy et al., 2012). Genes with a count-per-Million (CPM) less than 1 in more than 2 samples were excluded. The differentially expressed genes at KCl induced conditions with a p < 0.05 were selected as BRG1-regulated genes. The differences of transcripts per million (TPM) among WT, SA, and SE samples were compared as log2TPM(SA/WT) or log2TPM(SE/WT).
ChIP experiments and ChIP-seq analyses
ChIP experiments were performed as described previously (Shi et al., 2016; Zhan et al., 2011). Culture cortical neurons were treated with or without 50 mM KCl for one hour. Dounced tissue or dissociated cells were crosslinked with PFA or double crosslinked with DSG (Pierce), and nucleic acids were sonicated into fragments of 200–500 bp. Antibodies used were against BRG1/BRM (J1) (Khavari et al., 1993), H3K27Ac (Abcam, ab4729), RNA pol II (Abcam, ab817), GATAD2B (NB100–60646, Novus Biologicals), HDAC2 (PA-1–861, Thermo Fisher Scientific), and STAG2 (a gift from Dr. Hongtao Yu, UTSW). J1 antibody has been used previously for BRG1 ChIP-seq analyses (Ho et al., 2009; Yu et al., 2013). Precipitated DNA was purified and subjected to either real-time PCR (Table S4) or next generation sequencing. NEBNext ChIP-Seq Sample Prep Master Mix Set 1 was used for library generation, and a Hiseq 2500 sequencer was used for sequencing at the UT Southwestern Medical Center Sequencing Core Facility. Short reads were mapped to UCSC reference mouse genome (GRCm38/mm10) with BWA aligner (Li and Durbin, 2009) and then SICER was used to detect the BRG1-binding regions (Shi et al., 2016; Zang et al., 2009). Default parameter settings with three 200-bp windows were used to calculate the enrichment of BRG1-binding regions. The corresponding input sample was used as control. Duplicate reads were removed before peak calling by SICER. Statistically significant peaks (FDR < 0.05) enriched in the BRG1-ChIP sample relative to its corresponding input sample were annotated for genomic location. H3K27Ac ChIP-seq data was downloaded from NCBI GEO:-GSE60192 (Malik et al., 2014), and aligned with UCSC reference mouse genome (GRCm38, Dec. 2011) with BWA, following peak calling by MACS2 (Zhang et al., 2008) with default parameter to call narrow peaks. DeepTools (Ramírez et al., 2016) was used to illustrate the reads distribution from ChIP-seq in the enhancers and their 10-kb flanking regions. BRG1 ChIP and H3K27Ac ChIP peaks were overlapping if there was a 1 bp overlap of the peak regions. The control random regions are of the average BRG1 peak size and the same number as BRG1 peaks that are randomly selected in the genome.
Immunoprecipitation and western blot
Cortical tissues or cultured cortical cells were harvested and lysed in buffer A (25 mM Tris, pH 7.5, 25 mM KCl, 5 mM MgCl, 0.1% NP-40, 10% glycerol). Nuclear pellets were resuspended in RIPA buffer (25 mM Tris, pH 7.5, 150 mM NaCl, 0.05% SDS, 1% Triton X-100, 0.5% SDC) to prepare nuclear extracts. The anti-BRG1 J1 antibody was pre-incubated with Protein A-coated magnetic Dynabeads (Invitrogen) before adding to nuclear extracts. Samples were incubated at 4°C overnight, beads were washed with RIPA buffer four times. Precipitated proteins were eluted by boiling in 2X Sample Buffer (Bio-Rad) before SDS-PAGE and western blot analysis. Protease and phosphatase inhibitors were used in all immunoprecipitation buffers. For immunoblotting, cell lysates or precipitated proteins were separated on SDS-PAGE gels. Antibodies used were against BRG1 (G7, Santa Cruz Biotechnology), BRG1/BRM (J1), SMC3 (E3, Santa Cruz Biotechnology), GAPDH (G9545, Sigma), BAF53b (Wu et al., 2007), BAF170 (Wang et al., 1996), GATAD2B (NB100–60646,Novus Biologicals), HDAC2 (PA-1–861, Thermo Fisher Scientific), H3S10P (#9701, Cell Signaling). HRP-conjugated (Jackson Immunology) or IRDye-conjugated secondary antibodies (LI-COR) were used in western blot. Rabbit BRG1-S1382 phospho-specific antibody was generated against peptide antigen VDYSD(pS)LTEKQ by Ab-Mart (Shanghai). The specificity of the antibody was confirmed with BRG1 S1382 mutants (Figure S5C).
ATPase activity assay
ATPase activity was measured using a high-sensitivity colorimetric ATPase assay kit following the manufacturer’s instruction (Innova Biosciences). Briefly, affinity-purified BAF complexes as described above were suspended in 25 mM Tris, pH 7.5 and incubated with the reaction buffer (100 mM Tris, pH 7.4, 5 mM MgCl2, and 1 mM ATP) in the presence of 0.5 µg plasmid DNA for 15 min at 37°C. The reaction was stopped by adding the PiColorLock mix. The amount of inorganic phosphate released was quantified colorimetrically at 620 nm.
Affinity purification of BAF complexes and mass spectrometry analysis
Nuclear extracts are prepared as describe above from 6-div mouse cortical neurons with or without KCl treatment for 1 h. J1 antibody or rabbit IgG control was cross-linked to GE FastFlow Sepharose Protein A beads as previously described (Lessard et al., 2007). BAF complexes were affinity-purified from nuclear extracts (600 µg) using 20 µg antibody-conjugated beads. Protein complexes were eluted with 0.1 M acetic acid. After silver staining (LC6070, Invitrogen) to confirm purity, the eluted BAF complexes and associated proteins from gel slices were subjected to LC-MS/MS, which was performed by PTM Biolabs. Protein mixes were digested in the gel with trypsin, and peptides were extracted. The peptides were separated and then analyzed using a Q Exactive Plus Hybrid Quadrupole-Orbitrap mass spectrometer (ThermoFisher Scientific). The resulting MS/MS data were processed using the Mascot search engine (version 2.3) (Perkins et al., 1999). Tandem mass spectra were searched against SwissProt database concatenated with reverse decoy database. Trypsin/P was specified as cleavage enzyme allowing up to 3 missing cleavages. Mass error was set to 10 ppm for precursor ions and 0.02 Da for fragment ions. Phosphorylations of Ser, Thr, and Tyr were specified as variable modifications. An FDR of less than 1% was used to filter the identified peptides. All the other parameters in Mascot were set to default values. The purification yielded 272 proteins in the basal condition and 305 proteins in depolarized neurons. Numbers of BRG1 peptide identified were 277 and 270, respectively (Table S2). Numbers of all phosphorylated peptides identified were 35 and 40, respectively (Table S3).
Chromosome conformation capture
Chromosome conformation capture (3C) experiments were performed as described previously (Naumova et al., 2012). Extracts of cultured neurons or homogenized mouse cortices collected at post-natal day 5 (P5) were crosslinked with 1% formaldehyde for 10 min; the reaction was stopped by addition of 0.125 M glycine. Crosslinked cells were lysed within buffer (10 mM Tris, pH 8.0, 10 mM NaCl, 0.2% NP-40) containing protease inhibitor for 15 min at 4°C. Nuclei were resuspended in the 1× NEB restriction enzyme buffer, and 1% SDS was added. After incubating for 10 min at 65°C, 1% Triton X-100 was added. Samples were digested with SacI for c-Fos analysis or with BgIII and NcoI for Arc analysis at 37°C overnight. The restriction enzyme was inactivated with 1.6% SDS for 30 min at 65◦C followed by incubation in 1% Triton X-100 for 1 h at 37°C. Ligation was performed using T4 DNA ligase for 4 h at 16°C and then 30 min at room temperature. Samples were treated with 200 µg proteinase K at 65°C overnight for reverse crosslinking, followed by RNase treatment (1 µg/ml) for 1 h at 37°C. After phenol-chloroform extraction and ethanol precipitation, DNA was analyzed by qPCR using primers described previously (Joo et al., 2016) (Table S4). Two bacterial artificial chromosome clones containing the c-Fos and Arc genes were used as negative controls (Joo et al., 2016).
ATAC-sequencing
Cultured wild-type and Brg1cko cortical neurons with or without KCl treatment for 1 h were harvested in PBS. Neurons (50,000 cells/sample, n = 2 in each condition) were lysed with lysis buffer (10 mM Tris, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40). The transposition reaction was performed using Tn5 transposase from the Nextra kit (Illumina, FC-121–1030) at 37°C for 30 min. Transposed DNA samples were purified using a QIAGEN MinElute Kit (catalog no. 28004) followed by PCR amplification. Sequencing libraries were generated using Ad1_noMX with barcoded primers and were amplified (7–9 cycles) before DNA purification and concentration. All samples were sequenced using a Hiseq 2500 sequencer with 75-bp paired end reads and 60 million reads were obtained for each library. The in-house script was developed to remove the adaptor and extract the genomics sequences from raw sequencing reads. Bowtie 2 (Langmead and Salzberg, 2012) was used to align all reads to UCSC reference mouse genome (GRCm38/mm10), and open-chromatin regions were called by MACS2 (Zhang et al., 2008) with parameter of ‘‘callpeak–nomodel–shift −100–extsize 200.’’
Behavioral tests
All experiments in this study were performed in the Behavior Core Facility and approved by the IACUC at UT Southwestern Medical Center. Mice were housed with food and water available ad libitum with a 12-h light/dark cycle, and all behavior testing occurring during the light cycle. Behavioral tests were conducted by testing less stressful behaviors before more stressful ones. The sequence of behavior tests was light/dark box test, followed by elevated maze test, and then Y-maze swimming reversal learning test.
Light/Dark box test:
Mice were placed in the dark chamber of a custom-made light/dark box and allowed to habituate for 2 min. After opening of the divider separating the dark side from the light side, mice were allowed to freely explore both chambers for 10 min while monitored from above by a video camera connected to a computer running video tracking software (Ethovision 3.0, Noldus) to measure the time, frequency, and activity (travel distance) in each chamber. Each chamber of the box was 25 × 26 cm with 1,700 lux on the light side and ~0.1 lux on the dark side.
Elevated maze test:
Mice were placed in the center of a white Plexiglas elevated plus maze (each arm 33 cm long and 5 cm wide with 15-cm-high black Plexiglas walls on closed arms) and allowed to explore for 5 min. The test was conducted in dim white light (~7 lux). Data on duration of time spent in each arm and entering frequencies were recorded and analyzed using the CleverSys TopScan software.
Y maze reversal learning:
The Y maze (each arm 7 cm wide and 20 cm long) was filled with water (approximately 24°C) and a small amount of non-toxic paint. One arm of the maze was designated the Start Arm, and the other two arms the Choice Arms. One of the Choice Arms had a platform located at the end, submerged 1–2 cm below the water level. Mice were placed into the Start Arm of the maze and allowed to swim to locate the hidden platform. Once the mouse found the platform, it was removed from the maze. Mice that did not locate the platform within 60 s were gently guided to the platform and then removed from the maze. Mice were placed briefly on a paper towel to dry after removal from the maze. Each mouse was placed in the maze 15 times per day with 30 min between trials. The mice were trained for 3 days. On the next day, the platform location was moved to the opposite arm, and each mouse was tested an additional 15 times. The number of times the mouse found the platform each day was recorded. During the experiment, certain mice remained immobile in the water unless stimulated. We defined immobile mice as those that floated but did not swim for longer than 30 s after being put in water for any trial. Immuno-fluorescent staining of c-FOS was performed using a c-FOS antibody (9F6, Cell Signaling) on brain cryosections of mice that had performed six trials in 1 h. c-FOS-positive cells were counted from paraventricular nucleus and cortical regions from three mice (3 sections/brain), and averages are reported. ARG expression were analyzed by qRT-PCR using cortices dissected from mice that had performed six trials at 1 h and 5 h afterward.
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise specified, at least three independent experiments were performed and each condition was analyzed in triplicate. Bar graphs shown are representative experiments. Dot graphs represent results from individual samples. Data are expressed as means ± standard deviation. Statistical analysis was performed by either analysis of variance with ANOVA post hoc t test for multiple comparisons or a two-tailed unpaired Student’s t test. A p value of < 0.05 was considered significant. Statistical details of experiments can be found in the figure legends.
Supplementary Material
Highlights.
BRG1 binds neuronal activity-induced enhancers and regulates enhancer activities
Neuronal activities induce BRG1 phosphorylation at S1382, which depends on CAMKII
BRG1 phosphorylation regulates its interactions with transcription co-factors
BRG1 phosphorylation modulates inducibilities of c-Fos enhancers and ARGs in vivo
ACKNOWLEDGMENTS
We thank Dr. Taekyung Kim for providing 3C reagents, Dr. Hongtao Yu for providing the anti-STAG2 antibody, and Dr. Shari Birnbaum and the UTSW Rodent Behavior Facility for aiding in the behavior tests. We thank Huaxia Dong for technical help and Michael Zhan for reading the manuscript. This work was supported by grants from the NIH (R01NS096068 and R21NS104596 to J.W.) and Welch Foundation (I-1940–20170325 to J.W.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109357.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alexander JM, Hota SK, He D, Thomas S, Ho L, Pennacchio LA, and Bruneau BG (2015). Brg1 modulates enhancer activation in mesoderm lineage commitment. Development 142, 1418–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alver BH, Kim KH, Lu P, Wang X, Manchester HE, Wang W, Haswell JR, Park PJ, and Roberts CW (2017). The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat. Commun. 8, 14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornelöv S, Reynolds N, Xenophontos M, Gharbi S, Johnstone E, Floyd R, Ralser M, Signolet J, Loos R, Dietmann S, et al. (2018). The Nucleosome Remodeling and Deacetylation Complex Modulates Chromatin Structure at Sites of Active Transcription to Fine-Tune Gene Expression. Mol. Cell 71, 56–72.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose DA, Donahue G, Reinberg D, Shiekhattar R, Bonasio R, and Berger SL (2017). RNA Binding to CBP Stimulates Histone Acetylation and Transcription. Cell 168, 135–149.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA, Crump NT, Hazzalin CA, Liszczak G, Yuan H, et al. (2010). Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem. Biol. 17, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken AP, Brien GL, and Verrijzer CP (2019). Dangerous liaisons: interplay between SWI/SNF, NuRD, and Polycomb in chromatin regulation and cancer. Genes Dev 33, 936–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Brechalov AV, Valieva ME, Georgieva SG, and Soshnikova NV (2016). [PHF10 isoforms are phosphorylated in the PBAF mammalian chromatin remodeling complex]. Mol. Biol. (Mosk.) 50, 320–326. [DOI] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, and Greenberg ME (2003). Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 302, 885–889. [DOI] [PubMed] [Google Scholar]
- Chen LF, Lin YT, Gallegos DA, Hazlett MF, Gómez-Schiavon M, Yang MG, Kalmeta B, Zhou AS, Holtzman L, Gersbach CA, et al. (2019). Enhancer Histone Acetylation Modulates Transcriptional Bursting Dynamics of Neuronal Activity-Inducible Genes. Cell Rep 26, 1174–1188.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. ; DDD Study; Homozygosity Mapping Collaborative for Autism; UK10K Consortium (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisseroth K, Mermelstein PG, Xia H, and Tsien RW (2003). Signaling from synapse to nucleus: the logic behind the mechanisms. Curr. Opin. Neurobiol 13, 354–365. [DOI] [PubMed] [Google Scholar]
- Dephoure N, Zhou C, Villén J, Beausoleil SA, Bakalarski CE, Elledge SJ, and Gygi SP (2008). A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 105, 10762–10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, and Greenberg ME (2013). Activity-dependent neuronal signalling and autism spectrum disorder. Nature 493, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Müller S, Pawson T, et al. (2012). Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 149, 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, Griffith EC, Hu LS, Chen C, and Greenberg ME (2006). Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science 311, 1008–1012. [DOI] [PubMed] [Google Scholar]
- Foster KS, McCrary WJ, Ross JS, and Wright CF (2006). Members of the hSWI/SNF chromatin remodeling complex associate with and are phosphorylated by protein kinase B/Akt. Oncogene 25, 4605–4612. [DOI] [PubMed] [Google Scholar]
- Gallo FT, Katche C, Morici JF, Medina JH, and Weisstaub NV (2018). Immediate Early Genes, Memory and Psychiatric Disorders: Focus on c-Fos, Egr1 and Arc. Front. Behav. Neurosci 12, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Lukin K, Ramírez J, Fields S, Lopez D, and Hagman J (2009). Opposing effects of SWI/SNF and Mi-2/NuRD chromatin remodeling complexes on epigenetic reprogramming by EBF and Pax5. Proc. Natl. Acad. Sci. USA 106, 11258–11263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstenberger BS, Trzupek JD, Tallant C, Fedorov O, Filippakopoulos P, Brennan PE, Fedele V, Martin S, Picaud S, Rogers C, et al. (2016). Identification of a Chemical Probe for Family VIII Bromodomains through Optimization of a Fragment Hit. J. Med. Chem 59, 4800–4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren C, Kjaergaard S, Bak M, Hansen C, El-Schich Z, Anderson CM, Henriksen KF, Hjalgrim H, Kirchhoff M, Bijlsma EK, et al. (2012). Corpus callosum abnormalities, intellectual disability, speech impairment, and autism in patients with haploinsufficiency of ARID1B. Clin. Genet 82, 248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Wu Z, Tian Y, Yu Z, Yu J, Wang X, Li J, Liu B, and Xu Y (2020). Structure of nucleosome-bound human BAF complex. Science 367, 875–881. [DOI] [PubMed] [Google Scholar]
- Helsmoortel C, Vulto-van Silfhout AT, Coe BP, Vandeweyer G, Rooms L, van den Ende J, Schuurs-Hoeijmakers JH, Marcelis CL, Willemsen MH, Vissers LE, et al. (2014). A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat. Genet 46, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, Jothi R, Ronan JL, Cui K, Zhao K, and Crabtree GR (2009). An embryonic stem cell chromatin remodeling complex, esBAF, is an essential component of the core pluripotency transcriptional network. Proc. Natl. Acad. Sci. USA 106, 5187–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges C, Kirkland JG, and Crabtree GR (2016). The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb. Perspect. Med 6, a026930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo JY, Schaukowitch K, Farbiak L, Kilaru G, and Kim TK (2016). Stimulus-specific combinatorial functionality of neuronal c-fos enhancers. Nat. Neurosci 19, 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C (2019). Diverse compositions and functions of chromatin remodeling machines in cancer. Sci. Transl. Med 11, eaay1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, et al. (2010). Mediator and cohesin connect gene expression and chromatin architecture. Nature 467, 430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khavari PA, Peterson CL, Tamkun JW, Mendel DB, and Crabtree GR (1993). BRG1 contains a conserved domain of the SWI2/SNF2 family necessary for normal mitotic growth and transcription. Nature 366, 170–174. [DOI] [PubMed] [Google Scholar]
- Kim TK, and Shiekhattar R (2015). Architectural and Functional Commonalities between Enhancers and Promoters. Cell 162, 948–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. (2010). Widespread transcription at neuronal activity-regulated enhancers. Nature 465, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A, Arakawa N, and Hirano H (2014). Mass spectrometric analysis of the phosphorylation levels of the SWI/SNF chromatin remodeling/tumor suppressor proteins ARID1A and Brg1 in ovarian clear cell adenocarcinoma cell lines. J. Proteome Res 13, 4959–4969. [DOI] [PubMed] [Google Scholar]
- Kueng S, Hegemann B, Peters BH, Lipp JJ, Schleiffer A, Mechtler K, and Peters JM (2006). Wapl controls the dynamic association of cohesin with chromatin. Cell 127, 955–967. [DOI] [PubMed] [Google Scholar]
- Kwon SJ, Park JH, Park EJ, Lee SA, Lee HS, Kang SW, and Kwon J (2015). ATM-mediated phosphorylation of the chromatin remodeling enzyme BRG1 modulates DNA double-strand break repair. Oncogene 34, 303–313. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard J, Wu JI, Ranish JA, Wan M, Winslow MM, Staahl BT, Wu H, Aebersold R, Graef IA, and Crabtree GR (2007). An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron 55, 201–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao L, McClatchy DB, Park SK, Xu T, Lu B, and Yates JR 3rd. (2008). Quantitative analysis of brain nuclear phosphoproteins identifies developmentally regulated phosphorylation events. J. Proteome Res 7, 4743–4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Liu H, Chen X, Kirby M, Brown PO, and Zhao K (2001). Regulation of CSF1 promoter by the SWI/SNF-like BAF complex. Cell 106, 309–318. [DOI] [PubMed] [Google Scholar]
- Liu NQ, Maresca M, van den Brand T, Braccioli L, Schijns MMGA, Teunissen H, Bruneau BG, Nora EP, and de Wit E (2021). WAPL maintains a cohesin loading cycle to preserve cell-type-specific distal gene regulation. Nat. Genet 53, 100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik AN, Vierbuchen T, Hemberg M, Rubin AA, Ling E, Couch CH, Stroud H, Spiegel I, Farh KK, Harmin DA, and Greenberg ME (2014). Genome-wide identification and characterization of functional neuronal activity-dependent enhancers. Nat. Neurosci 17, 1330–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning CE, Williams ES, and Robison AJ (2017). Reward Network Immediate Early Gene Expression in Mood Disorders. Front. Behav. Neurosci 11, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashtalir N, Suzuki H, Farrell DP, Sankar A, Luo J, Filipovski M, D’Avino AR, St Pierre R, Valencia AM, Onikubo T, et al. (2020). A Structural Model of the Endogenous Human BAF Complex Informs Disease Mechanisms. Cell 183, 802–817.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, and Smyth GK (2012). Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 40, 4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P, Yang F, Liu T, Mani DR, Petyuk VA, Gillette MA, Clauser KR, Qiao JW, Gritsenko MA, Moore RJ, et al. (2014). Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol. Cell. Proteomics 13, 1690–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchardt C, Reyes JC, Bourachot B, Leguoy E, and Yaniv M (1996). The hbrm and BRG-1 proteins, components of the human SNF/SWI complex, are phosphorylated and excluded from the condensed chromosomes during mitosis. EMBO J 15, 3394–3402. [PMC free article] [PubMed] [Google Scholar]
- Nasipak BT, Padilla-Benavides T, Green KM, Leszyk JD, Mao W, Konda S, Sif S, Shaffer SA, Ohkawa Y, and Imbalzano AN (2015). Opposing calcium-dependent signalling pathways control skeletal muscle differentiation by regulating a chromatin remodelling enzyme. Nat. Commun 6, 7441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumova N, Smith EM, Zhan Y, and Dekker J (2012). Analysis of long-range chromatin interactions using Chromosome Conformation Capture. Methods 58, 192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nord AS, and West AE (2020). Neurobiological functions of transcriptional enhancers. Nat. Neurosci 23, 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla-Benavides T, Nasipak BT, Paskavitz AL, Haokip DT, Schnabl JM, Nickerson JA, and Imbalzano AN (2017). Casein kinase 2-mediated phosphorylation of Brahma-related gene 1 controls myoblast proliferation and contributes to SWI/SNF complex composition. J. Biol. Chem 292, 18592–18607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla-Benavides T, Haokip DT, Yoon Y, Reyes-Gutierrez P, Rivera-Pérez JA, and Imbalzano AN (2020). CK2-Dependent Phosphorylation of the Brg1 Chromatin Remodeling Enzyme Occurs during Mitosis. Int. J. Mol. Sci 21, 923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJ, Creasy DM, and Cottrell JS (1999). Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567. [DOI] [PubMed] [Google Scholar]
- Qiu Z, and Ghosh A (2008). A calcium-dependent switch in a CREST-BRG1 complex regulates activity-dependent gene expression. Neuron 60, 775–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44 (W1), W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesley SNA, La Marca JE, Deans AJ, Mckenzie L, Suryadinata R, Burke P, Portela M, Wang H, Bernard O, Sarcevic B, and Richardson HE (2018). Phosphorylation of Drosophila Brahma on CDK-phosphorylation sites is important for cell cycle regulation and differentiation. Cell Cycle 17, 1559–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santen GW, Aten E, Sun Y, Almomani R, Gilissen C, Nielsen M, Kant SG, Snoeck IN, Peeters EA, Hilhorst-Hofstee Y, et al. (2012). Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat. Genet 44, 379–380. [DOI] [PubMed] [Google Scholar]
- Schaukowitch K, Joo JY, Liu X, Watts JK, Martinez C, and Kim TK (2014). Enhancer RNA facilitates NELF release from immediate early genes. Mol. Cell 56, 29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen P, Vivas P, Dechassa ML, Mooney AM, Poirier MG, and Bartholomew B (2013). The SnAC domain of SWI/SNF is a histone anchor required for remodeling. Mol. Cell. Biol 33, 360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Wang Q, Gu J, Xuan Z, and Wu JI (2016). SMARCA4/Brg1 coordinates genetic and epigenetic networks underlying Shh-type medulloblastoma development. Oncogene 35, 5746–5758. [DOI] [PubMed] [Google Scholar]
- Sif S, Stukenberg PT, Kirschner MW, and Kingston RE (1998). Mitotic inactivation of a human SWI/SNF chromatin remodeling complex. Genes Dev 12, 2842–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simone C, Forcales SV, Hill DA, Imbalzano AN, Latella L, and Puri PL (2004). p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat. Genet 36, 738–743. [DOI] [PubMed] [Google Scholar]
- Sinnamon JR, Torkenczy KA, Linhoff MW, Vitak SA, Mulqueen RM, Pliner HA, Trapnell C, Steemers FJ, Mandel G, and Adey AC (2019). The accessible chromatin landscape of the murine hippocampus at single-cell resolution. Genome Res 29, 857–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokpor G, Xie Y, Rosenbusch J, and Tuoc T (2017). Chromatin Remodeling BAF (SWI/SNF) Complexes in Neural Development and Disorders. Front. Mol. Neurosci 10, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son EY, and Crabtree GR (2014). The role of BAF (mSWI/SNF) complexes in mammalian neural development. Am. J. Med. Genet. C. Semin. Med. Genet 166C, 333–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Shin J, Zhong C, Wang S, Roychowdhury P, Lim J, Kim D, Ming GL, and Song H (2017). Neuronal activity modifies the chromatin accessibility landscape in the adult brain. Nat. Neurosci 20, 476–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsurusaki Y, Okamoto N, Ohashi H, Kosho T, Imai Y, Hibi-Ko Y, Kaname T, Naritomi K, Kawame H, Wakui K, et al. (2012). Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat. Genet 44, 376–378. [DOI] [PubMed] [Google Scholar]
- Tyssowski KM, DeStefino NR, Cho JH, Dunn CJ, Poston RG, Carty CE, Jones RD, Chang SM, Romeo P, Wurzelmann MK, et al. (2018). Different Neuronal Activity Patterns Induce Different Gene Expression Programs. Neuron 98, 530–546.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Houdt JK, Nowakowska BA, Sousa SB, van Schaik BD, Seuntjens E, Avonce N, Sifrim A, Abdul-Rahman OA, van den Boogaard MJ, Bottani A, et al. (2012). Heterozygous missense mutations in SMARCA2 cause Nicolaides-Baraitser syndrome. Nat. Genet 44, 445–449, S1. [DOI] [PubMed] [Google Scholar]
- Vierbuchen T, Ling E, Cowley CJ, Couch CH, Wang X, Harmin DA, Roberts CWM, and Greenberg ME (2017). AP-1 Transcription Factors and the BAF Complex Mediate Signal-Dependent Enhancer Selection. Mol. Cell 68, 1067–1082.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Xue Y, Zhou S, Kuo A, Cairns BR, and Crabtree GR (1996). Diversity and specialization of mammalian SWI/SNF complexes. Genes Dev 10, 2117–2130. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wong RH, Tang T, Hudak CS, Yang D, Duncan RE, and Sul HS (2013). Phosphorylation and recruitment of BAF60c in chromatin remodeling for lipogenesis in response to insulin. Mol. Cell 49, 283–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenderski W, Wang L, Krokhotin A, Walsh JJ, Li H, Shoji H, Ghosh S, George RD, Miller EL, Elias L, et al. (2020). Loss of the neural-specific BAF subunit ACTL6B relieves repression of early response genes and causes recessive autism. Proc. Natl. Acad. Sci. USA 117, 10055–10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, and Greenberg ME (2011). Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb. Perspect. Biol 3, a005744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RR, Kwon YG, Taing M, Lawrence DS, and Edelman AM (1998). Definition of optimal substrate recognition motifs of Ca2+-calmodulin-dependent protein kinases IV and II reveals shared and distinctive features. J. Biol. Chem 273, 3166–3172. [DOI] [PubMed] [Google Scholar]
- Wong RO, and Ghosh A (2002). Activity-dependent regulation of dendritic growth and patterning. Nat. Rev. Neurosci 3, 803–812. [DOI] [PubMed] [Google Scholar]
- Wu JI (2012). Diverse functions of ATP-dependent chromatin remodeling complexes in development and cancer. Acta Biochim. Biophys. Sin. (Shanghai) 44, 54–69. [DOI] [PubMed] [Google Scholar]
- Wu JI, Lessard J, Olave IA, Qiu Z, Ghosh A, Graef IA, and Crabtree GR (2007). Regulation of dendritic development by neuron-specific chromatin remodeling complexes. Neuron 56, 94–108. [DOI] [PubMed] [Google Scholar]
- Wu JI, Lessard J, and Crabtree GR (2009). Understanding the words of chromatin regulation. Cell 136, 200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Wong J, Moreno GT, Young MK, Côté J, and Wang W (1998). NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol. Cell 2, 851–861. [DOI] [PubMed] [Google Scholar]
- Yamada T, Yang Y, Hemberg M, Yoshida T, Cho HY, Murphy JP, Fioravante D, Regehr WG, Gygi SP, Georgopoulos K, and Bonni A (2014). Promoter decommissioning by the NuRD chromatin remodeling complex triggers synaptic connectivity in the mammalian brain. Neuron 83, 122–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Yamada T, Hill KK, Hemberg M, Reddy NC, Cho HY, Guthrie AN, Oldenborg A, Heiney SA, Ohmae S, et al. (2016). Chromatin remodeling inactivates activity genes and regulates neural coding. Science 353, 300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap EL, and Greenberg ME (2018). Activity-Regulated Transcription: Bridging the Gap between Neural Activity and Behavior. Neuron 100, 330–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Chen Y, Kim B, Wang H, Zhao C, He X, Liu L, Liu W, Wu LM, Mao M, et al. (2013). Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell 152, 248–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang C, Schones DE, Zeng C, Cui K, Zhao K, and Peng W (2009). A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 25, 1952–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, Shi X, Zhang Z, Chen Y, and Wu JI (2011). Dual role of Brg chromatin remodeling factor in Sonic hedgehog signaling during neural development. Proc. Natl. Acad. Sci. USA 108, 12758–12763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, Cao M, Yoo AS, Zhang Z, Chen L, Crabtree GR, and Wu JI (2015). Generation of BAF53b-Cre transgenic mice with pan-neuronal Cre activities. Genesis 53, 440–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Cao M, Chang CW, Wang C, Shi X, Zhan X, Birnbaum SG, Bezprozvanny I, Huber KM, and Wu JI (2015). Autism-Associated Chromatin Regulator Brg1/SmarcA4 Is Required for Synapse Development and Myocyte Enhancer Factor 2-Mediated Synapse Remodeling. Mol. Cell. Biol 36, 70–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the next generation sequencing raw data reported in this paper is NCBI Gene Expression Omnibus: GSE174585. No new code was generated in this study.