

ABSTRACT
OXA-48-type β-lactamases are now routinely encountered in bacterial infections caused by carbapenem-resistant Enterobacterales. These enzymes are of high and growing clinical significance due to the importance of carbapenems in treatment of health care-associated infections by Gram-negative bacteria, the wide and increasing dissemination of OXA-48 enzymes on plasmids, and the challenges posed by their detection. OXA-48 confers resistance to penicillin (which is efficiently hydrolyzed) and carbapenem antibiotics (which is more slowly broken down). In addition to the parent enzyme, a growing array of variants of OXA-48 is now emerging. The spectrum of activity of these variants varies, with some hydrolyzing expanded-spectrum oxyimino-cephalosporins. The growth in importance and diversity of the OXA-48 group has motivated increasing numbers of studies that aim to elucidate the relationship between structure and specificity and establish the mechanistic basis for β-lactam turnover in this enzyme family. In this review, we collate recently published structural, kinetic, and mechanistic information on the interactions between clinically relevant β-lactam antibiotics and inhibitors and OXA-48 β-lactamases. Collectively, these studies are starting to form a detailed picture of the underlying bases for the differences in β-lactam specificity between OXA-48 variants and the consequent differences in resistance phenotype. We focus specifically on aspects of carbapenemase and cephalosporinase activities of OXA-48 β-lactamases and discuss β-lactamase inhibitor development in this context. Throughout the review, we also outline key open research questions for future investigation.
KEYWORDS: OXA-48, antibiotic resistance, β-lactamases
INTRODUCTION
Antimicrobial resistance has been recognized globally as one of the most serious threats to modern medicine. According to the 2014 UK Review on Antimicrobial Resistance, antibiotic-resistant bacterial infections are predicted to result in 10 million deaths annually by 2050 if no preventative measures are taken (1). β-Lactams are the single most prescribed antibiotic class, accounting for over half of all antibiotic prescriptions in human patients (2); thus, the consequences of widespread resistance to these agents are especially severe. Resistance against carbapenems is a particular concern, as these drugs are the most recently introduced and potent class of β-lactams; they are, for example, favored treatments for opportunistic infections of secondary care patients by Gram-negative bacteria resistant to other agents (3). Furthermore, the continuing weakness of the antibacterial pipeline means that alternative treatments are limited (4). This existing threat is highlighted by the Centers for Disease Control and Prevention, which classified carbapenem-resistant Enterobacterales as an urgent antibiotic resistance threat in the United States in 2019 (5).
Since the initial introduction of β-lactam antibiotics in the 1940s, bacteria have developed many different mechanisms to bypass their effect; these include changes in expression levels of porins and efflux pumps, target modification through gene acquisition or mutation, and enzymatic drug modification (6). In Gram-negative bacteria, β-lactamase enzymes are the main resistance mechanism against β-lactam antibiotics. β-Lactamases modify the antibiotic by hydrolytic cleavage of the β-lactam amide bond (7); as β-lactams work by binding to penicillin-binding proteins (PBPs) and disrupting bacterial cell wall biosynthesis (8, 9), degrading the β-lactam pharmacophore renders these antibiotics inactive. Over 4,500 β-lactamases have now been identified (see www.bldb.eu for details) (10), with the continuing explosion of genomic data driving further discovery of new enzymes from both environmental and clinical sources.
According to the Ambler classification, β-lactamases are divided into four groups: classes A, C, and D are serine β-lactamases (SBLs), which utilize an active site serine nucleophile to hydrolyze β-lactams via a covalent acyl-enzyme intermediate, while class B metallo-β-lactamases (MBLs) utilize zinc cofactors to activate a water molecule to undertake antibiotic inactivation (7). Within SBLs, class D β-lactamases form a structurally diverse group of enzymes, which were first identified as having enhanced hydrolytic activity toward semisynthetic penicillins, such as oxacillin, and reduced activity toward penicillin (rates compared against class A β-lactamases) (11, 12). Subsequently, they were named as oxacillinases, or OXAs for short. OXA β-lactamases include five recognized subgroups of carbapenem-hydrolyzing enzymes: four of these, namely, OXA-23-like, OXA-24/40-like, OXA-51-like, and OXA-58-like β-lactamases, are largely restricted to Acinetobacter baumannii, while OXA-48-like β-lactamases are most commonly encountered in the Enterobacterales (13–21). Additionally, some OXA enzymes (OXA-2 and OXA-10) classified as narrow-spectrum β-lactamases have demonstrated comparable rates of carbapenem hydrolysis to recognized carbapenem-hydrolyzing OXAs, which could imply that most OXAs can (to some extent) be considered carbapenemases (22). OXA-48 β-lactamases are now among the most common carbapenemases (23) and are often coproduced with other β-lactamases (MBLs or extended-spectrum β-lactamases [ESBLs]) (24). For an in-depth overview of the global epidemiology of OXA-48 β-lactamase-producing pathogens, we refer the reader to recent reviews by Bush and Bradford (25) and Pitout et al. (26).
Even though OXA-48 β-lactamases are not closely related in sequence to other class D β-lactamases (less than 50% amino acid identity), their sequences include three active site motifs that are broadly conserved within class D enzymes (27). Motif I (SXXK) includes the nucleophilic Ser70 and the catalytically important Lys73, which needs to be carboxylated for efficient hydrolysis to take place (Fig. 1) (27, 28). Motifs II and III are in the vicinity of these key catalytic residues and include residues Ser118-Val119-Val120 and Lys208-Thr209-Gly210, respectively, in OXA-48-like enzymes. Additionally, the Ω-loop (residues 143 to 165) and β5-β6 loop (residues 213 to 218) bordering the active site seem to be important determinants of OXA-48 activity, as discussed below. According to the β-lactamase database (10), at least 15 plasmid-encoded OXA-48 β-lactamases have been identified and validated (with further variants chromosomally encoded mainly in different Shewanella species [29, 30]). These variants differ from wild-type OXA-48 by certain amino acid substitutions or deletions. Selected key family members along with their hydrolytic profiles are listed in Fig. 1.
FIG 1.

Structure of OXA-48. The cartoon shows unliganded OXA-48 (PDB ID 6P96) (46) with selected elements of the structure highlighted. The three conserved motifs within class D β-lactamases are shown in blue shades, the Ω-loop in yellow, and the β5-β6 loop in orange. Selected OXA-48 variants are listed according to their primary hydrolysis phenotype, and their amino acid substitutions or deletions are highlighted in the corresponding color in the amino acid sequence. Carbapenemases show efficient imipenem hydrolysis and some activity toward other carbapenem substrates. ESBL-like β-lactamases show only weak activity against all carbapenems and activity against expanded-spectrum oxyimino-cephalosporins.
As mentioned above, OXA-48 enzymes degrade a variety of β-lactam antibiotics, including ampicillin and oxacillin (more efficiently than, e.g., temocillin) (27, 31) and, perhaps most notably, the “last-resort antibiotics” carbapenems (Fig. 2) (32). However, there are large phenotypic variations within the enzyme family. Compared to the parent OXA-48 enzyme, some variants have enhanced carbapenemase activity (such as OXA-162 [33] and OXA-181 [34]), while others have expanded their hydrolysis profile to better accommodate expanded-spectrum oxyimino-cephalosporins (such as OXA-163 [35] and OXA-405 [36]). OXA-48 carbapenemases tend to favor imipenem over other carbapenems and display only low-level meropenem and ertapenem hydrolysis (Table 1 and Supplemental Data Set S1). Weak carbapenem hydrolysis can complicate diagnosis and treatment of bacterial infections involving OXA-48 producers, as their activity can be below the detection limit of clinical tests but still sufficient to confer resistance, especially in strains with reduced antibiotic permeability (37). OXA-48 itself shows various activities against cephalosporins; e.g., cephalothin and cefotaxime are inactivated readily, whereas minimal (or no) activity is measured against ceftazidime and cefepime. Further enzyme kinetic data for OXA-48 and key variants are collated in Data Set S1 in the supplemental material.
FIG 2.

β-Lactam antibiotics as substrates for OXA-48. Examples of penicillin, cephalosporin, and carbapenem antibiotics (left, middle, and right, respectively) which are generally ineffective against OXA-48 producers (red box) and which can be used to treat OXA-48-producing infections (green box). Notably, activity profiles vary within the OXA-48 family, e.g., the ESBL-like OXA-163 has acquired activity against expanded-spectrum oxyimino-cephalosporins (ceftazidime).
TABLE 1.
Kinetic parameters for OXA-48, OXA-163, OXA-181, and OXA-232 with different β-lactam antibioticsa
| Drug | OXA-48b |
OXA-163 |
OXA-181 |
OXA-232 |
|||||
|---|---|---|---|---|---|---|---|---|---|
| kcat (s–1) | Km (μM) | kcat (s–1) | Km (μM) | kcat (s–1) | Km (μM) | kcat (s–1) | Km (μM) | ||
| Imipenem | 5 | 13 | 0.03 | 520 | 7.5 | 13 | 0.2 | 9 | |
| Meropenem | 0.07 | 10 | >0.1 | >2,000 | 0.1 | 70 | 0.03 | 100 | |
| Ertapenem | 0.13 | 100 | 0.05 | 130 | 0.2 | 100 | 0.04 | 110 | |
| Doripenem | —c | —c | NH | NH | 0.04 | 55 | 0.005 | 10 | |
| Ceftazidime | NH | NH | 8 | >1,000 | — | — | >0.6 | >1,000 | |
| Cefotaxime | >9 | >900 | 10 | 45 | >62 | >1,000 | >6.5 | >1,000 | |
| Cephalothin | 44 | 195 | 3 | 10 | 13 | 250 | 13 | 125 | |
| Benzylpenicillin | —d | —d | 23 | 13 | 444 | 90 | 125 | 60 | |
| Ampicillin | 955 | 400 | 23 | 315 | 218 | 170 | 132 | 220 | |
| Temocillin | 0.3 | 45 | NH | NH | 0.3 | 60 | 0.03 | 60 | |
| Oxacillin | 130 | 95 | 34 | 90 | 90 | 80 | 156 | 130 | |
Presented values are taken from reference 31, and more comprehensive data of enzyme kinetics are provided in the supplemental material. NH, no hydrolysis; —, no values found.
Data from reference 33 do not show doripenem hydrolysis by OXA-48, but kinetic data from reference 22 indicate weak doripenem hydrolysis.
Kinetic data from reference 32 indicate that benzylpenicillin is hydrolyzed by OXA-48.
General hydrolysis mechanism.
In SBLs, the overall hydrolysis reaction consists of two parts, acylation followed by deacylation (Fig. 3) (7). After initial formation of the noncovalent Michaelis complex, the β-lactamase is acylated by the antibiotic, resulting in covalent bond formation between Ser70 and the carbonyl carbon of the β-lactam ring. This covalent acyl-enzyme structure is hydrolyzed in the deacylation step, in which an active site water molecule (the so-called deacylating water) acts as the nucleophile to attack the acyl-enzyme carbonyl. Both acylation and deacylation involve formation of short-lived tetrahedral intermediate (TI) structures. For OXA-48-like β-lactamases, deacylation was shown to be rate limiting for carbapenem breakdown (38).
FIG 3.

Hydrolysis mechanism of OXA-48 β-lactamases. Starting from the formation of a Michaelis complex for a general carbapenem substrate (1), the substrate is acylated (tetrahedral intermediate formation in 1 → 2), which yields a covalent acyl-enzyme structure (3). The bound antibiotic is subsequently deacylated (4 and tetrahedral deacylation intermediate 5), resulting in the final hydrolysis product (6).
As depicted in Fig. 3, both acylation and deacylation involve a negatively charged general base. For class A β-lactamases, this residue is largely accepted to be Glu166 (39, 40), but for OXA enzymes, the general base is a carboxylated lysine (Lys73 in OXA-48 numbering) (27, 41). This posttranslational carboxylation is needed for efficient hydrolysis to take place, as mutating Lys73 results in enzymes incapable of substrate turnover (28). The degree of carboxylation increases with pH, and preparation of catalytically competent enzymes can be ensured by adding a suitable CO2 source for carboxylation (bicarbonate), even though atmospheric CO2 may also be enough (42). This carboxylation is reversible, and it has been monitored with 19F nuclear magnetic resonance (NMR) spectroscopy in the presence of different inhibitors to understand how (de)carboxylation contributes to enzyme inhibition (43). The results indicate that Lys73 is carboxylated to a lesser extent with some covalently bound inhibitors (like avibactam), which may contribute to more efficient inhibition.
Carbapenemase activity.
OXA-48 enzymes are carbapenemases or, more specifically, imipenemases with weak turnover rates for other carbapenems, such as meropenem and ertapenem (Table 1 and Supplemental Data Set S1). Based on the structural information originally derived from other carbapenem-hydrolyzing OXAs (44), carbapenemase activity in class D β-lactamases was hypothesized to originate from a hydrophobic bridge spanning the active site (Phe110 and Met221 for OXA-23 and Tyr112 and Met223 for OXA-24). However, structural comparisons between OXA-48 and other OXA carbapenemases show OXA-48 to be lacking this hydrophobic bridge (27), which implies that the OXA-48 group has evolutionally diverged from other class D β-lactamases and acquired carbapenemase activity by other means (Fig. 4). Fortunately, within the last several years a plethora of new crystal structures of OXA-48s complexed with carbapenems have been released, and new mechanistic knowledge has been derived from them (all currently available structures of OXA-48 enzymes in the Protein Data Bank [www.rcsb.org/] are listed in Supplemental Table S1).
FIG 4.

Divergent active sites of carbapenem-hydrolyzing OXA enzymes. Active sites of OXA-48 (PDB ID 6P96, left) (46) and OXA-23 (PDB ID 4K0X, right) (112) highlight the missing hydrophobic bridge in OXA-48 with respect to other class D carbapenemases. In OXA-23, the hydrophobic bridge is formed by residues Phe110 and Met221, while the corresponding residues in OXA-48 are Ile102 and Thr213, which leave the active site more open. Additionally, residues forming the so-called “deacylating water channel” are also highlighted in sticks (V120 and L158 for OXA-48 and V128 and L166 for OXA-23).
The first carbapenem acyl-enzyme structure of OXA-48 (with imipenem) was released in 2018 (PDB identifier [ID] 5QB4) alongside multiple structures with small inhibitor fragments (45). From 2019 onward, further acyl-enzyme structures have been deposited with imipenem (PDB IDs 6P97, 6PTU, and 7KH9) (38, 46, 47), meropenem (PDB IDs 6P98, 6PT1, and 7KHQ) (38, 46, 47), doripenem (PDB IDs 6P9C and 6PXX) (46, 48), ertapenem (PDB ID 6P99) (46), and faropenem (PDB ID 6PSG) (47). Additionally, two acyl-enzyme structures of inactivated OXA-163 (K73A) with imipenem and meropenem are available (PDB IDs 7KHZ and 7KHY, respectively) (38). Common features in these structures include a covalent bond between Ser70 and the substrate and hydrogen bonds between Thr209/Arg250 and the carbapenem C-3 carboxylate (Fig. 5). The carbonyl oxygen of the cleaved β-lactam ring is positioned in the oxyanion hole formed by the backbone amides of Ser70 and Tyr211; active site interactions in selected crystallized carbapenem acyl-enzyme complexes are presented in Fig. 6 and 7. Carbapenem “tail” groups (C-2 substituents) are not anchored by any strong interactions, which implies that they are dynamic and do not need to adopt any one specific orientation. This likely disorder was also inspected by Papp-Wallace et al., who further refined previously deposited imipenem and doripenem complexes (PDB IDs 5QB4 and 6P9C, respectively) (48). Their analysis of the re-refined structures supports the presence of a covalent bond between Ser70 and the antibiotic, but observation of weak or absent density for the pyrroline ring and C-2 tail groups indicates disorder (i.e., multiple conformations) for these regions. In addition to previously mentioned covalent complexes, a structure of OXA-48 with hydrolyzed imipenem has also been published (PDB ID 6PK0) (47). Noncovalently bonded hydrolyzed imipenem forms interactions with Thr209 and Arg250 similar to those observed in the acyl-enzyme, and the newly formed C-7 carboxylate group is hydrogen bonded to Ser70, Lys73, and Tyr211 (Fig. 6). Although the deacylating water is not present in any acyl-enzyme structure, the orientation of hydrolyzed imipenem (specifically coordination of the C-7 carboxylate to Ser70 and Lys73) indicates the possible position of the deacylating water molecule prior to deacylation.
FIG 5.

Carbapenem, cephalosporin, and diazabicyclooctanone (DBO) scaffolds with atom numbering. The 6α-hydroxyethyl group (C-6 substituent) in the carbapenem scaffold is shown in red.
FIG 6.

Carbapenem complexes of OXA-48. (Top left) Imipenem acyl-enzyme (PDB ID 6P97) (46); interactions with active site residues are highlighted. The imipenem pyrroline ring is modeled as the Δ2 tautomer. (Top middle) Imipenem acyl-enzyme (PDB ID 6PTU) (47), with the pyrroline ring as the (R)-Δ1 tautomer. (Top right) Hydrolyzed imipenem (PDB ID 6PK0) (47), with the pyrroline ring as the (S)-Δ1 tautomer. (Bottom left) Doripenem acyl-enzyme (PDB ID 6P9C) (46), with the pyrroline ring as the Δ2 tautomer. (Bottom right) Doripenem acyl-enzyme (PDB ID 6PXX) (48), with the pyrroline ring as the (R)-Δ1 tautomer.
FIG 7.

Further carbapenem complexes of OXA-48. Acyl-enzyme structures with meropenem (left; PDB ID 6P98) (46) and faropenem (right; PDB ID 6PSG) (47). The pyrroline ring is present as the Δ2 tautomer in both structures.
In OXA-48 enzymes, the basis for carbapenemase activity has been attributed to the presence of the β5-β6 loop bordering the active site, as, for example, engineering this loop from OXA-48 into the noncarbapenemase OXA-10 changes its phenotype to hydrolyze imipenem at higher rates than native OXA-48 (49). The specific role of Arg214 (in the β5-β6 loop) was studied by comparing hydrolysis kinetics and crystal structures of OXA-181 and OXA-232, the difference between these two variants being residue 214 (Arg in OXA-181 and Ser in OXA-232) (50). OXA-181 is a slightly better carbapenemase than OXA-48 (34), while OXA-232 has decreased carbapenem hydrolysis rates but has also acquired weak activity against ceftazidime (Table 1) (51). The authors suggest that the presence of Arg214 is crucial for carbapenem hydrolysis by OXA-48, as it aids in the formation of a productive binding pose for imipenem. Replacing this arginine with a negatively charged residue (Glu) results in poor affinity, which was reasoned to be due to an unproductive binding pose of imipenem (both hypotheses based on molecular docking). Similar results were obtained by Dabos et al., who substituted the β5-β6 loop of OXA-18 into OXA-48 (52). Steady-state kinetics of the OXA-48 loop18 variant showed decreased ampicillin and imipenem hydrolysis and elevated ceftazidime hydrolysis. The importance of the β5-β6 loop for the hydrolysis profile indicated by these studies is further emphasized by the decrease in imipenem hydrolysis and increase in ceftazidime hydrolysis in OXA-163 (Table 1) (31, 35), in which the loop is partially deleted (Fig. 1). Pre-steady-state kinetics indicate that the loss of efficient imipenemase activity in OXA-163 is due to decreased deacylation rates (38). However, even though the β5-β6 loop is evidently important for carbapenem hydrolysis, the specific origin of imipenemase activity in OXA-48 enzymes (e.g., over meropenem hydrolysis) remains to be investigated. The presence of the 1β-methyl group, e.g., in meropenem and doripenem (instead of the 1β-proton in imipenem), has been suggested to impair hydrolysis, as this methyl group might prevent deacylation by disfavoring rotation of the carbapenem 6α-hydroxyethyl moiety (attached to C-6 [Fig. 5]), which would, in turn, prohibit the nucleophilic attack (47). In all OXA-48/carbapenem crystal structures (excluding 5QB4), the 6α-hydroxyethyl side chain adopts a similar orientation where its methyl group points toward Leu158 and Arg214 and values for the C-7–C-6–C–O dihedral angle are between 147° and 192° (Fig. 7). However, for hydrolyzed imipenem, this orientation has changed, and the methyl group points out of the active site toward bulk solvent (with the same dihedral angle being between 275° and 292° depending on the protein chain). As the 6α-hydroxyethyl group is likely able to rotate in the acyl-enzyme, verifying the extent of its influence on positioning and movement of the deacylating water remains an important aspect for future mechanistic studies.
The pyrroline ring of carbapenem acyl enzymes can exist as two different tautomers: Δ2 or Δ1, the latter of which also has two stereoisomers, (R)-Δ1 and (S)-Δ1 (Fig. 8). For class A β-lactamases, the Δ2 tautomer has been proposed to be the catalytically competent form (53, 54) and the Δ1 to not deacylate efficiently (potentially due to displacement of the deacylating water from the active site [55] or loss of stabilizing interactions with the oxyanion hole [56]). The same has been suggested for class D enzymes when comparing the doripenem complex of carbapenem-hydrolyzing OXA-24 against carbapenem-inhibited OXA-1 (57). The tautomeric form can be identified in crystal structures with sufficiently strong electron density for the ligand, as the pyrroline ring C-2–sulfur bond present in all carbapenems is planar (sp2 hybridized) in the case of the Δ2 form and sp3 hybridized for the Δ1 form. For previous class D β-lactamases complexed with carbapenems, all three tautomers have been observed (57, 58). In the case of OXA-48, the Δ2 form was assigned in the first deposited imipenem complex (45), and the same tautomer was subsequently observed for the meropenem, imipenem, doripenem, and ertapenem acyl enzymes published by Smith et al. (structures prepared by soaking crystals of apo-OXA-48 with 50 mM carbapenem solution over timescales between 30 s and 10 min) (46). The same authors inspected the possibility of accommodating ligands in the active site in the Δ1 form by superimposition of their structures onto OXA-23 with (R)-Δ1 and (S)-Δ1 ligands. They suggest that the formation of the (S)-Δ1 tautomer of meropenem is feasible, while the (R)-Δ1 conformer would clash sterically with Tyr211. Shortly after the publication of these carbapenem acyl enzymes, a new structure of deacylation-deficient OXA-48 (Lys73Ala) in complex with doripenem was released (also prepared using crystal soaking) (48). The doripenem acyl-enzyme was observed as both (R)-Δ1 and (S)-Δ1 tautomers (Fig. 6), and only a partial salt bridge with Arg250 is formed, which most likely prevents any severe steric clashes between doripenem and Tyr211 for either tautomer. In further structures deposited by Akhtar et al., different carbapenems have different tautomers present: the meropenem acyl-enzyme is in the Δ2 form (as depicted for another crystal structure in Fig. 7), imipenem and faropenem are found as (R)-Δ1 (Fig. 6 and 7), and the imipenem hydrolysis product is found as the (S)-Δ1 tautomer (Fig. 6; structures prepared by soaking OXA-48 crystal with a solution containing the ligand for 30 min or, for the OXA-48 imipenem product complex, for 2 h) (47). Characterization of the enzyme-hydrolyzed products by NMR spectroscopy implies that for OXA-48 (as well as all other tested SBLs and MBLs), the preferred hydrolysis product would be in either the Δ2 or (R)-Δ1 form, but deducing the exact enzyme-catalyzed reaction product was not feasible due to the ability of released products to undergo tautomerization in solution (59).
FIG 8.

Mechanism for carbapenem side product formation by OXA-48. The pyrroline ring in carbapenem substrates can undergo Δ2 → Δ1 tautomerization (7 → 8) postacylation. In addition to the general hydrolysis mechanism (7 → 10), 1β-methyl carbapenems such as meropenem can form a 1β-lactone product (8 → 9), which has been suggested to be mainly in the Δ1 form (60).
In addition to the generic hydrolysis mechanism of serine β-lactamases, OXA-48 enzymes were shown to possess an additional mechanism for carbapenem breakdown that involves the formation of a β-lactone product, as illustrated in Fig. 8 (60, 61). Starting from the acyl-enzyme, the β-lactone is suggested to form by intramolecular cyclization, where the hydroxyl group of the carbapenem 6α-hydroxyethyl side chain donates a proton to the carboxylated lysine (Lys73) and attacks the same electrophilic C-7 carbon as in deacylation. This results in formation of a four-membered lactone ring, which is structurally close to the original β-lactam ring and capable of reacting further to give (unidentified) reaction products. Interestingly, β-lactone formation by OXA-48 appears to be carbapenem dependent, as it was observed only for 1β-methyl carbapenems (such as meropenem, doripenem, and ertapenem) and not for carbapenems with a 1β-proton (imipenem and biapenem). The reason for this dependence on the presence of the 1β substituent was studied by simulating the dynamics of OXA-1 (one 100-ns simulation) and suggested to be due to more favorable conformational sampling of the 6α-hydroxyethyl side chain: with a 1β-methyl group, bound carbapenems formed closer interactions with the carboxylated lysine, which would aid in proton transfer from the hydroxyl group to the lysine carboxylate oxygen (60). More recently, however, lactone formation was shown to also depend upon the structure of the active site: OXA-519 (Val120Leu variant of OXA-48) demonstrated both an increase in the proportion of the lactone product and generated lactones from both 1β-proton and 1β-methyl carbapenems (61).
Cephalosporinase activity.
While OXA-48 is considered of particular importance as a result of its carbapenemase activity, there are variations in hydrolytic phenotypes between different OXA-48 variants. OXA-48 itself does hydrolyze some cephalosporin antibiotics, such as cephalothin and cefotaxime, but shows no significant hydrolysis of the expanded-spectrum oxyimino-cephalosporin ceftazidime or the fourth-generation cephalosporin cefepime (31). However, variants such as OXA-163 and OXA-405 (that contain partial deletions in the β5-β6 loop) are capable of hydrolyzing ceftazidime, at the expense of efficient imipenem breakdown (Fig. 1, Table 1, and Supplemental Table S1) (35, 36). Interestingly, their hydrolysis rates for other carbapenems (such as meropenem) seem to be on the same low level as for OXA-48 (further kinetic information is collated in Supplemental Data Set S1).
In 2019, the structure of an OXA-48 (Pro68Ala) ceftazidime acyl-enzyme was deposited (PDB ID 6Q5F) (Fig. 9) (62); this single point mutant was obtained by passage of a laboratory OXA-48 producer strain against increasing concentrations of ceftazidime. Comparison of this OXA-48 structure with previously deposited OXA/ceftazidime complexes (OXA-225 and OXA-160 [PDB IDs 4X55 and 4X56, respectively]) (63) shows that ceftazidime exhibits a different binding pose in OXA-48 than observed in the OXA-23 or OXA-24/40 variants, the difference being in the orientation of the C-7 substituents (carboxypropyl oxyimino and thiazole groups [Fig. 5]). Another distinct feature in the OXA-48/ceftazidime structure was the lack of interpretable electron density for the Ω-loop (including residues Leu158 and Asp159 [Fig. 1]). The authors suggested ceftazidime binding to displace Arg214, which, in turn, results in a distorted (and thus flexible) Ω-loop; the Pro68Ala mutation might then contribute to Ω-loop distortion by increasing flexibility of the active site. Molecular dynamics simulations and quantum mechanics/molecular mechanics (QM/MM) reaction modeling of ceftazidime deacylation by OXA-48, OXA-163, and OXA-181 suggest that in addition to the β5-β6 loop and Arg214, Leu158 could also play an important role in determining the efficiency of ceftazidime turnover (64). The orientation of Leu158 was observed to correlate with active site hydration, and an increase in water molecules in the active site was observed to impair deacylation efficiency in OXA-48. Additionally, the study proposed that distorting the Ω-loop, as is implied by the absence of electron density for this region in the OXA-48 ceftazidime crystal structure, would fully open the active site to bulk water and diminish deacylation rates. Although some consideration has been given to the routes by which the water molecule necessary for deacylation may enter the active site (46, 58), the importance of active site hydration to the activity of OXA-48 β-lactamases (or of SBLs in general) has to date not been extensively discussed in the literature.
FIG 9.

Cephalosporin acyl-enzyme complexes of OXA-48. Hydrogen bonds between the substrate and active site residues are highlighted with dashed lines. (Left) Ceftazidime (PDB ID 6Q5F) (62); (middle) cefotaxime (PDB ID 6PQI) (47); (right) cefoxitin (PDB ID 6PT5) (47).
In addition to ceftazidime, structures of OXA-48 acyl-enzyme complexes with cefotaxime and cefoxitin have also been determined (PDB IDs 6PT5 and 6PQI for cefoxitin and cefotaxime, respectively) (47). Cefotaxime has a binding pose similar to that of ceftazidime, where the thiazole ring orients to make stacking interactions with Tyr211 and the oxyimino group occupies a pocket between residues Leu158, Thr213, and Arg214 (Fig. 9). Unlike the ceftazidime complexes, the Ω-loop remains ordered, as found in the apoenzyme, and the salt bridge between Asp159 and Arg214 is preserved. This is most likely due to the smaller size of the cefotaxime C-7 methoxyimino group than of the equivalent carboxypropyl oxyimino group of ceftazidime. In the case of cefoxitin, the thiophene ring is rotated toward Leu158, breaking the Asp159-Arg214 salt bridge. Low cefotaxime hydrolysis rates are hypothesized to be due to limited access of potential deacylating water molecules to the active site, while cefoxitin hydrolysis (kcat > 0.05 s−1 and Km > 200 μM [see the supplemental material]) is essentially hindered by the presence of its 7-α-methoxy group, which would sterically clash with any active site water molecules (47). Additionally, carboxylation of Lys73 could lead to further steric clashes with the 7-α-methoxy group, which could increase preference for lysine decarboxylation in the presence of cefoxitin (Lys73 is decarboxylated in the crystal structure).
OXA-48 inhibitors.
A common strategy for treating challenging, β-lactam-resistant bacterial infections is to prescribe a β-lactam antibiotic together with a β-lactamase inhibitor (65, 66). FDA-approved antibiotic/inhibitor combinations include amoxicillin-clavulanate, piperacillin-tazobactam, ceftazidime-avibactam, and meropenem-vaborbactam (65–68). In general, OXA-48 β-lactamases are not susceptible to traditional β-lactamase inhibitors like sulbactam, tazobactam, and clavulanate (except for some exceptions like OXA-163) (69). Of the new-generation β-lactamase inhibitors, avibactam (70, 71) shows efficacy against OXA-48 (72, 73). Avibactam belongs to the diazabicyclooctanone (DBO) class and exhibits broad-spectrum inhibition of SBLs. The ceftazidime-avibactam combination specifically shows promise as an effective therapy against OXA-48 producers in both in vitro testing and clinical practice (24, 69, 74–76). Compared with other OXAs, it appears that DBOs such as avibactam inhibit OXA-48 better than enzymes with more hydrophobic active site residues (77). Several crystal structures of OXA-48 with covalently bound avibactam all show very similar binding poses for the acyl-enzyme (PDB IDs 6Q5B [62], 4WMC [78], 4S2J [42], 4S2K [42], and 4S2N [42]), with the carbamate carbonyl positioned in the oxyanion hole (analogous to the position of the ester carbonyl carbon in β-lactam antibiotics) and the sulfonate group positioned toward motif II and Arg250 (Fig. 10). The amide group of avibactam is positioned toward Leu158 on the Ω-loop. Based on the published OXA-48/avibactam structures, the presence of avibactam seems to favor Lys73 decarboxylation: for structures crystallized at pH 6.5 or 7.5 (PDB IDs 4S2J and 4S2K), no carboxylation was observed, and at pH 8.5, only partial occupancy of the carboxylate was seen in two out of four monomers in the asymmetric unit (PDB ID 4S2N) (42). Partial carboxylation of Lys73 was also observed in another study, in which only two out of eight monomers displayed electron density for carboxylated Lys73 (PDB ID 4WMC) (78).
FIG 10.
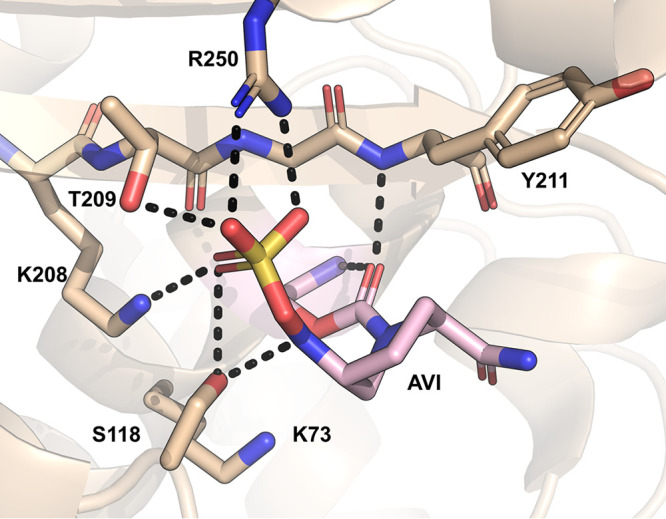
Crystal structure of the avibactam-OXA-48 acyl-enzyme at pH 7.5 (PDB ID 4S2K) (42).
Inhibition kinetics indicate that avibactam readily acylates OXA-48 but that its recyclization to release intact avibactam happens very slowly (while no analogue for the “standard” β-lactam ring-opened hydrolysis product is observed) (78). In acylation, the C-7–N-6 bond in the five-membered ring structure is broken (as opposed to the C-7–N-1 bond [Fig. 5]), likely due to the N-6–sulfate moiety being a better leaving group than the N-1–R group (42). At least two different reaction mechanisms for avibactam with OXA-48 have been proposed in the literature (Fig. 11). King et al. proposed a general mechanism for all SBLs, which involves a decarboxylated, neutral Lys73 acting as a general base in acylation; Lys73 would then subsequently protonate the N-6 ring nitrogen via Ser118 (42). Recyclization occurs as the reverse reaction (Fig. 11, pathway 1). This mechanism was based on the preference for Lys73 to be decarboxylated in the presence of avibactam. Additionally, mutational studies of the class A ESBL CTXM-15 identified Lys73 to be the most likely general base in avibactam acylation (42). Since decarboxylated Lys73 was observed to form a hydrogen bond with Ser118 (Fig. 10), it is possible it has a similar role in class D and class A SBLs.
FIG 11.

Two proposed reaction pathways for the avibactam inhibition mechanism with OXA-48. (Left) Pathway 1, based on a proposed “universal” avibactam reaction scheme for SBLs (42). Neutral Lys73 is suggested to act as a general base in acylation and recyclization, while Ser118 (de)protonates the ring nitrogen. (Right) Pathway 2, where carboxylated Lys73 is proposed to act as the general base in acylation and as the general acid in recyclization (78). Ser118 has the same role as in pathway 1, except it donates a proton to Lys208 instead of Lys73 during recyclization.
The second proposed mechanism for avibactam inhibition in Fig. 11 (pathway 2) was suggested by Lahiri et al.; in this case, Lys73 is indicated to be carboxylated for the whole reaction cycle (78). Carboxylated Lys73 acts as the general base in acylation, and Lys208 protonates N-6 via Ser118. Recyclization takes place similarly but in reverse, where N-6 first donates a proton back to Lys208 via Ser118, and Lys73 acts as a general acid protonating Ser70. As the authors also observed decarboxylation of Lys73 in the presence of avibactam, they attribute the slow avibactam recyclization rates to Lys73 decarboxylation, which hinders reactivity. In addition to these crystal structures, decarboxylation of Lys73 in the presence of covalently bound avibactam has also been measured using NMR spectroscopy (43). The authors observed that Lys73 favors the decarboxylated form when OXA-48 is complexed with avibactam (or the related DBO inhibitors relebactam and zidebactam). The extent of Lys73 decarboxylation in reactions of OXA-48 with DBOs and its exact mechanistic role remain unclear.
To study the possible emergence of resistance to avibactam, OXA-48 producers were passaged against a combination of ceftazidime and avibactam (62). Resistance was observed to develop as a result of two amino acid substitutions: Pro68Ala (as discussed above in “Cephalosporinase activity”), and Tyr211Ser. The catalytic efficiency of ceftazidime turnover increased >10-fold and >20-fold for the single- and double-substitution variants, respectively. Inhibitory activity of avibactam stayed on the same level as for OXA-48 for the Pro68Ala variant, but for Pro68Ala/Tyr211Ser, the activity of avibactam decreased >5-fold. Tyr211 is known to be a key residue in stabilizing tetrahedral intermediates in β-lactam hydrolysis through the formation of an oxyanion hole (together with the backbone amide of Ser70). Additionally, Tyr211 was suggested to possibly aid in the formation of a Michaelis complex. Notably, however, the observed evolutionary trajectory toward ceftazidime-avibactam resistance comes at a fitness cost, as the enzyme thermostability is reduced and the primary hydrolysis phenotype (carbapenemase/penicillinase) is compromised (62).
Other β-lactamase inhibitors with a DBO scaffold include relebactam, nacubactam, zidebactam, durlobactam (previously ETX2514), ARX-1796, and the investigational compound BOS-752 (Fig. 12). The relebactam/imipenem combination has been approved for clinical use, but this inhibitor does not effectively inhibit OXA-48; measured MICs for carbapenems do not change (or change only slightly) in the presence of relebactam (79–81). Based on MICs, zidebactam combined with cefepime shows inhibitory activity against OXA-48 (82). This is due to OXA-48 inhibition by cefepime, as in vitro kinetics indicate that zidebactam on its own does not inhibit OXA-48 (83). Similarly, nacubactam inhibits class A and C SBLs, but in vitro data on its activity against OXA-48 are sparse. In MIC tests, bacterial isolates expressing OXA-48 were susceptible to aztreonam/nacubactam and cefepime/nacubactam, but the potentiation of antibiotic activity by nacubactam was concluded to be mainly due to inhibition of coexpressed ESBLs and AmpC β-lactamases (84). Durlobactam was originally developed to combat infections involving OXA enzymes in Acinetobacter baumannii (77), and this compound inhibits OXA-48 effectively irreversibly (as well as class A and C SBLs): in MIC tests, durlobactam restored imipenem potency against OXA-48-producing pathogens better than avibactam (85). Durlobactam is currently in phase III clinical trials in combination with sulbactam (86). New β-lactamase inhibitors utilizing the DBO scaffold have been synthesized by replacing the avibactam C-2 carboxamide (Fig. 5) with new functional groups (83, 87). The size of the C-2 substituent appears to correlate with β-lactamase inhibitory activity: new DBO compounds with larger C-2 groups (with respect to avibactam) have approximately an order of magnitude lower on-rates and higher off-rates for OXA-48 (87). However, the studied derivatives with larger C-2 substituents inhibit PBPs in bacterial cells. OXA-48 complexes with avibactam derivatives (PDB IDs 5FAQ, 5FAS, and 5FAT) (87) show essentially the same binding pose as observed for avibactam, the main differences being in the respective C-2 substituents. Avibactam itself has poor oral bioavailability, but the avibactam prodrug ARX-1796 can be administered orally and subsequently metabolized in the body to produce avibactam (88). ARX-1796 differs from avibactam through the addition of a neopentyl ester protecting group on the N-6 sulfate moiety. Recent data show DBO inhibitory activity toward OXA-48 to be dependent upon the N-6 substituent, as replacing the durlobactam N-6 sulfate with fluoroacetate reduces potency but can form the basis for an orally available therapy (89). Another investigational β-lactamase inhibitor in the DBO group is BOS-752, which has a third ring fused to the DBO scaffold (making it a dioxotriazatricyclohendecane) (90). BOS-752 does not possess antibacterial activity on its own, but combined with piperacillin it lowered measured MICs against bacterial strains producing SBLs, including OXA-48 (90).
FIG 12.

Examples of β-lactamase inhibitors in different inhibitor classes. β-Lactam ring-based inhibitors, diazabicyclooctanones, and boronates each block divided into investigational compounds (top), compounds in clinical trials (middle), and inhibitors approved in clinical use (bottom). Inhibitors in red and italics do not effectively inhibit OXA-48; inhibitors in black show inhibitory activity.
In addition to DBOs, other β-lactamase inhibitors currently in clinical development include mechanism-based β-lactam inhibitors and boronic acid compounds (Fig. 12). An example of a β-lactam inhibitor is enmetazobactam, which is a penicillanic acid sulfone currently developed in combination with cefepime (91, 92). This combination was found to be effective against OXA-48 producers, but the efficacy is most likely again attributable to the activity of cefepime and not to efficient inhibition by enmetazobactam, which is active primarily against ESBLs (92, 93). On the other hand, boronates show promise as broad-spectrum β-lactamase inhibitors. In particular, cyclic boronates can act as analogues of the tetrahedral acylation transition state of SBLs (94), and they have potential for at least moderate activity against MBLs (95, 96). The first boronic acid inhibitor approved in clinical use was vaborbactam (originally RPX7009) (97), which is currently administered in combination with meropenem (98, 99). Vaborbactam is a monocyclic boronic acid compound showing inhibition mainly against class A and C SBLs, and it is not able to effectively inhibit OXA-48 based on both biochemical data and MIC measurements (potency of meropenem not restored) (79, 100). Further boronic acid derivatives developed as β-lactamase inhibitors include taniborbactam (VNRX-5133), which is a bicyclic boronate (101). Based on both in vitro and whole-cell assay data, taniborbactam exhibits pan-β-lactamase inhibition (i.e., is able to inhibit all four Ambler classes), including moderate inhibition of OXA-48 (with a 50% inhibitory concentration [IC50] of approximately 0.54 μM) (102). Taniborbactam is currently in clinical development in combination with cefepime (103). Another potent bicyclic boronate with ultrabroad-spectrum β-lactamase inhibition is the compound QPX7728, which can efficiently inhibit carbapenem-resistant Enterobacterales and restore the potency of meropenem against OXA-48 (104, 105). QPX7728 entered phase I clinical trials in December 2020 (106, 107). VNRX-7145, which is orally bioavailable, also demonstrates OXA-48 inhibition and entered phase I clinical trials in 2020 combined with ceftibuten (108–110). In addition to boronates, other cyclic compounds mimicking the tetrahedral intermediate (such as phosphonates, sulfonates, and sulfonamides) may also provide a source of future inhibitors, but these are yet to be explored in detail (94). Growing appreciation of the clinical importance of OXA-48 has also motivated exploration of other routes to inhibitors, such as the use of DNA-encoded libraries, but these, too, remain at an early stage (111).
Conclusions.
Carbapenem-hydrolyzing Enterobacterales are classified as an urgent global threat to modern medicine, while OXA-48 β-lactamases are endemic in some regions (especially Turkey and the Mediterranean) and continue to disseminate. In general, OXA-48 enzymes convey penicillin and low-level carbapenem resistance; their weak carbapenem hydrolysis often complicates diagnosis and subsequent treatment of infections involving OXA-48 producers. Most variants within the OXA-48 family are imipenemases with low turnover rates for other carbapenems and resist established mechanism-based β-lactam inhibitors. However, certain variants (such as OXA-163 and OXA-405) have acquired a more ESBL-like hydrolysis profile with activity against expanded-spectrum oxyimino-cephalosporins (such as ceftazidime) and significantly decreased imipenemase activity. The extent to which further evolution of the OXA-48 scaffold toward genuinely broad-spectrum activity is possible remains to be established.
Recent crystallographic efforts have yielded structures of acyl-enzyme complexes of OXA-48 not only with clinically relevant carbapenem and cephalosporin substrates but also with new-generation DBO inhibitors (avibactam). These supply much new information regarding the interactions of substrates and inhibitors with the OXA-48 active site, including the importance of active site structure (specifically the Ω-loop), hydration, and, with respect to carbapenems, rearrangements such as tautomerization and lactone formation that occur after β-lactam cleavage. The origin of preferential activity toward imipenem over other carbapenems, however, remains to be verified. Importantly, structural data for other OXA-48-like enzymes have started to emerge, too, which is important to increase understanding of how substitutions affect specificity across the enzyme group. Combining knowledge from biochemical characterization and X-ray crystallography as well as atomistic computational modeling will likely lead to a detailed picture of the origin of activity and specificity in OXA-48 enzymes, ultimately benefitting design of inhibitors effective against this widespread and variable β-lactamase family.
ACKNOWLEDGMENTS
Viivi H. A. Hirvonen is supported by the UK Medical Research Council (MR/N0137941/1 for the GW4 BioMed DTP awarded to the Universities of Bath, Bristol, Cardiff and Exeter). Marc W. van der Kamp is a BBSRC David Phillips Fellow and thanks the Biotechnology and Biological Sciences Research Council for funding (BB/M026280/1).
Footnotes
Supplemental material is available online only.
aac.00184-21-s0001.xlsx (57.4KB, xlsx)
aac.00184-21-s0002.pdf (191.4KB, pdf)
Contributor Information
Viivi H. A. Hirvonen, Email: viivi.hirvonen@bristol.ac.uk.
James Spencer, Email: jim.spencer@bristol.ac.uk.
Marc W. van der Kamp, Email: marc.vanderkamp@bristol.ac.uk.
REFERENCES
- 1.O’Neill J. 2014. Review on antimicrobial resistance. Antimicrobial resistance: tackling a crisis for the health and wealth of nations. Review on Antimicrobial Resistance, London, United Kingdom. [Google Scholar]
- 2.Klein EY, Van Boeckel TP, Martinez EM, Pant S, Gandra S, Levin SA, Goossens H, Laxminarayan R. 2018. global increase and geographic convergence in antibiotic consumption between 2000 and 2015. Proc Natl Acad Sci U S A 115:E3463–E3470. 10.1073/pnas.1717295115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Papp-Wallace KM, Endimiani A, Taracila MA, Bonomo RA. 2011. Carbapenems: past, present, and future. Antimicrob Agents Chemother 55:4943–4960. 10.1128/AAC.00296-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peri AM, Doi Y, Potoski BA, Harris PNA, Paterson DL, Righi E. 2019. Antimicrobial treatment challenges in the era of carbapenem resistance. Diagn Microbiol Infect Dis 94:413–425. 10.1016/j.diagmicrobio.2019.01.020. [DOI] [PubMed] [Google Scholar]
- 5.CDC. 2019. Antibiotic resistance threats in the United States. US Department of Health and Human Services, Atlanta, GA. [Google Scholar]
- 6.Alekshun MN, Levy SB. 2007. Molecular mechanisms of antibacterial multidrug resistance. Cell 128:1037–1050. 10.1016/j.cell.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 7.Tooke CL, Hinchliffe P, Bragginton EC, Colenso CK, Hirvonen VHA, Takebayashi Y, Spencer J. 2019. β-Lactamases and β-lactamase inhibitors in the 21st century. J Mol Biol 431:3472–3500. 10.1016/j.jmb.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. 2008. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev 32:234–258. 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 9.Zapun A, Contreras-Martel C, Vernet T. 2008. Penicillin-binding proteins and β-lactam resistance. FEMS Microbiol Rev 32:361–385. 10.1111/j.1574-6976.2007.00095.x. [DOI] [PubMed] [Google Scholar]
- 10.Naas T, Oueslati S, Bonnin RA, Dabos ML, Zavala A, Dortet L, Retailleau P, Iorga BI. 2017. Beta-Lactamase Database (BLDB)—structure and function. J Enzyme Inhib Med Chem 32:917–919. 10.1080/14756366.2017.1344235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Datta N, Kontomichalou P. 1965. Penicillinase synthesis controlled by infectious R factors in Enterobacteriaceae. Nature 208:239–241. 10.1038/208239a0. [DOI] [PubMed] [Google Scholar]
- 12.Hedges RW, Datta N, Kontomichalou P, Smith JT. 1974. Molecular specificities of R factor-determined β-lactamases: correlation with plasmid compatibility. J Bacteriol 117:56–62. 10.1128/JB.117.1.56-62.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liapis E, Pantel A, Robert J, Nicolas-Chanoine M-H, Cavalie L, van der Mee-Marquet N, de Champs C, Aissa N, Eloy C, Blanc V, Guyeux C, Hocquet D, Lavigne J-P, Bertrand X, ONERBA . 2014. Molecular epidemiology of OXA-48 producing Klebsiella pneumoniae in France. Clin Microbiol Infect 20:O1121–O1123. 10.1111/1469-0691.12727. [DOI] [PubMed] [Google Scholar]
- 14.Lascols C, Peirano G, Hackel M, Laupland KB, Pitout JD. 2013. Surveillance and molecular epidemiology of Klebsiella pneumoniae isolates that produce carbapenemases: first report of OXA-48-like enzymes in North America. Antimicrob Agents Chemother 57:130–136. 10.1128/AAC.01686-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palacios-Baena ZR, Oteo J, Conejo C, Larrosa MN, Bou G, Fernandez-Martinez M, Gonzalez-Lopez JJ, Pintado V, Martinez-Martinez L, Merino M, Pomar V, Mora-Rillo M, Rivera MA, Oliver A, Ruiz-Carrascoso G, Ruiz-Garbajosa P, Zamorano L, Bautista V, Ortega A, Morales I, Pascual A, Campos J, Rodriguez-Bano J, Geih G, GEIH-GEMARA (SEIMC) and REIPI Group for CPE . 2016. Comprehensive clinical and epidemiological assessment of colonisation and infection due to carbapenemase-producing Enterobacteriaceae in Spain. J Infect 72:152–160. 10.1016/j.jinf.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 16.Dortet L, Poirel L, Al Yaqoubi F, Nordmann P. 2012. NDM-1, OXA-48 and OXA-181 carbapenemase-producing Enterobacteriaceae in Sultanate of Oman. Clin Microbiol Infect 18:E144–E148. 10.1111/j.1469-0691.2012.03796.x. [DOI] [PubMed] [Google Scholar]
- 17.Wang S, Zhao SY, Xiao SZ, Gu FF, Liu QZ, Tang J, Guo XK, Ni YX, Han LZ. 2016. Antimicrobial resistance and molecular epidemiology of Escherichia coli causing bloodstream infections in three hospitals in Shanghai, China. PLoS One 11:e0147740. 10.1371/journal.pone.0147740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bouguenoun W, Bakour S, Bentorki AA, Al Bayssari C, Merad T, Rolain JM. 2016. Molecular epidemiology of environmental and clinical carbapenemase-producing Gram-negative bacilli from Hospitals in Guelma, Algeria: multiple genetic lineages and first report of OXA-48 in Enterobacter cloacae. J Glob Antimicrob Resist 7:135–140. 10.1016/j.jgar.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 19.Zowawi HM, Sartor AL, Balkhy HH, Walsh TR, Al Johani SM, AlJindan RY, Alfaresi M, Ibrahim E, Al-Jardani A, Al-Abri S, Al Salman J, Dashti AA, Kutbi AH, Schlebusch S, Sidjabat HE, Paterson DL. 2014. Molecular characterization of carbapenemase-producing Escherichia coli and Klebsiella pneumoniae in the countries of the Gulf Cooperation Council: dominance of OXA-48 and NDM producers. Antimicrob Agents Chemother 58:3085–3090. 10.1128/AAC.02050-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee CR, Lee JH, Park KS, Kim YB, Jeong BC, Lee SH. 2016. Global dissemination of carbapenemase-producing Klebsiella pneumoniae: epidemiology, genetic context, treatment options, and detection methods. Front Microbiol 7:895. 10.3389/fmicb.2016.00895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Duin D, Doi Y. 2017. The global epidemiology of carbapenemase-producing Enterobacteriaceae. Virulence 8:460–469. 10.1080/21505594.2016.1222343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Antunes NT, Lamoureaux TL, Toth M, Stewart NK, Frase H, Vakulenko SB. 2014. Class D β-lactamases: are they all carbapenemases? Antimicrob Agents Chemother 58:2119–2125. 10.1128/AAC.02522-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karlowsky JA, Lob SH, Kazmierczak KM, Badal RE, Young K, Motyl MR, Sahm DF. 2017. In vitro activity of imipenem against carbapenemase-positive Enterobacteriaceae isolates collected by the SMART Global Surveillance Program from 2008 to 2014. J Clin Microbiol 55:1638–1611. 10.1128/JCM.02316-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Jonge BL, Karlowsky JA, Kazmierczak KM, Biedenbach DJ, Sahm DF, Nichols WW. 2016. In vitro susceptibility to ceftazidime-avibactam of carbapenem-nonsusceptible Enterobacteriaceae isolates collected during the INFORM global surveillance study (2012 to 2014). Antimicrob Agents Chemother 60:3163–3169. 10.1128/AAC.03042-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bush K, Bradford PA. 2020. Epidemiology of β-lactamase-producing pathogens. Clin Microbiol Rev 33:e00047-19. 10.1128/CMR.00047-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pitout JDD, Peirano G, Kock MM, Strydom KA, Matsumura Y. 2019. The global ascendency of OXA-48-type carbapenemases. Clin Microbiol Rev 33:e00102-19. 10.1128/CMR.00102-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Docquier JD, Calderone V, De Luca F, Benvenuti M, Giuliani F, Bellucci L, Tafi A, Nordmann P, Botta M, Rossolini GM, Mangani S. 2009. Crystal structure of the OXA-48 β-lactamase reveals mechanistic diversity among class D carbapenemases. Chem Biol 16:540–547. 10.1016/j.chembiol.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 28.Golemi D, Maveyraud L, Vakulenko S, Samama J-P, Mobashery S. 2001. Critical involvement of a carbamylated lysine in catalytic function of class D β-lactamases. Proc Natl Acad Sci U S A 98:14281–14285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zong Z. 2012. Discovery of bla(OXA-199), a chromosome-based bla(OXA-48)-like variant, in Shewanella xiamenensis. PLoS One 7:e48280. 10.1371/journal.pone.0048280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dabos L, Jousset AB, Bonnin RA, Fortineau N, Zavala A, Retailleau P, Iorga BI, Naas T. 2018. Genetic and Biochemical Characterization of OXA-535, a Distantly Related OXA-48-Like β-Lactamase. Antimicrob Agents Chemother 62:e01198-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oueslati S, Nordmann P, Poirel L. 2015. Heterogeneous Hydrolytic Features for OXA-48-like β-lactamases. J Antimicrob Chemother 70:1059–1063. 10.1093/jac/dku524. [DOI] [PubMed] [Google Scholar]
- 32.Poirel L, Heritier C, Tolun V, Nordmann P. 2004. Emergence of oxacillinase-mediated resistance to imipenem in Klebsiella pneumoniae. Antimicrob Agents Chemother 48:15–22. 10.1128/aac.48.1.15-22.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kasap M, Torol S, Kolayli F, Dundar D, Vahaboglu H. 2013. OXA-162, a novel variant of OXA-48 displays extended hydrolytic activity towards imipenem, meropenem and doripenem. J Enzyme Inhib Med Chem 28:990–996. 10.3109/14756366.2012.702343. [DOI] [PubMed] [Google Scholar]
- 34.Potron A, Nordmann P, Lafeuille E, Al Maskari Z, Al Rashdi F, Poirel L. 2011. Characterization of OXA-181, a carbapenem-hydrolyzing class D β-lactamase from Klebsiella pneumoniae. Antimicrob Agents Chemother 55:4896–4899. 10.1128/AAC.00481-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poirel L, Castanheira M, Carrer A, Rodriguez CP, Jones RN, Smayevsky J, Nordmann P. 2011. OXA-163, an OXA-48-related class D β-lactamase with extended activity toward expanded-spectrum cephalosporins. Antimicrob Agents Chemother 55:2546–2551. 10.1128/AAC.00022-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dortet L, Oueslati S, Jeannot K, Tande D, Naas T, Nordmann P. 2015. Genetic and biochemical characterization of OXA-405, an OXA-48-type extended-spectrum β-lactamase without significant carbapenemase activity. Antimicrob Agents Chemother 59:3823–3828. 10.1128/AAC.05058-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hrabak J, Chudackova E, Papagiannitsis CC. 2014. Detection of carbapenemases in Enterobacteriaceae: a challenge for diagnostic microbiological laboratories. Clin Microbiol Infect 20:839–853. 10.1111/1469-0691.12678. [DOI] [PubMed] [Google Scholar]
- 38.Stojanoski V, Hu L, Sankaran B, Wang F, Tao P, Prasad BVV, Palzkill T. 2021. Mechanistic basis of OXA-48-like β-lactamases’ hydrolysis of carbapenems. ACS Infect Dis 7:445–460. 10.1021/acsinfecdis.0c00798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hermann JC, Ridder L, Mulholland AJ, Höltje H-D. 2003. Identification of Glu166 as the general base in the acylation reaction of class a β-lactamases through QM/MM modeling. J Am Chem Soc 125:9590–9591. 10.1021/ja034434g. [DOI] [PubMed] [Google Scholar]
- 40.Hermann JC, Ridder L, Holtje HD, Mulholland AJ. 2006. Molecular mechanisms of antibiotic resistance: QM/MM modelling of deacylation in a class A β-lactamase. Org Biomol Chem 4:206–210. 10.1039/B512969A. [DOI] [PubMed] [Google Scholar]
- 41.Maveyraud L, Golemi-Kotra D, Ishiwata A, Meroueh O, Mobashery S, Samama J-P. 2002. High-resolution X-ray structure of an acyl-enzyme species for the class D OXA-10 β-lactamase. J Am Chem Soc 124:2461–2465. 10.1021/ja016736t. [DOI] [PubMed] [Google Scholar]
- 42.King DT, King AM, Lal SM, Wright GD, Strynadka NC. 2015. Molecular mechanism of avibactam-mediated β-lactamase inhibition. ACS Infect Dis 1:175–184. 10.1021/acsinfecdis.5b00007. [DOI] [PubMed] [Google Scholar]
- 43.van Groesen E, Lohans CT, Brem J, Aertker KMJ, Claridge TDW, Schofield CJ. 2019. 19F NMR monitoring of reversible protein post-translational modifications: class D β-lactamase carbamylation and inhibition. Chem Eur J 25:11837–11841. 10.1002/chem.201902529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Santillana E, Beceiro A, Bou G, Romero A. 2007. Crystal structure of the carbapenemase OXA-24 reveals insights into the mechanism of carbapenem hydrolysis. Proc Natl Acad Sci U S A 104:5354–5359. 10.1073/pnas.0607557104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Akhter S, Lund BA, Ismael A, Langer M, Isaksson J, Christopeit T, Leiros HS, Bayer A. 2018. A focused fragment library targeting the antibiotic resistance enzyme—oxacillinase-48: synthesis, structural evaluation and inhibitor design. Eur J Med Chem 145:634–648. 10.1016/j.ejmech.2017.12.085. [DOI] [PubMed] [Google Scholar]
- 46.Smith CA, Stewart NK, Toth M, Vakulenko SB. 2019. Structural insights into the mechanism of carbapenemase activity of the OXA-48 β-lactamase. Antimicrob Agents Chemother 63:e01202-19. 10.1128/AAC.01202-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Akhtar A, Pemberton OA, Chen Y. 2020. Structural basis for substrate specificity and carbapenemase activity of OXA-48 class D β-lactamase. ACS Infect Dis 6:261–271. 10.1021/acsinfecdis.9b00304. [DOI] [PubMed] [Google Scholar]
- 48.Papp-Wallace KM, Kumar V, Zeiser ET, Becka SA, van den Akker F. 2019. Structural analysis of the OXA-48 carbapenemase bound to a “poor” carbapenem substrate, doripenem. Antibiotics (Basel) 8:145. 10.3390/antibiotics8030145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Luca F, Benvenuti M, Carboni F, Pozzi C, Rossolini GM, Mangani S, Docquier JD. 2011. Evolution to carbapenem-hydrolyzing activity in noncarbapenemase class D β-lactamase OXA-10 by rational protein design. Proc Natl Acad Sci U S A 108:18424–18429. 10.1073/pnas.1110530108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oueslati S, Retailleau P, Marchini L, Berthault C, Dortet L, Bonnin RA, Iorga BI, Naas T. 2020. Role of arginine 214 in the substrate specificity of OXA-48. Antimicrob Agents Chemother 64:e02329-19. 10.1128/AAC.02329-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Potron A, Rondinaud E, Poirel L, Belmonte O, Boyer S, Camiade S, Nordmann P. 2013. Genetic and biochemical characterisation of OXA-232, a carbapenem-hydrolysing class D β-lactamase from Enterobacteriaceae. Int J Antimicrob Agents 41:325–329. 10.1016/j.ijantimicag.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 52.Dabos L, Zavala A, Bonnin RA, Beckstein O, Retailleau P, Iorga BI, Naas T. 2020. Substrate specificity of OXA-48 after β5-β6 loop replacement. ACS Infect Dis 6:1032–1043. 10.1021/acsinfecdis.9b00452. [DOI] [PubMed] [Google Scholar]
- 53.Taibi P, Mobashery S. 1995. Mechanism of turnover of imipenem by the TEM β-lactamase revisited. J Am Chem Soc 117:7600–7605. 10.1021/ja00134a003. [DOI] [Google Scholar]
- 54.Fonseca F, Chudyk EI, Van der Kamp MW, Correia A, Mulholland AJ, Spencer J. 2012. The basis for carbapenem hydrolysis by class A β-lactamases: a combined investigation using crystallography and simulations. J Am Chem Soc 134:18275–18285. 10.1021/ja304460j. [DOI] [PubMed] [Google Scholar]
- 55.Tremblay LW, Fan F, Blanchard JS. 2010. Biochemical and structural characterization of Mycobacterium tuberculosis β-lactamase with the carbapenems ertapenem and doripenem. Biochemistry 49:3766–3773. 10.1021/bi100232q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kalp M, Carey PR. 2008. Carbapenems and SHV-1 β-lactamase form different acyl-enzyme populations in crystals and solution. Biochemistry 47:11830–11837. 10.1021/bi800833u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schneider KD, Ortega CJ, Renck NA, Bonomo RA, Powers RA, Leonard DA. 2011. Structures of the class D carbapenemase OXA-24 from Acinetobacter baumannii in complex with doripenem. J Mol Biol 406:583–594. 10.1016/j.jmb.2010.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith CA, Antunes NT, Stewart NK, Toth M, Kumarasiri M, Chang M, Mobashery S, Vakulenko SB. 2013. Structural basis for carbapenemase activity of the OXA-23 β-lactamase from Acinetobacter baumannii. Chem Biol 20:1107–1115. 10.1016/j.chembiol.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lohans CT, Freeman EI, Groesen EV, Tooke CL, Hinchliffe P, Spencer J, Brem J, Schofield CJ. 2019. Mechanistic insights into β-lactamase-catalysed carbapenem degradation through product characterisation. Sci Rep 9:13608. 10.1038/s41598-019-49264-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lohans CT, van Groesen E, Kumar K, Tooke CL, Spencer J, Paton RS, Brem J, Schofield CJ. 2018. A new mechanism for β-lactamases: class D enzymes degrade 1β-methyl carbapenems through lactone formation. Angew Chem Int Ed 57:1282–1285. 10.1002/anie.201711308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aertker KMJ, Chan HTH, Lohans CT, Schofield CJ. 2020. Analysis of β-lactone formation by clinically observed carbapenemases informs on a novel antibiotic resistance mechanism. J Biol Chem 295:16604–16613. 10.1074/jbc.RA120.014607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fröhlich C, Sorum V, Thomassen AM, Johnsen PJ, Leiros HS, Samuelsen O. 2019. OXA-48-mediated ceftazidime-avibactam resistance is associated with evolutionary trade-offs. mSphere 4:e00024-19. 10.1128/mSphere.00024-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mitchell JM, Clasman JR, June CM, Kaitany KC, LaFleur JR, Taracila MA, Klinger NV, Bonomo RA, Wymore T, Szarecka A, Powers RA, Leonard DA. 2015. Structural basis of activity against aztreonam and extended spectrum cephalosporins for two carbapenem-hydrolyzing class D β-lactamases from Acinetobacter baumannii. Biochemistry 54:1976–1987. 10.1021/bi501547k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hirvonen VHA, Mulholland AJ, Spencer J, Van der Kamp MW. 2020. Small changes in hydration determine cephalosporinase activity of OXA-48 β-lactamases. ACS Catal 10:6188–6196. 10.1021/acscatal.0c00596. [DOI] [Google Scholar]
- 65.Drawz SM, Bonomo RA. 2010. Three decades of β-lactamase inhibitors. Clin Microbiol Rev 23:160–201. 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Toussaint KA, Gallagher JC. 2015. β-Lactam/β-lactamase inhibitor combinations: from then to now. Ann Pharmacother 49:86–98. 10.1177/1060028014556652. [DOI] [PubMed] [Google Scholar]
- 67.Tehrani KHME, Martin NI. 2018. β-Lactam/β-lactamase inhibitor combinations: an update. Medchemcomm 9:1439–1456. 10.1039/C8MD00342D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhanel GG, Lawson CD, Adam H, Schweizer F, Zelenitsky S, Lagace-Wiens PR, Denisuik A, Rubinstein E, Gin AS, Hoban DJ, LynchJP, III, Karlowsky JA. 2013. Ceftazidime-avibactam: a novel cephalosporin/β-lactamase inhibitor combination. Drugs 73:159–177. 10.1007/s40265-013-0013-7. [DOI] [PubMed] [Google Scholar]
- 69.Stewart A, Harris P, Henderson A, Paterson D. 2018. Treatment of infections by OXA-48-producing Enterobacteriaceae. Antimicrob Agents Chemother 62:e01195-18. 10.1128/AAC.01195-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Kern G, Walkup GK, Fisher SL. 2012. Avibactam is a covalent, reversible, non-β-lactam β-lactamase inhibitor. Proc Natl Acad Sci U S A 109:11663–11668. 10.1073/pnas.1205073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang DY, Abboud MI, Markoulides MS, Brem J, Schofield CJ. 2016. The road to avibactam: the first clinically useful non-β-lactam working somewhat like a β-lactam. Future Med Chem 8:1063–1084. 10.4155/fmc-2016-0078. [DOI] [PubMed] [Google Scholar]
- 72.Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Durand-Reville TF, Lahiri S, Thresher J, Livchak S, Gao N, Palmer T, Walkup GK, Fisher SL. 2013. Kinetics of avibactam inhibition against class A, C, and D β-lactamases. J Biol Chem 288:27960–27971. 10.1074/jbc.M113.485979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aktaş Z, Kayacan C, Oncul O. 2012. In vitro activity of avibactam (NXL104) in combination with β-lactams against Gram-negative bacteria, including OXA-48 β-lactamase-producing Klebsiella pneumoniae. Int J Antimicrob Agents 39:86–89. 10.1016/j.ijantimicag.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 74.Sousa A, Perez-Rodriguez MT, Soto A, Rodriguez L, Perez-Landeiro A, Martinez-Lamas L, Nodar A, Crespo M. 2018. Effectiveness of ceftazidime/avibactam as salvage therapy for treatment of infections due to OXA-48 carbapenemase-producing Enterobacteriaceae. J Antimicrob Chemother 73:3170–3175. 10.1093/jac/dky295. [DOI] [PubMed] [Google Scholar]
- 75.Kazmierczak KM, Bradford PA, Stone GG, de Jonge BLM, Sahm DF. 2018. In vitro activity of ceftazidime-avibactam and aztreonam-avibactam against OXA-48-carrying Enterobacteriaceae isolated as part of the International Network for Optimal Resistance Monitoring (INFORM) global surveillance program from 2012 to 2015. Antimicrob Agents Chemother 62:e00592-18. 10.1128/AAC.00592-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vasoo S, Cunningham SA, Cole NC, Kohner PC, Menon SR, Krause KM, Harris KA, De PP, Koh TH, Patel R. 2015. In vitro activities of ceftazidime-avibactam, aztreonam-avibactam, and a panel of older and contemporary antimicrobial agents against carbapenemase-producing Gram-negative bacilli. Antimicrob Agents Chemother 59:7842–7846. 10.1128/AAC.02019-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Durand-Reville TF, Guler S, Comita-Prevoir J, Chen B, Bifulco N, Huynh H, Lahiri S, Shapiro AB, McLeod SM, Carter NM, Moussa SH, Velez-Vega C, Olivier NB, McLaughlin R, Gao N, Thresher J, Palmer T, Andrews B, Giacobbe RA, Newman JV, Ehmann DE, de Jonge B, O’Donnell J, Mueller JP, Tommasi RA, Miller AA. 2017. ETX2514 is a broad-spectrum β-lactamase inhibitor for the treatment of drug-resistant Gram-negative bacteria including Acinetobacter baumannii. Nat Microbiol 2:17104. 10.1038/nmicrobiol.2017.104. [DOI] [PubMed] [Google Scholar]
- 78.Lahiri SD, Mangani S, Jahic H, Benvenuti M, Durand-Reville TF, De Luca F, Ehmann DE, Rossolini GM, Alm RA, Docquier JD. 2015. Molecular basis of selective inhibition and slow reversibility of avibactam against class D carbapenemases: a structure-guided study of OXA-24 and OXA-48. ACS Chem Biol 10:591–600. 10.1021/cb500703p. [DOI] [PubMed] [Google Scholar]
- 79.Lomovskaya O, Sun D, Rubio-Aparicio D, Nelson K, Tsivkovski R, Griffith DC, Dudley MN. 2017. Vaborbactam: spectrum of β-lactamase inhibition and impact of resistance mechanisms on activity in Enterobacteriaceae. Antimicrob Agents Chemother 61:e01443-17. 10.1128/AAC.01443-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schmidt-Malan SMS, Mishra AJ, Mushtaq A, Brinkman CL, Patel R. 2018. In vitro activity of imipenem-relebactam and ceftolozane-tazobactam against resistant Gram-negative bacilli. Antimicrob Agents Chemother 62:e00533-18. 10.1128/AAC.00533-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tselepis L, Langley GW, Aboklaish AF, Widlake E, Jackson DE, Walsh TR, Schofield CJ, Brem J, Tyrrell JM. 2020. In vitro efficacy of imipenem-relebactam and cefepime-AAI101 against a global collection of ESBL-positive and carbapenemase-producing Enterobacteriaceae. Int J Antimicrob Agents 56:105925. 10.1016/j.ijantimicag.2020.105925. [DOI] [PubMed] [Google Scholar]
- 82.Livermore DM, Mushtaq S, Warner M, Vickers A, Woodford N. 2017. In vitro activity of cefepime/zidebactam (WCK 5222) against Gram-negative bacteria. J Antimicrob Chemother 72:1373–1385. 10.1093/jac/dkw593. [DOI] [PubMed] [Google Scholar]
- 83.Papp-Wallace KM, Nguyen NQ, Jacobs MR, Bethel CR, Barnes MD, Kumar V, Bajaksouzian S, Rudin SD, Rather PN, Bhavsar S, Ravikumar T, Deshpande PK, Patil V, Yeole R, Bhagwat SS, Patel MV, van den Akker F, Bonomo RA. 2018. Strategic approaches to overcome resistance against Gram-negative pathogens using β-lactamase inhibitors and β-lactam enhancers: activity of three novel diazabicyclooctanes WCK 5153, zidebactam (WCK 5107), and WCK 4234. J Med Chem 61:4067–4086. 10.1021/acs.jmedchem.8b00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Livermore DM, Mushtaq S, Warner M, Woodford N. 2015. Activity of OP0595/β-lactam combinations against Gram-negative bacteria with extended-spectrum, AmpC and carbapenem-hydrolysing β-lactamases. J Antimicrob Chemother 70:3032–3041. 10.1093/jac/dkv239. [DOI] [PubMed] [Google Scholar]
- 85.Shapiro AB, Gao N, Jahic H, Carter NM, Chen A, Miller AA. 2017. Reversibility of covalent, broad-spectrum serine β-lactamase inhibition by the diazabicyclooctenone ETX2514. ACS Infect Dis 3:833–844. 10.1021/acsinfecdis.7b00113. [DOI] [PubMed] [Google Scholar]
- 86.Entasis Therapeutics. 2020. Study to evaluate the efficacy and safety of intravenous sulbactam-ETX2514 in the treatment of patients with infections caused by Acinetobacter Baumannii-calcoaceticus complex (ATTACK). https://clinicaltrials.gov/ct2/show/NCT03894046. Accessed 7 July 2020.
- 87.King AM, King DT, French S, Brouillette E, Asli A, Alexander JA, Vuckovic M, Maiti SN, ParrTR, Jr, Brown ED, Malouin F, Strynadka NC, Wright GD. 2016. Structural and kinetic characterization of diazabicyclooctanes as dual inhibitors of both serine-β-lactamases and penicillin-binding proteins. ACS Chem Biol 11:864–868. 10.1021/acschembio.5b00944. [DOI] [PubMed] [Google Scholar]
- 88.Gordon EM, Duncton MAJ, Gallop MA. 2018. Orally absorbed derivatives of the β-lactamase inhibitor avibactam. Design of novel prodrugs of sulfate containing drugs. J Med Chem 61:10340–10344. 10.1021/acs.jmedchem.8b01389. [DOI] [PubMed] [Google Scholar]
- 89.Durand-Réville TF, Comita-Prevoir J, Zhang J, Wu X, May-Dracka TL, Romero JAC, Wu F, Chen A, Shapiro AB, Carter NM, McLeod SM, Giacobbe RA, Verheijen JC, Lahiri SD, Sacco MD, Chen Y, O’Donnell JP, Miller AA, Mueller JP, Tommasi RA. 2020. Discovery of an orally available diazabicyclooctane inhibitor (ETX0282) of class A, C, and D serine β-lactamases. J Med Chem 63:12511–12525. 10.1021/acs.jmedchem.0c00579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Reck F, Bermingham A, Blais J, Casarez A, Colvin R, Dean CR, Furegati M, Gamboa L, Growcott E, Li C, Lopez S, Metzger L, Nocito S, Ossola F, Phizackerley K, Rasper D, Shaul J, Shen X, Simmons RL, Tang D, Tashiro K, Yue Q. 2019. IID572: a new potentially best-in-class β-lactamase inhibitor. ACS Infect Dis 5:1045–1051. 10.1021/acsinfecdis.9b00031. [DOI] [PubMed] [Google Scholar]
- 91.Papp-Wallace KM, Bajaksouzian S, Bonomo RA, R Bethel C, Caillon J, Rutter JD, Reghal A, Jacqueline C. 2019. Beyond piperacillin-tazobactam: cefepime and AAI101 as a potent β-lactam–β-lactamase inhibitor combination. Antimicrob Agents Chemother 63:e00105-19. 10.1128/AAC.00105-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bernhard F, Odedra R, Sordello S, Cardin R, Franzoni S, Charrier C, Belley A, Warn P, Machacek M, Knechtle P. 2020. Pharmacokinetics-pharmacodynamics of enmetazobactam combined with cefepime in a neutropenic murine thigh infection model. Antimicrob Agents Chemother 64:e00078-20. 10.1128/AAC.00078-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bush K, Bradford PA. 2019. Interplay between β-lactamases and new β-lactamase inhibitors. Nat Rev Microbiol 17:295–306. 10.1038/s41579-019-0159-8. [DOI] [PubMed] [Google Scholar]
- 94.Cahill ST, Cain R, Wang DY, Lohans CT, Wareham DW, Oswin HP, Mohammed J, Spencer J, Fishwick CW, McDonough MA, Schofield CJ, Brem J. 2017. Cyclic boronates inhibit all classes of β-lactamases. Antimicrob Agents Chemother 61:e02260-16. 10.1128/AAC.02260-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Brem J, Cain R, Cahill S, McDonough MA, Clifton IJ, Jimenez-Castellanos JC, Avison MB, Spencer J, Fishwick CW, Schofield CJ. 2016. Structural basis of metallo-β-lactamase, serine-β-lactamase and penicillin-binding protein inhibition by cyclic boronates. Nat Commun 7:12406. 10.1038/ncomms12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Langley GW, Cain R, Tyrrell JM, Hinchliffe P, Calvopina K, Tooke CL, Widlake E, Dowson CG, Spencer J, Walsh TR, Schofield CJ, Brem J. 2019. Profiling interactions of vaborbactam with metallo-β-lactamases. Bioorg Med Chem Lett 29:1981–1984. 10.1016/j.bmcl.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hecker SJ, Reddy KR, Totrov M, Hirst GC, Lomovskaya O, Griffith DC, King P, Tsivkovski R, Sun D, Sabet M, Tarazi Z, Clifton MC, Atkins K, Raymond A, Potts KT, Abendroth J, Boyer SH, Loutit JS, Morgan EE, Durso S, Dudley MN. 2015. Discovery of a cyclic boronic acid β-lactamase inhibitor (RPX7009) with utility vs class A serine carbapenemases. J Med Chem 58:3682–3692. 10.1021/acs.jmedchem.5b00127. [DOI] [PubMed] [Google Scholar]
- 98.Cho JC, Zmarlicka MT, Shaeer KM, Pardo J. 2018. Meropenem/vaborbactam, the first carbapenem/β-lactamase inhibitor combination. Ann Pharmacother 52:769–779. 10.1177/1060028018763288. [DOI] [PubMed] [Google Scholar]
- 99.Lee Y, Kim J, Trinh S. 2019. Meropenem–vaborbactam (Vabomere™): another option for carbapenem-resistant Enterobacteriaceae. P T 44:110–113. [PMC free article] [PubMed] [Google Scholar]
- 100.Tsivkovski R, Lomovskaya O. 2020. Biochemical activity of vaborbactam. Antimicrob Agents Chemother 64:e01935-19. 10.1128/AAC.01935-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu B, Trout REL, Chu GH, McGarry D, Jackson RW, Hamrick JC, Daigle DM, Cusick SM, Pozzi C, De Luca F, Benvenuti M, Mangani S, Docquier JD, Weiss WJ, Pevear DC, Xerri L, Burns CJ. 2020. Discovery of taniborbactam (VNRX-5133): a broad-spectrum serine- and metallo-β-lactamase inhibitor for carbapenem-resistant bacterial infections. J Med Chem 63:2789–2801. 10.1021/acs.jmedchem.9b01518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Krajnc A, Brem J, Hinchliffe P, Calvopina K, Panduwawala TD, Lang PA, Kamps J, Tyrrell JM, Widlake E, Saward BG, Walsh TR, Spencer J, Schofield CJ. 2019. Bicyclic boronate VNRX-5133 inhibits metallo- and serine-β-lactamases. J Med Chem 62:8544–8556. 10.1021/acs.jmedchem.9b00911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.VenatoRx Pharmaceuticals, Inc. 2020. Safety and efficacy study of cefepime/VNRX-5133 in patients with complicated urinary tract infections. https://clinicaltrials.gov/ct2/show/NCT03840148. Accessed 7 July 2020.
- 104.Nelson K, Rubio-Aparicio D, Sun D, Dudley M, Lomovskaya O. 2020. In vitro activity of the ultra-broad-spectrum β-lactamase inhibitor QPX7728 against carbapenem-resistant Enterobacterales (CRE) with varying intrinsic and acquired resistance mechanisms. Antimicrob Agents Chemother 64:e00757-20. 10.1128/AAC.00757-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tsivkovski R, Totrov M, Lomovskay O. 2020. Biochemical characterization of QPX7728, a new ultrabroad-spectrum β-lactamase inhibitor of serine and metallo-β-lactamases. Antimicrob Agents Chemother 64:e00130-20. 10.1128/AAC.00130-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Qpex Biopharma, Inc. 2020. P1 single and multiple ascending dose (SAD/MAD) study of IV QPX7728 alone and combined with QPX2014 in NHV. https://clinicaltrials.gov/ct2/show/NCT04380207. Accessed 7 July 2020.
- 107.Business Wire. 2021. Qpex Biopharma initiates phase 1 clinical trial of QPX7728 for drug-resistant bacterial infections. https://www.businesswire.com/news/home/20201203005300/en/Qpex-Biopharma-Initiates-Phase-1-Clinical-Trial-of-QPX7728-for-Drug-Resistant-Bacterial-Infections. Accessed 2 February 2021.
- 108.Papp-Wallace KM. 2019. The latest advances in β-lactam/β-lactamase inhibitor combinations for the treatment of Gram-negative bacterial infections. Expert Opin Pharmacother 20:2169–2184. 10.1080/14656566.2019.1660772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.VenatoRx Pharmaceuticals, Inc. 2021. VNRX-7145 SAD/MAD safety and PK in healthy adult volunteers. https://clinicaltrials.gov/ct2/show/NCT04243863. Accessed 25 February 2021.
- 110.VenatoRx Pharmaceuticals, Inc. 2021. Ceftibuten/VNRX-7145. https://www.venatorx.com/ceftibuten-vnrx-7145/. Accessed 25 February 2021.
- 111.Taylor DM, Anglin J, Park S, Ucisik MN, Faver JC, Simmons N, Jin Z, Palaniappan M, Nyshadham P, Li F, Campbell J, Hu L, Sankaran B, Prasad BVV, Huang H, Matzuk MM, Palzkill T. 2020. Identifying oxacillinase-48 carbapenemase inhibitors using DNA-encoded chemical libraries. ACS Infect Dis 6:1214–1227. 10.1021/acsinfecdis.0c00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kaitany KC, Klinger NV, June CM, Ramey ME, Bonomo RA, Powers RA, Leonard DA. 2013. Structures of the class D carbapenemases OXA-23 and OXA-146: mechanistic basis of activity against carbapenems. Antimicrob Agents Chemother 57:4848–4855. 10.1128/AAC.00762-13. [DOI] [PMC free article] [PubMed] [Google Scholar]