

Abstract
Pulmonary arterial hypertension (PAH) is a devastating disease that involves pulmonary vasoconstriction, small vessel obliteration, large vessel thickening and obstruction, and development of plexiform lesions. PAH vasculopathy leads to progressive increases in pulmonary vascular resistance, right heart failure, and ultimately, premature death. Besides other cell types that are known to be involved in PAH pathogenesis (e.g. smooth muscle cells, fibroblasts, and leukocytes), recent studies demonstrate a crucial role of endothelial cells (ECs) in the initiation and progression of PAH. The EC-specific role in PAH is multi-faceted and impacts upon numerous pathophysiological processes including vasoconstriction, inflammation, coagulation, metabolism, and oxidative/nitrative stress, as well as cell viability, growth, and differentiation. In this review, we describe how EC dysfunction and cell signaling regulate the pathogenesis of PAH. We also highlight areas of research that warrant attention in future studies, and discuss potential molecular signaling pathways in ECs that could be targeted therapeutically in the prevention and treatment of PAH.
Keywords: Endothelial cells, pulmonary arterial hypertension, pulmonary hypertension, vascular remodeling
1. Introduction
Pulmonary hypertension (PH) is defined as an increase in mean pulmonary arterial pressure of >20 mmHg from the normal range of 10 to 20 mmHg at rest, as assessed by right heart catheterization [1]. PH is a heterogenous cardiopulmonary disease, which is divided into five groups, including pulmonary arterial hypertension (PAH, group I), PH due to left heart disease, PH due to lung disease and/or hypoxia, chronic thromboembolic PH, and PH with unclear multifactorial mechanisms [2]. PAH, including idiopathic PAH (IPAH), is characterized by a progressive rise in pulmonary vascular resistance and occlusive vascular remodeling, which leads to right heart failure and premature death. The histopathological features of PAH include intima and media thickening, muscularization of distal pulmonary arteries, vascular occlusion, and complex plexiform lesions [3–5]. Some of these histological features are also present in other groups of PH, but to a lesser extent. Despite major advances in the field over recent years, the underlying molecular mechanisms of obliterative vascular remodeling remain largely unknown. Current therapies are based on concepts of endothelial dysfunction developed in almost 3 decades ago targeting the endothelin, nitric oxide (NO), and prostacyclin pathways, and do not address the fundamental disease-modifying mechanisms. These have only resulted in a modest improvement in morbidity and mortality, and therefore, the ultimate treatment remains lung transplantation [6–8].
The healthy endothelial monolayer lining the inner wall of blood vessels regulates the flux of fluid, proteins and blood cells across the vessel wall into parenchymal tissue, maintains vascular tone and integrity as well as exerts anti-thrombotic and anti-inflammatory influences on the vascular bed [9]. However, endothelial cells (ECs) are damaged and/or dysfunctional in PAH patients [10–13]. Factors that can cause EC injury include hypoxia, toxins, inhibition of survival signaling (e.g. VEGF antagonists), recreational drug use, inflammatory cytokines, as well as pathological shear stress and fluid mechanics in the pulmonary circulation raised by left to right shunts in congenital heart disease. Shear stress is of particular importance in the pathogenesis of PAH, given the dramatic changes in arterial pressure and fluid dynamics that occur in the pulmonary circulation of PAH patients. Many characteristics of PAH are consequences of dysfunctional EC signaling; these characteristics include pulmonary inflammation and coagulation, oxidative/nitrative stress, altered vascular cell viability (e.g. apoptosis-resistance), proliferation, metabolic shift, and accumulations of inflammatory cells and fibroblasts [14–16]. Mice with Egln1 (encoding prolyl-4 hydroxylase-2, PHD2) deletion in ECs and bone marrow cells exhibit unprecedented severe PH recapitulating many features of clinical PAH including occlusive vascular remodeling and right heart failure [17], supporting the critical role of EC dysfunction in the pathogenesis of PAH (Figure 1).). It has been proposed that EC phenotypic changes contribute to the onset of PAH, for example in the cases of smoking-induced lung EC apoptosis or inherited epigenetic EC dysfunction [18–20]. On the other hand, dysfunctional EC phenotypes can manifest in parallel with PAH or after the onset of PAH, for example in the instances of chronic vascular inflammation or proliferative vasculopathy following anti-angiogenic therapy [21, 22]. In other words, the precise timing of the phenotypic changes from healthy to dysfunctional endothelium during the pathogenesis of PAH is unclear. The evolution of PAH-associated EC phenotypes likely depends on multiple variables such as the EC phenotype and subpopulation being studied, the disease type and severity, and the patient’s genetic inheritance and demographics including age, sex, and other co-existing PAH risk factors.
Figure 1.
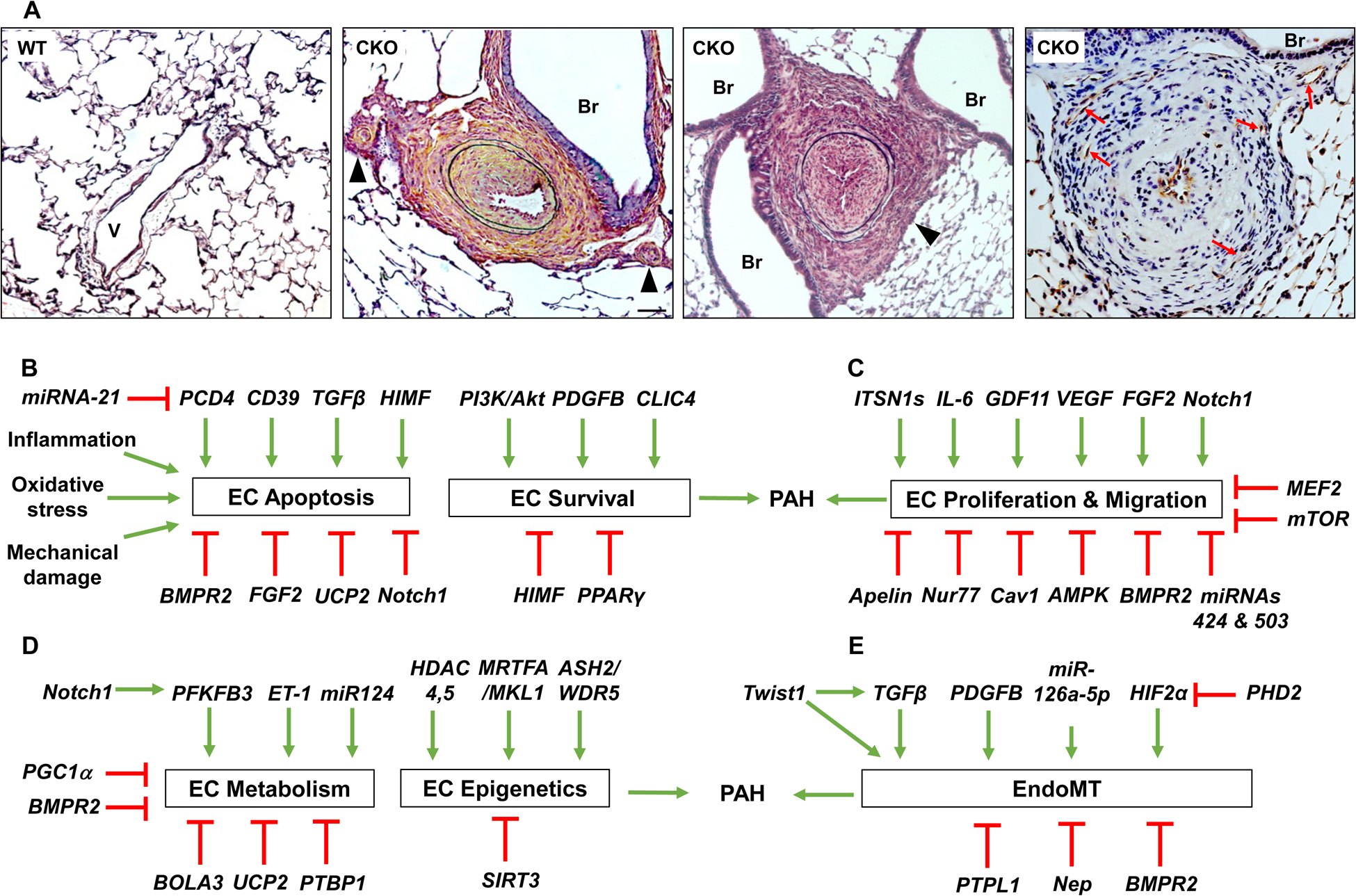
(A) PHD2 deficiency in endothelial cells and hematopoietic cells induces obliterative vascular remodeling recapitulating the histopathological features of clinical PAH. Representative micrographs of Russel-Movat pentachrome staining demonstrating thickening of the intima, medial, and adventitial, and occlusion of the large and small vessels (black arrowheads) in 3.5 mo. old Egln1Tie2Cre mice (Middle 2). Br, bronchus; V, vessel. Scale bar: 50 μm. Anti-CD31 immunohistochemistry showing multiple-channel lesions positive for the endothelial marker CD31 (red arrows) (Right). Scale bar: 50 μm. Figure adapted with permission from Dai, et al. [17]. (B-E) Factors regulating endothelial cell functions contributing to pulmonary vascular remodeling and PAH. (B) Factors regulating pulmonary EC apoptosis and survival in the pathogenesis of PAH. (C) Factors regulating pulmonary EC proliferation and migration contributing to PAH. (D) Factors affecting pulmonary EC metabolism (glycolysis switch) and epigenetic regulation. (E) Factors regulating EndoMT contributing to PAH. Abbreviations: AMPK, AMP-activated protein kinase; BMPR2, bone morphogenic protein receptor 2; Cav1, caveolin 1; CLIC4, chloride intracellular channel 4; FGF2, fibroblast growth factor 2; GDF11, growth differentiation factor 11; GrzB, granzyme B; HIF2α, hypoxia-inducible factor 2α; HIMF, hypoxia-induced mitogenic factor; IL, interleukin; ITSN 1s, intersectin 1 short; MEF2, myocyte enhancer factor 2; miRNA, micro RNA; mTOR, mammalian target of rapamycin; Nep, neprilysin; PCD4, programmed cell death 4; PDGFB, platelet-derived growth factor-BB; PHD2, prolyl hydroxylase 2; PI3K, phosphoinositide 3-kinase; PPARγ, peroxisome proliferator-activated receptor γ; PTPL1, protein tyrosine phosphatase; TGFβ, transforming growth factor β; VEGF, vascular endothelial growth factor.
Although current treatments for PAH can reduce disease symptoms and delay clinical worsening in PAH patients, a therapy or combination of therapies that prevents the onset of PAH or completely alleviates the disease is lacking. This review will describe the abnormal endothelial signaling pathways that contribute to the initiation and development of PAH and describe how a dysfunctional endothelium regulates PAH pathogenesis and progression. We will also highlight areas of research that could ultimately support the development of EC-targeted therapies against PAH and identify future studies that could improve understanding of obliterative vascular remodeling and PAH pathogenesis.
2. Pulmonary EC Phenotypes
The early stage of PAH development involves EC injury and apoptosis, while apoptosis-resistant ECs emerge later as PAH progresses [23–25]. In a transgenic mouse model with Fas-induced EC apoptosis, PH and pulmonary arteriopathy are observed, providing direct evidence that lung EC damage acts as a trigger to initiate PAH [26]. In the late stages of PAH, hyperproliferative and apoptosis-resistant ECs predominate, contributing to the formation of plexiform lesions [24, 25, 27, 28]. In distal pulmonary arteries of lungs from IPAH patients, there are increased numbers of proliferating ECs and decreased numbers of apoptotic ECs [29]. These observations are also seen in vitro, with pulmonary ECs from IPAH patients exhibiting increased proliferation and reduced sensitivity to apoptosis [29, 30]. In the end stage of PAH, there is evidence of endothelial senescence. A switch from proliferative to senescent vascular phenotype contributes to the loss of reversibility of PAH [31]. Dysfunctional EC signaling also results in increased coagulability and decreased EC integrity, which contribute to the development of PAH [32]. PAH pathogenesis is often associated with aberrant EC barrier integrity and IPAH patients commonly demonstrate a hypercoagulable phenotype [33]. Additionally, it is increasingly being recognized that alterations in multiple metabolic and epigenetic pathways are driving the development of PAH [34]. Survival and hyperproliferation of the PAH endothelium requires increased glutamine metabolism through the tricarboxylic acid cycle (TCA) and PAH patients exhibit systemic and lung-specific changes in glutamine metabolism, with PAH lung vasculature taking up more glutamine than healthy controls [35].
However, It is important to note here, that pulmonary ECs comprise of separate subpopulations of ECs, including proximal pulmonary artery ECs (PAECs) and distal microvascular ECs, which may be subject to different injurious stimuli and mechanical forces according to their position in the pulmonary vasculature [36, 37]. Moreover, the alveolar endothelium can be resolved by single cell transcriptomics into at least two specialized capillary EC phenotypes characterized by expression or apelin and its receptor, respectively, that play specialized role in gas exchange and repair[38]. This EC heterogeneity could therefore affect severity of aberrant EC phenotypes observed in PAH, including EC proliferative potential. This section provides an overview of EC-expressed factors that control the aberrant EC phenotypes seen in PAH (Figure 1).
2.1. Factors affecting EC survival
PAH is hereditable in about 10% of cases, and the vast majority of hereditary PAH patients harbor heterozygous mutations in BMPR2 [39]. Loss of BMPR2 is an initiating factor for PAH [39], heterozygous Bmpr2 knockout causes EC injury and persistent PH in mice [40, 41], and genetic ablation of Bmpr2 in pulmonary ECs predisposes to PH [42, 43]. BMPR2 mediates pro-survival signaling in pulmonary artery ECs (PAECs) [41, 44]. Overexpression of mutant BMPR2 in human PAECs increases susceptibility to apoptosis [39]. Thus, as in the experimental models [26], EC apoptosis appears to represent a potential initiating mechanism in the pathogenesis of human PAH as well. Meanwhile, the BMPR2 ligand, BMP9, prevents EC apoptosis and enhances EC monolayer integrity, and inhibits PH induced by BMPR2 mutations, monocrotaline (MCT)-, or Sugen/hypoxia (SuHx) [45]. Furthermore, the BMPR2 activator, FK506, improves endothelial function, inhibits apoptosis and reverses PH in hypoxic mice [46]. However, another study has demonstrated that inhibition of BMP9 signaling partially protects against experimental PH [47]. These data suggest that the TGF-beta/BMP pathway is highly complex and the effects of BMP9 may depend on other factors [48]. It has also been shown that BMPR2 mediates the transcriptional complex between PPARγ and β-catenin in PAECs [49]. Apelin is a downstream target of this complex. Apelin-deficient PAECs are prone to apoptosis and promote PA smooth muscle cell (PASMC) proliferation [49]. Apelin treatment can increase CD39 ATPase enzymatic activity in PAECs [50] whereas repression of CD39 in vitro results in an ATP-enriched environment, that acts as a phenotypic switch promoting apoptosis-resistance in PAECs via the P2Y11 receptor [50]. Indeed, genetic deletion of CD39 [51] and the apelin/APJ system [52] augments hypoxia-induced PH in mice. Other factors including CLIC4, PDGF-B and HIMF also regulate PAEC survival and PH development [53–57].
2.2. Factors affecting EC proliferation
In later stages of PAH, EC proliferation is a dominant feature leading to complex arterial remodeling. Several pathways have been shown involved in this transition. The loss of PPARγ is associated with PAH development [58, 59]. Inhibition of PPARγ in PAECs upregulates expression of cell cycle genes, worsens VEGF-induced EC barrier dysfunction [60], and attenuates the migration and angiogenic capacity of pulmonary microvascular ECs [61]. PPARγ also maintains EC homeostasis via UBR5/ATMIN-mediated DNA repair [62]. Accordingly, endothelial deletion of PPARγ induces spontaneous PH and impairs recovery from hypoxia-induced PH in mice [63]. The role of endothelial PHD2 in the development of PAH has recently been studied [17, 64, 65]. Mice with Tie2Cre-mediated disruption of PHD2 in ECs and hematopoietic cells exhibit severe PH, and occlusive vascular remodeling [17]. Marked increases in EC proliferation are seen in the pulmonary vascular lesions of these mice. In IPAH patients, PHD2 expression is diminished in ECs of the occlusive vascular lesions. PHD2 deficiency-induced PAH is mediated by endothelial activation of HIF-2α [17, 64, 66], which alters the expression of many of the PAH-causing factors. Genetic deletion of endothelial HIF-2a inhibits PH development in hypoxic mice [66, 67]. Pharmacological inhibition of HIF-2α inhibits PH in experimental mouse and rat models and promotes survival [66, 68]. HIF2A mutation is identified in IPAH patients [69] and mice with the mutation exhibit PH [70], thus, HIF-2α is emerging as a promising target of PAH therapy.
Caveolin1 expression is markedly decreased in pulmonary vascular ECs of IPAH patients [71, 72]. Inheritable mutations have been reported in CAV1 in PAH patients [73], and Cav1−/− mice develop PH [74], while re-expression of Caveolin1 in endothelium rescues PH in Cav1−/− mice [75]. Treatment with Cavtrin, a Caveolin1 mimic peptide, inhibits EC proliferation and promotes apoptosis [76] whereas Caveolin1 deficiency induces PAEC proliferation [72]. Consistently, disruption of Cav1 in ECs augments hypoxia-induced PH [72]. mTOR [77] and Nur 77 [78] are also negative regulators of PAEC proliferation and protective against PH development.
A number of other factors are also involved in PAEC proliferation contributing to the pathogenesis of PAH. For example, Granzyme B cleaves intersectin-1s, generating a N-terminal fragment which enhances EC proliferation [79]. IL-6 stimulates EC proliferation and increases endothelin 1 expression in ECs [80, 81]. p130Cas may modulate PAEC migration and proliferation by acting as an amplifier of receptor tyrosine kinase downstream signals [82]. Upregulation of GDF11 enhances the aberrant angiogenesis and proliferation in PAECs induced by hypoxia or VEGF treatment [83]. Endothelial dysfunction is strongly associated with oxidative and nitrative stress and the anti-oxidant, TEMPOL or MitoQ decreases migration and proliferation of ECs [84]. Inhibition of reactive oxygen species (ROS)-induced calcium entry also attenuates EC migration and proliferation [84].
Van der Feen et al showed that the loss of reversibility of the pulmonary arterial remodeling in a congenital heart disease PAH model induced by MCT and aortocaval shunt is related to an EC phenotypic switch from proliferation to senescence [31]. Cultured pulmonary ECs from PAH patients are more prone to becoming senescent in response to shear stress and the senescent cells are more sensitive to senolytic ABT263-induced apoptosis. Treatment of end-stage PH rats with ABT263 to target vascular cell senescence reversed the hemodynamic and structural changes. These studies demonstrate a new way to reverse end-stage PAH.
2.3. Factors affecting both EC survival and proliferation
Studies have also identified several factors that affect both EC proliferation and survival. For example, PAECs from IPAH patients exhibit increased FGF2 expression. Disruption of FGF2 signaling normalizes IPAH EC sensitivity to apoptosis and proliferation [29]. Apelin also regulates EC survival and proliferation [49]. Kim et al. described a miRNA-dependent association between apelin and FGF2 in PAECs [85] in which Apelin deficiency in PAECs leads to increased expression of FGF2 as a result of decreased expression of miR-424 and miR-503 mediated by MEF2. MEF2 activity is impaired in PAECs from IPAH patients due to excessive nuclear accumulation of class IIa histone deacetylases, HDAC4 and HDAC5. Indeed, pharmacological inhibition of class IIa HDACs restored MEF2 activity, decreasing cell migration and proliferation in PAECs and rescued experimental PH. A recent study shows endostatin, a cleavage product of Col18A1, inhibits EC migration via ID1/TSP-1/CD36 signaling and proliferation and apoptosis through CD36 and CD47 [86]. Elevated serum endostatin is associated with increased mortality and disease severity in PAH and a COL18A1 variant is associated with survival difference in PAH patients [87]. In a separate study, Notch1 is shown to increase PAEC proliferation and inhibit apoptosis, and pharmacological inhibition of Notch1 reduced PH in SuHx rats [88]. However, genetic deletion of endothelial Notch1 in mice worsens hypoxia-induced PH possibly by increasing EC monolayer vulnerability [89], demonstrating that the relationship between EC survival and proliferation in PAH is complex.
2.4. Factors affecting EC activation and thrombogenicity
P-selectin is a pro-coagulant factor that is present on pulmonary ECs and platelets, and its expression reflects the extent of pulmonary EC injury [90]. Increased levels of P-selectin appear in the plasma of PAH patients, which can be decreased by infusion of the vasodilator, prostacyclin [90]. Furthermore, soluble P-selectin levels in PAH patients are associated with EC dysfunction [91]. vWF (another pro-coagulant factor) levels are also increased in the plasma [92] and pulmonary ECs [93] of PAH patients correlating with risk of death [94, 95], as well as with EC damage and dysfunction [92]. These studies suggest that P-selectin and vWF could act as prognostic markers in PAH, for example to predict EC dysfunction and likelihood of disease onset or progression [96]. Intriguingly, the plasma levels of thrombomodulin, an anti-coagulant factor are decreased in PAH patients [90, 97], and could be restored by infusion of the vasodilators, prostacyclin [90] or tadalafil [97]. Thus, these reports suggest that the pulmonary vascular endothelium in PAH patients is prothrombogenic with increased expression of pro-coagulant molecules and decreased expression of anti-coagulant factor.
2.5. Factors affecting EC metabolism and epigenetics
Abnormal metabolism, especially aerobic glycolysis or the Warburg effect, has been proposed as an important pathogenic mechanism in the development of PAH. Pulmonary vascular ECs from PAH patients rely heavily on glycolysis (a shift from oxidative phosphorylation) for increased growth [98–101]. PFKFB is a key regulator of glycolysis. Mice with EC-targeted Pfkfb3 deficiency exhibits attenuated PH or slowed PH progression with less EC inflammation and leukocyte recruitment to the lungs [102]. BMPR2-mediated Notch activation increases mitochondrial mass and expression of PFKFB3, which is necessary for citrate-dependent acetylation of H3K27 leading to expression of Notch1 target genes such as c-Myc and thus EC proliferation [89]. Overexpression of miR-124 or knockdown of PTBP1 restores normal levels of proliferation and glycolysis in ECs from PAH patients [103]. BMPR2 positively regulates miR-124 expression in ECs which targets PTBP1. Increased PTBP1 expression results in alternative splicing of pyruvate kinase muscle isoforms 1 and 2 (PKM1 and 2) leading to increased PKM2 expression. Thus, BMPR2 mutation or deficiency increases EC glycolysis via the miR-124/PTBP1/PKM2 signaling [103]. Endothelin 1/eNOS signaling is also involved in the glycolytic shift [104]. Endothelin 1 disrupts carnitine homeostasis and mitochondrial bioenergetics which correlate with uncoupled eNOS redistribution from the plasma membrane to the mitochondria. The glycolytic switch appears to be dependent on mitochondrial-derived ROS that activates HIF signaling [104].
Studies also show that BOLA3 regulates glycolysis and mitochondrial respiration [105]. Bola3 knockdown in mice or BOLA3 mutations in human decreases glycine cleavage system protein H, and thus enhances intracellular glycine. Bola3 deficiency enhances EC proliferation, survival, and vasoconstriction leading to PH. Iron-sulfur deficiency and changes in electron transport/cellular respiration have also been demonstrated in PAH via deficiencies in ISCU signaling [106]. White et al showed in mouse and human vascular and endothelial tissues that miR-210 level was elevated in PAH samples, accompanied by reduced ISCU1/2 and iron-sulfur integrity [106]. In mice, miR-210 repressed ISCU1/2 and enhanced PH. Conversely, mice deficient in EC-specific miR-210 showed increased ISCU1/2 levels and were resistant to PH, while ISCU1/2 knockdown promoted PH. Thus, the miR-210-ISCU1/2 axis causes iron-sulfur deficiency and PH. [106]. Other miRNAs that have been shown to regulate PH-associated dysfunctional phenotypes in ECs include miR-126 and −140–5p [107, 108] Although the mechanisms through which each miRNA regulates PH remain incompletely understood, it is possible that miRNAs regulate EC PH phenotypes in an endocrine manner [109].
PGC1α is a master regulator of cellular metabolism and mitochondrial biogenesis [110]. Reduced PGC1α expression in PAECs by hypoxia leads to decreased oxidative metabolism, mitochondrial function, and ROS generation, as well as increased ATP formation and eNOS phosphorylation, while upregulated PGC1α restores mitochondria function. Another study demonstrates that Ucp2 is also involved in EC mitochondria function [111]. Cobalt chloride treatment (which mimics hypoxia) of Ucp2-deficient ECs increases mitophagy and decreases mitochondrial biogenesis. Thus, the loss of endothelial Ucp2 leads to inadequate mitochondrial biosynthesis which may cause EC apoptosis.
Epigenetic mechanisms have also been shown to be important in the regulation of EC metabolism. Delivery of glutamine carbon into the TCA cycle is increased in ECs with BMPR2 mutations, which is required for endothelial survival in PAH, the maintenance of energetics, and the hyperproliferative phenotype [35]. The strict requirement for glutamine is driven by the loss of deacetylase sirtuin 3 activity. Preservation of sirtuin 3 function restores glutamine metabolism and prevents PH [35]. It has also been shown that vascular stiffness activates glutaminolysis to drive PH [112]. In the MCT-induced PH rat model, pharmacologic targeting of pulmonary vascular stiffness and YAP-dependent mechano-transduction modulated glutaminolysis, pulmonary vascular proliferation, and PH. Furthermore, pharmacologic targeting of glutaminase reduced MCT-induced PH progression [112].
Additionally, PAH ECs exhibit altered DNA methylation in many of the genes related to lipid metabolism including ABCA1 [113]. In rats, treatment with an agonist of ABCA1 reduces MCT-induced PH. Histone methylation in ECs is also involved in PH development [114]. MRTFA/MKL1 regulates expression of cell adhesion molecules including ICAM1 and VCAM1 through recruitment of H3K4 methyltransferase to the promoters and Mrtfa−/− mice inhibits hypoxia-induced PH with decreased expression of cell adhesin molecules. Endothelial-specific knockdown of ASH2 and WDR5, 2 components of the H3K4 methyltransferase complex, reduces hypoxic PH in mice [114]. As described above, increased nuclear accumulation of HDAC4 and HDAC5 is also observed in PAH ECs, which impairs MEF2 activity leading to decreased miR-424 and miR-503 expression and increased EC proliferation [85, 115].
2.6. Factors affecting EC dedifferentiation
Under pathological conditions, ECs may undergo mesenchymal cell transition (EndoMT). Previous studies provide circumstantial evidence that EndoMT may contribute to PAH directly, by EC transformation into smooth muscle-like cells with higher proliferative and migratory potential or indirectly, through paracrine effects on vascular intimal and medial proliferation [116]. A recent study employing genetic lineage tracing demonstrates that EndoMT didn’t contribute to neointimal formation in a chronic inflammatory PH mouse model, but rather this resulted from a subpopulation of Notch3-expressing SMCs, a finding which raises questions about the direct contribution of EndoMT to PAH pathogenesis[117]. EndoMT markers are observed in complex vascular lesions in PAH patients and rats with BMPR2 mutation [116]. In normal PAECs, BMPR2 knockdown leads to increased expression of HMGA1 and EndoMT markers. The expression of EndoMT markers can be largely reversed by double knockdown of BMPR2 and HMGA1 or slug [118]. Also, Rapamycin treatment inhibits expression of EndoMT markers, improves PH in BMPR2 mutant rats, and decreases human PAEC migration [116]. In lungs of Egln1Tie2Cre mice, EndoMT marker expression is increased along with SNAI1/2 in a HIF2α-dependent manner [65]. In IPAH lung ECs, PHD2 is downregulated, HIF2α expression is increased, and expression of EndoMT markers is enhanced [65]. Future studies using genetic lineage tracing approaches in various animal models of severe PH, such as Egln1Tie2Cre mice and SuHx rats, are warrantied to investigate the role of EndoMT in occlusive vascular remodeling and the pathogenesis of severe PAH.
3. Pulmonary EC Crosstalk with SMCs
Heightened vasoconstrictor activity or reduced vasodilator activity contribute to PAH [119–122] and multiple EC-derived factors including endothelin 1, NO, and prostacyclins regulate vascular tone. A key early component of PAH pathogenesis involves SMC vasoconstriction in response to increased endothelin 1, reduced NO bioavailability, and low prostacyclins. Paracrine factors released from pulmonary ECs may also regulate SMC survival, proliferation, and their functional phenotype, i.e., contractile versus synthetic, possibly contributing to the emergence of apoptosis-resistant hyperproliferative SMCs as PAH progresses [23–25], and ultimately remodeling of the pulmonary vasculature. This section provides an overview of EC-dependent mechanisms that control the aberrant SMC phenotypes seen in PAH.
3.1. EC regulation of SMC vasomotion
Endothelium-dependent pulmonary vasodilator signaling involves three main pathways: endothelium-derived hyperpolarizing factor (EDHF), NO, and prostacyclins. EDHF requires activation of calcium-sensitive potassium channels and cytochrome metabolites [123]. Impaired NO synthesis and bioavailability has been described in PH animal models and PAH patients [124–128]. In experimental studies, a wide variety of treatments that increase eNOS activity directly or indirectly have been shown to attenuate PH [129–134] and the evidence that NO signaling plays a crucial role in PAH is reviewed in detail elsewhere [135]. Prostacyclins are also potent vasodilators that are generated by vascular ECs as well as SMCs and EPCs. The efficacy of prostacyclins for the treatment of PAH patients is well established [136, 137]. Endothelin 1, predominantly expressed in ECs, is a potent vasoconstrictor that play an important role in the pathogenesis of PAH [138–140], as evidenced by its marked upregulation, particular associated with complex arterial lesions, in lungs from patients with PAH [140]. Hypoxia-induced PH, for example, is suppressed in EC-specific Edn1 knockout mice [141]. This and many other studies have led to the development of drugs that target the vasoconstrictive actions of endothelin 1 and this area of research has been thoroughly reviewed by others [142–144]. Endothelial-derived oxidative/nitrative stress, e.g., secondary to Caveolin1 deficiency in ECs [71, 127] is another vasoconstriction mechanism which induces PKG tyrosine nitration leading to impairment of NO signaling due to a reduction in PKG activity, thereby inducing vasoconstriction and vascular remodeling [127, 145, 146]. Accordingly, PKG nitration is a prominent feature of IPAH lungs [127, 147], and targeting endothelial nitrative stress-induced PKG dysfunction may represent a novel therapeutic strategy for PAH treatment.
3.2. EC regulation of SMC proliferation, migration, and survival
Culture of PASMCs in medium conditioned by IPAH ECs results in increased proliferation [148]. PAECs release a variety of growth factors and chemokines including PDGF-B, CXCL12, FGF2, MIF, and endothelin-1 that stimulate PASMC proliferation and pulmonary vascular remodeling [17, 63, 85, 149–153], likely through the transcription factor FoxM1 [154]. Genetic deletion of Foxm1 in SMCs prevents hypoxia-induced PH in mice and pharmacological inhibition of FoxM1 inhibits severe PH in experimental PH models [154]. FoxO1 is a negative regulator of SMC proliferation in response to some of the angiocrine factors [155]. Apoptosis-resistant ECs from PAH patients also release miRNA1-95-5p to promote SMC proliferation via HIF1α and Smad7 [156]. AMPK expression is decreased in PAECs from PAH patients [157]. Endothelial AMPK deficiency augments hypoxia-induced PASMC proliferation through phosphorylation and stabilization of ACE2 which increases eNOS-derived NO bioavailability and reduces PH [158].
It has been shown that several factors released from pulmonary ECs can induce SMC migration. In IPAH patients, CC chemokine ligand (CCL) 2 release by pulmonary ECs is enhanced [159]. PASMCs from IPAH patients exhibit greater migratory and proliferative responses to CCL2. CXCL12 is another potent chemokine derived from ECs which may play an important role in promoting SMC migration contributing to vascular remodeling [17].
EC-specific gene transfer of Indoleamine-2,3-dioxygenase attenuates PH in preclinical models [160]. Specifically, EC-derived Indoleamine-2,3-dioxygenase promotes PASMC apoptosis via depolarization of mitochondrial transmembrane potential and inhibits PASMC proliferation in a paracrine mechanism which remains to be elucidated [160]. In response to injury, apoptotic ECs release TGFβ1 and VEGF which induce SMC proliferation [161]. Thus, EC death induced by inflammation and proinflammatory cytokines could activate SMC proliferation leading to progression of pulmonary vascular remodeling and PAH. TPT1 (also called TCTP) is a potent anti-apoptotic factor that has been implicated in malignant cell transformation. TPT1 is released by ECs undergoing apoptosis in apoptotic nanovesicles, which are taken up by SMCs directly inducing SMC apoptosis-resistance and growth dysregulation [162–164].
3.3. SMC regulation of EC proliferation
Recent studies provide some intriguing findings about SMC regulation of EC proliferation. Activation of Notch1 by BMPR2 leads to EC proliferation in SMC-EC co-cultures that is mediated by direct SMC-EC contact [89]. BMPR2 is required by both cell types to produce collagen IV to activate integrin-linked kinase leading to stabilization of presenilin 1 and activation of Notch1 which maintains the EC proliferative capacity by increasing mitochondrial mass and inducing PFKFB3. EC-targeted deletion of Notch1 in mice worsens hypoxia-induced PH in association with impaired EC proliferation and regeneration, and thus loss of precapillary arteries [89]. This study provides direct evidence that SMC promotes EC proliferation and regeneration to maintain monolayer integrity and vascular homeostasis in response to injury. miR-143–3p released from SMC exosomes is another mechanism promoting EC migration and proliferation [165]. However, in this case EC proliferation is pathological since inhibition of miR143–3p reduces hypoxic PH in mice [165].
4. Pulmonary EC Crosstalk with Non-SMCs
Besides the direct effects of EC injury and dysfunction in the pathogenesis of PAH, crosstalk of ECs with SMCs and non-SMCs is increasingly recognized to play an important role in PAH progression. PAH is characterized by fibro-proliferative changes in the adventitia and immune cell accumulation in pathologically remodeled pulmonary vessels [27, 166]. (Myo)fibroblasts and inflammatory leukocytes are recruited to the lung through EC-dependent signaling mechanisms [166]. Several proinflammatory adhesion molecules and proinflammatory cytokines are abundantly expressed in activated ECs in experimental PH models and in the lungs of IPAH patients, which leads to inflammatory cell binding and recruitment [167]. Infiltrating inflammatory cells release cytokines including IL-1β and TNF-α, which activate ECs to express adhesion molecules, chemokines, and cytokines and promote EC proliferation and death. Furthermore, crosstalk between ECs and other non-SMCs such as pericytes or T-cells can contribute to the pathogenesis of PAH. In this section, signaling mechanisms that occur in PAH pathogenesis between ECs and non-SMCs are described.
4.1. Inflammatory cells and immune (T-)cells
Accumulation of inflammatory cells in vascular lesions is a characteristic feature of clinical PAH. The expression levels of proinflammatory adhesion molecules such as ICAM1, VCAM1, and E-selectin, are markedly elevated in the pulmonary vascular endothelium of IPAH patients and in cultured ECs from IPAH patients [167]. Activated ECs release GM-CSF [168], CCL2 [159], CXCL12 [17, 169], CTGF [170], IL-6 [80], and leptin [171, 172] to promote leukocyte recruitment and accumulation. The accumulated leukocytes release other factors such as macrophage-derived LTB4 [173] to induce PAEC apoptosis and T-cell lymphocyte-derived MIF [167] to induce the proinflammatory phenotype of ECs and the further recruitment of inflammatory cells. In contrast, regulatory T cells function to limit endothelial injury and inflammation. VEGFR2 inhibition with SU5416 alone induces severe PH with pulmonary EC apoptosis in T-cell-deficient rats, and in a sub-strain of ‘hyper-responder’ Sprague-Dawley rats [174]. Immune reconstituted nude rats exhibit limited lung perivascular inflammation and EC apoptosis and attenuated PH [175].
4.2. Pericytes
Pericyte numbers are increased in PAH and pericyte-EC crosstalk also contributes to pulmonary vascular remodeling in PAH [176]. IPAH ECs promote pericyte migration via release of FGF2 and IL-6 and proliferation by FGF2 [176]. EC-specific disruption of PHD2 increases pericyte coverage of pulmonary arteries [177]. In contrast, pericytes induce the expression of Wnt5a in normal ECs which promotes the recruitment of pericytes thereby stabilizing the distal arteriolar bed, but not ECs derived from PAH patients [178]. Accordingly, pulmonary microvascular ECs from PAH patients have a reduced capacity to recruit pericytes. EC-targeted deletion of Wnt5a reduces microvessel pericyte coverage and induces vessel loss resulting in persistent PH and right heart failure after cessation of hypoxia. Thus, endothelium Wnt5a plays an important role in pericyte recruitment and microvessel stabilization [178]. Additionally, PAH pericytes have increased levels of pyruvate dehydrodenase kinase 4 (PDK4) [179], correlating with their reduced mitochondrial metabolism, higher rates of glycolysis, and hyperproliferation, while reducing PDK4 restores pericyte mitochondrial metabolism, and cell proliferation, and enhances EC-pericyte interactions stabilizing small vessels [179]. Thus, genes that regulate pericyte-EC interactions could represent novel therapeutic targets to prevent small vessel loss in PAH.
4.3. Fibroblasts
EC-fibroblast crosstalk also plays a pathogenic role in PAH. As mentioned above, ECs may undergo EndoMT to become fibroblast-like cells. ECs also secrete factors such as Endothelin-1, PDGF and CXCL12 [17, 180] to induce fibroblast migration/recruitment, and proliferation. Furthermore, ECs can release factors such as Endothelin-1 and IL-6 to induce fibroblast differentiation to myofibroblasts [181, 182], which are highly proliferative, proinflammatory, invasive, and producing collagen and other extracellular matrix proteins and variety factors contributing to pulmonary vascular remodeling [166, 183]. Adventitial fibroblasts contribute to pulmonary vascular remodeling through several mechanisms. For example, accumulation of myofibroblasts increases extracellular matrix stiffness which leads to activation of PAEC proliferation [183]. (Myo)fibroblasts-derived MMP2 and MMP9 and 15-hydroxyeicosatetraenoic acid (15-HETE) could induce EC proliferation [184]. Fibroblast-released thrombospondin-1 could destabilize EC-EC interaction [185] leading to injured endothelium which contributes pulmonary vascular remodeling.
4.4. Endothelial progenitor cells (EPCs)
EPC markers were markedly increased in remodeled arteries from PAH patients, particularly in plexiform lesions that display increased SDF1 expression [186]. Circulating angiogenic EPC numbers are also increased in PAH patients and EPCs from PAH patients with BMPR2 mutations have a hyperproliferative phenotype with impaired vascularization, suggesting that dysfunction of circulating EPCs contribute to PAH vascular remodeling [186]. Clinical studies provide evidence of beneficial effects of EPC transplantation to PAH patients [187–189]. EPC-conditioned medium inhibits EC apoptosis via VEGF-A or VEGF-B and EC proliferation by VEGF-A or IL-8 [180]. Treatment with EPCs or EPC-conditioned medium improves pulmonary artery relaxation suggesting paracrine mechanisms are promoting vasoprotection, such as through the release of prostacyclin and cAMP [190]. This is consistent with work showing that the therapeutic potential of EPCs can be enhanced by the transfection of eNOS, another important paracrine signaling pathway [134]. This study also provides evidence that eNOS-transfected EPCs can induce regeneration of lung microvasculature. Together, these studies demonstrate that EPCs can attenuate pulmonary vascular remodeling and PAH development through paracrine mechanisms.
5. Future Perspectives
Despite major advances in the understanding of the pathophysiology of PAH, these mechanistic insights have been translated into approved therapies for PAH patients in only a limited number of instances, mainly related to the three major endothelial vasoactive pathways, and although such therapies can reduce the symptoms of PAH, they do not prevent progression or cure the disease. The 5-year mortality of PAH are still as high as 40% [7, 191]. Here we summarize the recent findings on the role of ECs in the pathogenesis of PAH (Figure 2). Novel therapeutic advances could occur from an improved understanding of EC-mediated mechanisms that regulate PAH (Table 1, Table 2). In this regard, many of the EC signaling pathways described above now represent novel therapeutic targets for the prevention or treatment of PAH (Table 3). Novel PAH treatments could aim to: (a) inhibit EC injury/apoptosis in the early stages of disease and promote EC regeneration and repair; (b) inhibit apoptosis-resistant EC hyperproliferation, (c) target the EC secretome; (d) inhibit EndoMT; or (e) target EC-derived oxidative/nitrative stress. The ideal targets are those nodal signaling molecules regulating multiple pathways in the pathogenesis of PAH. For example, endothelial PHD2/HIF-2α regulates EC release of PDGF-B, SDF1, Endothelin-1, Apelin among others and affect BMPR2 signaling, Caveolin1 as well as PKG expression [17, 64–68]. This option is particularly appealing given that HIF-2α inhibitors are already in clinical trials of renal cancer patients [192–194]. Similarly, targeting the mechanisms that regulate metabolic reprogramming of pulmonary ECs may represent a novel therapeutic approach [35, 102, 105]. Likewise, the epigenetic manipulation of key pathways and miRNAs [85, 103, 113] could also represent a new therapeutic strategy. In terms of specifically targeting lung ECs in the treatment of PAH, several therapeutic options deserve attention in future experimental and clinical studies. It may be possible to deliver PAH treatments in nanoscale delivery vehicles that target the lung endothelium through coating with antibodies or receptors that enhance EC uptake and retention. For example, small molecule inhibitors or other pharmacological agents could be encapsulated in nanoparticles that have been coated with anti-vWF or anti-CD31 antibodies. Endothelial-enriched nanoparticles can be employed to deliver siRNA oligoes to disrupt gene expression in vascular ECs [195]. Our unpublished data (manuscript under revision) show a novel nanoparticle can efficiently deliver plasmid DNA targeting the vascular ECs with high genome editing efficiency, which holds great potential for non-viral gene therapy in PAH by genome-editing-mediated gene disruption and correction of genetic mutations, etc.
Figure 2: Cross-talk between pulmonary endothelial cells and other cells leads to obliterative pulmonary vascular remodeling and progressive vasoconstriction and thereby PAH.
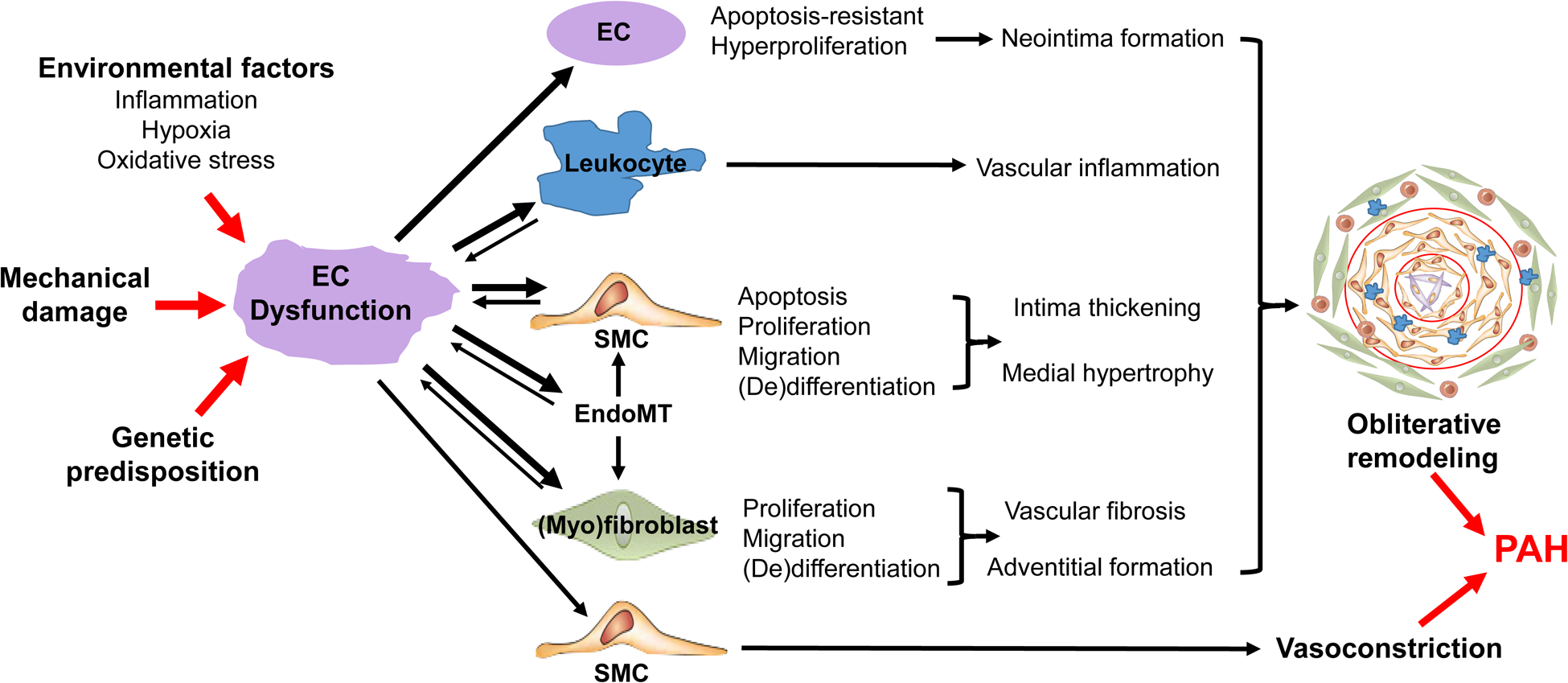
Various environmental factors, mechanical damage, and genetic predisposition converge on pulmonary vascular ECs leading to EC injury and dysfunction which affect EC activation, survival, proliferation, migration, metabolic and epigenetic status resulting in EC-apoptosis-resistant hyperproliferation, EndoMT, and releases of vascular tone modulators, angiocrine factors, cytokine and chemokines which mediate crosstalk between ECs and SMCs, leukocytes, (myo)fibroblasts. Together, EC dysfunction induces obliterative pulmonary vascular remodeling and vasoconstriction resulting in PAH. Thus, targeting altered EC signalings will provide novel effective therapeutic approaches for inhibiting/reversing obliterative vascular remodeling and PAH. Abbreviations: EC, endothelial cell; EndoMT, endothelial-to-mesenchymal transition; PH, pulmonary hypertension; SMC, smooth muscle cell.
Table 1:
Genes regulating endothelial cell signaling pathways and PAH
| Gene (Pro + or anti − PH) | Expression Change in PH | PH Phenotypes | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Rat | Mu | Human | Rat | Mu | ||||||
| LEC | LEC | WL | LEC | MCT | SuHx | Basal | MCT | Hx | SuHx | |
| ↓ [113] | Agonist ↓ [113] | |||||||||
| ↓ [157] | ↓ [157] | EC KO ↑ [157] | ||||||||
| ↓ [41, 42] | ↓ [43] | LEC KO ↑ [42] | Het KO ↑ [40] | EC KO ↑ [41] | ||||||
| ↓ [49, 52] | ↓ [49] | Admin ↓ [49] | Hom KO ↑ [52] | |||||||
| ↓ [105] | ↓ [105] | LEC KO ↑ [105] | LEC OE ↓ [105] | |||||||
| ↓ [75] | ↓ [71, 72] | Hom KO ↑ [74] | EC KO ↑ [72] | |||||||
| ↑ [159] | ||||||||||
| ↑ [167] | Inhib ↓ [167] | |||||||||
| ↓ [51] | Hom KO ↑ [51] | |||||||||
| ↑ [53] | ↑ [53] | Hom KO ↓ [53] | ||||||||
| ↑ [170] | EC KO ↓ [170] | |||||||||
| ↑ [154] | EC KO ↓ [154] | |||||||||
| ↑ [172] | ↑ [172] | EC OE ↑ [172] | ||||||||
| ↑ [140] | OE ↑ [139] | Inhib ↓ [138] | OE ↑ [139] | |||||||
| ↑ [153] | Inhib ↓ [153] | |||||||||
| ↑ [67] | ↑ [68] | Inhib ↓ [68] | Inhib ↓ [68] | LEC KO ↓ [67] | ||||||
| ↓ [160] | EC KO ↑ [160] | |||||||||
| ↑ [114] | Hom KO ↓ [114] | |||||||||
| ↓ [77] | Admin ↓ [77] | |||||||||
| ↓ [115] | Agonist ↓ [115] | Agonist ↓ [115] | ||||||||
| ↑ [89] | ↑ [88] | Inhib ↓ [88] | EC KO ↑ [89] | |||||||
| ↓ [78] | ↓ [78] | Agonist ↓ [78] | ||||||||
| ↑ [82] | ↑ [82] | ↑ [82] | Inhib ↓ [82] | Inhib ↓ [82] | ||||||
| ↑ [54] | ||||||||||
| ↑ [102] | ↑ [102] | ↑ [102] | Inhib ↓ [102] | Het / EC KO ↓ [102] | ||||||
| ↓ [17, 64] | ↓ [17] | EC & HC KO ↑ [17] | ||||||||
| Hom KO ↑ [146] | ||||||||||
| ↓ [58] | EC KO ↑ [63] | |||||||||
| ↑ [163] | ↑ [163] | |||||||||
Multiple EC genes are expressed differentially in animal models of PH and human PH and have been shown to positively (+) or negatively (−) regulate PH. Abbreviations: ABCA, ATP binding cassette subfamily A member; Admin, administration; AMPK, AMP-activated protein kinase; BMPR, bone morphogenic protein receptor; BOLA, bolA family member; Cav, caveolin; CCL, CC ligand; CLIC, chloride intracellular channel; CM, conditioned media; CTGF, connective tissue growth factor; CXCL, C-X-C motif chemokine; CypA, cyclophilin A; EC, endothelial cell; Exp, expression; FGF, fibroblast growth factor; HC, hematopoietic cell; Het, heterozygous; HIF, hypoxia-inducible factor; Hom, homozygous; Hx, hypoxia; Inhib, inhibitor; IDO, indoleamine 2,3-dioxygenase; IPAH, idiopathic pulmonary arterial hypertension; KO, knockout; LEC, lung endothelial cells; MCT, monocrotaline; MEF, myocyte enhancer factor; MRTFA, myocardin-related transcription factor A; mTOR, mammalian target of rapamycin; Mu, mouse; Mut, mutation; OE, overexpression; PAH, pulmonary arterial hypertension; PFKFB, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase; PH, pulmonary hypertension; PHD, prolyl hydroxylase; PKG, protein kinase G; PPAR, peroxisome proliferator-activated receptor; SuHx, sugen/hypoxia; TCTP, translationally controlled tumor protein; VEGFR, vascular endothelial growth factor receptor; WL, whole lung.
Table 2:
Targeting endothelial signaling pathways to reduce PH in experimental studies
| EC Phenotype targeted | Therapy | Mechanism of Action | Species | Model | Ref |
|---|---|---|---|---|---|
| Vasoconstriction | eNOS gene transfer | eNOS upregulation | Rabbit | Flow | [130] |
| Fluvastatin | eNOS uncoupling | Rat | Hypoxia | [133] | |
| LU135252 | Endothelin A receptor inhibition | Rat | MCT | [138] | |
| Inflammation | PDTC | NFκB inhibition | Rat | MCT | [197] |
| MnTMPyP | Superoxide scavenging | Mouse | Cav1 loss | [127] | |
| Leptin receptor | Leptin neutralization | Mouse | Hypoxia | [171] | |
| ISO-1 | MIF inhibition | Rat | MCT | [167] | |
| Anti-CD74 antibody | CD74 inhibition | Rat | MCT | [167] | |
| Anti-GMCSF antibody | GM-CSF neutralization | Mouse | Hypoxia | [168] | |
| Coagulation | Thrombomodulin | Thrombin binding | Rat | MCT | [198] |
| Acute EC Apoptosis | BMP9 protein | BMP signaling agonist | Mouse | MCT, Sugen/hypoxia, BMPR2 loss | [45] |
| CAG-miRNA-21 | PCD4 downregulation | Mouse | Sugen/hypoxia | [54] | |
| SD-208 | TGFβ inhibition | Rat | MCT | [199] | |
| EC Survival | Simvastatin | HMG-CoA reductase inhibition | Rat | Sugen/hypoxia | [30] |
| EC Proliferation | mTOR adenovirus | mTOR overexpression | Mouse | Hypoxia | [77] |
| MC1568 | MEF2 restoration | Rat | MCT, Sugen | [115] | |
| SMC Proliferation | IDO gene transfer | SMC apoptosis inducer | Mouse | Hypoxia | [160] |
| FGF2 siRNA or SU5402 | FGF2 inhibition | Rat | MCT | [153] | |
| EC Dysfunction | C76 | HIF2α inhibition | Rat, mouse | MCT, Sugen/hypoxia, PHD2 loss | [68] |
| EC Dedifferentiation | R05–3335 | Runx1 inhibition | Mouse | MCT, Sugen/hypoxia | [200] |
| Altered EC metabolism | 2-hydroxy-benzylamine | Lipid peroxidation product scavenger | Mouse | BMP2 loss | [35] |
| EC Cross-talk | Thiostrepton | FoxM1 inhibition in SMCs | Rat, mouse | MCT, Sugen/hypoxia, PHD2 loss | [154] |
| Bestatin | LTA4H inhibition in macrophages | Rat | Sugen/hypoxia | [173] |
Multiple EC signaling pathways have been targeted to reduce PH in preclinical studies. Abbreviations: BMP, bone morphogenic protein; Cav, caveolin; EC, endothelial cell; eNOS, endothelial nitric oxide synthase; FGF, fibroblast growth factor; FoxM1, forkhead box M1; GM-CSF, granulocyte macrophage colony-stimulating factor; HIF, hypoxia-inducible factor; HMG-CoA, β-hydroxy β-methylglutaryl-CoA; IDO, indoleamine-2,3-dioxygenase; LTA4H, leukotriene A4 hydrolase; MCT, monocrotaline; MEF, myocyte enhancer factor; MIF, macrophage migration inhibitory factor; mTOR, mammalian target of rapamycin; NFκB, nuclear factor κB; PCD, programmed cell death; PDTC, pyrrolidine dithiocarbamate PH, pulmonary hypertension; SMC, smooth muscle cell; TGF, transforming growth factor.
Table 3:
Recent PAH clinical trials targeting endothelial signaling pathways
| EC Phenotype targeted | Therapy | Mechanism of Action | Design | Duration | Identifier(s) |
|---|---|---|---|---|---|
| Altered EC metabolism | Dichloroacetate | Inhibits PDK | Phase I | 16weeks | NCT01083524 |
| Metformin | Increases glucose uptake | Phase I | 12weeks | NCT03349775 | |
| Ranolazine | Reduces intracellular calcium | Phase I-IV RCTs | 12–26weeks | NCT02829034 | |
| Trimetazidine | Inhibits oxidation of free fatty acids | Phase II/III | 12weeks | NCT03273387 | |
| Altered EC metabolism, Inflammation | Bardoxolone methyl | Nrf2/NFkB pathway | Phase II/III RCTs | 16weeks-5years | NCT02036970 |
| Inflammation | Anakinra | Inhibits IL-1 receptor | Phase I/II | 14days-4weeks | NCT01479010 |
| Tocilizumab | Inhibits IL-6 binding | Phase II | 24weeks | NCT02676947 | |
| Ubenimex | Inhibits aminopeptidases | Phase II | 24weeks-1year | NCT02736149 | |
| Vasoconstriction | EPC-induced eNOS gene transfer | eNOS gene transfer | Phase II | 24weeks | NCT03001414 |
Multiple EC signaling pathways are being targeted by therapies that are currently in clinical trials of PAH. Abbreviations: EC, endothelial cell; eNOS, endothelial nitric oxide synthase; EPC, endothelial progenitor cell; IL, interleukin; NFκB, nuclear factor κB; Nrf, nuclear factor erythroid 2–related factor; PDK, pyruvate dehydrogenase kinase; RCT, randomized controlled trials.
Given the heterogeneity in EC phenotypes [196], future studies should define the subpopulations of ECs in the pathogenesis of PAH employing single cell RNA sequencing analysis coupled with genetic lineage tracing and depletion studies. Future research could also employ, for instance, computational modeling techniques to improve understanding of the relationships between these heterogeneous EC subpopulations and their interaction with neighboring cell types. Similarly, the potential role of EndoMT in the mechanisms of obliterative vascular remodeling shall also be defined with similar genetic lineage tracing approaches. Although PAH has an inflammatory component, it is unclear whether inflammation is a cause or consequence of this disease. Future studies should aim to determine whether and how inflammation triggers EC dysfunction contributing to vascular remodeling. Future studies could assess the timing of the EC phenotypic changes in relation to PAH pathogenesis and progression by employing extensive time-course experiments or longitudinal experiments of labeled ECs from healthy to diseased states. Our knowledge about the mechanisms of vascular fibrosis in PAH is also limited; thus, studying the role of EC dysfunction in vascular fibrosis is another important research direction. Moreover, targeting EC-derived oxidative/nitrative stress may provide a novel therapeutic approach for treatment of PAH [132, 145].
6. Conclusions
Dysfunctional EC signaling pathways tightly regulate multiple aspects of PAH, including pulmonary vascular tone, inflammation, coagulation, metabolism, and remodeling. Given the increasingly large body of evidence demonstrating that ECs are crucial mediators of PAH initiation and progression, novel therapies for PAH could aim to target multiple aspects of EC dysfunction and EC signaling, especially those nodal signaling molecules regulating multiple pathways in the pathogenesis of PAH. Improved understanding of the EC signaling pathways responsible for the initiation and progression of PAH will facilitate the development of effective treatments for PAH.
Funding
This work was supported in part by NIH grants R01HL123957, R01HL133951, R01HL140409, and R01HL148810 to Y.Y.Z and by an American Heart Association Career Development Award (19CDA34500000) to C.E.E.
Footnotes
Conflicts of Interest
None.
References
- 1.Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG, Souza R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019: 53(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Krishna Kumar R, Landzberg M, Machado RF, Olschewski H, Robbins IM, Souza R. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013: 62(25 Suppl): D34–41. [DOI] [PubMed] [Google Scholar]
- 3.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest 2012: 122(12): 4306–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, Jessup M, Grizzle WE, Aldred MA, Cool CD, Tuder RM. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med 2012: 186(3): 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rabinovitch M. Pathobiology of pulmonary hypertension. Annu Rev Pathol 2007: 2: 369–399. [DOI] [PubMed] [Google Scholar]
- 6.Farber HW. The status of pulmonary arterial hypertension in 2008. Circulation 2008: 117(23): 2966–2968. [DOI] [PubMed] [Google Scholar]
- 7.Sutendra G, Michelakis ED. Pulmonary arterial hypertension: challenges in translational research and a vision for change. Sci Transl Med 2013: 5(208): 208sr205. [DOI] [PubMed] [Google Scholar]
- 8.McLaughlin VV, Shah SJ, Souza R, Humbert M. Management of pulmonary arterial hypertension. J Am Coll Cardiol 2015: 65(18): 1976–1997. [DOI] [PubMed] [Google Scholar]
- 9.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol 2007: 7(10): 803–815. [DOI] [PubMed] [Google Scholar]
- 10.Cella G, Bellotto F, Tona F, Sbarai A, Mazzaro G, Motta G, Fareed J. Plasma markers of endothelial dysfunction in pulmonary hypertension. Chest 2001: 120(4): 1226–1230. [DOI] [PubMed] [Google Scholar]
- 11.Hughes R, Tong J, Oates C, Lordan J, Corris PA. Evidence for systemic endothelial dysfunction in patients and first-order relatives with pulmonary arterial hypertension. Chest 2005: 128(6 Suppl): 617S. [DOI] [PubMed] [Google Scholar]
- 12.Peled N, Bendayan D, Shitrit D, Fox B, Yehoshua L, Kramer MR. Peripheral endothelial dysfunction in patients with pulmonary arterial hypertension. Respir Med 2008: 102(12): 1791–1796. [DOI] [PubMed] [Google Scholar]
- 13.Wolff B, Lodziewski S, Bollmann T, Opitz CF, Ewert R. Impaired peripheral endothelial function in severe idiopathic pulmonary hypertension correlates with the pulmonary vascular response to inhaled iloprost. Am Heart J 2007: 153(6): 1088 e1081–1087. [DOI] [PubMed] [Google Scholar]
- 14.Loscalzo J. Endothelial dysfunction in pulmonary hypertension. N Engl J Med 1992: 327(2): 117–119. [DOI] [PubMed] [Google Scholar]
- 15.Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation 2004: 109(2): 159–165. [DOI] [PubMed] [Google Scholar]
- 16.Huertas A, Perros F, Tu L, Cohen-Kaminsky S, Montani D, Dorfmuller P, Guignabert C, Humbert M. Immune dysregulation and endothelial dysfunction in pulmonary arterial hypertension: a complex interplay. Circulation 2014: 129(12): 1332–1340. [DOI] [PubMed] [Google Scholar]
- 17.Dai Z, Li M, Wharton J, Zhu MM, Zhao YY. Prolyl-4 Hydroxylase 2 (PHD2) Deficiency in Endothelial Cells and Hematopoietic Cells Induces Obliterative Vascular Remodeling and Severe Pulmonary Arterial Hypertension in Mice and Humans Through Hypoxia-Inducible Factor-2alpha. Circulation 2016: 133(24): 2447–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petrusca DN, Van Demark M, Gu Y, Justice MJ, Rogozea A, Hubbard WC, Petrache I. Smoking exposure induces human lung endothelial cell adaptation to apoptotic stress. Am J Respir Cell Mol Biol 2014: 50(3): 513–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Napoli C, Benincasa G, Loscalzo J. Epigenetic Inheritance Underlying Pulmonary Arterial Hypertension. Arterioscler Thromb Vasc Biol 2019: 39(4): 653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miyagawa T, Taniguchi T, Saigusa R, Fukayama M, Takahashi T, Yamashita T, Hirabayashi M, Miura S, Nakamura K, Yoshizaki A, Sato S, Asano Y. Fli1 deficiency induces endothelial adipsin expression, contributing to the onset of pulmonary arterial hypertension in systemic sclerosis. Rheumatology (Oxford) 2020: 59(8): 2005–2015. [DOI] [PubMed] [Google Scholar]
- 21.Hu Y, Chi L, Kuebler WM, Goldenberg NM. Perivascular Inflammation in Pulmonary Arterial Hypertension. Cells 2020: 9(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winter MP, Sharma S, Altmann J, Seidl V, Panzenböck A, Alimohammadi A, Zelniker T, Redwan B, Nagel F, Santer D, Stieglbauer A, Podesser B, Sibilia M, Helbich T, Prager G, Ilhan-Mutlu A, Preusser M, Lang IM. Interruption of vascular endothelial growth factor receptor 2 signaling induces a proliferative pulmonary vasculopathy and pulmonary hypertension. Basic Res Cardiol 2020: 115(6): 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levy M, Maurey C, Celermajer DS, Vouhe PR, Danel C, Bonnet D, Israel-Biet D. Impaired apoptosis of pulmonary endothelial cells is associated with intimal proliferation and irreversibility of pulmonary hypertension in congenital heart disease. J Am Coll Cardiol 2007: 49(7): 803–810. [DOI] [PubMed] [Google Scholar]
- 24.Masri FA, Xu W, Comhair SA, Asosingh K, Koo M, Vasanji A, Drazba J, Anand-Apte B, Erzurum SC. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 2007: 293(3): L548–554. [DOI] [PubMed] [Google Scholar]
- 25.Sakao S, Tatsumi K, Voelkel NF. Endothelial cells and pulmonary arterial hypertension: apoptosis, proliferation, interaction and transdifferentiation. Respir Res 2009: 10: 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goldthorpe H, Jiang JY, Taha M, Deng Y, Sinclair T, Ge CX, Jurasz P, Turksen K, Mei SH, Stewart DJ. Occlusive lung arterial lesions in endothelial-targeted, fas-induced apoptosis transgenic mice. Am J Respir Cell Mol Biol 2015: 53(5): 712–718. [DOI] [PubMed] [Google Scholar]
- 27.Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol 1994: 144(2): 275–285. [PMC free article] [PubMed] [Google Scholar]
- 28.Lee SD, Shroyer KR, Markham NE, Cool CD, Voelkel NF, Tuder RM. Monoclonal endothelial cell proliferation is present in primary but not secondary pulmonary hypertension. J Clin Invest 1998: 101(5): 927–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tu L, Dewachter L, Gore B, Fadel E, Dartevelle P, Simonneau G, Humbert M, Eddahibi S, Guignabert C. Autocrine fibroblast growth factor-2 signaling contributes to altered endothelial phenotype in pulmonary hypertension. Am J Respir Cell Mol Biol 2011: 45(2): 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taraseviciene-Stewart L, Scerbavicius R, Choe KH, Cool C, Wood K, Tuder RM, Burns N, Kasper M, Voelkel NF. Simvastatin causes endothelial cell apoptosis and attenuates severe pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2006: 291(4): L668–676. [DOI] [PubMed] [Google Scholar]
- 31.van der Feen DE, Bossers GPL, Hagdorn QAJ, Moonen JR, Kurakula K, Szulcek R, Chappell J, Vallania F, Donato M, Kok K, Kohli JS, Petersen AH, van Leusden T, Demaria M, Goumans MTH, De Boer RA, Khatri P, Rabinovitch M, Berger RMF, Bartelds B. Cellular senescence impairs the reversibility of pulmonary arterial hypertension. Sci Transl Med 2020: 12(554). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chaouat A, Weitzenblum E, Higenbottam T. The role of thrombosis in severe pulmonary hypertension. Eur Respir J 1996: 9(2): 356–363. [DOI] [PubMed] [Google Scholar]
- 33.Tournier A, Wahl D, Chaouat A, Max JP, Regnault V, Lecompte T, Chabot F. Calibrated automated thrombography demonstrates hypercoagulability in patients with idiopathic pulmonary arterial hypertension. Thromb Res 2010: 126(6): e418–422. [DOI] [PubMed] [Google Scholar]
- 34.Ranchoux B, Harvey LD, Ayon RJ, Babicheva A, Bonnet S, Chan SY, Yuan JX, Perez VJ. Endothelial dysfunction in pulmonary arterial hypertension: an evolving landscape (2017 Grover Conference Series). Pulm Circ 2018: 8(1): 2045893217752912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Egnatchik RA, Brittain EL, Shah AT, Fares WH, Ford HJ, Monahan K, Kang CJ, Kocurek EG, Zhu S, Luong T, Nguyen TT, Hysinger E, Austin ED, Skala MC, Young JD, Roberts LJ 2nd, Hemnes AR, West J, Fessel JP. Dysfunctional BMPR2 signaling drives an abnormal endothelial requirement for glutamine in pulmonary arterial hypertension. Pulm Circ 2017: 7(1): 186–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim C, Seedorf GJ, Abman SH, Shepherd DP. Heterogeneous response of endothelial cells to insulin-like growth factor 1 treatment is explained by spatially clustered sub-populations. Biology open 2019: 8(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stevens T. Molecular and cellular determinants of lung endothelial cell heterogeneity. Chest 2005: 128(6 Suppl): 558s–564s. [DOI] [PubMed] [Google Scholar]
- 38.Gillich A, Zhang F, Farmer CG, Travaglini KJ, Tan SY, Gu M, Zhou B, Feinstein JA, Krasnow MA, Metzger RJ. Capillary cell-type specialization in the alveolus. Nature 2020: 586(7831): 785–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frump A, Prewitt A, de Caestecker MP. BMPR2 mutations and endothelial dysfunction in pulmonary arterial hypertension (2017 Grover Conference Series). Pulm Circ 2018: 8(2): 2045894018765840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song Y, Coleman L, Shi J, Beppu H, Sato K, Walsh K, Loscalzo J, Zhang YY. Inflammation, endothelial injury, and persistent pulmonary hypertension in heterozygous BMPR2-mutant mice. Am J Physiol Heart Circ Physiol 2008: 295(2): H677–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Diebold I, Hennigs JK, Miyagawa K, Li CG, Nickel NP, Kaschwich M, Cao A, Wang L, Reddy S, Chen PI, Nakahira K, Alcazar MA, Hopper RK, Ji L, Feldman BJ, Rabinovitch M. BMPR2 preserves mitochondrial function and DNA during reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension. Cell Metab 2015: 21(4): 596–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hong KH, Lee YJ, Lee E, Park SO, Han C, Beppu H, Li E, Raizada MK, Bloch KD, Oh SP. Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation 2008: 118(7): 722–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rhodes CJ, Im H, Cao A, Hennigs JK, Wang L, Sa S, Chen PI, Nickel NP, Miyagawa K, Hopper RK, Tojais NF, Li CG, Gu M, Spiekerkoetter E, Xian Z, Chen R, Zhao M, Kaschwich M, Del Rosario PA, Bernstein D, Zamanian RT, Wu JC, Snyder MP, Rabinovitch M. RNA Sequencing Analysis Detection of a Novel Pathway of Endothelial Dysfunction in Pulmonary Arterial Hypertension. Am J Respir Crit Care Med 2015: 192(3): 356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Teichert-Kuliszewska K, Kutryk MJ, Kuliszewski MA, Karoubi G, Courtman DW, Zucco L, Granton J, Stewart DJ. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ Res 2006: 98(2): 209–217. [DOI] [PubMed] [Google Scholar]
- 45.Long L, Ormiston ML, Yang X, Southwood M, Graf S, Machado RD, Mueller M, Kinzel B, Yung LM, Wilkinson JM, Moore SD, Drake KM, Aldred MA, Yu PB, Upton PD, Morrell NW. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat Med 2015: 21(7): 777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spiekerkoetter E, Tian X, Cai J, Hopper RK, Sudheendra D, Li CG, El-Bizri N, Sawada H, Haghighat R, Chan R, Haghighat L, de Jesus Perez V, Wang L, Reddy S, Zhao M, Bernstein D, Solow-Cordero DE, Beachy PA, Wandless TJ, Ten Dijke P, Rabinovitch M. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest 2013: 123(8): 3600–3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tu L, Desroches-Castan A, Mallet C, Guyon L, Cumont A, Phan C, Robert F, Thuillet R, Bordenave J, Sekine A, Huertas A, Ritvos O, Savale L, Feige JJ, Humbert M, Bailly S, Guignabert C. Selective BMP-9 Inhibition Partially Protects Against Experimental Pulmonary Hypertension. Circ Res 2019: 124(6): 846–855. [DOI] [PubMed] [Google Scholar]
- 48.Ormiston ML, Godoy RS, Chaudhary KR, Stewart DJ. The Janus Faces of Bone Morphogenetic Protein 9 in Pulmonary Arterial Hypertension. Circ Res 2019: 124(6): 822–824. [DOI] [PubMed] [Google Scholar]
- 49.Alastalo TP, Li M, Perez Vde J, Pham D, Sawada H, Wang JK, Koskenvuo M, Wang L, Freeman BA, Chang HY, Rabinovitch M. Disruption of PPARγ/β-catenin-mediated regulation of apelin impairs BMP-induced mouse and human pulmonary arterial EC survival. J Clin Invest 2011: 121(9): 3735–3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Helenius MH, Vattulainen S, Orcholski M, Aho J, Komulainen A, Taimen P, Wang L, de Jesus Perez VA, Koskenvuo JW, Alastalo TP. Suppression of endothelial CD39/ENTPD1 is associated with pulmonary vascular remodeling in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 2015: 308(10): L1046–1057. [DOI] [PubMed] [Google Scholar]
- 51.Visovatti SH, Hyman MC, Goonewardena SN, Anyanwu AC, Kanthi Y, Robichaud P, Wang J, Petrovic-Djergovic D, Rattan R, Burant CF, Pinsky DJ. Purinergic dysregulation in pulmonary hypertension. Am J Physiol Heart Circ Physiol 2016: 311(1): H286–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chandra SM, Razavi H, Kim J, Agrawal R, Kundu RK, de Jesus Perez V, Zamanian RT, Quertermous T, Chun HJ. Disruption of the apelin-APJ system worsens hypoxia-induced pulmonary hypertension. Arterioscler Thromb Vasc Biol 2011: 31(4): 814–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wojciak-Stothard B, Abdul-Salam VB, Lao KH, Tsang H, Irwin DC, Lisk C, Loomis Z, Stenmark KR, Edwards JC, Yuspa SH, Howard LS, Edwards RJ, Rhodes CJ, Gibbs JS, Wharton J, Zhao L, Wilkins MR. Aberrant chloride intracellular channel 4 expression contributes to endothelial dysfunction in pulmonary arterial hypertension. Circulation 2014: 129(17): 1770–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.White K, Dempsie Y, Caruso P, Wallace E, McDonald RA, Stevens H, Hatley ME, Van Rooij E, Morrell NW, MacLean MR, Baker AH. Endothelial apoptosis in pulmonary hypertension is controlled by a microRNA/programmed cell death 4/caspase-3 axis. Hypertension 2014: 64(1): 185–194. [DOI] [PubMed] [Google Scholar]
- 55.Li L, Xu M, Li X, Lv C, Zhang X, Yu H, Zhang M, Fu Y, Meng H, Zhou J. Platelet-derived growth factor-B (PDGF-B) induced by hypoxia promotes the survival of pulmonary arterial endothelial cells through the PI3K/Akt/Stat3 pathway. Cell Physiol Biochem 2015: 35(2): 441–451. [DOI] [PubMed] [Google Scholar]
- 56.Yamaji-Kegan K, Takimoto E, Zhang A, Weiner NC, Meuchel LW, Berger AE, Cheadle C, Johns RA. Hypoxia-induced mitogenic factor (FIZZ1/RELMalpha) induces endothelial cell apoptosis and subsequent interleukin-4-dependent pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2014: 306(12): L1090–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Johns RA, Takimoto E, Meuchel LW, Elsaigh E, Zhang A, Heller NM, Semenza GL, Yamaji-Kegan K. Hypoxia-Inducible Factor 1alpha Is a Critical Downstream Mediator for Hypoxia-Induced Mitogenic Factor (FIZZ1/RELMalpha)-Induced Pulmonary Hypertension. Arterioscler Thromb Vasc Biol 2016: 36(1): 134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ameshima S, Golpon H, Cool CD, Chan D, Vandivier RW, Gardai SJ, Wick M, Nemenoff RA, Geraci MW, Voelkel NF. Peroxisome proliferator-activated receptor gamma (PPARgamma) expression is decreased in pulmonary hypertension and affects endothelial cell growth. Circ Res 2003: 92(10): 1162–1169. [DOI] [PubMed] [Google Scholar]
- 59.Hansmann G, Wagner RA, Schellong S, Perez VA, Urashima T, Wang L, Sheikh AY, Suen RS, Stewart DJ, Rabinovitch M. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation 2007: 115(10): 1275–1284. [DOI] [PubMed] [Google Scholar]
- 60.Tian J, Smith A, Nechtman J, Podolsky R, Aggarwal S, Snead C, Kumar S, Elgaish M, Oishi P, Goerlach A, Fratz S, Hess J, Catravas JD, Verin AD, Fineman JR, She JX, Black SM. Effect of PPARgamma inhibition on pulmonary endothelial cell gene expression: gene profiling in pulmonary hypertension. Physiol Genomics 2009: 40(1): 48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vattulainen-Collanus S, Akinrinade O, Li M, Koskenvuo M, Li CG, Rao SP, de Jesus Perez V, Yuan K, Sawada H, Koskenvuo JW, Alvira C, Rabinovitch M, Alastalo TP. Loss of PPARγ in endothelial cells leads to impaired angiogenesis. J Cell Sci 2016: 129(4): 693–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li CG, Mahon C, Sweeney NM, Verschueren E, Kantamani V, Li D, Hennigs JK, Marciano DP, Diebold I, Abu-Halawa O, Elliott M, Sa S, Guo F, Wang L, Cao A, Guignabert C, Sollier J, Nickel NP, Kaschwich M, Cimprich KA, Rabinovitch M. PPARγ Interaction with UBR5/ATMIN Promotes DNA Repair to Maintain Endothelial Homeostasis. Cell Rep 2019: 26(5): 1333–1343.e1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guignabert C, Alvira CM, Alastalo TP, Sawada H, Hansmann G, Zhao M, Wang L, El-Bizri N, Rabinovitch M. Tie2-mediated loss of peroxisome proliferator-activated receptor-gamma in mice causes PDGF receptor-beta-dependent pulmonary arterial muscularization. Am J Physiol Lung Cell Mol Physiol 2009: 297(6): L1082–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kapitsinou PP, Rajendran G, Astleford L, Michael M, Schonfeld MP, Fields T, Shay S, French JL, West J, Haase VH. The Endothelial Prolyl-4-Hydroxylase Domain 2/Hypoxia-Inducible Factor 2 Axis Regulates Pulmonary Artery Pressure in Mice. Mol Cell Biol 2016: 36(10): 1584–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tang H, Babicheva A, McDermott KM, Gu Y, Ayon RJ, Song S, Wang Z, Gupta A, Zhou T, Sun X, Dash S, Wang Z, Balistrieri A, Zheng Q, Cordery AG, Desai AA, Rischard F, Khalpey Z, Wang J, Black SM, Garcia JGN, Makino A, Yuan JX. Endothelial HIF-2alpha contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am J Physiol Lung Cell Mol Physiol 2018: 314(2): L256–L275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hu CJ, Poth JM, Zhang H, Flockton A, Laux A, Kumar S, McKeon B, Mouradian G, Li M, Riddle S, Pugliese SC, Brown RD, Wallace EM, Graham BB, Frid MG, Stenmark KR. Suppression of HIF2 signalling attenuates the initiation of hypoxia-induced pulmonary hypertension. Eur Respir J 2019: 54(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cowburn AS, Crosby A, Macias D, Branco C, Colaco RD, Southwood M, Toshner M, Crotty Alexander LE, Morrell NW, Chilvers ER, Johnson RS. HIF2alpha-arginase axis is essential for the development of pulmonary hypertension. Proc Natl Acad Sci U S A 2016: 113(31): 8801–8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dai Z, Zhu MM, Peng Y, Machireddy N, Evans CE, Machado R, Zhang X, Zhao YY. Therapeutic Targeting of Vascular Remodeling and Right Heart Failure in PAH with HIF-2alpha Inhibitor. Am J Respir Crit Care Med 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gale DP, Harten SK, Reid CD, Tuddenham EG, Maxwell PH. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2 alpha mutation. Blood 2008: 112(3): 919–921. [DOI] [PubMed] [Google Scholar]
- 70.Tan Q, Kerestes H, Percy MJ, Pietrofesa R, Chen L, Khurana TS, Christofidou-Solomidou M, Lappin TR, Lee FS. Erythrocytosis and pulmonary hypertension in a mouse model of human HIF2A gain of function mutation. J Biol Chem 2013: 288(24): 17134–17144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Achcar RO, Demura Y, Rai PR, Taraseviciene-Stewart L, Kasper M, Voelkel NF, Cool CD. Loss of caveolin and heme oxygenase expression in severe pulmonary hypertension. Chest 2006: 129(3): 696–705. [DOI] [PubMed] [Google Scholar]
- 72.Oliveira SDS, Chen J, Castellon M, Mao M, Usha Raj J, Comhair S, Erzurum S, Silva CLM, Machado RF, Bonini MG, Minshall RD. Injury-Induced Shedding of Extracellular Vesicles Depletes Endothelial Cells of Cav-1 (Caveolin-1) and Enables TGF-beta (Transforming Growth Factor-beta)-Dependent Pulmonary Arterial Hypertension. Arterioscler Thromb Vasc Biol 2019: ATVBAHA118312038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Austin ED, Ma L, LeDuc C, Berman Rosenzweig E, Borczuk A, Phillips JA 3rd, Palomero T, Sumazin P, Kim HR, Talati MH, West J, Loyd JE, Chung WK. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet 2012: 5(3): 336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhao YY, Liu Y, Stan RV, Fan L, Gu Y, Dalton N, Chu PH, Peterson K, Ross J Jr., Chien KR. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci U S A 2002: 99(17): 11375–11380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Murata T, Lin MI, Huang Y, Yu J, Bauer PM, Giordano FJ, Sessa WC. Reexpression of caveolin-1 in endothelium rescues the vascular, cardiac, and pulmonary defects in global caveolin-1 knockout mice. J Exp Med 2007: 204(10): 2373–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xu H, Zhang L, Chen W, Xu J, Zhang R, Liu R, Zhou L, Hu W, Ju R, Lee C, Lu W, Kumar A, Li X, Tang Z. Inhibitory effect of caveolin-1 in vascular endothelial cells, pericytes and smooth muscle cells. Oncotarget 2017: 8(44): 76165–76173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li L, Wang X, Wang L, Qu L, Zhu X, Li M, Dang X, Li P, Gao Y, Peng Z, Pan L, Wan L. Mammalian target of rapamycin overexpression antagonizes chronic hypoxia-triggered pulmonary arterial hypertension via the autophagic pathway. Int J Mol Med 2015: 36(1): 316–322. [DOI] [PubMed] [Google Scholar]
- 78.Kurakula K, Sun XQ, Happé C, da Silva Goncalves Bos D, Szulcek R, Schalij I, Wiesmeijer KC, Lodder K, Tu L, Guignabert C, de Vries CJM, de Man FS, Vonk Noordegraaf A, Ten Dijke P, Goumans MJ, Bogaard HJ. Prevention of progression of pulmonary hypertension by the Nur77 agonist 6-mercaptopurine: role of BMP signalling. Eur Respir J 2019: 54(3). [DOI] [PubMed] [Google Scholar]
- 79.Patel M, Predescu D, Tandon R, Bardita C, Pogoriler J, Bhorade S, Wang M, Comhair S, Hemnes AR, Chen J, Machado R, Husain A, Erzurum S, Predescu S. A novel p38 mitogen-activated protein kinase/Elk-1 transcription factor-dependent molecular mechanism underlying abnormal endothelial cell proliferation in plexogenic pulmonary arterial hypertension. J Biol Chem 2013: 288(36): 25701–25716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Van Hung T, Emoto N, Vignon-Zellweger N, Nakayama K, Yagi K, Suzuki Y, Hirata K. Inhibition of vascular endothelial growth factor receptor under hypoxia causes severe, human-like pulmonary arterial hypertension in mice: potential roles of interleukin-6 and endothelin. Life Sci 2014: 118(2): 313–328. [DOI] [PubMed] [Google Scholar]
- 81.Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, Duroux P, Galanaud P, Simonneau G, Emilie D. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med 1995: 151(5): 1628–1631. [DOI] [PubMed] [Google Scholar]
- 82.Tu L, De Man FS, Girerd B, Huertas A, Chaumais MC, Lecerf F, François C, Perros F, Dorfmüller P, Fadel E, Montani D, Eddahibi S, Humbert M, Guignabert C. A critical role for p130Cas in the progression of pulmonary hypertension in humans and rodents. Am J Respir Crit Care Med 2012: 186(7): 666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yu X, Chen X, Zheng XD, Zhang J, Zhao X, Liu Y, Zhang H, Zhang L, Yu H, Zhang M, Ma C, Hao X, Zhu D. Growth Differentiation Factor 11 Promotes Abnormal Proliferation and Angiogenesis of Pulmonary Artery Endothelial Cells. Hypertension 2018: 71(4): 729–741. [DOI] [PubMed] [Google Scholar]
- 84.Wang EL, Jia MM, Luo FM, Li T, Peng JJ, Luo XJ, Song FL, Yang JF, Peng J, Liu B. Coordination between NADPH oxidase and vascular peroxidase 1 promotes dysfunctions of endothelial progenitor cells in hypoxia-induced pulmonary hypertensive rats. Eur J Pharmacol 2019: 857: 172459. [DOI] [PubMed] [Google Scholar]
- 85.Kim J, Kang Y, Kojima Y, Lighthouse JK, Hu X, Aldred MA, McLean DL, Park H, Comhair SA, Greif DM, Erzurum SC, Chun HJ. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat Med 2013: 19(1): 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Goyanes AM, Moldobaeva A, Marimoutou M, Varela LC, Wang L, Johnston LF, Aladdin MM, Peloquin GL, Kim BS, Damarla M, Suresh K, Sato T, Kolb TM, Hassoun PM, Damico RL. Functional Impact of Human Genetic Variants of COL18A1/Endostatin on Pulmonary Endothelium. Am J Respir Cell Mol Biol 2020: 62(4): 524–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Damico R, Kolb TM, Valera L, Wang L, Housten T, Tedford RJ, Kass DA, Rafaels N, Gao L, Barnes KC, Benza RL, Rand JL, Hamid R, Loyd JE, Robbins IM, Hemnes AR, Chung WK, Austin ED, Drummond MB, Mathai SC, Hassoun PM. Serum endostatin is a genetically determined predictor of survival in pulmonary arterial hypertension. Am J Respir Crit Care Med 2015: 191(2): 208–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dabral S, Tian X, Kojonazarov B, Savai R, Ghofrani HA, Weissmann N, Florio M, Sun J, Jonigk D, Maegel L, Grimminger F, Seeger W, Savai Pullamsetti S, Schermuly RT. Notch1 signalling regulates endothelial proliferation and apoptosis in pulmonary arterial hypertension. Eur Respir J 2016: 48(4): 1137–1149. [DOI] [PubMed] [Google Scholar]
- 89.Miyagawa K, Shi M, Chen PI, Hennigs JK, Zhao Z, Wang M, Li CG, Saito T, Taylor S, Sa S, Cao A, Wang L, Snyder MP, Rabinovitch M. Smooth Muscle Contact Drives Endothelial Regeneration by BMPR2-Notch1-Mediated Metabolic and Epigenetic Changes. Circ Res 2019: 124(2): 211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sakamaki F, Kyotani S, Nagaya N, Sato N, Oya H, Satoh T, Nakanishi N. Increased plasma P-selectin and decreased thrombomodulin in pulmonary arterial hypertension were improved by continuous prostacyclin therapy. Circulation 2000: 102(22): 2720–2725. [DOI] [PubMed] [Google Scholar]
- 91.Tanaseanu C, Tudor S, Tamsulea I, Marta D, Manea G, Moldoveanu E. Vascular endothelial growth factor, lipoporotein-associated phospholipase A2, sP-selectin and antiphospholipid antibodies, biological markers with prognostic value in pulmonary hypertension associated with chronic obstructive pulmonary disease and systemic lupus erithematosus. Eur J Med Res 2007: 12(4): 145–151. [PubMed] [Google Scholar]
- 92.Collados MT, Sandoval J, Lopez S, Masso FA, Paez A, Borbolla JR, Montano LF. Characterization of von Willebrand factor in primary pulmonary hypertension. Heart Vessels 1999: 14(5): 246–252. [DOI] [PubMed] [Google Scholar]
- 93.Muller AM, Skrzynski C, Skipka G, Muller KM. Expression of von Willebrand factor by human pulmonary endothelial cells in vivo. Respiration 2002: 69(6): 526–533. [DOI] [PubMed] [Google Scholar]
- 94.Lopes AA, Maeda NY, Bydlowski SP. Abnormalities in circulating von Willebrand factor and survival in pulmonary hypertension. Am J Med 1998: 105(1): 21–26. [DOI] [PubMed] [Google Scholar]
- 95.Kawut SM, Horn EM, Berekashvili KK, Widlitz AC, Rosenzweig EB, Barst RJ. von Willebrand factor independently predicts long-term survival in patients with pulmonary arterial hypertension. Chest 2005: 128(4): 2355–2362. [DOI] [PubMed] [Google Scholar]
- 96.Collados MT, Borbolla JR. Prognostic value of von Willebrand factor concentrations in pulmonary hypertension. Chest 2000: 118(4): 1225–1226. [DOI] [PubMed] [Google Scholar]
- 97.Maeda NY, Clave MM, Bydlowski SP, Lopes AA. Decreased circulating thrombomodulin is improved by tadalafil therapy in hypoxemic patients with advanced pulmonary arterial hypertension. Thromb Res 2016: 146: 15–19. [DOI] [PubMed] [Google Scholar]
- 98.Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, Janocha AJ, Masri FA, Arroliga AC, Jennings C, Dweik RA, Tuder RM, Stuehr DJ, Erzurum SC. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci U S A 2007: 104(4): 1342–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Marsboom G, Wietholt C, Haney CR, Toth PT, Ryan JJ, Morrow E, Thenappan T, Bache-Wiig P, Piao L, Paul J, Chen CT, Archer SL. Lung 18F-fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension. Am J Respir Crit Care Med 2012: 185(6): 670–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhao L, Ashek A, Wang L, Fang W, Dabral S, Dubois O, Cupitt J, Pullamsetti SS, Cotroneo E, Jones H, Tomasi G, Nguyen QD, Aboagye EO, El-Bahrawy MA, Barnes G, Howard LS, Gibbs JS, Gsell W, He JG, Wilkins MR. Heterogeneity in lung (18)FDG uptake in pulmonary arterial hypertension: potential of dynamic (18)FDG positron emission tomography with kinetic analysis as a bridging biomarker for pulmonary vascular remodeling targeted treatments. Circulation 2013: 128(11): 1214–1224. [DOI] [PubMed] [Google Scholar]
- 101.Fijalkowska I, Xu W, Comhair SA, Janocha AJ, Mavrakis LA, Krishnamachary B, Zhen L, Mao T, Richter A, Erzurum SC, Tuder RM. Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol 2010: 176(3): 1130–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cao Y, Zhang X, Wang L, Yang Q, Ma Q, Xu J, Wang J, Kovacs L, Ayon RJ, Liu Z, Zhang M, Zhou Y, Zeng X, Xu Y, Wang Y, Fulton DJ, Weintraub NL, Lucas R, Dong Z, Yuan JX, Sullivan JC, Meadows L, Barman SA, Wu C, Quan J, Hong M, Su Y, Huo Y. PFKFB3-mediated endothelial glycolysis promotes pulmonary hypertension. Proc Natl Acad Sci U S A 2019: 116(27): 13394–13403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Caruso P, Dunmore BJ, Schlosser K, Schoors S, Dos Santos C, Perez-Iratxeta C, Lavoie JR, Zhang H, Long L, Flockton AR, Frid MG, Upton PD, D’Alessandro A, Hadinnapola C, Kiskin FN, Taha M, Hurst LA, Ormiston ML, Hata A, Stenmark KR, Carmeliet P, Stewart DJ, Morrell NW. Identification of MicroRNA-124 as a Major Regulator of Enhanced Endothelial Cell Glycolysis in Pulmonary Arterial Hypertension via PTBP1 (Polypyrimidine Tract Binding Protein) and Pyruvate Kinase M2. Circulation 2017: 136(25): 2451–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sun X, Kumar S, Sharma S, Aggarwal S, Lu Q, Gross C, Rafikova O, Lee SG, Dasarathy S, Hou Y, Meadows ML, Han W, Su Y, Fineman JR, Black SM. Endothelin-1 induces a glycolytic switch in pulmonary arterial endothelial cells via the mitochondrial translocation of endothelial nitric oxide synthase. Am J Respir Cell Mol Biol 2014: 50(6): 1084–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yu Q, Tai YY, Tang Y, Zhao J, Negi V, Culley MK, Pilli J, Sun W, Brugger K, Mayr J, Saggar R, Saggar R, Wallace WD, Ross DJ, Waxman AB, Wendell SG, Mullett SJ, Sembrat J, Rojas M, Khan OF, Dahlman JE, Sugahara M, Kagiyama N, Satoh T, Zhang M, Feng N, Gorcsan J 3rd, Vargas SO, Haley KJ, Kumar R, Graham BB, Langer R, Anderson DG, Wang B, Shiva S, Bertero T, Chan SY. BOLA (BolA Family Member 3) Deficiency Controls Endothelial Metabolism and Glycine Homeostasis in Pulmonary Hypertension. Circulation 2019: 139(19): 2238–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.White K, Lu Y, Annis S, Hale AE, Chau BN, Dahlman JE, Hemann C, Opotowsky AR, Vargas SO, Rosas I, Perrella MA, Osorio JC, Haley KJ, Graham BB, Kumar R, Saggar R, Saggar R, Wallace WD, Ross DJ, Khan OF, Bader A, Gochuico BR, Matar M, Polach K, Johannessen NM, Prosser HM, Anderson DG, Langer R, Zweier JL, Bindoff LA, Systrom D, Waxman AB, Jin RC, Chan SY. Genetic and hypoxic alterations of the microRNA-210-ISCU1/2 axis promote iron-sulfur deficiency and pulmonary hypertension. EMBO Mol Med 2015: 7(6): 695–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yuan Y, Shen C, Zhao SL, Hu YJ, Song Y, Zhong QJ. MicroRNA-126 affects cell apoptosis, proliferation, cell cycle and modulates VEGF/TGF-β levels in pulmonary artery endothelial cells. Eur Rev Med Pharmacol Sci 2019: 23(7): 3058–3069. [DOI] [PubMed] [Google Scholar]
- 108.Rothman AM, Arnold ND, Pickworth JA, Iremonger J, Ciuclan L, Allen RM, Guth-Gundel S, Southwood M, Morrell NW, Thomas M, Francis SE, Rowlands DJ, Lawrie A. MicroRNA-140–5p and SMURF1 regulate pulmonary arterial hypertension. J Clin Invest 2016: 126(7): 2495–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhao J, Florentin J, Tai YY, Torrino S, Ohayon L, Brzoska T, Tang Y, Yang J, Negi V, Woodcock CC, Risbano MG, Nouraie SM, Sundd P, Bertero T, Dutta P, Chan SY. Long Range Endocrine Delivery of Circulating miR-210 to Endothelium Promotes Pulmonary Hypertension. Circ Res 2020: 127(5): 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ye JX, Wang SS, Ge M, Wang DJ. Suppression of endothelial PGC-1alpha is associated with hypoxia-induced endothelial dysfunction and provides a new therapeutic target in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 2016: 310(11): L1233–1242. [DOI] [PubMed] [Google Scholar]
- 111.Haslip M, Dostanic I, Huang Y, Zhang Y, Russell KS, Jurczak MJ, Mannam P, Giordano F, Erzurum SC, Lee PJ. Endothelial uncoupling protein 2 regulates mitophagy and pulmonary hypertension during intermittent hypoxia. Arterioscler Thromb Vasc Biol 2015: 35(5): 1166–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bertero T, Oldham WM, Cottrill KA, Pisano S, Vanderpool RR, Yu Q, Zhao J, Tai Y, Tang Y, Zhang YY, Rehman S, Sugahara M, Qi Z, Gorcsan J 3rd, Vargas SO, Saggar R, Saggar R, Wallace WD, Ross DJ, Haley KJ, Waxman AB, Parikh VN, De Marco T, Hsue PY, Morris A, Simon MA, Norris KA, Gaggioli C, Loscalzo J, Fessel J, Chan SY. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J Clin Invest 2016: 126(9): 3313–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hautefort A, Chesné J, Preussner J, Pullamsetti SS, Tost J, Looso M, Antigny F, Girerd B, Riou M, Eddahibi S, Deleuze JF, Seeger W, Fadel E, Simonneau G, Montani D, Humbert M, Perros F. Pulmonary endothelial cell DNA methylation signature in pulmonary arterial hypertension. Oncotarget 2017: 8(32): 52995–53016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chen D, Yang Y, Cheng X, Fang F, Xu G, Yuan Z, Xia J, Kong H, Xie W, Wang H, Fang M, Gao Y, Xu Y. Megakaryocytic leukemia 1 directs a histone H3 lysine 4 methyltransferase complex to regulate hypoxic pulmonary hypertension. Hypertension 2015: 65(4): 821–833. [DOI] [PubMed] [Google Scholar]
- 115.Kim J, Hwangbo C, Hu X, Kang Y, Papangeli I, Mehrotra D, Park H, Ju H, McLean DL, Comhair SA, Erzurum SC, Chun HJ. Restoration of impaired endothelial myocyte enhancer factor 2 function rescues pulmonary arterial hypertension. Circulation 2015: 131(2): 190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Péchoux C, Bogaard HJ, Dorfmüller P, Remy S, Lecerf F, Planté S, Chat S, Fadel E, Houssaini A, Anegon I, Adnot S, Simonneau G, Humbert M, Cohen-Kaminsky S, Perros F. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015: 131(11): 1006–1018. [DOI] [PubMed] [Google Scholar]
- 117.Steffes LC, Froistad AA, Andruska A, Boehm M, McGlynn M, Zhang F, Zhang W, Hou D, Tian X, Miquerol L, Nadeau K, Metzger RJ, Spiekerkoetter E, Kumar ME. A Notch3-Marked Subpopulation of Vascular Smooth Muscle Cells Is the Cell of Origin for Occlusive Pulmonary Vascular Lesions. Circulation 2020: 142(16): 1545–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hopper RK, Moonen JR, Diebold I, Cao A, Rhodes CJ, Tojais NF, Hennigs JK, Gu M, Wang L, Rabinovitch M. In Pulmonary Arterial Hypertension, Reduced BMPR2 Promotes Endothelial-to-Mesenchymal Transition via HMGA1 and Its Target Slug. Circulation 2016: 133(18): 1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Brett SJ, Simon J, Gibbs R, Pepper JR, Evans TW. Impairment of endothelium-dependent pulmonary vasodilation in patients with primary pulmonary hypertension. Thorax 1996: 51(1): 89–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Reddy VM, Wong J, Liddicoat JR, Johengen M, Chang R, Fineman JR. Altered endothelium-dependent responses in lambs with pulmonary hypertension and increased pulmonary blood flow. Am J Physiol 1996: 271(2 Pt 2): H562–570. [DOI] [PubMed] [Google Scholar]
- 121.Matsukura Y, Washizu M, Kondo M, Motoyoshi S, Itoh A, Nakajyo S, Shimizu K, Urakawa N. Decreased pulmonary arterial endothelium-dependent relaxation in heartworm-infected dogs with pulmonary hypertension. Am J Vet Res 1997: 58(2): 171–174. [PubMed] [Google Scholar]
- 122.Martinez-Lemus LA, Hester RK, Becker EJ, Jeffrey JS, Odom TW. Pulmonary artery endothelium-dependent vasodilation is impaired in a chicken model of pulmonary hypertension. Am J Physiol 1999: 277(1 Pt 2): R190–197. [DOI] [PubMed] [Google Scholar]
- 123.Morio Y, Homma N, Takahashi H, Yamamoto A, Nagaoka T, Sato K, Muramatsu M, Fukuchi Y. Activity of endothelium-derived hyperpolarizing factor is augmented in monocrotaline-induced pulmonary hypertension of rat lungs. J Vasc Res 2007: 44(4): 325–335. [DOI] [PubMed] [Google Scholar]
- 124.Yildiz P, Oflaz H, Cine N, Erginel-Unaltuna N, Erzengin F, Yilmaz V. Gene polymorphisms of endothelial nitric oxide synthase enzyme associated with pulmonary hypertension in patients with COPD. Respir Med 2003: 97(12): 1282–1288. [DOI] [PubMed] [Google Scholar]
- 125.Xu W, Kaneko FT, Zheng S, Comhair SA, Janocha AJ, Goggans T, Thunnissen FB, Farver C, Hazen SL, Jennings C, Dweik RA, Arroliga AC, Erzurum SC. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J 2004: 18(14): 1746–1748. [DOI] [PubMed] [Google Scholar]
- 126.Ghosh S, Gupta M, Xu W, Mavrakis DA, Janocha AJ, Comhair SA, Haque MM, Stuehr DJ, Yu J, Polgar P, Naga Prasad SV, Erzurum SC. Phosphorylation inactivation of endothelial nitric oxide synthesis in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 2016: 310(11): L1199–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhao YY, Zhao YD, Mirza MK, Huang JH, Potula HH, Vogel SM, Brovkovych V, Yuan JX, Wharton J, Malik AB. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. J Clin Invest 2009: 119(7): 2009–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Duarte JD, Kansal M, Desai AA, Riden K, Arwood MJ, Yacob AA, Stamos TD, Cavallari LH, Zamanian RT, Shah SJ, Machado RF. Endothelial nitric oxide synthase genotype is associated with pulmonary hypertension severity in left heart failure patients. Pulm Circ 2018: 8(2): 2045894018773049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Khoo JP, Zhao L, Alp NJ, Bendall JK, Nicoli T, Rockett K, Wilkins MR, Channon KM. Pivotal role for endothelial tetrahydrobiopterin in pulmonary hypertension. Circulation 2005: 111(16): 2126–2133. [DOI] [PubMed] [Google Scholar]
- 130.Zhang F, Wu S, Lu X, Wang M, Liu M. Gene transfer of endothelial nitric oxide synthase attenuates flow-induced pulmonary hypertension in rabbits. Ann Thorac Surg 2008: 85(2): 581–585. [DOI] [PubMed] [Google Scholar]
- 131.Sun X, Ku DD. Allicin in garlic protects against coronary endothelial dysfunction and right heart hypertrophy in pulmonary hypertensive rats. Am J Physiol Heart Circ Physiol 2006: 291(5): H2431–2438. [DOI] [PubMed] [Google Scholar]
- 132.Sun X, Ku DD. Rosuvastatin provides pleiotropic protection against pulmonary hypertension, right ventricular hypertrophy, and coronary endothelial dysfunction in rats. Am J Physiol Heart Circ Physiol 2008: 294(2): H801–809. [DOI] [PubMed] [Google Scholar]
- 133.Murata T, Kinoshita K, Hori M, Kuwahara M, Tsubone H, Karaki H, Ozaki H. Statin protects endothelial nitric oxide synthase activity in hypoxia-induced pulmonary hypertension. Arterioscler Thromb Vasc Biol 2005: 25(11): 2335–2342. [DOI] [PubMed] [Google Scholar]
- 134.Zhao YD, Courtman DW, Deng Y, Kugathasan L, Zhang Q, Stewart DJ. Rescue of monocrotaline-induced pulmonary arterial hypertension using bone marrow-derived endothelial-like progenitor cells: efficacy of combined cell and eNOS gene therapy in established disease. Circ Res 2005: 96(4): 442–450. [DOI] [PubMed] [Google Scholar]
- 135.Klinger JR, Abman SH, Gladwin MT. Nitric oxide deficiency and endothelial dysfunction in pulmonary arterial hypertension. Am J Respir Crit Care Med 2013: 188(6): 639–646. [DOI] [PubMed] [Google Scholar]
- 136.Mubarak KK. A review of prostaglandin analogs in the management of patients with pulmonary arterial hypertension. Respir Med 2010: 104(1): 9–21. [DOI] [PubMed] [Google Scholar]
- 137.Sitbon O, Channick R, Chin KM, Frey A, Gaine S, Galiè N, Ghofrani HA, Hoeper MM, Lang IM, Preiss R, Rubin LJ, Di Scala L, Tapson V, Adzerikho I, Liu J, Moiseeva O, Zeng X, Simonneau G, McLaughlin VV. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med 2015: 373(26): 2522–2533. [DOI] [PubMed] [Google Scholar]
- 138.Prie S, Leung TK, Cernacek P, Ryan JW, Dupuis J. The orally active ET(A) receptor antagonist (+)-(S)-2-(4,6-dimethoxy-pyrimidin-2-yloxy)-3-methoxy-3,3-diphe nyl-propionic acid (LU 135252) prevents the development of pulmonary hypertension and endothelial metabolic dysfunction in monocrotaline-treated rats. J Pharmacol Exp Ther 1997: 282(3): 1312–1318. [PubMed] [Google Scholar]
- 139.Satwiko MG, Ikeda K, Nakayama K, Yagi K, Hocher B, Hirata K, Emoto N. Targeted activation of endothelin-1 exacerbates hypoxia-induced pulmonary hypertension. Biochem Biophys Res Commun 2015: 465(3): 356–362. [DOI] [PubMed] [Google Scholar]
- 140.Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, Kimura S, Masaki T, Duguid WP, Stewart DJ. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med 1993: 328(24): 1732–1739. [DOI] [PubMed] [Google Scholar]
- 141.Kelland NF, Bagnall AJ, Morecroft I, Gulliver-Sloan FH, Dempsie Y, Nilsen M, Yanagisawa M, Maclean MR, Kotelevtsev YV, Webb DJ. Endothelial ET(B) limits vascular remodelling and development of pulmonary hypertension during hypoxia. J Vasc Res 2010: 47(1): 16–22. [DOI] [PubMed] [Google Scholar]
- 142.Miyagawa K, Emoto N. Current state of endothelin receptor antagonism in hypertension and pulmonary hypertension. Ther Adv Cardiovasc Dis 2014: 8(5): 202–216. [DOI] [PubMed] [Google Scholar]
- 143.Pullamsetti SS, Schermuly RT. Endothelin receptor antagonists in preclinical models of pulmonary hypertension. Eur J Clin Invest 2009: 39Suppl 2: 3–13. [DOI] [PubMed] [Google Scholar]
- 144.Attina T, Camidge R, Newby DE, Webb DJ. Endothelin antagonism in pulmonary hypertension, heart failure, and beyond. Heart 2005: 91(6): 825–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Evans CE, Zhao YY. Molecular Basis of Nitrative Stress in the Pathogenesis of Pulmonary Hypertension. Adv Exp Med Biol 2017: 967: 33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhao YD, Cai L, Mirza MK, Huang X, Geenen DL, Hofmann F, Yuan JX, Zhao YY. Protein kinase G-I deficiency induces pulmonary hypertension through Rho A/Rho kinase activation. Am J Pathol 2012: 180(6): 2268–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Aggarwal S, Gross CM, Rafikov R, Kumar S, Fineman JR, Ludewig B, Jonigk D, Black SM. Nitration of tyrosine 247 inhibits protein kinase G-1alpha activity by attenuating cyclic guanosine monophosphate binding. J Biol Chem 2014: 289(11): 7948–7961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Eddahibi S, Guignabert C, Barlier-Mur AM, Dewachter L, Fadel E, Dartevelle P, Humbert M, Simonneau G, Hanoun N, Saurini F, Hamon M, Adnot S. Cross talk between endothelial and smooth muscle cells in pulmonary hypertension: critical role for serotonin-induced smooth muscle hyperplasia. Circulation 2006: 113(15): 1857–1864. [DOI] [PubMed] [Google Scholar]
- 149.Perros F, Montani D, Dorfmüller P, Durand-Gasselin I, Tcherakian C, Le Pavec J, Mazmanian M, Fadel E, Mussot S, Mercier O, Hervé P, Emilie D, Eddahibi S, Simonneau G, Souza R, Humbert M. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2008: 178(1): 81–88. [DOI] [PubMed] [Google Scholar]
- 150.Katayose D, Ohe M, Yamauchi K, Ogata M, Shirato K, Fujita H, Shibahara S, Takishima T. Increased expression of PDGF A- and B-chain genes in rat lungs with hypoxic pulmonary hypertension. Am J Physiol 1993: 264(2 Pt 1): L100–106. [DOI] [PubMed] [Google Scholar]
- 151.Lannér MC, Raper M, Pratt WM, Rhoades RA. Heterotrimeric G proteins and the platelet-derived growth factor receptor-beta contribute to hypoxic proliferation of smooth muscle cells. Am J Respir Cell Mol Biol 2005: 33(4): 412–419. [DOI] [PubMed] [Google Scholar]
- 152.ten Freyhaus H, Dagnell M, Leuchs M, Vantler M, Berghausen EM, Caglayan E, Weissmann N, Dahal BK, Schermuly RT, Ostman A, Kappert K, Rosenkranz S. Hypoxia enhances platelet-derived growth factor signaling in the pulmonary vasculature by down-regulation of protein tyrosine phosphatases. Am J Respir Crit Care Med 2011: 183(8): 1092–1102. [DOI] [PubMed] [Google Scholar]
- 153.Izikki M, Guignabert C, Fadel E, Humbert M, Tu L, Zadigue P, Dartevelle P, Simonneau G, Adnot S, Maitre B, Raffestin B, Eddahibi S. Endothelial-derived FGF2 contributes to the progression of pulmonary hypertension in humans and rodents. J Clin Invest 2009: 119(3): 512–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Dai Z, Zhu MM, Peng Y, Jin H, Machireddy N, Qian Z, Zhang X, Zhao YY. Endothelial and Smooth Muscle Cell Interaction via FoxM1 Signaling Mediates Vascular Remodeling and Pulmonary Hypertension. Am J Respir Crit Care Med 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Savai R, Al-Tamari HM, Sedding D, Kojonazarov B, Muecke C, Teske R, Capecchi MR, Weissmann N, Grimminger F, Seeger W, Schermuly RT, Pullamsetti SS. Pro-proliferative and inflammatory signaling converge on FoxO1 transcription factor in pulmonary hypertension. Nat Med 2014: 20(11): 1289–1300. [DOI] [PubMed] [Google Scholar]
- 156.Zeng Z, Yao J, Li Y, Xue Y, Zou Y, Shu Z, Jiao Z. Anti-apoptosis endothelial cell-secreted microRNA-195–5p promotes pulmonary arterial smooth muscle cell proliferation and migration in pulmonary arterial hypertension. J Cell Biochem 2018: 119(2): 2144–2155. [DOI] [PubMed] [Google Scholar]
- 157.Omura J, Satoh K, Kikuchi N, Satoh T, Kurosawa R, Nogi M, Otsuki T, Kozu K, Numano K, Suzuki K, Sunamura S, Tatebe S, Aoki T, Sugimura K, Miyata S, Hoshikawa Y, Okada Y, Shimokawa H. Protective Roles of Endothelial AMP-Activated Protein Kinase Against Hypoxia-Induced Pulmonary Hypertension in Mice. Circ Res 2016: 119(2): 197–209. [DOI] [PubMed] [Google Scholar]
- 158.Zhang J, Dong J, Martin M, He M, Gongol B, Marin TL, Chen L, Shi X, Yin Y, Shang F, Wu Y, Huang HY, Zhang J, Zhang Y, Kang J, Moya EA, Huang HD, Powell FL, Chen Z, Thistlethwaite PA, Yuan ZY, Shyy JY. AMP-activated Protein Kinase Phosphorylation of Angiotensin-Converting Enzyme 2 in Endothelium Mitigates Pulmonary Hypertension. Am J Respir Crit Care Med 2018: 198(4): 509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sanchez O, Marcos E, Perros F, Fadel E, Tu L, Humbert M, Dartevelle P, Simonneau G, Adnot S, Eddahibi S. Role of endothelium-derived CC chemokine ligand 2 in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2007: 176(10): 1041–1047. [DOI] [PubMed] [Google Scholar]
- 160.Xiao Y, Christou H, Liu L, Visner G, Mitsialis SA, Kourembanas S, Liu H. Endothelial indoleamine 2,3-dioxygenase protects against development of pulmonary hypertension. Am J Respir Crit Care Med 2013: 188(4): 482–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sakao S, Taraseviciene-Stewart L, Wood K, Cool CD, Voelkel NF. Apoptosis of pulmonary microvascular endothelial cells stimulates vascular smooth muscle cell growth. Am J Physiol Lung Cell Mol Physiol 2006: 291(3): L362–368. [DOI] [PubMed] [Google Scholar]
- 162.Sirois I, Raymond MA, Brassard N, Cailhier JF, Fedjaev M, Hamelin K, Londono I, Bendayan M, Pshezhetsky AV, Hébert MJ. Caspase-3-dependent export of TCTP: a novel pathway for antiapoptotic intercellular communication. Cell Death Differ 2011: 18(3): 549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lavoie JR, Ormiston ML, Perez-Iratxeta C, Courtman DW, Jiang B, Ferrer E, Caruso P, Southwood M, Foster WS, Morrell NW, Stewart DJ. Proteomic analysis implicates translationally controlled tumor protein as a novel mediator of occlusive vascular remodeling in pulmonary arterial hypertension. Circulation 2014: 129(21): 2125–2135. [DOI] [PubMed] [Google Scholar]
- 164.Ferrer E, Dunmore BJ, Hassan D, Ormiston ML, Moore S, Deighton J, Long L, Yang XD, Stewart DJ, Morrell NW. A Potential Role for Exosomal Translationally Controlled Tumor Protein Export in Vascular Remodeling in Pulmonary Arterial Hypertension. Am J Respir Cell Mol Biol 2018: 59(4): 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Deng L, Blanco FJ, Stevens H, Lu R, Caudrillier A, McBride M, McClure JD, Grant J, Thomas M, Frid M, Stenmark K, White K, Seto AG, Morrell NW, Bradshaw AC, MacLean MR, Baker AH. MicroRNA-143 Activation Regulates Smooth Muscle and Endothelial Cell Crosstalk in Pulmonary Arterial Hypertension. Circ Res 2015: 117(10): 870–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Stenmark KR, Yeager ME, El Kasmi KC, Nozik-Grayck E, Gerasimovskaya EV, Li M, Riddle SR, Frid MG. The adventitia: essential regulator of vascular wall structure and function. Annu Rev Physiol 2013: 75: 23–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Le Hiress M, Tu L, Ricard N, Phan C, Thuillet R, Fadel E, Dorfmuller P, Montani D, de Man F, Humbert M, Huertas A, Guignabert C. Proinflammatory Signature of the Dysfunctional Endothelium in Pulmonary Hypertension. Role of the Macrophage Migration Inhibitory Factor/CD74 Complex. Am J Respir Crit Care Med 2015: 192(8): 983–997. [DOI] [PubMed] [Google Scholar]
- 168.Sawada H, Saito T, Nickel NP, Alastalo TP, Glotzbach JP, Chan R, Haghighat L, Fuchs G, Januszyk M, Cao A, Lai YJ, Perez Vde J, Kim YM, Wang L, Chen PI, Spiekerkoetter E, Mitani Y, Gurtner GC, Sarnow P, Rabinovitch M. Reduced BMPR2 expression induces GM-CSF translation and macrophage recruitment in humans and mice to exacerbate pulmonary hypertension. J Exp Med 2014: 211(2): 263–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Costello CM, McCullagh B, Howell K, Sands M, Belperio JA, Keane MP, Gaine S, McLoughlin P. A role for the CXCL12 receptor, CXCR7, in the pathogenesis of human pulmonary vascular disease. Eur Respir J 2012: 39(6): 1415–1424. [DOI] [PubMed] [Google Scholar]
- 170.Pi L, Fu C, Lu Y, Zhou J, Jorgensen M, Shenoy V, Lipson KE, Scott EW, Bryant AJ. Vascular Endothelial Cell-Specific Connective Tissue Growth Factor (CTGF) Is Necessary for Development of Chronic Hypoxia-Induced Pulmonary Hypertension. Front Physiol 2018: 9: 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Huertas A, Tu L, Gambaryan N, Girerd B, Perros F, Montani D, Fabre D, Fadel E, Eddahibi S, Cohen-Kaminsky S, Guignabert C, Humbert M. Leptin and regulatory T-lymphocytes in idiopathic pulmonary arterial hypertension. Eur Respir J 2012: 40(4): 895–904. [DOI] [PubMed] [Google Scholar]
- 172.Xue C, Sowden M, Berk BC. Extracellular Cyclophilin A, Especially Acetylated, Causes Pulmonary Hypertension by Stimulating Endothelial Apoptosis, Redox Stress, and Inflammation. Arterioscler Thromb Vasc Biol 2017: 37(6): 1138–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Tian W, Jiang X, Tamosiuniene R, Sung YK, Qian J, Dhillon G, Gera L, Farkas L, Rabinovitch M, Zamanian RT, Inayathullah M, Fridlib M, Rajadas J, Peters-Golden M, Voelkel NF, Nicolls MR. Blocking macrophage leukotriene b4 prevents endothelial injury and reverses pulmonary hypertension. Sci Transl Med 2013: 5(200): 200ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Jiang B, Deng Y, Suen C, Taha M, Chaudhary KR, Courtman DW, Stewart DJ. Marked Strain-Specific Differences in the SU5416 Rat Model of Severe Pulmonary Arterial Hypertension. Am J Respir Cell Mol Biol 2016: 54(4): 461–468. [DOI] [PubMed] [Google Scholar]
- 175.Tamosiuniene R, Tian W, Dhillon G, Wang L, Sung YK, Gera L, Patterson AJ, Agrawal R, Rabinovitch M, Ambler K, Long CS, Voelkel NF, Nicolls MR. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ Res 2011: 109(8): 867–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ricard N, Tu L, Le Hiress M, Huertas A, Phan C, Thuillet R, Sattler C, Fadel E, Seferian A, Montani D, Dorfmuller P, Humbert M, Guignabert C. Increased pericyte coverage mediated by endothelial-derived fibroblast growth factor-2 and interleukin-6 is a source of smooth muscle-like cells in pulmonary hypertension. Circulation 2014: 129(15): 1586–1597. [DOI] [PubMed] [Google Scholar]
- 177.Wang S, Zeng H, Xie XJ, Tao YK, He X, Roman RJ, Aschner JL, Chen JX. Loss of prolyl hydroxylase domain protein 2 in vascular endothelium increases pericyte coverage and promotes pulmonary arterial remodeling. Oncotarget 2016: 7(37): 58848–58861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yuan K, Shamskhou EA, Orcholski ME, Nathan A, Reddy S, Honda H, Mani V, Zeng Y, Ozen MO, Wang L, Demirci U, Tian W, Nicolls MR, de Jesus Perez VA. Loss of Endothelium-Derived Wnt5a Is Associated With Reduced Pericyte Recruitment and Small Vessel Loss in Pulmonary Arterial Hypertension. Circulation 2019: 139(14): 1710–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Yuan K, Shao NY, Hennigs JK, Discipulo M, Orcholski ME, Shamskhou E, Richter A, Hu X, Wu JC, de Jesus Perez VA. Increased Pyruvate Dehydrogenase Kinase 4 Expression in Lung Pericytes Is Associated with Reduced Endothelial-Pericyte Interactions and Small Vessel Loss in Pulmonary Arterial Hypertension. Am J Pathol 2016: 186(9): 2500–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Xia L, Fu GS, Yang JX, Zhang FR, Wang XX. Endothelial progenitor cells may inhibit apoptosis of pulmonary microvascular endothelial cells: new insights into cell therapy for pulmonary arterial hypertension. Cytotherapy 2009: 11(4): 492–502. [DOI] [PubMed] [Google Scholar]
- 181.Shi-Wen X, Chen Y, Denton CP, Eastwood M, Renzoni EA, Bou-Gharios G, Pearson JD, Dashwood M, du Bois RM, Black CM, Leask A, Abraham DJ. Endothelin-1 promotes myofibroblast induction through the ETA receptor via a rac/phosphoinositide 3-kinase/Akt-dependent pathway and is essential for the enhanced contractile phenotype of fibrotic fibroblasts. Mol Biol Cell 2004: 15(6): 2707–2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Gallucci RM, Lee EG, Tomasek JJ. IL-6 modulates alpha-smooth muscle actin expression in dermal fibroblasts from IL-6-deficient mice. J Invest Dermatol 2006: 126(3): 561–568. [DOI] [PubMed] [Google Scholar]
- 183.Thenappan T, Chan SY, Weir EK. Role of extracellular matrix in the pathogenesis of pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 2018: 315(5): H1322–h1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Liu Y, Zhang H, Yan L, Du W, Zhang M, Chen H, Zhang L, Li G, Li J, Dong Y, Zhu D. MMP-2 and MMP-9 contribute to the angiogenic effect produced by hypoxia/15-HETE in pulmonary endothelial cells. J Mol Cell Cardiol 2018: 121: 36–50. [DOI] [PubMed] [Google Scholar]
- 185.Labrousse-Arias D, Castillo-González R, Rogers NM, Torres-Capelli M, Barreira B, Aragonés J, Cogolludo Á, Isenberg JS, Calzada MJ. HIF-2α-mediated induction of pulmonary thrombospondin-1 contributes to hypoxia-driven vascular remodelling and vasoconstriction. Cardiovasc Res 2016: 109(1): 115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Toshner M, Voswinckel R, Southwood M, Al-Lamki R, Howard LS, Marchesan D, Yang J, Suntharalingam J, Soon E, Exley A, Stewart S, Hecker M, Zhu Z, Gehling U, Seeger W, Pepke-Zaba J, Morrell NW. Evidence of dysfunction of endothelial progenitors in pulmonary arterial hypertension. AmJ Respir Crit Care Med 2009: 180(8): 780–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Granton J, Langleben D, Kutryk MB, Camack N, Galipeau J, Courtman DW, Stewart DJ. Endothelial NO-Synthase Gene-Enhanced Progenitor Cell Therapy for Pulmonary Arterial Hypertension: The PHACeT Trial. Circ Res 2015: 117(7): 645–654. [DOI] [PubMed] [Google Scholar]
- 188.Wang XX, Zhang FR, Shang YP, Zhu JH, Xie XD, Tao QM, Zhu JH, Chen JZ. Transplantation of autologous endothelial progenitor cells may be beneficial in patients with idiopathic pulmonary arterial hypertension: a pilot randomized controlled trial. J Am Coll Cardiol 2007: 49(14): 1566–1571. [DOI] [PubMed] [Google Scholar]
- 189.Zhu JH, Wang XX, Zhang FR, Shang YP, Tao QM, Zhu JH, Chen JZ. Safety and efficacy of autologous endothelial progenitor cells transplantation in children with idiopathic pulmonary arterial hypertension: open-label pilot study. Pediatr Transplant 2008: 12(6): 650–655. [DOI] [PubMed] [Google Scholar]
- 190.Jiang DM, Han J, Zhu JH, Fu GS, Zhou BQ. Paracrine effects of bone marrow-derived endothelial progenitor cells: cyclooxygenase-2/prostacyclin pathway in pulmonary arterial hypertension. PLoS One 2013: 8(11): e79215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Gall H, Felix JF, Schneck FK, Milger K, Sommer N, Voswinckel R, Franco OH, Hofman A, Schermuly RT, Weissmann N, Grimminger F, Seeger W, Ghofrani HA. The Giessen Pulmonary Hypertension Registry: Survival in pulmonary hypertension subgroups. J Heart Lung Transplant 2017: 36(9): 957–967. [DOI] [PubMed] [Google Scholar]
- 192.Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, Homayoun F, Ma Y, Patel N, Yell P, Hao G, Yousuf Q, Joyce A, Pedrosa I, Geiger H, Zhang H, Chang J, Gardner KH, Bruick RK, Reeves C, Hwang TH, Courtney K, Frenkel E, Sun X, Zojwalla N, Wong T, Rizzi JP, Wallace EM, Josey JA, Xie Y, Xie XJ, Kapur P, McKay RM, Brugarolas J. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016: 539(7627): 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Courtney KD, Ma Y, Diaz de Leon A, Christie A, Xie Z, Woolford L, Singla N, Joyce A, Hill H, Madhuranthakam AJ, Yuan Q, Xi Y, Zhang Y, Chang J, Fatunde O, Arriaga Y, Frankel AE, Kalva S, Zhang S, McKenzie T, Reig Torras O, Figlin RA, Rini BI, McKay RM, Kapur P, Wang T, Pedrosa I, Brugarolas J. HIF-2 Complex Dissociation, Target Inhibition, and Acquired Resistance with PT2385, a First-in-Class HIF-2 Inhibitor, in Patients with Clear Cell Renal Cell Carcinoma. Clin Cancer Res 2020: 26(4): 793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Xu R, Wang K, Rizzi JP, Huang H, Grina JA, Schlachter ST, Wang B, Wehn PM, Yang H, Dixon DD, Czerwinski RM, Du X, Ged EL, Han G, Tan H, Wong T, Xie S, Josey JA, Wallace EM. 3-[(1S,2S,3R)-2,3-Difluoro-1-hydroxy-7-methylsulfonylindan-4-yl]oxy-5-fluorobenzonitrile (PT2977), a Hypoxia-Inducible Factor 2α (HIF-2α) Inhibitor for the Treatment of Clear Cell Renal Cell Carcinoma. J Med Chem 2019: 62(15): 6876–6893. [DOI] [PubMed] [Google Scholar]
- 195.Dahlman JE, Barnes C, Khan O, Thiriot A, Jhunjunwala S, Shaw TE, Xing Y, Sager HB, Sahay G, Speciner L, Bader A, Bogorad RL, Yin H, Racie T, Dong Y, Jiang S, Seedorf D, Dave A, Sandu KS, Webber MJ, Novobrantseva T, Ruda VM, Lytton-Jean AKR, Levins CG, Kalish B, Mudge DK, Perez M, Abezgauz L, Dutta P, Smith L, Charisse K, Kieran MW, Fitzgerald K, Nahrendorf M, Danino D, Tuder RM, von Andrian UH, Akinc A, Schroeder A, Panigrahy D, Kotelianski V, Langer R, Anderson DG. In vivo endothelial siRNA delivery using polymeric nanoparticles with low molecular weight. Nat Nanotechnol 2014: 9(8): 648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Saygin D, Tabib T, Bittar HET, Valenzi E, Sembrat J, Chan SY, Rojas M, Lafyatis R. Transcriptional profiling of lung cell populations in idiopathic pulmonary arterial hypertension. Pulm Circ 2020: 10(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Huang J, Kaminski PM, Edwards JG, Yeh A, Wolin MS, Frishman WH, Gewitz MH, Mathew R. Pyrrolidine dithiocarbamate restores endothelial cell membrane integrity and attenuates monocrotaline-induced pulmonary artery hypertension. Am J Physiol Lung Cell Mol Physiol 2008: 294(6): L1250–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Yamada Y, Maruyama J, Zhang E, Okada A, Yokochi A, Sawada H, Mitani Y, Hayashi T, Suzuki K, Maruyama K. Effect of thrombomodulin on the development of monocrotaline-induced pulmonary hypertension. J Anesth 2014: 28(1): 26–33. [DOI] [PubMed] [Google Scholar]
- 199.Zaiman AL, Podowski M, Medicherla S, Gordy K, Xu F, Zhen L, Shimoda LA, Neptune E, Higgins L, Murphy A, Chakravarty S, Protter A, Sehgal PB, Champion HC, Tuder RM. Role of the TGF-beta/Alk5 signaling pathway in monocrotaline-induced pulmonary hypertension. Am J Respir Crit Care Med 2008: 177(8): 896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Liang OD, So EY, Egan PC, Goldberg LR, Aliotta JM, Wu KQ, Dubielecka PM, Ventetuolo CE, Reginato AM, Quesenberry PJ, Klinger JR. Endothelial to haematopoietic transition contributes to pulmonary arterial hypertension. Cardiovasc Res 2017: 113(13): 1560–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]