

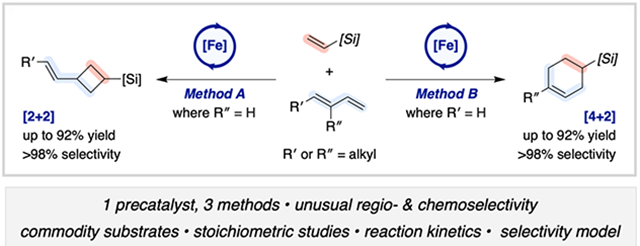
Abstract
The selective, intermolecular, homodimerization and cross-cycloaddition of vinylsilanes with unbiased 1,3-dienes, catalyzed by a pyridine-2,6-diimine (PDI) iron complex is described. In the absence of a diene coupling partner, vinylsilane hydroalkenylation products were obtained chemoselectively with unusual head-to-head regioselectivity (up to >98% purity, 98:2 E/Z). In the presence of a 4- or 2-substituted diene coupling partner, under otherwise identical reaction conditions, formation of value-added [2+2]- and [4+2]-cycloadducts, respectively, was observed. The chemoselectivity profile was distinct from that observed for analogous α-olefin dimerization and cross-reactions with 1,3-dienes. Mechanistic studies conducted with well-defined, single-component precatalysts (MePDI)Fe(L2) (where MePDI = 2,6-(2,6-Me2-C6H3N═CMe)2C5H3N; L2 = butadiene or 2(N2)) provided insights into the kinetic and thermodynamic factors contributing to the substrate-controlled regioselectivity for both the homodimerization and cross cycloadditions. Diamagnetic iron diene and paramagnetic iron olefin complexes were identified as catalyst resting states, were characterized by in situ NMR and Mössbauer spectroscopic studies, and were corroborated with DFT calculations. Stoichiometric reactions and computational models provided evidence for a common mechanistic regime where competing steric and orbital-symmetry requirements dictate the regioselectivity of oxidative cyclization. Although distinct chemoselectivity profiles were observed in cross-cycloadditions with the vinylsilane congeners of α-olefins, these products arose from metallacycles with the same connectivity. The silyl substituents ultimately governed the relative rates of β-H elimination and C–C reductive elimination to dictate final product formation.
Keywords: cycloaddition, diene, iron, metallacycles, silicon, vinylsilane
Graphical Abstract
Introduction
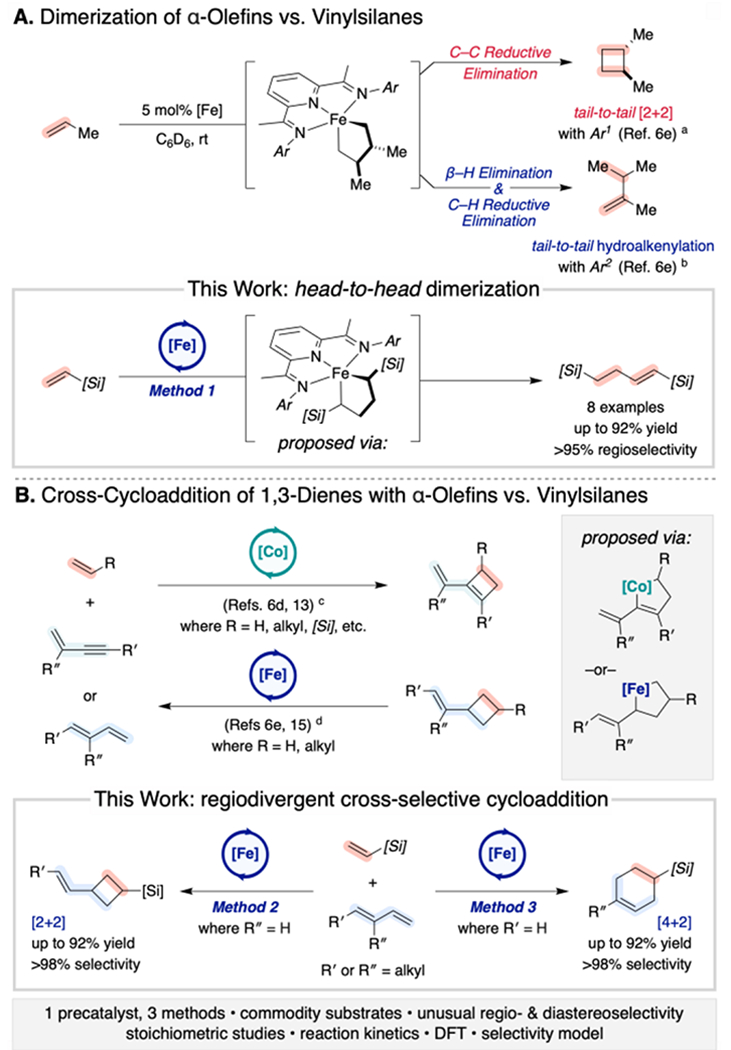
Metallacycle formation has emerged as a versatile strategy for controlling chemo- and site-selectivity in metal-mediated and metal-catalyzed transformations.1 Oxidative cyclization of multiple unsaturated coupling partners at a metal center enables access to cyclic and/or linear products following reductive elimination, β-elimination, or cycloreversion.2 For example, the exceptional chemoselectivity in the chromium-catalyzed trimerization of ethylene to 1-hexene, which is practiced industrially on kilo tons per annum (kta) scales,3 arises from selective β-elimination from a chromacycloheptane.4 The utility of this atom-economical approach for generating structural complexity in a single synthetic step has also motivated its application with electronically biased coupling partners to generate hetero- and carbocycles of interest for small-molecule synthesis (e.g. pyrones, pyridones, pyridines, benzene derivatives).1b,5 These formal cycloaddition products are generally distinct from those that can be accessed through thermal, photochemical, or Lewis acid catalyzed processes alone. Additionally, polarized substrates offer a direct handle for tuning regioselectivity.1b,5 However, electronically neutral or unbiased substrates such as alkene, diene, and alkyne feedstocks pose challenges for control of chemo- and especially regioselectivity.6
Our group and others have recently introduced iron,6e,7 cobalt,8 and nickel9-catalyzed processes for the [2+2]-cyclodimerization of unsaturated hydrocarbons to generate cyclobutane or cyclobutene products. These rigid carbocycles are scaffolds of interest for the synthesis of small-molecule targets ranging from pharmaceuticals and fragrances to fuels and materials.10 Mechanistic studies of both intra- and intermolecular olefin cyclodimerization have provided support for a mechanism involving oxidative cyclization to form a substituted metallacyclopentane prior to C–C bond-forming reductive elimination (Scheme 1A).11 With related catalysts derived from these Earth-abundant metals, metallacyclic intermediates have been observed and/or implicated in cross-[2+2] reactions between alkenes and alkynes,6d,12 enynes,6c,6d,13 allenes,14 and conjugated dienes15 (Scheme 1B) as well as in hydroalkenylations of alkenes and conjugated dienes.16–17
Scheme 1. Contrasting Reactivity of α-olefins and Vinylsilanes in Iron- and Cobalt-Catalyzed Cycloaddition Reactions.
a Ref. 6e; Ar1 = 2,6-(c-C5H9)2-C6H3 b Ref. 6e; Ar2 = 2-tBu-C6H4 c Refs. 6d, 13 d Refs. 6e, 15. [Si] = silyl
As part of an ongoing program toward the development of non-canonical cycloaddition reactions to upgrade abundant hydrocarbons, we have sought to manipulate the reactivity of iron metallacycles to achieve bond constructions with unique regioselectivity patterns using unactivated substrates.6e,6g,15,17e As part of these efforts, exposure of vinyltrimethylsilane to standard conditions for iron-catalyzed α-olefin cyclodimerization resulted in the formation of the linear head-to-head dimer rather than the corresponding cyclobutane or more typically observed tail-to-tail products (Scheme 1A). This anomalous product is consistent with β-H elimination and C–H reductive elimination from a metallacyclopentane intermediate with both SiMe3 groups proximal (α) to the iron center. Oxidative cyclizations and migratory insertions of alkynyl silanes have previously been demonstrated to favor placement of the silyl substituent α to early transition metals in stoichiometric and catalytic reactions.18 This “α-[Si] effect” has, in turn, been attributed to orbital symmetry requirements, electrostatic polarization, and the possibility of C–Si agostic interactions (Scheme S1, see Supporting Information).18–21 While similar preferences have not been typically observed with mid to late transition metals, this result suggested that pyridine-2,6-diimine (PDI) supported iron complexes may exhibit a kinetic and/or thermodynamic preference for α-silicon incorporation. Given the availability of varied vinylsilanes, which are employed as (co)monomers in α-olefin polymerizations,22 as well as the versatility of organosilicon groups toward further synthetic manipulation, evaluation of their potential to access unique regioselectivity patterns through iron-catalyzed cycloaddition reactions was explored.
Here we describe the development of three [(PDI)Fe]-catalyzed methods for the chemo- and regioselective elaboration of vinylsilanes: (i) head-to-head vinylsilane dimerization, (ii) cross-[2+2]-cycloaddition with 4-substituted 1,3-dienes, and (iii) cross-[4+2]-cycloaddition with 2-substituted 1,3-dienes (Scheme 1). Each case enabled access to unique scaffolds, including relatively unactivated Diels–Alder products that are inaccessible with conventional methods,23 with potential for strategic applications in the silicones industry as well as in small-molecule synthesis. Mechanistic studies including stoichiometric reactions, kinetic measurements, in situ Mössbauer spectroscopy, and DFT analyses provide support for pathways in which competing steric and orbital-symmetry requirements between the vinylsilane and 1,3-diene coupling partners coupled with β-[Si]-suppressed β-H elimination give rise to the unusual selectivity profiles.
RESULTS AND DISCUSSION
Head-to-Head Vinylsilane Dimerization.
Our study began with re-examination of the iron-catalyzed homocoupling of vinylsilanes using [(MePDI)Fe(N2)]2(μ2-N2) (where MePDI = 2,6-(2,6-Me2-C6H3N═CMe)2C5H3N) as the precatalyst. As reported previously, exposure of a benzene-d6 solution of the iron precatalyst (2.5 mol% dimer, 5 mol% [Fe]) to an excess of propylene (1a) resulted in near complete (96%) consumption of 1a within 48 hours at ambient temperature (~23 °C) and formation of a mixture of dimerization products including [2+2]-cycloadduct 2a and the tail-to-tail dimer 3a (Scheme 2A).6e Neither the corresponding head-to-head dimer (4a) nor head-to-tail dimer 5a was detected by NMR spectroscopy. By contrast, exposure of vinyltrimethylsilane (6a) to the same reaction conditions resulted in negligible conversion. Conducting the reaction in neat vinylsilane produced (32% yield after 48 h) but clean formation of the head-to-head dimer 9a; [2+2]-cycloadduct 7a, tail-to-tail dimer 8a, and head-to-tail dimer 10a were not observed (Scheme 2B). Control reactions conducted without iron or the PDI ligand resulted in no conversion or the formation of an intractable product mixture, respectively. It is useful at this point to explain the labeling scheme used throughout the manuscript. Each of the products arising either from hydrovinylation or cycloaddition is labeled with a number referring to the carbon skeleton followed by a letter signaling the vinylsilane from which it was derived.
Scheme 2. Inverted Regioselectivity for Vinylsilane Dimerization.
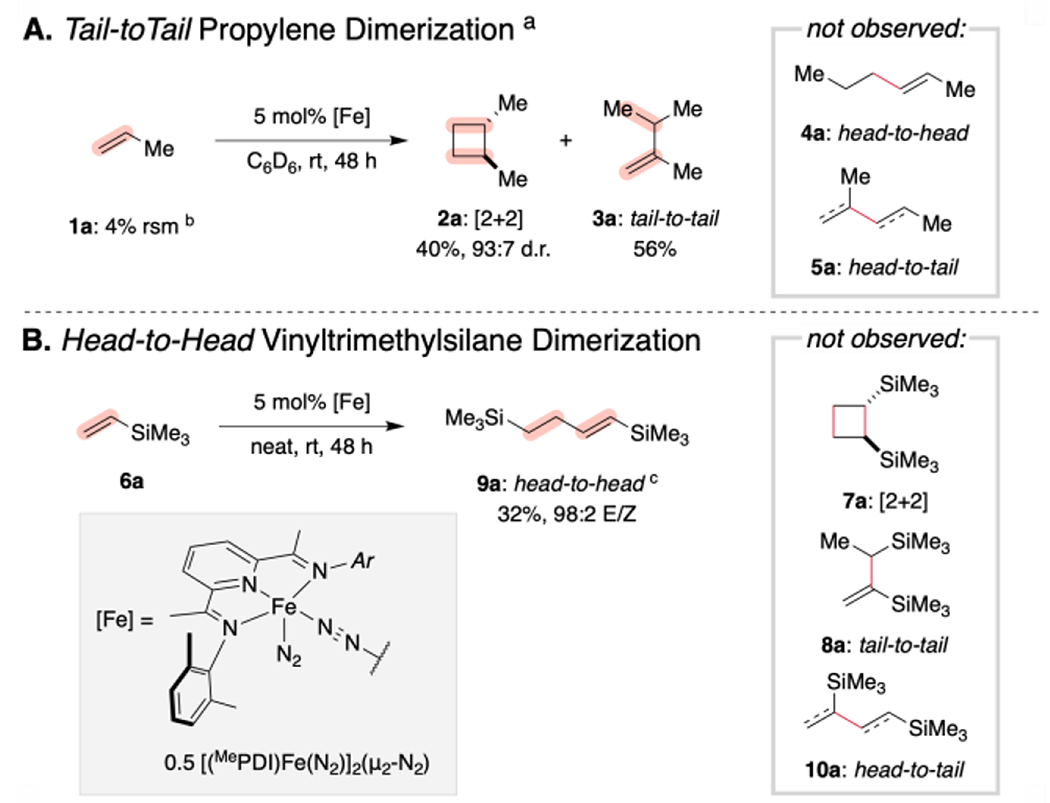
a Ref. 6e. b rsm = remaining starting material c Neither 7a nor any other regio- or stereoisomeric [2+2] cycloaddition products were observed.
Homocoupling of various vinylsilanes and vinylsiloxanes was examined, and clean conversion to the head-to-head dimer (9) was observed in all cases (Scheme 3). The chemoselectivity was insensitive to both steric and electronic variation of the silyl group and preferential formation products with (E)-olefin geometry was uniform. The low yield (32%) for 9c was a likely result of the dilute reaction conditions. Notably, 1,3-divinyltetramethyldisiloxane (6g), a commodity chemical used widely as a crosslinking agent in the silicones industry,22a,24 formed C–C bonds from both termini to afford macrocyclic product 9g, which exhibits the same head-to-head orientation within each repeat unit. The macrocycle was assigned as the 21-membered cyclic trimer on the basis of the parent mass observed by atmospheric pressure chemical ionization (APCI) mass-spectrometric analysis under low fragmentation conditions and corroborated by diffusion ordered NMR spectroscopy (DOSY) in chloroform-d. This viscous, slippery oil bears resemblance to the silicone surfactants in consumer products such as soaps and cosmetics.24
Scheme 3. Substrate Scope for Iron-Catalyzed Vinylsilane Dimerization.a.
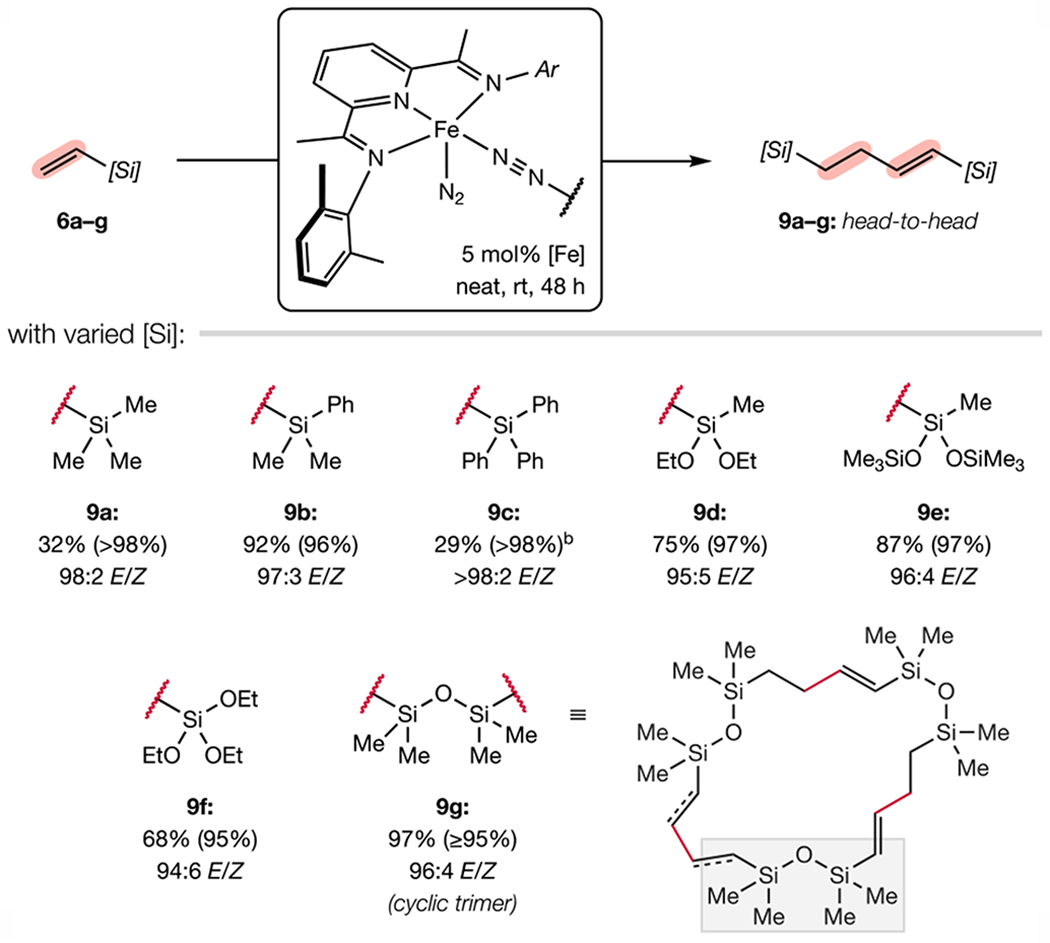
a Reactions conducted on 0.5 mmol scale. Isolated yields reported (with selectivity for the head-to-head hydroalkenylation product in parentheses). b Reaction conducted in toluene.
Despite the general efficacy of the vinylsilane coupling, the homologous allyltrimethylsilane (1b) underwent isomerization to trimethyl(prop-1-en-1-yl)silane, rather than homocoupling, upon exposure to [(MePDI)Fe(N2)]2(μ2-N2). Furthermore, t-butyl-ethylene (1c) and 3,3,3-trifluoropropylene (1d), which were examined as steric mimics of 6a, failed to undergo coupling reactions upon exposure to [(MePDI)Fe(N2)]2(μ2-N2), even at elevated temperature (50 °C), at which point decomposition of the iron precatalyst and elemental iron deposition was observed. Given that these substrate-based probes did not prove mechanistically informative, additional experiments were conducted to elucidate the basis for the chemo- and regioselectivity of the (PDI)Fe-catalyzed dimerization of vinylsilanes.
A series of stoichiometric reactions was undertaken. Addition of trimethylvinylsilane (6a), dimethylphenylvinylsilane (6b), or diethoxymethylvinylsilane (6d) to a solution of [(MePDI)Fe(N2)]2(μ2-N2) (2 equiv of vinylsilane relative to Fe) in benzene-d6 resulted in an immediate color change from red-brown to yellow-brown and afforded the corresponding iron bis(vinylsilane) complex in >98% conversion within 15 minutes at ambient temperature (Scheme 4). In each case, the 1H NMR spectrum was consistent with that expected for a paramagnetic, effectively C2v-symmetric complex with paramagnetically shifted resonances ranging in chemical shift from δ 170 to −110 ppm. A representative, solution-state magnetic moment of 2.8 μB was measured for (MePDI)Fe(η2-6a)2 at 23 °C (benzene-d6, Evans method), consistent with the spin-only value for two unpaired electrons and an S = 1 ground state. However, the bis(vinylsilane) complexes proved unstable to vacuum, and attempts to isolate or crystallize them, even in the presence of excess vinylsilane, resulted in re-isolation of [(MePDI)Fe(N2)]2(μ2-N2). Based on these results and prior studies with α-olefins,11 it is likely that (MePDI)Fe(η2-6a)2, (MePDI)Fe(η2-6a), and [(MePDI)Fe(N2)]2(μ2-N2) are in equilibrium under catalytic conditions. A zero-field 57Fe Mössbauer spectrum (80 K, toluene glass) was collected following exposure of [(MePDI)Fe(N2)]2(μ2-N2) (0.5 equiv) to excess trimethylvinylsilane (6a, 10 equiv) in toluene at ambient temperature to generate (MePDI)Fe(6a)2 in situ (Figure 1). The spectrum was fit with two quadrupole doublets, where the major component exhibited parameters (92%: δ = 0.66 mm/s, |ΔE| = 1.04 mm/s) in accord with those reported previously for isolable (κ2-PDI)Fe olefin complexes assigned as the Fe(I) oxidation state.11 These parameters also agreed with those predicted for (κ2-MePDI)Fe(η2-6a)2 using full-molecule density functional theory (δ = 0.73 mm/s, |ΔE| = 1.72 mm/s; Figure 2).25, 26
Scheme 4.
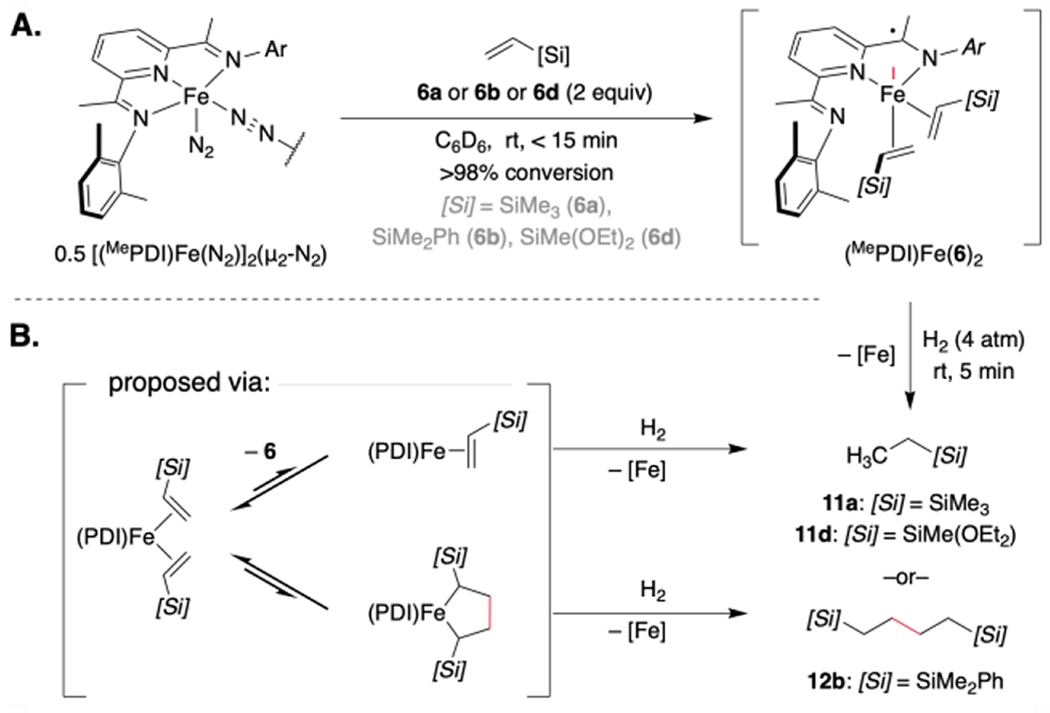
Synthesis and Hydrogenolysis of (MePDI)Fe(vinylsilane)2 complexes.
Figure 1.
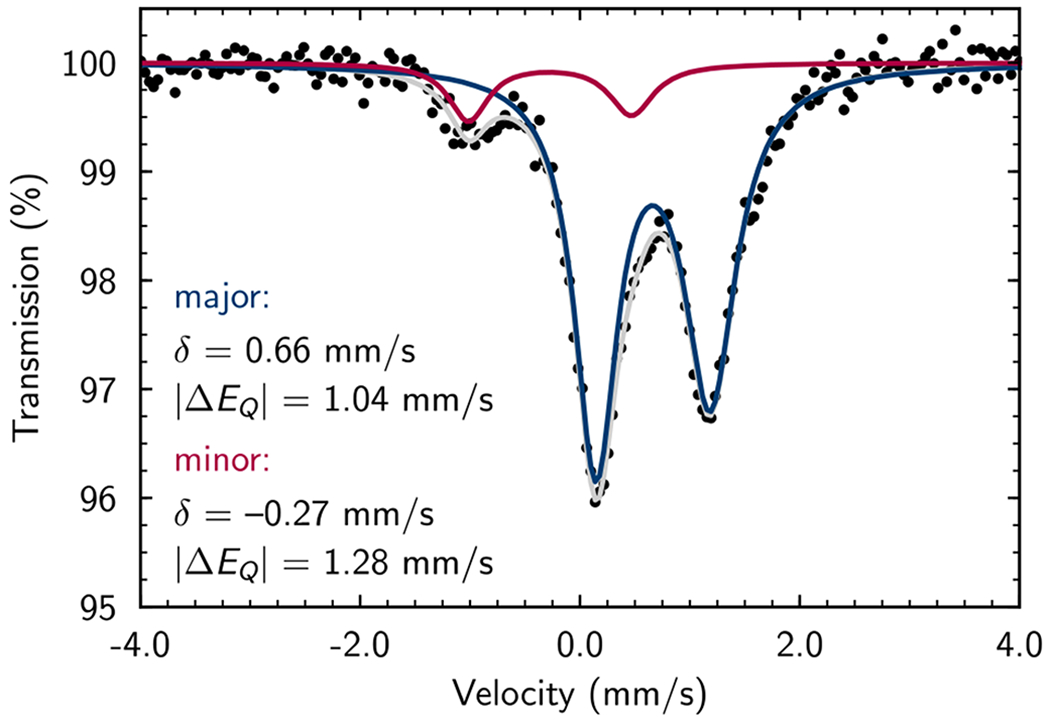
Solution, zero-field 57Fe Mössbauer spectrum (toluene glass, 80 K) of (MePDI)Fe(6a)2 generated in situ. Simulated spectra plotted for the major (92%, blue) and minor (12%, red) components with the listed parameters, along with the net spectrum (grey).
Figure 2.
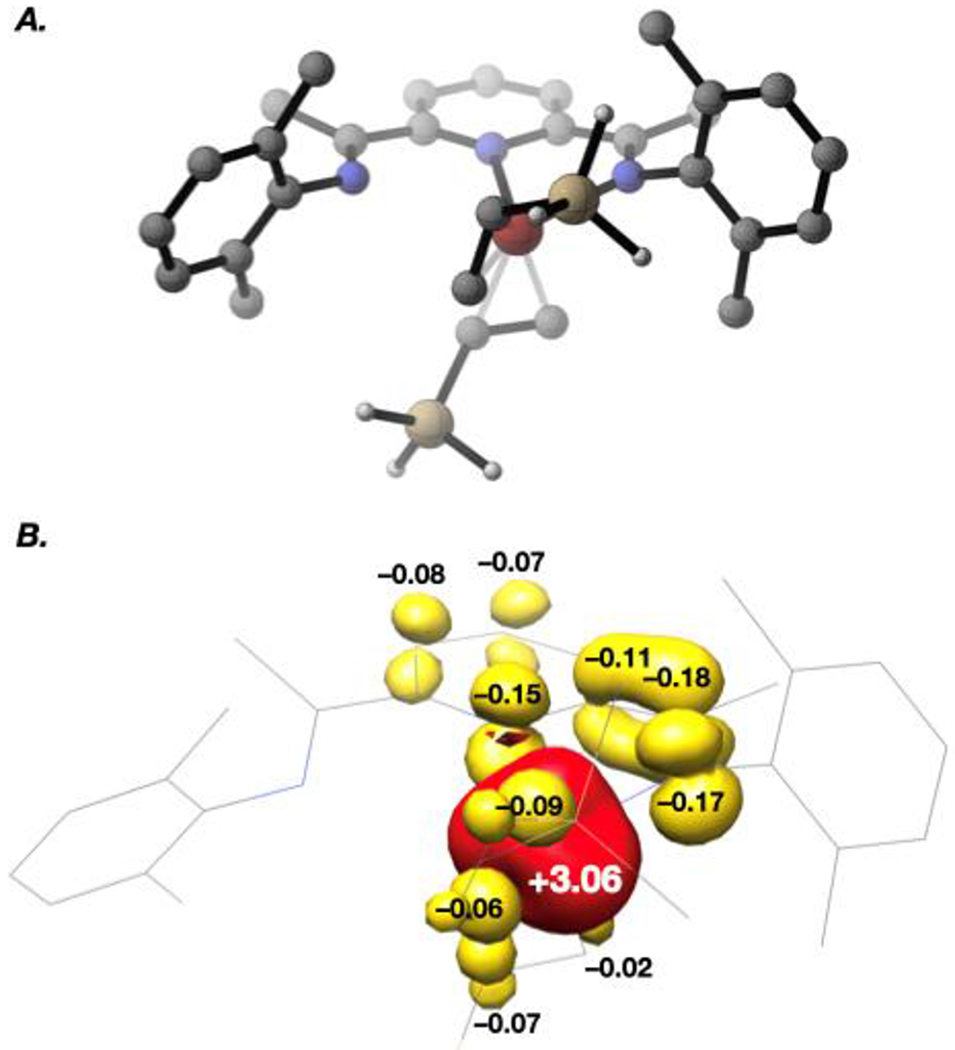
(A) Lowest-energy optimized structure and (B) spin-density plot for (MePDI)Fe(6a)2, computed at the B3LYP level of density functional theory with the ZORA-def2-TZVP(-f) basis set augmented by SARC/J terms for Fe, N, and C atoms in the primary coordination sphere, and the ZORA-def2-SVP basis set augmented with SARC/J terms for all other atoms. Hydrogen atoms omitted for clarity. H = white, C = gray, N = blue, Fe = tan, Fe = red-orange, positive spin density = red, negative spin density = yellow.
Given the challenges associated with isolation of the iron vinylsilane complexes, alternative strategies were pursued to establish their connectivity and reactivity. Hydrogenolysis of (MePDI)Fe(η2-6b)2 furnished the head-to-head coupled alkane 12b as the exclusive organic product (Scheme 4B). However, exposure of (MePDI)Fe(η2-6a)2 and (MePDI)Fe(η2-6d)2 to 4 atm of H2 instead resulted in direct hydrogenation to yield alkylsilanes 11a and 11d, respectively. While both (a) direct hydrogenolysis of a transient metallacycle (Scheme 4B) and (b) hydrogen-induced alkene dimerization are precedented, the divergent outcomes observed with (MePDI)Fe(η2-6b)2 versus (MePDI)Fe(η2-6a)2 and (MePDI)Fe(η2-6d)2 suggested that a metal hydride was most likely not responsible for the observed catalytic dimerization.27 Regardless, the combined results indicated that the (PDI)-supported iron complex exhibited a pronounced and consistent α-Si preference in the C–C bond-forming step.
Cross-[2+2]-Cycloadditions with 4-Substituted-1,3-Dienes.
Given the evidence for an α-[Si] preference in metallocycle formation and ultimately vinylsilane dimerization, we were motivated to explore whether a similar effect could be applied to alter the regioselectivity of other (PDI)Fe-catalyzed coupling reactions. [(MePDI)Fe(N2)]2(μ2-N2) serves as an active and selective precatalyst for the cross-[2+2]-cycloaddition of (E)-piperylene (13a) with ethylene and α-olefins, yielding substituted propenylcyclobutanes (14; Scheme 5A).15b The 1,3-cyclobutane substitution pattern has been attributed to the combination of maximizing orbital overlap (dz2→π*)19 and avoiding steric encumbrance from geminal substituents during selectivity-determining olefin coordination or oxidative cyclization. Although silylalkynes have been reported to exhibit inverted π* polarization relative to their aliphatic counterparts (vide supra), NBO analyses of the orbital coefficients for representative vinylsilanes (6a–d,f) revealed only negligible polarization, comparable in magnitude and sign to that of α-olefins (see Supporting Information).28 Accordingly, vinylsilanes proved to be excellent substrates for (PDI)Fe-catalyzed cross-[2+2]-cycloadditions with (E)-piperylene (Scheme 5B). No change in regioselectivity was observed relative to the pure hydrocarbon case. While the diastereoselectivity proved modest, high chemoselectivity for the [2+2]-products (17aa–ga) and exclusive reaction with the (E)-diene isomers was observed, even when using (E/Z) substrate mixtures across a variety of vinylsilane and 4-substituted diene coupling partners (Scheme 6). For clarity, the cross-cycloaddition products are each labeled with a number referring to the carbon skeleton and two letters indicating the vinylsilane (first letter) and diene (second letter) from which they were derived.
Scheme 5. Conserved Chemoselectivity for Cross-Cycloadditions with 4-Substituted-1,3-Dienes.
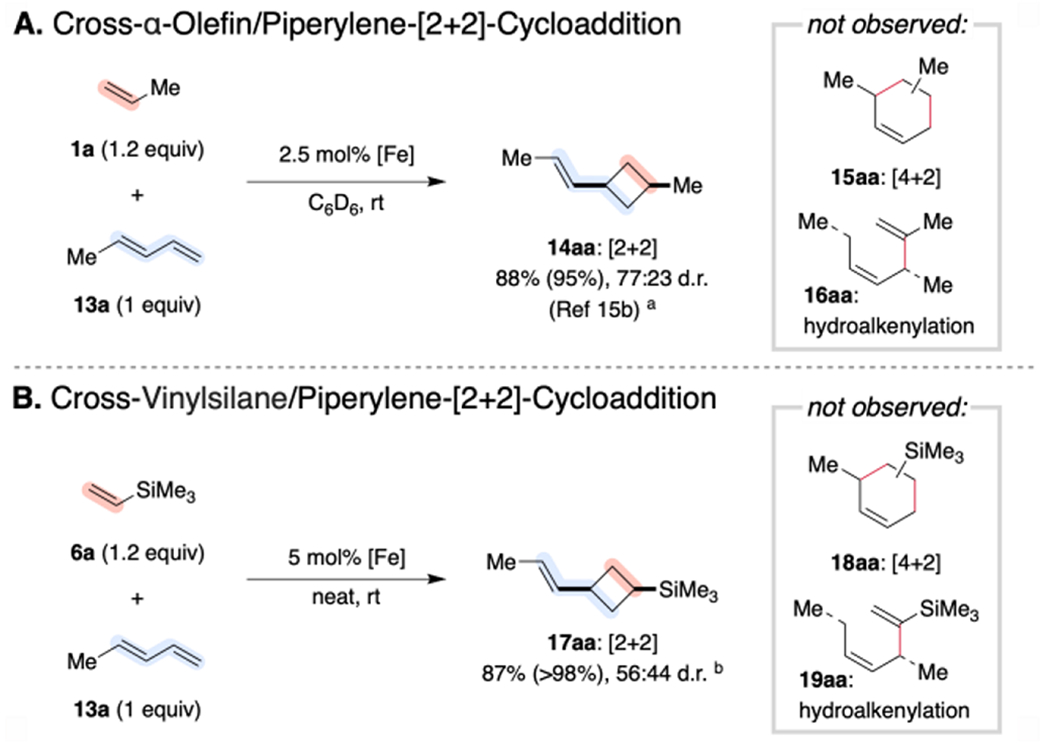
a Conducted on 1.0 mmol scale with 1.25 mol% [(Me(Et)PDI)Fe(N2)]2(μ2-N2). Yield (and selectivity for [2+2]-cycloadduct 14aa) determined by integration of diagnostic 1H NMR signals relative to an internal standard. See Ref. 15b. b Conducted on 1.0 mmol scale with 2.5 mol% [(MePDI)Fe(N2)]2(μ2-N2). Isolated yield reported (with selectivity for the [2+2]-cycloadduct 17aa product in parentheses).
Scheme 6. Substrate Scope for Iron-Catalyzed Cross-[2+2]-Cycloaddition of Vinylsilanes with 4-Substituted-1,3-Dienes.a.
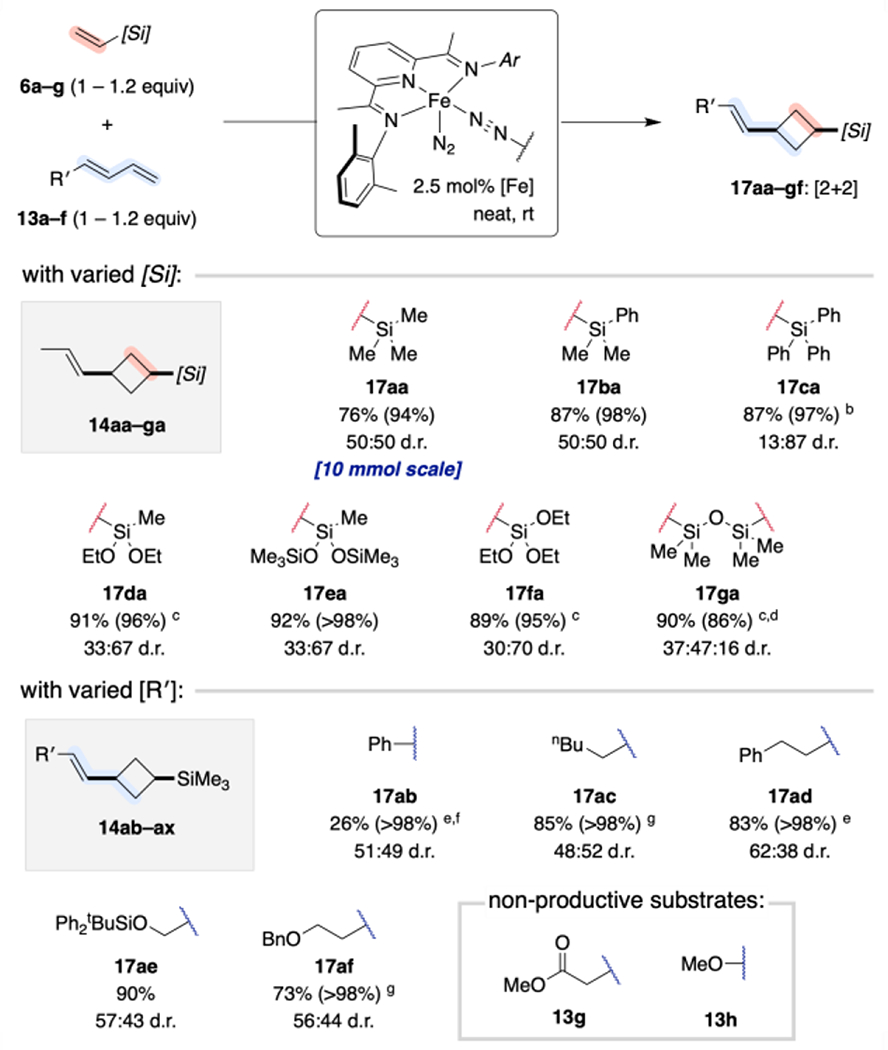
a Reactions conducted on 1.0 mmol scale unless indicated otherwise. Isolated yields reported (with selectivity for the [2+2]-cycloadduct 17aa product in parentheses). b Reaction conducted in toluene. c With 5 mol% [Fe] and 2.4 equiv 13a relative to 6g. d Mass balance (14%) comprised of the [2+2] cycloadduct where only one of the two vinyl groups of 6g reacted (60:40 d.r.) e With 1 mol% (MePDI)Fe(butadiene) f Mass balance comprised of the adduct resulting from thermal [4+2]-cyclodimerization of 13b f With 2.5 mol% (MePDI)Fe(butadiene)
Cross-[4+2]-Cycloadditions with 2-Substituted-1,3-Dienes.
Despite the high chemoselectivity observed in (PDI)Fe-catalyzed cross-[2+2]-cycloadditions with piperylene and other 4-substituted dienes, the method has previously been shown to be highly sensitive to the position of the diene substituent. For example, exposing precatalyst [(MePDI)Fe(N2)]2(μ2-N2) to an equimolar mixture of isoprene (13i) and ethylene (1e) afforded the hydrovinylation product 16ie (instead of [2+2] adduct 14ie) as the exclusive organic product.15a Subjecting representative α-olefin/myrcene (13j) combinations to analogous conditions generated mixtures of hydroalkenylation products 16aj–dg along with [2+2]-cycloadducts 14aj–dg (Scheme 7A). A more specialized ligand (iPr(TB)PDI) has also been identified to achieve high [2+2]-chemoselectivity in these cases.6e
Scheme 7. Inverted Chemoselectivity for Cross-Cycloadditions of Vinylsilanes with 2-Substituted-1,3-Dienes.a.
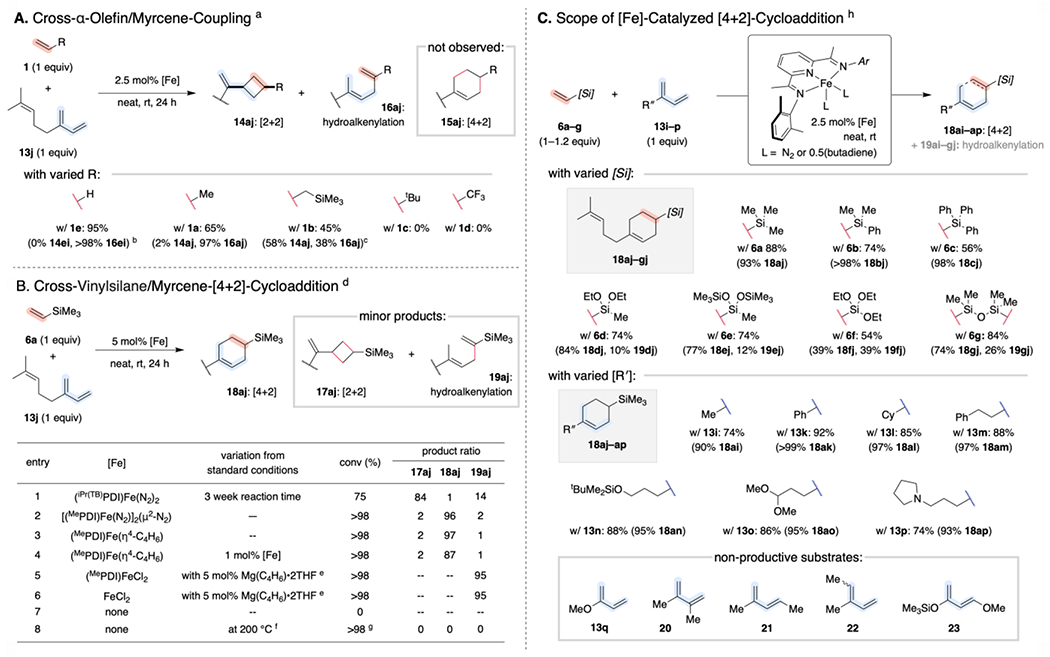
a Reactions conducted on 0.25 or 0.5 mmol scale. Combined isolated yields reported (with selectivity for each product listed in parentheses). b Reaction conducted in C6D6 using isoprene (13i) in place of myrcene (13j) in C6D6. Yield (and selectivity for hydrovinylation product 16ei) determined by integration of diagnostic 1H NMR signals relative to an internal standard. c With 5 mol% [Fe] d Reactions conducted on 0.25 mmol scale. Conversion and product ratio determined after 24 h by gas chromatographic analysis relative to mesitylene, which was added as an internal standard at the end of the reaction. e 5 mol% Mg(butadiene)•2THF added as an in situ reductant. f Reaction performed on a 1.9 mmol scale at 200 °C for 24 h with 5 mg BHT. g Complex mixture of decomposition products. h Reactions conducted on 1.0 mmol scale. Combined isolated yields reported (with selectivity for each product listed in parentheses).
Like its α-olefin counterparts, vinyltrimethylsilane proved to be a competent (albeit sluggish) substrate for (iPr(TB)PDI)Fe-catalyzed cross-[2+2]-cycloaddition with myrcene (Scheme 7B, entry 1). However, we were intrigued as to whether vinylsilane–myrcene coupling would exhibit an analogous selectivity profile using the more readily accessed (MePDI) ligand. Remarkably, exposure of an equimolar mixture of vinyltrimethylsilane (6a) and myrcene (13j) to [(MePDI)Fe(N2)]2(μ2-N2) yielded [4+2]-cycloadduct 18aj rather than corresponding [2+2]-product 17aj (Scheme 7B, entry 2). Ligands with larger 2,6-aryl substituents, e.g. (iPrPDI)Fe(N2)]2(μ2-N2), resulted in lower levels of chemoselectivty. Butadiene complex (MePDI)Fe(η4-butadiene) afforded 18aj with effectively identical selectivity to [(MePDI)Fe(N2)]2(μ2-N2) (Scheme 7B, entry 3) and exhibited improved shelf-stability. The two precatalysts were employed interchangeably in subsequent studies to extend this finding. With either precatalyst, the preference for [4+2]-cycloaddition with vinyltrimethylsilane was consistent with a variety of 2-aryl- and 2-alkyl-substituted 1,3-dienes (Scheme 7C). However, neither polar nor disubstituted dienes (13q, 20–23) afforded any cross products under analogous conditions (even upon heating to 50 °C); trace amounts of vinylsilane dimerization product 9a were instead detected after 24 hours. These results are remarkable in that they produce formal Diels–Alder products with unactivated substrates that are not kinetically competent for thermal cycloaddition by a pericyclic mechanism (Scheme 7B, entry 8).
Similar reactivity patterns were observed with varied vinylsilane and vinylsiloxane coupling partners, where the [4+2]-cycloadducts with 13j were the major products in all cases examined (Scheme 7C). Exclusive [4+2]-chemoselectivity was observed with alkyl- and aryl-substituted vinylsilanes 6a–c; however, competitive formation of the analogous 1,4-hydroalkenylation products (19dj–gj) was observed with vinylsiloxanes 6d–g. The decreased chemoselectivity observed with vinylsiloxane substrates was hypothesized to arise from increased flexibility of the resulting metallacycle (vide infra). However, in control experiments, hydroalkenylation product 16aj also formed cleanly upon exposure to a combination of FeCl2 and Mg(butadiene)•2THF, even in the absence of added ligand (Scheme 7B, entry 6).29 Although all substrates were dried over CaH2, degassed, and filtered through alumina prior to use, the presence of trace impurities in the vinylsiloxanes that could degrade the (MePDI)Fe(L)2 precatalyst and result in a competitive “ligand-free” background reaction cannot be excluded.
Potential Mechanistic Bases for [4+2] vs. [2+2] Selectivity.
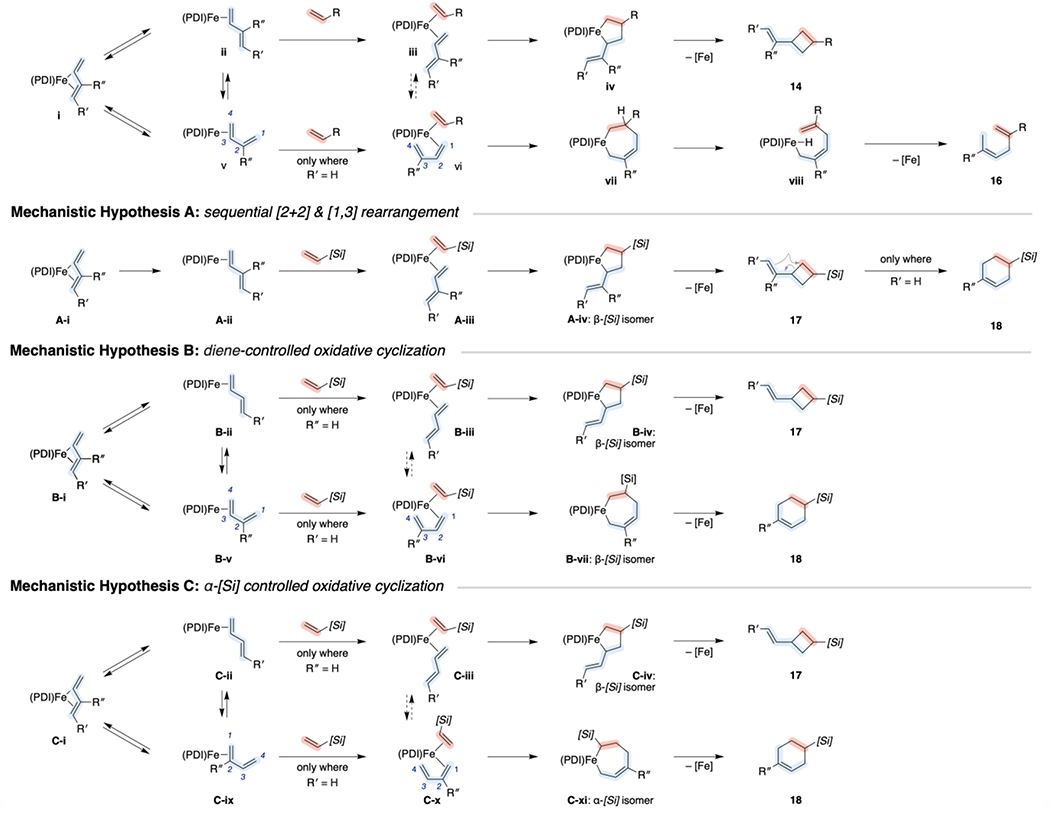
To inform the development of new synthetic methods and strategies for catalyst-control, the basis for the unusual chemoselectivity observed for the [(PDI)Fe]-catalyzed cycloaddition with the specific combination of vinylsilane and 2-substituted-1,3-diene substrates was of interest. In comparison to the mechanisms described previously to account for [(PDI)Fe]-catalyzed cross-reactions with α-olefins (Scheme 8, Mechanistic Hypothesis 0), three distinct possibilities were considered (Scheme 8), as follows:
Scheme 8.
Mechanistic Hypotheses Accounting for Divergent Chemoselectivity of Vinylsilane/Diene Cycloadditions.
Mechanistic Hypothesis A:
As outlined for the formation of 14 from α-olefin/diene cycloaddition, the [(MePDI)Fe]-catalyzed vinylsilane/diene cycloaddition proceeds through metallacyclopentane A-iv to afford [2+2]-product 17 regardless of diene substitution pattern. However, only the [2+2]-cycloadducts arising from 2-substitued dienes and vinylsilanes undergo rapid thermal or Lewis-acid promoted rearrangement to 18 on the timescale of the cycloaddition.
Mechanistic Hypothesis B:
As outlined for the formation of 14 and 16 from α-olefin/diene coupling, the [(MePDI)Fe]-catalyzed vinylsilane/diene cycloadditions proceed through metallacycles B-iv and B-vii with [Si] substituents distal (β) to [Fe]. Oxidative cyclization from the s-trans or s-cis diene isomer is dictated predominantly by the diene substitution pattern. 4-Subsituted dienes are biased to react through an s-trans conformation (B-iii), giving rise to metallacyclopentane B-iv. By contrast, 2-substituted dienes may react through an s-cis conformation (B-vi) giving rise to metallacycloheptene B-vii. The flexibility of either resulting metallacycle (with or without β-silyl substituents) then determines the relative rates of C–C reductive elimination giving rise to 17 and 18 versus β-H elimination and C–H reductive elimination giving rise to 19 (where the latter case is not depicted). This mechanistic pathway would require that the α-[Si] effect is negligible in the presence of 1,3-dienes.
Mechanistic Hypothesis C:
If the α-[Si] effect is competitive with the directing effects of 1,3-dienes, oxidative cyclization from vinylsilane complex C-x occurs selectively with [Si] proximal (α) to [Fe]. This inverted regioselectivity is accessible only with 2-substituted dienes and requires reaction through the 1,2-coordianted s-cis diene complexes C-ix and C-x, giving rise to metallacycloheptene C-xi, which is poised for C–C reductive elimination to afford 18. However, reaction through this s-cis conformation is not energetically feasible with 4-substituted dienes, obviating the α-Si effect and forcing reaction through the metallacyclopentane intermediate C-iv en route to 17.
Mechanistic hypotheses B and C may also be modified to describe related scenarios in which olefin coordination or reductive elimination, rather than oxidative cyclization, are selectivity-determining.
Stability of Independently Synthesized 17bi.
To probe the viability of mechanistic hypothesis A, a representative example of the posited intermediate, a [2+2]-cycloadduct, was independently prepared (see Supporting Information). This isopropenylcyclobutane (17bi) was exposed to a battery of conditions to probe for kinetically accessible paths for thermal or Lewis acid-promoted rearrangement (Scheme 9). Notably the cyclobutane was stable to prolonged heating in benzene-d6 (up to 80 °C), exposure to [(MePDI)Fe(N2)]2(μ2-N2) at ambient temperature or 50 °C, or treatment with a variety of Lewis acids (including deliberately decomposed [(MePDI)Fe(N2)]2(μ2-N2)). While 17bi decomposed to an intractable mixture of products upon exposure to AlCl3, formation of the putative rearrangement product, 18bi, was not observed in any case. Taken together, these results eliminate mechanistic hypothesis A.
Scheme 9.
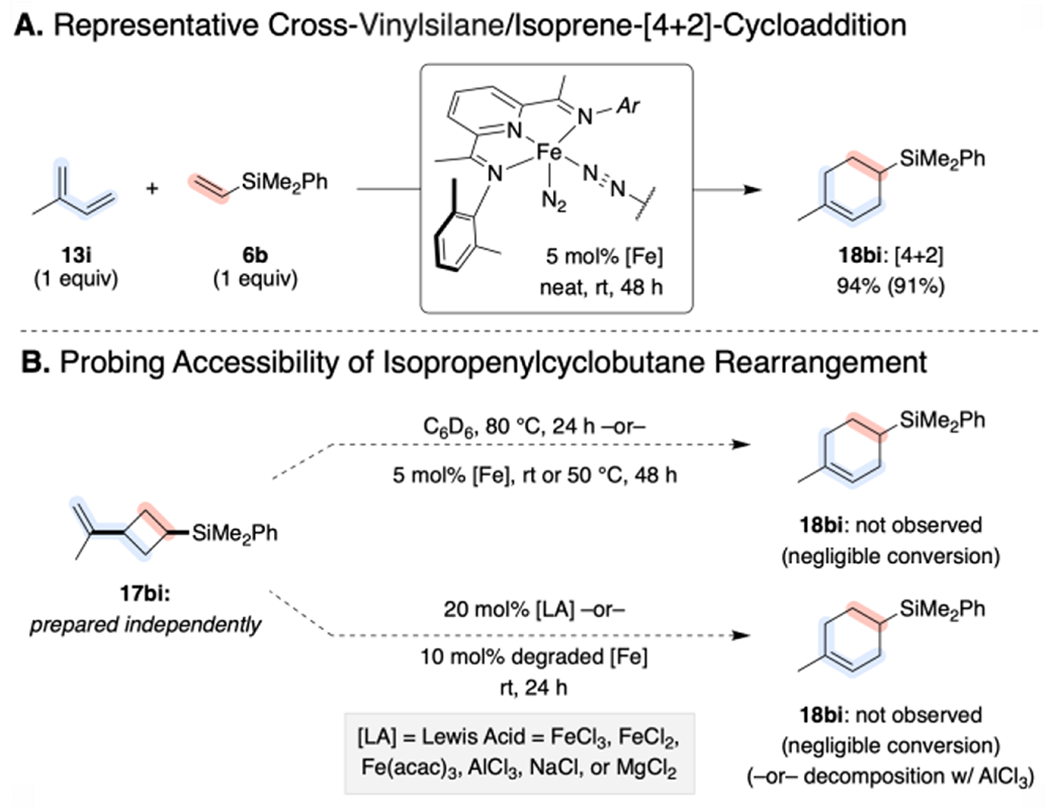
Probing the Viability of Mechanistic Hypothesis A.
Observation of Catalytically Relevant Iron Complexes.
To distinguish between mechanistic hypotheses B and C, the identity of the catalyst resting state and speciation in the context of the iron-catalyzed [4+2] cycloaddition reaction was studied. As reported previously, (MePDI)Fe(η4-isoprene) is formed readily in solution from [(MePDI)Fe(N2)]2(μ2-N2) upon addition of isoprene (13i).15b Similarly, a diamagnetic, C1 symmetric complex assigned as (MePDI)Fe(η4-13j) was observed within two hours upon treatment of a benzene-d6 solution of [(MePDI)Fe(N2)]2(μ2-N2) with myrcene (13j, 2 equiv per dimer, 1 equiv per [Fe]) at ambient temperature (~23 °C; Scheme 10). This complex proved unstable to vacuum or crystallization under an N2 atmosphere.
Scheme 10.
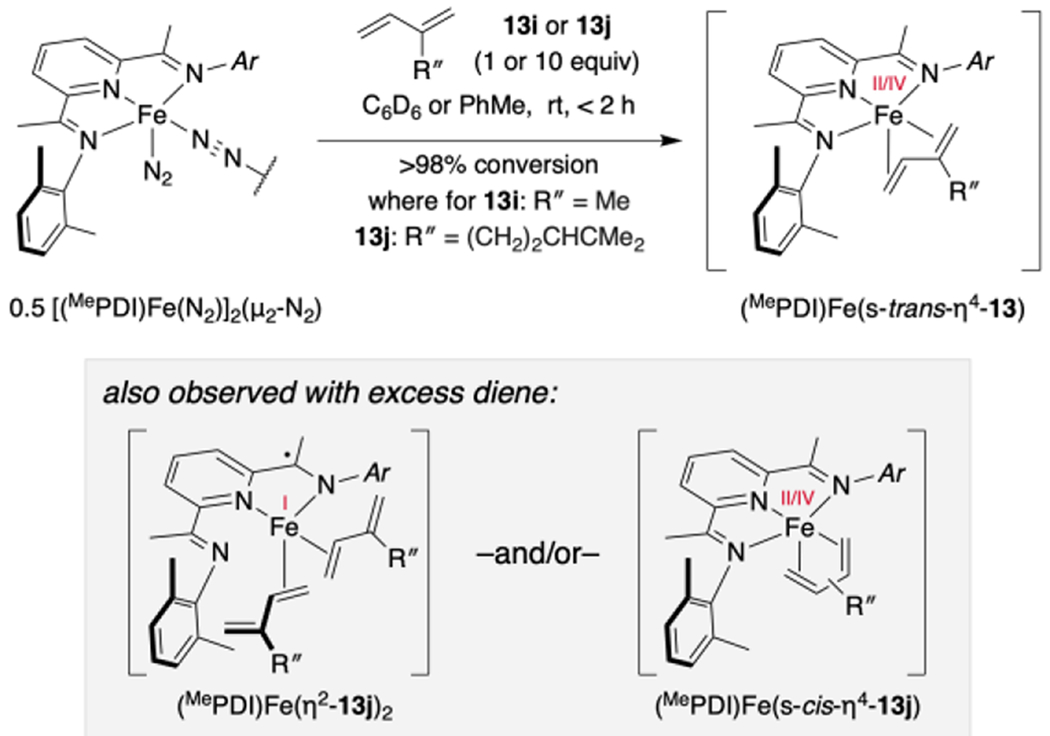
Synthesis of (MePDI)Fe(diene) Complexes.
The zero-field 57Fe Mössbauer spectrum (80 K, toluene glass) was recorded following exposure of [(MePDI)Fe(N2)]2(μ2-N2) (0.5 equiv) to excess myrcene (13j, 10 equiv) in toluene at ambient temperature. Two partially overlapping signals were observed and fit to quadrupole doublets (Figure 3). The major component (63%) exhibited parameters (δmajor = 0.24 mm/s, |ΔE|major = 1.32 mm/s) in accord with those reported previously for isolable (PDI)Fe(s-trans-η4-diene) complexes in the solid state15 and was assigned as (MePDI)Fe(s-trans-η4-13j). The minor component (37%) exhibited parameters (δminor = 0.52 mm/s, |ΔE|minor = 0.82 mm/s) similar to κ2-pyridine(diimine)11 and iminopyridine iron(I) s-cis-η4 complexes6g as well as α-diimine and iminopyridine iron(I) bis(olefin) complexes.6f,6g,15b The minor species was thus tentatively assigned as either (MePDI)Fe(η2-13j)2 or (MePDI)Fe(s-cis-η4-13j). Both assignments were corroborated with full-molecule DFT predictions (see Supporting Information); however, alternative possibilities cannot be excluded based on these data alone.
Figure 3.
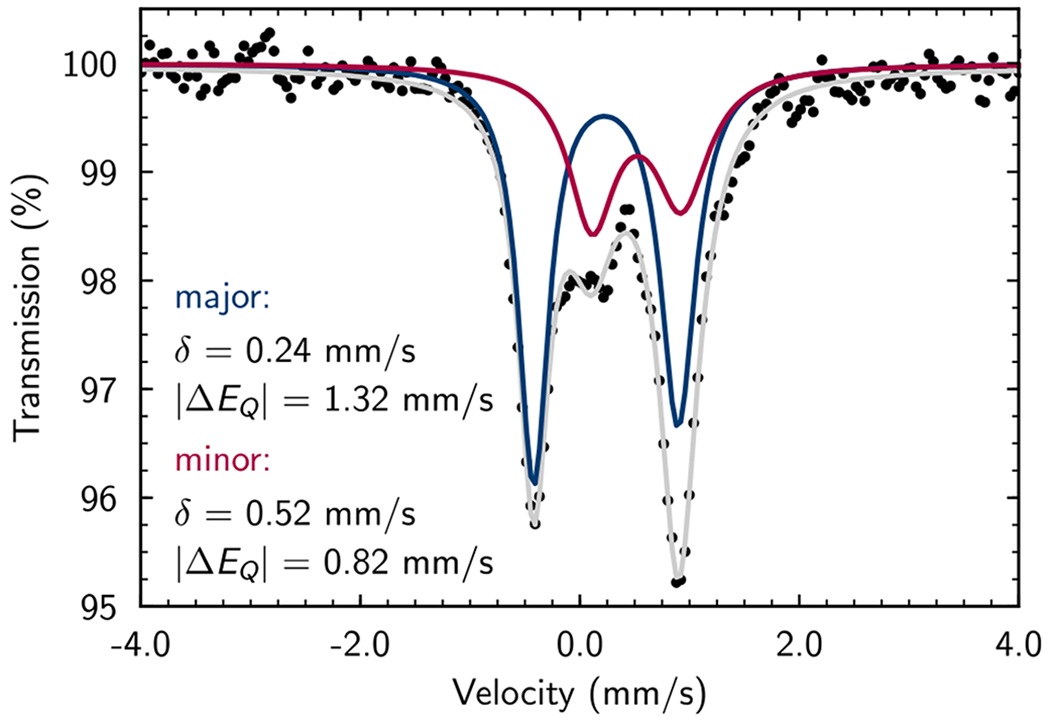
Solution-state, zero-field 57Fe Mössbauer spectrum (toluene glass, 80 K) of (MePDI)Fe(13j)n (n = 1,2) generated in situ. Simulated spectra plotted for the major (63%, blue) and minor (37%, red) components with the listed parameters, along with the net spectrum (grey).
With diagnostic spectroscopic data in hand for both (MePDI)Fe(η4-13) and (MePDI)Fe(η2-6)2 complexes, iron speciation in the presence of both coupling partners was examined. A mixture of [(MePDI)Fe(N2)]2(μ2-N2), dimethylphenylvinylsilane (6b), and isoprene (13i) was prepared in benzene-d6 in a 1:4:2 ratio and examined by 1H NMR spectroscopy (Scheme 11). An equilibrium mixture of both complexes as well as free 13i and 6b was observed, with an equilibrium constant favoring isoprene binding over the vinylsilane (Keq = 10.8 at 23 °C). Additionally, both complexes were detected under catalytic conditions (e.g. 5 mol% [Fe]). The proposed metallacycle was not observed spectroscopically, which was consistent with previous findings from our group.15 Taken together, these data suggest that multiple diene coordination modes and electronic structures of pyridine(diimine) iron 1,3-diene and vinylsilane species are accessible under catalytically relevant conditions but that (MePDI)Fe(η4-13) predominates.30
Scheme 11.
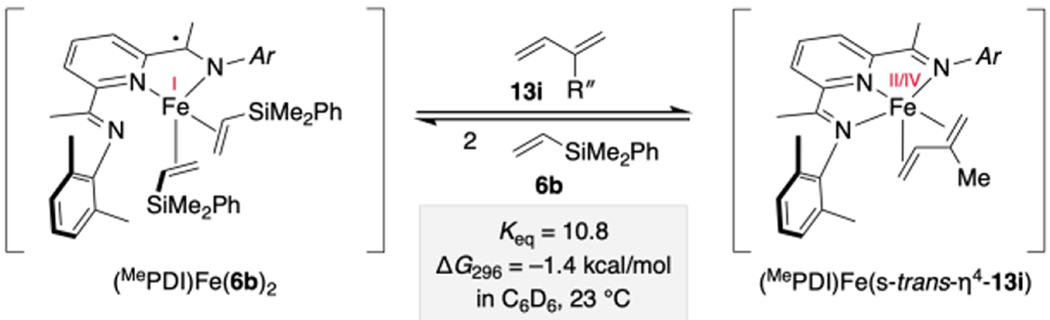
Relative Stability of Representative (MePDI)Fe(vinylsilane)2 and (MePDI)Fe(isoprene) Complexes.
Kinetic Analysis.
The rate-determining steps in the [(PDI)Fe]-catalyzed intra- and intermolecular olefin [2+2]-cyclodimerization and cross-[2+2]-cycloaddition are highly system dependent.11,13b In an effort to identify the possible rate- and selectivity-determining step(s) in cross-[4+2]-cycloaddition of vinylsilanes and 1,3-dienes, reaction progress kinetic analysis was performed for the cycloaddition of vinyldimethphenylsilane (6b) with myrcene (13j) using [(MePDI)Fe(N2)]2(μ2-N2) as the precatalyst.31 Although preparative reactions were generally conducted in neat substrate, concentrated stock solutions of catalyst and substrate were employed for the kinetic measurements while maintaining a constant total volume. Under these conditions, the chemoselectivity for [4+2] cycloaddition remained high, and the disappearance of vinylsilane 6b and formation of [4+2]-cycloadduct 16bi were monitored readily over the entire course of the reaction by gas chromatographic analysis of aliquots removed from the reaction mixture at regular timepoints. Select cases were validated by 1H NMR spectroscopic measurements. Runs initiated at different concentrations of vinylsilane 6b but with the same excess concentration of 13i afforded concentration versus time data that overlaid graphically, indicating that negligible catalyst decomposition or product inhibition occurred over the course of the reactions (Figure 4A).
Figure 4.
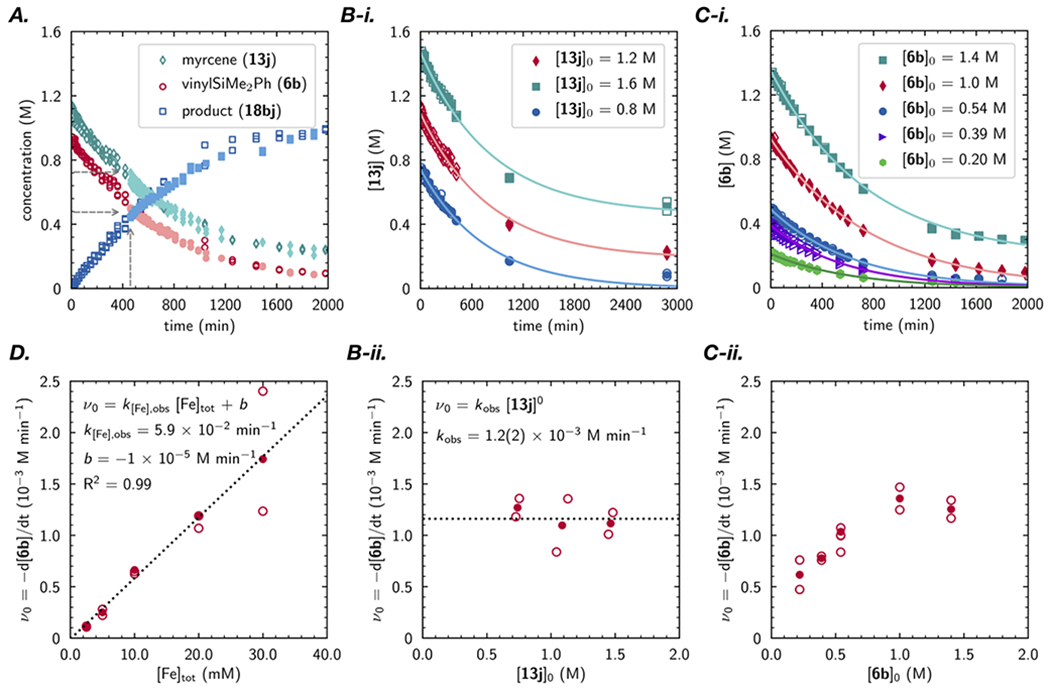
Kinetic analysis of cross-vinylsilane/diene-[4+2]-cycloaddition. (A.) Rate profiles for trials conducted with a ‘same excess’ of diene (dark open markers vs. light filled markers) but a different initial concentration. (B.) Rate dependence on diene 13j concentration. (C.) Rate dependence on vinylsilane 6b. (D.) Rate dependence on [Fe]tot. See supporting information for details.
For all of the kinetic measurements performed, the profiles of rate versus substrate concentration were linear, signaling a net first-order dependence on the vinylsilane substrate concentration. Correspondingly, effective rate constants were readily obtained from fitting the concentration versus time data to an exponential, first-order rate law. Given the well-behaved kinetic regime and the long reaction times necessary to reach full conversion with the most dilute reaction concentrations, the orders in [5b], [13j], and [Fe]tot were determined using the method of initial rates, varying the initial concentration of each component while maintaining that of all others. This established no rate dependence on diene [13i] (Figure 4B) and a saturation-dependence in vinylsilane [6b] (Figure 4C) with typical conditions falling in the first-order regime. A strictly first-order dependence on [Fe]tot was observed (Figure 4D). The rate law was determined to be rate = k[Fe][vinylsilane]. Taken in combination with the stoichiometric studies, these kinetic data are consistent with a mechanism in which either vinylsilane coordination to a resting state iron diene complex (at low concentration) or oxidative cyclization (at high concentration) is rate-determining. Given that these are the first effectively irreversible steps involving both substrates, this indicates that either vinylsilane coordination or oxidative cyclization is also regioselectivity-determining. Although C–C reductive elimination cannot be ruled out as rate- and regioselectivity-determining based on kinetic data alone, this possibility is disfavored by the stoichiometric experiments.
Selectivity Model and Analogy to α-Olefins.
Given the evidence implicating olefin coordination and/or oxidative cyclization as selectivity determining, the relative energies of intermediates B-vi/B-vii (β-[Si]) vs. C-x/C-xi (α-[Si]) (Scheme 8) were assessed using full-molecule density functional theory.32 Although the complicated electronic structures of the relevant iron complexes would necessitate in-depth multiconfigurational analysis for rigorous analysis of the full reaction coordinate,33 the ground-state DFT structures were evaluated to provide qualitative understanding of the regioisomeric candidate metallacycles. In the lowest energy conformers identified for each pair of intermediates, the β-[Si] disposition was preferred. These ground state calculations suggest that there is no intrinsic preference for α-[Si] incorporation. To the extent that the transition state for oxidative cyclization resembles the incipient metallacycle, these computational models support mechanistic hypothesis B.
Further experimental support for mechanistic hypothesis B follows from reexamination of the product mixtures formed from (MePDI)Fe-catalyzed α-olefin/myrcene coupling (Scheme 7A). In these cases, the formation of hydroalkenylation products (16) reports on the intermediacy of metallacycloheptene vii (Scheme 8). Furthermore, the connectivity of 16 was analogous to that of the hydroalkenylation side-products (19dj–gj) generated using vinylsiloxane substrates, which likely formed from the analogous metallacycloheptene B-vii.34 The hydroalkenylation isomers that would arise via metallacycloheptene C-xi were not observed. Although this metallacyclic intermediate cannot be conclusively ruled out, our findings support that it is not part of the mechanistic pathway. The parallel regioselectivity trends are suggestive of a shared mechanistic pathway in which any α-[Si] effect is negligible. In the presence of a conjugated diene, the diene substitution pattern alone controls metallacycle formation. In this case, the difference in [4+2]-cycloaddition versus hydroalkenylation chemoselectivity arises from the facility of β-H elimination and C–H reductive elimination relative to C–C reductive elimination. We hypothesize that the highly substituted β-SiR3 groups impart enhanced conformational rigidity in the intermediate metallacycle, thereby preventing the orbital overlap necessary for β-H elimination and instead favoring C–C reductive elimination.
CONCLUSIONS
In summary, three types of [(PDI)Fe]-catalyzed carbon-carbon bond forming reactions for the chemo- and regioselective elaboratio of vinylsilanes that are distinct from analogous processes with pure hydrocarbons. Specifically: (i) head-to-head vinylsilane dimerization, (ii) cross-[2+2]-cycloaddition with 4-substituted 1,3-dienes, and (iii) cross-[4+2]-cycloaddition with 2-substituted 1,3-dienes were achieved with selectivity derived from substrate control and with commodity reagents yield products with potential applications in silicones and small-molecule synthesis. Stoichiometric reactions, kinetic measurements, in situ Mössbauer spectroscopy, and DFT analyses provided insights into the origin of the observed selectivities. In contrast to α-olefin coupling partners, vinylsilanes exhibit distinct stereoelectronic preferences in some cases and specifically, suppress β-H elimination from key metallacyclic intermediates. Nonetheless, addition of a silyl group was insufficient for perturbing the regioselectivity of metallacycle formation in the presence of 1,3-dienes. These insights shed light on the interplay of ligand and substrate control in the development of selective chemistry that leverages metallacyclic intermediates to upgrade unsaturated coupling partners.
Supplementary Material
ACKNOWLEDGMENT
C.R.K. thanks the NIH for a Ruth L. Kirschstein National Research Service Award (F32 GM126640). The authors thank Dr. Megan Mohadjer Beromi (Princeton), Dr. Samantha Yruegas (Princeton), Dr. Jonathan Darmon (Princeton), and Andreu Tortajada Navarro (Institut Català d’Investigació Química) for helpful discussion as well as Rachel Macaulay (Princeton) for synthetic assistance. Financial support and a gift of piperylene was provided by Firmenich.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental details, compound characterization data, kinetic data, computational methods and results (PDF)
REFERENCES
- 1.a) Rosenthal U; Burlakov VV; Bach MA; Beweries T Five-Membered Metallacycles of Titanium and Zirconium – Attractive Compounds for Organometallic Chemistry and Catalysis. Chem. Soc. Rev 2007, 36, 5, 719–728 [DOI] [PubMed] [Google Scholar]; b) Thakur A; Louie J Advances in Nickel-Catalyzed Cycloaddition Reactions To Construct Carbocycles and Heterocycles. Acc. Chem. Res 2015, 48, 2354–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crabtree RH The Organometallic Chemistry of the Transition Metals, 5th Ed. Wiley: Hoboken, 2009; pp 172–175. [Google Scholar]
- 3.a) Schmidt R; Griesbaum K; Behr A; Biedenkapp D; Voges H-W; Garbe D; Paetz C; Collin G; Mayer D; Höke H Hydrocarbons. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: Chichester, U.K., 2014; pp. 1–74 [Google Scholar]; b) Farrell LM Developments in Linear Alpha Olefin (LAO) Comonomer Technologies for Polyethylene. Technical Report for ChemSystems PERP Program, Nexant: 2012 [Google Scholar]; c) Ring K-L; deGuzman M Chemical Economics Handbook: Linear Alpha-Olefins. Technical Report; IHS Markit: 2017; https://www.ihs.com/products/linear-alpha-olefins-chemical-economics-handbook.html [Google Scholar]
- 4.a) Briggs JR The Selective Trimerization of Ethylene to Hex-1-ene. J. Chem. Soc. Chem. Commun 1989, 674–675 [Google Scholar]; b) Dixon JT; Green MJ; Hess FH; Morgan DH Advances in Selective Ethylene Trimerisation – a Critical Overview. J. Organomet. Chem 2004, 689, 3641–3668 [Google Scholar]; c) McGuinness D,S Olefin Oligomerization via Metallacycles: Dimerization, Trimerization, Tetramerization, and Beyond. Chem. Rev 2011, 111, 2321–2341 [DOI] [PubMed] [Google Scholar]; d) Agapie T Selective Ethylene Oligomerization: Recent Advances in Chromium Catalysis and Mechanistic Investigations. Coord. Chem. Rev 2011, 255, 861–880. [Google Scholar]
- 5.a) Lautens M; Klute W; Tam W Transition Metal-Mediated Cycloaddition Reactions. Chem. Rev 1996, 96, 49–92 [DOI] [PubMed] [Google Scholar]; b) Wang Z Reppe Alkyne Cyclotrimerization. In Comprehensive Organic Named Reactions and Reagents; Wang Z, Ed.; Wiley, 2010; pp. 2345–2351. [Google Scholar]
- 6.Select examples of state-of-the-art methods for the synthesis of (a) pyrroles, (b) cyclopentenes, (c/d) cyclobutenes, (e) cyclobutanes, and (f/g) cyclooctadienes using electronically unbiased coupling partners:; a) Gilbert ZW; Hue RJ; Tonks IA Catalytic Formal [2+2+1] Synthesis of Pyrroles from Alkynes and Diazenes via TiII/TiIV Redox Catalysis. Nature Chem. 2016, 8, 63–68 [DOI] [PubMed] [Google Scholar]; b) Zhou Y-Y; Uyeda C Catalytic Reductive [4 + 1]-Cycloadditions of Vinylidenes and Dienes. Science, 2019, 363, 857–862 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Pagar VV; RajanBabu TV Tandem Catalysis for Asymmetric Coupling of ERthylene and Enynes to Functionalized Cyclobutanes. Science, 2018, 361, 68–72 [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Parsutkar MM; Pagar VV; RajanBabu TV Catalytic Enantioselective Synthesis of Cyclobutenes from Alkynes and Alkenyl Derivatives. J. Am. Chem. Soc 2019, 141, 15367–15377 [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Hoyt JM; Schmidt VA; Tondreau AM; Chirik PJ Science, 2015, 349, 960–963 [DOI] [PubMed] [Google Scholar]; f) Lee H; Campbell MG; Hernández Sánchez R; Börgel J; Raynaud J; Parker SE; Ritter T Mechanistic Insight Into High-Spin Iron(I)-Catalyzed Butadiene Dimerization. Organometallics, 2016, 35, 2923–2929 [Google Scholar]; g) Kennedy CR; Zhang H; Macaulay RL; Chirik PJ Regio- and Diastereoselective Iron-Catalyzed [4+4]-Cycloaddition of 1,3-Dienes. J. Am. Chem. Soc 2019, 141, 8557–8573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Regioselective, Intramolecular [Fe]-Catalyzed [2+2]-Cycloaddition of Alkenes:; Bouwkamp MW; Bowman AC; Lobkovsky E; Chirik PJ J. Am. Chem. Soc 2006, 128, 13340–13341. [DOI] [PubMed] [Google Scholar]
- 8.Regioselective, Intramolecular [Co]-Catalyzed [2+2]-Cycloaddition of Alkenes:; Schmidt VA; Hoyt JM; Margulieux GW; Chirik PJ Cobalt-Catalyzed [2π + 2π] Cycloadditions of Alkenes: Scope, Mechanism, and Elucidation of Electronic Structure of Catalytic Intermediates. J. Am. Chem. Soc 2015, 137, 7903–7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Regioselective, [Ni]-catalyzed [2+2]-cyclodimerizations:; a) Huang D-J; Cheng C-H [2+2] Dimerization of Norbornadiene and its Derivatives in the Presence of Nickel Complexes and Zinc Metal. J. Organomet. Chem 1995, 490, C1–C7; [Google Scholar]; b) Saito S; Hirayama K; Kabuto C; Yamamoto Y Nickel(0)-Catalyzed [2+2] Annulation of Electron-Deficient Allenes. Highly Regioselective Synthesis of Cyclobutanes. J. Am. Chem. Soc 2000, 122, 10776–10780 [Google Scholar]; c) Canellas S; Montgomery J; Pericas MA Nickel-Catalyzed Reductive [2+2] Cycloaddition of Alkynes. J. Am. Chem. Soc 2018, 140, 17349–17355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Namyslo JC; Kaufmann DE The Application of Cyclobutane Derivatives in Organic Synthesis. Chem. Rev 2003, 103, 1485–1537 [DOI] [PubMed] [Google Scholar]; b) Sergeiko A; Poroikov VV; Hanus LO; Dembitsky VM Cyclobutane-Containing Alkaloids: Origin, Synthesis, and Biological Activities. Open Med. Chem. J 2008, 2, 26–37 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Morris DM; Quintana RL; Harvey BG High-Performance Jet Fuels Derived from Bio-Based Alkenes by Iron-Catalyzed [2+2] Cycloaddition. Chem. Sus. Chem 2019, 12, 1646–1652 [DOI] [PubMed] [Google Scholar]; d) Harvey BG; Morris D Alkyl cyclobutane fuels. US 10,604,711, 2020; e) Chen Z; Mercer JAM; Zhu X; Romaniuk JAH; Pfattner R; Cegelski L; Martinez TJ; Burns NZ; Xia Y Mechanochemical Unzipping of Insulating Polyladderene to Semiconducting Polyacetylene. Science, 2017, 357, 475–479 [DOI] [PubMed] [Google Scholar]; f) Su JK; Feist JD; Yang J; Mercer JAM; Romaniuk JAH; Chen Z; Cegelski L; Burns NZ; Xia Y Synthesis and Mechanochemical Activation of Ladderene–Norbornene Block Copolymers. J. Am. Chem. Soc 2018, 140, 12388–12391 [DOI] [PubMed] [Google Scholar]; g) Beromi MM; Kennedy CR; Younker JM; Carpenter AE; Mattler SJ; Throckmorton JA; Chirik PJ Iron Catalyzed Synthesis and Chemical Recycling of Telechelic, 1,3-Enchained Oligocyclobutanes. ChemRxiv, 2020, DOI: 10.26434/chemrxiv.11994489.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Hoyt JM; Sylvester KT; Semproni SP; Chirik PJ Synthesis and Electronic Structure of Bis(imino)pyridine Iron Metallacyclic Intermediates in Iron-Catalyzed Cyclization Reactions. J. Am. Chem. Soc 2013, 135, 4862–4877 [DOI] [PubMed] [Google Scholar]; b) Joannou MV; Hoyt JM; Chirik PJ Investigations into the Mechanism of Inter- and Intramolecular Iron-Catalyzed [2 + 2] Cycloaddition of Alkenes. J. Am. Chem. Soc 2020, 142, 5314–5330. [DOI] [PubMed] [Google Scholar]
- 12.Cross-[2+2]-cycloaddition between alkenes and alkynes:; a) Chao KC; Rayabarapu DK; Wang C-C; Cheng C-H Cross [2 + 2] Cycloaddition of Bicyclic Alkenes with Alkynes Mediated by Cobalt Complexes: A Facile Synthesis of Cyclobutene Derivatives. J. Org. Chem 2001, 66, 8804–8810 [DOI] [PubMed] [Google Scholar]; b) Hilt G; Paul A; Treutwein J Cobalt Catalysis at the Crossroads: Cobalt-Catalyzed Alder–Ene Reaction versus [2 + 2] Cycloaddition. Org. Lett 2010, 12, 1536–1539 [DOI] [PubMed] [Google Scholar]; c) Ding W, Yoshikai N, Cobalt-Catalyzed Intermolecular [2+2] Cycloaddition Between Alkynes and Allenes. Angew. Chem. Int. Ed 2019, 58, 2500–2504 [DOI] [PubMed] [Google Scholar]; d) Huang D-J; Rayabarapu DK; Li L-P; Sambaiah T; Cheng C-H Nickel-Catalyzed [2+2] Cycloaddition of Alkynes with Activated Cyclic Alkenes: Synthesis and Novel Ring Expansion Studies of Cyclobutene Products. Chem. Eur. J 2000, 6, 3706–3713 [DOI] [PubMed] [Google Scholar]; e) Abulimiti A; Nishimura A; Ohashi M; Ogoshi S Nickel-catalyzed [2+2] Cycloaddition Reaction of Bulky Enones with Simple Alkynes. The Effect of Bulkiness of Substituent Attached at β-Carbon. Chem. Lett 2013, 42, 904–905 [Google Scholar]; f) Qin H; Chen J; Li K; He Z; Zhou Y; Fan B Nickel-Catalyzed Asymmetric [2+2] Cycloaddition Reaction of Hetero-Bicyclic Alkenes with Internal Alkynes. Chem. Asian J 2018, 13, 2431–2434. [DOI] [PubMed] [Google Scholar]
- 13.Cross-[2+2]-Cycloaddition Between Alkenes and Enynes:; a) Treutwein J; Hilt G Cobalt-Catalyzed [2+2] Cycloaddition. Angew. Chem. Int. Ed 2008, 47, 6811–6813 [DOI] [PubMed] [Google Scholar]; b) Nishimura A; Tamai E; Ohashi M; Ogoshi S Synthesis of Cyclobutenes and Allenes by Cobalt-Catalyzed Cross-Dimerization of Simple Alkenes with 1,3-Enynes. Chem. Eur. J 2014, 20, 6613–6617 [DOI] [PubMed] [Google Scholar]; c) Nishimura A; Ohashi M; Ogoshi S Nickel-Catalyzed Intermolecular [2+2] Cycloaddition of Conjugated Enynes with Alkenes. J. Am. Chem. Soc 2012, 134, 15692–15695 [DOI] [PubMed] [Google Scholar]; d) Kumar R; Tamai E; Ohnishi A; Nishimura A; Hoshimoto Y; Ohashi M; Ogoshi S Nickel-Catalyzed Enantioselective Synthesis of Cyclobutenes via [2+2] Cycloaddition of α,β-Unsaturated Carbonyls with 1,3-Enynes. Synthesis, 2016, 48, 2789–2794. [Google Scholar]
- 14.Cross-[2+2]-Cycloaddition Between Alkenes and Allenes:; Noucti NN; Alexanian EJ Stereoselective Nickel-Catalyzed [2+2] Cycloadditions of Ene-Allenes. Angew. Chem., Int. Ed 2015, 54, 5447–5450. [DOI] [PubMed] [Google Scholar]
- 15.Cross-[2+2]-cycloaddition between alkenes and dienes:; a) Russell SK; Lobkovsky E; Chirik PJ J. Am. Chem. Soc 2011, 133, 8858–8861 [DOI] [PubMed] [Google Scholar]; b) Kennedy CR; Zhong H; Joannou MV; Chirik PJ Pyridine(diimine) Iron Diene Complexes Relevant to Catalytic [2+2]-Cycloaddition Reactions. Adv. Synth. Catal 2020, 362, 404–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reviews:; a) RajanBabu TV In Comprehensive Asymmetric Catalysis; Jacobsen EN, Pfaltz A, Yamamto H, Eds.; Springer: Berlin, 1999; pp 417–427 [Google Scholar]; b) RajanBabu TV In Pursuit of an Ideal C-C Bond-Forming Reaction: Development and Applications of the Hydrovinylation of Olefins. Synlett, 2009, 853–885 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hilt G Hydrovinylation Reactions - Atom-Economic Transformations with Steadily Increasing Synthetic Potential. Eur. J. Org. Chem 2012, 4441–4451 [Google Scholar]; d) Hirano M Recent Advances in the Catalytic Linear Cross-Dimerizations. ACS Catal. 2019, 9, 1408–1430. [Google Scholar]
- 17.Select examples since 2015:; a) Timsina YN; Sharma RK; RajanBabu TV Cobalt-Catalysed Asymmetric Hydrovinylation of 1,3-Dienes. Chem. Sci 2015, 6, 3994–4008 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Biswas S; Page JP; Dewese KR; RajanBabu TV Asymmetric Catalysis with Ethylene. Synthesis of Functionalized Chiral Enolates. J. Am. Chem. Soc 2015, 137, 14268–14271 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Jing SM; Balasanthiran V; Pagar V; Gallucci JC; RajanBabu TV Catalytic Enantioselective Hetero-Dimerization of Acrylates and 1,3-Dienes. J. Am. Chem. Soc 2017, 139, 18034–18043 [DOI] [PMC free article] [PubMed] [Google Scholar]; d) McNeill E; Ritter T 1,4-Functionalization of 1,3-Dienes With Low-Valent Iron Catalysts. Acc. Chem. Res 2015, 48, 2330–2343 [DOI] [PubMed] [Google Scholar]; e) Schmidt VA; Kennedy CR; Bezdek MJ; Chirik PJ Selective [1,4]-Hydrovinylation of 1,3-Dienes with Unactivated Olefins Enabled by Iron Diimine Catalysts. J. Am. Chem. Soc 2018, 140, 3443–3453 [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Gray M; Hines MT; Parsutkar MM; Wahlstrom AJ; Brunelli NA; RajanBabu TV Mechanism of Cobalt-Catalyzed Heterodimerization of Acrylates and 1,3-Dienes. A Potential Role of Cationic Cobalt(I) Intermediates. ACS Catal. 2020, 10, 4337–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Sturla SJ; Buchwald SL Monocyclopentadienyltitanium Aryloxide Complexes: Preparation, Characterization, and Application in Cyclization Reactions. Organometallics, 2002, 21, 739–748 [Google Scholar]; b) Miller AD; McBee JL; Tilley TD Mechanism of Reversible Alkyne Coupling at Zirconocene: Ancillary Ligand Effects. J. Am. Chem. Soc 2008, 130, 4992–4999 [DOI] [PubMed] [Google Scholar]; c) Zirngast M; Marschner C; Baumgartner J Group 4 Metallocene Complexes of Tris(trimethylsilyl)silylacetylene and Related Alkynes. Organometallics, 2008, 27, 2570–2583 [Google Scholar]; d) Ren S; Qiu Z; Xie Z Reaction of Zirconocene–Carboryne with Alkenes: Synthesis and Structure of Zirconacyclopentanes with a Carborane Auxiliary, Organometallics, 2012, 31, 4435–4441 [Google Scholar]; e) Altenburger K, Baumann W , Spannenberg A, Arndt P; Rosenthal U Coordination Behavior and Reactivity of α-Heterosubstituted Trimethylsilylalkynes at Titano- and Zirconocene. Eur. J. Inorg. Chem 2014, 5948–5957 [Google Scholar]; f) Desnoyer AN; See XY; Tonks IA Diverse Reactivity of Diazatitanacyclohexenes: Coupling Reactions of 2H-Azirines Mediated by Titanium(II). Organometallics, 2018, 37, 4327–4331 [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Chiu H-C; Tonks IA Trimethylsilyl-Protected Alkynes as Selective Cross-Coupling Partners in Titanium-Catalyzed [2+2+1] Pyrrole Synthesis. Angew. Chem. Int. Ed 2018, 57, 6090–6094 [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Bando M; Nakajima K; Song Z; Takahashi T Metal-Dependent Regioselective Homocoupling of Stannyl- and Alkyl-Substituted Alkynes on Group 4 Elements. Formation of Unsymmetrical Titanacyclopentadienes and Symmetrical Zircona-Cyclopentadienes. Dalton Trans. 2019, 48, 13912–13915 [DOI] [PubMed] [Google Scholar]; i) Urrego-Riveros S; Ramirez y Medina I-M; Duvinage D; Lork E; Sönnichsen FD; Staubitz A Negishi’s Reagent Versus Rosenthal’s Reagent in the Formation of Zirconacyclopentadienes. Chem. Eur. J 2019, 25, 13318–13328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.For an orbital symmetry treatment:; Stockis A; Hoffmann R Metallacyclopentanes and Bisolefin Complexes. J. Am. Chem. Soc 1980, 102, 2952–2962. [Google Scholar]
- 20.On the stereoelectronics of vinylsilanes:; a) Overman LE; Burk RM The Importance of Vinylsilane Stereochemistry and σ - π Stabilization in Iminium Ion-Vinylsilane Cyclizations. A Short Total Synthesis of the Alkaloid (−) –epielwesine. Tetrahedron Lett. 1984, 25, 5739–5742 [Google Scholar]; b) Fleming I; Dunogués J; Smithers R The Electrophilic Substitution of Allylsilanes and Vinylsilanes. Org. React 1989, 37, 57–575 [Google Scholar]
- 21.For discussion of agostic interactions invoked with Si–H/C bonds in analogy to C–H agostics in polymerization catalysis:; a) Grubbs RH; Coates GW α-Agostic Interactions and Olefin Insertion in Metallocene Polymerization Catalysts. Acc. Chem. Res 1996, 29, 85–93 [Google Scholar]; b) Hieringer W; Eppinger J; Anwander R; Herrmann WA C2-Symmetric Ansa-Lanthanidocene Complexes. Theoretical Evidence for a Symmetric Ln⋯(SiH) β-Diagostic Interaction. J. Am. Chem. Soc 2000, 122, 11983–11994 [Google Scholar]; c) Clark DL; Gordon JC; Hay PJ; Martin RL; Poli R DFT Study of Tris(bis(trimethylsilyl)methyl)lanthanum and Samarium. Organometallics, 2002, 21, 5000–5006 [Google Scholar]; d) Sadow AD; Tilley TD Cationic Hafnium Silyl Complexes and Their Enhanced Reactivity in σ-Bond Metathesis Processes with Si–H and C–H Bonds. J. Am. Chem. Soc 2003, 125, 9462–9475 [DOI] [PubMed] [Google Scholar]; e) Perrin L; Maron L; Eisenstein O; Lappert MF γ Agostic C–H or β agostic Si–C bonds in La{CH(SiMe3)2}3? A DFT Study of the Role of the Ligand. New J. Chem 2003, 27, 121–127 [Google Scholar]; f) Lamač M; Spannenberg A; Baumann W; Jiao H; Fischer F; Hansen S; Arndt P; Rosenthal U Si–H Bond Activation of Alkynylsilanes by Group 4 Metallocene Complexes. J. Am. Chem. Soc 2010, 132, 4369–4380. [DOI] [PubMed] [Google Scholar]
- 22.a) Rösch L; John P; Reitmeier R Silicon Compounds, Organic. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley, 2000; pp. 637–674 [Google Scholar]; b) Schöbel A; Winkenstette M; Anselment TMJ; Rieger B Copolymerization of Alkenes and Polar Monomers by Early and Late Transition Metal Catalysts. In Polymer Science: A Comprehensive Reference, Volume 3, Science Direct: 2012, pp 779–823 [Google Scholar]; c) Chen Z; Brookhart M Exploring Ethylene/Polar Vinyl Monomer Copolymerizations Using Ni and Pd α-Diimine Catalysts. Acc. Chem. Res 2018, 51, 1831–1839 [DOI] [PubMed] [Google Scholar]; d) Walsh DJ; Hyatt MG; Miller SA; Guironnet D Recent Trends in Catalytic Polymerizations. ACS Catal. 2019, 9, 11153–11188. [Google Scholar]
- 23.a) Bartlett PD; Schueller KE Cycloaddition. VIII. Ethylene as a Dienophile. A Minute Amount of 1,2-Cycloaddition of Ethylene to Butadiene. J. Am. Chem. Soc 1968, 90, 6071–6077 [Google Scholar]; b) Lang J Electronic Effects in the Intramolecular Vinylsilane Diels-Alder reaction. 2. A Practical Synthesis of Difunctional Organosilane Reagents and their Application to Cycloaddition Reactions. PhD Dissertation, State University of New York at Stony Brook, Stony Brook, NY, 1999. [DOI] [PubMed] [Google Scholar]
- 24.Moretto H; Schulze M; Wagner G Silicones. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley, 200; pp. 675–712. [Google Scholar]
- 25.The unrestricted Kohn–Sham and broken-symmetry BS(3,1) inputs both converged to a BS(3,1) solution.
- 26.The minor component (13%) exhibited parameters (δ = −0.27 mm/s, |ΔE| = 1.48 mm/s) in agreement with the corresponding Fe(III) metallacycles, although insufficient additional data was available to allow for a definitive assignment.
- 27.However, addition of D2 to (MePDI)Fe(η2-CH2=C(H)SiMe2Ph)2 resulted in deuterium incorporation at all aliphatic carbon positions as well as at the imine groups of the PDI. Reduced (PDI)Fe complexes are known to undergo ligand cyclometallation and deuterium scrambling, which complicate any deuterium labelling studies to potentially differentiate between the two mechanisms. See ref. 11b.
- 28.Calculations were performed in Gaussian using the M06-2x level of density functional theory (DFT) with the 6-311++g(d,p) basis set and implicit solvent correction (CPCM, cyclohexane).
- 29.The analogous control experiments conducted with 1,3-dienes and α-olefins (not vinylsilanes) does not afford hydrovinylation products.
- 30.Attempts to monitor the catalytic reaction by freeze-quench 57Fe Mössbauer spectroscopy studies proved unsuccessful. The resulting spectra contained hallmarks of multiple species that could not be calibrated reliably against any known standard PDI iron compound.
- 31.a) Blackmond DG Reaction Progress Kinetic Analysis: A Powerful Methodology for Mechanistic Studies of Complex Catalytic Reactions. Angew. Chem. Int. Ed 2005, 44, 4302–4320 [DOI] [PubMed] [Google Scholar]; b) Blackmond DG Kinetic Profiling of Catalytic Organic Reactions as a Mechanistic Tool. J. Am. Chem. Soc 2015, 137, 10852–10866. [DOI] [PubMed] [Google Scholar]
- 32.Calculations were performed using the B3LYP level of density functional theory with the def2-TZVP(-f) basis set augmented by SARC/J terms for Fe, N, and C atoms in the primary coordination sphere, and the def2-SVP basis set augmented with SARC/J terms for all other atoms.
- 33.Hu L; Chen H Substrate-Dependent Two-State Reactivity in Iron-Catalyzed Alkene [2+2] Cycloaddition Reactions. J. Am. Chem. Soc 2017, 139, 15564–15567. [DOI] [PubMed] [Google Scholar]
- 34.We further hypothesize that vinylsiloxane substituents are less conformationally constrained and more remote than the corresponding silane substituents, thus leading to their reduced chemoselectivity under experimental conditions. No cross-reaction was observed with t-butyl-ethylene (1c) or 3,3,3-trifluoropropylene (1d), preventing direct experimental evaluation of this hypothesis.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.