

Nephrologists are increasingly including genetic diagnosis into clinical practice as sequencing costs come down, availability improves, and the list of kidney disease genes becomes more complete. Multiple studies suggest that around 10% of the adult ESKD population and 30% of pediatric cohorts have an identifiable genetic kidney disease (1–3). One study found that genetic diagnosis provided new clinical insight in nearly 75% of solved cases by identifying, reclassifying, or specifying disease etiology or informing prognostication, treatment, or transplant decisions (1). Many nephrologists have little experience with genetic testing, and thus, they may have uncertainty about whom to test, what test to select, and what to expect from the genetic results.
Kidney disease phenotypes can be caused by mutations in any of hundreds of genes. The exons, which are protein-encoding regions of genes that make up only 1% of the whole genome known as the “exome,” are estimated to carry at least 85% of disease-causing variants (4). Next generation sequencing (NGS), also known as massively parallel sequencing, can evaluate all >18,000 genes or a targeted list of genes. NGS can be done on the whole genome, whole exome, or a selection of genes within the exome; these are known as whole-genome sequencing (WGS), whole-exome sequencing (WES), or targeted NGS, respectively (Figure 1A). Because of their efficient multiplexing, these methods provide a clear advantage over Sanger sequencing individual PCR amplicons for all but a small number of indications. Sequencing of individual variants, such as a known familial variant or specific risk allele, or a short list of genes specified by the phenotype does not require NGS, but the list of genes to evaluate does not need to be long before NGS becomes more cost effective. Large deletions or duplications also known as copy number variations are not detected by individual gene sequencing or standard NGS analysis. Additional analyses of WGS data can identify these well, but the segmented data of short sequences provided by WES have limited sensitivity and specificity for this analysis. For this reason, additional testing, such as multiplex ligation probe-dependent amplification (MLPA), may be performed as an adjunctive test in cases in which WES does not find a definitive variant, and for now, this remains the cost-effective approach over WGS.
Figure 1.
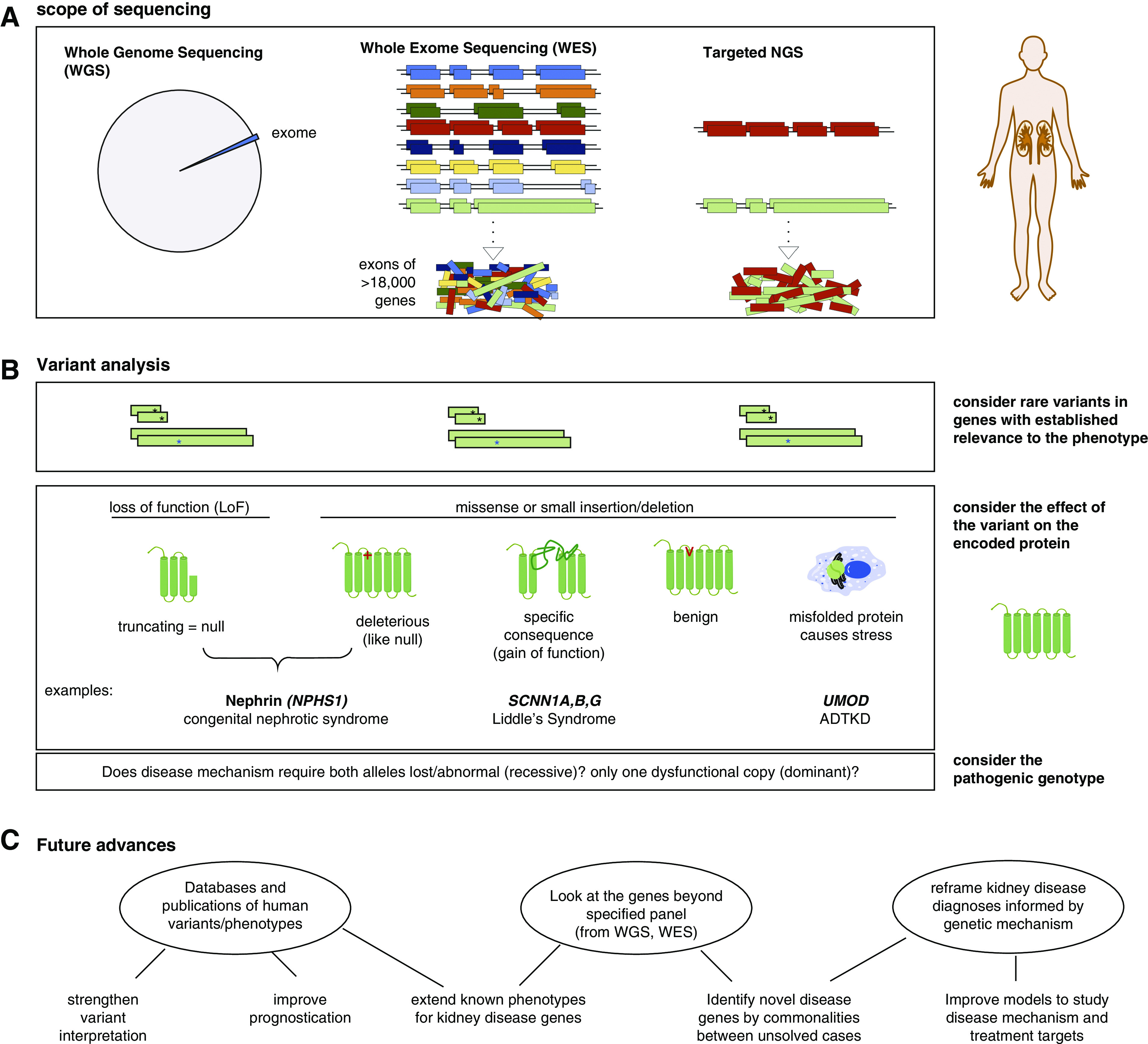
Next generation sequencing (NGS) applied to kidney disease analysis. (A) NGS can be applied to the whole genome (whole-genome sequencing [WGS]), the exome (whole-exome sequencing [WES]), or specific genes (targeted NGS). (B) Variant analysis of a gene panel proceeds similarly regardless of scope of the sequencing. A “pathogenic” variant must be more rare in the general population than the rare kidney phenotype and either encode a premature truncation of the protein or have an established causative association with disease in public databases. Other rare nontruncating variants can be considered “likely pathogenic” on the basis of frequency and predictive algorithms that consider whether the affected amino acids have remained unchanged (“conserved”) through species evolution and are in critical protein domains or other regulatory features. Beyond the consequence of the variant on the protein, interpretation of its effect must include knowledge of the disease mechanism and whether monoallelic (heterozygous) or biallelelic (homozygous or compound heterozygous) mutation is needed for pathogenesis. ADTKD, autosomal dominant tubulointerstitial kidney disease. (C) Applying genetic analysis to kidney disease evaluation has several potential future benefits.
NGS is available in many academic institutional laboratories, and it is increasingly available by commercial entities to evaluate genes implicated in kidney disease. An important yet surprisingly variable component to genetic analysis is the interpretation after sequencing and variant calling are complete. Standards on what constitutes “pathogenic” versus “likely pathogenic” versus “variant of unknown significance” are published by the American College of Medical Genetics (5). However, copy number variation analysis is not always included, and genetic kidney disease mechanisms are diverse. For example, only mutations in UMOD that cause specific misfolding of the encoded protein Uromodulin (Tamm–Horsfall protein) cause autosomal dominant tubulointerstitial kidney disease, not those that might be labeled as “pathogenic” because they truncate the protein (Figure 1B). Gene lists in recent published studies have ranged from 207 to 625 genes (6). Lists are often subdivided into gene panels for specific phenotype groups, such as cystic kidney disease, nephrotic syndrome, GN, or congenital abnormalities, in order to focus attention on genes with mutation that would be most likely to explain the disease in question. Some institutions have invested the resources to design NGS capture reagents specific for the gene panels of interest in order to achieve higher sensitivity in those genes with targeted NGS over WES. Many others do WES and then, only consider the selected panel of genes. This provides the opportunity to retrospectively consider other genes subsequently implicated in kidney disease and if the patient is consented accordingly, to contribute WES data from unsolved cases to researchers for novel gene discovery (Figure 1C).
In Kidney360, Wilson et al. (7) report their findings from physician-ordered clinical genetic testing referrals for kidney disease during a 4-year period at Washington University in St. Louis, Missouri. The Genomics and Pathology Services carried out WES on 324 cases during this time and evaluated genes on phenotype-specific gene panels. This consisted of 224 cases tested for genetic causes of atypical hemolytic uremic syndrome (aHUS) and 100 for other panels: nephrotic syndrome (n=56 cases evaluated for 34 genes), cystic kidney disease/nephronophthisis (n=26 cases evaluated for 23 genes), Alport syndrome (n=13 cases evaluated for three genes), or a custom panel (n=5 cases). The cohort was 57% white, 16% black, 8% Hispanic, 2% Asian, and 15% other. The average age was 30 years but ranged from infancy to later adulthood, and only a minority had known family history. For cases without pathogenic variants by WES on the aHUS panel testing, MLPA was run to assess CFHR gene cluster rearrangements.
This study provides valuable insights into what is realistically found with clinically obtained genetic testing by representing one institution’s experience. The cohort contains one of the largest groups tested for aHUS studied to date. In contrast to genetic research cohorts with defined phenotypic inclusion criteria, this cohort has many cases where the ordering physician requested the genetic testing for an unclear clinical diagnosis, often pediatric ESKD. As such, learning from each case is more valuable than considering the percentage of cases solved. The likelihood of an individual patient receiving a genetic diagnosis depends on the specificity of the phenotype, including potential extrarenal manifestations, age of diagnosis, and presence of family history. Indeed, compared with another large study of clinically confirmed aHUS, this study found fewer cases with variants in the gene panel, although similar incidence of CFHR1-CFHR3 homozygous rearrangements (11% versus 12%) (8). The percentage of cases given a genetic diagnosis may increase in the next decade as many variants of unknown significance are found in kidney disease genes in published studies, and future variant interpretation can be informed by past experience and subsequent discovery of novel disease genes (Figure 1C). Recently, journals have required submission of published genetic variants to public databases.
After initial WES-based analyses of the five phenotype-specific gene panels in which they found pathogenic or likely pathogenic variants in 66 of 324 (20%) cases, the authors considered whether broadening the gene panel to consider 309 gene associated with kidney disease regardless of specified phenotype would provide a more informative clinical results. They report several interesting findings and describe that this improved their “diagnostic yield” from 20% to nearly 30%. However, these percentages seem to include cases with heterozygous carrier status or genetic risk alleles, not just those with causative genetic diagnosis, and thus, they may be quite misleading. For example, the 66 includes individuals with a single pathogenic allele in recessive disease genes, including Nephrin (NPHS1), Podocin (NPHS2), Fibrocystin (PKHD1), NPHP4, and RPGRIP1L. If these individual pathogenic variants are expected to be part of a pathogenic genotype that explains disease, they must either be homozygous or accompanied by another mutation or deletion not reported due to the modality of testing. A well curated and regularly updated centralized nephrology gene list that includes both the inheritance pattern and allele type—loss of function versus gain of function or other specific alteration—for which each gene is implicated as pathogenic would help streamline these efforts.
Among those unsolved by the aHUS panel plus MLPA, extending analysis to 309 genes found that two cases had heterozygous ADAMTS13 variants indicating carrier status for thrombotic thrombocytopenia purpura, one case had genetically defined recessive Alport syndrome, and one case had congenital acute renal failure with recessive loss of the REN gene encoding renin. The underlying pathogenic variants in PKD1 aa of ordering this test. These included a 26-year-old patient with GN found to have a pathogenic EYA1 genotype for branchiotorenal syndrome, which is characterized by hearing loss and congenital renal hypoplasia with incomplete penetrance, and an infant with a likely pathogenic recessive genotype in a calcium oxalate nephrolithiasis-associated gene SLC26A1. It would be of interest to know the perspective of the ordering clinicians regarding the benefit of these expected and unexpected genetic findings and whether in retrospect there are additional clinical findings to support the relevance of the genetic diagnosis to the kidney phenotype in question.
In summary, the study by Wilson et al. (7) reports the genetic findings and clinical history for a large cohort representative of current usage of clinical genetic testing in kidney disease. Increasing use of clinical genetic testing with public database entry will allow for increasing ability to interpret the consequences of genetic variants and define the cause of kidney disease phenotypes in ways most amenable to mechanism research. Further, analysis of WES data from large numbers of unsolved cases, if collaborations and appropriate patient consent were obtained, stands to advance further gene discovery. Clinical genetic testing is not without challenges, but with careful analysis and collaboration, it has the potential to accelerate progress in our understanding of the mechanistic basis of kidney disease and development of targets for treatments that could alter the landscape of both inherited and acquired kidney disease.
Disclosures
The author has nothing to disclose.
Funding
This work was supported by National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grant K08 DK119642; PKD Foundation research grant 217G18a; and a Robert E. Leet and Clara Guthrie Patterson Trust mentored research award.
Acknowledgments
The content of this article reflects the personal experience and views of the author and should not be considered medical advice or recommendations. The content does not reflect the views or opinions of the American Society of Nephrology (ASN) or Kidney360. Responsibility for the information and views expressed herein lies entirely with the author(s).
Author Contributions
W. Besse conceptualized the study, was responsible for funding acquisition, wrote the original draft, and reviewed and edited the manuscript.
Footnotes
See related article, “Beyond Panel-Based Testing: Exome Analysis Increases Sensitivity for Diagnosis of Genetic Kidney Disease” on pages 772–780.
References
- 1.Groopman EE, Marasa M, Cameron-Christie S, Petrovski S, Aggarwal VS, Milo-Rasouly H, Li Y, Zhang J, Nestor J, Krithivasan P, Lam WY, Mitrotti A, Piva S, Kil BH, Chatterjee D, Reingold R, Bradbury D, DiVecchia M, Snyder H, Mu X, Mehl K, Balderes O, Fasel DA, Weng C, Radhakrishnan J, Canetta P, Appel GB, Bomback AS, Ahn W, Uy NS, Alam S, Cohen DJ, Crew RJ, Dube GK, Rao MK, Kamalakaran S, Copeland B, Ren Z, Bridgers J, Malone CD, Mebane CM, Dagaonkar N, Fellström BC, Haefliger C, Mohan S, Sanna-Cherchi S, Kiryluk K, Fleckner J, March R, Platt A, Goldstein DB, Gharavi AG: Diagnostic utility of exome sequencing for kidney disease. N Engl J Med 380: 142–151, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mallett A, Patel C, Salisbury A, Wang Z, Healy H, Hoy W: The prevalence and epidemiology of genetic renal disease amongst adults with chronic kidney disease in Australia. Orphanet J Rare Dis 9: 98, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Connaughton DM, Hildebrandt F: Personalized medicine in chronic kidney disease by detection of monogenic mutations. Nephrol Dial Transplant 35: 390–397, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, Bakkaloğlu A, Ozen S, Sanjad S, Nelson-Williams C, Farhi A, Mane S, Lifton RP: Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A 106: 19096–19101, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee : Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mallett AJ, McCarthy HJ, Ho G, Holman K, Farnsworth E, Patel C, Fletcher JT, Mallawaarachchi A, Quinlan C, Bennetts B, Alexander SI: Massively parallel sequencing and targeted exomes in familial kidney disease can diagnose underlying genetic disorders. Kidney Int 92: 1493–1506, 2017 [DOI] [PubMed] [Google Scholar]
- 7.Wilson PC, Love-Gregory L, Corliss M, McNulty S, Heusel JW, Gaut JP: Beyond panel-based testing: Exome analysis increases sensitivity for diagnosis of genetic kidney disease. Kidney360 1: 771–779, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bu F, Zhang Y, Wang K, Borsa NG, Jones MB, Taylor AO, Takanami E, Meyer NC, Frees K, Thomas CP, Nester C, Smith RJH: Genetic analysis of 400 patients refines understanding and implicates a new gene in atypical hemolytic uremic syndrome. J Am Soc Nephrol 29: 2809–2819, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]