

Abstract
Amines such as 1,2,3,4-tetrahydroisoquinoline undergo redox-neutral annulations with ortho-(nitromethyl)benzaldehyde. Benzoic acid acts as a promoter in these reactions, which involve concurrent amine α-C–H bond and N–H bond functionalization. Subsequent removal of the nitro group provides access to tetrahydroprotoberberines not accessible via typical redox-annulations. Also reported are decarboxylative annulations of ortho-(nitromethyl)benzaldehyde with proline and pipecolic acid.
Keywords: C–H bond functionalization, redox-neutral, redox-annulation, denitration, decarboxylative annulation
Graphical Abstract
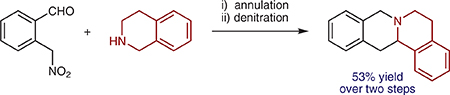
New methods for the C–H bond functionalization of amines and their derivatives continue to be developed at a rapid pace.1,2 However, few approaches have emerged that are compatible with unprotected secondary amines while at the same time enabling α-C–H bond functionalization with concurrent C−N bond formation.1m,o Particularly attractive in this regard are redox-annulations of cyclic amines, which allow for the rapid formation of polycyclic amines from simple starting materials (Scheme 1). Water is the only byproduct in these reactions. Examples of this type of transformation include condensations of amines with ortho-aminobenzaldehydes to provide aminals (Scheme 1a, X = NR),3 and related, carboxylic-acid-catalyzed transformations involving α-C–O and α-C–S bond formation.4 Redox-annulations that achieve α-C–C bond formation with orthotolualdehyde derivatives require the presence of at least one electron-withdrawing group on the ortho-methyl group.5 In addition, activation of an ortho-methyl group has been achieved with heteroaryl substrates (Scheme 1b)6 and highly electron-deficient o-tolualdehydes (Scheme 1c).7–10 Here, we report the first redox-annulations of amines with ortho-(nitromethyl)benzaldehydes (Scheme 1d). In these reactions, the nitro group acts as a traceless activator as it can be removed in a subsequent step. The overall strategy represents an attractive new pathway to members and analogues of the tetrahydroprotoberberine family of natural products.11
Scheme 1.

Examples of amine redox-annulations and present work
ortho-(Nitromethyl)benzaldehyde (1a)12 and 1,2,3,4-tetrahydroisoquinoline (THIQ) were selected as the model substrates in the initial evaluation of the proposed redox-annulation. Key optimization experiments are summarized in Table 1. While conditions used in other redox-annulations (reflux in toluene with benzoic acid as a promoter) provided the target product 2a in substantial amounts, improved results were obtained under microwave conditions. The maximum yield of 76% was achieved in a reaction that was performed in dichloroethane solvent at 150 °C for 5 min (entry 4). The reactions exhibited low but variable diastereoselectivities. We suspected that the two diastereomers of 2a may interconvert under the reaction conditions by means of a retro-nitro-Mannich/nitro-Mannich sequence with little thermodynamic preference for either diastereomer. Indeed, while accompanied by some decomposition, exposure of diastereomerically pure 2a to the reaction conditions led to the recovery of 2a as a nearly 1:1 mixture of diastereomers (Scheme 2).
Table 1.
Reaction Developmenta
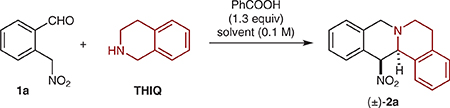 | ||||||
|---|---|---|---|---|---|---|
| Entry | THIQ (equiv) | Solvent | T (°C) | Time (min) | Yield (%) | dr |
| 1 | 1.3 | PhMe | reflux | 60 | 56 | 1:1 |
| 2 | 1.3 | DCE | reflux | 60 | 58 | 1.3:1 |
| 3b | 1.3 | PhMe | 150 | 5 | 61 | 1.1:1 |
| 4b | 1.3 | DCE | 150 | 5 | 76 | 1:1 |
| 5b | 2.0 | DCE | 150 | 5 | 61 | 1.1:1 |
| 6b | 1.3 | DCE | 100 | 5 | 71 | 1.4:1 |
| 7b | 1.3 | DCE | 100 | 15 | 75 | 1.2:1 |
Reactions were performed on a 0.25 mmol scale. All yields correspond to isolated yields. The dr was determined by 1H NMR analysis after purification.
Performed under microwave irradiation.
Scheme 2.

Equilibration experiment
We then turned our attention to the denitration step (Table 2). Following some optimization, conditions similar to those developed by Carreira and co-workers were found to be efficient in removing the nitro group,13 providing product 3a in up to 70% yield (entry 6).
Table 2.
Optimization of the Denitration Stepa
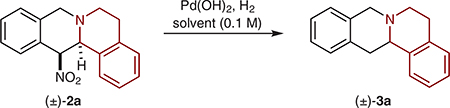 | ||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | H2 (atm) | Additive (equiv) | T (°C) | Time (h) | Yield (%) |
| 1 | EtOH | 10.2 | – | 85 | 4.5 h | 57 |
| 2 | EtOH | 10.2 | – | rt | 4.5 h | trace |
| 3b | EtOH | 1 | – | 85 | 4.5 h | trace |
| 4b | EtOH | 10.2 | – | 85 | 24 h | 54 |
| 5 | PhMe | 10.2 | – | 85 | 4.5 h | 54 |
| 6 | PhMe | 10.2 | AcOH (1.0) | 85 | 4.5 h | 70 |
| 7 | PhMe | 10.2 | AcOH (2.0) | 85 | 4.5 h | 26 |
Reactions were performed on a 0.25 mmol scale. All yields correspond to isolated yields.
Reaction was performed on a 0.15 mmol scale.
The annulation/denitration sequence was applied to a number of substituted tetrahydroisoquinolines (Scheme 3). Moderate to good yields were achieved in the individual steps with acceptable overall yields. Gratifyingly, 1-aryl tetrahydroisoquinolines with electronically diverse substituents also readily participated in redox-annulations to provide the corresponding sterically congested products as essentially single diastereomers in reasonable yields (Scheme 4). A related tetrahydro-β-carboline also participated in the reaction but provided the annulation product in significantly lower yield.
Scheme 3.

Evaluation of substituted tetrahydroisoquinolines
Scheme 4.

Formation of sterically congested tetrahydroprotoberberine analogues
Unfortunately, the products shown in Scheme 4 were not amenable to denitration under the reaction conditions employed above. However, removal of the nitro group was readily achieved with tributyltin hydride (Scheme 5).14
Scheme 5.

Denitration of a sterically congested annulation product
Despite significant experimentation, less activated amines such as pyrrolidine and piperidine did not participate in redox-annulations with ortho-(nitromethyl)benzaldehyde (1a). However, as has been shown in a number of related reactions,15,16 the corresponding decarboxylative reactions in which proline and pipecolic acid are used in place of pyrrolidine and piperidine provided annulation products in good yields (Scheme 6). Denitration under Carreira conditions was also successful.
Scheme 6.

Decarboxylative annulation/denitration
In conclusion, we have achieved the first traceless redox-annulations of amines using a substrate with an activating nitro group that can be subsequently removed. This strategy provides access to products that are not readily available by using conventional synthetic approaches.
Starting materials, reagents, and solvents were purchased from commercial sources and used as received unless stated otherwise. 1,2,3,4-Tetrahydroisoquinoline was freshly distilled prior to use. L-Proline, L/D-pipecolic acid, 2,2′-(diazene-1,2-diyl)bis(2-methylpropanenitrile), and tributyltin hydride were used as received. HPLC grade 1,2-dichloroethane (DCE) was purchased from Sigma–Aldrich and was used without further purification. Purification of reaction products was carried out by flash column chromatography using Sorbent Technologies Standard Grade silica gel (60 Å, 230–400 mesh). Analytical thin-layer chromatography was performed on EM Reagent 0.25 mm silica gel 60 F254 plates. Visualization was accomplished with UV light and Dragendorff–Munier stains, followed by heating. 1H NMR spectra were recorded with a Bruker 400 MHz or Bruker 600 MHz instrument and chemical shifts are reported in ppm using the solvent as an internal standard (CDCl3 at 7.26 ppm). Data are reported as app = apparent, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, comp = complex, br = broad; coupling constant(s) in Hz. Proton-decoupled carbon nuclear magnetic resonance spectra (13C NMR) spectra were recorded with a Bruker 400 MHz or Bruker 600 MHz instrument and chemical shifts are reported in ppm using the solvent as an internal standard (CDCl3 at 77.16 ppm). Diastereomeric ratios of the products were determined by 1H NMR analysis of the purified products. Accurate mass data (ESI) was obtained with Agilent 1260 Infinity II LC/MSD using MassWorks 5.0 from CERNO bioscience.17 Reactions under microwave irradiation were conducted with a Biotage Initiator+, SW version: 4.1.4 build 11991.
1-Phenyl-1,2,3,4-tetrahydroisoquinoline,18a 1-(4-fluorophenyl)-1,2,3,4-tetrahydroisoquinoline,18b 1-(4-chlorophenyl)-1,2,3,4-tetrahydroisoquinoline,18b 1-(4-bromophenyl)-1,2,3,4-tetrahydroisoquinoline,18c 1-(4-(trifluoromethyl)phenyl)-1,2,3,4-tetrahydroisoquinoline,18d 1-(4-methoxyphenyl)-1,2,3,4-tetrahydroisoquinoline,18b 1-(p-tolyl)-1,2,3,4-tetrahydroisoquinoline,18b 1-(m-tolyl)-1,2,3,4-tetrahydroisoquinoline,18e 1-(4-bromophenyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole,18f 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline,18g 5,6,7,8-tetrahydro-[1,3]dioxolo[4,5-g]isoquinoline,18h 5-methyl-1,2,3,4-tetrahydroisoquinoline,18i and 2-(nitromethyl)benzaldehyde18j were prepared according to reported procedures and their published characterization data matched our own in all respects.
13-Nitro-5,8,13,13a-tetrahydro-6H-isoquinolino[3,2-a]isoquinoline (2a)
2-(Nitromethyl)benzaldehyde (1a) (41.3 mg, 0.25 mmol, 1 equiv), 1,2,3,4-tetrahydroisoquinoline (41.5 μL, 0.33 mmol, 1.3 equiv), and benzoic acid (40.3 mg, 0.33 mmol, 1.3 equiv) were added to a microwave vial charged with a stir bar. Dichloroethane (2.5 mL) was added and the microwave vial was sealed. The vial was stirred until complete dissolution of the solids and then placed in the microwave, followed by heating for 5 minutes at 150 °C with the instrument set to low absorption. The reaction mixture was neutralized with sat. NaHCO3 (15 mL) and extracted with EtOAc (3 × 15 mL). The combined organic layers were dried over Na2SO4. The solvent was removed under reduced pressure and the crude residue was purified by silica gel chromatography using hexanes containing EtOAc (0–15%), yielding 2a as a mixture of diastereomers with a dr of 1:1.
Yield: 76% (53.3 mg); brown oil; Rf = 0.16 (hexanes/EtOAc 90:10 v/v).1H NMR (600 MHz, CDCl3): δ = 7.47 (dd, J = 7.7, 1.3 Hz, 0.5 H), 7.43–7.35 (comp, 1 H), 7.34–7.10 (comp, 6 H), 7.04–6.96 (m, 0.5 H), 6.17 (d, J = 3.3 Hz, 0.5 H), 5.90 (d, J = 8.6 Hz, 0.5 H), 4.76 (d, J = 8.6 Hz, 0.5 H), 4.38 (dd, J = 15.8, 1.3 Hz, 0.5 H), 4.26 (d, J = 15.3 Hz, 0.5 H), 4.20 (d, J = 3.3 Hz, 0.5 H), 3.96 (d, J = 15.8 Hz, 0.5 H), 3.79 (d, J = 15.3 Hz, 0.5 H), 3.33–3.19 (comp, 1 H), 3.08–2.96 (comp, 2 H), 2.92–2.85 (m, 0.5 H), 2.77–2.69 (m, 0.5 H).
13C NMR (151 MHz, CDCl3): δ = 136.4, 136.4, 134.7, 134.7, 134.1, 130.0, 129.6, 129.6, 129.5, 129.3, 128.6, 127.8, 127.7, 127.4, 127.3, 127.2, 127.0, 127.0, 126.9, 126.6, 126.3, 126.0, 125.7, 90.1, 87.0, 63.3, 62.1, 57.8, 56.7, 50.8, 48.0, 29.3, 29.2.
HRMS (ESI): m/z [M + H]+ calcd for C17H17N2O2: 281.1285; found: 281.1655. Spectral Accuracy: 98.8%.
General Procedure A
2-(Nitromethyl)benzaldehyde (1a) (82.6 mg, 0.5 mmol, 1 equiv), amine (0.65 mmol, 1.3 equiv), and benzoic acid (79.4 mg, 0.65 mmol, 1.3 equiv) were added to a microwave vial charged with a stir bar. Dichloroethane (5.0 mL) was added and the microwave vial was sealed. The vial was stirred until complete dissolution of the solids and placed in the microwave, followed by heating for 5 minutes at 150 °C with the instrument set to low absorption. The reaction mixture was neutralized with sat. NaHCO3 (20 mL) and extracted with EtOAc (3 × 20 mL). The combined organic layers were dried over Na2SO4. The solvent was removed under reduced pressure and the crude residue was purified by silica gel chromatography. The product was used directly in the next step.
General Procedure B
The annulation product obtained according to General Procedure A was added to a reaction vial charged with acetic acid (1.0 equiv) and a stir bar. Toluene (5.0 mL) was added followed by 20% wt. Pd(OH)2/C (66.7 mg). The reaction vial was placed in a bomb and back filled with H2 (5×). H2 was added to the bomb until the internal pressure reached 150 PSI. The reaction mixture was heated at 85 °C for 4.5 hours. The reaction mixture was then allowed to cool to r.t., followed by removal of the solvent under reduced pressure. The crude mixture was purified by silica gel chromatography followed by treatment with sat. NaHCO3 (15 mL) and extraction with EtOAc (3 × 15 mL). The combined organic layers were dried over Na2SO4. The solvent was removed under reduced pressure yielding the final product.
General Procedure C
2-(Nitromethyl)benzaldehyde (1a) (41.3 mg, 0.25 mmol, 1 equiv), amine (0.50 mmol, 2.0 equiv), and benzoic acid (40.3 mg, 0.33 mmol, 1.3 equiv) were added to a microwave vial charged with a stir bar. Dichloroethane (2.5 mL) was added and the microwave vial was sealed. The vial was stirred until complete dissolution of the solids and placed in the microwave, followed by heating for 15 minutes at 115 °C with the microwave set to low absorption. The reaction mixture was neutralized with sat. NaHCO3 (15 mL) and extracted with EtOAc (3 × 15 mL). The combined organic layers were dried over Na2SO4. The solvent was removed under reduced pressure and the crude residue was purified by silica gel chromatography.
13-Nitro-13a-phenyl-5,8,13,13a-tetrahydro-6H-isoquinolino-[3,2-a]isoquinoline (2e)
By following General Procedure C, compound (±)-2e was obtained from aldehyde 1a (41.3 mg, 0.25 mmol, 1equiv) and 1-phenyl-1,2,3,4-tetrahydroisoquinoline (104.g mg, 0.5 mmol, 2.0 equiv). Hexanes containing EtOAc (0–10%) was used as the eluent for silica gel chromatography.
Yield: 70% (62.4 mg) and a > 20:1 diastereomeric ratio; white solid; Rf = 0.13 (hexanes/EtOAc 95:5 v/v).
1H NMR (600 MHz, CDCl3): δ = 7.58 (dd, J = 7.6, 1.5 Hz, 1 H), 7.39–7.34 (comp, 2 H), 7.25–7.16 (comp, 3 H), 7.15–7.09 (comp, 4 H), 7.03 (dd, J = 8.0, 1.2 Hz, 1 H), 6.83–6.78 (comp, 2 H), 6.59 (s, 1 H), 3.93 (d, J = 16.3 Hz, 1 H), 3.39–3.26 (comp, 3 H), 3.07–3.00 (m, 1 H), 2.91–2.84 (m, 1 H).
13C NMR (151 MHz, CDCl3): δ = 139.6, 136.9, 136.8, 136.0, 129.6, 129.2, 128.9, 128.5, 128.4, 128.3, 127.8, 127.6, 127.5, 127.3, 126.8, 126.2, 91.5, 65.8, 52.3, 45.6, 29.5.
HRMS (ESI): m/z [M + H]+ calcd for C23H21N2O2: 357.1598; found: 357.1589. Spectral Accuracy: 97.3%.
13a-(4-Fluorophenyl)-13-nitro-5,8,13,13a-tetrahydro-6H-isoquinolino[3,2-a]isoquinoline (2f)
By following General Procedure C, compound (±)-2f was obtained from aldehyde 1a (41.3 mg, 0.25 mmol, 1 equiv) and 1-(4-fluorophenyl)-1,2,3,4-tetrahydroisoquinoline (113.g mg, 0.5 mmol, 2.0 equiv). Hexanes containing EtOAc (0–10%) was used as the eluent for silica gel chromatography.
Yield: 64% (59.9 mg) and a > 20:1 diastereomeric ratio; off-white solid; Rf = 0.30 (hexanes/EtOAc 90:10 v/v).
1H NMR (600 MHz, CDCl3): δ = 7.56 (dd, J = 7.7, 1.4 Hz, 1 H), 7.38 (app td, J = 7.5, 1.5 Hz, 1 H), 7.34 (app td, J = 7.5, 1.4 Hz, 1 H), 7.23–7.11 (comp, 4 H), 7.00 (dd, J = 7.9, 1.3 Hz, 1 H), 6.82 (app t, J = 8.7 Hz, 2 H), 6.79–6.74 (comp, 2 H), 6.53 (s, 1 H), 3.94 (d, J = 16.3 Hz, 1 H), 3.37–3.27 (comp, 2 H), 3.21 (app td, J = 11.6, 3.3 Hz, 1 H), 3.03 (ddd, J = 11.8, 6.0, 1.9 Hz, 1 H), 2.85 (app dt, J = 15.6, 2.7 Hz, 1 H).
13C NMR (151 MHz, CDCl3): δ = 161.8 (d, JC–F = 247.8 Hz), 136.7, 136.0, 135.4 (d, JC–F = 3.2 Hz), 130.2 (d, JC–F = 7.8 Hz), 129.8, 129.0, 128.9, 128.4, 128.2, 127.6, 127.5, 126.9, 126.3, 114.7 (d, JC–F = 21.0 Hz), 91.5, 65.4, 52.2, 45.5, 29.5.
HRMS (ESI): m/z [M + H]+ calcd for C23H20FN2O2: 375.1503; found: 375.1379. Spectral Accuracy: 97.4%.
13a-(4-Chlorophenyl)-13-nitro-5,8,13,13a-tetrahydro-6H-isoquinolino[3,2-a]isoquinoline (2g)
By following General Procedure C, compound (±)-2g was obtained from aldehyde 1a (41.3 mg, 0.25 mmol, 1 equiv) and 1-(4-chlorophenyl)-1,2,3,4-tetrahydroisoquinoline (121.9 mg, 0.5 mmol, 2.0 equiv). Hexanes containing EtOAc (0–15%) was used as the eluent for silica gel chromatography.
Yield: 66% (64.5 mg) and > 20:1 diastereomeric ratio; off-white solid; Rf = 0.52 (hexanes/EtOAc 80:20 v/v).
1H NMR (600 MHz, CDCl3): δ = 7.56 (dd, J = 7.6, 1.4 Hz, 1 H), 7.39–7.33 (comp, 2 H), 7.29–7.11 (comp, 6 H), 7.06–6.96 (m, 1 H), 6.76–6.71 (comp, 2 H), 6.52 (s, 1 H), 3.95 (d, J = 16.3 Hz, 1 H), 3.38–3.27 (comp, 2 H), 3.22 (app td, J = 11.6, 3.2 Hz, 1 H), 3.05 (dd, J = 12.0, 5.8 Hz, 1 H), 2.85 (d, J = 15.5 Hz, 1 H).
13C NMR (151 MHz, CDCl3): δ = 138.1, 136.6, 136.4, 136.0, 133.6, 129.8, 129.8, 129.0, 128.8, 128.4, 128.2, 128.0, 127.6, 127.6, 126.8, 126.3, 91.3, 65.5, 52.2, 45.6, 29.4.
HRMS (ESI): m/z [M + H]+ calcd for C23H20ClN2O2: 391.1208; found: 391.1429. Spectral Accuracy: 97.2%.
13a-(4-Bromophenyl)-13-nitro-5,8,13,13a-tetrahydro-6H-isoquinolino[3,2-a]isoquinoline (2h)
By following General Procedure C, compound (±)-2h was obtained from aldehyde 1a (41.3 mg, 0.25 mmol, 1 equiv) and 1-(4-bromophenyl)-1,2,3,4-tetrahydroisoquinoline (144.1 mg, 0.5 mmol, 2.0 equiv). Hexanes containing EtOAc (0–10%) was used as the eluent for silica gel chromatography.
Yield: 71% (77.3 mg) and > 20:1 diastereomeric ratio; off-white solid; Rf = 0.27 (hexanes/EtOAc 90:10 v/v).
1H NMR (600 MHz, CDCl3): δ = 7.58 (dd, J = 7.7, 1.4 Hz, 1 H), 7.42–7.35 (comp, 2 H), 7.33–7.25 (comp, 2 H), 7.25–7.08 (comp, 4 H), 7.08–7.00 (m, 1 H), 6.72–6.67 (comp, 2 H), 6.54 (s, 1 H), 3.97 (d, J = 16.3 Hz, 1 H), 3.41–3.29 (comp, 2 H), 3.25 (app td, J = 11.6, 3.2 Hz, 1 H), 3.07 (ddd, J = 12.0, 6.0, 2.0 Hz, 1 H), 2.87 (app dt, J = 15.4, 2.6 Hz, 1 H).
13C NMR (151 MHz, CDCl3): δ = 138.6, 136.6, 136.3, 136.0, 131.0, 130.1, 129.8, 129.0, 128.8, 128.4, 128.2, 127.6, 127.6, 126.8, 126.3, 121.8, 91.2, 65.5, 52.2, 45.5, 29.4.
HRMS (ESI): m/z [M + H]+ calcd for C23H20BrN2O2: 435.0703; found: 435.0610. Spectral Accuracy: 98.1%.
13-Nitro-13a-(4-(trifluoromethyl)phenyl)-5,8,13,13a-tetrahydro-6H-isoquinolino[3,2-a]isoquinoline (2i)
By following General Procedure C, compound (±)-2i was obtained from aldehyde 1a (41.3 mg, 0.25 mmol, 1 equiv) and 1-(4-(trifluoromethyl)phenyl)-1,2,3,4-tetrahydroisoquinoline (138.6 mg, 0.5 mmol, 2.0 equiv). Hexanes containing EtOAc (0–10%) was used as the eluent for silica gel chromatography.
Yield: 69% (73.2 mg) and > 20:1 diastereomeric ratio; off-white solid; Rf = 0.30 (hexanes/EtOAc 90:10 v/v).
1H NMR (600 MHz, CDCl3): δ = 7.58 (dd, J = 7.6, 1.5 Hz, 1 H), 7.43–7.32 (comp, 4 H), 7.24–7.17 (comp, 2 H), 7.14 (app ddt, J = 6.5, 4.6, 2.1 Hz, 2 H), 6.99 (dd, J = 7.9, 1.2 Hz, 1 H), 6.94 (d, J = 8.3 Hz, 2 H), 6.57 (s, 1 H), 3.97 (d, J = 16.4 Hz, 1 H), 3.40–3.30 (comp, 2 H), 3.26 (app td, J = 11.5, 3.0 Hz, 1 H), 3.18–3.05 (m, 1 H), 2.89 (dd, J = 15.7, 2.9 Hz, 1 H).
13C NMR (151 MHz, CDCl3): δ = 143.7, 136.5, 136.0, 129.9, 129.7 (q, JC–F = 32.6 Hz), 129.2, 128.8, 128.7, 128.5, 128.2, 127.7, 126.9, 126.4, 124.8 (q, JC–F = 3.8 Hz), 123.9 (q, JC–F = 272.4 Hz), 91.1, 65.6, 52.2, 45.6, 29.4.
HRMS (ESI): m/z [M + H]+ calcd for C24H20F3N2O2: 425.1471; found: 425.1820. Spectral Accuracy: 97.5%.
13a-(4-Methoxyphenyl)-13-nitro-5,8,13,13a-tetrahydro-6H-isoquinolino[3,2-a]isoquinoline (2j)
By following General Procedure C, compound (±)-2j was obtained from aldehyde 1a (41.3 mg, 0.25 mmol, 1 equiv) and 1-(4-methoxyphenyl)-1,2,3,4-tetrahydroisoquinoline (119.7 mg, 0.5 mmol, 2.0 equiv). Hexanes containing EtOAc (0–10%) was used as the eluent for silica gel chromatography.
Yield: 40% (38.6 mg) and > 20:1 diastereomeric ratio; off-white solid; Rf = 0.19 (hexanes/EtOAc 90:10 v/v).
1H NMR (600 MHz, CDCl3): δ = 7.60–7.54 (m, 1 H), 7.39–7.32 (comp, 2 H), 7.21–7.09 (comp, 4 H), 7.07–6.97 (m, 1 H), 6.73–6.68 (comp, 2 H), 6.68–6.62 (comp, 2 H), 6.54 (s, 1 H), 3.91 (d, J = 16.1 Hz, 1 H), 3.71 (s, 3 H), 3.39–3.27 (comp, 2 H), 3.23 (app td, J = 11.6, 3.2 Hz, 1 H), 3.00 (ddd, J = 11.7, 5.9, 1.9 Hz, 1 H), 2.84 (app dt, J = 15.4, 2.6 Hz, 1 H).
13C NMR (151 MHz, CDCl3): δ = 158.7, 137.2, 136.9, 136.0, 131.6, 129.8, 129.6, 129.2, 128.9, 128.4, 128.3, 127.5, 127.3, 126.8, 126.1, 113.0, 91.7, 65.5, 55.2, 52.3, 45.5, 29.6.
HRMS (ESI): m/z [M + H]+ calcd for C24H23N2O3: 387.1703; found: 387.1899. Spectral Accuracy: 98.8%.
13-Nitro-13a-(p-tolyl)-5,8,13,13a-tetrahydro-6H-isoquinolino-[3,2-a]isoquinoline (2k)
By following General Procedure C, compound (±)-2k was obtained from aldehyde 1a (41.3 mg, 0.25 mmol, 1 equiv) and 1-(p-tolyl)-1,2,3,4-tetrahydroisoquinoline (111.7 mg, 0.5 mmol, 2.0 equiv). Hexanes containing EtOAc (0–10%) was used as the eluent for silica gel chromatography.
Yield: 61% (56.5 mg) and > 20:1 diastereomeric ratio; off-white solid; Rf = 0.32 (hexanes/EtOAc 90:10 v/v).
1H NMR (600 MHz, CDCl3): δ = 7.57 (d, J = 7.9, 1.2 Hz, 1 H), 7.40–7.31 (comp, 2 H), 7.24–7.15 (comp, 2 H), 7.12 (ddd, J = 9.5, 7.1, 1.9 Hz, 2 H), 7.03 (d, J = 7.9, 1.2 Hz, 1 H), 6.94 (d, J = 8.2 Hz, 2 H), 6.70–6.65 (comp, 2 H), 6.57 (s, 1 H), 3.91 (d, J = 16.2 Hz, 1 H), 3.39–3.24 (comp, 3 H), 3.05–2.99 (m, 1 H), 2.85 (dd, J = 15.3, 3.0 Hz, 1 H), 2.24 (s, 3 H).
13C NMR (151 MHz, CDCl3): δ = 137.3, 137.1, 136.9, 136.5, 136.0, 129.5, 129.3, 128.9, 128.5, 128.5, 128.4, 128.3, 127.4, 127.2, 126.8, 126.1, 91.62, 65.7, 52.3, 45.56, 29.6, 21.0.
HRMS (ESI): m/z [M + H]+ calcd for C24H23N2O2: 371.1759; found: 371.1935. Spectral Accuracy: 97.5%.
13-Nitro-13a-(m-tolyl)-5,8,13,13a-tetrahydro-6H-isoquinolino-[3,2-a]isoquinoline (2l)
By following General Procedure C, compound (±)-2l was obtained from aldehyde 1a (41.3 mg, 0.25 mmol, 1 equiv) and 1-(m-tolyl)-1,2,3,4-tetrahydroisoquinoline (111.7 mg, 0.5 mmol, 2.0 equiv). Hexanes containing EtOAc (0–10%) was used as the eluent for silica gel chromatography.
Yield: 60% (55.6 mg) and > 20:1 diastereomeric ratio; off-white solid; Rf = 0.28 (hexanes/EtOAc 90:10 v/v).
1H NMR (600 MHz, CDCl3): δ = 7.57 (dd, J = 7.6, 1.5 Hz, 1 H), 7.38–7.32 (comp, 2 H), 7.23–7.16 (comp, 2 H), 7.12 (app ddt, J = 6.4, 4.5, 2.2 Hz, 2 H), 7.07–6.96 (comp, 3 H), 6.66–6.54 (comp, 3 H), 3.92 (d, J = 16.2 Hz, 1 H), 3.39 (d, J = 16.2 Hz, 1 H), 3.35–3.27 (comp, 2 H), 3.09–3.01 (m, 1 H), 2.92–2.83 (m, 1 H), 2.15 (s, 3 H).
13C NMR (151 MHz, CDCl3): δ = 139.6, 137.3, 136.9, 136.8, 135.9, 129.4, 129.4, 129.2, 128.8, 128.5, 128.3, 128.3, 127.5, 127.3, 127.2, 126.7, 126.0, 125.3, 91.5, 65.7, 52.3, 45.5, 29.5, 21.8.
HRMS (ESI): m/z [M + H]+ calcd for C24H23N2O2: 371.1754; found: 371.2009. Spectral Accuracy: 98.2%.
13b-(4-Bromophenyl)-14-nitro-5,7,8,13,13b,14-hexahydroindolo-[2′,3′:3,4]pyrido[1,2-b]isoquinoline (2m)
2-(Nitromethyl)benzaldehyde (82.6 mg, 0.5 mmol, 1 equiv), 1-(4-bro-mophenyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole (327.2 mg, 1.0 mmol, 2.0 equiv), and benzoic acid (79.4 mg, 0.65 mmol, 1.3 equiv) were added to a microwave vial charged with a stir bar. Dichloroethane (5.0 mL) was added and the microwave vial was sealed. The vial was stirred and placed in the microwave, followed by heating for 15 minutes at 115 °C with the microwave set to low absorption. The reaction mixture was neutralized with sat. NaHCO3 (20 mL) and extracted with EtOAc (3 × 20 mL). The combined organic layers were dried over Na2SO4. The solvent was removed under reduced pressure and the crude residue purified by silica gel chromatography using hexanes containing EtOAc (0–15%) as the eluent, yielding 2m.
Yield: 24% (56.9 mg) and > 20:1 diastereomeric ratio; pale-green solid; Rf = 0.40 (hexanes/EtOAc 80:20 v/v).
1H NMR (600 MHz, CDCl3): δ = 7.83 (s, 1 H), 7.59 (d, J = 7.7 Hz, 1 H), 7.49–7.41 (comp, 2 H), 7.38 (app td, J = 7.5, 1.3 Hz, 1 H), 7.32–7.26 (comp, 4 H), 7.24–7.19 (comp, 2 H), 6.73 (d, J = 8.3 Hz, 2 H), 6.51 (s, 1 H), 4.08 (d, J = 16.3 Hz, 1 H), 3.48 (d, J = 16.3 Hz, 1 H), 3.22 (app td, J = 12.7, 11.9, 3.9 Hz, 1 H), 3.13 (app ddt, J = 16.4, 10.7, 4.4 Hz, 2 H), 2.98–2.91 (m, 1 H).
13C NMR (151 MHz, CDCl3): δ = 137.0, 131.5, 130.9, 130.0, 129.9, 128.6, 128.1, 127.8, 127.0, 126.4, 122.9, 119.9, 118.9, 113.4, 111.5, 90.0, 63.8, 51.5, 46.7, 21.3.
HRMS (ESI): m/z [M + H]+ calcd for C25H21BrN3O2: 474.0812; found: 474.0616. Spectral Accuracy: 97.7%.
5,8,13,13a-Tetrahydro-6H-isoquinolino[3,2-a]isoquinoline (3a)
By following General Procedures A and B, compound (±)-3a was obtained from aldehyde 1a (82.6 mg, 0.5 mmol, 1 equiv) and 1,2,3,4-tetrahydroisoquinoline (81.7 μL, 0.65 mmol, 1.3 equiv). Hexanes containing EtOAc (0–20%) was used as the eluent for silica gel chromatography. Characterization data for 3a match literature reports in all respects.19a,19b
Yield: 53% (62.4 mg) over two steps; yellow solid; Rf = 0.39 (hexanes/EtOAc 70:30 v/v).
1H NMR (400 MHz, CDCl3): δ = 7.30 (d, J = 7.0 Hz, 1 H), 7.26–7.14 (comp, 6 H), 7.10 (dd, J = 6.5, 2.7 Hz, 1 H), 4.06 (d, J = 14.9 Hz, 1 H), 3.85–3.67 (comp, 2 H), 3.48–3.36 (m, 1 H), 3.32–3.15 (comp, 2 H), 2.96 (ddd, J = 16.3, 11.3, 1.8 Hz, 1 H), 2.85–2.75 (m, 1 H), 2.72–2.62 (m, 1 H).
13C NMR (101 MHz, CDCl3): δ = 136.0, 134.6, 134.6, 134.5, 129.0, 128.9, 126.4, 126.3, 126.3, 126.2, 126.0, 125.6, 60.0, 58.7, 51.3, 36.8, 29.6.
HRMS (ESI): m/z [M + H]+ calcd for C17H18N: 236.1434; found: 236.1526. Spectral Accuracy: 98.6%.
4-Methyl-5,8,13,13a-tetrahydro-6H-isoquinolino[3,2-a]isoquinoline (3b)
By following General Procedures A and B, compound (±)-3b was obtained from aldehyde 1a (82.6 mg, 0.5 mmol, 1 equiv) and 5-methyl1,2,3,4-tetrahydroisoquinoline (95.7 mg, 0.65 mmol, 1.3 equiv). Hexanes containing EtOAc (0–10%) was used as the eluent for silica gel chromatography.
Yield: 47% (58.6 mg) over two steps; white solid; Rf = 0.25 (hexanes/EtOAc 90:10 v/v).
1H NMR (600 MHz, CDCl3): δ = 7.28–7.17 (comp, 5 H), 7.13–7.10 (comp, 2 H), 4.09 (d, J = 14.9 Hz, 1 H), 3.87–3.69 (comp, 2 H), 3.42 (dd, J = 16.3, 4.1 Hz, 1 H), 3.27 (ddd, J = 11.5, 5.8, 2.2 Hz, 1 H), 3.10–2.88 (comp, 2 H), 2.77 (app dt, J = 16.5, 2.9 Hz, 1 H), 2.67 (app td, J = 11.4, 3.8 Hz, 1 H), 2.31 (s, 3 H).
13C NMR (151 MHz, CDCl3): δ = 138.0, 136.3, 134.6, 134.4, 133.2, 128.8, 127.6, 126.4, 126.2, 125.9, 125.9, 123.3, 60.1, 58.8, 51.2, 36.9, 27.1, 19.4.
HRMS (ESI): m/z [M + H]+ calcd for C18H20N: 250.1590; found: 250.1705. Spectral Accuracy: 99.0%.
2,3-Dimethoxy-5,8,13,13a-tetrahydro-6H-isoquinolino[3,2-a]isoquinoline (3c)
By following General Procedures A and B, compound (±)-3c was obtained from aldehyde 1a (82.6 mg, 0.5 mmol, 1 equiv) and 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline (125.6 mg, 0.65 mmol, 1.3 equiv). Hexanes containing EtOAc (0–40%) was used as the eluent for silica gel chromatography. Characterization data for 3c match a literature report in all respects.19c
Yield: 34% (50.2 mg) over two steps; white solid; Rf = 0.14 (hexanes/EtOAc 75:25 v/v).
1H NMR (600 MHz, CDCl3): δ = 7.20–7.12 (comp, 3 H), 7.11–7.06 (m, 1 H), 6.75 (s, 1 H), 6.62 (s, 1 H), 4.04 (d, J = 14.9 Hz, 1 H), 3.90 (s, 3 H), 3.87 (s, 3 H), 3.78–3.73 (m, 1 H), 3.70–3.62 (m, 1 H), 3.34 (dd, J = 16.2, 3.9 Hz, 1 H), 3.20–3.12 (comp, 2 H), 2.98–2.87 (m, 1 H), 2.73–2.60 (comp, 2 H).
13C NMR (151 MHz, CDCl3): δ = 147.6, 147.6, 134.4, 129.7, 128.8, 126.7, 126.4, 126.2, 126.0, 111.4, 108.6, 59.6, 58.6, 56.2, 55.9, 51.4, 36.8, 29.0.
HRMS (ESI): m/z [M + H]+ calcd for C19H22NO2: 296.1645; found: 296.1739. Spectral Accuracy: 98.6%.
5,8,13,13a-Tetrahydro-6H-[1,3]dioxolo[4,5-g]isoquinolino[3,2-a]isoquinoline (3d)
By following General Procedures A and B, compound (±)-3d was obtained from aldehyde 1a (82.6 mg, 0.5 mmol, 1 equiv) and 5,6,7,8-tetrahydro[1,3]dioxolo[4,5-g]isoquinoline (115.2 mg, 0.65 mmol, 1.3 equiv). Hexanes containing EtOAc (0–20%) was used as the eluent for silica gel chromatography. Characterization data for 3d match a literature report in all respects.19b
Yield: 38% (53.1 mg) over two steps; white solid; Rf = 0.28 (hexanes/EtOAc 75:25 v/v).
1H NMR (600 MHz, CDCl3): δ = 7.21–7.13 (comp, 3 H), 7.11–7.05 (m, 1 H), 6.76 (s, 1 H), 6.60 (s, 1 H), 5.92–5.91 (comp, 2 H), 4.03 (d, J = 14.9 Hz, 1 H), 3.75 (d, J = 14.9 Hz, 1 H), 3.62 (dd, J = 11.2, 4.0 Hz, 1 H), 3.29 (dd, J = 16.2, 4.0 Hz, 1 H), 3.18–3.09 (comp, 2 H), 2.95–2.87 (m, 1 H), 2.71–2.58 (comp, 2 H).
13C NMR (151 MHz, CDCl3): δ = 146.3, 146.1, 134.4, 134.3, 130.8, 128.8, 127.8, 126.4, 126.2, 126.0, 108.5, 105.6, 100.9, 60.0, 58.6, 51.4, 36.9, 29.6.
HRMS (ESI): m/z [M + H]+ calcd for C18H18NO2: 280.1332; found: 280.1565. Spectral Accuracy: 99.1%.
13a-Phenyl-5,8,13,13a-tetrahydro-6H-isoquinolino[3,2-a]isoquinoline (3e)
Compound (±)-2e (71.3 mg, 0.20 mmol. 1.0 equiv), and AIBN (9.9 mg, 0.06 mmol, 0.3 equiv) was added to benzene (2.0 mL) and stirred until complete dissolution. Tributyltin hydride (80.9 μL, 0.3 mmol, 1.5 equiv) was then added and the reaction mixture was heated under reflux for 1 hour. The reaction mixture was extracted with 1 M HCl (3 × 10 mL) and the combined aqueous layers were basified with 1 M NaOH. The aqueous layer was back extracted with EtOAc (3 × 15 mL) and the combined organic layers were dried over Na2SO4. The solvent was removed under reduced pressure and the crude residue was purified by silica gel chromatography using hexanes containing EtOAc (0–5%) as the eluent yielding 3e.
Yield: 72% (44.8 mg); white solid; Rf = 0.33 (hexanes/EtOAc 95:5 v/v).
1H NMR (600 MHz, CDCl3): δ = 7.25–7.13 (comp, 10 H), 7.06 (app td, J = 7.4, 1.7 Hz, 1 H), 6.98 (d, J = 7.5 Hz, 1 H), 6.78 (dd, J = 7.9, 1.3 Hz, 1 H), 3.71–3.55 (comp, 3 H), 3.44 (d, J = 17.5 Hz, 1 H), 3.28–3.23 (m, 1 H), 3.17 (ddd, J = 11.8, 8.3, 4.7 Hz, 1 H), 3.09 (app dt, J = 11.9, 5.3 Hz, 1 H), 3.02 (app dt, J = 15.8, 4.8 Hz, 1 H).
13C NMR (151 MHz, CDCl3): δ = 134.5, 134.2, 133.4, 129.8, 128.9, 128.9, 128.4, 128.2, 127.9, 127.8, 126.9, 126.5, 126.4, 126.0, 126.0, 126.0, 62.5, 53.5, 46.5, 36.2, 29.9.
HRMS (ESI): m/z [M + H]+ calcd for C23H22N: 312.1747; found: 312.1787. Spectral Accuracy: 97.4%.
1,2,3,5,10,10a-Hexahydropyrrolo[1,2-b]isoquinoline (4)
By following General Procedures A and B, compound (±)-4 was obtained from aldehyde 1a (82.6 mg, 0.5 mmol, 1 equiv) and L-proline (74.8 mg, 0.65 mmol, 1.3 equiv). Dichloromethane containing MeOH (0–10%) was used as the eluent for silica gel chromatography. Characterization data for 4 match a literature report in all respects.19e
Yield: 52% (45.0 mg) over two steps; colorless oil; Rf = 0.13 (CH2Cl2/ MeOH 96:4 v/v).
1H NMR (600 MHz, CDCl3): δ = 7.13–7.10 (comp, 3 H), 7.11–7.05 (m, 1 H), 4.16 (d, J = 14.6 Hz, 1 H), 3.47 (d, J = 14.6 Hz, 1 H), 3.30 (app td, J = 8.7, 2.5 Hz, 1 H), 3.01 (dd, J = 15.9, 3.9 Hz, 1 H), 2.78–2.71 (m, 1 H), 2.42–2.36 (m, 1 H), 2.31 (app q, J = 8.8 Hz, 1 H), 2.12 (dddd, J = 12.3, 9.8, 6.8, 4.2 Hz, 1 H), 1.95 (app ddtd, J = 12.7, 11.2, 8.6, 4.2 Hz, 1 H), 1.89–1.79 (m, 1 H), 1.58 (dddd, J = 12.3, 11.3, 9.8, 6.8 Hz, 1 H).
13C NMR (151 MHz, CDCl3): δ = 135.0, 134.9, 129.1, 126.7, 126.3, 125.8, 60.8, 55.9, 54.8, 36.0, 31.1, 21.7.
HRMS (ESI): m/z [M + H]+ calcd for C12H16N: 174.1277; found: 174.1276. Spectral Accuracy: 99.4%.
1,3,4,6,11,11a-Hexahydro-2H-pyrido[1,2-b]isoquinoline (5)
By following General Procedures A and B, compound (±)-5 was obtained from aldehyde 1a (82.6 mg, 0.5 mmol, 1 equiv) and L/D-pipecolic acid (84.0 mg, 0.65 mmol, 1.3 equiv). Dichloromethane containing MeOH (0–4%) was used as the eluent for silica gel chromatography. Characterization data for 5 match a literature report in all respects.19d
Yield: 47% (44.0 mg) over two steps; white solid; Rf = 0.18 in EtOAc.
1H NMR (600 MHz, CDCl3): δ = 7.12–7.08 (comp, 2 H), 7.06–7.03 (m, 1 H), 7.02–6.98 (m, 1 H), 3.86 (d, J = 15.1 Hz, 1 H), 3.39 (d, J = 15.1 Hz, 1 H), 3.12–3.05 (m, 1 H), 2.90–2.62 (comp, 2 H), 2.25 (app tt, J = 10.2, 4.2 Hz, 1 H), 2.12 (app td, J = 11.4, 4.2 Hz, 1 H), 1.88–1.76 (comp, 2 H), 1.76–1.67 (comp, 2 H), 1.42–1.32 (comp, 2 H).
13C NMR (151 MHz, CDCl3): δ = 134.3, 134.0, 128.1, 126.2, 126.0, 125.6, 58.4, 58.4, 56.2, 36.8, 33.7, 25.9, 24.3.
HRMS (ESI): m/z [M + H]+ calcd for C13H18N: 188.1434; found: 188.1383. Spectral Accuracy: 99.2%.
Supplementary Material
Acknowledgment
We thank Dr. Ion Ghiviriga (University of Florida) for assistance with NMR experiments.
Funding Information
Financial support from the NIH–NIGMS (Grant R01GM101389) is gratefully acknowledged. We further acknowledge the National Science Foundation (grant # 1828064 to K.A.A.) and the University of Florida for funding the purchase of the X-ray equipment.
Footnotes
Supporting Information
Supporting information for this article is available online at https://doi.org/10.1055/s-0040-1706004.
References
- (1).(a) Selected recent reviews on amine C–H functionalization, including redox-neutral approaches: Campos KR Chem. Soc. Rev 2007, 36, 1069. [DOI] [PubMed] [Google Scholar]; (b) Jazzar R; Hitce J; Renaudat A; Sofack-Kreutzer J; Baudoin O Chem. Eur. J 2010, 16, 2654. [DOI] [PubMed] [Google Scholar]; (c) Yeung CS; Dong VM Chem. Rev 2011, 111, 1215. [DOI] [PubMed] [Google Scholar]; (d) Mitchell EA; Peschiulli A; Lefevre N; Meerpoel L; Maes BU W. Chem. Eur. J 2012, 18, 10092. [DOI] [PubMed] [Google Scholar]; (e) Jones KM; Klussmann M Synlett 2012, 23, 159. [Google Scholar]; (f) Peng B; Maulide N Chem. Eur. J 2013, 19, 13274. [DOI] [PubMed] [Google Scholar]; (g) Girard SA; Knauber T; Li C-J Angew. Chem. Int. Ed 2014, 53, 74. [DOI] [PubMed] [Google Scholar]; (h) Haibach MC; Seidel D Angew. Chem. Int. Ed 2014, 53, 5010. [DOI] [PubMed] [Google Scholar]; (i) Wang L; Xiao J Adv. Synth. Catal 2014, 356, 1137. [Google Scholar]; (j) Vo C-VT; Bode JW J. Org. Chem 2014, 79, 2809. [DOI] [PubMed] [Google Scholar]; (k) Seidel D Org. Chem. Front 2014, 1, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Qin Y; Lv J; Luo S Tetrahedron Lett 2014, 55, 551. [Google Scholar]; (m) Seidel D Acc. Chem. Res 2015, 48, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Beatty JW; Stephenson CR J. Acc. Chem. Res 2015, 48, 1474. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Mahato S; Jana CK Chem. Rec 2016, 16, 1477. [DOI] [PubMed] [Google Scholar]; (p) Qin Y; Zhu L; Luo S Chem. Rev 2017, 117, 9433. [DOI] [PubMed] [Google Scholar]; q) Cheng M-X; Yang S-D Synlett 2017, 28, 159. [Google Scholar]; (r) Chu JCK; Rovis T Angew. Chem. Int. Ed 2018, 57, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]; (s) Gonnard L; Guérinot A; Cossy J Tetrahedron 2019, 75, 145. [Google Scholar]; (t) Liu S; Zhao Z; Wang Y Chem. Eur. J 2019, 25, 2423. [DOI] [PubMed] [Google Scholar]; (u) Antermite D; Bull JA Synthesis 2019, 51, 3171. [Google Scholar]; (v) Trowbridge A; Walton SM; Gaunt MJ Chem. Rev 2020, 120, 2613. [DOI] [PubMed] [Google Scholar]
- (2).(a) Recent examples of mechanistically diverse amine C–H bond functionalization reactions: Zhao Z; Luo Y; Liu S; Zhang L; Feng L; Wang Y Angew. Chem. Int. Ed 2018, 57, 3792. [DOI] [PubMed] [Google Scholar]; (b) Wang F; Rafiee M; Stahl SS Angew. Chem. Int. Ed 2018, 57, 6686. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Greßies S; Klauck FJR; Kim JH; Daniliuc CG; Glorius F Angew. Chem. Int. Ed 2018, 57, 9950. [DOI] [PubMed] [Google Scholar]; (d) Griffiths RJ; Kong WC; Richards SA; Burley GA; Willis MC; Talbot EP A. Chem. Sci 2018, 9, 2295. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Idiris FIM; Majeste CE; Craven GB; Jones CR Chem. Sci 2018, 9, 2873. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Li S-S; Lv X; Ren D; Shao C-L; Liu Q; Xiao J Chem. Sci 2018, 9, 8253. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Maier AFG; Tussing S; Zhu H; Wicker G; Tzvetkova P; Flörke U; Daniliuc CG; Grimme S; Paradies J Chem. Eur. J 2018, 24, 16287. [DOI] [PubMed] [Google Scholar]; (h) Mori K; Isogai R; Kamei Y; Yamanaka M; Akiyama TJ Am. Chem. Soc 2018, 140, 6203. [DOI] [PubMed] [Google Scholar]; (i) Shang M; Chan JZ; Cao M; Chang Y; Wang Q; Cook B; Torker S; Wasa MJ Am. Chem. Soc 2018, 140, 10593. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Lennox AJJ; Goes SL; Webster MP; Koolman HF; Djuric SW; Stahl SS J. Am. Chem. Soc 2018, 140, 11227. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Zhang J; Park S; Chang SJ Am. Chem. Soc 2018, 140, 13209. [DOI] [PubMed] [Google Scholar]; (l) Nauth AM; Schechtel E; Dören R; Tremel W; Opatz TJ Am. Chem. Soc 2018, 140, 14169. [DOI] [PubMed] [Google Scholar]; (m) Jiang H-J; Zhong X-M; Yu J; Zhang Y; Zhang X; Wu Y-D; Gong L-Z Angew. Chem. Int. Ed 2019, 58, 1803. [DOI] [PubMed] [Google Scholar]; (n) Ashley MA; Yamauchi C; Chu JCK; Otsuka S; Yorimitsu H; Rovis T Angew. Chem. Int. Ed 2019, 58, 4002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Guin S; Dolui P; Zhang X; Paul S; Singh VK; Pradhan S; Chandrashekar HB; Anjana SS; Paton RS; Maiti D Angew. Chem. Int. Ed 2019, 58, 5633. [DOI] [PubMed] [Google Scholar]; (p) Whitehurst WG; Blackwell JH; Hermann GN; Gaunt MJ Angew. Chem. Int. Ed 2019, 58, 9054. [DOI] [PubMed] [Google Scholar]; (q) Ma Y; Yao X; Zhang L; Ni P; Cheng R; Ye J Angew. Chem. Int. Ed 2019, 58, 16548. [DOI] [PubMed] [Google Scholar]; (r) Grainger R; Heightman TD; Ley SV; Lima F; Johnson CN Chem. Sci 2019, 10, 2264. [DOI] [PMC free article] [PubMed] [Google Scholar]; (s) Vasu D; Fuentes de Arriba AL; Leitch JA; de Gombert A; Dixon DJ Chem. Sci 2019, 10, 3401. [DOI] [PMC free article] [PubMed] [Google Scholar]; (t) Asako S; Ishihara S; Hirata K; Takai KJ Am. Chem. Soc 2019, 141, 9832. [DOI] [PubMed] [Google Scholar]; (u) Lin W; Zhang K-F; Baudoin O Nat. Catal 2019, 2, 882. [DOI] [PMC free article] [PubMed] [Google Scholar]; (v) Chan JZ; Chang Y; Wasa M Org. Lett 2019, 21, 984. [DOI] [PMC free article] [PubMed] [Google Scholar]; (w) Zhou L; Shen Y-B; An X-D; Li X-J; Li S-S; Liu Q; Xiao J Org. Lett 2019, 21, 8543. [DOI] [PubMed] [Google Scholar]; (x) Kataoka M; Otawa Y; Ido N; Mori K Org. Lett 2019, 21, 9334. [DOI] [PubMed] [Google Scholar]; (y) Lee M; Adams A; Cox PB; Sanford MS Synlett 2019, 30, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]; (z) Kapoor M; Chand-Thakuri P; Maxwell JM; Liu D; Zhou H; Young MC Synlett 2019, 30, 519. [Google Scholar]; (aa) Ohmatsu K; Suzuki R; Furukawa Y; Sato M; Ooi T ACS Catal 2020, 10, 2627. [Google Scholar]; (ab) Roque JB; Kuroda Y; Jurczyk J; Xu L-P; Ham JS; Göttemann LT; Roberts CA; Adpressa D; Saurí J; Joyce LA; Musaev DG; Yeung CS; Sarpong R ACS Catal 2020, 10, 2929. [DOI] [PMC free article] [PubMed] [Google Scholar]; (ac) Rand AW; Yin H; Xu L; Giacoboni J; Martin-Montero R; Romano C; Montgomery J; Martin R ACS Catal 2020, 10, 4671. [Google Scholar]; (ad) Liu W; Babl T; Röther A; Reiser O; Davies HM L. Chem. Eur. J 2020, 26, 4236. [DOI] [PMC free article] [PubMed] [Google Scholar]; (ae) Verma P; Richter JM; Chekshin N; Qiao JX; Yu J-QMM; Koronkiewicz B; Chen S; Houk KN; Mayer JM; Ellman JA J. Am. Chem. Soc 2020, 142, 8194. [DOI] [PMC free article] [PubMed] [Google Scholar]; (ag) Feng K; Quevedo RE; Kohrt JT; Oderinde MS; Reilly U; White MC Nature 2020, 580, 621. [DOI] [PMC free article] [PubMed] [Google Scholar]; (ah) Sarver PJ; Bacauanu V; Schultz DM; DiRocco DA; Lam Y.-h.; Sherer EC; MacMillan DWC. Nat. Chem 2020, 12, 459. [DOI] [PubMed] [Google Scholar]; (ai) McManus JB; Onuska NPR; Jeffreys MS; Goodwin NC; Nicewicz DA Org. Lett 2020, 22, 679. [DOI] [PubMed] [Google Scholar]; (aj) Oeschger R; Su B; Yu I; Ehinger C; Romero E; He S; Hartwig J Science 2020, 368, 736. [DOI] [PMC free article] [PubMed] [Google Scholar]; (ak) Short MA; Blackburn JM; Roizen JL Synlett 2020, 31, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3) (a).Zhang C; De C K; Mal R; Seidel DJ Am. Chem. Soc 2008, 130, 416. [DOI] [PubMed] [Google Scholar]; (b) Zheng L; Yang F; Dang Q; Bai X Org. Lett 2008, 10, 889. [DOI] [PubMed] [Google Scholar]; (c) Dieckmann A; Richers MT; Platonova AY; Zhang C; Seidel D; Houk KN J. Org. Chem 2013, 78, 4132. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Richers MT; Deb I; Platonova AY; Zhang C; Seidel D Synthesis 2013, 45, 1730. [PMC free article] [PubMed] [Google Scholar]
- (4) (a).Richers MT; Breugst M; Platonova AY; Ullrich A; Dieckmann A; Houk KN; Seidel DJ Am. Chem. Soc 2014, 136, 6123. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jarvis CL; Richers MT; Breugst M; Houk KN; Seidel D Org. Lett 2014, 16, 3556. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mahato S; Haque MA; Dwari S; Jana CK RSC Adv 2014, 4, 46214. [Google Scholar]
- (5) (a).Ma L; Seidel D Chem. Eur. J 2015, 21, 12908. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Paul A; Chandak HS; Ma L; Seidel D Org. Lett 2020, 22, 976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6) (a).Li J; Qin C; Yu Y; Fan H; Fu Y; Li H; Wang W Adv. Synth. Catal 2017, 359, 2191. [Google Scholar]; (b) Li J; Fu Y; Qin C; Yu Y; Li H; Wang W Org. Biomol. Chem 2017, 15, 6474. [DOI] [PubMed] [Google Scholar]; (c) Zhu Z; Seidel D Org. Lett 2017, 19, 2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Paul A; Adili A; Seidel D Org. Lett 2019, 21, 1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Additional examples of amine redox-annulations: Zhang C; Das D; Seidel D Chem. Sci 2011, 2, 233. [Google Scholar]; (b) Kang Y; Chen W; Breugst M; Seidel DJ Org. Chem 2015, 80, 9628. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chen W; Seidel D Org. Lett 2016, 18, 1024. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhu Z; Lv X; Anesini JE; Seidel D Org. Lett 2017, 19, 6424. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhu Z; Chandak HS; Seidel D Org. Lett 2018, 20, 4090. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Liu Y; Wu J; Jin Z; Jiang H Synlett 2018, 29, 1061. [Google Scholar]
- (9).(a) For detailed discussions on the mechanisms of these transformations, see references: Xue X; Yu A; Cai Y; Cheng J-P Org. Lett 2011, 13, 6054. [DOI] [PubMed] [Google Scholar]; (b) Ma L; Paul A; Breugst M; Seidel D Chem. Eur. J. 2016, 22, 18179; [DOI] [PMC free article] [PubMed] [Google Scholar]; see also refs 1m, 3c, 4a, 4b, and 8b.
- (10).(a) Examples of redox-neutral α-C–H bond annulations of secondary amines that likely involve a pericyclic step: Grigg R; Nimal Gunaratne HQ; Henderson D; Sridharan V Tetrahedron 1990, 46, 1599. [Google Scholar]; (b) Soeder RW; Bowers K; Pegram LD; Cartaya-Marin CP Synth. Commun 1992, 22, 2737. [Google Scholar]; (c) Grigg R; Kennewell P; Savic V; Sridharan V Tetrahedron 1992, 48, 10423. [Google Scholar]; (d) Deb I; Seidel D Tetrahedron Lett 2010, 51, 2945. [Google Scholar]; (e) Kang Y; Richers MT; Sawicki CH; Seidel D Chem. Commun 2015, 51, 10648. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Cheng Y-F; Rong H-J; Yi C-B; Yao J-J; Qu J Org. Lett 2015, 17, 4758. [DOI] [PubMed] [Google Scholar]; (g) Yang Z; Lu N; Wei Z; Cao J; Liang D; Duan H; Lin YJ Org. Chem 2016, 81, 11950. [DOI] [PubMed] [Google Scholar]; (h) Rong H-J; Cheng Y-F; Liu F-F; Ren S-J; Qu JJ Org. Chem 2017, 82, 532. [DOI] [PubMed] [Google Scholar]; (i) Purkait A; Roy SK; Srivastava HK; Jana CK Org. Lett 2017, 19, 2540. [DOI] [PubMed] [Google Scholar]
- (11) (a).Chrzanowska M; Rozwadowska MD Chem. Rev 2004, 104, 3341. [DOI] [PubMed] [Google Scholar]; (b) Grycova L; Dostal J; Marek R Phytochemistry 2007, 68, 150. [DOI] [PubMed] [Google Scholar]; (c) Bhadra K; Kumar GS Med. Res. Rev 2011, 31, 821. [DOI] [PubMed] [Google Scholar]; (d) Yu J; Zhang Z; Zhou S; Zhang W; Tong R Org. Chem. Front 2018, 5, 242. [Google Scholar]
- (12) (a).Enders D; Wang C; Bats JW Synlett 2009, 1777. [Google Scholar]; (b) Enders D; Hahn R; Atodiresei I Adv. Synth. Catal 2013, 355, 1126. [Google Scholar]; (c) Hahn R; Jafari E; Raabe G; Enders D Synthesis 2015, 47, 472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Fessard TC; Motoyoshi H; Carreira EM Angew. Chem. Int. Ed 2007, 46, 2078. [DOI] [PubMed] [Google Scholar]
- (14).Ono N; Miyake H; Kamimura A; Hamamoto I; Tamura R; Kaji A Tetrahedron 1985, 41, 4013. [Google Scholar]
- (15).(a) Decarboxylative annulations: Cohen N; Blount JF; Lopresti RJ; Trullinger DP J. Org. Chem 1979, 44, 4005. [Google Scholar]; (b) Tang M; Tong L; Ju L; Zhai W; Hu Y; Yu X Org. Lett. 2015, 17, 5180. [DOI] [PubMed] [Google Scholar]; (c) Kang Y; Seidel D Org. Lett 2016, 18, 4277. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wu J.-s.; Jiang H.-j.; Yang J.-g.; Jin Z.-n.; Chen D.-b. Tetrahedron Lett 2017, 58, 546. [Google Scholar]; (e) Paul A; Thimmegowda NR; Galani Cruz T; Seidel D Org. Lett 2018, 20, 602; [DOI] [PMC free article] [PubMed] [Google Scholar]; see also references 3a, 3b, 3d, 5b, 6c, 8a, 8e, and 14..
- (16).(a) Selected reviews on decarboxylative coupling reactions not limited to amino acids: Rodriguez N; Goossen LJ Chem. Soc. Rev 2011, 40, 5030. [DOI] [PubMed] [Google Scholar]; (b) Xuan J; Zhang Z-G; Xiao W-J Angew. Chem. Int. Ed 2015, 54, 15632. [DOI] [PubMed] [Google Scholar]; (c) Patra T; Maiti D Chem. Eur. J 2017, 23, 7382. [DOI] [PubMed] [Google Scholar]; (d) Wei Y; Hu P; Zhang M; Su W Chem. Rev 2017, 117, 8864. [DOI] [PubMed] [Google Scholar]; (e) Rahman M; Mukherjee A; Kovalev IS; Kopchuk DS; Zyryanov GV; Tsurkan MV; Majee A; Ranu BC; Charushin VN; Chupakhin ON; Santra S Adv. Synth. Catal 2019, 361, 2161. [Google Scholar]
- (17) (a).Kind T; Fiehn O BMC Bioinformatics 2007, 8, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang Y; Gu M Anal. Chem 2010, 82, 7055. [DOI] [PubMed] [Google Scholar]
- (18).(a) Starting material synthesis: Ghislieri, D; Green AP; Pontini M; Willies SC; Rowles I; Frank A; Grogan G; Turner NJ J. Am. Chem. Soc 2013, 135, 10863. [DOI] [PubMed] [Google Scholar]; (b) Gray NM; Cheng BK; Mick SJ; Lair CM; Contreras PC J. Med. Chem 1989, 32, 1242. [DOI] [PubMed] [Google Scholar]; (c) Ji Y; Wang J; Chen M; Shi L; Zhou Y Chin. J. Chem 2018, 36, 139. [Google Scholar]; (d) Tamayo NA; Bo Y; Gore V; Ma V; Nishimura N; Tang P; Deng H; Klionsky L; Lehto SG; Wang W; Youngblood B; Chen J; Correll TL; Bartberger MD; Gavva NR; Norman MH J. Med. Chem 2012, 55, 1593. [DOI] [PubMed] [Google Scholar]; (e) Ji Y; Shi L; Chen M-W; Feng G-S; Zhou Y-GZ; Sun Y; Wang L; Chen X; Sun Y; Lin L; Tang Y; Li F; Chen D Tetrahedron Lett 2019, 60, 800. [Google Scholar]; (g) Schönbauer D; Sambiagio C; Noël T; Schnürch M Beilstein J. Org. Chem 2020, 16, 809. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Cutter PS; Miller R; Schore NE Tetrahedron 2002, 58, 1471. [Google Scholar]; (i) Bailey DM; Degrazia CG; Lape HE; Frering R; Fort D; Skulan TJ Med. Chem 1973, 16, 151. [DOI] [PubMed] [Google Scholar]; See also ref 12b.
- (19) (a).Kraus GA; Wu TA Tetrahedron 2010, 66, 569. [Google Scholar]; (b) Dai-Ho G; Mariano PS J. Org. Chem 1988, 53, 5113. [Google Scholar]; (c) Orito K; Satoh Y; Nishizawa H; Harada R; Tokuda M Org. Lett 2000, 2, 2535. [DOI] [PubMed] [Google Scholar]; (d) Azzena UJ Chem. Soc., Perkin Trans 1 2002, 360. [Google Scholar]; (e) Lahm G; Stoye A; Opatz TJ Org. Chem 2012, 77, 6620. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.