

Kinesin-1 uses a membrane-induced curvature-sensitive amphipathic helix to bind directly to cargo membranes.
Abstract
The cargo-binding capabilities of cytoskeletal motor proteins have expanded during evolution through both gene duplication and alternative splicing. For the light chains of the kinesin-1 family of microtubule motors, this has resulted in an array of carboxyl-terminal domain sequences of unknown molecular function. Here, combining phylogenetic analyses with biophysical, biochemical, and cell biology approaches, we identify a highly conserved membrane-induced curvature-sensitive amphipathic helix within this region of a subset of long kinesin light-chain paralogs and splice isoforms. This helix mediates the direct binding of kinesin-1 to lipid membranes. Membrane binding requires specific anionic phospholipids, and it contributes to kinesin-1–dependent lysosome positioning, a canonical activity that, until now, has been attributed exclusively the recognition of organelle-associated cargo adaptor proteins. This leads us to propose a protein-lipid coincidence detection framework for kinesin-1–mediated organelle transport.
INTRODUCTION
While ancestral reconstructions suggest that early eukaryotes were already capable of kinesin-mediated microtubule transport, the kinesin repertoire has been elaborated throughout eukaryotic evolution through gene duplication and alternative splicing (1, 2). How this complexity subsequently confers broader functional capacity often remains unclear. This question is particularly pertinent for the kinesin-1 family that uses a remarkably versatile cargo recognition mechanism to enable the binding and transport of protein and ribonuclear protein complexes, viruses, microtubules, and many different membrane-bound organelles (MBOs) (3–6). This mechanism comprises an interlinked and cooperative network of cargo binding and autoregulatory elements within the motor adenosine triphosphatase bearing kinesin heavy chains (KHCs) and kinesin light chains (KLCs) (7–9).
The number of KLC genes varies between species; humans and mice have four paralogous KLCs (KLC1 to KLC4) (10–19). KLC pairs combine with one of three KHC dimers (encoded by the KIF5A, -B, or -C paralogs) to form an array of kinesin-1 heterotetramers with different cell and tissue expression profiles and cargo-binding properties (20–24). The physiological importance of kinesin-1 composition is underscored by a range of different neurological diseases that are reported to be contributed to or caused by dysregulation of particular KHCs and KLCs. These include loss of KLC4 function in hereditary spastic paraplegia; up-regulation of KLC1 isoform E (KLC1E) in Alzheimer’s disease (AD); and up-regulation of KLC2 in spastic paraplegia, optic atrophy, and neuropathy (SPOAN) syndrome (25–28).
KLCs recognize MBO-associated cargo adaptor proteins using their highly conserved tetratricopeptide repeat (KLCTPR) domains. Recent structural studies have revealed the molecular details of this motor-cargo interface that can engage both leucine-zipper and short linear peptide motif (SLiM) features in adaptor proteins. These adaptors act as a regulatory tether between motor and cargo that couples cargo binding with motor activity and transport (8, 29–33). KLC amino acid sequences mainly diverge in their C-terminal domains (KLCCTD: defined here as any protein sequences after the end of KLCTPR). KLCCTDs help to direct MBO targeting (34–36) and enable the phosphorylation-dependent regulation of kinesin-1 activity in axonal transport (37, 38). The molecular basis for these activities is not understood.
Here, we describe the discovery and characterization of an evolutionarily conserved, membrane-induced, amphipathic helix (AH) within the C-terminal domains (CTDs) of a subset of long KLCs that mediates the direct phospholipid-dependent binding of kinesin-1 to membranes and facilitates the transport of lysosomes.
RESULTS
In silico sequence and structural analysis reveals an AH within the long KLCs
A comparison of KLCCTD primary sequences and their predicted secondary structures indicated the presence of a lone α helix within an otherwise unstructured domain of the long KLC1 splice isoforms (KLC1D and KLC1E, encoded by exon 16) and KLC2 and KLC4, but not the short KLC1, isoforms (KLC1A, KLC1B, KLC1C, KLC1F, and KLC1G) (Fig. 1, A and B). The predicted helix in KLC1D/E is also found in the KLC1I, KLC1J, KLC1K, KLC1M, KLC1N, and KLC1O variants that are not considered in the present study (16). Further characterization of this sequence suggested that the helix, if formed, would have a hydrophobic face and a net positively charged face (i.e., an AH) (Fig. 1C). A shorter sequence that retains some similar hydrophobic and charged amino acids encoded by exon 14 (skipped in KLC1D/E) is found in KLC1B and KLC1C (Fig. 1B). Further complexity arises in the case of KLC3; a structure-based alignment revealed that, unlike the other paralogs, KLC3TPR terminates within TPR5, and so it lacks the final sixth TPR (fig. S1). However, the predicted AH is still present after the truncated KLCTPR domain (Fig. 1C and fig. S1).
Fig. 1. Long KLC paralogs and isoforms carry an amphipathic helix within their C-terminal domain.
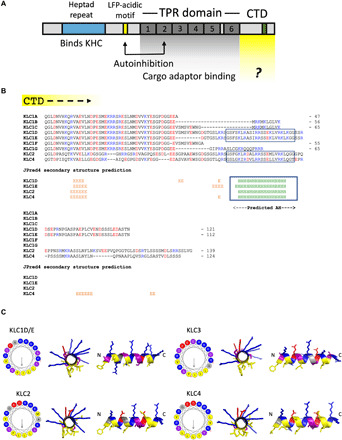
(A) Domain architecture of the KLCs. LFP, leucine-phenylalanine-proline. (B) Manually edited PROMALS3D-based alignment and JPred4 secondary structure predictions for the CTDs of mouse KLC1 to KLC4. Lengths are indicated from a conserved post-KLCTPR QG(L/T)D sequence. H, helix; E, extended. (C) Visualization of the AH in KLC1 to KLC4 using HeliQuest and PyMOL.
We identified the same AH feature in vertebrate (chicken, frog, and zebrafish) KLC orthologs (fig. S2). Kinesin-1 associates with membranes in sea urchins [Stronglyocentrotus purpuratus (SP)] (39), and we identified the AH within long KLC isoforms, SPKLC1 to SPKLC3, but not the short SPKLC4 (fig. S2) (15). BLASTp searches of the National Center for Biotechnology Information (NCBI) nonredundant (nr) database indicated its presence in many other deuterostome KLCs (fig. S3A). The AH was not apparent in the relatively short fly [Drosophila melanogaster; (19)] or worm [Caenorhabditis elegans; (18)] orthologs; this is despite the fact that their KLCs have very well-conserved KLCTPR domains that enables clear definition of the KLCCTD (fig. S2). Further analysis using HHpred failed to reveal any significant similarity (E value of <1) to any protein in these species (40). However, further database analysis and secondary structure prediction did identify closely related AH sequences in the KLCCTD of other protostomes, including Brachiopoda and Annelida, as well as nonbilaterian animals, including Cnidaria, Ctenophora, and Placozoa (fig. S3B), suggesting that the AH motif is evolutionarily conserved.
Phylogenetic analysis indicates that the AH is an ancestral KLC feature
A KLC phylogeny (fig. S4) indicated that the four paralogous KLC protein coding genes of human and mouse arose from gene duplications on the gnathostome stem, with KLC1 to KLC4 being represented in the various classes that make up Eugnathostomata: Mammalia, Eureptillia, Amphibia, Actinopterygii, and Chondrichthyes. The multiple KLC homologs of Petromyzon appear to be the result of independent, lineage-specific duplications, with the same mechanism resulting in C. elegans klc-1 and klc-2. The presence of clear KLC orthologs in animals and choanoflagellates, but not in more distant opisthokonts, indicates that the KLC gene originated in their common choanozoan ancestor. Paralogs of KLC are present in some fungi, Viridiplantae, and members of the Stramenopila, Alveolata, and Rhizaria clade of eukaryotes. As for D. melanogaster and C. elegans, the AH region was not detected in sequences of the wider Ecdysozoa. This can be explained by secondary loss because the AH motif is present in nonbilaterian holozoan sequences, including choanoflagellates. The onset of alternative splicing as a means of producing AH-containing and AH-lacking protein variants in mice and humans likely arose on the agnathan stem lineage because the deep-branching agnathan Petromyzon (sea lamprey) has isoforms displaying the short and long form, whereas more distantly related genomes do not.
Together, these data show that the KLC protein family can be divided into short forms that lack, and long forms that carry, an evolutionarily well-conserved ≈20 amino acid predicted AH within their CTD. In general, amino acids on the hydrophobic face are highly conserved, whereas the abundance, position, and polarity of charges vary on the hydrophilic face but retain the overall basicity. Serine residues occur frequently, but their number and position vary (Fig. 1C and figs. S2 and S3).
The AH peptide adopts a helical conformation in the presence of model membranes
A synthetic peptide encompassing this sequence from KLC1D/E was analyzed in vitro using circular dichroism (CD) spectroscopy (Fig. 2A). In aqueous solution, the KLC1D/E peptide was unstructured. However, in the presence of the zwitterionic membrane mimetic dodecylphosphocholine (DPC) micelles, the CD spectrum changed to one characteristic of a highly folded (71% helicity) α helix (Fig. 2A). To test this further, a control peptide with two aliphatic residues replaced by proline (IL/PP KLC1D/E) was designed to prevent helical folding. This gave a CD spectrum expected for a random coil, both in the absence and presence of DPC micelles (Fig. 2A). The helical folding of the parent KLC1D/E peptide was not induced with the nonionic detergents n-dodecyl β-d-maltoside (DDM), octyl β-d-glucopyranoside (OG), or the zwitterionic detergent lauryldimethylamine oxide (LDAO), indicating that specific electrostatic protein-lipid interactions may be important for AH folding (fig. S5A).
Fig. 2. The AH peptide folds into a helix in the presence of model membranes.
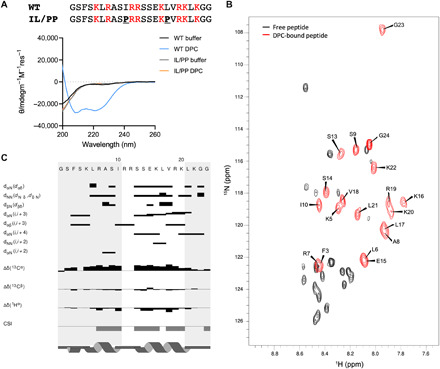
(A) Peptide sequences and their CD spectra in the absence or presence of 0.35% DPC. (B) Overlaid 15N HSQCs of the unbound (black) and DPC-bound (red with assignments) peptides. ppm, parts per million. (C) NOE/chemical shift index (CSI) plot of the DPC-bound peptide sequence.
Next, we used nuclear magnetic resonance (NMR) to examine conformational changes in the peptide in more detail (Fig. 2, B and C, and fig. S5, B and C). The one-dimensional (1D) 1H NMR spectrum of the peptide in buffer was typical of a random coil with very little dispersion in either the amide or methyl regions (fig. S5B). However, upon titration with perdeuterated DPC, the peptide chemical shifts became sharper and more dispersed, saturating at around 0.7% DPC. This was particularly evident in the amide region of spectrum indicative of folding in the presence of DPC (fig. S5B). This was confirmed by collecting 1H-15N heteronuclear single-quantum coherence (HSQC) datasets at natural 15N abundance (Fig. 2B). In the presence of deuterated DPC, the 1H-1H nuclear Overhauser effect spectroscopy (NOESY) and total correlation spectroscopy (TOCSY) spectra of the peptide gave sufficiently dispersed HN and Hα resonances to allow a NOE-based sequential assignment of the spectra (Fig. 2B). Backbone NH resonances could be assigned for ≈80% of the peptide with missing assignments at the N terminus and in the central R11/R12 region (Fig. 2B). Analysis of the 2D NOESY spectra identified a number of dNN(i,i + 1) and dαN(i,i + 1) throughout the peptide backbone along with several dαN(i,i + 3) and dαN(i,i + 4) NOE correlations. These are diagnostic of α-helical conformation (Fig. 2C). In particular, a number dαN(i,i + 3) and dαN(i,i + 4) NOEs between residues S13 and K20 indicated clearly that this region is an α helix. Although there were distinct dNN(i,i + 1), dαN(i,i + 1), and dαN(i,i + 3) NOEs in the N terminus of the peptide, this region was less well defined due to a greater overlap in the NH-Hα region of the 2D NOESY spectra. In addition to the NOE analysis, the 13Cα/Cβ and Hα backbone chemical shifts for the assigned residues were extracted using a natural abundance 1H-13C HSQC. The subsequent chemical shift index analysis of the chemical shifts for the DPC-bound peptide were again consistent with an α helix for two stretches of residues, K5-I10 and S13-K20, as observed in the NOESY spectra (Fig. 2C).
The AH mediates KLCCTD binding to lipid membranes
To evaluate the capacity of KLCCTD to bind membranes, we assembled liposomes [small unilamellar vesicle (SUV) and large unilamellar vesicle (LUV)] from Folch fraction I brain lipids and analyzed the capacity of KLCCTDs (fig. S6A) to associate with them using cosedimentation assays. We focused on the KLC1 paralog for these liposome-binding assays because of the availability of natural isoform controls. Purified recombinant KLC1ECTD that contains the predicted AH (long), but not KLC1BCTD (short), bound to LUVs in a liposome concentration–dependent manner (Fig. 3, A and B). Binding of KLC1ECTD to SUVs was significantly greater than to LUVs, indicating that the interaction is sensitive to membrane curvature (Fig. 3B). In a separate experiment, as expected, KLC1ACTD also failed to associate with membranes (fig. S6B). A single helix-disrupting proline mutation (I560P in full-length protein; I/P mutant) in the hydrophobic face of KLC1ECTD AH was sufficient to eliminate any detectable membrane interaction (Fig. 3, C and D). Furthermore, amine cross-linking by bis(sulfosuccinimidyl)suberate (BS3), in the absence or presence of LUVs and SUVs (Fig. 3E), resulted in the AH- and liposome-dependent formation of KLC1ECTD oligomers with electrophoretic mobility corresponding to dimers, trimers, and tetramers, as well as higher-order assemblies. Membranes were also remodeled because analysis of (non–cross-linked) samples by cryo–electron microscopy revealed extensive AH-dependent membrane bending and vesicle clustering, as well as the accumulation of small vesicles (Fig. 3F). We concluded that KLC1ECTD binds directly to membranes, and this interaction requires the AH.
Fig. 3. The AH is required for KLC1ECTD binding to liposomes.
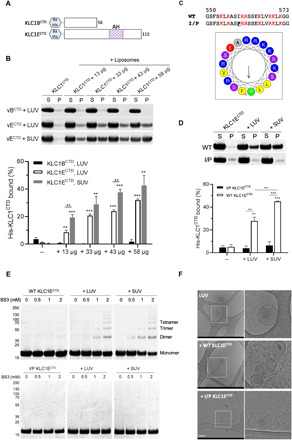
(A) Representation of the His-tagged KLC1B/ECTD constructs. (B) Cosedimentation analysis of His-KLC1B/ECTD with Folch fraction I LUV or SUV. Samples were analyzed by SDS-PAGE/Coomassie (top) and quantified by densitometry (bottom). Means ± SEM of at least three experiments. **P < 0.01 and ***P < 0.001 compared to the sample without liposomes or as indicated in the graph. (C) Sequence comparison and visualization of the AH in the I560P KLC1ECTD mutant. (D) Cosedimentation analysis of WT or I/P His-KLC1ECTD with Folch fraction I LUV or SUV. Means ± SEM of at least three experiments. **P < 0.01 and ***P < 0.001 compared to the sample without liposomes or as indicated in the graph. (E) BS3 cross-linking assay showing oligomers of WT, but not I/P mutant His-KLC1ECTD upon incubation with Folch fraction I LUV or SUV. (F) Cryo–electron microscopy observation of Folch fraction I LUV in the absence or presence of WT or I/P mutant KLC1ECTD. Scale bar, 200 nm.
Long KLCCTDs membrane binding is dependent on specific anionic phospholipids
Liposomes of defined composition were prepared from pure lipids. No binding was observed with either wild-type (WT) or the helix-disrupting mutant KLC1ECTD upon incubation with liposomes consisting of PC, PC:PE (7:3), or PC:PE:PS (5:3:2) (Fig. 4A) (where PC is phosphatidylcholine, PE is phosphatidylethanolamine, and PS is phosphatidylserine), suggesting that an additional component is required. We considered phosphatidic acid (PA) to be a good candidate because KLC1 (along with KIF5B) has been identified recently in a proteomic screen for PA-binding proteins (41). WT KLC1ECTD, but not the helix-disrupting mutant, bound to PC:PE:PA (5:3:2) liposomes, although to a slightly lesser extent than to the Folch fraction liposomes (Fig. 4A). Moreover, KLC1BCTD showed no detectable binding to PA-containing liposomes (fig. S6C).
Fig. 4. Long KLCCTDs bind to specific anionic phospholipids.
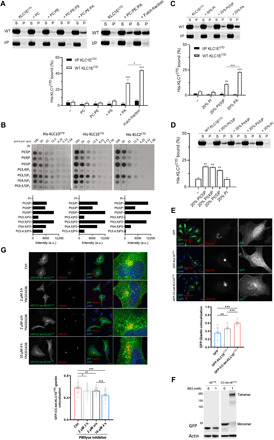
(A) Cosedimentation analysis of WT or I/P His-KLC1ECTD incubated with PC, PC:PE (7:3), PC:PE:PS (5:3:2), PC:PE:PA (5:3:2), or Folch fraction I LUV. (B) Binding of the indicated long KLCCTDs to phosphoinositides in a protein lipid overlay assay. Quantitation is provided at the 50 pmol per spot. a.u., arbitrary units. (C) Cosedimentation analysis of WT or I/P His-KLC1ECTD incubated with PC:PE:PI, PC:PE:PI(5)P, or PC:PE:PA (5:3:2) LUV. (D) Cosedimentation analysis of WT His-KLC1ECTD incubated with PC:PE:PI(3)P, PC:PE:PI(4)P, PC:PE:PI(5)P, or PC:PE:PI (5:3:2) LUV. Means ± SEM of at least three experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to the sample without liposomes or as indicated in the graph. (E) Immunofluorescence analysis (top) showing localization of GFP, GFP-KLC1ECTD, and GFP-CC-tet-KLC1ECTD. Golgi is stained with anti-giantin antibody. Representative Pearson’s correlation from one experiment and ≈40 cells (bottom). (F) BS3 cross-linking assay showing monomeric or tetrameric expression of KLC1ECTD in lysates of HeLa cells transfected with GFP-KLC1ECTD or GFP-CC-tet-KLC1ECTD. (G) GFP-CC-tet-KLC1ECTD-giantin colocalization in cells treated with the PIKfyve inhibitor YM201636 as indicated (top). Representative Pearson’s correlation from one experiment and ≈40 cells (bottom). Means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001. Scale bars, 20 μm. DAPI, 4′,6-diamidino-2-phenylindole.
Together, these data above show that KLC1ECTD binds to PA in an AH-dependent manner. However, as PA only comprises a small component of Folch fraction I liposomes, and even at 20% as used in our pure lipid assays was insufficient to match the degree of binding, we considered the possibility that KLC1ECTD has a broader capacity for acidic phospholipid recognition. Consistent with this, long KLCCTDs (KLC1D, KLC1E, and KLC2) bound to phosphoinositides on membrane strips with some preference for PI(5)P and PI(3,5)P2; moderate association with PI(3)P, PI(4)P, and PI(4,5)P2; very little binding to PI(3,4)P2 and PI(3,4,5)P3; and no detectable interaction with PI (Fig. 4B). The short variants lacking the AH (KLC1A/B/CCTD) did not interact with any phosphoinositide (fig. S6D). As these strips present the lipid outside of the context of a bilayer, we performed liposome-binding assays with one of the top binding candidates, PI(5)P (Fig. 4C). Consistent with the PIP array data, KLC1ECTD was also capable of interacting with PI(5)P, but not PI, although to a lesser extent than with the same amount of PA (Fig. 4C). Again, the helix-disrupting mutant showed no binding (Fig. 4C). We extended this analysis to PI(3)P and PI(4)P. Consistent with the PIP array data, KLC1CTD bound to PI(3)P, PI(4)P, and PI(5)P; however, the preference for PI(5)P was not apparent (Fig. 4D). We concluded that the AH is essential for KLC1CTD-mediated membrane interaction, it shows a requirement and some selectivity for specific anionic phospholipids, and those properties extend to the closely related sequences in KLC2CTD.
To explore these interactions in cells, we compared the subcellular localization of green fluorescent protein (GFP) and GFP-KLC1ECTD in HeLa cells (Fig. 4E). Whereas GFP was diffusely distributed throughout the cytoplasm and nucleus, GFP-KLC1ECTD signal was slightly enriched in the perinuclear region. It partially colocalized with antibody staining for the Golgi marker giantin (Fig. 4E). We anticipated that the forced oligomerization of GFP-KLC1ECTD might increase avidity and enhance target membrane association. To test this, we introduced the sequence for a de novo–designed coiled-coil tetramer between GFP and KLC1ECTD (GFP-CC-tet-KLC1ECTD) (42). The mobility of the fusion proteins from HeLa cell lysates was monitored by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting (Fig. 4F). Confirming the functionality of the coiled-coil sequence, a band shift to the expected molecular weight of an SDS-resistant tetramer was only observed with the GFP-CC-tet-KLC1ECTD after BS3 treatment (Fig. 4F). As predicted, the perinuclear membrane association became more pronounced with GFP-CC-tet-KLC1ECTD, as determined by the giantin colocalization analysis of the GFP signal (Fig. 4E). This was AH dependent because incorporation of the I/P AH mutant in GFP-CC-tet-KLC1ECTD or its truncation in GFP-CC-tet-KLC1ACTD resulted in fluorescence signal diffusely distributed throughout the cytoplasm and nucleus (fig. S7). Although the GFP-CC-tet-KLC1ECTD fluorescence signal overlapped with giantin, there was clearly recruitment to other perinuclear compartments (Fig. 4E), suggesting that KLC1ECTD-organelle recruitment is not specific for the Golgi and appears consistent with the quite broad acidic phospholipid-binding properties observed in vitro.
However, as our vitro analysis showed that KLCCTDs are capable of binding to PI(5)P and PI(3,5)P2, and PIKfyve inhibition modifies the motility of lysosomes, a canonical kinesin-1 cargo (43, 44), we used the PIKfyve kinase inhibitor YM201636 to reduce 5′ phosphorylation. This led to the redistribution of GFP-CC-tet-KLC1ECTD to more dispersed puncta (Fig. 4G) and a time- and inhibitor concentration–dependent reduction in the colocalization of GFP-CC-tet-KLC1ECTD with giantin. Thus, consistent with the membrane-binding properties observed in vitro using recombinant protein (Fig. 4, A to D), KLC1ECTD localization is sensitive to membrane phosphoinositide composition in cells.
Long KLCCTD-dependent membrane association of kinesin-1 is phosphoinositide sensitive and is important for lysosome positioning
To determine how membrane binding by isolated KLCCTD extends to the complete tetrameric holoenzyme, we focused on endogenously expressed kinesin-1 in biochemical assays, first seeking to establish the KLC1 and KLC2 expression profile in HeLa cells. These were transfected with plasmids to express full-length hemagglutinin (HA)–tagged KLC1A, KLC1E, and KLC2 and compared to nontransfected cells by SDS-PAGE and Western blot analysis. An anti-HA antibody detected the overexpressed proteins, and, consistent with their molecular weights, HA-KLC1E and HA-KLC2 migrated more slowly than HA-KLC1A (Fig. 5A). Anti-KLC1 antibodies detected both overexpressed isoforms of KLC1 with similar efficiency, but not KLC2, and also a slightly faster migrating band in all samples that was confirmed to be an endogenous KLC1 isoform by short interfering RNA (siRNA) depletion of KLC1 (Fig. 5A). Similarly, anti-KLC2 antibodies specifically detected overexpressed HA-KLC2, but not HA-KLC1, and a slightly faster migrating band that was confirmed as endogenous KLC2 by siRNA knockdown (Fig. 5A). Together, these data indicate that most KLC1 in HeLa cells is a short isoform. In addition, the approximately equal expression of the HA-tagged proteins and difference in band intensity between endogenous and overexpressed proteins for the KLC1- and KLC2-specific antibodies led us to conclude that HeLa cells express similar amounts of KLC1 and KLC2 (roughly 40:60).
Fig. 5. Membrane association of kinesin-1 is mediated by KLCCTD and is phosphoinositide sensitive.
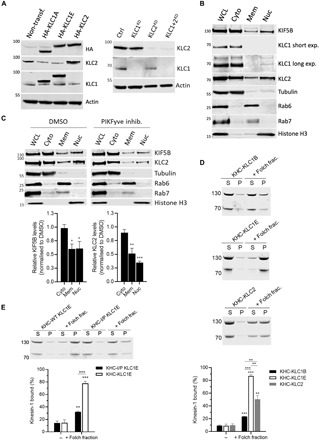
(A) KLC1/2 expression in HeLa cells transfected with HA-KLC1A/1E/2 or nontransfected cells (left) or after siRNA knockdown of KLC1/2 (right) was analyzed by Western blot. (B) Fractionation and Western blot analysis of endogenous KIF5B and KLC1/2. (C) Fractionation analysis of cells treated with dimethyl sulfoxide (DMSO) or PIKfyve inhibitor YM201636 (10 μM, 4 hours). Means ± SEM of at least three experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to the sample treated with DMSO. (D) Cosedimentation assay and analysis by SDS-PAGE/Commassie staining using purified KHC-KLC1B/1E/2 and Folch fraction I LUV. (E) Cosedimentation assay and analysis by SDS-PAGE/Commassie staining using purified KHC-KLC1E or –I/P KLC1E and Folch fraction I LUV. Means ± SEM of at least three experiments. **P < 0.01 and ***P < 0.001 compared to the sample without liposomes or as indicated in the graphs.
To compare the localization of the endogenous KLC paralogs, HeLa cell extracts were fractionated. As well as in the cytosolic fraction (containing β-tubulin), KLC2 was found in the membrane (containing Rab6 and Rab7) and nuclear (containing histone H3) fractions (Fig. 5B). In contrast, the shorter KLC1 was almost exclusively found in the cytosolic fraction (very weak bands in nuclear fraction on long exposure) and not at all in the membrane fraction, consistent with a specific role for the long KLCs membrane association. The major KHC paralog in HeLa, KIF5B, was found in all three fractions (Fig. 5B). Confirming a role for phosphoinositides in directing membrane association, treatment of cells with PIKfyve inhibitor reduced the amount KLC2 and KIF5B in the membrane and nuclear fractions (Fig. 5C).
To ask whether KLCCTD is necessary and sufficient for direct membrane targeting of kinesin-1, KHC-KLC1B, KHC-KLC1E, and KHC-KLC2 complexes were isolated from 293T cells by HaloTag purification and size exclusion chromatography (9). Consistent with a direct role in membrane binding of the holoenzyme, tetramers containing the long KLC1E or KLC2 sequences bound significantly better to Folch fraction I liposomes in cosedimentation assays compared to the short KLC1B isoform (Fig. 5D). In a separate experiment, I/P mutant KHC-KLC1E significantly reduced liposome binding (Fig. 5E), confirming a role for the AH in tetramer-membrane association.
To explore the functional role for the AH and its relationship to KLCTPR-mediated SLiM interactions, we turned to a lysosomal transport model. Lysosome distribution in HeLa is controlled by kinesin-1 through a well-defined cargo adaptor axis, BORC-Arl8-SKIP-KLC2-KIF5B, where SifA and kinesin interacting protein (SKIP) acts as the cargo adaptor protein, binding KLCTPR and KHC using the W-acidic class of SLiM (29, 45–50). siRNA knockdown of KLC2 caused reduction in the proportion of peripheral lysosomes compared to control cells (Fig. 6, A and B). A typical WT distribution profile characterized by many lysosomes in the cytoplasm outside the perinuclear region and accumulations at the cell periphery could be restored by expression of WT KLC2, but not KLC2, with a helix-disrupting mutation (L550P, equivalent to I560P in KLC1E). This confirms a role for the AH-membrane binding in kinesin-1–mediated lysosome positioning (Fig. 6, C and D). Similarly, expression of the long KLC1E, but not the short KLC1A, isoform restored cytoplasmic and peripheral lysosome accumulations (Fig. 6, C and D). Furthermore, the defect in transport associated with the L550P mutation was comparable to that observed for a tryptophan-acidic class SLiM-binding KLC2 mutant (N287L) (Fig. 6E). Together, these data indicate that the long AH-containing paralogs have some functional redundancy in lysosome transport (consistent with their shared ancestry) and that AH-membrane binding is as important as SLiM-mediated cargo adaptor interactions.
Fig. 6. Long KLCs are required for kinesin-1–mediated lysosome positioning.
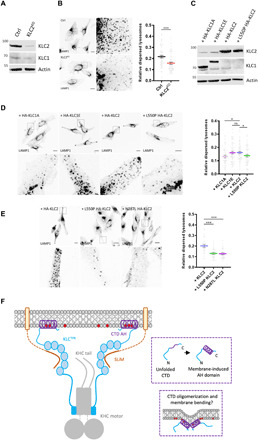
(A) Western blot analysis of KLC2 expression in HeLa cells after siRNA knockdown of KLC2. (B) Lysosome distribution in control or KLC2 knockdown cells. Lysosome dispersion was determined by measuring perinuclear and whole-cell LAMP1 signal fluorescence. Means ± SEM of at least three experiments. ***P < 0.001. (C) KLC1/2 expression in KLC2 knockdown HeLa cells transfected with HA-KLC1A/1E/2/L550P 2 was analyzed by Western blot. (D) Lysosome distribution in KLC2 knockdown cells transfected with HA-KLC1A/1E/2/L550P 2. Means ± SEM with samples pooled from three independent experiments. *P < 0.05. (E) Lysosome distribution in KLC2 knockdown cells transfected with HA-KLC2 or N287L/L550P variants. Means ± SEM with samples pooled from three independent experiments. ***P < 0.001. (F) Model showing representation of kinesin-1 cargo membrane association by the KLCs. The membrane-induced helical folding and membrane remodeling capacity of the KLCCTD helix are also represented. ns, not significant.
DISCUSSION
The mechanistic basis for the critical role that the KLCCTDs play in organelle transport has proved elusive. This is intimately linked with the question of why, particularly in vertebrates, the evolution of intracellular transport has favored gene duplication and alternative splicing events that have resulted in so many KLCs that differ mainly in their CTD. The identification here of a membrane-induced AH within a subset of long KLCCTDs provides an explanation, showing that the role of these sequences is to confer a direct membrane binding capacity on the motor complex. Functionally, this brings the kinesin-1 family in line with the kinesin-3 family members that also incorporate classical membrane binding structures and play key roles in vesicle and organelle transport. However, in the case of kinesin-3, these are globular Pleckstrin homology and Phox homology domains (51). To the best of our knowledge, this is the first report of a membrane-binding/induced AH, a feature more typically associated with small guanosine triphosphatases, vesicular coats, and their accessory proteins, being used directly by a microtubule motor protein. The potential to attribute a relative increase in KLC1E or KLC2 expression to modification of the direct membrane properties of the kinesin-1 pool offers a pathway to understanding their reported roles in AD and SPOAN syndrome, respectively (25, 28).
Given the well-established role of cargo adaptor proteins in motor recruitment and activation (7, 8), including at the lysosome (29, 45, 47), we favor a coat-like coincidence detection framework to explain our observations (Fig. 6F). In this model, cargo adaptor proteins bind to KLCTPR and membranes to KLCCTD. It will be important to sequence these protein and lipid interactions correctly. Previous work has indicated that the KLCTPR-adaptor binding gates access to cargo-binding features of the KHC tail (46). It seems reasonable to suggest that the KLCCTD-membrane interaction may function higher in this hierarchy to locally concentrate kinesin-1 at sites where it is available to engage cargo adaptor proteins. The nature of the final transport competent assembly will depend on the adaptor system because kinesin-1 cargo adaptors can be integral or peripheral membrane proteins (8). The complex role of the KHC tail merits further study. Overlapping, mostly basic, coiled-coil–containing KHC sequences have been shown to functionally bind to membranes, adaptor proteins, and microtubules. In several cases, there is evidence of sequence specificity; in others, binding may be mostly electrostatic (41, 46, 52–59). The direct, AH-dependent binding of the KLCs to membranes containing specific anionic phospholipids shown here fits well within a broader model of cooperative sequence/structure specific and electrostatic elements effecting KHC- and KLC-dependent cargo recognition and motor activation for diverse transport functions.
It is likely that phosphorylation will emerge as a key KLC membrane-binding regulator; CKII and GSK3β have been shown to phosphorylate KLC2CTD to negatively regulate axonal transport, and a serine residue within AH itself (S542 in mouse) as well as another at S579 in KLC2CTD are phosphorylated by AMP-activated protein kinase to inhibit axonal PI3K trafficking (37, 38). We posit that sequence differences between the long KLCCTDs could offer the kinesin-1 family different sensitivity to particular kinases and phosphatases. A report of binding of 14-3-3 proteins to phosphorylated KLC2CTD suggests that an AH/CTD-sequestration hypothesis is also worth considering as another level of regulation (60).
What of the short KLCs? Our phylogenetic analysis suggests that membrane binding by the AH is an ancestral animal KLC feature. Counterintuitively, it appears that the loss of the AH rather than its acquisition may be associated with diversification of function. Previous reports have implicated KLC1B and KLC1C isoforms in rough endoplasmic reticulum and mitochondrial targeting despite lacking the exon 16 sequence encoded by the AH (16, 34, 35). However, both isoforms share some sequences with limited homology to the second half of the AH (encoded by exon 14). Although helical properties of this 10–amino acid sequence were not confidently predicted by our in silico analysis, if folded, the resulting structure would be amphipathic. Whether it has a membrane/lipid-binding capacity that was not detected in our assays is clearly worth further investigation. More generally, given their de facto positioning at the motor-organelle interface, there is a good case for a wider exploration of the membrane-binding properties of the long and short KLCCTDs throughout the wider KLC family. Specifically, it will be important to consider why the AH sequence was lost (relatively recently in evolutionary terms) in Ecdysozoa, including D. melanogaster and C. elegans, and if KLC intrinsic or extrinsic compensatory mechanisms have emerged. In D. melanogaster, the single kinesin-1 (one KHC gene and one KLC gene) carries out diverse activities including microtubule-microtubule sliding, RNA transport, and organelle transport (6, 61, 62). It will be interesting to ask whether the range of KLC transcripts and CTD sequences in vertebrates are an example of functional diversity created and maintained by gene duplication and alternative splicing, where short KLC1 isoforms might be primarily used for the transport of nonmembranous cargos such as microtubules or ribonuclear protein complexes (63).
In conclusion, the identification of an AH in the cargo-binding mechanism of the kinesin-1 motor should lead to a step change in the view of how the family is classified, and cargoes are recognized by this central player in intracellular transport. It points to a direct role for membrane biology in KLC-dependent vesicle and organelle recognition that may be as important as cargo adaptor protein interaction.
MATERIALS AND METHODS
Reagents and antibodies
DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine), bovine liver phosphatidylinositol (PI), brain PS, and egg PA were purchased from Avanti Polar Lipids. PI 3-phosphate diC16 [PI(3)P], PI 4-phosphate diC16 [PI(4)P], and PI 5-phosphate diC16 [PI(5)P] were obtained from Echelon Biosciences. Folch fraction I and egg PC were purchased from Sigma-Aldrich. YM201636 PIKfyve inhibitor was purchased from Cayman Chemical. A cell fractionation kit was obtained from Cell Signaling Technology. Nontargeting control siRNA was obtained from Horizon Discovery, and human KLC1/2 siRNAs were from Santa Cruz Biotechnology. The following primary antibodies were used: horseradish peroxidase (HRP)–conjugated anti-His6 (71841, Novagen), anti-GFP for immunoblotting (3E1, Roche), anti-actin (AC-74, Sigma-Aldrich), anti–β-tubulin (AA2, Sigma-Aldrich), anti–histone H3 (Abcam), anti-giantin (PRB-114C, Covance), anti-HA (HA-7, Sigma-Aldrich), anti-KLC1 ([EPR12441(B)], Abcam), anti-KLC2 (Abcam), anti-KIF5B (Abcam), anti-LAMP1 (lysosome-associated membrane protein 1) (D2D11, Cell Signaling Technology), anti-Rab7 (D95F2, Cell Signaling Technology), and anti-Rab6 (D37C7, Cell Signaling Technology). Alexa 568– and Alexa 633–conjugated anti-mouse or anti-rabbit secondary antibodies were from Thermo Fisher Scientific.
Peptides and plasmids
KLC1D/E synthetic peptides used for CD or NMR measurements were purchased from GenScript (>98% purity). Sequences were as follows: WT KLC1D/E, GSFSKLRASIRRSSEKLVRKLKGG; IL/PP KLC1D/E, GSFSKLRASPRRSSEKPVRKLKGG. Peptides were dissolved in water and stored at −20°C until use.
KLC1 isoforms were generated by synthetic DNA synthesis and restriction enzyme cloning using a mouse KLC1A clone as a starting point. KLC1A coding sequence corresponding to NM_008450.2 was in vector CB6-HA (N-terminal HA epitope tag, mammalian expression) flanked Eco RI and Bam HI restriction enzyme sites. A unique Nhe I restriction endonuclease site was added to the KLC1A sequence by site-directed mutagenesis at 1162 to 1167 by introducing a silent GCCTCC to GCTAGC mutation. This encodes an Ala-Ser sequence within the TPR domain. As the KLC1 isoforms only differ at the C terminus of the protein, this allowed the synthesis of a series of DNAs (GenScript) that could then be inserted between the newly created Nhe I site and the existing C-terminal Eco RI site. These sequences were identical to NM_001025358.2 → NP_001020529.2 for KLC1B, NM_001025359.2 → NP_001020530.2 for KLC1C, NM_001025360.2 → NP_001020531.2 for KLC1D, and NM_001025361.2 → NP_001020532.2 for KLC1E, with the exception of KLC1D/E where an unwanted Nhe I site in the sequence encoding the CTD was removed before synthesis by introducing a silent mutation. For expression of the CTDs in bacteria, the DNAs encoding the CTD sequence commencing at Q496 (of QGLD sequence) were amplified by polymerase chain reaction (PCR) and subcloned into the Nde I/Xho I sites of pET28a. The KLC2 sequence used in this study corresponded to NM_008451.3 → NP_032477.2 (annotated KLC2 isoform 1 in the NCBI database) and was cloned into the CB6-HA vector using Not I and Eco RI restriction endonuclease sites. The CTD sequence was amplified from Q481 of QGLD sequence and subcloned into pET28a as for KLC1 CTDs. The I560P helix disrupting KLC1E mutant was constructed by PCR-based site-directed mutagenesis using the following primers: forward, 5′-CAAACTCCGGGCTTCCCCTAGACGCAGCAGTGAG-3′; reverse, 5′-CTCACTGCTGCGTCTAGGGGAAGCCCGGAGTTTG-3′. The L550P helix-disrupting KLC2 mutant was constructed by PCR-based site-directed mutagenesis using the following primers: forward, 5′-GCTCCGGGATGCTCCGAGACGCAGCAGTGAG-3′; reverse, 5′-CTCACTGCTGCGTCTCGGAGCATCCCGGAGC-3′. GFP-tagged KLC1ECTD was cloned from KLC1ECTD complementary DNA (cDNA) into a cytomegalovirus-driven expression vector (CB6-GFP) for expression in mammalian cells. For the expression of GFP-CC-tet-KLC1E/ACTD, cDNAs encompassing a de novo–designed tetrameric coiled coil (39) and KLC1E/ACTD were synthesized by Eurofins Genomics (pEX-A128 vector) and subcloned into CB6-GFP. The coiled-coil amino acid sequence was GELAAIKQELAAIKKELAAIKWELAAIKQ. The I560P helix-disrupting GFP-CC-tet-KLC1ECTD mutant was constructed by PCR-based site-directed mutagenesis using the following primers: forward, 5′-CCAAACTTCGCGCTAGTCCACGGAGAAGTTCCGAG-3′; reverse, 5′-CTCGGAACTTCTCCGTGGACTAGCGCGAAGTTTGG-3′. All plasmids were verified by DNA sequencing.
Bacterial protein expression and purification
His-tagged KLCCTDs were expressed in Escherichia coli BL21(DE3) cells. Single colonies were picked and grown at 37°C overnight. Small-scale overnight bacterial cultures were used to inoculate two 1-liter cultures that were incubated at 37°C until they reached an optical density at 600 nm of 0.6 to 0.8. The temperature was then lowered to 16°C, and protein synthesis was induced by the addition of 300 μM isopropyl b-d-1-thiogalactopyranoside for 16 hours. Cells were harvested by centrifugation at 4000g for 30 min at 4°C and resuspended in 25 mM Hepes buffer (pH 7.4), 500 mM NaCl, 20 mM imidazole, and 5 mM β-mercaptoethanol supplemented with protease inhibitor cocktail (Roche). Cell lysis was accomplished by sonication. Insoluble material was sedimented by centrifugation at 13,500g for 30 min at 4°C before loading on His-trap FF columns (GE Healthcare) preequilibrated with lysis buffer. Proteins were eluted with an imidazole gradient and fractions containing the target protein collected using an AKTA Prime system (GE Life Sciences). Protein quality was analyzed by Coomassie gel analysis. Protein concentration was typically determined by band densitometry using a bovine serum albumin (BSA) standard curve due to the low molar extinction coefficient of the proteins. Proteins were snap-frozen and stored at −80°C.
Purification of HaloTag protein from mammalian cells
The day before transfection, 5 15-cm dishes were seeded with 4.5 × 106 293T cells. These were transfected with 20 μg of KHC and KLC expression plasmid (KIF5C/KLC2-TEV-Halo, HA-KLC1B/KLC1E/I/PKLC1E/KIF5C) using the ProFection Mammalian Transfection System (Promega) as per the manufacturer’s instructions. Cells were left to express the proteins for 48 hours. Cells were then washed with phosphate-buffered saline (PBS) and gently scrapped in 2.5 ml of lysis buffer [50 mM tris (pH 7.5), 150 mM NaCl, 1 mM dithiothreitol (DTT), 1% Triton X-100, 0.1% sodium deoxycholate, 1 mM MgCl2, and 0.1 mM adenosine 5′-diphosphate (ADP) supplemented with protease inhibitor cocktail (Promega)]. Cells were placed on a rocker for 15 min at room temperature before being diluted in three volumes of Halo buffer [50 mM Hepes (pH 7.5), 150 mM NaCl, 1 mM DTT, 0.005% IGEPAL CA-630, 1 mM MgCl2, and 0.1 mM ADP]. The lysate was sonicated for 2 min, 30 s, then pulse for 5 s on, and 10 s off, at 40% amplitude (Vibra Sonics). Insoluble material was pelleted by centrifugation at 10,000g for 30 min at 4°C. The soluble fraction was incubated with 2.5 ml of HaloLink resin slurry (Promega, preequilibrated in Halo buffer) and allowed to incubate on a roller for 90 min at room temperature. Resin was washed in batch by three repeated rounds of centrifugation (1500g, 5 min, 4°C) and resuspension in Halo buffer. After the final wash, liquid was removed as much as possible, and HaloTEV (Tobacco Etch Virus) protease (300 units, Promega) was added to liberate protein. TEV cleavage proceeded for 60 min at room temperature. The soluble material was separated from the resin by use of a spin filter (Thermo Fisher Scientific) and centrifugation (5000g, 5 min, 4°C). Insoluble material was removed by centrifugation (20,000g, 5 min, 4°C), and the sample was further purified by size exclusion chromatography on a Superdex 200 Increase (10/300) column (GE) preequilibrated in 20 mM Hepes (pH 7.5), 150 mM NaCl, 1 mM MgCl2, 0.1 mM ADP, and 5 mM β-mercaptoethanol. The resulting purified protein was snap-frozen in liquid nitrogen and stored at −80°C until use.
Circular dichroism
CD spectra were recorded with a 1-mm path length cell at 20°C using a Jasco 815 spectropolarimeter (Jasco, MD, USA) fitted with a Peltier temperature controller, and baseline-corrected. KLC1D/E synthetic peptides were diluted at 57 mM in PBS buffer (pH 7.4) in the absence or presence of DPC (0.35%, Anatrace), DDM (0.05%, Anatrace), OG (2%, Sigma-Aldrich), or LDAO (0.05%; Anatrace). Spectra were recorded from 260 to 190 nm with a data pitch and bandwidth of 1 nm and a scan speed of 100 nm/min. An average of three runs was recorded for each peptide sample. Fraction helicity (%) was calculated by (%) = 100(([θ]222 − [θ]coil) / (−42,500(1 − (3/n) − [θ]coil)), where [θ]coil = 640 − 45 T = −260° cm2 dmol−1 res−1 at 20°C and n is the number of peptide bonds (64).
NMR spectroscopy
1D proton spectra of free KLC1D/E synthetic peptide titrated with increasing concentrations of DPC were acquired at ∼300 mM in PBS buffer supplemented with 10% D2O at 298 K. The DPC-bound peptide sample was prepared by dissolving lyophilized peptide in PBS supplemented with 7% D2O with 0.7% perdeuterated d38-DPC (Cambridge Isotope Laboratories) at a final concentration of ~2 mM. Standard 2D NMR spectra [NOESY, TOCSY, correlation spectroscopy (COSY), 1H-15N HSQC, and 1H-13C HSQC] were acquired at 298 K. NOESY spectra had mixing times of 80, 120, and 250 ms. To guide assignment of the bound peptide, standard 2D NMR spectra (NOESY, TOCSY, COSY, 1H-15N HSQC and 1H-13C HSQC) were also acquired for the free peptide at 278 K. The signals of the free peptide at the lower temperature were better dispersed and sharper than at 298 K, allowing partially assignment (fig. S5C). All data were collected on a Bruker Avance III HD 700-MHz spectrometer (Bruker, MA, USA) equipped with a 1.7-mm inverse triple resonance micro-cryoprobe using standard pulse sequences from the Bruker library. NMR data were processed in NMRPipe (65) or TopSpin 3.6 and analyzed using CCPNMR analysis v 2.4.2 (66). Dextran sulfate sodium was used as a chemical shift standard.
Liposome preparation and cosedimentation assays
The appropriate lipids were mixed in organic solution, and the solvent was evaporated to dryness under an N2 stream. The sample was then kept under N2 stream for an additional 30 min to remove solvent traces. The lipids were swollen in 25 mM Hepes (pH 7.4), 150 mM NaCl, and 1 mM DTT (Sigma-Aldrich) to obtain multilamellar vesicles (MLVs) at 1 mg/ml. LUVs were prepared from MLVs. They were subjected to 10 freeze/thaw cycles and then extruded using 0.2-μm pore size Nuclepore filters (Whatman). SUVs were obtained by sonicating MLVs with a probe tip sonicator (MSE Soniprep 150, MSE, UK) for 10 min (10 s on, 10 s off) on ice. For cosedimentation assays, 43 μg (unless otherwise specified) of liposomes was incubated with 5 or 0.2 μM of the indicated KLCCTD or tetrameric kinesin-1, respectively (100 μl, final volume), for 30 min and spun down at 170,000g in a Beckman TLA-100 rotor. Supernatant and pellet fractions were collected and analyzed by SDS-PAGE and Coomassie stain.
PIP arrays
PIP arrays (Echelon Biosciences) were used according to the manufacturer’s instructions. Briefly, strips were blocked with 3% fatty acid–free BSA (Sigma-Aldrich) in PBS buffer containing 0.1% Tween 20 overnight at 4°C before incubation with the indicated protein (1 μg/ml) for 1 hour at room temperature. PIP strips were rinsed thoroughly, and protein binding was visualized using an HRP-conjugated anti-His antibody and enhanced chemiluminiscence reagents (Geneflow). Spot intensities were detected on films (GE Healthcare).
BS3 cross-linking assays
BS3 cross-linker (2 mg; Thermo Fisher Scientific) was dissolved in Milli-Q water immediately before use, and 0 to 2 mM were added to the corresponding sample. The reaction mixture was incubated at room temperature for 30 min before quenching with SDS sample buffer for an additional 15 min. Samples were heated at 95°C for 10 min followed by SDS-PAGE analysis.
Cryo–electron microscopy
Forty-three micrograms of liposomes was incubated in the presence or absence of 5 μM His-KLC1ECTD for 30 min. Lacey carbon film grids (EM Resolutions) were glow-discharged for 30 s in a Leica ACE 600 Coater system (Leica Microsystems, Heidelberg, Germany). Five microliters of sample was pipetted onto the grids, and the sample was plunge-frozen in liquid ethane using a Leica GP Plunge freezing device (Leica Microsystems, Heidelberg, Germany). The grids were then transferred under liquid nitrogen into a Gatan 626 Cryotransfer system (Gatan, CA, USA) and imaged at ≤−170°C in a FEI Tecnai 20 LaB6 Scanning Transmission Electron Microscope (FEI, OR, USA) operating at 200 kV and using low-dose imaging conditions. Images were acquired on a FEI CETA 16 M complementary metal-oxide semiconductor camera (FEI, OR, USA).
Cell culture, transfection, and immunoblotting
HeLa and 293T cells were grown in high-glucose Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich) and 1% penicillin/streptomycin (Gibco) at 37°C with 5% CO2. HeLa transient transfections were performed using Effectene transfection reagent (QIAGEN) according to the manufacturer’s instructions. The efficacy of cell transfection was checked using fluorescence microscopy or Western blotting. For immunoblotting, cells grown on six-well plates were initially washed with ice-cold PBS and then lysed with ice-cold radioimmunoprecipitation assay (100 μl per well) buffer consisting of 50 mM Hepes (pH7.5), 150 mM NaCl, 0.1% NP40, 0.5% sodium deoxycholate, and 0.1% SDS supplemented with protease inhibitor cocktail. The homogenates were incubated on ice for 15 min and then cleared by centrifugation at 12,000g for 15 min at 4°C. Supernatants were collected as soluble fractions. Protein samples were resolved on NuPAGE 4 to 12% precast gels (Invitrogen), transferred onto polyvinylidene difluoride membranes, blocked in 5% milk in TBS-T [20 mM tris, 0.25 M NaCl, and 0.1% Tween-20 (pH 7.5) with HCl], and probed with the indicated primary and secondary antibodies (listed above) followed by detection using the Odyssey Infrared Scanning System (LI-COR Biosciences).
Immunofluorescence and cell imaging
HeLa cells were seeded on coverslips in six-well plates before transfection. Twenty-four hours after transfection, cells were fixed with 4% formaldehyde (Thermo Fisher Scientific) for 15 min or ice-cold methanol for 5 min. Fixed cells were blocked in 3% BSA and 1% fetal calf serum (Sigma-Aldrich) buffer and incubated for 30 min with primary and secondary antibodies (listed above) diluted in blocking buffer. Cells were 4′,6-diamidino-2-phenylindole–stained (Thermo Fisher Scientific) and placed cell side down in FluorSave reagent (Calbiochem). Confocal microscopy was carried out using a Leica SP5-AOBS confocal laser scanning microscope attached to a Leica DM I6000 inverted epifluorescence microscope (Leica Microsystems, Heidelberg, Germany). A 63×, 1.4 numerical aperture oil immersion objective (Plan Apochromat BL; Leica Biosystems) and the standard SP5 system acquisition software and detector were used. Image analysis was performed using ImageJ. For colocalization studies, Pearson’s correlation coefficient was calculated from ∼40 cells per condition using ImageJ.
siRNA transfection
In HeLa cells, siRNA transfection was carried out through reverse transfection protocol with HiPerFect (QIAGEN). Six microliters of 10 μM siRNAs targeting human siKLC1 or siKLC2 was mixed with 12 μl of HiPerFect in high-glucose DMEM (Sigma-Aldrich) and combined with 1 × 105 cells per well of a six-well plate. Samples were collected 72 hours after the transfection. For rescue experiments, corresponding mouse KLC1/2 cDNAs (near identical proteins) were transiently transfected so that the siRNA would not recognize the targeting sequence.
Sequence and secondary structure analysis
Sequence alignment of the KLCCTDs was performed using PROMALS3D (67) or Clustal Omega using the MegAlign Pro application of the DNASTAR Lasergene suite as indicted. Some alignments were further manually edited as indicated in the figure legends. The analysis and characterization of the predicted secondary structure were performed using JPred4 (68) and HeliQuest (69). Helical 3D models were prepared with PyMol (Schrödinger).
KLC phylogeny
Orthologs from ENSEMBL (PMID 31691826) for KLC1 to KCL4 were used as a starting point, followed by sequence similarity searching with BLAST to identify KLC homologs in a wider range of species. Sequences were aligned using the l-INS-I mode in MAFFT (70), and IQ-TREE2 (71) was used to infer phylogenies under the best-fitting model, selected by Bayesian information criterion (BIC). An iterative process of tree inference was used to classify homologous sequences as orthologs and paralogs and to identify species-specific duplications. For the final tree, the best-fit model was JTT + R6 under the BIC, Akaike information criterion (AIC), and corrected AIC (AICc) criteria.
Statistical analysis
Data are presented as means ± SEM and were analyzed by t test using Prism 8 (GraphPad Software, CA); *P < 0.05, **P < 0.01, and ***P < 0.001.
Acknowledgments
We are grateful for the assistance and access to equipment at the Wolfson Bioimaging Facility and NMR Facility at the University of Bristol and BrisSynBio, a BBSRC/EPSRC-funded Synthetic Biology Research Centre (BB/L01386X/1). We are grateful to J. Carlton (King’s College London/Francis Crick Institute) for the assistance with pilot experiments associated with this work. Funding: This work was funded by the Biotechnology and Biosciences Research Council (BB/ S000917/1 and BB/S000828/1). M.P.D. is a Lister Institute of Preventative Medicine Prize Fellow. J.A.C is supported by the EPSRC Bristol Center for Doctoral Training in Chemical Synthesis. E.R.R.M. is supported by a Royal Society Enhancement Award to T.A.W. Y.Y.Y. was also supported by the Biotechnology and Biosciences Research Council (BB/L006774/1). Author contributions: Z.A., J.F.W., C.W., E.R.R.M., J.M., Y.Y.Y., and J.A.C. performed the experiments, analyzed the data, and edited the draft manuscript. T.A.W., R.A.S., M.P.C., and D.N.W. analyzed the data and edited the draft manuscript. Z.A. and M.P.D. wrote the original draft. R.A.S. and M.P.D. obtained the funding that supported this work. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/31/eabg6636/DC1
REFERENCES AND NOTES
- 1.Wickstead B., Gull K., Richards T. A., Patterns of kinesin evolution reveal a complex ancestral eukaryote with a multifunctional cytoskeleton. BMC Evol. Biol. 10, 110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vale R. D., The molecular motor toolbox for intracellular transport. Cell 112, 467–480 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Hirokawa N., Noda Y., Tanaka Y., Niwa S., Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol. 10, 682–696 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Vale R. D., Reese T. S., Sheetz M. P., Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell 42, 39–50 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dodding M. P., Way M., Coupling viruses to dynein and kinesin-1. EMBO J. 30, 3527–3539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu W., Gelfand V. I., Moonlighting motors: kinesin, dynein, and cell polarity. Trends Cell Biol. 27, 505–514 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verhey K. J., Hammond J. W., Traffic control: regulation of kinesin motors. Nat. Rev. Mol. Cell Biol. 10, 765–777 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Cross J. A., Dodding M. P., Motor-cargo adaptors at the organelle-cytoskeleton interface. Curr. Opin. Cell Biol. 59, 16–23 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Yip Y. Y., Pernigo S., Sanger A., Xu M., Parsons M., Steiner R. A., Dodding M. P., The light chains of kinesin-1 are autoinhibited. Proc. Natl. Acad. Sci. U.S.A. 113, 2418–2423 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakagawa T., Tanaka Y., Matsuoka E., Kondo S., Okada Y., Noda Y., Kanai Y., Hirokawa N., Identification and classification of 16 new kinesin superfamily (KIF) proteins in mouse genome. Proc. Natl. Acad. Sci. U.S.A. 94, 9654–9659 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cyr J. L., Pfister K. K., Bloom G. S., Slaughter C. A., Brady S. T., Molecular genetics of kinesin light chains: Generation of isoforms by alternative splicing. Proc. Natl. Acad. Sci. U.S.A. 88, 10114–10118 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cabeza-Arvelaiz Y., Shih L. C., Hardman N., Asselbergs F., Bilbe G., Schmitz A., White B., Siciliano M. J., Lachman L. B., Cloning and genetic characterization of the human kinesin light-chain (KLC) gene. DNA Cell Biol. 12, 881–892 (1993). [DOI] [PubMed] [Google Scholar]
- 13.Beushausen S., Kladakis A., Jaffe H., Kinesin light chains: Identification and characterization of a family of proteins from the optic lobe of the squid Loligo pealii. DNA Cell Biol. 12, 901–909 (1993). [DOI] [PubMed] [Google Scholar]
- 14.Lamerdin J. E., Stilwagen S. A., Ramirez M. H., Stubbs L., Carrano A. V., Sequence analysis of the ERCC2 gene regions in human, mouse, and hamster reveals three linked genes. Genomics 34, 399–409 (1996). [DOI] [PubMed] [Google Scholar]
- 15.Wedaman K. P., Knight A. E., Kendrick-Jones J., Scholey J. M., Sequences of sea urchin kinesin light chain isoforms. J. Mol. Biol. 231, 155–158 (1993). [DOI] [PubMed] [Google Scholar]
- 16.McCart A. E., Mahony D., Rothnagel J. A., Alternatively spliced products of the human kinesin light chain 1 (KNS2) gene. Traffic 4, 576–580 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Rahman A., Friedman D. S., Goldstein L. S., Two kinesin light chain genes in mice. Identification and characterization of the encoded proteins. J. Biol. Chem. 273, 15395–15403 (1998). [DOI] [PubMed] [Google Scholar]
- 18.Fan J., Amos L. A., Kinesin light chain isoforms in Caenorhabditis elegans. J. Mol. Biol. 240, 507–512 (1994). [DOI] [PubMed] [Google Scholar]
- 19.Gauger A. K., Goldstein L. S., The Drosophila kinesin light chain. Primary structure and interaction with kinesin heavy chain. J. Biol. Chem. 268, 13657–13666 (1993). [PubMed] [Google Scholar]
- 20.Niclas J., Navone F., Hom-Booher N., Vale R. D., Cloning and localization of a conventional kinesin motor expressed exclusively in neurons. Neuron 12, 1059–1072 (1994). [DOI] [PubMed] [Google Scholar]
- 21.Xia C., Rahman A., Yang Z., Goldstein L. S., Chromosomal localization reveals three kinesin heavy chain genes in mouse. Genomics 52, 209–213 (1998). [DOI] [PubMed] [Google Scholar]
- 22.Kanai Y., Okada Y., Tanaka Y., Harada A., Terada S., Hirokawa N., KIF5C, a novel neuronal kinesin enriched in motor neurons. J. Neurosci. 20, 6374–6384 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Boer S. R., You Y. M., Szodorai A., Kaminska A., Pigino G., Nwabuisi E., Wang B., Estrada-Hernandez T., Kins S., Brady S. T., Morfini G., Conventional kinesin holoenzymes are composed of heavy and light chain homodimers. Biochemistry 47, 4535–4543 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gyoeva F. K., Sarkisov D. V., Khodjakov A. L., Minin A. A., The tetrameric molecule of conventional kinesin contains identical light chains. Biochemistry 43, 13525–13531 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Morihara T., Hayashi N., Yokokoji M., Akatsu H., Silverman M. A., Kimura N., Sato M., Saito Y., Suzuki T., Yanagida K., Kodama T. S., Tanaka T., Okochi M., Tagami S., Kazui H., Kudo T., Hashimoto R., Itoh N., Nishitomi K., Yamaguchi-Kabata Y., Tsunoda T., Takamura H., Katayama T., Kimura R., Kamino K., Hashizume Y., Takeda M., Transcriptome analysis of distinct mouse strains reveals kinesin light chain-1 splicing as an amyloid-beta accumulation modifier. Proc. Natl. Acad. Sci. U.S.A. 111, 2638–2643 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sleigh J. N., Rossor A. M., Fellows A. D., Tosolini A. P., Schiavo G., Axonal transport and neurological disease. Nat. Rev. Neurol. 15, 691–703 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Bayrakli F., Poyrazoglu H. G., Yuksel S., Yakicier C., Erguner B., Sagiroglu M. S., Yuceturk B., Ozer B., Doganay S., Tanrikulu B., Seker A., Akbulut F., Ozen A., Per H., Kumandas S., Torun Y. A., Bayri Y., Sakar M., Dagcinar A., Ziyal I., Hereditary spastic paraplegia with recessive trait caused by mutation in KLC4 gene. J. Hum. Genet. 60, 763–768 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Melo U. S., Macedo-Souza L. I., Figueiredo T., Muotri A. R., Gleeson J. G., Coux G., Armas P., Calcaterra N. B., Kitajima J. P., Amorim S., Olávio T. R., Griesi-Oliveira K., Coatti G. C., Rocha C. R. R., Martins-Pinheiro M., Menck C. F. M., Zaki M. S., Kok F., Zatz M., Santos S., Overexpression of KLC2 due to a homozygous deletion in the non-coding region causes SPOAN syndrome. Hum. Mol. Genet. 24, 6877–6885 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pernigo S., Lamprecht A., Steiner R. A., Dodding M. P., Structural basis for kinesin-1:cargo recognition. Science 340, 356–359 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pernigo S., Chegkazi M. S., Yip Y. Y., Treacy C., Glorani G., Hansen K., Politis A., Bui S., Dodding M. P., Steiner R. A., Structural basis for isoform-specific kinesin-1 recognition of Y-acidic cargo adaptors. eLife 7, e38362 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cockburn J. J. B., Hesketh S. J., Mulhair P., Thomsen M., O’Connell M. J., Way M., Insights into kinesin-1 activation from the crystal structure of KLC2 bound to JIP3. Structure 26, 1486–1498.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verhey K. J., Meyer D., Deehan R., Blenis J., Schnapp B. J., Rapoport T. A., Margolis B., Cargo of kinesin identified as JIP scaffolding proteins and associated signaling molecules. J. Cell Biol. 152, 959–970 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cross J. A., Chegkazi M. S., Steiner R. A., Woolfson D. N., Dodding M. P., Fragment-linking peptide design yields a high-affinity ligand for microtubule-based transport. Cell Chem. Biol. 10.1016/j.chembiol.2021.03.010 (2021). [DOI] [PubMed] [Google Scholar]
- 34.Wozniak M. J., Allan V. J., Cargo selection by specific kinesin light chain 1 isoforms. EMBO J. 25, 5457–5468 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khodjakov A., Lizunova E. M., Minin A. A., Koonce M. P., Gyoeva F. K., A specific light chain of kinesin associates with mitochondria in cultured cells. Mol. Biol. Cell 9, 333–343 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gyoeva F. K., Bybikova E. M., Minin A. A., An isoform of kinesin light chain specific for the Golgi complex. J. Cell Sci. 113, 2047–2054 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Amato S., Liu X., Zheng B., Cantley L., Rakic P., Man H.-Y., AMP-activated protein kinase regulates neuronal polarization by interfering with PI 3-kinase localization. Science 332, 247–251 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morfini G., Szebenyi G., Elluru R., Ratner N., Brady S. T., Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 21, 281–293 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wright B. D., Henson J. H., Wedaman K. P., Willy P. J., Morand J. N., Scholey J. M., Subcellular localization and sequence of sea urchin kinesin heavy chain: Evidence for its association with membranes in the mitotic apparatus and interphase cytoplasm. J. Cell Biol. 113, 817–833 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Söding J., Biegert A., Lupas A. N., The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Z., Zhang F., He J., Wu P., Tay L. W. R., Cai M., Nian W., Weng Y., Qin L., Chang J. T., McIntire L. B., Paolo G. D., Xu J., Peng J., Du G., Binding of PLD2-generated phosphatidic acid to KIF5B promotes MT1-MMP surface trafficking and lung metastasis of mouse breast cancer cells. Dev. Cell 43, 186–197.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fletcher J. M., Boyle A. L., Bruning M., Bartlett G. J., Vincent T. L., Zaccai N. R., Armstrong C. T., Bromley E. H. C., Booth P. J., Brady R. L., Thomson A. R., Woolfson D. N., A basis set of de novo coiled-coil peptide oligomers for rational protein design and synthetic biology. ACS Synth. Biol. 1, 240–250 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Tsuruta F., Dolmetsch R. E., PIKfyve mediates the motility of late endosomes and lysosomes in neuronal dendrites. Neurosci. Lett. 605, 18–23 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Li X., Rydzewski N., Hider A., Zhang X., Yang J., Wang W., Gao Q., Cheng X., Xu H., A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat. Cell Biol. 18, 404–417 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pu J., Guardia C. M., Keren-Kaplan T., Bonifacino J. S., Mechanisms and functions of lysosome positioning. J. Cell Sci. 129, 4329–4339 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanger A., Yip Y. Y., Randall T. S., Pernigo S., Steiner R. A., Dodding M. P., SKIP controls lysosome positioning using a composite kinesin-1 heavy and light chain-binding domain. J. Cell Sci. 130, 1637–1651 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosa-Ferreira C., Munro S., Arl8 and SKIP act together to link lysosomes to kinesin-1. Dev. Cell 21, 1171–1178 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dodding M. P., Mitter R., Humphries A. C., Way M., A kinesin-1 binding motif in vaccinia virus that is widespread throughout the human genome. EMBO J. 30, 4523–4538 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keren-Kaplan T., Bonifacino J. S., ARL8 relieves SKIP autoinhibition to enable coupling of lysosomes to kinesin-1. Curr. Biol. 31, 540–554.e5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pu J., Schindler C., Jia R., Jarnik M., Backlund P., Bonifacino J. S., BORC, a multisubunit complex that regulates lysosome positioning. Dev. Cell 33, 176–188 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Siddiqui N., Straube A., Intracellular cargo transport by kinesin-3 motors. Biochemistry (Mosc) 82, 803–815 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seiler S., Kirchner J., Horn C., Kallipolitou A., Woehlke G., Schliwa M., Cargo binding and regulatory sites in the tail of fungal conventional kinesin. Nat. Cell Biol. 2, 333–338 (2000). [DOI] [PubMed] [Google Scholar]
- 53.Blasius T. L., Cai D., Jih G. T., Toret C. P., Verhey K. J., Two binding partners cooperate to activate the molecular motor kinesin-1. J. Cell Biol. 176, 11–17 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Skoufias D. A., Cole D. G., Wedaman K. P., Scholey J. M., The carboxyl-terminal domain of kinesin heavy chain is important for membrane binding. J. Biol. Chem. 269, 1477–1485 (1994). [PubMed] [Google Scholar]
- 55.Lu W., Lakonishok M., Gelfand V. I., Kinesin-1-powered microtubule sliding initiates axonal regeneration in Drosophila cultured neurons. Mol. Biol. Cell 26, 1296–1307 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Navone F., Niclas J., Hom-Booher N., Sparks L., Bernstein H. D., McCaffrey G., Vale R. D., Cloning and expression of a human kinesin heavy chain gene: Interaction of the COOH-terminal domain with cytoplasmic microtubules in transfected CV-1 cells. J. Cell Biol. 117, 1263–1275 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Setou M., Seog D.-H., Tanaka Y., Kanai Y., Takei Y., Kawagishi M., Hirokawa N., Glutamate-receptor-interacting protein GRIP1 directly steers kinesin to dendrites. Nature 417, 83–87 (2002). [DOI] [PubMed] [Google Scholar]
- 58.Du W., Su Q. P., Chen Y., Zhu Y., Jiang D., Rong Y., Zhang S., Zhang Y., Ren H., Zhang C., Wang X., Gao N., Wang Y., Sun L., Sun Y., Yu L., Kinesin 1 drives autolysosome tubulation. Dev. Cell 37, 326–336 (2016). [DOI] [PubMed] [Google Scholar]
- 59.Barry J., Xu M., Gu Y., Dangel A. W., Jukkola P., Shrestha C., Gu C., Activation of conventional kinesin motors in clusters by Shaw voltage-gated K+ channels. J. Cell Sci. 126, 2027–2041 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ichimura T., Wakamiya-Tsuruta A., Itagaki C., Taoka M., Hayano T., Natsume T., Isobe T., Phosphorylation-dependent interaction of kinesin light chain 2 and the 14-3-3 protein. Biochemistry 41, 5566–5572 (2002). [DOI] [PubMed] [Google Scholar]
- 61.Brendza R. P., Serbus L. R., Duffy J. B., Saxton W. M., A function for kinesin I in the posterior transport of oskar mRNA and staufen prote. Science 289, 2120–2122 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jolly A. L., Kim H., Srinivasan D., Lakonishok M., Larson A. G., Gelfand V. I., Kinesin-1 heavy chain mediates microtubule sliding to drive changes in cell shape. Proc. Natl. Acad. Sci. U.S.A. 107, 12151–12156 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fukuda Y., Pazyra-Murphy M. F., Silagi E. S., Tasdemir-Yilmaz O. E., Li Y., Rose L., Yeoh Z. C., Vangos N. E., Geffken E. A., Seo H.-S., Adelmant G., Bird G. H., Walensky L. D., Marto J. A., Dhe-Paganon S., Segal R. A., Binding and transport of SFPQ-RNA granules by KIF5A/KLC1 motors promotes axon survival. J. Cell Biol. 220, e202005051 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Scholtz J. M., Qian H., York E. J., Stewart J. M., Baldwin R. L., Parameters of helix-coil transition theory for alanine-based peptides of varying chain lengths in water. Biopolymers 31, 1463–1470 (1991). [DOI] [PubMed] [Google Scholar]
- 65.Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A., NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 66.Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., Laue E. D., The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins 59, 687–696 (2005). [DOI] [PubMed] [Google Scholar]
- 67.Pei J., Kim B. H., Grishin N. V., PROMALS3D: A tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 36, 2295–2300 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Drozdetskiy A., Cole C., Procter J., Barton G. J., JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 43, W389–W394 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gautier R., Douguet D., Antonny B., Drin G., HELIQUEST: A web server to screen sequences with specific alpha-helical properties. Bioinformatics 24, 2101–2102 (2008). [DOI] [PubMed] [Google Scholar]
- 70.Katoh K., Standley D. M., MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Minh B. Q., Schmidt H. A., Chernomor O., Schrempf D., Woodhams M. D., von Haeseler A., Lanfear R., IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/31/eabg6636/DC1