

Abstract
Epoxyeicosatrienoic acids (EETs) are lipid signaling molecules formed from arachidonic acid by the action of CYP450s. EETs cause vasodilation, exert anti-inflammatory effects, and enhance insulin secretion and sensitivity in rodents. EETs are metabolized to less active DHETs by soluble epoxide hydrolase (sEH), and 14,15-DHET has been reported to be increased in patients with primary aldosteronism, but the effects of aldosterone on EET concentrations and sEH activity are unknown.
We tested the hypothesis that treatment of primary aldosteronism would alter EET concentrations in humans. We studied nine patients with primary aldosteronism before and at least three months after unilateral adrenalectomy (six) or treatment with a mineralocorticoid (MR) antagonist (three). Circulating total EET concentrations increased to 18.5±12.6 from 11.9±5.9 nM with treatment of primary aldosteronism (P=0.027). Among the regioisomers of EETs, 14,15-EET significantly increased after treatment, whereas 11,12-EET and 8,9 EET were unaltered. Total DHET concentrations and specific regioisomers of DHET did not change significantly. Circulating total EETs (ρ=−0.52, P=0.026), 14,15-EET (ρ=−0.56, P=0.015) and 11,12-EET (ρ=−0.48, P=0.042) correlated inversely with aldosterone. We also assessed EETs after a 12-hour aldosterone or vehicle infusion in a randomized cross-over study in healthy volunteers. Plasma EETs were similar after 12-hour aldosterone or vehicle infusion. Lastly, three-day infusion of aldosterone in mice decreased EET concentrations in adipose and muscle and increased sEH expression as determined by molar ratio of DHETs to EETs and soluble epoxide hydrolase (Ephx2) mRNA expression in adipose tissue. These studies suggest that prolonged exposure to increased aldosterone decreases EET concentrations.
Keywords: aldosterone, mineralocorticoids, CYP450 eicosanoids, primary aldosteronism, hypertension, high blood pressure, insulin resistance
Graphical Abstract
Introduction
Epoxyeicosatrienoic acids (EETs) are lipid signaling molecules formed from arachidonic acid by the action of cytochrome P450s (predominately CYP2C and CYP2J).1 EETs cause vasodilation, exert anti-inflammatory effects, and enhance insulin secretion and sensitivity in rodent models.2–6 EETs are hydrolyzed to their less-active diols, dihydroxyeicosatrienoic acids (DHETs), by soluble epoxide hydrolase (sEH).7, 8 Thus, factors that reduce EET concentrations, such as increases in sEH activity, may be expected to promote vasoconstriction, inflammation and insulin resistance.
Increased sEH expression and decreased EET concentrations contribute to hypertension and end-organ damage during activation of the renin-angiotensin-aldosterone system in rodent models. For example, Ang II infusion increases expression of sEH in the heart and kidney of rats,9, 10 and treatment of human endothelial cells with angiotensin II (Ang II) increases sEH expression via the binding of c-Jun to AP-1 sites in the sEH promoter.11 Conversely, administration of an sEH inhibitor or stable EET analog reduces Ang II-induced injury.12–14 Whether increased sEH activity or decreased EETs contribute to the cardiovascular effects of excess aldosterone has not been studied extensively. sEH deficient mice are protected against hypertension and renal inflammation in a deoxycorticosterone acetate/high salt-induced model of mineralocorticoid receptor (MR)-mediated hypertension.15 In addition, whereas aldosterone causes sodium retention by activating the epithelial sodium channel (ENaC), EETs inhibit ENaC.16 In a single study in humans, Liu et al reported that serum 14,15-DHET concentrations are increased in patients with primary aldosteronism compared to patients with essential hypertension and correlate with aldosterone concentrations.17 The authors inferred that 14,15-DHET concentrations reflected EET production, but did not measure EET concentrations or sEH activity.
This study tested the hypothesis that aldosterone alters EET concentrations in humans. To determine the contribution of endogenous aldosterone to EET concentrations, we measured circulating EET concentrations in patients with primary aldosteronism before and after surgical resection of an aldosterone-secreting adenoma or medical treatment with an MR antagonist. We measured EET regioisomers using a highly specific mass spectrometric assay. To further assess the mechanism for changes in EET concentrations, we measured sEH activity in plasma. We next measured the effect of overnight infusion of exogenous aldosterone on plasma EET concentrations in a double-blind, placebo-controlled crossover study in healthy men and women. We also assessed the effect of sustained three-day treatment with aldosterone on EET concentrations in liver, adipose, and muscle in mice.
Methods
This article adheres to the American Heart Association Journals implementation of the Transparency and Openness Promotion Guidelines. The data that support the findings of this study are available from the corresponding author on reasonable request.
Human Study 1: Effect of treatment of primary aldosteronism on plasma EET concentrations
Patients with a primary aldosteronism were enrolled at Vanderbilt University Medical Center (VUMC) and the Brigham and Women’s Hospital in a study designed to determine the effect of reducing excess aldosterone on insulin secretion and sensitivity (NCT02362308). The details of the protocol are provided elsewhere.18 In brief, patients age 18 to 70 years with primary aldosteronism were identified prospectively by physician referral and medical record review. After informed consent was obtained, participants underwent a screening history and physical exam and were included if they met the following criteria for primary aldosteronism: 1) screening plasma aldosterone ≥ 15 ng/dL or aldosterone-to-renin ratio of ≥ 40 (or ≥ 30 if on ACE inhibitor) and 2) confirmatory testing by either failure to suppress aldosterone to <7 ng/dL after intravenous saline infusion or urinary aldosterone excretion ≥ 12 μg/day while urine sodium excretion ≥200 mmol/day. Females of child-bearing potential were excluded unless they were willing to use adequate birth-control and undergo pregnancy testing throughout the study. Other exclusion criteria included prior diagnosis of diabetes or use of anti-diabetic medication, potassium >5.5 mmol/L, sodium <135 mmol/L, allergy to study medications, or major cardiovascular, renal, hepatic, pulmonary, neurologic, or psychiatric disorders.
Participants were studied after withdrawal of any MR antagonist or amiloride for six weeks prior to definitive treatment, and again three to twelve months after unilateral adrenalectomy (six patients) or initiation of MR antagonist therapy (three patients). To standardize sodium intake on each study day, patients were provided a calorie-controlled weight-maintenance diet containing 160 mmol/d sodium 100 mmol/d potassium, and 1000 mg/d calcium under the guidance of study dieticians. On the 5th and 7th day of diet we conducted hyperglycemic and euglycemic clamps, respectively. One participant consented only for the hyperglycemic clamp. Body composition was measured by dual energy X-ray absorptiometry (DEXA) once during each study period, with a GE Lunar model IDXA Absorptiometer. Blood for measurement of plasma EETs was collected prior to the initiation of the hyperglycemic and hyperinsulinemic clamps.
Human Study 2: Effect of overnight infusion of aldosterone on plasma EET concentrations
Healthy volunteers were enrolled at VUMC in a study designed to assess the effect of acute aldosterone infusion on glucose-stimulated insulin secretion (NCT00732160) and described previously.19 Individuals with a body mass index ≥30 kg/m2, fasting glucose >110 mg/dL, anti-diabetic medication use, serum triglycerides >150 mg/dL, total cholesterol >200 mg/dL, supine blood pressure >130/85 mmHg, recent glucocorticoid therapy, renal insufficiency, serum potassium < 3.5 mEq/L, or any serious medical condition were excluded. Participants were provided a 20 mmol/d sodium, calorie-controlled diet for nine days and a 160 mmol/d sodium diet for seven days. Diets were similar in caloric content from carbohydrate (56%), fat (27%), and protein (17%) and were controlled for potassium (80 mEq/d) and calcium (10 mEq/d).
Participants presented to the Vanderbilt Clinical Research Center in the evenings of the sixth and eighth days of the low-sodium diet and of the fourth and sixth days of the high-sodium diet. Beginning at 10 pm, they underwent a 12.5-hour, overnight infusion of either aldosterone (0.7 μg/kg/h in 5% dextrose water, Professional Compounding Corporation of America) or vehicle in a randomized, double-blind, crossover fashion during both low and high salt intake. Hyperglycemic clamps were performed during the last two hours of each infusion. Serum potassium was monitored, and potassium supplementation was administered as necessary to maintain the serum potassium greater than 3.7 mEq/L prior to the initiation of the clamp. The procedure and data from the clamp studies have been reported previously. For this study, we analyzed EETs in plasma samples that were collected for this purpose at the end of aldosterone and vehicle infusion during low (N=3) and high (N=16) salt intake. Because there was no significant effect of salt intake and the number of samples collected during low salt intake was small, we analyzed samples collected during low and high salt intake together.
Mouse study: Effect of three-day administration of aldosterone on tissue EET concentrations
All experiments were approved by the Vanderbilt Institutional Animal Care and Use Committee, and NIH principle of laboratory animal care were followed. Aldosterone (1.5 µg/day, Sigma-Aldrich; n=8) or vehicle (0.9% sodium chloride, 1.25% DMSO; n=8) was infused via osmotic minipump (Alzet Model 1003D, Alza, Palo Alto, CA) in 20–25 g male wild-type C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME, USA). After minipump implantation, mice were provided standard chow and free access to water. After three days mice were euthanized under anesthesia, and tissues were then collected and snap frozen in liquid nitrogen (gonadal adipose, liver, and gastrocnemius muscle). Tissues were stored at −80°C until analysis, and then EET and DHET regioisomers were analyzed.
Laboratory Analysis
Plasma steroid concentrations were measured by liquid chromatography-tandem mass spectrometry at the University of Michigan as previously described.20, 21 Blood for plasma glucose was immediately centrifuged and analyzed using the glucose oxidase method (YSI 2300 STAT Plus Glucose Analyzer YSI Life Sciences; Yellow Springs, IL). Blood was collected in EDTA and plasma was separated and stored at −80ºC until assayed. Aprotinin was added to plasma for measurement of insulin and C-peptide by radioimmunoassay. Insulin, C-peptide, and ACTH were determined by RIA in the Vanderbilt Diabetes Research Translational Center Hormone Assay laboratory.
Plasma EETs, DHETs, epoxy-12Z-octadecenoic acids (EpOMEs), and dihydroxy-12Z-octadecenoic acids (DiHOMEs) were quantified using ultra-performance liquid chromatography/tandem mass spectrometry with select reaction monitoring (SRM) on a triple quadrupole mass spectrometer equipped with an electrospray source (ESI) operated in the negative ion-mode.16
Plasma sEH activity was measured by the hydration of pharmacologic concentrations of EETs or EpOMEs to their respective DHETs or DiHOMEs. Activity was performed in 50 µl of human plasma with 50 µl of 0.1M NaPi (PH 7.4) containing 0.1 mg/ml BSA or 100 µl of plasma. After pre-warming the plasma to 37°C, 50 µM (1 mM, 5 µl EtOH) of EET or EpOME was added. Samples were then incubated for 10 min at 37°C in a shaking water bath. Reactions were stopped by the addition of 5 ml of AcOEt containing 0.5% AcOH (v/v), followed by addition of 2 ng of D11-DHETs or D4-DiHOMEs (internal standards) and 1 ml of 0.15M KCl, and centrifugation at 3000 X g for 1 min. The organic phase was separated from the mixture and evaporated to dryness under a N2 stream in a 37°C water bath. Each sample was dissolved in a mixture of 15 mM-aqueous AcONH4 (PH 8.3): CH3CN (1:1, v/v), followed by centrifugation at 23,000 X g for 3 min. The supernatant (10 µL) was injected onto a Waters Acquity UPLC coupled with a TSQ Vantage Mass Spectrometer for product- DHETs or DiHOMEs -quantification as previously described.16, 22
Total RNA was extracted from adipose tissues of treatment and control groups using TRIzol Reagent (Invitrogen) followed by clean up with RNeasy Mini Kit (Qiagen). RNA was reverse transcribed to cDNA using iScript™ Reverse Transcription Supermix (BIO-RAD). qPCR was performed to evaluate Ephx2 mRNA expression level with GAPDH as a housekeeping control. Gene expression was evaluated by comparing the mRNA expression of the gene of interest with GAPDH utilizing the comparative CT method for Bio-Rad qPCR system.23 Primer sequences are provided in Table S1.
Statistical analysis
Continuous variables are presented as mean ± standard deviation, unless otherwise indicated. Within-patient comparisons were made using the Wilcoxon signed rank test, and unpaired comparisons were made using Wilcoxon rank sum test. In patients with primary aldosteronism, two pretreatment measurements of EETs were averaged (one before each clamp study), as were post-treatment measurements. Correlations between measures were assessed using Spearman’s rho (ρ). To account for multiple comparisons, we set false discovery rate (FDR) at 0.05 using the FDR-controlling procedures by Benjamini and Hochberg within a group of endpoints.24 All statistical analyses were conducted using R (version ≥ 4.0).25 A two-sided p-value less than 0.05 was considered significant.
Results
Effect of treatment of primary aldosteronism on plasma EETs
Among nine patients with primary aldosteronism, six were treated with adrenalectomy and three were treated by administration of a MR antagonist. The characteristics of these patients appear in Table S2 and have been published previously.18 Treatment significantly decreased serum aldosterone and 11-deoxycorticosterone, whereas serum potassium trended higher (Table S3). Android body fat increased significantly after treatment (Table S3).
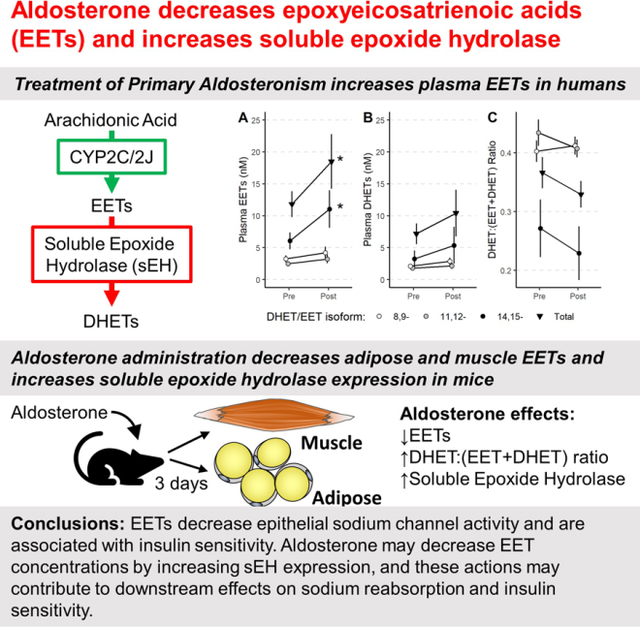
Plasma total EETs (18.5±12.6 vs 11.8±5.9 nmol/L; P=0.027) and 14,15-EETs (11.0±8.6 vs 6.1±3.7 nmol/L; P=0.12) were significantly increased following treatment compared to prior to treatment of primary aldosteronism (Figure 1). DHET concentrations did not change significantly after treatment. There was no difference in EET concentrations after treatment between those who underwent adrenalectomy and those who were treated with an MR antagonist (Figure S2, total plasma EETs 16.2±10.3 vs 23.1±17.9 nM in adrenalectomized patients and in patients treated with an MR antagonist; P=0.64).
Figure 1: Plasma 14,15- and Total EETs increased after treatment of primary aldosteronism.
Plasma total (▼), 14,15- (●), 11,12- (●), and 8,9 (○)-epoxyeicosatrienoic acid (EET, A) and dihydroxyeicosatrienoic acid (DHET, B) concentrations before (Pre) and three to twelve months after (Post) treatment of primary aldosteronism. Soluble epoxide hydrolase (sEH) activity was estimated indirectly by the molar ratios of individual and total DHET/(EET+DHET) (C). Data for individual EET concentrations appear in Figure S1.
To determine whether the increase in EET concentrations observed following treatment of primary aldosteronism resulted from decreased sEH activity, we measured plasma sEH activity by incubation of plasma with pharmacological concentrations of EETs and EpOMEs to determine the rate of hydrolysis to their respective DHET and DiHOME products. We also calculated the molar ratio of each DHET to EET + DHET isomer in plasma [DHET:(EET+DHET)]. There was no effect of treatment on the ex vivo hydrolysis of 14,15-EET, 11,12-EET, 12,13-EpOME, or 9,10-EpOME (Table S4). The hydrolysis of exogenous 8,9-EET to 8,9-DHET was significantly increased following treatment (0.80±0.03 vs 0.66±0.03 pmol/mL/min; P=0.027), but the molar ratio of endogenous 8,9-DHET to 8,9-(EET+DHET) (0.7±0.2 vs 0.7±0.1, P=0.82) and other EETs were not significantly changed (Table S5, total DHET:(EET+DHET) ratio shown in Figure 1C). Thus, a decrease in the activity of sEH, the primary mediator of EET hydrolysis, does not appear to mediate the increase in EET concentrations.
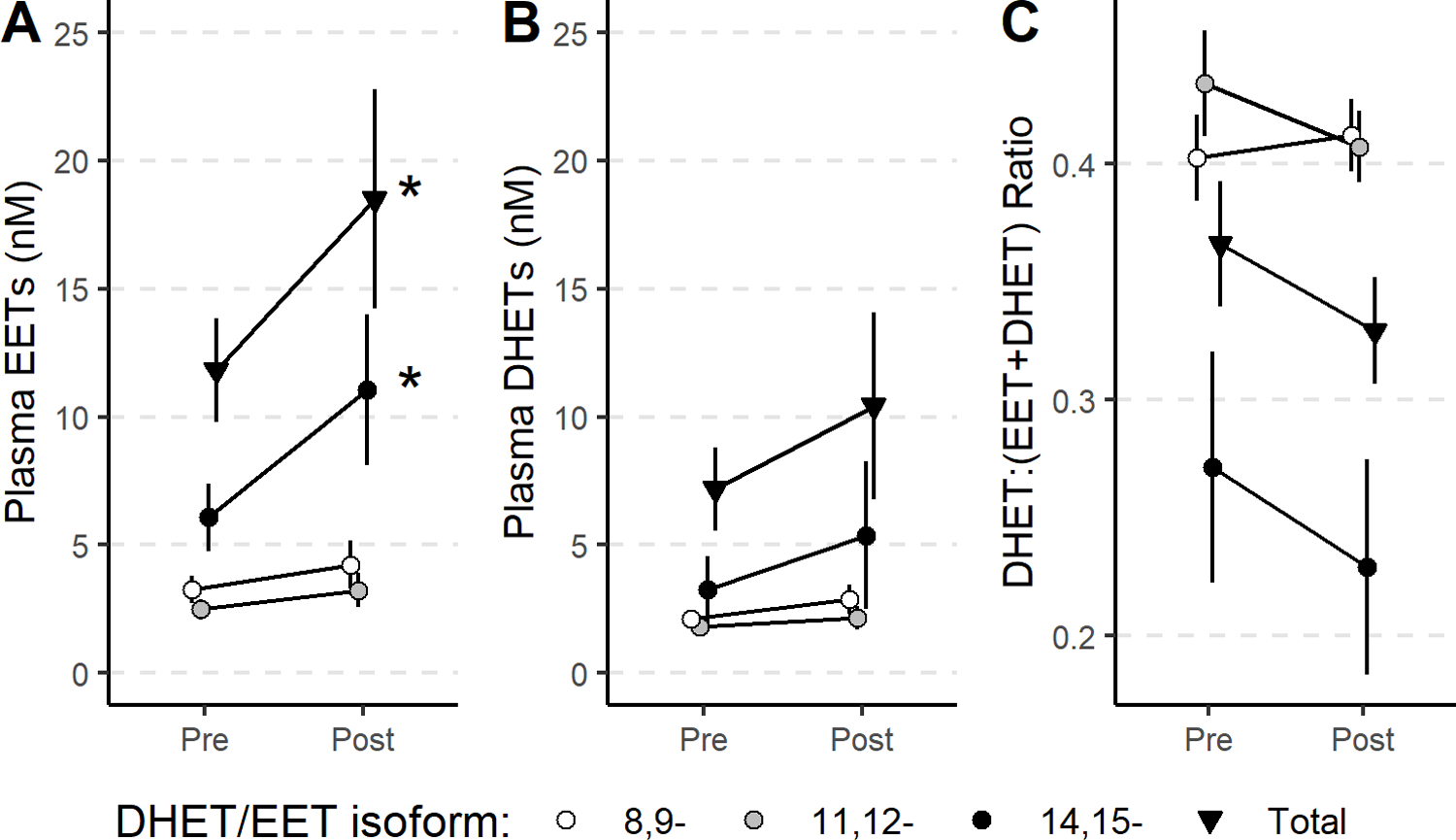
We next assessed the relationship between EET concentrations and aldosterone concentrations measured before and after treatment. Aldosterone correlated inversely with 14,15-EET (ρ=0.56, P=0.015), 11,12-EET (ρ=0.48, P=0.042), and total-EETs (ρ=0.52, P=0.026) (Figure 2). Total EETs and all regioisomers correlated inversely with plasma cortisol and with ACTH concentrations, but these correlations were not significant after adjustment for multiple comparisons. There was also no correlation between any EET isomer and other steroids measured (Table S6).
Figure 2: Plasma aldosterone inversely correlates with EETs in participants with primary aldosteronism.
Correlation between plasma aldosterone and plasma 8,9-EET (A), 11,12-EET (B), 14,15-EET (C), and total EETs (D) concentrations in participants with primary aldosteronism.
There was no relationship between body weight or body composition and aldosterone. Weight correlated inversely with 14,15-EET and total EETs (Table 1). Fat mass measured by DEXA, correlated inversely with 14,15-EET, and % android fat correlated inversely with 14,15-DHET:(EET+DHET) ratio. During the hyperinsulinemic clamp, glucose infusion rate (GIR) a measure of insulin sensitivity, correlated with 14,15-EET (ρ=0.58, P=0.036), 8,9-EET (ρ=0.56, P=0.036), and total EETs (ρ=0.64, P=0.027) (Figure S2).
Table 1:
Correlation between epoxyeicosatrienoic acid (EET) concentrations and measures of body composition or steroids
| Measure | 14,15-EET | 11,12-EET | 8,9-EET | Total EETs | 14,15-DHET:(DHET+EET) |
|---|---|---|---|---|---|
| Body Weight |
ρ=−0.72 P=0.01 |
ρ=−0.52 P=0.12 |
ρ=−0.44 P=0.20 |
ρ=−0.66 P=0.030 |
ρ=−0.28 P=0.52 |
| Fat mass |
ρ=−0.67 P=0.028 |
ρ=−0.46 P=0.19 |
ρ=−0.45 P=0.20 |
ρ=−0.61 P=0.056 |
ρ=−0.45 P=0.20 |
| % Android Fat |
ρ=−0.6 P=0.057 |
ρ=−0.51 P=0.13 |
ρ=−0.55 P=0.099 |
ρ=−0.6 P=0.057 |
ρ=−0.67 P=0.028 |
| Corticosterone |
ρ=−0.11 P=0.83 |
ρ=−0.2 P=0.67 |
ρ=−0.24 P=0.59 |
ρ=−0.14 P=0.77 |
ρ=−0.1 P=0.85 |
| Cortisol |
ρ=−0.49 P=0.14 |
ρ=−0.53 P=0.11 |
ρ=−0.52 P=0.12 |
ρ=−0.54 P=0.10 |
ρ=−0.4 P=0.28 |
| MHigh-dose Insulin |
ρ=0.6 P=0.093 |
ρ=0.48 P=0.22 |
ρ=0.58 P=0.11 |
ρ=0.65 P=0.057 |
ρ=0.69 P=0.044 |
P-values are adjusted according to Benjamini and Hochberg. ρ indicates Spearman’s rank correlation ρ. ACTH indicates adrenocorticotropic hormone, and M indicates glucose infusion rate during high-dose insulin infusion, the primary measure of insulin sensitivity during hyperinsulinemic clamp.
Effect of acute overnight infusion of aldosterone on plasma EETs
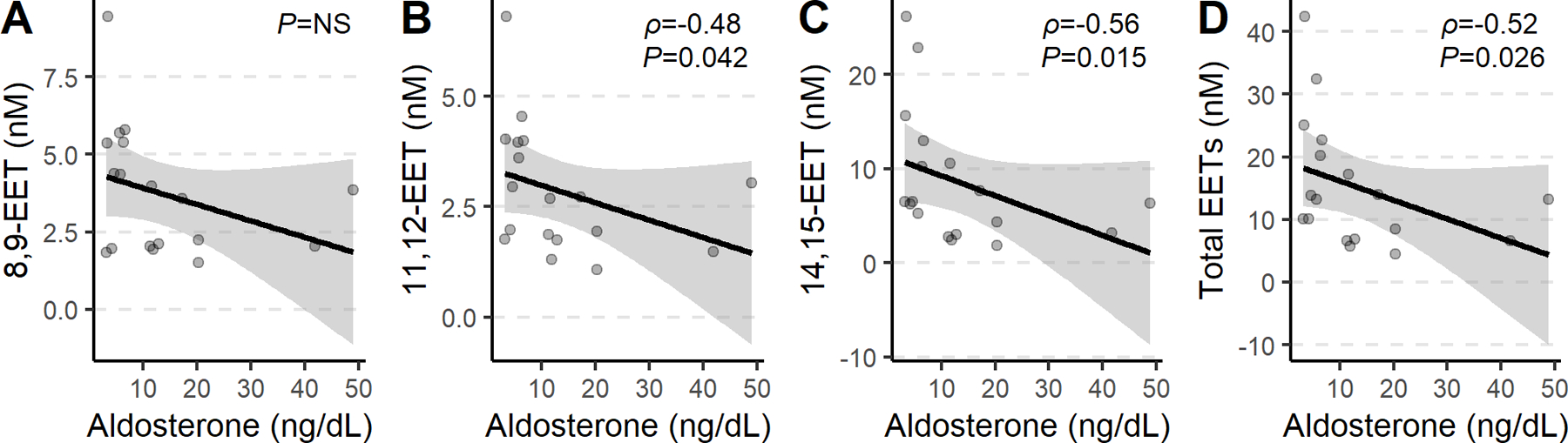
Plasma aldosterone increased to 48.26±15.50 from 9.35±7.23 ng/mL ng/mL (p<0.001) after overnight infusion of aldosterone and did not increase significantly after overnight infusion of vehicle (to 11.17±11.22 from 8.10±5.87 ng/mL). Plasma 14,15-EET tended to be lower during aldosterone infusion compared to during placebo (Figure 3), but this difference was not significant (P=0.08). There was no significant effect of acute aldosterone infusion on any of the other EET isomers or on total plasma EETs.
Figure 3: Plasma EETs, DHETs, and ratios in participants receiving aldosterone infusions.
Plasma total (▼), 14,15- (●), 11,12- (●), and 8,9 (○)-epoxyeicosatrienoic acid (EET, A), DHET concentrations (B) and DHET:EET+DHET ratio (C) did not change significantly after 12.5-hour overnight infusion of aldosterone versus vehicle in healthy volunteers.
Effect of three-day infusion of aldosterone on tissue DHETs and EETs in mice
EET, DHET, EPOME, and DiHOME concentrations were similar in liver from mice treated with aldosterone and vehicle (Table 2). In contrast, the concentration of 8,9-DHET, the molar ratio of DHETs to EETs, and the molar ratio of DiHOME to EPOME were all significantly increased in adipose tissue from aldosterone-treated mice compared to controls. Aldosterone significantly increased soluble epoxide hydrolase in adipose tissue, as determined by the expression of Ephx2 mRNA (4.24±1.75 vs 1.71±1.31 AU, P=0.03, Figure S3).
Table 2:
Effect of aldosterone infusion on tissue EETs, DHET, and molar ratio in mice
| Tissue | Treatment | 14,15 EET | 11,12 EET | 8,9 EET | TOTAL EETs | 14,15- DHET:(EET+DHET) |
12,13- DiHOME:(EpOME+DiHOME) |
|---|---|---|---|---|---|---|---|
| Adipose | Control | 79.9±20.7 | 36.8±13.0 | 49.7±23.7 | 157.2±44.7 | 0.261±0.079 | 0.194±0.055 |
| Aldosterone | 72.0±17.7 | 22.6±9.1 | 56.6±26.7 | 148.3±21.6 | 0.392±0.118* | 0.273±0.076* | |
| Liver | Control | 4917±1884 | 2155±703 | 1461±409 | 8533±2899 | 0.118±0.049 | 0.015±0.006 |
| Aldosterone | 5391±1464 | 2417±906 | 1462±464 | 9270±2588 | 0.102±0.022 | 0.013±0.004 | |
| Muscle | Control | 51.3±6.6 | 49.4±16.8 | 27.3±7.1 | 128.1±18.1 | 0.091±0.012 | 0.128±0.012 |
| Aldosterone | 44.6±6.6* | 25.4±7.6** | 25.5±9.0 | 95.5±16.3** | 0.086±0.020 | 0.116±0.021 | |
Wilcoxon rank sum test. Results are expressed as mean±SD. The units for EETS and DHETs are pmol/g tissue.
p<0.05
p<0.01 versus control.
n=8 in each of the vehicle- treated and aldosterone-treated groups
In muscle, 11,12-DHET and total DHET were significantly increased, while 14,15-, 11,12-, and total EET concentrations were significantly decreased in aldosterone-treated mice compared to controls. The molar ratio of DHET to EET was also increased in the muscle of aldosterone-treated mice compared to controls.
Discussion
EETs act as vasodilatory and anti-natriuretic factors, which generally oppose the pro-hypertensive effects of aldosterone. The factors which regulate endogenous EET production and hydrolyzation in humans are largely unknown, and the effect of aldosterone on EET concentrations has not been reported previously. Our novel findings show that circulating plasma EET concentrations increase following treatment of primary aldosteronism, suggesting that chronic elevation of endogenous aldosterone decreases EETs. Supporting studies in mice demonstrate that these changes are accompanied by increased expression of sEH and hydrolyzation of EETs to their inactive DHETs.
The significantly increased EET concentrations in patients treated with an MR antagonist as well as those treated with adrenalectomy suggests that aldosterone decreases EETs through an MR-dependent mechanism, but further studies are needed to test this hypothesis. The increase in EETs with treatment of primary aldosteronism was driven primarily by an increase in 14,15-EET and was seen in patients treated either by surgical resection of an aldosterone-producing tumor or by MR antagonism. Other alterations, such as increased renin and Ang II concentrations after successful treatment of primary aldosteronism could also contribute to increased EET concentrations. For example, Ang II increases adrenal zona glomerulosa cell EET and DHET production which contributes to increased adrenal blood flow, and this vascular response is prevented by inhibition of EET synthesis or administration of the EET antagonist 14,15-EEZE 26 Ang II has not been demonstrated to affect EET production in other tissues or circulating EETs, but has been shown to increase Ephx2 expression which could counteract these effects.11 Plasma cortisol concentrations exceed aldosterone concentrations by 100 to 1000-fold, which can activate the MR in many tissues due to a similar affinity of the MR for cortisol and aldosterone. In aldosterone-sensitive tissues, conversion of cortisol to the MR-inactive cortisone by 11β-hydroxysteroid dehydrogenase confers specificity for aldosterone.27 In the present study, treatment of primary aldosteronism reduced circulating aldosterone and 11-deoxycorticosterone concentrations without affecting cortisol concentrations. Nevertheless, we also observed an inverse relationship between ACTH, 11-deoxycortisol and cortisol and EETs. Taken together, these data suggest that activation of the MR by either aldosterone or cortisol decreases plasma EET concentrations.
Increased adrenal steroids and MR activation may decrease EET concentrations either by decreasing EET production from arachidonic acid via CYP2C and CYP2J or by increasing hydrolysis of EETs to DHETs by sEH. We did not detect a change in plasma sEH activity in humans, measured by the ex vivo conversion of EETs or EpOMEs to their DHET or DiHOME products, after treatment of primary aldosteronism. To the contrary, there was a significant increase in 8,9-EET epoxide hydrolase activity. Because sEH is a more specific hydrolase against 14,15- and 11,12 EET,28 the change in 8,9 EET hydrolase activity may reflect changes to a yet-to-be-determined hydrolase enzyme or accumulation of a compound which specifically impairs 8,9-epoxide hydrolase activity. We also did not detect a change in the molar ratio of any DHET to EET isoform with treatment of primary aldosteronism or acute infusion of aldosterone overnight. In contrast, aldosterone increased the molar ratio of DHETs to EETs or DiHOMEs to EpOMEs in mice and increased mRNA expression of adipose Ephx2. Further studies are needed to determine the effects of aldosterone on the balance of EET production and metabolism in humans.
Our studies suggest that aldosterone impairs EET metabolism chronically, but not acutely. In the only prior cross-sectional clinical study to assess the effect of chronic elevated aldosterone excess, 14,15-DHET concentrations determined by ELISA positively correlated with aldosterone concentrations and aortic calcification in patients with primary aldosteronism, but EETs were not measured.17 Our colleagues also demonstrated that plasma aldosterone correlates positively with plasma total DHETs in participants with hypertension that is salt-resistant, but not in those that are salt-sensitive.29 Because they also demonstrated that dietary sodium altered circulating and urinary EETs and DHETs, we provided a diet with constant dietary sodium in the current study. The cross-over design of these studies also avoids the potential confounding effects of obesity and medication use, which were similar during both treatment periods. While decreasing aldosterone concentrations or giving an MR antagonist for at least three months resulted in increased circulating EET concentrations, we did not find a significant effect of acute (12.5-hour) overnight infusion of aldosterone on plasma EET concentrations in healthy participants, even though circulating aldosterone concentrations were significantly increased. Treatment of primary aldosteronism could increase EET concentrations via an indirect mechanism other than decreased aldosterone concentrations or decreased MR activation. More likely, our findings indicate that chronic exposure to aldosterone is necessary to induce decreased EET production. In this regard, total EET concentrations trended downward after overnight aldosterone infusion. Unfortunately, it is not feasible to infuse aldosterone for longer periods in humans and other maneuvers to increase aldosterone chronically also increase Ang II or affect volume status. Instead, we assessed the effect of a three-day exposure to exogenous aldosterone on EET concentrations and sEH activity in mice, which provides a longer time period but avoids the confounding effects of hypertension or prolonged hypokalemia. Aldosterone administration for three days decreases EET concentrations in adipose tissue and muscle, but not in liver.
Animal studies demonstrate that EETs contribute to hypertension via effects in the vasculature and kidney. In the kidney, 11,12-EET and 14,15-EET synthesized in the cortical collecting duct in response to high salt intake inhibit ENaC and enhance sodium excretion.30 In the kidney, 11,12-EET and 14,15-EET inhibit ENaC in the collecting duct.31 Disruption of the arachidonic acid epoxygenase Cyp2c44 in mice results in decreased EET concentrations and the development of ENaC-dependent hypertension in response to a high sodium or potassium diet.30 In the vasculature, EETs act as endothelium-derived relaxing factors and regulate resistance vessel tone. In the adrenal vasculature, Ang II increases adrenal cortical blood flow via an EET-dependent mechanism.26, 32 Reduced EET concentrations are associated with endothelial dysfunction in patients with hypertension, and Cyp2c44 disruption impairs endothelium-dependent vasodilation.31, 33 The therapeutic value of increasing EETs in humans requires further research, however, and currently available drugs have not been reported to increase EETs. Inhibition of EET hydrolysis with a sEH inhibitor improves bradykinin-induced arterial vasodilatation, but sEH inhibitors are not yet approved for clinical use.34 EET analogues have beneficial effects in pre-clinical studies, but have not yet been studied in humans.31 The current study demonstrates that reducing aldosterone or blocking the MR in patients with mineralocorticoid excess could provide an alternative approach to increasing EETs in humans.
Circulating EET concentrations are decreased in rodent models of obesity and have been previously associated with insulin sensitivity. Consistent with prior rodent studies,35 we found an inverse relationship between weight, fat mass, and android fat with circulating EET concentrations. The glucose infusion rate during hyperinsulinemic clamps, a marker of insulin sensitivity, correlated positively with circulating EET concentrations. This relationship is consistent with previously reported association with insulin sensitivity as measured by hyperglycemic clamp or intravenous glucose tolerance tests.33, 36 These data are also consistent with rodent models of obesity indicating that decreased EETs or genetic Cyp2c44 deletion are associated with insulin resistance.33, 37 Administration of an sEH inhibitor or an EET mimetic can increase insulin sensitivity in rodents, but the effects in humans are unknown.38–40 EETs may increase insulin sensitivity by acutely improving insulin-stimulated capillary recruitment and tissue perfusion, or chronically by increasing tissue capillary density.41
Increasing EETs is an attractive therapeutic approach which could confer cardiovascular benefit by increasing insulin sensitivity and reducing blood pressure. The inverse relationship between EETs and aldosterone, and prior research in rodents suggests that EETs may oppose the actions of aldosterone in humans.
Perspectives
Inappropriately elevated aldosterone concentrations in patients with hypertension has been associated with increased morbidity and mortality. EETs are lipid signaling molecules formed from arachidonic acid by CYP450s that cause vasodilation and exert anti-inflammatory effects. We report for the first time that treatment of primary aldosteronism either by adrenalectomy or MR antagonism increases circulating EETs in humans. Consistent with these results, treatment of mice with aldosterone decreases EETs and increases the expression of sEH, the enzyme hydrolyzing EETs to their less active metabolites DHETs.
Supplementary Material
Novelty and Significance.
- What is new?
- Chronically elevated endogenous aldosterone, but not acute administration of aldosterone overnight, decrease circulating EETs.
- Circulating EETs correlated negatively with plasma aldosterone and insulin sensitivity.
- Aldosterone administration decreases EETs and increases soluble epoxide hydrolase mRNA expression in adipose tissue in mice.
- What is relevant?
- Aldosterone actions may be mediated in part by promoting hydrolyzation of the vasodilatory and anti-natriuretic EETs.
- Summary:
- Treatment of primary aldosteronism by either surgical adrenalectomy or mineralocorticoid receptor antagonism increases plasma EETs, which may confer cardiovascular and metabolic benefits.
Sources of Funding:
This project was supported by NIH grants DK117875 (JML, NJB) DK096994, DK081662 (JML), DK109116 (AFT), K24HL103845 (GKA), DK DK119212 (AP), TR002243, TR002541, Harvard Catalyst, and Doris Duke Charitable Foundation grant 2019087 (AFT), by an ADA grant 1-19-IBS-282 (AP); and by a VA merit review 1I01BX002025 (AP). The project described was supported by the National Center for Research Resources, Grant UL1 RR024975-01, and is now at the National Center for Advancing Translational Sciences, Grant 2 UL1 TR000445-06. This work utilized the core(s) of the Vanderbilt Diabetes Research and Training Center funded by grant DK020593 from the National Institute of Diabetes and Digestive and Kidney Disease. This work utilized the core(s) of the Vanderbilt Diabetes Research and Training Center funded by grant DK020593 from the National Institute of Diabetes and Digestive and Kidney Disease. AP is the recipient of a Veteran Affairs Senior Research Career Scientist Award.
Footnotes
Disclosures:
JML: reports consultant relationship with Selenity Therapeutics and Mineralys.
KG: none
DSW: none
GKA: reports consultant relationship with Pfizer Pharmaceuticals.
AFT: none
HN: none
CY: none
CS: none
AP: none
NJB: none
References:
- 1.Capdevila JH, Falck JR. The arachidonic acid monooxygenase: From biochemical curiosity to physiological/pathophysiological significance. J Lipid Res 2018;59:2047–2062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Proctor KG, Falck JR, Capdevila J. Intestinal vasodilation by epoxyeicosatrienoic acids: Arachidonic acid metabolites produced by a cytochrome p450 monooxygenase. Circ. Res 1987;60:50–59 [DOI] [PubMed] [Google Scholar]
- 3.Gebremedhin D, Ma YH, Falck JR, Roman RJ, VanRollins M, Harder DR. Mechanism of action of cerebral epoxyeicosatrienoic acids on cerebral arterial smooth muscle. The American journal of physiology. 1992;263:H519–525 [DOI] [PubMed] [Google Scholar]
- 4.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome p450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Romashko M, Schragenheim J, Abraham NG, McClung JA. Epoxyeicosatrienoic acid as therapy for diabetic and ischemic cardiomyopathy. Trends Pharmacol Sci 2016;37:945–962 [DOI] [PubMed] [Google Scholar]
- 6.Luria A, Bettaieb A, Xi Y, Shieh GJ, Liu HC, Inoue H, Tsai HJ, Imig JD, Haj FG, Hammock BD. Soluble epoxide hydrolase deficiency alters pancreatic islet size and improves glucose homeostasis in a model of insulin resistance. Proc. Natl. Acad. Sci. U. S. A 2011;108:9038–9043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res 2000;87:992–998 [DOI] [PubMed] [Google Scholar]
- 8.Morisseau C, Hammock BD. Epoxide hydrolases: Mechanisms, inhibitor designs, and biological roles. Annu Rev Pharmacol Toxicol 2005;45:311–333 [DOI] [PubMed] [Google Scholar]
- 9.Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin ii hypertension. Hypertension (Dallas, Tex. : 1979). 2002;39:690–694 [DOI] [PubMed] [Google Scholar]
- 10.Ai D, Pang W, Li N, Xu M, Jones PD, Yang J, Zhang Y, Chiamvimonvat N, Shyy JY, Hammock BD, Zhu Y. Soluble epoxide hydrolase plays an essential role in angiotensin ii-induced cardiac hypertrophy. Proc. Natl. Acad. Sci. U. S. A 2009;106:564–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ai D, Fu Y, Guo D, Tanaka H, Wang N, Tang C, Hammock BD, Shyy JY, Zhu Y. Angiotensin ii up-regulates soluble epoxide hydrolase in vascular endothelium in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A 2007;104:9018–9023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cervenka L, Huskova Z, Kopkan L, Kikerlova S, Sedlakova L, Vanourkova Z, Alanova P, Kolar F, Hammock BD, Hwang SH, Imig JD, Falck JR, Sadowski J, Kompanowska-Jezierska E, Neckar J. Two pharmacological epoxyeicosatrienoic acid-enhancing therapies are effectively antihypertensive and reduce the severity of ischemic arrhythmias in rats with angiotensin ii-dependent hypertension. Journal of hypertension. 2018;36:1326–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Varcabova S, Huskova Z, Kramer HJ, Hwang SH, Hammock BD, Imig JD, Kitada K, Cervenka L. Antihypertensive action of soluble epoxide hydrolase inhibition in ren-2 transgenic rats is mediated by suppression of the intrarenal renin-angiotensin system. Clinical and experimental pharmacology & physiology. 2013;40:273–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neckar J, Kopkan L, Huskova Z, Kolar F, Papousek F, Kramer HJ, Hwang SH, Hammock BD, Imig JD, Maly J, Netuka I, Ostadal B, Cervenka L. Inhibition of soluble epoxide hydrolase by cis-4-[4-(3-adamantan-1-ylureido)cyclohexyl-oxy]benzoic acid exhibits antihypertensive and cardioprotective actions in transgenic rats with angiotensin ii-dependent hypertension. Clinical science (London, England : 1979). 2012;122:513–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manhiani M, Quigley JE, Knight SF, Tasoobshirazi S, Moore T, Brands MW, Hammock BD, Imig JD. Soluble epoxide hydrolase gene deletion attenuates renal injury and inflammation with doca-salt hypertension. Am. J. Physiol. Renal Physiol 2009;297:F740–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Capdevila JH, Pidkovka N, Mei S, Gong Y, Falck JR, Imig JD, Harris RC, Wang W. The cyp2c44 epoxygenase regulates epithelial sodium channel activity and the blood pressure responses to increased dietary salt. The Journal of biological chemistry. 2014;289:4377–4386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu P, Zhang S, Gao J, Lin Y, Shi G, He W, Touyz RM, Yan L, Huang H. Downregulated serum 14, 15-epoxyeicosatrienoic acid is associated with abdominal aortic calcification in patients with primary aldosteronism. Hypertension. 2018;71:592–598 [DOI] [PubMed] [Google Scholar]
- 18.Adler GK, Murray GR, Turcu AF, Nian H, Yu C, Solorzano CC, Manning R, Peng D, Luther JM. Primary aldosteronism decreases insulin secretion and increases insulin clearance in humans. Hypertension. 2020;75:1251–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luther JM, Byrne LM, Yu C, Wang TJ, Brown NJ. Dietary sodium restriction decreases insulin secretion without affecting insulin sensitivity in humans. The Journal of clinical endocrinology and metabolism. 2014;99:E1895–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turcu AF, Nanba AT, Chomic R, Upadhyay SK, Giordano TJ, Shields JJ, Merke DP, Rainey WE, Auchus RJ. Adrenal-derived 11-oxygenated 19-carbon steroids are the dominant androgens in classic 21-hydroxylase deficiency. Eur J Endocrinol 2016;174:601–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turcu AF, Wannachalee T, Tsodikov A, Nanba AT, Ren J, Shields JJ, O’Day PJ, Giacherio D, Rainey WE, Auchus RJ. Comprehensive analysis of steroid biomarkers for guiding primary aldosteronism subtyping. Hypertension. 2020;75:183–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Savas U, Wei S, Hsu MH, Falck JR, Guengerich FP, Capdevila JH, Johnson EF. 20-hydroxyeicosatetraenoic acid (hete)-dependent hypertension in human cytochrome p450 (cyp) 4a11 transgenic mice: Normalization of blood pressure by sodium restriction, hydrochlorothiazide, or blockade of the type 1 angiotensin ii receptor. The Journal of biological chemistry. 2016;291:16904–16919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods. 2001;25:402–408 [DOI] [PubMed] [Google Scholar]
- 24.Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B-Statistical Methodology. 1995;57:289–300 [Google Scholar]
- 25.R Core Team. R: A language and environment for statistical computing. 2020
- 26.Shah AJ, Kriska T, Gauthier KM, Falck JR, Campbell WB. Effect of angiotensin ii and acth on adrenal blood flow in the male rat adrenal gland in vivo. Endocrinology. 2018;159:217–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Funder JW, Pearce PT, Smith R, Smith AI. Mineralocorticoid action: Target tissue specificity is enzyme, not receptor, mediated. Science (New York, N.Y.). 1988;242:583–585 [DOI] [PubMed] [Google Scholar]
- 28.Decker M, Adamska M, Cronin A, Di Giallonardo F, Burgener J, Marowsky A, Falck JR, Morisseau C, Hammock BD, Gruzdev A, Zeldin DC, Arand M. Eh3 (abhd9): The first member of a new epoxide hydrolase family with high activity for fatty acid epoxides. J. Lipid Res 2012;53:2038–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elijovich F, Milne GL, Brown NJ, Laniado-Schwartzman M, Laffer CL. Two pools of epoxyeicosatrienoic acids in humans: Alterations in salt-sensitive normotensive subjects. Hypertension. 2018;71:346–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun P, Antoun J, Lin DH, Yue P, Gotlinger KH, Capdevila J, Wang WH. Cyp2c44 epoxygenase is essential for preventing the renal sodium absorption during increasing dietary potassium intake. Hypertension (Dallas, Tex. : 1979). 2012;59:339–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Imig JD. Epoxyeicosatrienoic acids, hypertension, and kidney injury. Hypertension. 2015;65:476–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kopf PG, Gauthier KM, Zhang DX, Falck JR, Campbell WB. Angiotensin ii regulates adrenal vascular tone through zona glomerulosa cell-derived eets and dhets. Hypertension. 2011;57:323–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gangadhariah MH, Dieckmann BW, Lantier L, Kang L, Wasserman DH, Chiusa M, Caskey CF, Dickerson J, Luo P, Gamboa JL, Capdevila JH, Imig JD, Yu C, Pozzi A, Luther JM. Cytochrome p450 epoxygenase-derived epoxyeicosatrienoic acids contribute to insulin sensitivity in mice and in humans. Diabetologia 2017;60:1066–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang L, Cheriyan J, Gutterman DD, Mayer RJ, Ament Z, Griffin JL, Lazaar AL, Newby DE, Tal-Singer R, Wilkinson IB. Mechanisms of vascular dysfunction in copd and effects of a novel soluble epoxide hydrolase inhibitor in smokers. Chest 2017;151:555–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sodhi K, Puri N, Inoue K, Falck JR, Schwartzman ML, Abraham NG. Eet agonist prevents adiposity and vascular dysfunction in rats fed a high fat diet via a decrease in bach 1 and an increase in ho-1 levels. Prostaglandins Other Lipid Mediat 2012;98:133–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramirez CE, Shuey MM, Milne GL, Gilbert K, Hui N, Yu C, Luther JM, Brown NJ. Arg287gln variant of ephx2 and epoxyeicosatrienoic acids are associated with insulin sensitivity in humans. Prostaglandins Other Lipid Mediat 2014;113–115:38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sodhi K, Inoue K, Gotlinger KH, Canestraro M, Vanella L, Kim DH, Manthati VL, Koduru SR, Falck JR, Schwartzman ML, Abraham NG. Epoxyeicosatrienoic acid agonist rescues the metabolic syndrome phenotype of ho-2-null mice. J. Pharmacol. Exp. Ther 2009;331:906–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raffaele M, Bellner L, Singh SP, Favero G, Rezzani R, Rodella LF, Falck JR, Abraham NG, Vanella L. Epoxyeicosatrienoic intervention improves nafld in leptin receptor deficient mice by an increase in pgc1alpha-ho-1-pgc1alpha-mitochondrial signaling. Exp. Cell Res 2019;380:180–187 [DOI] [PubMed] [Google Scholar]
- 39.Sodhi K, Inoue K, Gotlinger KH, Canestraro M, Vanella L, Kim DH, Manthati VL, Koduru SR, Falck JR, Schwartzman ML, Abraham NG. Epoxyeicosatrienoic acid agonist rescues the metabolic syndrome phenotype of ho-2-null mice. J. Pharmacol. Exp. Ther 2009;331:906–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Skepner JE, Shelly LD, Ji C, Reidich B, Luo Y. Chronic treatment with epoxyeicosatrienoic acids modulates insulin signaling and prevents insulin resistance in hepatocytes. Prostaglandins Other Lipid Mediat 2011;94:3–8 [DOI] [PubMed] [Google Scholar]
- 41.Shim CY, Kim S, Chadderdon S, Wu M, Qi Y, Xie A, Alkayed NJ, Davidson BP, Lindner JR. Epoxyeicosatrienoic acids mediate insulin-mediated augmentation in skeletal muscle perfusion and blood volume. Am. J. Physiol. Endocrinol. Metab 2014;307:E1097–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.