

Abstract
The healthy lung was long thought of as sterile, but recent advances using molecular sequencing approaches have detected bacteria at low levels. Healthy lung bacteria largely reflect communities present in the upper respiratory tract that enter the lung via microaspiration, which is balanced by mechanical and immune clearance and likely involves limited local replication. The nature and dynamics of the lung microbiome, therefore, differ from those of ecological niches with robust self-sustaining microbial communities. Aberrant populations (dysbiosis) have been demonstrated in many pulmonary diseases not traditionally considered microbial in origin, and potential pathways of microbe-host crosstalk are emerging. The question now is whether and how dysbiotic microbiota contribute to initiation or perpetuation of injury. The fungal microbiome and virome are less well studied. This Review highlights features of the lung microbiome, unique considerations in studying it, examples of dysbiosis in selected disease, emerging concepts in lung microbiome–host interactions, and critical areas for investigation.
Introduction
It is estimated that the human body contains roughly equal numbers of human and bacterial cells (1), and over the past decade studies of the human microbiome have expanded from the gut and other microbe-rich environments to sites previously considered sterile, such as the bladder (2), and the lung, the focus of this Review. Although the lungs were known to harbor microbes in acute infections and chronic suppurative diseases such as bronchiectasis and cystic fibrosis (CF), microbial communities in the healthy lung were only recently described through culture-independent methods (3–5). It is not surprising that the lung is not sterile, given its contiguity with the microbe-rich upper respiratory tract. However, the healthy lung possesses exceedingly low microbial biomass, and the nature of the lung microbiome differs markedly in quantity and dynamics from that of body habitats with robust microbiomes, such as the gut, skin, mouth, and vagina.
Many respiratory illnesses not traditionally considered microbially driven have now been linked to lung microbiota that differ from those of healthy people (“dysbiosis”), although research has only begun to address causality and potential mechanisms. While investigators differ in precisely how the term is interpreted and employed, “dysbiosis” refers broadly to disruption of microbial homeostasis with compositional or functional deviation from normal or optimal in a particular niche (6, 7). In the lung, consensus has not yet emerged on what defines dysbiosis; here we use the term to refer to deviation from microbiota seen in healthy people without lung disease, recognizing that there may be optimal microbial communities in certain disease states, such as CF, that nonetheless differ from health. Importantly, given the exceedingly low microbial burden of the normal lung, dysbiosis includes not just changes in relative abundances (proportions) of microbiome constituents within a community, but changes in total microbial content as well.
Lung microbiome publications are increasing exponentially, and, similarly to how microbiome research in other sites matured, the key questions now extend beyond “who is there?” to: What are they doing? How do they impact the host? What are the mechanisms? And can we harness them therapeutically?
Approach to studying the lung microbiome
Analytic approaches.
In contrast to culture methods that focus on individual organisms of a priori interest, microbiome studies use sequence-based approaches that capture entire communities. The most widely used approach employs PCR amplification and sequencing of a region shared by all members of a group that can then be used to infer identity, such as the 16S rRNA gene of bacteria and archaea (the latter of uncertain importance in the lung) and the 18S rRNA gene or internal transcribed spacer (ITS) regions of fungi (8). Sequencing of the 16S rRNA gene typically targets one or more of the nine hypervariable regions (V1–V9) that differ across taxa. While efficient, this approach is limited in species-level identification, a deficit given the wide range in biological activities between species and even strains. Full-length 16S rRNA gene sequencing provides greater taxonomic definition (9, 10) and has been applied on a limited basis in the lung (11, 12), but short-read 16S rRNA gene variable region sequencing remains most common. Regardless of methodology, 16S rRNA gene profiling is largely restricted to descriptive correlations between taxonomy and host phenotype.
Another approach is shotgun metagenomic sequencing, in which all the DNA in a sample is sequenced. Although the high proportion of host-derived DNA present in lung samples is a challenge, shotgun sequencing is being increasingly used in lung studies (13–17). Shotgun metagenomics provides taxonomic composition and microbiome gene content, which enumerates the community’s functional potential. It can annotate bacterial strains and includes information regarding carriage of antibiotic resistance or virulence genes. Shotgun metagenomics concurrently identifies bacterial and fungal constituents, and also viruses, which do not share any sequence tags comparable to bacteria and fungi.
Both 16S rRNA gene sequencing and shotgun sequencing are associated with reads that do not align to reference databases (18). Furthermore, there may be low-abundance taxa with functional impact, such as Erysipelotrichaceae, that are present below the limits of detection or difficult to distinguish from background contaminant sequences (19). Notably, neither targeted sequencing nor shotgun metagenomics differentiates between live and dead bacteria. Distinguishing viability is particularly relevant in the lung, where a balance exists between microbial entry and clearance, and some diseases are associated with abundant relic DNA, such as CF (20, 21). Also, shotgun metagenomics details the functional potential of a microbial community based on gene content but not its actual activity.
Beyond shotgun metagenomics, metatranscriptomics (RNA sequencing) and metabolomics (small-molecule analysis) depend on living cells and may better reflect functional activity (13). Finally, sequence-based methods are complementary to culture, which has high sensitivity for targeted organisms (22). Thus, molecular approaches are not inherently better than culture, and combining culture and sequencing approaches may provide optimal community resolution in specimens dominated by human DNA and/or low-abundance taxa (23), and also provide an opportunity to study the specific organisms.
Sequence-based studies typically report taxa in terms of relative abundances, that is, proportionality within a community that totals 100%. However, it is essential to consider relative enrichment of specific taxa in the context of the total microbial burden, particularly in the lung, where the microbial burden varies over many orders of magnitude. Indeed, bacterial burden may be as important as composition, as described in studies where total bacterial burden in bronchoalveolar lavage fluid (BALF) predicted chronic lung allograft dysfunction or death following lung transplantation (24), progression of interstitial pulmonary fibrosis (25, 26), and outcomes during critical illness (27).
Sampling the lung microbiome.
Sputum and BALF are common approaches to lung microbiome sampling and are associated with distinct biases. Sputum is noninvasive and can be collected serially but represents a mix of upper and lower respiratory tract (URT and LRT). This admixture is especially problematic in states in which the lung microbial burden is low, though sputum may better reflect the lung in conditions like CF in which the LRT microbial burden is high (28). There is little consensus regarding how well microbial communities in spontaneous and induced sputum correlate (29, 30). While bronchoscope passage through the URT means that BALF may contain carryover (particularly of periglottic material; refs. 3, 31), it is less influenced by contamination than sputum but requires an invasive procedure. Still, careful analysis is necessary to distinguish lung versus other sources. The use of whole versus host cell–depleted BALF varies by study, though cell-depleted BALF often shows loss of α-diversity, a marker of how many different taxa are present and how evenly they are distributed, likely from removal of cell-associated bacteria (32). Other sampling options with both advantages and limitations include endotracheal aspirates, used extensively in intubated patients (33–35); protected specimen brushings (36, 37); bronchial biopsies (38, 39); exhaled breath condensates (40, 41); and tissue from explanted lungs (42–44).
The low microbial biomass of lung specimens except in highly colonized or purulent conditions makes lung samples exquisitely susceptible to contamination from the URT during collection, and also from background sequences derived from equipment, kits, and reagents, which are sterile but not necessarily DNA free (3, 45, 46). Artifactual sequences can also result from the sequencing process itself (47). Many BALF samples from healthy people have very low bacterial content and a high relative abundance of taxa characteristic of background controls (e.g., bronchoscope channel wash prior to the procedure), indicating a paucity of authentic lung microbiota (48, 49). A point of some controversy is whether taxa suspected as contaminants should be removed before analysis (8, 45). In shotgun sequencing, a large portion of reads in low-microbial-content samples will correspond with human as opposed to microbial sequences, impeding the capture of microbial constituents. Thus, executing and interpreting lung microbiome studies requires careful attention to stringent sampling and the distinguishing of authentic signals from both URT contamination and background artifact.
The healthy lung microbiome
Nature of the lung microbiome in health.
Traditional teaching was that healthy lungs were sterile, meaning the airways below the vocal cords (glottis) and distal to the alveoli, but studies using rigorous sampling revealed that bacterial sequences are usually detectable in the lung (3, 37, 50). However, bacterial density is orders of magnitude lower in the lung than in the URT, and the community composition closely matches the URT, especially the periglottic region. Firmicutes and Bacteroidetes are the most common phyla, while Prevotella, Streptococcus, and Veillonella are the most common genera (3, 51). The nearly identical composition of two contiguous communities, one of which has high microbial biomass (URT) and one of which has low (lung), suggests that the lung microbiota is derived passively from the URT through microaspiration, which occurs even in health (52). The lung microbiome, therefore, is largely transient and composed of URT-derived immigrants that are cleared through mucociliary and innate immune mechanisms. Some of these bacteria are viable and transcriptionally and metabolically active (13, 53). Bacterial levels are greater in more proximal lung regions, and modest regional differences exist as well, suggesting differential clearance and, potentially, limited local replication (37). Whether there are true long-term, self-sustaining bacterial residents is unclear. Thus, the nature and the dynamics of the healthy lung microbiome differ from those in sites such as gut, skin, and vaginal and oral cavities, which have robust, self-sustaining microbial communities.
This work has led to a healthy lung microbiome model that accounts for a steady-state low-level microbial community as a consequence of immigration from the URT, clearance, and limited local replication, sometimes termed the “adapted island model” (37). This concept is supported by murine studies modeling aspiration, in which the bacterial RNA metatranscriptome signal disappeared from the lung within a day while DNA sequences persisted for several days (13). This model is useful as a basis for understanding disease states in which functional or structural defects result in changes to one or more of the parameters of entry, clearance, and local replication (Figure 1).
Figure 1. Ecological dynamics of the lung microbiome.
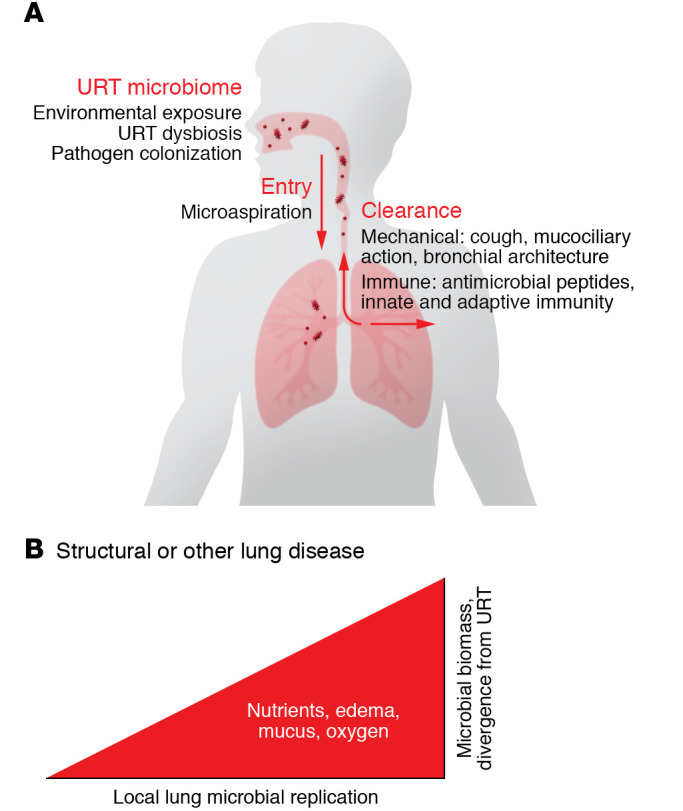
The lung microbiome is determined by the balance between immigration from the URT and elimination via immune and mechanical processes. In healthy lungs, local microbial replication in the airways and lung parenchyma is limited. In lung disease, structural disturbances and alterations to the local environment favor microbial growth, which predominantly determines lung microbial composition.
Source of lung microbiota.
Since the URT is the initial source of lung microbiota, dysbiosis of the URT microbiome may impact the lung microbiome. Cigarette smoking is a principal driver of lung disease, and there are modest differences in the URT microbiome of healthy smokers versus nonsmokers (54, 55). However, studies have failed to show significant differences in lung microbial communities between healthy smokers and nonsmokers (55). One possibility is that URT microbiome changes associated with smoking are modest, and effects are not detectable after the attenuation that occurs along the URT-LRT gradient. In contrast, smoking alters the lung microbiome in mice (56); this may be detectable because of the controlled nature of animal models. The impact of other environmental exposures on the lung microbiome is understudied. For example, lung microbiota in people highly exposed to indoor air pollution are enriched in Streptococcus and Neisseria (57). Further, URT dysbiosis may be an underappreciated driver of lung dysbiosis, as recently described in patients with end-stage lung disease and following lung transplantation (58).
In contrast, hematogenous sources for the lung microbiome seem limited to disease states. Lung infection from hematogenous seeding occurs commonly in bacteremia and fungemia. Sequences characteristic of gut bacteria are found in BALF in acute respiratory distress syndrome (ARDS), likely reflecting gut translocation and hematogenous spread; whether this process contributes to local inflammation in ARDS is unknown (59). Lung enrichment with Tropheryma whipplei, typically considered a gut bacteria, is seen in asymptomatic people with HIV infection, though its presence has unclear consequences (60).
Functional impact of the lung microbiome in health.
The gut microbiome is central in regulating both gut mucosal and systemic immune development and function (61–63). In the lung, emerging data suggest that, although scant, the microbiota even in health regulates immune tone (Figure 2). Healthy people can be grouped into individuals whose BALF microbiota resembles oral communities, indicating a URT-derived microbiome, and individuals with BALF that resembles background controls, suggesting an absence of (or below-detectable) authentic lung microbiota (64). People whose BALF was enriched in oral commensals exhibited a Th17/neutrophilic mucosal immune profile with less robust innate immunity. This phenotype was associated with distinct metabolic profiles, suggesting a potential mechanism of regulation. Similarly, community composition and bacterial diversity in mouse lungs correlated inversely with the inflammatory cytokines IL-1α and IL-4 (65). While inflammation is an obvious component of acute infection, these data indicate that a key frontier is to understand how the microbiome regulates lung immune function in health and nonpurulent diseases. Both live and dead bacteria could modulate immune function, via metabolite production by live organisms, and pattern recognition receptors by both.
Figure 2. Nature and potential impact of the lung microbiome in health and disease.
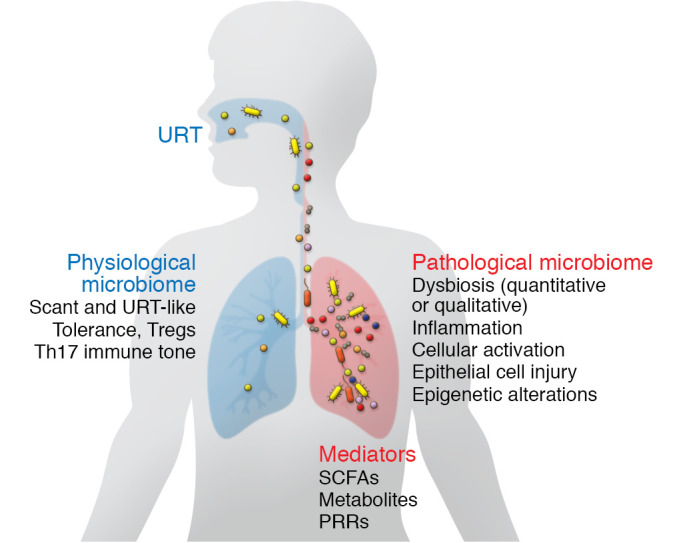
In healthy lungs, the bacterial communities (colored shapes) have low biomass and are largely derived passively from the URT. Low-biomass communities associate with a balanced immune tone that promotes mucosal regulatory T cells (Tregs) and Th17 readiness. Tregs and Th17 cells may promote a basal-level immune tone that is necessary for clearance of encountered pathogens without pathological inflammatory responses. In disease, a dysbiotic microbiota may activate innate and adaptive immune responses that promote immunopathology. This inflammatory state can lead to epithelial fibroproliferation, promotion of immune cell immigration to the lung in response to cytokine and chemokine elaboration, and heightened Th2 immune tone, among other potential pathological mechanisms. Mediators include microbial metabolites such as short-chain fatty acids (SCFAs), as well as pattern recognition receptor (PRR) ligands by which even nonviable bacteria may trigger responses. The virome and mycobiome in health and lung disease remain understudied.
Studies in mice also suggest that lung microbiota play a role in local immune development and tolerance. In germ-free mice, lung inflammatory responses are exaggerated, and neonatal animals exhibit robust responses to allergens that subsequently dampen as the bacterial burden increases and regulatory T cells appear (66, 67). Mice treated with antibiotics have increased susceptibility to respiratory infection following pathogen exposure (68). While it can be difficult to distinguish direct effects of the lung microbiome from pathways involving gut microbiota in these models, a recent study showed that intratracheal instillation of oral commensals in a mouse model of microaspiration induced a Th17 response and resistance to Streptococcus pneumoniae infection (69). Thus, physiological levels of lung bacteria likely shape local immune responses.
The lung mycobiome and virome.
Compared with bacteria, the fungal and viral communities (termed “mycobiome” and “virome”) of the lung are understudied. Fungal analysis employs targeted ITS or 18S rRNA gene or shotgun metagenomic sequencing. Mycobiome studies are challenging because of the low fungal biomass of the lung, the low number of fungal taxa present, difficulty in DNA extraction from fungi, biases in 18S rRNA gene and ITS amplification, inconsistent nomenclature, and suboptimal annotation of fungal databases, as well as frequently robust fungal populations in the URT (70, 71). Ascomycota and Basidiomycota are the most commonly identified taxa, followed by the genera Candida, Saccharomyces, Penicillium, Cladosporium, and Fusarium (72–75). Whether the lung mycobiome contributes to normal physiology remains unknown.
Viruses do not share any conserved regions comparable to bacterial 16S or fungal 18S genes. Thus, most studies of viruses in the lungs use targeted detection of specific viruses, whereas comprehensive delineation of the virome requires shotgun sequencing. Studies of the lung virome have found Anelloviridae as the predominant eukaryotic virus, with sporadic detection of herpesviruses, papillomaviruses, retroviruses, and other respiratory viruses (76–78). Anelloviridae are DNA viruses with greater than 90% prevalence in adults and wide distribution in blood and tissues (79). They are under tonic immune control, since levels rise with immune dysfunction (76), but have not been linked to physiological effects on the host or disease (80). Bacteriophages are also common, particularly those associated with ororespiratory tract bacterial hosts.
Virome studies are further challenged by the high proportion of shotgun sequences that fail to align with anything in current databases (called “metagenomic dark matter”), raising the possibility of yet-undiscovered viruses. A newly recognized virus family was identified in metagenomic sequences from the respiratory tract of healthy and sick patients, named Redondoviridae for its circular genome (81). Redondoviridae was found to be the second most common DNA virus in respiratory tract metagenomic data sets and was elevated in the airway of critically ill compared with healthy people (81). As with Anelloviridae, it is unknown whether Redondoviridae play a role in lung health or pathogenesis.
The lung microbiome in disease
Dysbiosis of the lung microbiome can result from disruption in entry, clearance, or local replication. Greater entry can result from increased aspiration, and dysbiotic URT microbiota may have downstream impact on the lung. Indeed, it has long been known that colonization of the URT with respiratory pathogens including Streptococcus pneumoniae or Gram-negative bacteria often precedes pneumonia. Decreased clearance can result from immune dysfunction, sputum defects like in CF, architectural distortion as in bronchiectasis and fibrosis, and cough defects or physical barriers as in lung transplantation. Increased local replication may result from augmented nutrients due to mucous or edema fluid, as well as from the increase in steady-state bacterial populations caused by greater entry or deficient clearance. Lung dysbiosis is now described in many pulmonary diseases, but the fundamental unknown is whether dysbiosis initiates or perpetuates inflammation and/or injury, or whether it is a consequence and marker of disease. This section highlights insights in selected lung diseases not traditionally considered microbially driven; advances in other conditions (CF, lung transplantation, ARDS) have been addressed in recent disease-specific reviews (82–84).
Chronic obstructive pulmonary disease.
Chronic obstructive pulmonary disease (COPD) affects over 300 million people globally and is characterized by progressive irreversible airflow limitation associated with chronic bronchitis, small airway occlusion, inflammation, and emphysematous destruction of parenchyma (85–87). It is associated primarily with smoking, though additional factors are indoor smoke exposure, pollution, early-life respiratory infections, and genetics (87). Bacteria can be isolated by culture from sputum and BALF during exacerbations (88–90), and prophylactic macrolide antibiotics reduce exacerbations in advanced disease (91), implicating microbial drivers.
Studies applying sequence-based methods in COPD differ in subject and specimen type, and describe dysbiosis during both periods of stable COPD and acute exacerbations. During stable disease, relative abundance of Pseudomonas was higher and of Prevotella lower in bronchial wash in comparison with healthy smokers and nonsmokers (92). A large meta-analysis of sputum data sets reported enrichment of Haemophilus, Streptococcus, Moraxella, and Lactobacillus, and depletion of several genera, relative to healthy controls (93). In a cross-sectional study of 72 COPD patients, airflow limitation correlated with increased relative abundance in sputum of Pseudomonas and decreased Treponema (94). A prospective study of 101 patients found that greater disease severity was associated with increased Haemophilus and decreased Prevotella and Veillonella (95). Microbial profiles have also been associated with bronchodilator responsiveness and peak expiratory flow rate in the SPIROMICS cohort (96). Diversity is lower in stable COPD compared with healthy smokers and nonsmokers (92, 97) and is further decreased during acute exacerbations (98). Lower diversity of LRT specimens also correlated with decreased lung function and exacerbation frequency in stable COPD (5, 12, 99).
In 200 COPD patients followed prospectively, acute exacerbations were correlated strongly with viral infections and only modestly with bacterial microbiota, but exacerbation frequency was linked with higher relative abundances of several taxa (Bacillus, Escherichia, Lactobacillus, Moraxella, and Staphylococcus) as well as greater longitudinal variability (100). Sputum bacterial communities during exacerbations have also predicted outcome; the presence of Staphylococcus and the absence of Veillonella, along with decreased diversity, were associated with higher mortality in a cohort of 102 subjects (101). Similarly, Haemophilus- and Streptococcus-dominated sputa have been associated with greater mortality in a cross-sectional study of 252 subjects (102).
However, findings are not always consistent across studies. As highlighted in a recent study of 122 patients, interindividual differences may outweigh group differences, and the complexity of lung microbiota presents substantial challenges for use as a biomarker or for simple mechanistic interpretation (103).
Distinct lung microbiota have also been described across COPD inflammatory endotypes. An analysis of 1706 sputa from 510 subjects reported that neutrophilic inflammation was associated with two major community classes during both stability and exacerbations, one characterized by an URT-like bacterial community and one characterized by pathogens including Haemophilus, Moraxella, and Streptococcus (104). Neutrophilia correlated with Moraxella abundance in sputa in another study of 87 COPD patients (105) and with Haemophilus and Neisseria in a 43-subject cohort (97). Moreover, Haemophilus-predominated neutrophilic inflammation correlated with increased sputum IL-1β and TNF-α (104, 106) and with neutrophil extracellular trap formation (107). Neutrophilic inflammation also associated with decreased diversity relative to other inflammatory endotypes (108). These findings suggest that a pathogen-predominant community type may drive neutrophilic inflammation in COPD. In contrast, eosinophilic inflammation in COPD is associated with fewer clinical bacterial infections, lower bacterial burden, increased diversity, and lower prevalence of Haemophilus and Moraxella (94, 102, 104, 105, 108, 109). Compared with patients who had neutrophilic inflammation, patients with eosinophilic inflammation demonstrated relative enrichment in Gemella, Campylobacter, Capnocytophaga, and Granulicatella in an analysis of three cohorts (104), and a higher proportion of Bacteroidetes in a cohort of 73 COPD and 32 asthma patients (109). Thus, distinct mechanisms likely link the lung microbiome to inflammation in COPD endotypes.
Finally, provocative data implicate changes in metabolic capabilities of the lung microbiome during exacerbations using both inferred metagenomics (bacterial gene content predicted from taxonomic data) and shotgun metagenomic sequencing (16, 93). Thus, there may be important functional changes in the microbiome not evident from community composition alone.
Asthma.
Asthma affects more than 300 million people and is a heterogeneous disease causing recurrent episodes of largely reversible airflow obstruction. It is characterized by hyperplasia of epithelial and smooth muscle cells, fibrosis, increased mucus production, and type 2 inflammation (110). Risk factors include genetics, early-life respiratory illness (particularly respiratory syncytial virus; ref. 111), airborne environmental exposure, and atopic sensitization (112). The lung microbiota in asthma is enriched in Neisseria, Moraxella, Haemophilus, Porphyromonas, Fusobacterium, and Sphingomonadaceae, and depleted in Mogibacteriaceae and Lactobacillales (4, 36). A small study reported increased microbial diversity in sputum relative to healthy individuals (113), and bacterial communities of bronchial brushings correlate with airflow obstruction (36, 114).
Like COPD, asthma is characterized by neutrophilic or eosinophilic phenotypes, and the associated microbiome profiles are similar, including decreased diversity and increased frequency of pathogenic taxa in neutrophilic inflammation (115–117). For example, Haemophilus-high and Haemophilus-low clusters were seen in sputum of 141 subjects with both asthma and COPD (106). Further, total bacterial burden and proinflammatory cytokines were decreased in high (eosinophilic) relative to low (neutrophilic/mixed) type 2 inflammation among 50 asthmatic patients (117). This common inflammation-microbiome association is consistent with either a particular microbiome type driving mucosal inflammation or, conversely, microbiome profiles reflecting a response to inflammation (118). Finally, beyond the scope of this Review, abundant evidence supports a key role for the gut microbiome in aberrant immune responses in asthma (119).
Lung cancer.
Lung cancer accounts for almost one-quarter of all cancer deaths (120), with 82% attributed to cigarette smoking (121). Among never-smokers, lung cancer is associated with diseases characterized by chronic airway inflammation, including COPD, Chlamydia pneumoniae, Mycobacterium tuberculosis, and respiratory viruses (122–124), suggesting that the lung microbiome may play a role in cancer development and progression. Supporting this theory, germ-free rodents develop lung cancer less frequently (125, 126). Interest in the airway microbiome in lung cancer has also been spurred by advances in understanding the gut microbiome and colon cancer; it is now recognized that microbial composition and ecological patterns differ in patients with cancer, local mucosal biofilms are associated with genetic and epigenetic abnormalities, and animal models suggest that specific bacteria play key roles in the molecular process of colorectal carcinogenesis (127, 128).
Several reports have associated lung cancer with enrichment of specific taxa in sputum and BAL, including Granulicatella, Abiotrophia, Streptococcus, Veillonella, Megasphaera, and Selenomonas (17, 129, 130). Altered lung microbiome diversity in cancer specimens relative to nonmalignant matched tissue and cancer-free patient controls has also been described, although without uniformity in the directionality of that association (39, 131, 132). Unique bacterial profiles have also been associated with recurrence- and disease-free survival (39, 133), and with specific mutations in squamous cell carcinoma (131). Compositional differences in lower airway microbiota have also been seen in lower airway brushings between small cell and non–small cell lung cancer (134). Finally, altered bacterial diversity in lung tissue associates with advanced cancer stage, as well as with air pollution and pack-years of tobacco smoking, which are lung cancer risk factors (38).
Beyond associations, the questions of whether and how microbiome dysbiosis contributes to lung cancer pathogenesis remain unresolved. Enrichment with Streptococcus and Veillonella in airway brushings of cancer patients was associated with increased inflammatory cell infiltration and bronchial epithelial upregulation of PI3K/ERK signaling pathways (135), an early pathogenic event in non–small cell lung carcinoma (136). These taxa also upregulated these signaling pathways in airway epithelial cells in vitro. Exposure of mice to bacterial consortia isolated from a late-stage murine lung tumor accelerated tumor growth, which was mediated by myeloid cell cytokine secretion and activation of IL-17–producing γδ T cells in the lung mucosa (125). Thus, associations exist between lung cancer and lung microbiome communities, and provocative findings suggest the possibility of mechanistic contributions of the microbiome.
Interstitial lung disease.
The interstitial lung diseases comprise multiple entities, the most common being idiopathic pulmonary fibrosis (IPF). IPF is a progressive fibroproliferative process with poor survival and few effective treatments, characterized by proliferation of mesenchymal cells, collagen and extracellular matrix deposition, fibrosis, and distortion of pulmonary architecture. Studies report that BALF from IPF patients has higher bacterial load than that from healthy people; furthermore, both total bacterial burden and specific taxa (Streptococcus and Staphylococcus) associate with more rapid disease progression (25, 26, 137, 138). A mechanistic pathway was suggested by findings that a peptide fragment of Staphylococcus was elevated in IPF patients compared with controls, and intrapulmonary instillation of this peptide induced lung epithelial cell injury and features of IPF acute exacerbations in mice (139). Another rodent model employed bleomycin-induced fibrosis in conventional and germ-free mice, and showed that bacterial colonization did not affect fibrosis directly; rather, germ-free mice exhibited altered Th1/Th2 immune responses and increased survival, suggesting that microbiota may impact IPF progression through inflammatory rather than fibrotic pathways (26). However, human studies have not uniformly supported findings of increased bacterial content or have suggested considerable interpatient and intrapatient geographic heterogeneity in lung explants of advanced IPF (140, 141). Also, a recent interventional trial found no benefit from the antibiotic trimethoprim-sulfamethoxazole, though the impact on microbiota was not assessed (142). Thus, it is unclear whether the microbiome has a causal role in lung injury, versus dysbiosis resulting from architectural distortion of IPF.
In one of the few lung virome studies, Anelloviridae were elevated in the BALF of patients with acute exacerbations compared with stable IPF (143), although cause versus consequence of inflammation in IPF exacerbations remains unclear.
From microbiome association to function
A principal pathway of gut microbiome influence on the host is through small-molecule metabolites, including short-chain fatty acids (SCFAs) (144). Many of these molecules are not produced by human metabolism and are thus markers of bacterial activity as well as potential mediators. SCFAs can directly modulate cell proliferation and differentiation, hormone secretion, metabolic homeostasis, and immune and inflammatory responses, and gut bacteria–derived butyrate even serves as a primary energy source for colon epithelial cells (145–148). Studies have begun to explore the role of SCFAs in microbiome-host crosstalk in the lung and relevant model systems. In vitro, butyrate impairs reactive oxygen species production, phagocytosis, and microbial killing by neutrophils (149). The presence of butyrate during differentiation of alveolar macrophages can induce cellular reprogramming and long-lasting antimicrobial activity (147). Similarly, the combination of propionate or butyrate with TNF-α synergistically increases IL-6 and CXCL8 production by primary human lung fibroblasts and airway smooth muscle cells, while acetate elicits a synergistic response to CXCL8 in airway smooth muscle cells (150). Thus, SCFAs may act as bioactive mediators of the lung microbiome, although relationships may be complex, as different types of lower airway cells respond differently to SCFAs (151).
In the sputum and BALF of CF patients, SCFAs are present in micromolar concentrations (145, 151). Concentrations of acetic acid correlate with IL-8 in CF BALF, and in vitro experiments suggest that SCFAs may mediate this increase in IL-8 via G protein–coupled receptors on airway epithelial cells (145). Similarly, the inferred metagenome of bronchial microbiota from people with asthma, sampled by protected bronchial brushings, was enriched in genes for pathways involved in the metabolism of SCFAs and amino acids (36). In contrast, inferred metagenomics of sputum indicated that butyrate production pathways were depleted in COPD (12).
Azithromycin is a broad-spectrum antibacterial that is used to prevent exacerbations or delay progression in lung diseases not traditionally considered microbe-driven, including COPD, asthma, and chronic lung allograft dysfunction following transplantation (152, 153). Recently, several studies investigated whether azithromycin might act by modifying lung microbiome communities, but results on community composition have been inconclusive (154–156). Alternatively, the antiinflammatory properties of azithromycin have been linked to modulation of bacterial metabolism, wherein production of glycolic acid and indole-3-acetate was increased following azithromycin treatment. These SCFAs reduced ex vivo secretion of TNF-α, IL-12 p40, IL-13, and CXCL1 by alveolar macrophages, and these cytokines were also reduced in BALF following azithromycin treatment of patients (157). Consequently, therapeutic interventions might modulate the composition of the lung microbiome or target its metabolic activity. Alternatively, SCFAs or other small-molecule microbially derived mediators might themselves be directly administered therapeutics, as is beginning to be investigated in the gut (158).
Notably, the microbiome as profiled taxonomically through 16S rRNA gene or shotgun metagenomic sequencing does not fully reflect the population of live, metabolically active organisms. Recent studies show that the microbial metagenome inferred from 16S rRNA gene taxonomy or assessed by shotgun sequencing differs from that derived from direct metatranscriptome analysis, which better correlated with bacterial metabolites (13). Animal studies suggest that this discrepancy reflects the mix of metabolically active (live) bacteria and dead (relic) bacteria caught in a snapshot of the dynamic entry/replication/clearance process. Of course, even nonviable bacteria may impact the lower respiratory tract through pattern recognition receptors that recognize the physical constituents of the organisms.
Finally, while this Review focuses on the respiratory tract microbiome, the gut microbiome likely also shapes the lung immune environment. Just as the gut microbiome plays a central role in the development and regulation of systemic immunity, emerging data suggest that a gut-lung axis also exists (159).
Knowledge gaps and future directions
The lung bacterial microbiome is altered in many diseases that are not traditionally considered infectious or microbial in etiology. However, the lung microbiome field is relatively young and requires more longitudinal, human intervention, and mechanistic animal studies to address causality. Another knowledge gap involves the lung mycobiome and virome, which are greatly understudied. Robust animal models will be needed to move the field forward and address the key question of directionality: Is the microbiome a cause (perpetuator) or a consequence (marker) of disease, or both? Can microbiome-targeted sequence-based technologies improve discrimination between health and disease or predict disease trajectory? Do environmental or other factors known to cause lung disease influence the lung microbiome, and is that a component of their mechanism of injury? What are the specific mechanisms by which the microbiome affects the host? Finally, this knowledge will provide groundwork for the most compelling questions: Can the microbiome be therapeutically manipulated in lung diseases, either by supplementation of communities with beneficial taxa; by selective reduction of proinflammatory or deleterious taxa or modification of their activities; or by direct use of microbially derived small molecules that have beneficial host effects?
Acknowledgments
This work was supported by NIH grants R33-HL137063 (to RGC) and KL2-TR001879 (to JEM), and a grant from the Cystic Fibrosis Foundation (to RGC and SAW).
Version 1. 08/02/2021
Electronic publication
Footnotes
Conflict of interest: RGC’s research laboratory has received support from OraSure Technologies Inc.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(15):e150473.https://doi.org/10.1172/JCI150473.
Contributor Information
Samantha A. Whiteside, Email: Samantha.Whiteside@Pennmedicine.upenn.edu.
John E. McGinniss, Email: John.McGinniss@pennmedicine.upenn.edu.
References
- 1.Sender R, et al. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14(8):e1002533. doi: 10.1371/journal.pbio.1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whiteside SA, et al. The microbiome of the urinary tract — a role beyond infection. Nat Rev Urol. 2015;12(2):81–90. doi: 10.1038/nrurol.2014.361. [DOI] [PubMed] [Google Scholar]
- 3.Charlson ES, et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184(8):957–963. doi: 10.1164/rccm.201104-0655OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hilty M, et al. Disordered microbial communities in asthmatic airways. PLoS One. 2010;5(1):e8578. doi: 10.1371/journal.pone.0008578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Erb-Downward JR, et al. Analysis of the lung microbiome in the ‘‘healthy’’ smoker and in COPD. PLoS One. 2011;6(2):e16384. doi: 10.1371/journal.pone.0016384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hooks KB, O’Malley MA. Dysbiosis and its discontents. mBio. 2017;8(5):e01492-17. doi: 10.1128/mBio.01492-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levy M, et al. Dysbiosis and the immune system. Nat Rev Immunol. 2017;17(4):219–232. doi: 10.1038/nri.2017.7. [DOI] [PubMed] [Google Scholar]
- 8.Carney SM, et al. Methods in lung microbiome research. Am J Respir Cell Mol Biol. 2020;62(3):283–299. doi: 10.1165/rcmb.2019-0273TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeong J, et al. The effect of taxonomic classification by full-length 16S rRNA sequencing with a synthetic long-read technology. Sci Rep. 2021;11(1):1727. doi: 10.1038/s41598-020-80826-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson JS, et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun. 2019;10(1):5029. doi: 10.1038/s41467-019-13036-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toma I, et al. Single-molecule long-read 16S sequencing to characterize the lung microbiome from mechanically ventilated patients with suspected pneumonia. J Clin Microbiol. 2014;52(11):3913–3921. doi: 10.1128/JCM.01678-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Z, et al. A refined view of airway microbiome in chronic obstructive pulmonary disease at species and strain-levels. Front Microbiol. 2020;11:1758. doi: 10.3389/fmicb.2020.01758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sulaiman I, et al. Functional lower airways genomic profiling of the microbiome to capture active microbial metabolism. Eur Respir J. doi: 10.1183/13993003.03434-2020. [published online January 14, 2021]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marotz CA, et al. Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome. 2018;6(1):42. doi: 10.1186/s40168-018-0426-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aogáin MM, et al. Metagenomics reveals a core macrolide resistome related to microbiota in chronic respiratory disease. Am J Respir Crit Care Med. 2020;202(3):433–447. doi: 10.1164/rccm.201911-2202OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Millares L, et al. Functional metagenomics of the bronchial microbiome in COPD. PLoS One. 2015;10(12):e0144448. doi: 10.1371/journal.pone.0144448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cameron SJS, et al. A pilot study using metagenomic sequencing of the sputum microbiome suggests potential bacterial biomarkers for lung cancer. PLoS One. 2017;12(5):e0177062. doi: 10.1371/journal.pone.0177062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas AM, Segata N. Multiple levels of the unknown in microbiome research. BMC Biol. 2019;17(1):48. doi: 10.1186/s12915-019-0667-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaakoush NO. Insights into the role of erysipelotrichaceae in the human host. Front Cell Infect Microbiol. 2015;5:84. doi: 10.3389/fcimb.2015.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Piva TC, et al. Extracellular DNA in sputum is associated with pulmonary function and hospitalization in patients with cystic fibrosis. Respir Med. 2020;172:106144. doi: 10.1016/j.rmed.2020.106144. [DOI] [PubMed] [Google Scholar]
- 21.Rogers GB, et al. The exclusion of dead bacterial cells is essential for accurate molecular analysis of clinical samples. Clin Microbiol Infect. 2010;16(11):1656–1658. doi: 10.1111/j.1469-0691.2010.03189.x. [DOI] [PubMed] [Google Scholar]
- 22.Sulaiman I, et al. Evaluation of the airway microbiome in nontuberculous mycobacteria disease. Eur Respir J. 2018;52(4):1800810. doi: 10.1183/13993003.00810-2018. [DOI] [PubMed] [Google Scholar]
- 23.Whelan FJ, et al. Culture-enriched metagenomic sequencing enables in-depth profiling of the cystic fibrosis lung microbiota. Nat Microbiol. 2020;5(2):379–390. doi: 10.1038/s41564-019-0643-y. [DOI] [PubMed] [Google Scholar]
- 24.Combs MP, et al. Lung microbiota predict chronic rejection in healthy lung transplant recipients: a prospective cohort study. Lancet Respir Med. 2021;9(6):601–612. doi: 10.1016/S2213-2600(20)30405-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Invernizzi R, et al. Bacterial burden in the lower airways predicts disease progression in idiopathic pulmonary fibrosis and is independent of radiological disease extent. Eur Respir J. 2020;55(4):1901519. doi: 10.1183/13993003.01519-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O’Dwyer DN, et al. Lung microbiota contribute to pulmonary inflammation and disease progression in pulmonary fibrosis. Am J Respir Crit Care Med. 2019;199(9):1127–1138. doi: 10.1164/rccm.201809-1650OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dickson RP, et al. Lung microbiota predict clinical outcomes in critically ill patients. Am J Respir Crit Care Med. 2020;201(5):555–563. doi: 10.1164/rccm.201907-1487OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu J, et al. Parallel analysis of cystic fibrosis sputum and saliva reveals overlapping communities and an opportunity for sample decontamination. mSystems. 2020;5(4):e00296–20. doi: 10.1128/mSystems.00296-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tangedal S, et al. Comparing microbiota profiles in induced and spontaneous sputum samples in COPD patients. Respir Res. 2017;18(1):164. doi: 10.1186/s12931-017-0645-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zemanick ET, et al. Assessment of airway microbiota and inflammation in cystic fibrosis using multiple sampling methods. Ann Am Thorac Soc. 2015;12(2):221–229. doi: 10.1513/AnnalsATS.201407-310OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Charlson ES, et al. Assessing bacterial populations in the lung by replicate analysis of samples from the upper and lower respiratory tracts. PLoS One. 2012;7(9):e42786. doi: 10.1371/journal.pone.0042786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dickson RP, et al. Cell-associated bacteria in the human lung microbiome. Microbiome. 2014;2:28. doi: 10.1186/2049-2618-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kelly BJ, et al. Composition and dynamics of the respiratory tract microbiome in intubated patients. Microbiome. 2016;4:7. doi: 10.1186/s40168-016-0151-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang WC, et al. Dynamics of the lung microbiome in intensive care patients with chronic obstructive pulmonary disease and community‑acquired pneumonia. Sci Rep. 2020;10(1):11046. doi: 10.1038/s41598-020-68100-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Panzer AR, et al. Lung microbiota is related to smoking status and to development of acute respiratory distress syndrome in critically ill trauma patients. Am J Respir Crit Care Med. 2018;197(5):621–631. doi: 10.1164/rccm.201702-0441OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Durack J, et al. Features of the bronchial bacterial microbiome associated with atopy, asthma, and responsiveness to inhaled corticosteroid treatment. J Allergy Clin Immunol. 2017;140(1):63–75. doi: 10.1016/j.jaci.2016.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dickson RP, et al. Spatial variation in the healthy human lung microbiome and the adapted island model of lung biogeography. Ann Am Thorac Soc. 2015;12(6):821–830. doi: 10.1513/AnnalsATS.201501-029OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu G, et al. Characterizing human lung tissue microbiota and its relationship to epidemiological and clinical features. Genome Biol. 2016;17(1):163. doi: 10.1186/s13059-016-1021-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peters BA, et al. The microbiome in lung cancer tissue and recurrence-free survival. Cancer Epidemiol Biomarkers Prev. 2019;28(4):731–740. doi: 10.1158/1055-9965.EPI-18-0966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glendinning L, et al. Microbiota in exhaled breath condensate and the lung. Appl Environ Microbiol. 2017;83(12):e00515-17. doi: 10.1128/AEM.00515-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.May AK, et al. A pilot study of the noninvasive assessment of the lung microbiota as a potential tool for the early diagnosis of ventilator-associated pneumonia. Chest. 2015;147(6):1494–1502. doi: 10.1378/chest.14-1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Melnik AV, et al. Molecular and microbial microenvironments in chronically diseased lungs associated with cystic fibrosis. mSystems. 2019;4(5):e00375-19. doi: 10.1128/mSystems.00375-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garg N, et al. Three-dimensional microbiome and metabolome cartography of a diseased human lung. Cell Host Microbe. 2017;22(5):705–716. doi: 10.1016/j.chom.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sze MA, et al. The lung tissue microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;185(10):1073–1080. doi: 10.1164/rccm.201111-2075OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salter SJ, et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014;12:87. doi: 10.1186/s12915-014-0087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim D, et al. Optimizing methods and dodging pitfalls in microbiome research. Microbiome. 2017;5(1):52. doi: 10.1186/s40168-017-0267-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Erb-Downward JR, et al. Critical relevance of stochastic effects on low-bacterial-biomass 16S rRNA gene analysis. mBio. 2020;11(3):e00258-20. doi: 10.1128/mBio.00258-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Segal LN, et al. Enrichment of lung microbiome with supraglottic taxa is associated with increased pulmonary inflammation. Microbiome. 2013;1(1):19. doi: 10.1186/2049-2618-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weyrich LS, et al. Laboratory contamination over time during low-biomass sample analysis. Mol Ecol Resour. 2019;19(4):982–996. doi: 10.1111/1755-0998.13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bassis CM, et al. Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. mBio. 2015;6(2):e00037-15. doi: 10.1128/mBio.00037-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dickson RP, et al. Bacterial topography of the healthy human lower respiratory tract. mBio. 2017;8(1):e02287-16. doi: 10.1128/mBio.02287-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gleeson K, et al. Quantitative aspiration during sleep in normal subjects. Chest. 1997;111(5):1266–1272. doi: 10.1378/chest.111.5.1266. [DOI] [PubMed] [Google Scholar]
- 53.Ren L, et al. Transcriptionally active lung microbiome and its association with bacterial biomass and host inflammatory status. mSystems. 2018;3(5):e00199-18. doi: 10.1128/mSystems.00199-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Charlson ES, et al. Disordered microbial communities in the upper respiratory tract of cigarette smokers. PLoS One. 2010;5(12):e15216. doi: 10.1371/journal.pone.0015216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morris A, et al. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med. 2013;187(10):1067–1075. doi: 10.1164/rccm.201210-1913OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang R, et al. Effects of smoking on the lower respiratory tract microbiome in mice. Respir Res. 2018;19(1):253. doi: 10.1186/s12931-018-0959-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rylance J, et al. Household air pollution and the lung microbiome of healthy adults in Malawi: a cross-sectional study. BMC Microbiol. 2016;16(1):182. doi: 10.1186/s12866-016-0803-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Simon-Soro A, et al. Upper respiratory dysbiosis with a facultative-dominated ecotype in advanced lung disease and dynamic change after lung transplant. Ann Am Thorac Soc. 2019;16(11):1383–1391. doi: 10.1513/AnnalsATS.201904-299OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dickson RP, et al. Enrichment of the lung microbiome with gut bacteria in sepsis and the acute respiratory distress syndrome. Nat Microbiol. 2016;1(10):16113. doi: 10.1038/nmicrobiol.2016.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lozupone C, et al. Widespread colonization of the lung by Tropheryma whipplei in HIV infection. Am J Respir Crit Care Med. 2013;187(10):1110–1117. doi: 10.1164/rccm.201211-2145OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157(1):121–141. doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hooper LV, et al. Interactions between the microbiota and the immune system. Science. 2012;336(6086):1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ivanov, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139(3):485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Segal LN, et al. Enrichment of the lung microbiome with oral taxa is associated with lung inflammation of a Th17 phenotype. Nat Microbiol. 2016;1:16031. doi: 10.1038/nmicrobiol.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dickson RP, et al. The lung microbiota of healthy mice are highly variable, cluster by environment, and reflect variation in baseline lung innate immunity. Am J Respir Crit Care Med. 2018;198(4):497–508. doi: 10.1164/rccm.201711-2180OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gollwitzer ES, et al. Lung microbiota promotes tolerance to allergens in neonates via PD-L1. Nat Med. 2014;20(6):642–647. doi: 10.1038/nm.3568. [DOI] [PubMed] [Google Scholar]
- 67.Herbst T, et al. Dysregulation of allergic airway inflammation in the absence of microbial colonization. Am J Respir Crit Care Med. 2011;184(2):198–205. doi: 10.1164/rccm.201010-1574OC. [DOI] [PubMed] [Google Scholar]
- 68.Brown RL, et al. The microbiota protects against respiratory infection via GM-CSF signaling. Nat Commun. 2017;8(1):1512. doi: 10.1038/s41467-017-01803-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu BG, et al. Episodic aspiration with oral commensals induces a MyD88-dependent, pulmonary T-helper cell type 17 response that mitigates susceptibility to Streptococcus pneumoniae. Am J Respir Crit Care Med. 2021;203(9):1099–1111. doi: 10.1164/rccm.202005-1596OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bittinger K, et al. Improved characterization of medically relevant fungi in the human respiratory tract using next-generation sequencing. Genome Biol. 2014;15(10):487. doi: 10.1186/s13059-014-0487-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tipton L, et al. The lung mycobiome in the next-generation sequencing era. Virulence. 2017;8(3):334–341. doi: 10.1080/21505594.2016.1235671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang C, et al. Fungal and bacterial microbiome dysbiosis and imbalance of trans‑kingdom network in asthma. Clin Transl Allergy. 2020;10:42. doi: 10.1186/s13601-020-00345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sharma A, et al. Associations between fungal and bacterial microbiota of airways and asthma endotypes. J Allergy Clin Immunol. 2019;144(5):1214–1227. doi: 10.1016/j.jaci.2019.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Soret P, et al. Respiratory mycobiome and suggestion of inter-kingdom network during acute pulmonary exacerbation in cystic fibrosis. Sci Rep. 2020;10(1):3589. doi: 10.1038/s41598-020-60015-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Charlson ES, et al. Lung-enriched organisms and aberrant bacterial and fungal respiratory microbiota after lung transplant. Am J Respir Crit Care Med. 2012;186(6):536–545. doi: 10.1164/rccm.201204-0693OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Young JC, et al. Viral metagenomics reveal blooms of anelloviruses in the respiratory tract of lung transplant recipients. Am J Transplant. 2015;15(1):200–209. doi: 10.1111/ajt.13031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lim YW, et al. Metagenomics and metatranscriptomics: windows on CF-associated viral and microbial communities. J Cyst Fibros. 2013;12(2):154–164. doi: 10.1016/j.jcf.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Willner D, et al. Metagenomic analysis of respiratory tract DNA viral communities in cystic fibrosis and non-cystic fibrosis individuals. PLoS One. 2009;4(10):e7370. doi: 10.1371/journal.pone.0007370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Focosi D, et al. Torquetenovirus: the human virome from bench to bedside. Clin Microbiol Infect. 2016;22(7):589–593. doi: 10.1016/j.cmi.2016.04.007. [DOI] [PubMed] [Google Scholar]
- 80.Freer G, et al. The virome and its major component, anellovirus, a convoluted system molding human immune defenses and possibly affecting the development of asthma and respiratory diseases in childhood. Front Microbiol. 2018;9:686. doi: 10.3389/fmicb.2018.00686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Abbas AA, et al. Redondoviridae, a family of small, circular DNA viruses of the human oro-respiratory tract associated with periodontitis and critical illness. Cell Host Microbe. 2019;25(5):719–729. doi: 10.1016/j.chom.2019.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Héry-Arnaud G, et al. The lung and gut microbiome: what has to be taken into consideration for cystic fibrosis? J Cyst Fibros. 2019;18(1):13–21. doi: 10.1016/j.jcf.2018.11.003. [DOI] [PubMed] [Google Scholar]
- 83.Martin-Loeches I, et al. The importance of airway and lung microbiome in the critically ill. Crit Care. 2020;24(1):537. doi: 10.1186/s13054-020-03219-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McGinniss JE, et al. The lung microbiome in lung transplantation. J Heart Lung Transplant. 2019;24(3):305–310. doi: 10.1016/j.healun.2021.04.014. [DOI] [PubMed] [Google Scholar]
- 85.Xie M, et al. Trends in prevalence and incidence of chronic respiratory diseases from 1990 to 2017. Respir Res. 2020;21(1):49. doi: 10.1186/s12931-020-1291-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rabe KF, Watz H. Chronic obstructive pulmonary disease. Lancet. 2017;389(10082):1931–1940. doi: 10.1016/S0140-6736(17)31222-9. [DOI] [PubMed] [Google Scholar]
- 87.Decramer M, et al. Chronic obstructive pulmonary disease. Lancet Respir Med. 2012;379(9823):1341–1351. doi: 10.1016/S0140-6736(11)60968-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sethi S. Bacteria in exacerbations of chronic obstructive pulmonary disease: phenomenon or epiphenomenon? Proc Am Thorac Soc. 2004;1(2):109–114. doi: 10.1513/pats.2306029. [DOI] [PubMed] [Google Scholar]
- 89.Papi A, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med. 2006;173(10):1114–1121. doi: 10.1164/rccm.200506-859OC. [DOI] [PubMed] [Google Scholar]
- 90.Sethi S, et al. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. N Engl J Med. 2002;347(7):465–471. doi: 10.1056/NEJMoa012561. [DOI] [PubMed] [Google Scholar]
- 91.Janjua S, et al. Prophylactic antibiotics for adults with chronic obstructive pulmonary disease: a network meta-analysis. Cochrane Database Syst Rev. 2021;1:Cd013198. doi: 10.1002/14651858.CD013198.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Einarsson GG, et al. Community dynamics and the lower airway microbiota in stable chronic obstructive pulmonary disease, smokers and healthy non-smokers. Thorax. 2016;71(9):795–803. doi: 10.1136/thoraxjnl-2015-207235. [DOI] [PubMed] [Google Scholar]
- 93.Wang Z, et al. Multi-omic meta-analysis identifies functional signatures of airway microbiome in chronic obstructive pulmonary disease. ISME J. 2020;14(11):2748–2765. doi: 10.1038/s41396-020-0727-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Millares L, et al. Relationship between the respiratory microbiome and the severity of airflow limitation, history of exacerbations and circulating eosinophils in COPD patients. BMC Pulm Med. 2019;19(1):112. doi: 10.1186/s12890-019-0867-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mayhew D, et al. Longitudinal profiling of the lung microbiome in the AERIS study demonstrates repeatability of bacterial and eosinophilic COPD exacerbations. Thorax. 2018;73(5):422–430. doi: 10.1136/thoraxjnl-2017-210408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Opron K, et al. Lung microbiota associations with clinical features of COPD in the SPIROMICS cohort. NPJ Biofilms Microbiomes. 2021;7(1):14. doi: 10.1038/s41522-021-00185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang Z, et al. Airway host-microbiome interactions in chronic obstructive pulmonary disease. Respir Res. 2019;20(1):113. doi: 10.1186/s12931-019-1085-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gupta S, et al. Comparative analysis of the alveolar microbiome in COPD, ECOPD, Sarcoidosis, and ILD patients to identify respiratory illnesses specific microbial signatures. Sci Rep. 2021;11(1):3963. doi: 10.1038/s41598-021-83524-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pragman AA, et al. Chronic obstructive pulmonary disease upper airway microbiota alpha diversity is associated with exacerbation phenotype: a case-control observational study. Respir Res. 2019;20(1):114. doi: 10.1186/s12931-019-1080-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bouquet J, et al. Microbial burden and viral exacerbations in a longitudinal multicenter COPD cohort. Respir Res. 2020;21(1):77. doi: 10.1186/s12931-020-01340-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Filho FSL, et al. Sputum microbiome is associated with 1-year mortality after chronic obstructive pulmonary disease hospitalizations. Am J Respir Crit Care Med. 2019;199(10):1205–1213. doi: 10.1164/rccm.201806-1135OC. [DOI] [PubMed] [Google Scholar]
- 102.Dicker AJ, et al. The sputum microbiome, airway inflammation, and mortality in chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2021;147(1):158–167. doi: 10.1016/j.jaci.2020.02.040. [DOI] [PubMed] [Google Scholar]
- 103.Leiten EO, et al. The airway microbiota and exacerbations of COPD. ERJ Open Res. 2020;6(3):00168-2020. doi: 10.1183/23120541.00168-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang Z, et al. Inflammatory endotype associated airway microbiome in COPD clinical stability and exacerbations — a multi-cohort longitudinal analysis. Am J Respir Crit Care Med. doi: 10.1164/rccm.202009-3448oc. [published online December 17, 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang Z, et al. Lung microbiome dynamics in COPD exacerbations. Eur Respir J. 2016;47(4):1034–1092. doi: 10.1183/13993003.00270-2016. [DOI] [PubMed] [Google Scholar]
- 106.Diver S, et al. Sputum microbiomic clustering in asthma and chronic obstructive pulmonary disease reveals a Haemophilus-predominant subgroup. Allergy. 2020;75(4):808–817. doi: 10.1111/all.14058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dicker AJ, et al. Neutrophil extracellular traps are associated with disease severity and microbiota diversity in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2018;141(1):117–127. doi: 10.1016/j.jaci.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang Z, et al. Sputum microbiome temporal variability and dysbiosis in chronic obstructive pulmonary disease exacerbations: an analysis of the COPDMAP study. Thorax. 2018;73(4):331–338. doi: 10.1136/thoraxjnl-2017-210741. [DOI] [PubMed] [Google Scholar]
- 109.Ghebre MA, et al. Biological exacerbation clusters demonstrate asthma and chronic obstructive pulmonary disease overlap with distinct mediator and microbiome profiles. J Allergy Clin Immunol. 2018;141(6):2027–2036. doi: 10.1016/j.jaci.2018.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mims JW. Asthma: definitions and pathophysiology. Int Forum Allergy Rhinol. 2015;5(suppl 1):S2–S6. doi: 10.1002/alr.21609. [DOI] [PubMed] [Google Scholar]
- 111.Mejias A, et al. Risk of childhood wheeze and asthma after respiratory syncytial virus infection in full-term infants. Pediatr Allergy Immunol. 2020;31(1):47–56. doi: 10.1111/pai.13131. [DOI] [PubMed] [Google Scholar]
- 112.Beasley R, et al. Risk factors for asthma: is prevention possible? Lancet. 2015;386(9998):1075–1085. doi: 10.1016/S0140-6736(15)00156-7. [DOI] [PubMed] [Google Scholar]
- 113.Marri PR, et al. Asthma-associated differences in microbial composition of induced sputum. J Allergy Clin Immunol. 2013;131(2):346–352. doi: 10.1016/j.jaci.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Denner DR, et al. Corticosteroid therapy and airflow obstruction influence the bronchial microbiome, which is distinct from that of bronchoalveolar lavage in asthmatic airways. J Allergy Clin Immunol. 2016;137(5):1398–1405. doi: 10.1016/j.jaci.2015.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Simpson JL, et al. Airway dysbiosis: haemophilus influenzae and Tropheryma in poorly controlled asthma. Eur Respir J. 2016;47(3):792–800. doi: 10.1183/13993003.00405-2015. [DOI] [PubMed] [Google Scholar]
- 116.Taylor SL, et al. Inflammatory phenotypes in patients with severe asthma are associated with distinct airway microbiology. J Allergy Clin Immunol. 2018;141(1):94–103. doi: 10.1016/j.jaci.2017.03.044. [DOI] [PubMed] [Google Scholar]
- 117.Durack J, et al. Distinct associations of sputum and oral microbiota with atopic, immunologic, and clinical features in mild asthma. J Allergy Clin Immunol. 2020;146(5):1016–1026. doi: 10.1016/j.jaci.2020.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Caverly LJ, et al. Past, present, and future research on the lung microbiome in inflammatory airway disease. Chest. 2019;156(2):376–382. doi: 10.1016/j.chest.2019.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Barcik W, et al. The role of lung and gut microbiota in the pathology of asthma. Immunity. 2020;52(2):241–255. doi: 10.1016/j.immuni.2020.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Siegel RL, et al. Cancer statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 121.Islami F, et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J Clin. 2018;68(1):1–54. doi: 10.3322/caac.21416. [DOI] [PubMed] [Google Scholar]
- 122.Mouronte-Roibás C, et al. COPD, emphysema and the onset of lung cancer. A systematic review. Cancer Lett. 2016;382(2):240–244. doi: 10.1016/j.canlet.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 123.Brenner DR, et al. Previous lung diseases and lung cancer risk: a pooled analysis from the International Lung Cancer Consortium. Am J Epidemiol. 2012;176(7):573–585. doi: 10.1093/aje/kws151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ahktar N, Bensal JG. Risk factors for lung cancer in nonsmoker. Curr Probl Cancer. 2017;41(5):328–339. doi: 10.1016/j.currproblcancer.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 125.Jin C, et al. Commensal microbiota promote lung cancer development via γδ T cells. Cell. 2019;176(5):998–1013. doi: 10.1016/j.cell.2018.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Schreiber H, et al. Induction of lung cancer in germfree, specific-pathogen-free, and infected rats by N-nitrosoheptamethyleneimine: enhancement by respiratory infection. J Natl Cancer Inst. 1972;49(4):1107–1114. [PubMed] [Google Scholar]
- 127.Wong SH, Yu J. Gut microbiota in colorectal cancer: mechanisms of action and clinical applications. Nat Rev Gastroenterol Hepatol. 2019;16(11):690–704. doi: 10.1038/s41575-019-0209-8. [DOI] [PubMed] [Google Scholar]
- 128.Allen J, Sears CL. Impact of the gut microbiome on the genome and epigenome of colon epithelial cells: contributions to colorectal cancer development. Genome Med. 2019;11(1):11. doi: 10.1186/s13073-019-0621-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hosgood HD, et al. The potential role of lung microbiota in lung cancer attributed to household coal burning exposures. Environ Mol Mutagen. 2014;55(8):643–651. doi: 10.1002/em.21878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lee SH, et al. Characterization of microbiome in bronchoalveolar lavage fluid of patients with lung cancer comparing with benign mass like lesions. Lung Cancer. 2016;102:89–95. doi: 10.1016/j.lungcan.2016.10.016. [DOI] [PubMed] [Google Scholar]
- 131.Greathouse KL, et al. Interaction between the microbiome and TP53 in human lung cancer. Genome Biol. 2018;19(1):123. doi: 10.1186/s13059-018-1501-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Liu HX, et al. Difference of lower airway microbiome in bilateral protected specimen brush between lung cancer patients with unilateral lobar masses and control subjects. Int J Cancer. 2018;142(4):769–778. doi: 10.1002/ijc.31098. [DOI] [PubMed] [Google Scholar]
- 133.Patnaik SK, et al. Lower airway bacterial microbiome may influence recurrence after resection of early-stage non-small cell lung cancer. J Thorac Cardiovasc Surg. 2020;161(2):419–429. doi: 10.1016/j.jtcvs.2020.01.104. [DOI] [PubMed] [Google Scholar]
- 134.Tsay JCJ, et al. Lower airway dysbiosis affects lung cancer progression. Cancer Discov. 2021;11(2):293–307. doi: 10.1158/2159-8290.CD-20-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tsay JCJ, et al. Airway microbiota is associated with upregulation of the PI3K pathway in lung cancer. Am J Respir Crit Care Med. 2018;198(9):1188–1198. doi: 10.1164/rccm.201710-2118OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gustafson AM, et al. Airway PI3K pathway activation is an early and reversible event in lung cancer development. Sci Transl Med. 2010;2(26):26ra5. doi: 10.1126/scitranslmed.3000251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Han MK, et al. Lung microbiome and disease progression in idiopathic pulmonary fibrosis: an analysis of the COMET study. Lancet Respir Med. 2014;2(7):548–556. doi: 10.1016/S2213-2600(14)70069-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Molyneaux PL, et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014;190(8):906–913. doi: 10.1164/rccm.201403-0541OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.D’Alessandro-Gabazza CN, et al. A Staphylococcus pro-apoptotic peptide induces acute exacerbation of pulmonary fibrosis. Nat Commun. 2020;11(1):1539. doi: 10.1038/s41467-020-15344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kitsios GD, et al. Microbiome in lung explants of idiopathic pulmonary fibrosis: a case-control study in patients with end-stage fibrosis. Thorax. 2018;73(5):481–484. doi: 10.1136/thoraxjnl-2017-210537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Valenzi E, et al. Topographic heterogeneity of lung microbiota in end-stage idiopathic pulmonary fibrosis: the Microbiome in Lung Explants-2 (MiLEs-2) study. Thorax. 2021;76(3):239–247. doi: 10.1136/thoraxjnl-2020-214770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wilson AM, et al. Effect of co-trimoxazole(trimethoprim-sulfamethoxazole) vs placebo on death, lung transplant, or hospital admission in patients with moderate and severe idiopathic pulmonary fibrosis: The EME-TIPAC randomized clinical trial. JAMA. 2020;324(22):2282–2291. doi: 10.1001/jama.2020.22960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wootton SC, et al. Viral infection in acute exacerbation of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183(12):1698–1702. doi: 10.1164/rccm.201010-1752OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Blacher E, et al. Microbiome-modulated metabolites at the interface of host immunity. J Immunol. 2017;198(2):572–580. doi: 10.4049/jimmunol.1601247. [DOI] [PubMed] [Google Scholar]
- 145.Mirkovic B, et al. The role of short-chain fatty acids, produced by anaerobic bacteria, in the cystic fibrosis airway. Am J Respir Crit Care Med. 2015;192(11):1314–1324. doi: 10.1164/rccm.201505-0943OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lin MY, et al. Redirection of epithelial immune responses by short-chain fatty acids through inhibition of histone deacetylases. Front Immunol. 2015;6:554. doi: 10.3389/fimmu.2015.00554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Schulthess J, et al. The short chain fatty acid butyrate imprints an antimicrobial program in macrophages. Immunity. 2019;50(2):432–445. doi: 10.1016/j.immuni.2018.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chang PV, et al. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U S A. 2014;111(6):2247–2252. doi: 10.1073/pnas.1322269111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Vinolo MAR, et al. Effects of short chain fatty acids on effector mechanisms of neutrophils. Cell Biochem Funct. 2009;27(1):48–55. doi: 10.1002/cbf.1533. [DOI] [PubMed] [Google Scholar]
- 150.Rutting S, et al. Short-chain fatty acids increase TNF-induced inflammation in primary human lung mesenchymal cells through the activation of p38 MAPK. Am J Physiol Lung Cell Mol Physiol. 2019;316(1):L157–L174. doi: 10.1152/ajplung.00306.2018. [DOI] [PubMed] [Google Scholar]
- 151.Ghorbani P, et al. Short-chain fatty acids affect cystic fibrosis airway inflammation and bacterial growth. Eur Respir J. 2015;46(4):1033–1045. doi: 10.1183/09031936.00143614. [DOI] [PubMed] [Google Scholar]
- 152.Li D, et al. Azithromycin prophylaxis after lung transplantation is associated with improved overall survival. J Heart Lung Transplant. 2020;39(12):1426–1434. doi: 10.1016/j.healun.2020.09.006. [DOI] [PubMed] [Google Scholar]
- 153.Albert RK, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med. 2011;365(8):689–698. doi: 10.1056/NEJMoa1104623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Spence CD, et al. Influence of azithromycin and allograft rejection on the post-lung transplant microbiota. J Heart Lung Transplant. 2020;39(2):176–183. doi: 10.1016/j.healun.2019.11.007. [DOI] [PubMed] [Google Scholar]
- 155.Chen YF, et al. The lung microbiota changes associated with inhaled corticosteroid and azithromycin in murine model of COPD. Eur Respir J. 2018;52(suppl 62):PA984 [Google Scholar]
- 156.Samson C, et al. Long-term effects of azithromycin in patients with cystic fibrosis. Respir Med. 2016;117:1–6. doi: 10.1016/j.rmed.2016.05.025. [DOI] [PubMed] [Google Scholar]
- 157.Segal LN, et al. Randomised, double-blind, placebo-controlled trial with azithromycin selects for anti-inflammatory microbial metabolites in the emphysematous lung. Thorax. 2017;72(1):13–22. doi: 10.1136/thoraxjnl-2016-208599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gill PA, et al. Review article: Short chain fatty acids as potential therapeutic agents in human gastrointestinal and inflammatory disorders. Aliment Pharmacol Ther. 2018;48(1):15–34. doi: 10.1111/apt.14689. [DOI] [PubMed] [Google Scholar]
- 159.Budden KF, et al. Emerging pathogenic links between microbiota and the gut-lung axis. Nat Rev Microbiol. 2017;15(1):55–63. doi: 10.1038/nrmicro.2016.142. [DOI] [PubMed] [Google Scholar]