

Keywords: adipose tissue resident macrophages, inflammation, insulin sensitivity, mitochondria, obesity
Abstract
Obesity is accompanied by numerous systemic and tissue-specific derangements, including systemic inflammation, insulin resistance, and mitochondrial abnormalities in skeletal muscle. Despite growing recognition that adipose tissue dysfunction plays a role in obesity-related disorders, the relationship between adipose tissue inflammation and other pathological features of obesity is not well-understood. We assessed macrophage populations and measured the expression of inflammatory cytokines in abdominal adipose tissue biopsies in 39 nondiabetic adults across a range of body mass indexes (BMI 20.5–45.8 kg/m2). Skeletal muscle biopsies were used to evaluate mitochondrial respiratory capacity, ATP production capacity, coupling, and reactive oxygen species production. Insulin sensitivity (SI) and β cell responsivity were determined from test meal postprandial glucose, insulin, c-peptide, and triglyceride kinetics. We examined the relationships between adipose tissue inflammatory markers, systemic inflammatory markers, SI, and skeletal muscle mitochondrial physiology. BMI was associated with increased adipose tissue and systemic inflammation, reduced SI, and reduced skeletal muscle mitochondrial oxidative capacity. Adipose-resident macrophage numbers were positively associated with circulating inflammatory markers, including tumor necrosis factor-α (TNFα) and C-reactive protein (CRP). Local adipose tissue inflammation and circulating concentrations of TNFα and CRP were negatively associated with SI, and circulating concentrations of TNFα and CRP were also negatively associated with skeletal muscle oxidative capacity. These results demonstrate that obese humans exhibit increased adipose tissue inflammation concurrently with increased systemic inflammation, reduced insulin sensitivity, and reduced muscle oxidative capacity and suggest that adipose tissue and systemic inflammation may drive obesity-associated metabolic derangements.
NEW AND NOTEWORTHY Adipose inflammation is proposed to be at the nexus of the systemic inflammation and metabolic derangements associated with obesity. The present study provides evidence to support adipose inflammation as a central feature of the pathophysiology of obesity. Adipose inflammation is associated with systemic and peripheral metabolic derangements, including increased systemic inflammation, reduced insulin sensitivity, and reduced skeletal muscle mitochondrial respiration.
INTRODUCTION
The rising prevalence of obesity in the United States (1) is accompanied by metabolic and cardiovascular derangements such as insulin resistance, type 2 diabetes, dyslipidemia, and cardiovascular disease (2). Central adiposity has emerged as a primary predictor of many metabolic abnormalities with obesity, including insulin resistance, type 2 diabetes, and atherosclerosis (3, 4), and adipose-derived inflammation is proposed to be at the nexus of central adiposity and many of the obesity-associated metabolic derangements. The hypertrophy of adipocytes and the accumulation of adipose-resident macrophages result in local inflammation and contribute to a proinflammatory secretome of adipose tissue, which is proposed to have profound effects on many aspects of metabolism, including insulin signaling and mitochondrial function.
Adipose tissue inflammation appears to have both local and systemic consequences. Local inflammation impairs adipocyte differentiation, exacerbating the proinflammatory environment (5) and inhibits insulin signaling within the adipose tissue (6), which along with skeletal muscle and liver, is one of three key insulin-sensitive tissues. Further, in addition to serving as an important energy storage reservoir, adipose tissue acts in an endocrine manner, releasing adipokines, cytokines, and lipids that affect peripheral tissues (6–11). Inasmuch, the secretome of inflamed adipose tissue can have profound effects on peripheral tissues. Adipose-derived inflammatory cytokines have been shown to inhibit insulin signaling and impair lipid and glucose metabolism in the liver and skeletal muscle, likely contributing to the insulin resistance associated with obesity (12). Inflammatory cytokines have also been shown to impair mitochondrial biogenesis and function in the skeletal muscle (13, 14), which may have important implications for metabolic health. However, despite some compelling evidence, there is lingering ambiguity regarding the role of adipose tissue inflammation in obesity-related metabolic derangements based on recent evidence that adipose inflammation and macrophage recruitment occur with obesity, but independently from insulin resistance (15, 16), and that acute adipose inflammation may actually be essential for the healthy expansion of adipose tissue (17). Efforts to understand potential links between adipose tissue biology and the associated metabolic derangements (e.g., reduced insulin sensitivity, altered mitochondrial function) will improve our understanding of the pathophysiology of obesity and help uncover new countermeasures for obesity-associated complications.
Obesity is associated with a state of chronic, systemic, low-grade inflammation which is postulated to arise from inflamed adipose tissue and is implicated in the pathophysiology of obesity. Although physiological and morphological changes to adipose tissue are coincident with obesity, the contribution of these obesity-associated changes to metabolic dysregulation remains an active area of investigation. This study was performed to evaluate the relationships between adipose tissue inflammation (macrophage burden, cytokine expression) and known metabolic impairments with obesity such as reduced insulin sensitivity, β cell function, and mitochondrial physiology in peripheral insulin sensitive tissues such as skeletal muscle. Toward this goal, we performed an extensive evaluation of adipose inflammation, including adipose-resident macrophage populations and gene and protein expression of inflammatory proteins, and of systemic inflammation in obese [body mass index (BMI) > 30 kg/m2] and nonobese (BMI < 30 kg/m2) men and women. In the same individuals, mitochondrial function was evaluated in skeletal muscle samples and whole body insulin sensitivity and β cell responsivity were assessed using a mixed meal challenge test. Within this cohort, we sought to determine the relationship between adipose tissue macrophage burden and inflammation and circulating systemic inflammation, and to assess the effects of adipose tissue inflammation and macrophage burden and systemic inflammation on whole body insulin responsiveness and skeletal muscle mitochondrial function. With increasing BMI, we observed increased adipose tissue macrophage burden and interleukin (IL)-6 concentration, elevated markers of systemic inflammation, reduced mitochondrial oxidative capacity in skeletal muscle, and reduced insulin sensitivity. Adipose tissue macrophage populations and cytokine concentrations were positively associated with the concentrations of multiple circulating inflammatory cytokines, including C-reactive protein (CRP) and tumor necrosis factor-α (TNFα), and were negatively associated with insulin sensitivity. Circulating concentrations of CRP were in turn negatively associated with insulin sensitivity and with mitochondrial oxidative capacity. These results support the notion that adipose-resident macrophages and adipose-derived cytokines may contribute to hallmark metabolic dysfunctions of obesity, including insulin resistance and mitochondrial dysfunction, through the effects of circulating inflammatory factors on distal tissues.
METHODS
Study Participants
Thirty-nine (23 F/16M) adults aged 30–55 yr with BMI ranging from 19–46 kg/m2 (Table 1) were recruited from the local community to participate in the study. Participants were weight-stable (self-reported; weight maintained ±2.5 kg in the previous 6 mo) and did not participate in structured exercise training (self-reported activity levels <30 min of exercise 3 times per week). Potential participants’ medical histories were reviewed, and participants were excluded if they had anemia, diagnosed diabetes, coronary artery or macrovascular disease, kidney disease, untreated thyroid disease, blood clotting disorders, or any disease or condition that would preclude participation in any study procedure or increase the risks of the study. Participants were excluded if their fasting plasma glucose was 126 mg/dL or higher at the time of screening. Pregnant or breastfeeding females were excluded from the study. All participants were nonsmokers, consumed <4 oz alcohol per day, and did not have any substance abuse disorders. Participants taking any medication that may affect the outcomes of the study or increase the risk of study procedures (e.g., warfarin group medications, metformin, tricyclic antidepressants, benzodiazepines, opiates, barbiturates, anticoagulants) were also excluded. Study participation consisted of a screening visit and an overnight visit, detailed below. The study was registered under Clinical Trial Number NCT02732509, and study procedures were approved by the Mayo Foundation Institutional Review Board and conformed to principles of the Declaration of Helsinki.
Table 1.
Study participant demographics, body composition, and physiological data
|
BMI < 30 kg/m2 (N = 19; 10F/9M) Mean (Range) |
BMI > 30 kg/m2 (N = 20; 13F/7M) Mean (Range) |
Correlation with BMI R (P Value) |
|
|---|---|---|---|
| Body Composition | |||
| BMI, kg/m2 | 25.1 (20.5–29.3) | 37.4 (30.7–45.8)* | |
| Age, yr | 37.32 (30–53) | 40.85 (30–55) | 0.247 (P = 0.130) |
| Mass, kg | 73.0 (50.8–94.0) | 107.0 (87.4–159.0)* | |
| Height, cm | 170.0 (154.7–184.9) | 169.1 (157.4–192.7) | |
| Body fat, % | 31.2 (21.5–42.6) | 47.6 (34.5–57.9)* | 0.861 (P < 0.001) |
| Body fat, kg | 21.8 (12.4–32.3) | 49.1 (31.5–73.5)* | 0.952 (P < 0.001) |
| Fat-free mass, kg | 50.6 (36.8–71.5) | 56.4 (39.5–85.2) | 0.385 (P = 0.015) |
| A:G ratio | 0.99 (0.55–1.44) | 1.21 (1.0–1.42)* | 0.554 (P < 0.001) |
| Adipocyte lipid content, µ/cell | 0.573 (0.076–0.987) | 1.212 (0.712–2.118)* | 0.756 (P < 0.001) |
| Blood Chemistry | |||
| Fasting glucose, mg/dL | 84.9 (74.5–92.7) | 83.7 (72.5–99.3) | 0.004 (P = 0.983) |
| Fasting insulin, µIU/mL | 3.36 (1.23–6.71) | 8.88 (2.37–19.60)* | 0.795 (P < 0.001) |
| Fasting TG, mg/dL | 74.4 (37.3–213.0) | 156.6 (55.7–472.0)* | 0.554 (P < 0.001) |
| TyG | 7.98 (7.28–9.2) | 8.64 (7.71–9.83)* | 0.5408 (P < 0.001) |
| Inflammatory Markers | |||
| WBC, × 109/mL | 6.1 (3.5–8.8) | 6.5 (4.6–9.4) | 0.174 (P = 0.289) |
| ESR, mm/h | 6.2 (0–20) | 9.5 (1–32) | 0.305 (P = 0.071) |
| CRP, mg/dL | 0.14 (0.01–0.69) | 0.58 (0.04–4.25)* | 0.671 (P < 0.001) |
| TNFα, pg/mL | 0.78 (0.31–1.60) | 0.98 (0.55–1.4)* | 0.458 (P = 0.003) |
| GM-CSF, pg/mL | 55.0 (5.2–268.7) | 36.9 (16.6–80.2) | –0.194 (P = 0.238) |
| IFNγ, pg/mL | 15.8 (3.3–23.8) | 14.9 (6.4–41.0) | 0.132 (P = 0.424) |
| IL-1β, pg/mL | 1.34 (0.28–2.08) | 1.27 (0.54–3.34) | 0.076 (P = 0.646) |
| IL-2, pg/mL | 4.07 (0.73–6.35) | 4.87 (1.24–24.04) | 0.199 (P = 0.223) |
| IL-4, pg/mL | 20.56 (4.75–58.33) | 21.66 (6.11–72.96) | 0.155 (P = 0.346) |
| IL-5, pg/mL | 1.46 (0.16–2.51) | 1.50 (0.36–3.94) | 0.256 (P = 0.116) |
| IL-6, pg/mL | 3.8 (0.8–9.2) | 5.8 (1.7–20.0)* | 0.218 (P = 0.182) |
| IL-8, pg/mL | 4.91 (1.94–8.96) | 5.01 (2.62–12.18) | 0.214 (P = 0.190) |
| IL-10, pg/mL | 7.07 (0.82–46.69) | 10.21 (0.73–99.22) | 0.067 (P = 0.683) |
| IL-12, pg/mL | 2.94 (0.04–7.91) | 2.35 (0.63–6.83) | –0.036 (P = 0.828) |
| IL-13, pg/mL | 4.46 (0.04–9.73) | 4.32 (0.59–10.75) | 0.150 (P = 0.362) |
| IL-17A, pg/mL | 12.78 (1.39–24.43) | 12.06 (1.46–43.11) | 0.124 (P = 0.451) |
| IL-23, pg/mL | 386.4 (48.4–1,087.6) | 325.0 (60.9–875.5) | 0.089 (P = 0.588) |
Thirty-nine participants (23 F/16 M) aged 30–55 yr with BMIs ranging from 20.5 to 45.8 participated in the study. Body composition was determined by dual energy x-ray absorptiometry. Adipocyte cell size (lipid content) was determined from subcutaneous adipose tissue biopsy samples (N = 34). Fasting blood samples were collected to assess fasting concentrations of insulin, glucose, triglycerides, the triglyceride-glucose index (TyG), and inflammatory cytokines. Data are presented as mean (range). *Significant difference (P < 0.05) between participants with BMI < 30 kg/m2 and those with BMI > 30 kg/m2, and the correlation coefficients (r) between BMI and body composition and physiological data are presented. A:G, android:gynoid; BMI, body mass index; CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; F, female; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFNγ, interferon-γ; IL, interleukin; M, male; TG, triglycerides; TNFα, tumor necrosis factor-α; TyG, triglyceride-glucose index; WBC, white blood count.
Screening Visit
Potential participants reported to the Mayo Clinic Clinical Research and Trials Unit (CRTU) following an overnight fast. Study procedures and risks were explained, and written informed consent was provided by all included subjects. Following consent, a blood sample was collected and complete blood counts with differentials, fasting glucose, serum creatinine, and thyroid stimulating hormone were measured by the Mayo Clinic Central Clinical Laboratory to determine subject eligibility. Body composition was measured by dual energy X-ray absorptiometry (DEXA; GE Lunar iDXA, GE Healthcare, Chicago, IL).
Inpatient Study Day
At least 1 wk after screening, participants were provided with three days of weight-maintaining meals by the CRTU Metabolic Kitchen. Caloric needs were determined using the Harris Benedict Equation (18), and the macronutrient distribution of the diet was ∼20% protein, 50% carbohydrate, and 30% fat. On the evening of the third day, subjects were admitted to the CRTU. After a meal at 1800, subjects remained fasted with ad libitum access to water. The following morning, a hand was placed in a Plexiglas box maintained at 55°C for the collection of arterialized blood from a retrograde intravenous catheter. At 0800, biopsies of the vastus lateralis muscle and subcutaneous abdominal adipose tissue were performed. At 1200, a mixed meal was consumed and blood samples were collected at intervals over the subsequent 5 h.
Mixed Meal Tolerance Test
Participants (N = 36) consumed a standardized mixed meal shake composed of 15% protein, 55% carbohydrate, and 30% fat, and comprising 35% of the individual’s resting energy expenditure. Arterialized blood was collected 30, 20, and 10 min before meal ingestion and at intervals over the 5 h following meal ingestion. Three participants were not able complete the meal challenge and all subsequent blood draws. At each time point, glucose was measured in freshly isolated plasma samples using a Yellow Springs Instrument (YSI) analyzer (YSI Inc, Yellow Springs, OH). EDTA plasma from each time point was stored at −80°C for the measurement of triglycerides using an enzymatic colorimetric test and insulin using a one-step immunoenzymatic Access Ultrasensitive Insulin assay by the Mayo Clinic Immunochemical Core Laboratory (ICL). Serum samples from each time point were stored at −80°C for subsequent measurement of C-peptide using the Roche 2-site immunometric assay with electrochemiluminescent detection by the ICL.
Insulin sensitivity (SI; N = 33) and β-cell responsivity (Φ; N = 30) were estimated using the oral glucose and C-peptide minimal models, respectively (19–22). The disposition index (DI: N = 30) was calculated as the product of SI and Φ (19). Individuals for which the minimal models could not be resolved were excluded from the analysis. Postprandial triglyceride responses were assessed using the incremental area under the curve (iAUC) and the change in triglyceride levels from baseline to peak postprandial triglyceride concentration (N = 36). The triglyceride-glucose index (TyG) was calculated from the fasting glucose and triglyceride concentrations using the equation ln{[triglyceride (mg/dL) · glucose (mg/dL)]/2} (23).
Adipose and Muscle Biopsies
Subcutaneous adipose tissue biopsies were collected (N = 34) from the lower abdominal region, two thirds of the distance from the iliac spine to the umbilicus, by small-needle liposuction with local anesthesia (1% lidocaine) and under sterile conditions (24). Tissue was transferred to a mesh strain and rinsed with 0.9% sodium chloride to remove excess blood. Blood clots and connective tissue were removed and tissue was processed for subsequent cell sizing, immunohistochemical (IHC) staining, gene, and protein measurements. For some participants, insufficient sample was collected for all assays. Ns for each assay are noted below. A percutaneous biopsy of the vastus lateralis muscle was performed (N = 39) under sterile conditions and local anesthesia (2% lidocaine) using a modified Bergstrom needle as previously described (25, 26). ∼100 mg of tissue was collected for measurements of mitochondrial function.
Adipose Tissue Cell Sizing
Adipocyte cell size was measured (N = 34) as previously described (27). Briefly, 1 mg/mL collagenase type II (Sigma, C-6885) was added to ∼100 mg of fresh adipose tissue in HEPES buffer (0.1 M HEPES, 0.12 M NaCl, 0.05 M KCl, 0.005 M glucose, 1.5% w/v BSA, 1 mM CaCl2, pH 7.4), Adipose tissue was digested in a 37° water bath with gentle agitation (115 rotations/min) for 10–20 min. Samples were subsequently centrifuged at 300 g for 5 min. A 50–150 µL aliquot of the adipocytes in the resultant top layer were transferred to 450 µL of 0.2% methylene blue in HEPES buffer and incubated in a 37° water bath for 15 min. Aliquots (5–10 µL) of the stained cell suspension were transferred to the wells of a 10-well glass slide and cover slipped. Adipocytes were visualized and photographed using a microscope (Labophot 2/2 A; Nikon Inc, Melville, NY) equipped with an eyepiece with a 10 mm scale reticle at ×10 magnification and a Nikon Coolpix 990 digital camera. Adipocyte size was determined using the Cell Counting and Analysis Program (previously AdCount, developed by the Mayo Clinic Biomedical Imaging Resource), which measures the area of individual adipocytes and computes adipocyte diameter and volume. The average volume of ≥300 fat cells for each participant was used to calculate mean adipocyte cell size, determined by the mean adipocyte lipid content (adipocyte lipid content = adipocyte volume × 0.915; the density of triolein) (24, 27).
Adipose Tissue Immunohistochemistry
Adipose tissue macrophage populations were evaluated by IHC (N = 31), as previously described (28). ∼350 mg of adipose tissue was fixed in 10% zinc formalin in the dark with gentle agitation for 24 h and then transferred to 1% formalin. Fixed samples were paraffin-embedded by the Mayo Clinic Histology Research Laboratory, Rochester, MN. Sequential 5 micron sections were mounted on slides and stained by the Mayo Clinic Pathology Research Core, Rochester, MN. Slides were stained with antibodies against cluster of differentiation (CD) 68 (Clone PG-M1, Dako, Carpinteria, CA) to assess total macrophages/monocytes, CD14 (Sigma-Aldrich, St. Louis, MO) to assess M1 pro-inflammatory macrophages, or CD206 (Clone 685645, R&D Systems, Minneapolis, MN) to assess M2 anti-inflammatory macrophages. IHC staining was performed online using the Leica Bond III Stainer (Leica, Buffalo, IL). Slides were dewaxed using Leica Bond Dewax and antigen retrieval was performed for 20 min using Leica Epitope Retrieval 1 (CD206) or Leica Epitope Retrieval 2 (CD14 and CD68). Primary antibodies were diluted 1:200 (CD68 and CD206) or 1:300 (CD14) in Leica Bond Antibody diluent. The Bond Polymer Refine Detection System (Leica), which includes hydrogen peroxidase block, post primary, polymer reagent, DAB chromogen and hematoxylin counterstain, was used for detection. Slides were rinsed with Leica 1’ Bond Wash Buffer between steps. Prior to DAB incubation, slides were rinsed with distilled water. Slides were then incubated in DAB (1:19 DAB:DAB buffer) for 10 min for immunostaining visualization and rinsed in distilled water. Slides were then counterstained for 5 min in Schmidt hematoxylin (1:1 hematoxylin:molecular biology grade water) and rinsed in 1´ Bond wash buffer and distilled water. Following IHC processing, slides were removed from the stainer, rinsed in water, and dehydrated using increasing concentrations of ethyl alcohol. Slides were then cleared with three changes of xylene and a permanent coverslip was placed using xylene-based medium.
Stained tissue slides were visualized using an Olympus BX43 light microscope and 10 to 12 randomly selected images per slide were taken at ×40 magnification. Positively stained macrophages and total adipocytes were counted for ≥10 images per slide by a trained observer blinded to the participant. Data are expressed as the number of positively stained macrophages per 100 adipocytes.
Quantitative RT-PCR of Adipose Tissue Transcripts
Adipose tissue expression of genes related to inflammation (CD68, CD163, CD206, adhesion G protein-coupled receptor E1 (ADGRE), monocyte chemoattractant protein 1 (MCP1), inducible nitric oxide synthase (iNOS), IL6, IL8, TNFα) and adipose tissue hormones [LEPD (leptin), ADIPOQ (adiponectin)] was assessed by quantitative (q)RT-PCR (N = 34). Briefly, total RNA was isolated from ∼50 mg frozen adipose tissue using the RNEasy fibrous tissue kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Spectrophotometric analysis (Nanodrop) was performed to assess the quantity and purity of the isolated RNA. The SuperScript III First-Strand Synthesis System for RT-PCR cDNA Synthesis Kit (Invitrogen, Carlsbad, CA) was used according to the manufacturer’s protocol to synthesize cDNA. The cDNA equivalent of 5 ng RNA was amplified in 384-well microtiter plates using a QuantStudio 7 cycler (Applied Biosystems, CA) with SYBR Green assays. Expression of beta-2-microglobulin (B2M) was assessed as a control, and samples were normalized with the ΔCt method using the geometrical mean of B2M. Data are expressed as the standardized Z-scores of the relative expression values. The gene-specific primers are presented in Table 2.
Table 2.
Primer sequences for quantitative RT-PCR of adipose tissue
| Gene | Primer 1 | Primer 2 |
|---|---|---|
| B2M | CCA GCG TAC TCC AAA GAT TCA | TGG ATG AAA CCC AGA CAC ATA G |
| CD68 | CCA TGT AGC TCA GGT AGA CAA C | CCA CCT GCT TCT CTC ATT CC |
| CD163 | CCA GAA CAC ATA TTC CCT CCA C | GCA TAG TAA CTG TAC TCA CCA ACA |
| CD206 | TCC ATC TTC CTT GTG TCA GC | GGT TTT GGA GTA ATA TTC ACT GTT CT |
| ADGRE | CAT CTT CCA CCC TTC ACA TCA | ACG GCT TCT CAG ATC CAA TC |
| MCP1 | GGC TGA GAC TAA CCC AGA AAC | GAA TGA AGG TGG CTG CTA TGA |
| INOS | GCA GCT CAG CCT GTA CT | CAC CAT CCT CTT TGC GAC A |
| IL6 | GGA GAC TTG CCT GGT GAA A | CTG GCT TGT TCC TCA CTA CTC |
| IL8 | CTT GGC AGC CTT CCT GAT TT | GGG TGG AAA GGT TTG GAG TAT G |
| TNFα | GGC TGA GAC TAA CCC AGA AAC | GAA TGA AGG TGG CTG CTA TGA |
| LEPD (LEPD) | AGA GTG GCT TAG AGG AGT CAG | TGG CTT CCA GGT ATC TCC A |
| ADIPOQ | GCA GGG GAA GCA GGA CT | TGC AGT CTG TGG TTC TGA TTC |
ADGRE, adhesion G protein-coupled receptor E1; ADIPOQ, adiponectin; B2M, beta-2-microglobulin; CD, cluster of differentiation; IL, interleukin; INOS, inducible nitric oxide synthase; IL, interleukin; LEPD, leptin; MCP1, monocyte chemoattractant protein 1; TNFα, tumor necrosis factor-α.
Adipose Tissue Lysate Cytokine Measurement
Adipose tissue lysates were generated from ∼75–100 mg adipose samples snap-frozen in liquid nitrogen and stored at −80° (N = 33). Tissue was homogenized in 4 µL/mg tissue radioimmunoprecipitation assay plus buffer (RIPA-P buffer: RIPA with 1% Triton X-100, 1 µM DL-Dithiothreitol and 1 Roche complete mini EDTA-free anti-protease table/10 mL RIPA) using Omni ceramic homogenization beads (Omni International, Kennesaw, GA) and an Omni bead ruptor (4°C, speed = 2.10, cycles = 2, time = 15 s, and delay = 10 s). The homogenate was then centrifuged at 1,000 g for 10 min at 4°C, and the whole tissue extract subnatant below the lipid cake was aspirated and transferred to a new tube. The Pierce BCA Protein Quantitation Kit (Thermo Scientific) was used to measure the whole-tissue extract protein concentration, and the protein content of all samples was normalized. Whole tissue protein extracts were then stored at −80°C until analysis. A Millipore MILLIPLEX Human High Sensitivity T Cell Discovery Array 14-Plex Assay (HDHSTC14) was performed by Eve Biotechnologies (Calgary, AB, Canada) to determine the concentration of 14 cytokines and immunomodulatory peptides in the whole tissue protein extracts.
Circulating Inflammatory Markers
Participant blood samples were assessed for the presence of circulating inflammatory markers (N = 39). The Mayo Clinic Central Clinic Laboratory measured the erythrocyte sedimentation rate (ESR) and white blood count (WBC) from blood collected in spray-coated EDTA vacutainers. Serum tumor necrosis factor α (TNFα) and interleukin (IL)-6 were measured by enzyme-linked immunosorbent assays (ELISA) and serum C-reactive protein (CRP) was measured by a latex particle enhanced immuno-turbidimetric assay by the ICL. EDTA plasma samples were analyzed by Eve Biotechnologies for the concentrations of 14 cytokines and immunomodulatory peptides using the Millipore MILLIPLEX Human High Sensitivity T Cell Discovery Array 14-Plex Assay (HDHSTC14).
Mitochondrial Function
Mitochondria were isolated from ∼100 mg of fresh muscle tissue for the assessment of mitochondrial respiration and reactive oxygen species (ROS) production, as previously described (29–31). Tissue suspended in Buffer A (100 mM KCl, 50 mM tris, 5 mM MgCl2, 1.8 mM ATP, and 1 mM EDTA, pH 7.2) was minced, incubated with protease medium, washed, and homogenized using a Potter-Elvehjem tissue grinder. Mitochondria were isolated from the homogenate by differential centrifugation, which included the extraction of myofibrillar components at 720 g followed by centrifugation of the resultant supernatant at 10,000 g to obtain a mitochondrial pellet. The mitochondrial pellet was washed and resuspended in a buffer containing 225 mM sucrose, 44 mM KH2PO4, 12.5 mM Mg acetate, and 6 mM EDTA at a volume of 4.2 µL buffer/mg tissue.
Mitochondrial oxidative capacity supported by carbohydrate-based substrates was assessed (N = 39) using an Oxygraph high resolution respirometer (Oroboros Instruments, Innsbruck, Austria). Briefly, 90 µL of the mitochondrial suspension were added to the Oxygraph chambers containing 2 mL MiRO5 respiration buffer (110 mM sucrose, 60 mM potassium lactobionate, 0.5 mM EGTA, 1 g/L BSA essentially fat free, 3 mM MgCl2, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, pH 7.1). A stepwise protocol was used to isolate electron flow through the respiratory chain complexes and assess oxygen flux (JO2) during respiratory states 1–4 in the presence of glutamate, malate and succinate (GMS), as previously described (30, 32, 33). The average oxygen flux rates were calculated for each state and corrected for background oxygen flux using the Datlab software (Oroboros Instruments). The respiratory control rate (RCR; ratio of State 3 respiration to State 4 respiration) was calculated (N = 37) as an indicator of mitochondrial coupling efficiency. The ADP:O ratio was assessed (N = 36) as an indicator of mitochondrial phosphorylation efficiency and was determined by measuring oxygen consumption in response to a non-saturating pulse of ADP (15 μM), as previously described (33). Mitochondrial ATP production was assessed fluorometrically (N = 30) as previously described (34, 35). Briefly, the mitochondrial suspension (10 µL) was added to a quartz cuvette containing 2 mL Buffer Z (110 mM K‐MES, 35 mM KCl, 1 mM EGTA, 5 mM K2HPO4, 3 mM MgCl2‐6H2O, and 5 mg/mL bovine serum albumin; pH 7.4, 295 mOsm). Buffer Z was supplemented with 1 U/mL hexokinase, 2.5 U/mL glucose-6-phosphate dehydrogenase (G6PDH), 2.5 mM NADP+, and 2.5 mM d‐Glucose, allowing for the conversion of ATP to NADPH through the hexokinase- and G6PDH-catalyzed sequential formation of glucose-6-phosphate, 6-phosphogluconate, and NADPH. The autofluorescence of NADPH was assessed continuously as an indicator of ATP production (Fluorolog 3; Horiba Jobin Yvon, Piscataway, NJ), and the rate of ATP production (JATP) during respiratory states 1–4 was subsequently determined using MatLab (MathWorks, Natick, MA). JO2 and ATP production were normalized to tissue wet weight to assess mitochondrial function at the tissue level, and to the protein content of the mitochondrial suspension, measured using the Pierce 660-nm Protein Assay, to assess mitochondrial function at the level of the organelle. To assess mitochondrial ROS production (N = 38), H2O2 flux was measured by fluorometric monitoring of the oxidation of Amplex Red in conjunction with the measurement of mitochondrial respiration using the Oxygraph O2K-Fluorescence LED2-Module, as previously described (29). H2O2 flux rates were normalized to oxygen consumption at the corresponding respiratory state. Although mitochondrial yield and membrane integrity following isolation were not evaluated in the present study due to the confounding effects of cytochrome c on Amplex Red measurements, we expect ∼30%–50% yield with high membrane integrity as previously described for this approach (30).
Statistical Analysis
Data were assessed for normality using the Shapiro-Wilks Normality test. Nonnormal variables were either log- or square root-transformed. Parametric tests were performed on raw or transformed normally distributed data, and nonparametric tests were performed on variables that did not meet the assumptions of normality after transformation. All data are presented in their original metrics. Stepwise multiple regression analyses were conducted to assess the unique contribution of adipose tissue inflammation and systemic inflammation on insulin sensitivity and mitochondrial function after controlling for BMI. Initial regression models assessed the bivariate associations of adipose tissue and systemic inflammation with measurements of insulin sensitivity and mitochondrial function, and secondary models included BMI as an additional predictive variable. To examine the effects of obesity, unpaired t tests (normally distributed data) or Wilcoxon Signed-Rank tests (nonnormally distributed data) were conducted to compare outcomes for individuals with BMI < 30 kg/m2 and individuals with BMI > 30 kg/m2. To assess the effects of inflammation on insulin sensitivity and mitochondrial function, participants were stratified into tertiles based on salient markers of adipose tissue inflammation (total macrophage number or adipose IL-6 concentration) and systemic inflammation (TNFα and CRP). Unpaired t tests or Wilcoxon Signed-Rank tests were conducted to compare outcomes for individuals in the top and bottom tertiles for each inflammatory marker. The top and bottom tertiles for each marker were also compared using a one-way analysis of covariance to control for BMI. Statistical significance was set a priori at P < 0.05 and the uncorrected P values were evaluated to evaluate associations between various markers of obesity and inflammation on mitochondrial function and insulin sensitivity. Analyses were performed using Statistical Package for the Social Sciences (v25, IBM, Armonk, NY) or GraphPad Prism (v8, GraphPad Software, San Diego, CA).
RESULTS
Participant Characteristics
Thirty-nine participants with BMIs 20.5 to 45.8 kg/m2 completed the study (Table 1). BMI was positively associated with body fat (% and mass), android to gynoid (A:G) ratio, adipocyte cell size (lipid content), fasting insulin and triglyceride concentrations, and the triglyceride-glucose index (TyG). In addition, participants with BMI > 30 had higher circulating concentrations of the inflammatory biomarkers CRP, TNFα, and IL-6 than participants with BMI < 30, and BMI was significantly positively associated with CRP and TNFα.
Adipose Tissue Inflammation Is Associated with Obesity and Systemic Inflammation
The number of adipose-resident macrophages per 100 adipocytes in the subcutaneous abdominal adipose tissue increased with BMI (Fig. 1, A–F). BMI was positively and significantly correlated with total (CD68+), M1-type (CD14+), and M2-type (CD206+) macrophages. Despite increased macrophage abundance in adipose tissue, there was no evidence of a phenotypic shift in macrophage populations with increasing BMI; the M1:M2 macrophage ratio was not different between individuals with BMI < 30 and individuals with BMI > 30, and there was no correlation between BMI and the M1:M2 macrophage ratio (Fig. 1G). Adipose tissue IL-6, a proinflammatory cytokine that promotes the infiltration of macrophages into the adipose tissue (36), was also significantly and positively associated with BMI (Fig. 1H). Targeted adipose gene expression measurements show transcriptional evidence of adipose tissue inflammation with increasing adiposity (Fig. 1I). The gene expression of immune cell markers (CD68, CD163, CD206, ADGRE) and the proinflammatory TNFα were higher in participants with BMI > 30 than in those with BMI < 30, as was gene expression of leptin (LEPD), a hormone with proinflammatory properties (37). Further, gene expression of adiponectin (ADIPOQ), a peptide that exhibits anti-inflammatory and insulin-sensitizing properties (38), was significantly lower in participants with BMI > 30. Precedent literature links adiposity with altered adipose tissue composition and physiology (15, 39), and the present data set adds to this with evidence that increasing BMI is associated with adipose tissue inflammation at the level of gene expression, IL-6 concentrations, and macrophage abundance.
Figure 1.
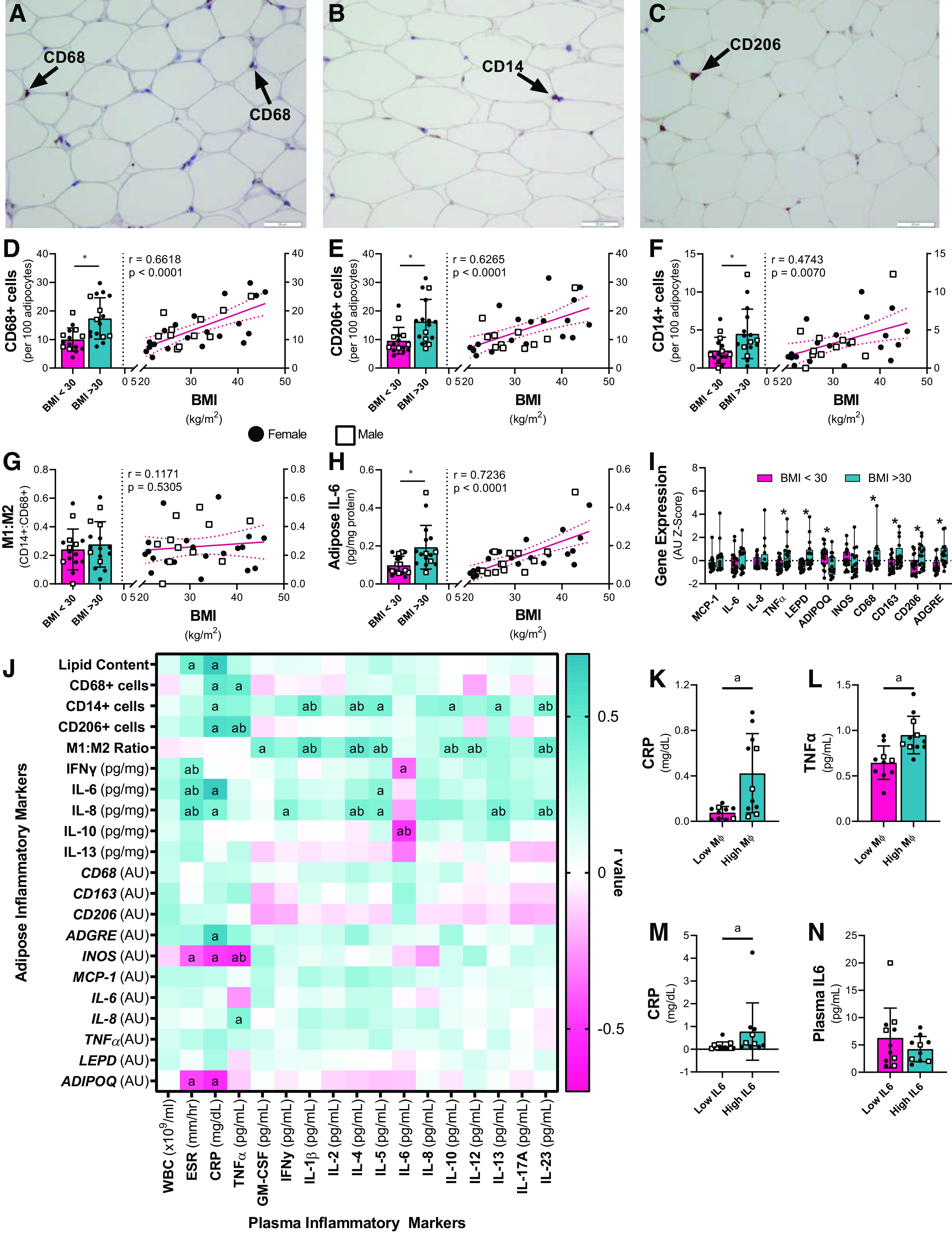
Adipose tissue resident macrophages and adipose tissue inflammatory cytokine concentrations increase with increasing BMI and are associated with circulating concentrations of inflammatory markers. Subcutaneous abdominal adipose tissue biopsies were collected under fasted conditions (N = 34) to assess (A–F) adipose tissue macrophage populations (N = 31). The number of (A, D) total macrophages (CD68+), (B, E) M1 proinflammatory macrophages (CD14+), and (C, F) M2 anti-inflammatory macrophages (CD206) per 100 adipocytes was determined by immunohistochemistry. A–C: representative immunohistochemistry images; arrows indicate positively stained cells. G: the ratio of M1:M2 adipose macrophages was calculated, and (H) the concentration of IL-6 in adipose tissue lysates was assessed (N = 33). Bivariate relationships between BMI and (D–G) adipose macrophage populations and (H) adipose IL-6 were assessed. Bar graphs are presented as mean and SD. *Significant difference between participants with BMI < 30 kg/m2 and those with BMI > 30 kg/m2, as determined by unpaired t tests or Wilcoxon Signed-Ranks tests. The bivariate correlations are presented as lines of best fit with 95% confidence intervals. The r- and p values represent correlation coefficients. Black circles: females; white squares: males. I: adipose tissue gene expression of proteins related to macrophages and inflammation were assessed by quantitative real-time PCR (N = 34). Data are presented as mean and SD of the gene expression Z-scores. *Significant difference (P < 0.05) between participants with BMI < 30 kg/m2 and those with BMI > 30 kg/m2 as determined by unpaired t tests or Wilcoxon Signed-Ranks tests. J: stepwise regression analyses assessing the associations between markers of adipose tissue inflammation and concentrations of systemic markers of inflammation were performed. Initial regression models assessed the bivariate correlations between adipose tissue inflammation and systemic inflammation, and secondary models included BMI as an additional predictive variable. a Significant (P < 0.05) bivariate correlations, and b significant associations after controlling for BMI. Presented r values represent the bivariate regression. The concentrations of circulating CRP and TNFα were compared between participants in the highest and lowest tertiles for (K, L) total CD68+ adipose-resident macrophage numbers, and (M, N) adipose tissue IL-6 concentrations using unpaired t tests and one-way analyses of covariance to control for BMI. aSignificant bivariate differences in cytokine concentrations, and b significant differences after controlling for BMI. ADGRE, adhesion G protein-coupled receptor E1; ADIPOQ, adiponectin; BMI, body mass index; CD, cluster of differentiation; CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFNγ, interferon-γ; IL, interleukin; INOS, inducible nitric oxide synthase; LEPD, leptin; MΦ, macrophage; MCP1, monocyte chemoattractant protein 1; TNFα, tumor necrosis factor-α; WBC, white blood count.
Obesity is associated with elevated circulating inflammatory markers. Consistent with this, we found increased inflammatory markers (CRP, TNFα) with increasing obesity, but more modest elevations in other circulating cytokines (Table 1). Many circulating inflammatory molecules are believed to originate from adipose tissue (6, 40). The bivariate regression analyses revealed significant associations between markers of inflammation in blood and macrophage populations and cytokine concentrations in adipose tissue (Fig. 1J). Total adipose-resident CD68+ macrophage number was positively associated with circulating CRP and TNFα, and the concentrations of CRP and TNFα were significantly lower in participants with low numbers of adipose resident macrophage numbers compared to those with high macrophage counts (Fig. 1, K and L). However, these relationships did not remain significant after controlling for BMI. ESR and CRP, common clinical markers of inflammation, were also positively associated with adipose macrophage numbers and proinflammatory cytokine concentrations (Fig. 1J). Participants with high concentrations of IL-6 in the adipose tissue had significantly higher circulating concentrations of CRP (Fig. 1M), but interestingly, high IL-6 concentrations in the adipose tissue did not correlate with circulating IL-6 concentrations (Fig. 1, J and N). The numbers of proinflammatory, M1-type CD14+ macrophages and the ratio of M1:M2 macrophages were also significantly and positively associated with the circulating concentrations of multiple pro- and anti-inflammatory cytokines, and many of these relationships remained significant even after controlling for BMI. The number of CD14+ macrophages per 100 adipocytes was associated with circulating pro-inflammatory CRP, IL-1β, and IL-23 concentrations, as well as the concentrations of anti-inflammatory cytokines IL-4, IL-5, IL-10, and IL-13. Although the M1:M2 ratio was not significantly associated with BMI, it was significantly and positively associated with circulating concentrations of proinflammatory cytokines GM-CSF, IL-1β, IL-12, and IL-23, and of anti-inflammatory cytokines IL-4, IL-5, and IL-10. Although multiple immune cell and inflammatory markers genes were elevated in adipose tissue of participants with BMI > 30, expression levels of genes related to inflammation and macrophage populations were not significantly associated with the majority of circulating cytokine concentrations. Taken together, these data show elevations in circulating inflammatory molecules and increased numbers of adipose-resident macrophages concomitant with increasing BMI. Furthermore, adipose tissue macrophage numbers and cytokine concentrations were positively associated with circulating inflammatory markers.
Increasing BMI and Inflammation Are Associated with Decreased Insulin Sensitivity
Glucose excursion was similar in participants with BMI < 30 and those with BMI > 30 in the hours following ingestion of the test meal (Fig. 2A); however, postprandial insulin and C-peptide concentrations were elevated in participants with higher BMIs (Fig. 2, B and C). Insulin sensitivity (SI), estimated from frequently-sampled postprandial glucose and insulin concentrations using the oral glucose minimal model, decreased with increasing BMI (Fig. 2D). β-cell responsivity (Φ), an indicator of β-cell function, was greater in participants with higher BMIs (Fig. 2, E–H). Basal (ΦB) and dynamic (ΦD) β-cell responsivity significantly increased with increasing BMI, and static (ΦS), and total (ΦTotal) β-cell responsivity exhibited similar trends, reflecting β-cell compensation that maintained postprandial glycemic control in participants with BMI > 30. However, static (DIs), dynamic (DID), and total (DITotal) disposition indexes, which reflect β-cell function adjusted for the prevailing level of insulin sensitivity and are an early indicator of inadequate β-cell compensation (41), were significantly lower in participants with BMI > 30 (Fig. 2, I–K). In agreement with the well-established association between insulin resistance and obesity (2), increasing BMI was associated with decreased insulin sensitivity measured in response to a mixed meal tolerance test designed to closely mimic the physiological response to a typical meal. Increasing BMI was also associated with greater postprandial triglyceride concentrations (Fig. 2, L–N), consistent with prior reports of impaired postprandial triglyceride clearance with obesity and metabolic syndrome (42).
Figure 2.

Insulin sensitivity decreases with increasing BMI. Following an overnight fast, participants (N = 36) consumed a mixed meal comprising 35% of the individual’s total daily energy expenditure. Blood samples were collected for the assessment of (A) glucose, (B) insulin, (C) C-peptide, and (L) triglycerides prior to meal ingestion, upon completion of the mixed meal (time = 0), and at scheduled intervals over the subsequent 5 h. (D) Insulin sensitivity (SI; N = 33), (E–H) β-cell responsivity (Φ; N = 30), and (I–K) disposition index (DI; N = 30) were calculated using the oral glucose and c-peptide minimal models. M: the postprandial incremental area under the curve of circulating triglycerides (iAUC; N = 36) and (N) the absolute change in triglycerides from baseline to peak triglyceride concentration (ΔTG; N = 36) were calculated. Bar graphs are presented as mean and SD. Black circles: females; white squares: males. *Significant difference between participants with BMI < 30 kg/m2 and those with BMI > 30 kg/m2, as determined by unpaired t tests or Wilcoxon Signed-Ranks tests. Correlations with BMI were assessed for each variable and are presented as lines of best fit with 95% confidence intervals. The r and P values represent the bivariate correlation coefficients. BMI, body mass index; DID, dynamic disposition index; DIS, static disposition index; DITotal, total disposition index; iAUC, triglyceride incremental area under the curve; SI, insulin sensitivity; TG, triglyceride; ΦB, basal β-cell responsivity; ΦD, dynamic β-cell responsivity; ΦS, static β-cell responsivity; ΦTotal, total β-cell responsivity; ΔTG, absolute change in triglycerides.
Both local adipose tissue inflammation (43) and systemic inflammation (44) have been reported to contribute to the development of insulin resistance. Therefore, correlations between measures of insulin sensitivity and adipose tissue (Fig. 3A) and systemic (Fig. 3H) inflammation were performed to examine associations between adipose tissue and systemic inflammation and various indices of metabolic health. Within the adipose tissue, the number of total CD68+ macrophages, anti-inflammatory CD206+ macrophages, and the concentration of IL-6 were associated with fasting insulin, SI and DITotal (Fig. 3A). In addition, when compared to participants with low numbers of adipose-resident CD68+ macrophages, participants with greater numbers of macrophages had higher fasting insulin, reduced SI, and lower DITotal (Fig. 3, B–D). Similarly, participants with high concentrations of IL-6 in the adipose tissue showed evidence of greater insulin resistance (Fig. 3, E–G), and the reductions in SI associated with high adipose IL-6 concentrations remained significant after controlling for BMI (Fig. 3, A and F). The expression of macrophage- and inflammation-related genes (CD68, CD163, CD206, ADGRE, MCP1, IL-8, TNFα) were also associated with indices of insulin sensitivity and β-cell responsivity. Circulating cytokine concentrations demonstrated less obvious associations with insulin sensitivity than did markers of adipose tissue inflammation (Fig. 3H). However, circulating concentrations of CRP and TNFα in plasma were significantly associated with elevated fasting insulin concentration and reduced SI and DITotal (Fig. 3, H–N). Interestingly, circulating TNFα concentrations and adipose tissue gene expression of TNFα showed significant positive correlations with fasting and postprandial triglyceride concentrations (Fig. 3, A and H). Overall, higher BMI was associated with decreased insulin sensitivity. While circulating concentrations of TNFα and CRP were associated with decreased insulin sensitivity, a greater number of adipose tissue inflammatory markers were associated with metabolic traits compared to systemic inflammatory markers.
Figure 3.

Insulin sensitivity is negatively associated with adipose tissue and systemic inflammation. Stepwise regression analyses assessing the associations between markers of (A) adipose tissue inflammation and (H) markers of systemic inflammation and insulin sensitivity, β-cell responsivity, disposition index, and triglyceride kinetics were performed. Initial regression models assessed the bivariate correlations between (A) adipose tissue inflammation and (B) systemic inflammation and measures of insulin sensitivity, and secondary models included BMI as an additional predictive variable. aSignificant (P < 0.05) bivariate correlations, and bsignificant associations after controlling for BMI. Presented r values represent the bivariate regression. Fasting insulin, SI, and DITotal were compared between participants in the highest and lowest tertiles for (B–D) total CD68+ adipose-resident macrophage numbers, (E–G) adipose tissue IL-6 concentrations, (I–K) circulating CRP concentrations, and (L–N) circulating TNFα. aSignificant bivariate differences in measures of insulin sensitivity as determined by unpaired t tests or Wilcoxon Signed-Rank tests, and bsignificant differences after controlling for BMI as determined by one-way analyses of covariance. Black circles: females; white squares: males. ADGRE, adhesion G protein-coupled receptor E1; ADIPOQ, adiponectin; BMI, body mass index; CD, cluster of differentiation; CRP, C-reactive protein; DID, dynamic disposition index; DIS, static disposition index; DITotal, total disposition index; ESR, erythrocyte sedimentation rate; GM-CSF, granulocyte-macrophage colony-stimulating factor; iAUC, triglyceride incremental area under the curve; IFNγ, interferon-γ; IL, interleukin; LEPD, leptin; NOS, inducible nitric oxide synthase; MΦ, macrophage; MCP-1, monocyte chemoattractant protein 1; SI, insulin sensitivity; TG, triglyceride; TNFα, tumor necrosis factor-α; WBC, white blood count; ΦB, basal β-cell responsivity; ΦD, dynamic β-cell responsivity; ΦS, static β-cell responsivity; ΦTotal, total β-cell responsivity; ΔTG, absolute change in triglycerides.
Mitochondrial Oxidative Capacity Decreases with Increasing BMI and Systemic Inflammation
Reduced skeletal muscle mitochondrial oxidative capacity has been reported with obesity and may contribute to the metabolic derangements associated with obesity (45). Accordingly, ADP-stimulated mitochondrial oxidative capacity (state 3 respiration) decreased with increasing BMI when normalized to muscle tissue wet weight (Fig. 4, A and C) and to mitochondrial protein abundance (Fig. 4, B and E). Despite reduced state 3 O2 consumption with increasing BMI, state 3 ATP production rate was similar across BMI categories and did not exhibit any significant association with BMI (Fig. 4, A, B, D, and F). This apparent maintenance of ATP production capacity with decreased O2 consumption capacity is suggestive of greater efficiency of oxidative phosphorylation with increasing BMI. Mitochondrial efficiency was evaluated from the respiratory control ratio (RCR, state 3 respiration/state 4 respiration), indicative of proton leak, and the ADP:O ratio, indicative of phosphorylation efficiency. Although RCR was not significantly associated with BMI (Fig. 4G), ADP:O was significantly and positively associated with BMI (Fig. 4H), suggesting that ADP phosphorylation was achieved with less O2 consumed in obese individuals. Elevated ROS production is associated with obesity and insulin-resistant skeletal muscle (46); however, no effects of obesity on the H2O2-emitting potential of mitochondrial isolated from skeletal muscle were observed in the present study (Fig. 4I).
Figure 4.

Skeletal muscle oxidative capacity decreases with increasing adiposity and is negatively associated with systemic inflammation. High-resolution respirometry was performed on mitochondria isolated from skeletal muscle biopsies of the vastus lateralis to assess (A–C, E) mitochondrial oxidative capacity (JO2; N = 39). Oxygen flux during respiratory States 1–4 and in the presence of FCCP and AA was normalized to (A, C) wet tissue weight or (B, E) mitochondrial protein content. A, B, D, and F: mitochondrial ATP production (JATP; N = 30) was assessed fluorometrically during State 3 respiration. G: mitochondrial coupling efficiency (RCR; N = 37) and (H) phosphorylation efficiency (ADP:O; N = 36) were calculated from respirometry measurements. I: mitochondrial reactive oxygen species (ROS) production (N = 38) was assessed fluorometrically by determining H2O2 emission during respiratory states 2 and 4 and was normalized to the oxygen consumption at the corresponding respiratory state. Bar graphs are presented as mean and SD. *Significant difference between participants with BMI < 30 kg/m2 and those with BMI > 30 kg/m2, determined by unpaired t tests or Wilcoxon Signed-Rank tests. Bivariate correlations between BMI and measures of mitochondrial function and efficiency are presented as lines of best fit with 95% confidence intervals. The r and P values represent the bivariate correlation coefficients. J: stepwise regression analyses assessing the associations between concentrations of circulating inflammatory proteins and measures of mitochondrial function and efficiency were performed. Initial regression models assessed the bivariate correlations between systemic inflammation and mitochondrial function, and secondary models included BMI as an additional predictive variable. aSignificant (P < 0.05) bivariate correlations and bSignificant associations after controlling for BMI. Presented r values represent the bivariate regression. State 3 respiration flux rates normalized to (K, M) wet tissue weight and (L, N) protein content were compared between participants in the highest and lowest tertiles for (K, L) circulating CRP concentrations, and (M, N) circulating TNFα. aSignificant bivariate differences in measures of mitochondrial oxidative capacity, as determined by unpaired t tests or Wilcoxon Signed-Rank tests. bSignificant differences after controlling for BMI using one-way analyses of covariance. Black circles: females; white squares: males. AA, antimycin A; BMI, body mass index; CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; FCCP, carbonylcyanide-4-(trifluoromethoxy)-phenylhydrazone, mitochondrial oxidative phosphorylation uncoupler; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFNγ, interferon-γ; IL, interleukin; JATP, ATP production rate; JO2, oxygen flux; RCR, respiratory control ratio; TNFα, tumor necrosis factor-α; WBC, white blood count.
Mitochondria have been considered to lie at the nexus of inflammation, not only contributing to systemic inflammation through reactive oxygen species production and the release of mitochondrial damage-associated molecular patterns (47, 48), but also undergoing morphological and physiological changes in response to inflammation (14, 49). Therefore, the associations between mitochondrial function and markers of systemic inflammation were assessed (Fig. 4J). State 3, ADP-stimulated respiration was negatively associated with ESR, CRP, IFNγ, and IL-8 when normalized to wet tissue weight and with CRP and TNFα when normalized to mitochondrial protein content, though these relationships were not significant after controlling for BMI (Fig. 4J). Compared to participants with high circulating concentrations of CRP, those participants with the lowest concentrations had significantly higher maximal State 3 respiration (Fig. 4, K and L), while no differences in respiration were observed between the extremes of circulating TNFα concentrations (Fig. 4, M and N). Maximal State 3 ADP-stimulated ATP production was positively associated with circulating levels of IL-4, and IL-5, two cytokines with anti-inflammatory properties, independent of BMI (Fig. 4J). Although mitochondrial ROS production was largely unassociated with markers of systemic inflammation, increasing State 4 H2O2 emission was associated with higher circulating concentrations of GM-CSF (Fig. 4J). Taken together, these results suggest reduced mitochondrial respiration with increasing BMI, and modest associations between circulating inflammatory molecules and mitochondrial function.
DISCUSSION
Obesity is associated with myriad morphological and physiological changes in adipose tissue, including the hypertrophy of adipocytes, the infiltration of proinflammatory macrophages, and reduced insulin sensitivity (3, 11, 15). No longer considered simply a site of energy storage, adipose tissue has emerged as an important endocrine and immune organ, and the pro-inflammatory secretome of the adipose tissue in obesity is postulated to have profound effects systemically and to play a critical role in the development of the metabolic derangements associated with obesity. Here we performed a cross-sectional analysis of adipose tissue inflammation, circulating inflammatory signals, whole-body postprandial glucose, insulin, c-peptide, and triglyceride kinetics, and skeletal muscle mitochondrial physiology in middle-aged adults across a wide BMI range. The potential contributions of adipose tissue macrophage burden and inflammatory cytokines and systemic inflammation to the insulin resistance and impaired skeletal muscle mitochondrial function observed in individuals with increasing BMI were investigated. We show that adipose macrophage populations and inflammatory proteins are associated with circulating inflammatory signals, including CRP and TNFα, and reduced whole body insulin sensitivity. In addition, increasing concentrations of circulating CRP and TNFα are associated with diminished skeletal muscle mitochondrial oxidative capacity.
Consistent with precedent literature (15, 50, 51), we observed adipocyte hypertrophy, greater numbers of adipose-resident macrophages, and higher concentrations of IL-6 in the subcutaneous adipose tissue with increasing BMI. Although we observed overall increases in adipose-resident macrophage numbers in participants with BMI > 30, we did not observe a shift in the ratio of pro-inflammatory CD14+ macrophages to anti-inflammatory CD206+ macrophages. Macrophages represent a highly heterogeneous population of cells with a relatively high degree of phenotypic plasticity, complicating the characterization of M1 and M2 macrophage populations (52). The characterization of M1 and M2 macrophages using a single cell surface marker may therefore have been insufficient to properly characterize the phenotypic profile of the adipose-resident macrophages. However, the overall increase in adipose-resident macrophages represents significant immune cell infiltration and the promotion of an inflammatory environment. Although the inflammatory adipose profile of individuals with obesity has been postulated to contribute to the metabolic derangements associated with obesity, recent evidence suggests that acute adipose inflammation is essential for the healthy expansion of adipose tissue (17). In mouse models, an inability to mount proinflammatory signaling processes impaired extracellular matrix remodeling and angiogenesis in the adipose tissue and led to glucose intolerance and systemic inflammation (17). Therefore, the observed elevations in markers of adipose inflammation may be reflective of processes involved in adipose tissue expansion and remodeling. However, only weight-stable individuals were included in the present study, so the observed adipose inflammation likely reflects a sustained inflammatory state of adipose tissue characteristic of obesity rather than an acute inflammatory response.
As adipose tissue comprises over half the body mass of some individuals and acts as both an endocrine and immune organ, changes in its function undoubtedly have systemic consequences. Inasmuch, adipose tissue and adipose-resident macrophage populations have been theorized to contribute to the systemic low-grade inflammation reported with increasing adiposity. In participants with BMI > 30, circulating concentrations of CRP, TNFα, and IL-6, all of which are pro-inflammatory proteins that have been shown to be produced in adipose tissue either by adipocytes themselves or by cells of the stromal vascular fraction (36, 53, 54), were elevated compared to participants with BMI < 30. While we did not find evidence to support adipose tissue as a source of circulating IL-6, circulating CRP concentrations were associated with both adipose-resident macrophage burden and adipose IL-6 concentrations. Although adipose tissue has been shown to contribute 10%–35% of the circulating levels of IL-6 (55), adipose tissue, and adipose tissue IL-6 concentrations in particular, have been hypothesized to play a significant role in driving circulating CRP concentrations (56, 57). In addition to being elevated in participants with obesity, circulating TNFα concentrations were associated with adipocyte macrophage numbers. Within the adipose tissue, TNFα is produced primarily by adipose-resident macrophages and has been shown to further exacerbate adipose tissue inflammation (9, 50, 58, 59), potentiating a vicious cycle of inflammation that may contribute to systemic inflammation (60, 61). Importantly, our observed associations between adipose inflammation and circulating inflammatory proteins do not establish the directionality of this relationship. Because inflammatory proteins such as TNFα can aggravate adipose tissue inflammation, it remains possible that systemic inflammation drives adipose tissue inflammation and macrophage polarization.
There is well-established evidence that obesity is associated with both adipose tissue inflammation and systemic inflammation. However, the causes of this inflammation and its consequences on the pathophysiology of obesity are current areas of investigation. Recent data suggest that insulin resistance may drive adipose tissue inflammation by promoting the recruitment of macrophages into the adipose tissue (16). In contrast, adipose tissue inflammation has been hypothesized to be a significant contributor to the insulin resistance associated with obesity. Pivotal work in animal models demonstrated that adipose tissue inflammation precedes the development of insulin resistance (10), and reducing the recruitment and abundance of adipose tissue macrophages improves insulin sensitivity in high fat fed mice, even in the absence of significant reductions in body mass (62). The mechanisms by which adipose tissue and systemic inflammation impair insulin signaling are likely multifactorial and include local adipose-specific and systemic processes. In our cohort of non-diabetic participants with normal fasting glucose, local IL-6 concentrations in adipose tissue were negatively associated with SI and DITotal. Chronic IL-6 exposure has been shown to impair insulin signaling in adipocytes through multiple pathways, including downregulated expression and activation of the insulin receptor-β subunit and increased expression of suppressor of cytokine signaling 3 (SOCS-3), a negative regulator of insulin signaling (63). In addition, circulating inflammatory mediators associated with adipose tissue inflammation may impair insulin signaling in peripheral tissues. In our cohort, both TNFα and CRP were positively associated with markers of adipose tissue inflammation, and negatively associated with insulin sensitivity. For the past 30 years, TNFα has been implicated in the development of insulin resistance, and it continues to be a target of interest for improving insulin sensitivity (8, 9, 12). In skeletal muscle, TNFα inhibits insulin-stimulated glucose uptake by inhibiting insulin receptor substrates-1 and -2-mediated phosphatidylinositol 3 kinase activation (64). As with TNFα, CRP has been shown to interfere with insulin signaling in peripheral tissues (65, 66). In rats, insulin-stimulated signaling cascades in the liver were significantly impaired by the administration of CRP (65). Interestingly, neither adipose tissue inflammation nor systemic inflammation appeared to be associated with ΦTotal, an index of β-cell function, indicating that adipose-derived and systemic inflammation and insulin secretion are dissociated. Overall, the observed relationships suggest local adipose-tissue specific and systemic effects of inflammation on insulin sensitivity.
Importantly, the associations observed in this study do not establish a causal relationship between inflammation and insulin resistance. Recent evidence suggests that although adipose inflammation and insulin resistance occur concurrently with increasing adiposity, adipose inflammation may not play a causative role in the development of insulin resistance (15, 17), and that insulin resistance may actually drive adipose tissue inflammation (16). However, anti-inflammatory agents have been shown to improve glucose control in people with type 2 diabetes (6, 67), making it difficult to ignore the potential role of adipose inflammation in the etiology of insulin resistance. Additional investigations are required to fully elucidate the causes and consequences of adipose tissue inflammation and its role in metabolic dysfunction. Elevated free fatty acids consequent to increased lipolysis (68) and derangements in adipose tissue mitochondrial function are proposed to be associated with inflammation and insulin resistance (69–72). These studies, alongside prior evidence of altered mitochondrial physiology in adipose tissue of obese individuals (73), provide strong rationale for continued investigation of the role of adipose tissue mitochondria in the development and progression of inflammation and insulin resistance in obesity. Overall, the strong associations between adipose inflammation and various metabolic parameters in the present cohort suggest that adipose tissue and metabolic health are closely intertwined.
Skeletal muscle mitochondrial derangements have also been implicated in the pathophysiology of obesity. Impaired mitochondrial function in skeletal muscle has been proposed to occur with obesity and to be an early primary event in the development of skeletal muscle insulin resistance (45, 74). Reduced mitochondrial function has been reported to result in increased intramyocellular lipid content and the accumulation of toxic lipids that impair insulin signaling (45, 75–78). Interestingly, although we observed reduced mitochondrial respiration with obesity, the phosphorylation efficiency was higher than that of individuals with BMI < 30, and ATP production rates were similar in individuals with BMI > 30 and those with BMI < 30. Although this improved efficiency may be a compensatory mechanism to preserve energy production, it may also contribute to the development of insulin resistance. In rats fed a high diet, increased mitochondrial efficiency preceded the development of insulin resistance. The authors postulate that the resultant reductions in substrate utilization that occur with increased mitochondrial efficiency may create a state of oversupply that contributes to insulin resistance (79–81). Another alternative viewpoint is that obesity may not alter mitochondrial efficiency, but increased mitochondrial efficiency may be an underlying component or contributor to a “thrifty phenotype” that may predispose some individuals to weight gain and associated metabolic disease (82). Mitochondrial-derived oxidative stress has been postulated to contribute to the insulin resistance, systemic inflammation, atherosclerosis, type 2 diabetes, and cardiovascular disease that accompany obesity (83, 84). However, although reduced skeletal muscle mitochondrial oxidative capacity was associated with increasing BMI, we did not observe concomitant increases in ROS production. The absence of any apparent increase in ROS production in obese individuals is in contrast to previous publications that convincingly implicate heightened ROS emissions in the etiology of insulin resistance with obesity and high-fat feeding (46). Several factors may explain these discrepant results, including the characteristics of the participant cohorts (severity of metabolic disease) and technical differences related to measurements in isolated mitochondria versus permeabilized muscle fibers. Despite the relatively modest reductions in mitochondrial function with increasing BMI in the present study, the decreased mitochondrial respiration and increased mitochondrial efficiency may have important implications for skeletal muscle insulin sensitivity.
The causes of the observed skeletal muscle mitochondrial derangements reported with obesity are unclear and likely multifactorial. Although physical inactivity is a likely contributor (85), the inflammatory environment may also contribute to these derangements (86). TNFα impairs mitochondrial biogenesis in adipose tissue and skeletal muscle (13) and attenuates the activity of pyruvate dehydrogenase, complex I, and complex II in cardiomyocytes (87). In macrophages, IFNγ activates aerobic glycolysis and promotes a reduction in oxidative phosphorylation (88). Because obesity is associated with increased circulating concentrations of inflammatory proteins, we sought to determine if circulating inflammatory markers contribute to the observed mitochondrial alterations. Only modest associations between circulating inflammatory molecule concentrations and measurements of mitochondrial function were observed. However, we did observe negative associations between mitochondrial oxidative capacity and circulating inflammatory proteins, including CRP, TNFα, IFNγ, and IL-8. In contrast, the anti-inflammatory cytokines IL-4 and IL-5 were positively associated with ATP production. In infection models, IL-4 has been shown to exert protective effects on TNFα- and IFNγ-induced mitochondrial damage (86). Although the specific mechanisms by which inflammation affect mitochondria remain unclear, from these data, we propose that the circulating cytokine milieu may have important consequences for skeletal muscle mitochondrial function in obesity. It is also possible that, even in the absence of increased ROS production, dysfunctional mitochondria contribute to systemic inflammation via mitochondrial DNA and damage associated molecular patterns (48). Although circulating inflammatory cytokines may directly affect skeletal muscle mitochondrial function, they may also impair mitochondrial function indirectly by promoting a local inflammatory environment within the skeletal muscle. Additional work is therefore required to examine local skeletal muscle-specific inflammation and to identify specific inflammatory pathways that contribute to the observed mitochondrial derangements.
In this study, we report significant adipose tissue inflammation, reduced insulin sensitivity, and impaired skeletal muscle mitochondrial oxidative capacity with increasing BMI. Further, adipose tissue inflammation was associated with circulating inflammatory markers, and both local and systemic inflammation were associated with reductions in insulin sensitivity and mitochondrial function. Although the current study is a carefully designed examination of these potentially interrelated physiological variables, longitudinal studies or large population-based cohorts representing individuals of similar BMIs but different levels of adipose inflammation are needed to fully parse the role of adipose inflammation in the pathophysiology of obesity. In addition, because obesity is associated with a significant number of physiological changes, there are multiple potential confounding factors, such as nonalcoholic fatty liver disease, for which we did not account. Although we did not evaluate liver fat content or hepatic enzymes, the fasting triglyceride-glucose index (TyG), which has been shown to be sensitive for detecting NAFLD (23) was elevated in obese individuals, suggesting that liver steatosis may be a factor. Habitual physical activity is another important lifestyle factor that strongly influences mitochondrial function and metabolic health (85). Although we made efforts to recruit only sedentary individuals for this study, we did not empirically measure free-living physical activity patterns, which could certainly influence the measured outcomes. Further, although sex did not appear to be a significant factor in the majority of the variables studied, it is important to note that the study included more females than males, and that the distribution of females and males across the BMI ranges was not equal.
In conclusion, the present study provides a thorough characterization of adipose tissue inflammation, systemic inflammation, insulin sensitivity, and skeletal muscle mitochondrial function in a cohort of individuals across a broad BMI range. Following prior implication of adipose tissue inflammation in the development of insulin resistance and mitochondrial dysfunction with obesity, this study evaluated the nuanced, interconnected relationships between these variables. Adipose inflammation, including increased macrophage burden and elevated IL-6 concentrations, was associated with systemic inflammation and insulin resistance, measured under highly physiological postprandial conditions. Systemic inflammation was also associated with impaired skeletal muscle mitochondrial oxidative capacity. Although these findings do not establish causal relationships between adipose tissue inflammation and metabolic derangements associated with obesity, they provide evidence supporting the role of adipose-resident macrophages and the pro-inflammatory secretome of adipose tissue in the pathophysiology of obesity. The results of this study support the hypothesis that adipose tissue inflammation may lie at the nexus of obesity and insulin resistance. As adipose inflammation appears to underlie or exacerbate many obesity-associated comorbidities, identifying the physiological contributors to its development is paramount to the identification of therapeutic targets for preserving insulin signaling and health in the population.
GRANTS
H. E. Kunz was supported by National Institute of Arthritis and Musculoskeletal and Skin Diseases for the Musculoskeletal Research Training Program (T32 AR056950). This project was supported by Grant Numbers DK116231 and UL1 TR002377 from the National Center for Advancing Translational Sciences and R01 AG054454 from the National Institute on Aging.
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
I.R.L. and C.R.H. conceived and designed research; H.E.K., M.D.J., I.R.L., C.R.H., K.J.G., N.M., X.Z., and Z.R. performed experiments; H.E.K., E.C.P., A.V., I.R.L., C.R.H., K.J.G., M.P., M.L., C.D.M., N.M., X.Z., and Z.R. analyzed data; H.E.K., M.D.J., A.V., I.R.L., C.R.H., K.J.G., M.P., and M.L. interpreted results of experiments; H.E.K. and I.R.L. prepared figures; H.E.K., E.C.P., and I.R.L. drafted manuscript; H.E.K., I.R.L., C.R.H., and K.J.G. edited and revised manuscript; H.E.K., E.C.P., M.D.J., A.V., I.R.L., C.R.H., K.J.G., M.P., M.L., C.D.M., N.M., X.Z., and Z.R. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Bobbie Soderberg and Vicky Wade for biopsy support and assistance. Work was also supported by the staff at the Mayo Clinic Clinical Research and Trials Unit, Immunochemical Core Laboratory, Histology Research Laboratory, and Pathology Research Core Laboratory. The authors also thank Dr. Michael Jensen’s laboratory, including Dr. Kelli Lytle, Lendia Zhou, and Deb Harteneck for assistance with the fat biopsies and processing. Dr. Sreekumaran Nair provided valuable clinical assistance, and his laboratory, including Katherine Klaus and Dawn Morse, provided guidance and laboratory expertise.
Present addresses: C. R. Hart, Air Force Research Laboratory, 711th Human Performance Wing, Wright Patterson Air Force Base, Dayton, Ohio; K. J. Gries, Department of Exercise and Sports Science, College of Health Professions, Marian University, Indianapolis, Indiana.
REFERENCES
- 1.Hales CM, Carroll MD, Fryar CD, Ogden CL. Prevalence of obesity and severe obesity among adults: United States, 2017–2018. NCHS Data Brief, no 360. Hyattsville, MD: National Center for Health Statistics, 2020. [PubMed] [Google Scholar]
- 2.Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest 106: 473–481, 2000. doi: 10.1172/JCI10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mundi MS, Karpyak MV, Koutsari C, Votruba SB, O'Brien PC, Jensen MD. Body fat distribution, adipocyte size, and metabolic characteristics of nondiabetic adults. J Clin Endocrinol Metab 95: 67–73, 2010. doi: 10.1210/jc.2009-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vague J. The degree of masculine differentiation of obesities: a factor determining predisposition to diabetes, atherosclerosis, gout, and uric calculous disease. Am J Clin Nutr 4: 20–34, 1956. doi: 10.1093/ajcn/4.1.20. [DOI] [PubMed] [Google Scholar]
- 5.Longo M, Zatterale F, Naderi J, Parrillo L, Formisano P, Raciti GA, Beguinot F, Miele C. Adipose tissue dysfunction as determinant of obesity-associated metabolic complications. Int J Mol Sci 20: 2358, 2019. doi: 10.3390/ijms20092358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zatterale F, Longo M, Naderi J, Raciti GA, Desiderio A, Miele C, Beguinot F. Chronic adipose tissue inflammation linking obesity to insulin resistance and type 2 diabetes. Front Physiol 10: 1607, 2019. doi: 10.3389/fphys.2019.01607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Skurk T, Alberti-Huber C, Herder C, Hauner H. Relationship between adipocyte size and adipokine expression and secretion. J Clin Endocrinol Metab 92: 1023–1033, 2007. doi: 10.1210/jc.2006-1055. [DOI] [PubMed] [Google Scholar]
- 8.Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest 95: 2409–2415, 1995. doi: 10.1172/JCI117936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259: 87–91, 1993. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 10.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112: 1821–1830, 2003. doi: 10.1172/JCI200319451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gastaldelli A, Gaggini M, DeFronzo RA. Role of adipose tissue insulin resistance in the natural history of type 2 diabetes: results from the San Antonio Metabolism Study. Diabetes 66: 815–822, 2017. doi: 10.2337/db16-1167. [DOI] [PubMed] [Google Scholar]
- 12.Kojta I, Chacińska M, Błachnio-Zabielska A. Obesity, bioactive lipids, and adipose tissue inflammation in insulin resistance. Nutrients 12: 1305, 2020. doi: 10.3390/nu12051305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valerio A, Cardile A, Cozzi V, Bracale R, Tedesco L, Pisconti A, Palomba L, Cantoni O, Clementi E, Moncada S, Carruba MO, Nisoli E. TNF-alpha downregulates eNOS expression and mitochondrial biogenesis in fat and muscle of obese rodents. J Clin Invest 116: 2791–2798, 2006. doi: 10.1172/jci28570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Horssen J, van Schaik P, Witte M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci Lett 710: 132931, 2019. doi: 10.1016/j.neulet.2017.06.050. [DOI] [PubMed] [Google Scholar]
- 15.Jia Q, Morgan-Bathke ME, Jensen MD. Adipose tissue macrophage burden, systemic inflammation, and insulin resistance. Am J Physiol Endocrinol Metab 319: E254–E264, 2020. doi: 10.1152/ajpendo.00109.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimobayashi M, Albert V, Woelnerhanssen B, Frei IC, Weissenberger D, Meyer-Gerspach AC, Clement N, Moes S, Colombi M, Meier JA, Swierczynska MM, Jeno P, Beglinger C, Peterli R, Hall MN. Insulin resistance causes inflammation in adipose tissue. J Clin Invest 128: 1538–1550, 2018. doi: 10.1172/JCI96139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wernstedt Asterholm I, Tao C, Morley TS, Wang QA, Delgado-Lopez F, Wang ZV, Scherer PE. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab 20: 103–118, 2014. doi: 10.1016/j.cmet.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harris JA, Benedict FG. A biometric study of human basal metabolism. Proc Natl Acad Sci USA 4: 370–373, 1918. doi: 10.1073/pnas.4.12.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cobelli C, Dalla Man C, Toffolo G, Basu R, Vella A, Rizza R. The oral minimal model method. Diabetes 63: 1203–1213, 2014. doi: 10.2337/db13-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cobelli C, Toffolo GM, Dalla Man C, Campioni M, Denti P, Caumo A, Butler P, Rizza R. Assessment of beta-cell function in humans, simultaneously with insulin sensitivity and hepatic extraction, from intravenous and oral glucose tests. Am J Physiol Endocrinol Metab 293: E1–E15, 2007. doi: 10.1152/ajpendo.00421.2006. [DOI] [PubMed] [Google Scholar]
- 21.Dalla Man C, Caumo A, Basu R, Rizza R, Toffolo G, Cobelli C. Minimal model estimation of glucose absorption and insulin sensitivity from oral test: validation with a tracer method. Am J Physiol Endocrinol Metab 287: E637–643, 2004. doi: 10.1152/ajpendo.00319.2003. [DOI] [PubMed] [Google Scholar]
- 22.Dalla Man C, Yarasheski KE, Caumo A, Robertson H, Toffolo G, Polonsky KS, Cobelli C. Insulin sensitivity by oral glucose minimal models: validation against clamp. Am J Physiol Endocrinol Metab 289: E954–E959, 2005. doi: 10.1152/ajpendo.00076.2005. [DOI] [PubMed] [Google Scholar]
- 23.Zhang S, Du T, Zhang J, Lu H, Lin X, Xie J, Yang Y, Yu X. The triglyceride and glucose index (TyG) is an effective biomarker to identify nonalcoholic fatty liver disease. Lipids Health Dis 16: 15, 2017. doi: 10.1186/s12944-017-0409-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tchoukalova YD, Koutsari C, Karpyak MV, Votruba SB, Wendland E, Jensen MD. Subcutaneous adipocyte size and body fat distribution. Am J Clin Nutr 87: 56–63, 2008. doi: 10.1093/ajcn/87.1.56. [DOI] [PubMed] [Google Scholar]
- 25.Lalia AZ, Dasari S, Johnson ML, Robinson MM, Konopka AR, Distelmaier K, Port JD, Glavin MT, Esponda RR, Nair KS, Lanza IR. Predictors of whole-body insulin sensitivity across ages and adiposity in adult humans. J Clin Endocrinol Metab 101: 626–634, 2016. doi: 10.1210/jc.2015-2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lalia AZ, Johnson ML, Jensen MD, Hames KC, Port JD, Lanza IR. Effects of dietary n-3 fatty acids on hepatic and peripheral insulin sensitivity in insulin-resistant humans. Diabetes Care 38: 1228–1237, 2015. doi: 10.2337/dc14-3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tchoukalova YD, Harteneck DA, Karwoski RA, Tarara J, Jensen MA. Quick, reliable, and automated method for fat cell sizing. J Lipid Res 44: 1795–1801, 2003. doi: 10.1194/jlr.D300001-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Morgan-Bathke M, Harteneck D, Jaeger P, Sondergaard E, Karwoski R, Espinosa De Ycaza A, Carranza-Leon BG, Faubion WA Jr, Oliveira AM, Jensen MD. Comparison of methods for analyzing human adipose tissue macrophage content. Obesity (Silver Spring) 25: 2100–2107, 2017. doi: 10.1002/oby.22012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krumschnabel G, Fontana-Ayoub M, Sumbalova Z, Heidler J, Gauper K, Fasching M, Gnaiger E. Simultaneous high-resolution measurement of mitochondrial respiration and hydrogen peroxide production. Methods Mol Biol 1264: 245–261, 2015. doi: 10.1007/978-1-4939-2257-4_22. [DOI] [PubMed] [Google Scholar]
- 30.Lanza IR, Nair KS. Functional assessment of isolated mitochondria in vitro. Methods Enzymol 457: 349–372, 2009. doi: 10.1016/S0076-6879(09)05020-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Makrecka-Kuka M, Krumschnabel G, Gnaiger E. High-resolution respirometry for simultaneous measurement of oxygen and hydrogen peroxide fluxes in permeabilized cells, tissue homogenate and isolated mitochondria. Biomolecules 5: 1319–1338, 2015. doi: 10.3390/biom5031319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kunz HE, Dasari S, Lanza IR. EPA and DHA elicit distinct transcriptional responses to high-fat feeding in skeletal muscle and liver. Am J Physiol Endocrinol Metab 317: E460–E472, 2019. doi: 10.1152/ajpendo.00083.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lanza IR, Blachnio-Zabielska A, Johnson ML, Schimke JM, Jakaitis DR, Lebrasseur NK, Jensen MD, Sreekumaran Nair K, Zabielski P. Influence of fish oil on skeletal muscle mitochondrial energetics and lipid metabolites during high-fat diet. Am J Physiol Endocrinol Metab 304: E1391–E1403, 2013. doi: 10.1152/ajpendo.00584.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gouspillou G, Rouland R, Calmettes G, Deschodt-Arsac V, Franconi JM, Bourdel-Marchasson I, Diolez P. Accurate determination of the oxidative phosphorylation affinity for ADP in isolated mitochondria. PLoS One 6: e20709, 2011. doi: 10.1371/journal.pone.0020709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lark DS, Torres MJ, Lin CT, Ryan TE, Anderson EJ, Neufer PD. Direct real-time quantification of mitochondrial oxidative phosphorylation efficiency in permeabilized skeletal muscle myofibers. Am J Physiol Cell Physiol 311: C239–245, 2016. doi: 10.1152/ajpcell.00124.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han MS, White A, Perry RJ, Camporez JP, Hidalgo J, Shulman GI, Davis RJ. Regulation of adipose tissue inflammation by interleukin 6. Proc Natl Acad Sci USA 117: 2751–2760, 2020. doi: 10.1073/pnas.1920004117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.La Cava A. Leptin in inflammation and autoimmunity. Cytokine 98: 51–58, 2017. doi: 10.1016/j.cyto.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Achari AE, Jain SK. Adiponectin, a therapeutic target for obesity, diabetes, and endothelial dysfunction. Int J Mol Sci 18: 1321, 2017. doi: 10.3390/ijms18061321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jocken JW, Langin D, Smit E, Saris WH, Valle C, Hul GB, Holm C, Arner P, Blaak EE. Adipose triglyceride lipase and hormone-sensitive lipase protein expression is decreased in the obese insulin-resistant state. J Clin Endocrinol Metab 92: 2292–2299, 2007. doi: 10.1210/jc.2006-1318. [DOI] [PubMed] [Google Scholar]
- 40.Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res 96: 939–949, 2005. doi: 10.1161/01.RES.0000163635.62927.34. [DOI] [PubMed] [Google Scholar]
- 41.Utzschneider KM, Prigeon RL, Faulenbach MV, Tong J, Carr DB, Boyko EJ, Leonetti DL, McNeely MJ, Fujimoto WY, Kahn SE. Oral disposition index predicts the development of future diabetes above and beyond fasting and 2-h glucose levels. Diabetes Care 32: 335–341, 2009. doi: 10.2337/dc08-1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lewis GF, OMeara NM, Soltys PA, Blackman JD, Iverius PH, Druetzler AF, Getz GS, Polonsky KS. Postprandial lipoprotein metabolism in normal and obese subjects: comparison after the vitamin A fat-loading test. J Clin Endocrinol Metab 71: 1041–1050, 1990. doi: 10.1210/jcem-71-4-1041. [DOI] [PubMed] [Google Scholar]
- 43.Wu H, Ballantyne CM. Metabolic inflammation and insulin resistance in obesity. Circ Res 126: 1549–1564, 2020. doi: 10.1161/CIRCRESAHA.119.315896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hotamisligil GS. Inflammation and metabolic disorders. Nature 444: 860–867, 2006. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 45.Sangwung P, Petersen KF, Shulman GI, Knowles JW. Mitochondrial dysfunction, insulin resistance, and potential genetic implications. Endocrinology 161: bqaa017, 2020. doi: 10.1210/endocr/bqaa017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW III, Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH, Neufer PD. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 119: 573–581, 2009. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zampino M, Brennan NA, Kuo PL, Spencer RG, Fishbein KW, Simonsick EM, Ferrucci L. Poor mitochondrial health and systemic inflammation? Test of a classic hypothesis in the Baltimore Longitudinal Study of Aging. Geroscience 42: 1175–1182, 2020. doi: 10.1007/s11357-020-00208-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grazioli S, Pugin J. Mitochondrial damage-associated molecular patterns: from inflammatory signaling to human diseases. Front Immunol 9: 832, 2018. doi: 10.3389/fimmu.2018.00832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abid H, Ryan ZC, Delmotte P, Sieck GC, Lanza IR. Extramyocellular interleukin-6 influences skeletal muscle mitochondrial physiology through canonical JAK/STAT signaling pathways. FASEB J 34: 14458–14472, 2020. doi: 10.1096/fj.202000965RR. [DOI] [PubMed] [Google Scholar]
- 50.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr.. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112: 1796–1808, 2003. doi: 10.1172/JCI200319246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sindhu S, Thomas R, Shihab P, Sriraman D, Behbehani K, Ahmad R. Obesity is a positive modulator of IL-6R and IL-6 expression in the subcutaneous adipose tissue: significance for metabolic inflammation. PLoS One 10: e0133494, 2015. doi: 10.1371/journal.pone.0133494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Castoldi A, Naffah de Souza C, Camara NO, Moraes- VP. The macrophage switch in obesity development. Front Immunol 6: 637, 2015. doi: 10.3389/fimmu.2015.00637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Calabro P, Chang DW, Willerson JT, Yeh ET. Release of C-reactive protein in response to inflammatory cytokines by human adipocytes: linking obesity to vascular inflammation. J Am Coll Cardiol 46: 1112–1113, 2005. doi: 10.1016/j.jacc.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 54.Fain JN, Bahouth SW, Madan AK. TNFalpha release by the nonfat cells of human adipose tissue. Int J Obes Relat Metab Disord 28: 616–622, 2004. doi: 10.1038/sj.ijo.0802594. [DOI] [PubMed] [Google Scholar]
- 55.FäLdt J, Wernstedt I, Fitzgerald SM, Wallenius K, BergströM GRAN, Jansson J-O. Reduced exercise endurance in interleukin-6-deficient mice. Endocrinology 145: 2680–2686, 2004. doi: 10.1210/en.2003-1319. [DOI] [PubMed] [Google Scholar]
- 56.Anty R, Bekri S, Luciani N, Saint-Paul MC, Dahman M, Iannelli A, Amor IB, Staccini-Myx A, Huet PM, Gugenheim J, Sadoul JL, Le Marchand-Brustel Y, Tran A, Gual P. The inflammatory C-reactive protein is increased in both liver and adipose tissue in severely obese patients independently from metabolic syndrome, Type 2 diabetes, and NASH. Am J Gastroenterol 101: 1824–1833, 2006. doi: 10.1111/j.1572-0241.2006.00724.x. [DOI] [PubMed] [Google Scholar]
- 57.Bastard JP, Jardel C, Delattre J, Hainque B, Bruckert E, Oberlin F. Evidence for a link between adipose tissue interleukin-6 content and serum C-reactive protein concentrations in obese subjects. Circulation 99: 2221–2222, 1999. doi: 10.1161/circ.99.16.2219/c. [DOI] [PubMed] [Google Scholar]
- 58.Suganami T, Nishida J, Ogawa Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: role of free fatty acids and tumor necrosis factor alpha. Arterioscler Thromb Vasc Biol 25: 2062–2068, 2005. doi: 10.1161/01.ATV.0000183883.72263.13. [DOI] [PubMed] [Google Scholar]
- 59.Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol 11: 85–97, 2011. doi: 10.1038/nri2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 72: 219–246, 2010. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 61.Zhu Q, An YA, Kim M, Zhang Z, Zhao S, Zhu Y, Asterholm IW, Kusminski CM, Scherer PE. Suppressing adipocyte inflammation promotes insulin resistance in mice. Mol Metab 39: 101010, 2020. doi: 10.1016/j.molmet.2020.101010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, Charo I, Leibel RL, Ferrante AW Jr.. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest 116: 115–124, 2006. [Erratum in J Clin Invest 116: 1457, 2006]. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lagathu C, Bastard JP, Auclair M, Maachi M, Capeau J, Caron M. Chronic interleukin-6 (IL-6) treatment increased IL-6 secretion and induced insulin resistance in adipocyte: prevention by rosiglitazone. Biochem Biophys Res Commun 311: 372–379, 2003. doi: 10.1016/j.bbrc.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 64.del Aguila LF, Claffey KP, Kirwan JP. TNF-alpha impairs insulin signaling and insulin stimulation of glucose uptake in C2C12 muscle cells. Am J Physiol Endocrinol Metab 276: E849–E855, 1999. doi: 10.1152/ajpendo.1999.276.5.E849. [DOI] [PubMed] [Google Scholar]
- 65.Xi L, Xiao C, Bandsma RH, Naples M, Adeli K, Lewis GF. C-reactive protein impairs hepatic insulin sensitivity and insulin signaling in rats: role of mitogen-activated protein kinases. Hepatology 53: 127–135, 2011. doi: 10.1002/hep.24011. [DOI] [PubMed] [Google Scholar]
- 66.Xu JW, Morita I, Ikeda K, Miki T, Yamori Y. C-reactive protein suppresses insulin signaling in endothelial cells: role of spleen tyrosine kinase. Mol Endocrinol 21: 564–573, 2007. doi: 10.1210/me.2006-0354. [DOI] [PubMed] [Google Scholar]
- 67.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest 116: 1793–1801, 2006. [Erratum in J Clin Invest 116: 2308, 2006]. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Morigny P, Houssier M, Mouisel E, Langin D. Adipocyte lipolysis and insulin resistance. Biochimie 125: 259–266, 2016. doi: 10.1016/j.biochi.2015.10.024. [DOI] [PubMed] [Google Scholar]
- 69.Heinonen S, Buzkova J, Muniandy M, Kaksonen R, Ollikainen M, Ismail K, Hakkarainen A, Lundbom J, Lundbom N, Vuolteenaho K, Moilanen E, Kaprio J, Rissanen A, Suomalainen A, Pietilainen KH. Impaired mitochondrial biogenesis in adipose tissue in acquired obesity. Diabetes 64: 3135–3145, 2015. doi: 10.2337/db14-1937. [DOI] [PubMed] [Google Scholar]
- 70.Heinonen S, Jokinen R, Rissanen A, Pietilainen KH. White adipose tissue mitochondrial metabolism in health and in obesity. Obes Rev 21: e12958, 2020. doi: 10.1111/obr.12958. [DOI] [PubMed] [Google Scholar]
- 71.Qatanani M, Tan Y, Dobrin R, Greenawalt DM, Hu G, Zhao W, Olefsky JM, Sears DD, Kaplan LM, Kemp DM. Inverse regulation of inflammation and mitochondrial function in adipose tissue defines extreme insulin sensitivity in morbidly obese patients. Diabetes 62: 855–863, 2013. doi: 10.2337/db12-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Soronen J, Laurila PP, Naukkarinen J, Surakka I, Ripatti S, Jauhiainen M, Olkkonen VM, Yki- JH. Adipose tissue gene expression analysis reveals changes in inflammatory, mitochondrial respiratory and lipid metabolic pathways in obese insulin-resistant subjects. BMC Med Genomics 5: 9, 2012. doi: 10.1186/1755-8794-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yin X, Lanza IR, Swain JM, Sarr MG, Nair KS, Jensen MD. Adipocyte mitochondrial function is reduced in human obesity independent of fat cell size. J Clin Endocrinol Metab 99: E209–E216, 2014. doi: 10.1210/jc.2013-3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Holloszy JO. “Deficiency” of mitochondria in muscle does not cause insulin resistance. Diabetes 62: 1036–1040, 2013. doi: 10.2337/db12-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med 350: 664–671, 2004. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Morino K, Petersen KF, Dufour S, Befroy D, Frattini J, Shatzkes N, Neschen S, White MF, Bilz S, Sono S, Pypaert M, Shulman GI. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest 115: 3587–3593, 2005. doi: 10.1172/JCI25151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes 55: S9–S15, 2006. doi: 10.2337/db06-S002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kelley DE, Goodpaster B, Wing RR, Simoneau JA. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am J Physiol Endocrinol Metab 277: E1130–1141, 1999. doi: 10.1152/ajpendo.1999.277.6.E1130. [DOI] [PubMed] [Google Scholar]
- 79.Crescenzo R, Bianco F, Coppola P, Mazzoli A, Cigliano L, Liverini G, Iossa S. Increased skeletal muscle mitochondrial efficiency in rats with fructose-induced alteration in glucose tolerance. Br J Nutr 110: 1996–2003, 2013. doi: 10.1017/S0007114513001566. [DOI] [PubMed] [Google Scholar]
- 80.Crescenzo R, Bianco F, Mazzoli A, Giacco A, Liverini G, Iossa S. Mitochondrial efficiency and insulin resistance. Front Physiol 5: 512, 2014. doi: 10.3389/fphys.2014.00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Crescenzo R, Bianco F, Mazzoli A, Giacco A, Liverini G, Iossa S. Skeletal muscle mitochondrial energetic efficiency and aging. IJMS 16: 10674–10685, 2015. doi: 10.3390/ijms160510674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Neel JV. Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? Am J Hum Genet 14: 353–362, 1962. [PMC free article] [PubMed] [Google Scholar]
- 83.Hulsmans M, Van Dooren E, Holvoet P. Mitochondrial reactive oxygen species and risk of atherosclerosis. Curr Atheroscler Rep 14: 264–276, 2012. doi: 10.1007/s11883-012-0237-0. [DOI] [PubMed] [Google Scholar]
- 84.Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal 20: 1126–1167, 2014. doi: 10.1089/ars.2012.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bilet L, Phielix E, van de Weijer T, Gemmink A, Bosma M, Moonen-Kornips E, Jorgensen JA, Schaart G, Zhang D, Meijer K, Hopman M, Hesselink MKC, Ouwens DM, Shulman GI, Schrauwen-Hinderling VB, Schrauwen P. One-leg inactivity induces a reduction in mitochondrial oxidative capacity, intramyocellular lipid accumulation and reduced insulin signalling upon lipid infusion: a human study with unilateral limb suspension. Diabetologia 63: 1211–1222, 2020. doi: 10.1007/s00125-020-05128-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Maiti AK, Sharba S, Navabi N, Forsman H, Fernandez HR, Linden SK. IL-4 protects the mitochondria against TNFα and IFNγ induced insult during clearance of infection with Citrobacter rodentium and Escherichia coli. Sci Rep 5: 15434, 2015. doi: 10.1038/srep15434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zell R, Geck P, Werdan K, Boekstegers P. TNF-alpha and IL-1 alpha inhibit both pyruvate dehydrogenase activity and mitochondrial function in cardiomyocytes: evidence for primary impairment of mitochondrial function. Mol Cell Biochem 177: 61–67, 1997. doi: 10.1023/a:1006896832582. [DOI] [PubMed] [Google Scholar]
- 88.Wang F, Zhang S, Jeon R, Vuckovic I, Jiang X, Lerman A, Folmes CD, Dzeja PD, Herrmann J. Interferon gamma induces reversible metabolic reprogramming of M1 macrophages to sustain cell viability and pro-inflammatory activity. EBioMedicine 30: 303–316, 2018. doi: 10.1016/j.ebiom.2018.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]