

Abstract
CFTR, a chloride channel and ion channel regulator studied mostly in epithelial cells, has been reported to participate in immune regulation and likely affect the risk of cancer development. However, little is known about the effects of CFTR on the differentiation and function of γδ T cells. In this study, we observed that CFTR was functionally expressed on the cell surface of γδ T cells. Genetic deletion and pharmacological inhibition of CFTR both increased IFN-γ release by peripheral γδ T cells and potentiated the cytolytic activity of these cells against tumor cells both in vitro and in vivo. Interestingly, the molecular mechanisms underlying the regulation of γδ T cell IFN-γ production by CFTR were either TCR dependent or related to Ca2+ influx. CFTR was recruited to TCR immunological synapses and attenuated Lck-P38 MAPK-c-Jun signaling. In addition, CFTR was found to modulate TCR-induced Ca2+ influx and membrane potential (Vm)-induced Ca2+ influx and subsequently regulate the calcineurin-NFATc1 signaling pathway in γδ T cells. Thus, CFTR serves as a negative regulator of IFN-γ production in γδ T cells and the function of these cells in antitumor immunity. Our investigation suggests that modification of the CFTR activity of γδ T cells may be a potential immunotherapeutic strategy for cancer.
Keywords: CFTR, γδ T cells, TCR, Membrane potential, Antitumor immunity
Subject terms: Interferons, Immunoediting
Introduction
Ion channels and transporters expressed in the plasma membrane of immune cells participate in the influx (Ca2+, Mg2+, Zn2+, and Na+) and efflux (K+ and Cl−) of ions to regulate the membrane potential and physiological cell functions, such as gene expression, apoptosis, proliferation, development, and migration.1–7 Mutations in Ca2+ release-activated channel and the Mg2+ channel MAGT1 lead to severe combined immunodeficiency disorders in humans.8–10 The membrane potential, which depends on electrical gradients across the cell membrane, is maintained and regulated by K+, Na+, and Cl− channels.11,12 Multiple investigations of immune cells involving membrane potential regulation have focused on K+ and TRPM4 channels; these channels transport K+ and Na+ and provide the driving force for Ca2+ influx following receptor activation that regulates the functions of many enzymes and transcription factors.13–17 However, the effects of Cl− and Cl− channels on membrane potential regulation and the subsequent functional impact on immune cells are still poorly elucidated.
Cystic fibrosis transmembrane conductance regulator (CFTR) is associated with the lethal genetic disease cystic fibrosis, which is mainly caused by bronchial inflammation, viscous secretions, and impaired mucociliary clearance, and is responsible for electrolyte and water transport in the apical membrane of the epithelial cells lining various tissues.18–21 CFTR is a unique Cl− channel due to its dual functions as a Cl− channel and a regulator of other ion channels and transporters.22 The opening of Cl− channels in immune cells triggers the efflux of Cl− ions caused by a relatively high [Cl−]i (~38 mM) and an equilibrium membrane potential (approximately −33 mV). Consequently, Cl− efflux depolarizes the membrane potential and inhibits Ca2+ influx by regulating voltage-gated Ca2+ channels.23 However, how CFTR regulates the membrane potential and subsequently affects Ca2+ influx in immune cells remains unclear.
As a regulator, CFTR modulates several membrane ion transporters in epithelial cells via protein–protein interactions, such as the epithelial Na+ channel,24 Ca2+-activated chloride channels,25 the outwardly rectifying chloride channel,26 and apical K+ channels in renal epithelial cells, including renal outer medullary potassium channel (ROMK) 1 and ROMK2.27 Recently, CFTR was reported to participate in immune regulation28–30 and potentially be a risk factor for cancer development.31 However, the functions and underlying mechanisms of CFTR in T cells have not yet been fully elucidated.
γδ T cells represent an unconventional innate-like T cell subset, which are different from αβ T cells by their unique characteristics, including their tissue-specific distribution, lower receptor variability, NK receptor expression, and non-MHC-restricted cytotoxicity against target cells.32,33 γδ T cells actively perform protective functions against tumors, viral infections, parasites, and bacterial infections.34 γδ T cells also participate in the development of autoimmune and inflammatory diseases via rapid production of proinflammatory cytokines.35 Thus, it is crucial to explore the molecular mechanisms that regulate γδ T cell effector functions. Effector γδ T cells are classified as either IFN-γ-producing cells or IL-17-producing cells and regulate immunity in divergent microenvironments. IFN-γ-producing γδ T cells control processes during herpes, influenza, and other viral infections and during bacterial infections.36,37 In our previous publications, we found that γδ T cells provided an early critical source of IFN-γ and played a protective role in antitumor immunity.38,39 Furthermore, we reported that the promoter region of the Ifng gene in γδ T cells was highly demethylated, which resulted in a rapid IFN-γ response upon activation by the transcription factors T-bet and Eomes.40 We recently demonstrated mTORC1-mediated IFN-γ production by γδ T cells. Due to the distinctive features of γδ T cells, several γδ T cell-based, small-sized clinical trials of tumor immunotherapy using either in vivo expansion or adoptive transfer have been conducted.41 Clinical trials led by our group using allogenic γδ T cells showed promising effects on solid tumors.42 Although TCR signal transduction from stimuli on the extracellular side to transcription factors in the nucleus of γδ T cells, including ligand recognition and intracellular signaling cassettes, has been reported,43,44 the detailed molecular pathways still need to be explored. The impacts of ion channels, especially Ca2+ channels, on αβ T cell functions have been well documented.45
In this regard, we wanted to explore the critical functions of CFTR as a Cl− channel and a regulator in γδ T cells. In our study, we discovered that CFTR was functionally expressed on the cell surface of γδ T cells. CFTR negatively regulated IFN-γ production and antitumor immunity in γδ T cells. Regarding the underlying molecular mechanism, we found that CFTR in γδ T cells modulated Vm-induced and TCR-stimulated Ca2+ influx and subsequently regulated the calcineurin-NFATc1 signaling pathway. In addition, CFTR was recruited to TCR immunological synapses (ISs) and participated in TCR signal transduction by attenuating Lck-P38 MAPK-c-Jun signaling. Our research systematically defined the role of CFTR in the activation of γδ T cells and the potential contribution of CFTR to antitumor immunity, which will contribute to the understanding of γδ T cell biology and increase awareness of the potential of CFTR in γδ T cell-mediated cancer immunotherapy.
Results
CFTR is expressed in γδ and CD4+ T cells and is polarized toward the IS via the cytoskeleton
To evaluate the relative mRNA expression levels of various Cl− channels, we first performed Q-PCR analysis of primary mouse γδ T cells and CD4+ T cells isolated from the spleen of C57BL/6 mice. We found that the transcripts of CFTR and chloride intracellular channels (CLICs) displayed relatively higher mRNA levels than those of other Cl− channels (Fig. S1A) and treatment with IAA94, which is a potent blocker of CLICs, did not alter IFN-γ production in either γδ T cells or CD4+ T cells (Fig. S1B). Due to the unique functions of CFTR among Cl− channels, we focused on CFTR in further investigations. We then confirmed the expression of CFTR (band B: core-glycosylated form; band C: fully glycosylated form) in primary mouse γδ T cells and CD4+ T cells by immunoblot analysis (Fig. S1C). Confocal microscopy showed that CFTR was predominantly and evenly distributed in the plasma membrane of resting γδ T cells and CD4+ T cells (Fig. S1D). CFTR−/− mice were used as a negative control for antibody specificity in the immunoblot and confocal analyses. Surprisingly, we found that endogenous CFTR and F-actin relocalized and polarized toward the IS (the arrowhead indicated in Fig. S1E) when γδ T cells were activated with anti-CD3/CD28-coated Dynabeads®, which was different than the distribution seen in resting γδ T cells (Fig. S1D). It has been reported that in polarized epithelial cells, the c-terminal PDZ binding motif of CFTR binds to PDZ domain-containing proteins, which interact with ezrin to tether this multiprotein complex to the apical actin cytoskeleton.46 In addition, actin and ezrin are involved in the formation of the IS.47 Given this information, we tested the localization of CFTR and ezrin in stimulated γδ T cells and found that ezrin was also polarized toward the IS region and colocalized with CFTR (Fig. S1F), suggesting that CFTR is recruited to the IS region through binding to the cytoskeletal complex when γδ T cells are activated. T cell activation drives morphological and functional changes in T cells, including large-scale accumulation of actin and signaling molecules, which form distinct supramolecular clusters termed the IS to stabilize receptor signal transduction.48 Thus, these results indicated the potential role of CFTR in γδ T cell activation.
CFTR modulates γδ T cell activation
To define the function of CFTR in γδ T cell activation, we analyzed γδ T cell properties in wild-type and CFTR−/− mice. By surface staining and FACS analysis, we observed that there was no difference in the quantity of γδ T cells between wild-type and CFTR−/− mice (Fig. 1a upper panel), suggesting that CFTR may not be significantly involved in γδ T cell development and maintenance. However, CFTR deficiency robustly elevated the size of the population of naturally activated (CD44highCD62Llow) γδ T cells (Fig. 1a lower panel), suggesting a negative role for CFTR in γδ T cell activation in vivo. Notably, although CFTR was also expressed in CD4+ T cells, there was no significant phenotype related to CD4+ T cell activation in CFTR−/− mice (Fig. 1b). To further confirm the contribution of CFTR to IS formation when γδ T cells are activated, we assessed the F-actin distribution by confocal microscopy after stimulating wild-type and CFTR−/− γδ T cells with anti-CD3/CD28-coated Dynabeads®. We observed that significantly more F-actin accumulated in the IS area upon stimulation in the CFTR−/− γδ T cells than in the wild-type γδ T cells (Fig. 1c). Given the cytoskeletal control of cell morphology and cell mechanical stiffness, we characterized the cell membrane by directly measuring Young’s modulus and the adhesion force with atomic force microscopy and membrane force analysis. As shown in Fig. 1d, the mean Young’s modulus of CFTR−/− γδ T cells was significantly higher than that of wild-type γδ T cells, which was consistent with our confocal results; in contrast, the mean adhesion force was decreased. Thus, the CFTR−/− γδ T cells were stiffer than the wild-type γδ T cells due to the increased F-actin accumulation in the IS region. These results collectively showed a stronger activation status in CFTR−/− γδ T cells.
Fig. 1.

CFTR deficiency resulted in increased activation and more actin accumulation in the immunological synapse (IS) of γδ T cells. Representative flow cytometry and statistical analyses of CD44 and CD62L expression in WT or CFTR−/− splenic γδ T cells (a) and CD4+ T cells (b) (n = 3, **p < 0.01). c Left: confocal microscopy of WT or CFTR−/− γδ T cells stimulated with Dynabeads® Mouse T-Activator CD3/CD28 and stained with a rabbit anti-Lck antibody followed by an Alexa Fluor 488-conjugated anti-rabbit IgG or with Alexa Fluor 594-conjugated phalloidin (lower panel, arrowheads indicate the immunosynapse area) (scale bar: 5 μm, BF: bright field); Right: quantification of actin accumulation at the IS contact area analyzed with ImageJ (n = 52 per group, ****p < 0.0001). d Atomic force microscopy of in vitro-expanded WT or CFTR−/− γδ T cells with analysis of Young’s modulus and the adhesion force (WT: n = 172, CFTR−/−: n = 184, ***p < 0.001)
CFTR−/− γδ T cells exhibit enhanced IFN-γ production and cytotoxicity against tumors in vitro and in vivo
We then hypothesized that the enhanced T cell activation in CFTR−/− mice might affect subsequent cytokine production. Since IFN-γ and IL-17 released by γδ T cells have been extensively studied and found to play distinct roles in many disease models, we assessed the alterations in the expression of these two cytokines between CFTR−/− γδ T cells and wild-type γδ T cells. By intracellular cytokine staining of splenocytes and peripheral lymph node cells ex vivo, we found that CFTR deficiency triggered a significant increase in IFN-γ production in γδ T cells (Fig. 2a) but not in CD4+ T cells (Fig. 2b), which was consistent with the impact of CFTR on γδ T cell activation (Fig. 1). In addition, peripheral IL-17 expression was not altered in CFTR−/− γδ T cells or CD4+ T cells (Fig. 2a, b), suggesting a unique mechanism of CFTR-mediated suppression of IFN-γ production in γδ T cells. In addition, we analyzed the phenotype of thymic γδ T cells in CFTR−/− mice and found no differences in CD44/CD69 expression or cytokine production between wild-type and CFTR−/− γδ T cells (Fig. S2), which suggested that CFTR played a regulatory role in the periphery rather than during thymic programming. To dissect the stage of γδ T cell differentiation or maintenance impacted by CFTR, we sorted naive and naturally active γδ T cells separately, polarized them toward the γδ T1 direction, and analyzed the terminally differentiated cells. The results showed that the γδ T1 cells derived from sorted wild-type or CFTR−/− naive cells did not exhibit differences (Fig. 2c, upper panel), indicating a redundant role for CFTR in naive γδ T cell differentiation, and the underlying mechanisms remain to be further studied. In contrast, CFTR−/− γδ T1 cells had a higher frequency than wild-type γδ T1 cells when derived from sorted activated cells (Fig. 2c, lower panel). These results indicated that CFTR played an important role in activated γδ T cell function. Collectively, these findings revealed that CFTR was a negative regulator of γδ T cell activation and IFN-γ production.
Fig. 2.
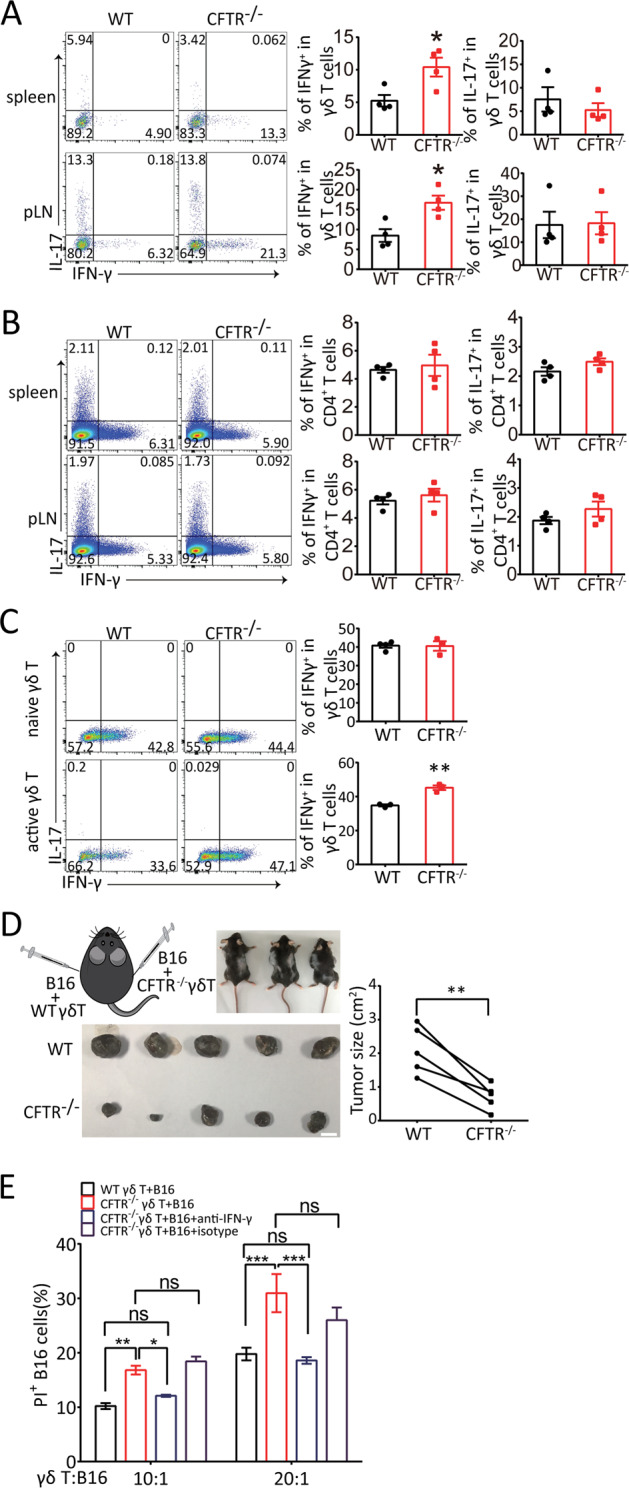
CFTR deficiency led to increased IFN-γ release and cytolytic ability against aggressive B16 melanoma. Representative flow cytometry analysis of IFN-γ and IL-17 production by WT or CFTR−/− peripheral γδ T cells (a) and CD4+ T cells (b) treated with PMA (50 ng/ml) and ionomycin (1 μg/ml) stimulation for 6 h is shown (n = 4, *p < 0.05). c Naive (CD44low) γδ T cells and active (CD44high) γδ T cells sorted from WT or CFTR−/− mouse splenocytes were polarized toward the γδ T1 subset for 4 days, and flow cytometry analysis of IFN-γ and IL-17 production by with WT or CFTR−/− γδ T cells treated with PMA (50 ng/ml) and ionomycin (1 μg/ml) stimulation for 6 h was performed (n = 3, **p < 0.01). d B16 cells (2 × 105 cells/mouse) were mixed with in vitro-expanded WT or CFTR−/− γδ T cells (0.5 × 105 cells/mouse) and s.c. injected into B6 TCR δ−/− mice (as shown in the schematic diagram in the left panel; n = 5 per group). On day 16 post tumor injection, tumors were isolated (left panel, scale bar: 1 cm), and tumor sizes were measured (right panel, paired t-test; **p < 0.01). e In vitro-expanded WT or CFTR−/− γδ T cells were cocultured with CFSE-labeled B16 cells at the indicated ratio, and an anti-IFN-γ antibody (10 µg/ml) was added to neutralize cytotoxicity, or an isotype control antibody was added. Six hours later, dead B16 cells were assessed by PI staining (***p < 0.001; **p < 0.01; *p < 0.05; ns not significant; n = 3)
In our previous studies, we found that γδ T cells, especially those in the Vγ4 subset, played a protective role in early tumor immune surveillance via IFN-γ and perforin production.39 Because γδ T cell activation and IFN-γ production were found to be enhanced in CFTR-deficient mice (Fig. 2a), we hypothesized that CFTR−/− γδ T cells may exert relatively strong antitumor immunity. To test this hypothesis, we first performed an in vitro killing assay by mixing γδ T cells and B16 melanoma tumor cells and then monitoring killing in real time. The B16 cells were labeled with calcein-AM and cocultured with the γδ T cells at a ratio of 1:5, and B16 viability was evaluated and quantified by assessing the fluorescence and density of calcein during a 6-h period. As shown in Fig. S3A, CFTR−/− γδ T cells displayed more rapid killing activity than wild-type γδ T cells. To test whether CFTR−/− γδ T cells have suppressive effects on tumors in vivo, we mixed B16 tumor cells with γδ T cells, which were expanded and sorted from wild-type or CFTR−/− mice, and separately inoculated the mixtures subcutaneously (s.c.) into the two flanks of B6 TCR δ−/− mice as described in our previous studies.39 Consistent with the in vitro results, the CFTR−/− γδ T cells showed significantly more powerful antitumor function than the wild-type γδ T cells (Fig. 2d). These results collectively demonstrated that CFTR might serve as a negative regulator in γδ T cell-mediated antitumor immunity, which was consistent with the enhanced IFN-γ production by CFTR−/− γδ T cells observed (Fig. 2a, c). To test whether the enhanced cytotoxicity of CFTR−/− γδ T cells against B16 tumor cells was due to elevated IFN-γ production, we blocked IFN-γ with an anti-IFN-γ antibody in the in vitro killing assay, and the results showed that blocking IFN-γ reduced the cytotoxic effect of CFTR−/− γδ T cells to wild-type levels (Fig. 2e), suggesting that the stronger antitumor function of CFTR−/− γδ T cells was dependent on elevated IFN-γ production. In addition, we performed an in vitro killing assay with EL4 lymphoma cells. As shown in Fig. S3B, CFTR−/− γδ T cells displayed enhanced cell cytotoxicity, which was mediated by IFN-γ, indicating that the killing ability of CFTR−/− γδ T cells was not B16 melanoma specific. Collectively, these results suggested that CFTR−/− γδ T cells had enhanced antitumor function mediated by upregulating IFN-γ expression.
CFTR inhibits γδ T cell IFN-γ production by regulating the TCR signaling cascade and Ca2+ influx
We demonstrated that CFTR deficiency resulted in increased activation and IFN-γ release in peripheral γδ T cells (Figs. 1a and 2a). We further investigated the molecular mechanism underlying CFTR-mediated γδ T cell activation. It is well known that the stimulation of T cells mediated by TCR signaling, which is critical for effective host responses to pathogens or tumors, requires the sequential activation and interaction of dozens of kinases and adapter proteins.49 Following the engagement of TCR, PLCγ-1/ZAP70/Lck form a TCR transduction complex to induce a cascade of events that spread through the membrane to the cytosol and the nucleus.50 Since the TCR signal transduction network requires protein–protein interactions, we first examined whether CFTR binds with TCR signaling molecules. Interestingly, coimmunoprecipitation analysis of wild-type γδ T cells showed that endogenous CFTR physically interacted with PLCγ-1, which bound to Lck and ZAP70, but this interaction did not occur in CFTR−/− γδ T cells (Fig. 3a). Furthermore, as shown in Fig. 3b, endogenous CFTR colocalized with the TCR transduction complex PLCγ-1/ZAP70/Lck in the IS area, but the PDZ binding motif in the c terminus of CFTR was not responsible for PLCγ-1 binding (Fig. S1G). Collectively, these results indicated that CFTR, when recruited to the IS region, could be a crucial signaling molecule in the proximal TCR transduction cascade.
Fig. 3.

CFTR participated in TCR signaling via interaction with the PLCγ-1–LCK–ZAP70 signaling complex and contributed to TCR-induced Ca2+ influx and membrane hyperpolarization in γδ T cells. a In vitro-expanded WT or CFTR−/− γδ T cells were immunoprecipitated with an anti-PLCγ-1 antibody, and the immunoprecipitates were immunoblotted with an antibody specific for PLCγ-1, CFTR, Lck, or ZAP70. b Confocal microscopy analysis of γδ T cells stimulated with Dynabeads® Mouse T-Activator CD3/CD28 and stained with a mouse anti-CFTR antibody followed by an Alexa Fluor 488-conjugated anti-mouse IgG or with an antibody to PLCγ-1, Lck, or ZAP70 followed by an Alexa Fluor 594-conjugated anti-rabbit IgG was performed (scale bar: 5 μm, BF: bright field). c A representative immunoblot analysis (from three independent experiments) of the indicated proteins in the whole-cell lysates of in vitro-expanded WT or CFTR−/− γδ T cells treated with soluble anti-CD3 (10 μg/ml) and anti-CD28 (1 μg/ml) antibody stimulation for 2, 5, 10, 15, or 30 min is shown. Band densities were quantified with ImageJ and ratios of indicated intensities were shown. act: actin. d ELISA analysis of IFN-γ production by in vitro-expanded and purified WT or CFTR−/− γδ T cells treated with SB203580 (10 μM) or PP2 (10 μM) for 12 h was performed (n = 3 for each group, ***p < 0.001). e The cytoplasmic Vm of WT or CFTR−/− γδ T (1 × 106 cells for each group) treated with soluble anti-CD3 (10 μg/ml) and anti-CD28 (1 μg/ml) antibody stimulation was measured, and the relative Vm in WT or CFTR−/− γδ T cells was quantified (n = 3, **p < 0.01). f Flow cytometry and statistical analyses of the relative phosphorylated NFATc1 level in in vitro-expanded WT or CFTR−/− γδ T cells treated with soluble anti-CD3 (10 μg/ml) and anti-CD28 (1 μg/ml) antibody stimulation for 30 min are shown (n = 3, ***p < 0.001). g The [Ca2+]i of WT or CFTR−/− γδ T cells (1 × 106 cells for each group) treated with soluble anti-CD3 (10 μg/ml) and anti-CD28 (1 μg/ml) antibody stimulation were measured, and the relative peak [Ca2+]i for the WT and CFTR−/− γδ T cells were quantified (n = 3, **p < 0.01)
We next aimed to evaluate the effect of CFTR deficiency on γδ T cell TCR signaling. We found no significant difference in the phosphorylation of PLCγ-1 between CFTR−/− γδ T cells and wild-type γδ T cells. However, CFTR deficiency in γδ T cells triggered robust increases in the phosphorylation levels of ZAP70, Lck, P38 MAPK, and c-Jun upon TCR stimulation (Fig. 3c). We then tested whether the augmented phosphorylation of TCR signaling molecules was the reason for the elevated IFN-γ production in CFTR−/− γδ T cells. Consistent with the results of the FACS analysis of CFTR−/− γδ T cells, we found that CFTR−/− γδ T cells produced higher levels of IFN-γ than did wild-type γδ T cells by ELISA. When the P38 MAPK antagonist SB203580 or the Lck inhibitor PP2 was added to cultures, no significant difference in IFN-γ production was found between WT and CFTR−/− γδ T cells, suggesting that Lck-P38 MAPK TCR signaling was involved in CFTR-regulated IFN-γ production (Fig. 3d). Together, these results suggested that CFTR negatively regulated TCR signal transduction and TCR-induced IFN-γ production.
Since CFTR modulates the membrane potential and TCR signaling, which subsequently regulate Ca2+ influx, we next investigated the contribution of CFTR to Vm-induced and TCR-stimulated Ca2+ influx with the Vm indicator DiBAC4(3) and Ca2+ indicator Fluo-4. As expected, CFTR−/− γδ T cells displayed a lower membrane potential than wild-type γδ T cells upon TCR stimulation (Fig. 3e). As shown in Fig. 3f, the phosphorylation level of NFATc1 was decreased in CFTR−/− γδ T cells, which indicated that NFATc1 was activated. In agreement with the data indicating that CFTR negatively regulated TCR transduction and NFATc1 activation, CFTR−/− γδ T cells exhibited a higher TCR-stimulated Ca2+ influx than wild-type γδ T cells (Fig. 3g).
CFTR inhibits IFN-γ production by regulating the membrane potential and Ca2+ influx in γδ T cells
To further confirm the contribution of CFTR channel activity to γδ T cell function, we treated wild-type γδ T cells with a CFTR-specific inhibitor. First, we evaluated whether CFTR was functional in sorted γδ T cells via the whole-cell patch clamp technique. We treated cells with the CFTR agonist forskolin and the CFTR-specific inhibitor GlyH101 and recorded cAMP-stimulated Cl− conductance. Indeed, surface expression of CFTR in γδ T cells led to a cAMP-stimulated Cl− current, which was blocked by GlyH101 (Fig. S1H). Next, we confirmed the modulation of CFTR channel activity-mediated IFN-γ production by treating wild-type γδ T cells derived from the mouse spleen with the CFTR-specific inhibitor CFTRinh172. In agreement with our in vivo results (Fig. 2), blocking CFTR activity and Cl− efflux enhanced IFN-γ expression in γδ T cells (Fig. 4a). In addition, CFTRinh172-treated γδ T cells exhibited markedly increased cytotoxicity against B16 melanoma cells in vitro (Fig. 4b) and in vivo (Fig. S3D). In addition, an in vitro killing assay with EL4 lymphoma cells showed that CFTRinh172-treated γδ T cells displayed enhanced cell cytotoxicity, but IAA94-treated γδ T cells did not, indicating the stronger killing ability of the CFTRinh172-treated γδ T cells and inability of CLICs to influence the cytotoxicity of γδ T cells (Fig. S3C). We also assessed TCR signaling when CFTR channel activity was blocked. We found that pharmacological inhibition of CFTR did not affect the activity of TCR signaling molecules (Fig. 4c), which suggested that CFTRinh172 did not interrupt the interaction between CFTR and TCR molecules and that the CFTRinh172-mediated increase in IFN-γ production was not due to an alteration in ZAP70-P38-c-Jun signaling.
Fig. 4.

Pharmacological inhibition of CFTR significantly increased IFN-γ release and cytotoxic activity in γδ T cells and triggered Vm-induced Ca2+ influx. a Flow cytometry analysis of splenic γδ T cells treated with PMA (50 ng/ml) and ionomycin (1 μg/ml) stimulation for 6 h in the presence of the CFTR inhibitor CFTRinh172 (5 μM) or vehicle DMSO was performed to detect IFN-γ and IL-17 production (n = 4, *p < 0.05). b In vitro-expanded γδ T cells were pretreated with the CFTR inhibitor CFTRinh172 (5 μM) or vehicle DMSO for 2 h and then cocultured with CFSE-labeled B16 cells at the indicated ratio. Six hours later, dead B16 cells were identified by PI staining (n = 3, *p < 0.05). c A representative immunoblot analysis (from three independent experiments) of the indicated proteins in the whole-cell lysate of in vitro-expanded γδ T cells stimulated in the presence of the CFTR inhibitor CFTRinh172 (5 μM) or vehicle DMSO with anti-CD3 (10 μg/ml) and anti-CD28 (1 μg/ml) antibodies for 2, 5, 10, 15, or 30 min is shown; band densities were quantified with ImageJ and ratios of indicated intensities were shown. act: actin. d The cytoplasmic Vm of γδ T cells treated with the CFTR inhibitor CFTRinh172 (10 μM) or vehicle DMSO (1 × 106 cells for each group) followed by gramicidin (10 μM) was measured, and the relative Vm in WT or CFTRinh172-treated γδ T cells was quantified (n = 3, **p < 0.01). e In vitro-expanded γδ T cells were pretreated with CFTRinh172 (10 μM) or the vehicle DMSO (1 × 106 cells for each group) for 2 h in Ca2+-free medium. Changes in the [Ca2+]i were monitored by flow cytometry, and the overlay of the Ca2+ influx profiles after the addition of CaCl2 (5 mM) to the extracellular medium in the presence of thapsigargin (1 μM) is shown. The relative peak [Ca2+]i in WT or CFTRinh172-treated γδ T cells was quantified (n = 3, *p < 0.05). f In vitro-expanded γδ T cells were treated with PMA (50 ng/ml) and ionomycin (1 μg/ml) for 6 h in the presence of the CFTR inhibitor CFTRinh172 (5 μM) or vehicle DMSO with the addition of FK506 and then evaluated to detect IFN-γ and IL-17 production (*p < 0.05, ns not significant, n = 3). g Flow cytometry analysis of the phosphorylated NFATc1 level in in vitro-expanded WT or CFTRinh172-treated γδ T cells stimulated with soluble anti-CD3 (10 μg/ml) and anti-CD28 (1 μg/ml) antibodies for 30 min was performed (n = 3, **p < 0.01). Dotted line: isotype control
We also observed that CFTRinh172 decreased the Vm and hyperpolarized γδ T cells (Fig. 4d) and that this cell hyperpolarization could be abolished by gramicidin, which was used as a positive control; furthermore, Ca2+ influx triggered by an elevated extracellular calcium concentration ([Ca2+]e) was significantly increased in CFTRinh172-treated γδ T cells treated with the sarcoplasmic endoplasmic reticulum Ca2+-ATPase pump inhibitor thapsigargin (Fig. 4e). These data suggested that CFTR might affect the Vm and Ca2+ influx by regulating an unknown Ca2+ channel(s) on the cell surface. Calcineurin is a calcium-dependent serine-threonine phosphatase that activates NFATc1, and we found that FK506, a calcineurin inhibitor, blocked CFTRinh172–induced IFN-γ production (Fig. 4f). In addition, the phosphorylation level of NFATc1 was decreased in CFTRinh172-treated γδ T cells (Fig. 4g), suggesting that the CFTR inhibitor, which triggered Ca2+ influx, led to calcineurin-NFATc1 activation and IFN-γ expression. These results collectively suggested that CFTR inhibited IFN-γ production by γδ T cells through its ion channel function.
CFTR affects IFN-γ production in human Vδ2 T cells and Vδ2 T cell-mediated cytotoxicity to human K562 leukemia cells
Vγ9Vδ2 γδ T cells, which are the dominant subset in human peripheral blood, are known for their high IFN-γ production and efficient antitumor activity.51 To explore the effect of CFTR on Vδ2 γδ T cells, we first evaluated the CFTR-mediated pharmacological inhibition of IFN-γ production in expanded Vδ2 γδ T cells from the peripheral blood of healthy donors. Consistent with our murine system, CFTR inhibition via CFTRinh172 significantly enhanced IFN-γ release (Fig. 5a); in addition, CFTRinh172-treated Vδ2 cells showed relatively high cytotoxicity to human K562 leukemia cells (Fig. 5b). Next, we used a lentiviral system to knockdown CFTR expression in Vδ2 γδ T cells and observed cytokine production. Before applying shRNA knockdown to Vδ2 γδ T cells, we measured the knockdown efficiencies of 4 different shRNA lentiviral particles. As shown in Fig. 5c, in GFP-positive Vδ2 γδ T cells, CFTR expression was decreased by approximately 50%, and similar to CFTR pharmacological inhibition, CFTR knockdown increased IFN-γ production upon anti-CD3/CD28 antibody stimulation. Collectively, these data indicated that CFTR negatively regulated human Vδ2 γδ T cell function.
Fig. 5.
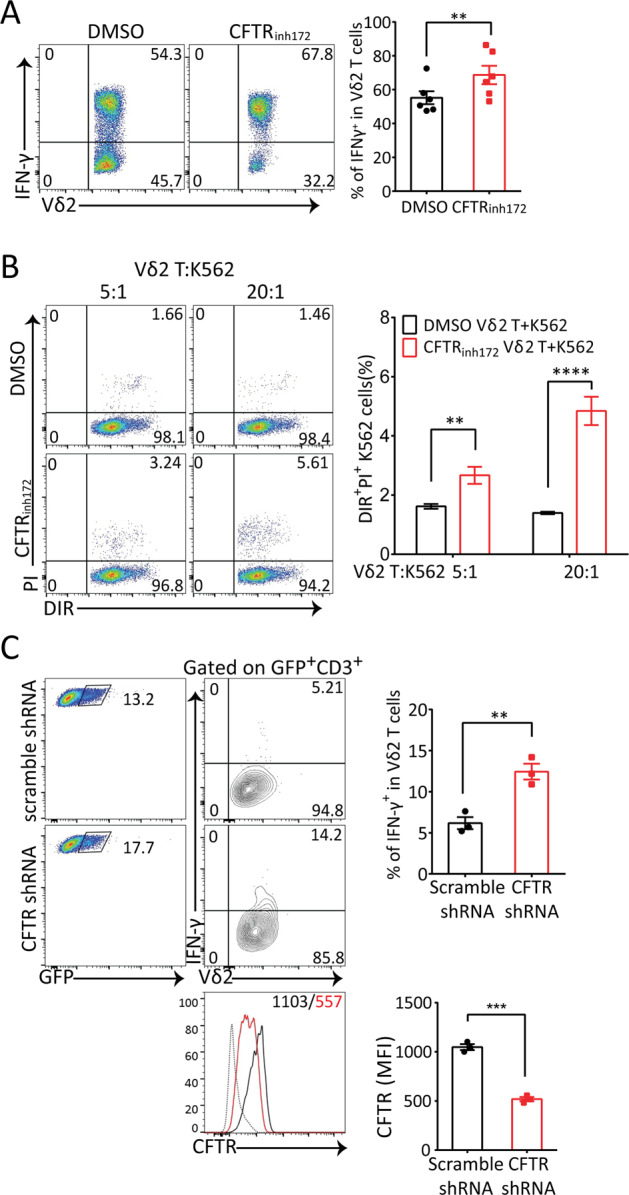
Pharmacological and genetic inhibition of CFTR both increased IFN-γ production in human Vδ2 T cells and Vδ2-mediated cytotoxicity to human K562 leukemia cells. a Flow cytometry analysis of in vitro-expanded human Vδ2 T cells treated with anti-CD3 (10 μg/ml) and anti-CD28 (1 μg/ml) antibody stimulation for 6 h in the presence of the CFTR inhibitor CFTRinh172 (5 μM) or vehicle DMSO was performed to detect IFN-γ production (n = 6, paired t-test, **p < 0.01). b In vitro-expanded human Vδ2 T cells were pretreated with the CFTR inhibitor CFTRinh172 (5 μM) or vehicle DMSO for 2 h and then cocultured with DIR-labeled K562 cells at the indicated ratio. Six hours later, dead K562 cells were identified by PI staining (n = 3, **p < 0.01, ****p < 0.0001). c Flow cytometry analysis of in vitro-expanded human Vδ2 T cells infected with scramble shRNA or CFTR-specific shRNA lentiviral particles was performed. After 72 h of infection, the cells were stimulated with 5 μg/ml plate-bound anti-human CD3 antibodies and 1 μg/ml soluble anti-human CD28 antibodies before IFN-γ production was detected (upper panels, n = 3, **p < 0.01, ***p < 0.001). The cells were stained with anti-CFTR and PE-anti-mouse IgM (polyclonal) antibodies and analyzed by FACS. Histograms of CFTR expression (gating on GFP+CD3+ cells) and the MFI of CFTR are shown (lower panels, black line/number: scramble shRNA; red line/number: CFTR-specific shRNA; dotted line: isotype control. n = 3, ***p < 0.001)
Discussion
γδ T cells play important roles in antitumor immunity.52 Our early studies demonstrated that γδ T cells rapidly produce IFN-γ at a much higher level than CD4 T cells, even when primed toward the Th1 program in vitro.53 The intrinsic mechanisms involve the expression of T-bet and Eomes and the epigenetically open status of the ifng gene.40,54 We further determined that γδ T cell-produced IFN-γ plays critical roles in antitumor immunity and the proinflammatory response in spinal cord injury.38,39,55 Subsequent studies by our group have shown that mTORC1-mediated metabolism and downstream signaling are required for IFN-γ production by γδ T cells.56 However, the regulation of IFN-γ remains unclear, especially the differences between γδ T cells and αβ T cells. Ion channels play critical roles in transmembrane ion homeostasis and have been found to play important functions in several types of immune cells.6 This is the first study to identify the ion channel CFTR as a negative regulator of IFN-γ production in γδ T cells and antitumor immunity.
We used genetically engineered KO mice/inhibitors and demonstrated that the chloride channel CFTR protein was a negative regulator of IFN-γ production in both mouse and human γδ T cells (Figs. 1, 2, and 5). This indicated that γδ T cells, although found to have innate features and be more effective than αβ T cells, could still be further strengthened by enhancing their activation through deactivation of negative molecules, including CFTR. Thus, our study identified a new member of the negative regulators of T cells, adding to PD-1 and CTLA-4, and provided potentially new targets for cancer immunotherapy based on specifically inhibiting CFTR activities. It is important to consider that several reports have shown contradictory effects of CFTR on different cancers, especially potentially protective effects on leukemia.57–60 In this study, we demonstrated that CFTR negatively regulated the cytotoxicity of γδ T cells to B16 melanoma cells and EL4 lymphoma cells. While recognizing the limitations of this research, especially studying CFTR function in γδ T cells via analysis of control of tumor cell lines, we believe that more mechanistic studies are necessary to define the precise role of CFTR in specific cell types in a particular cancer.
Another important finding of this study is that CFTR is a bifunctional molecule in γδ T cells (Fig. S4), with the nonchannel function being especially important (Fig. 3). Upon TCR activation, CFTR was recruited to the immunosynapse, bound to PLCγ-1 and directly attenuated Lck-P38 MAPK-c-Jun signaling. This further reminds us that ion channels may transduce previously unknown signals and perform additional functions in immune cells.
On the other hand, we found that CFTR modulated TCR-stimulated Ca2+ influx and Vm-induced Ca2+ influx and subsequently regulated the calcineurin-NFATc1 signaling pathway in γδ T cells (Fig. 4, Fig. S3). These findings further reinforced the relationships between CFTR and other ion channels. However, the potential candidate activated voltage-gated Ca2+ channels in this process have not yet been determined. Hence, CFTR, as an anion channel, needs to be researched further in regard to the comprehensive mechanism of immune cell function regulation.
As many previous studies have indicated, γδ and αβ T cells have distinct developmental and functional features.52 For instance, γδ TCRs recognize a distinct profile of ligands without MHC restriction rather than being activated by self MHC-presented pAgs like αβ T cells.52 In our previous studies, we also provided evidence that γδ T cells intrinsically produced more IFN-γ than αβ T cells in several conditions.53,54 In this study, we also found that CFTR−/− γδ T cells but not αβ T cells showed elevated IFN-γ production. This indicated that CFTR effectively suppressed IFN-γ production only in γδ T cells. The molecular basis of this discrepancy between γδ and αβ T cells is currently unknown. The possible explanations may involve the inability of CFTR to be recruited to the immunosynapse in αβ T cells, the compensatory effects of other ion channels, or the distinct machinery of signaling pathways in αβ T cells. Hence, it would be interesting to further investigate the distinct functions of CFTR in specific cell types, which could precisely determine the activity of CFTR in a particular environment.
In summary, this study defined CFTR as a negative regulator of IFN-γ production in γδ T cells and antitumor immunity. Our investigation suggested that modification of the CFTR activity of γδ T cells might provide a potential immunotherapeutic strategy for cancer. It is important to further study the detailed molecular mechanisms of CFTR in γδ T cells, particularly in relation to cell activation and quiescence and the transition between these states. Moreover, the regulation of the expression of CFTR and visualization of the spatial localization of CFTR in immune cells are still important areas that remain to be investigated.
Materials and methods
Mice and cell lines
C57BL/6J mice were obtained from Beijing HFK Bioscience Co., Ltd. (Beijing, China). B6.129P2-Cftrtm1Unc/J (CFTR−/−) mice, which have exon 10 replacement, were kindly provided by Dr. Pingbo Huang from the Hong Kong University of Science and Technology (Hong Kong). CFTR+/- breeders were set up, and CFTR+/+ and CFTR−/− littermates were used for experiments. B6.129P2-Tcrdtm1Mom/J (TCR δ−/−) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All animals were maintained under specific pathogen-free conditions and used at 6–8 weeks of age, and mouse protocols were approved by the Animal Experiment Committee of Jinan University. B16-F0 melanoma cells were purchased from ATCC (Manassas, VA) and were maintained in DMEM supplemented with 10% FBS. EL4 lymphoma cells and K562 leukemia cells were purchased from ATCC (Manassas, VA) and were maintained in RPMI 1640 medium supplemented with 10% FBS.
Antibodies and reagents
The following commercial antibodies were used in this study: a mouse monoclonal anti-CFTR C terminus antibody (clone 24-1, for immunoblotting) from R&D Systems (Minneapolis, MN); an anti-CFTR antibody (clone CF3, for immunofluorescence and FACS) from Novusbio (Littleton, CO); a monoclonal anti-GAPDH antibody (clone 71.1) from Sigma (St. Louis, MO); a β-Actin mouse monoclonal antibody (6G3) from Sungene Biotech (Tianjin, China); an anti-ZAP70 (99F2) rabbit mAb, an anti-Phospho-ZAP70 (Tyr319)/Syk (Tyr352) (65E4) rabbit mAb, an anti-PLCγ1 antibody, an anti-Phospho-PLCγ1 (Tyr783) (D6M9S) rabbit mAb, an anti-Lck antibody, an anti-p38 MAPK antibody, an anti-Phospho-p38 MAP kinase (Thr180/Tyr182) antibody, an anti-phospho-c-Jun (Ser73) (D47G9) XP® rabbit mAb, and an anti-Ezrin antibody from Cell Signaling Technology (Boston, MA); an anti-Lck (phospho Tyr394) rabbit polyclonal antibody from GeneTex (Irvine, CA); an anti-NFATc1 (phospho S237) antibody from Abcam (Cambridge, MA); an anti-mouse CD3 mAb (clone 145-2C11), an anti-mouse CD28 mAb (clone PV1), an anti-mouse TCR γδ mAb (clone UC7), an anti-mouse IFN-γ antibody (clone XMG1.2), a FITC-conjugated anti-mouse CD8 antibody, a PerCP-Cy5.5-conjugated anti-mouse CD4 antibody, a PE-conjugated anti-mouse TCR γδ mAb, a PE-conjugated anti-mouse CD44 mAb, an APC-conjugated anti-mouse CD62L mAb, a PE-Cy7-conjugated anti-mouse CD3 mAb, an APC-conjugated anti-mouse IL-17 mAb, and an APC-conjugated anti-mouse TNF-α mAb from Sungene Biotech (Tianjin, China); an Alexa Fluor 488-conjugated anti-mouse TCR γδ antibody and a Brilliant Violet 421™ anti-mouse IFN-γ antibody (clone XMG1.2) from BioLegend (San Diego, CA); and HRP-conjugated AffiniPure donkey anti-mouse and anti-rabbit secondary antibodies and Alexa Fluor 488- or Alexa Fluor 594-conjugated donkey anti-mouse and anti-rabbit IgGs from The Jackson ImmunoResearch Laboratory (West Grove, PA). All antibodies were used at the concentration recommended by the manufacturer. Protein A-agarose (sc-2001) was obtained from Santa Cruz (Dallas, TX). Anti-FLAG M2 magnetic beads (M8823) were obtained from Sigma (St. Louis, MO). The CFTR inhibitors CFTRinh172 and GlyH101, P38 MAPK inhibitor SB203580, Lck inhibitor PP2, calcineurin inhibitor FK506, and CLIC blocker IAA94 were purchased from Selleck (Houston, TX). The membrane potential probe DiBAC4(3) (Bis-(1,3-Dibutylbarbituric Acid) Trimethine Oxonol), intracellular calcium indicator Fluo-4, cell viability indicator Calcein-AM, and Dynabeads Mouse T-activator CD3/CD28 for mouse T cell activation were obtained from Thermo Fisher (Carlsbad, CA). A fixation/permeabilization solution kit was purchased from BD Biosciences (San Jose, CA).
Electrophysiology
In vitro-expanded and purified γδ T cells were subjected to whole-cell voltage clamp studies. The pipette solution contained (in mM): 140 CsCl, 2 MgCl2, 5 HEPES, 1 EGTA, 0.5 Li-ATP, 2 MgATP, and 10 glucose (pH 7.35). The bath solution contained (in mM): 170 Tris-Cl, 10 HEPES, 2.5 CaCl2, 1 MgCl2, and 15 glucose (pH 7.4). Reduction of [Cl−] in the external solution was performed by replacement of Tris-Cl with Tris-aspartate. the Patch pipettes had resistances of ~2–5 MΩ with these solutions. The procedure and data analysis were similar to those described in a previous protocol.22
Cell preparation and activation and intracellular cytokine staining
Mouse γδ T cells were expanded and sorted as previously described.39 For expansion of γδ T cells, day-6 γδ T cells were used, and the purity was checked (>99%). For analysis of TCR activation, γδ T cells from wild-type or CFTR−/− mice were activated with soluble anti-CD3 (10 µg/ml) and anti-CD28 (1 µg/ml) antibodies for the indicated times, followed by immunoblot analysis to assess the phosphorylation of PLCγ1, ZAP70 (Tyr319), Lck (Tyr394), p38 MAP kinase (Thr180/Tyr182), and c-Jun (Ser73). Intracellular cytokine staining was carried out following the protocols in our previous publication.61 Stained cells were analyzed with a BD FACSVerseTM flow cytometer (San Jose, CA), and the data were analyzed with FlowJo 10.0 software.
The use of blood from healthy adult blood donors was approved by the Institutional Review Boards of Jinan University, Guangzhou. Human Vδ2 T cells were expanded from PBMCs as previously described.51 For the expansion of Vδ2 T cells, day-12 Vδ2 T cells were used, and the purity was checked (>95%). For the detection of intracellular cytokines, Vδ2 T cells were stimulated for 6 h with 5 μg/ml plate-bound anti-human CD3 (clone OKT3, eBioscience) and 1 μg/ml soluble anti-human CD28 (clone CD28.2, eBioscience) antibodies in the presence of GolgiPlug™ (BD Biosciences, San Jose, CA). For intracellular staining, cells were stained with different cocktails of fluorochrome-conjugated monoclonal antibodies specific for CD3 (clones SK7 and UCHT1) or TNF-α (clone MAb11) from BD Biosciences (Heidelberg, Germany), or IFN-γ (clone B27) or TCR Vδ2 (clone B6) from BioLegend (London, UK).
In vitro γδ T1 cell priming
γδ+CD44low (naive) and γδ+CD44high (active) T cells were isolated from the spleen of wild-type or CFTR−/− mice. These cells were cultured (at a density of 1 × 106 cells per well) in flat-bottomed 24-well plates coated with anti-CD3 antibodies (10 μg/ml) in complete RPMI medium in the presence of anti-CD28 antibodies (1 μg/ml), recombinant mouse IL-12 (10 ng/ml), neutralizing anti-IL-4 antibodies (10 μg/ml), and recombinant mouse IL-2 (2 ng/ml). After 4 days, γδ T cells were stimulated for 6 h with PMA (50 ng/ml) and ionomycin (1 μg/ml) in the presence of GolgiPlug™. Intracellular cytokines were evaluated according to the manufacturer’s instructions (eBioscience).
In vitro killing assay
B16-F0 cells or EL4 cells were labeled with 2 µM CFSE. Then, the B16 cells or EL4 cells (8000 cells/well) were mixed with expanded γδ T cells at different ratios for 6 h and analyzed by flow cytometry. The killing capability was evaluated by measuring the percentage of dead B16 or EL4 cells (% of PI+CFSE+ cells). In addition, an anti-IFN-γ neutralizing antibody (10 μg/ml) was used to block IFN-γ-mediated cytotoxicity, and an isotype control antibody was added to exclude the side effects of the anti-IFN-γ antibody. K562 cells were stained with DiR (Invitrogen) and analyzed by flow cytometry after 6 h of coculture with autologous Vδ2 T cells at different ratios. The killing capability was evaluated by measuring the percentage of dead K562 cells (% of PI+DIR+ cells).
Tumor models
B16-F0 melanoma cells were mixed with expanded and purified γδ T cells and subcutaneously injected into TCR δ−/− mice. Tumor size was monitored as described previously.39
ELISA
In vitro-expanded and purified WT or CFTR−/− γδ T cells were treated with SB203580 or PP2 for 12 h, and the supernatants were collected for ELISA. The ELISA procedure followed the instructions of the IFN gamma Mouse Uncoated ELISA Kit (Thermo Fisher).
Real-time PCR for gene transcription
Total mRNA was isolated from cells using TRIzol® Reagent, and cDNAs were generated by reverse transcription-PCR using M-MLV reverse transcriptase with oligo(dT)15 and TaKaRa Taq®. Real-time PCR was performed following the instructions of TB Green Premix Ex Taq II from TaKaRa. The primers for mouse genes were: CFTR-F: GGCACTCCGGTTAAGTAACTC, CFTR-R: TGTCACTTGGTCGAATTTGTTCA; CLCA-1-F: CTAAACATCCGGTCTGCTAGACT, CLCA-1-R: ACCCGTGCGTACACAATCATC; CLCA-4-F: GAAAGAACACTGTCGAAGCAAC, CLCA-4-R: CAGTCTGGGATTTGTTTGGGATA; GABA-1-F: GGTCACAGTGAAAAACCGAATG, GABA-1-R: CGATGTCATCCGTGGTATAGCC; CLIC-1-F: CCCAGAGACTGTTCATGGTGC, CLIC-1-R: TCCCGAGGTGTTGGACTCA; CLIC-3-F: CCTGCTGTACGATGGGGATG, CLIC-3-R: GAAGGCAGAGAACTTGTGGAAG; CLIC-4-F: TTGTGCCCACCCAAGTACCTA, CLIC-4-R: CATTAGCCTCTGGTCTTGAGTTC; CLIC-5-F: GTGAAGGCTGGGATCGACG, CLIC-5-R: CCAGGTTGTGTAGATCGGCT; CLIC-6-F: CCCCAGGATGAGGCGATTG, CLIC-6-R: GTCCCTTCAACTCGGGTTCT; LRRC8a-F: GATTGCTGTCTTTGGAGGGAC, and LRRC8a-R: GGCAGGACTGTGGAGTTGG. The primers for the internal control gene β-Actin were Actb-F: AACAGTCCGCCTAGAAGCAC and Actb-R: CGTTGACATCCGTAAAGACC. Real-time PCR was performed on a BioRad CFX Connect cycler. The expression of genes encoding several Cl− channels in mouse γδ T cells/CD4+ T cells was compared after verification that the amplification efficiencies of the different target genes were similar.
shRNA knockdown of CFTR expression in human Vδ2 T cells
shRNA constructs (encoding the GFP gene) targeting CFTR were purchased from Era Biotech (Shanghai, China). The sequences were as follows: shRNA1: GCTCTATCGCGATTTATCTAG, shRNA2: CCTATGTGAGATACTTCAATA, shRNA3: GAACACATACCTTCGATATAT, and shRNA4: GCCAAATGACTGTCAAAGATC. The constructs were used to generate lentiviral particles in HEK293T cells. Day-5 Vδ2 T cells were infected with an shRNA lentivirus, and at 72 h post infection, infected Vδ2 T cells were used for experiments.
Plasmid constructs and transfection
CFTR and CFTR-ΔPDZ were amplified by PCR and inserted into the pcDNA3.1-Myc plasmid (Invitrogen) using the KpnI and NotI restriction enzyme sites. PLCγ-1 was amplified and inserted into the p3xFLAG-CMV-7.1 plasmid (Addgene) using the EcoRI and BamHI restriction enzyme sites. After verification by sequencing, the plasmids were cotransfected into HEK293T cells with Lipofectamine 3000 reagent (Invitrogen).
Coimmunoprecipitation
In vitro-expanded and purified γδ T cells (~1 × 107) were lysed in coimmunoprecipitation buffer (20 mM HEPES, pH 7.4; 175 mM NaCl; 1 mM EDTA; 1 mM DTT; 1 mM PMSF; 0.25% Nonidet P-40; 10% glycerol; and a protease inhibitor mixture (Roche Applied Science)), and the cell lysates were immunoprecipitated with 1 μg of anti-PLCγ1 antibody and protein A-agarose beads. The immunoprecipitates were washed with co-IP buffer three times, eluted with 1 × SDS loading buffer, and analyzed by western blotting. For transfected HEK293T cells, lysates were immunoprecipitated with anti-FLAG M2 magnetic beads overnight at 4 °C. After three washes, the samples were eluted with 1 × SDS loading buffer and resolved using SDS-PAGE gels.
Immunofluorescence
Cells were fixed by incubation in 4% paraformaldehyde for 10 min and permeabilized in 0.2 M NH4Cl/PBS plus 0.2% Triton X-100. After being blocked with 3% BSA/PBS, the cells were stained with an appropriate combination of primary antibodies, followed by staining with corresponding fluorophore-conjugated secondary antibodies. The cells were mounted on coverslips and examined under a confocal microscope (Leica). For analysis of the formation of the IS in γδ T cells, purified γδ T cells were treated with Dynabeads Mouse T-activator CD3/CD28 for 30 min, followed by immunostaining.
Measurement of the membrane potential and Ca2+ flux by flow cytometry
For determination of the membrane potential, cells were loaded with 2 µM DiBAC4(3) in phenol red-free RPMI 1640 medium at 37 °C and 5% CO2 for 60 min. The cells were then washed, stained with an APC-conjugated anti-TCR γ/δ antibody, and resuspended at a density of 1 × 106 cells per ml in complete RPMI 1640 medium. After baseline acquisition on an Accuri C6 flow cytometer, the cells were treated with a CFTR antagonist (CFTRinh172) or vehicle (0.1% DMSO), followed by treatment with 10 µM gramicidin. Data are presented as the DiBAC4(3) fluorescence (490 nm/516 nm) and were calculated for γδ T cells gated with FlowJo software. For measurement of Ca2+ flux, cells were loaded with 2 µM Fluo-4 in PBS at 37 °C and 5% CO2 for 30 min. The cells were then washed, stained with an APC-conjugated anti-TCR γ/δ antibody, and incubated in phenol red-free complete RPMI 1640 medium at 37 °C and 5% CO2 for 60 min. After baseline acquisition on an Accuri C6 flow cytometer, the cells were treated with a CFTR antagonist (CFTRinh172) or vehicle (0.1% DMSO), followed by the addition of 10 μg/ml anti-CD3 antibodies and 1 μg/ml anti-CD28 antibodies. Data are presented as the Fluo-4 fluorescence (494 nm/506 nm) and were calculated for γδ T cells gated with FlowJo software.
Atomic force microscopy
Wild-type and CFTR−/− γδ T cells were seeded in poly-l-lysine-coated dishes at a density of 1 × 106 cells per 2 ml of medium. The in situ approach was used to measure adhesion force and Young’s modulus by performing AFM and force analysis. The petri dishes with cells were directly transferred onto the AFM scanner stage for evaluation without any pretreatment, and whole measurements were conducted according to the protocols of previous studies.62
Statistics
Data are expressed as the mean ± SEM. Group sizes and the number of replications are stated in the figure legends. Statistical significance was evaluated using GraphPad Prism 6 for Windows (GraphPad). Tumor growth experiments and human Vδ2 T cell stimulation experiments were analyzed with paired t-tests. Flow cytometry, patch clamp, Q-PCR, actin quantification, AFM, Vm and Ca2+ influx recording, and stimulation experiments were analyzed using unpaired t-tests. Killing assay and ELISA results were analyzed using two-way ANOVA with a post hoc Bonferroni test. p values less than 0.05 were considered significant. The following annotations are used to show statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Supplementary information
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (31420103901 to Z.Y., 31830021 to Z.Y., 31970830 to J.H., 81702876 to X.L., 31500734 to Y.D., and 31700753 to G.C.), grants from the Guangzhou Municipal Science and Technology Bureau (201904010090 to J.H. and 201906010085 to X.L.), and a grant from the Health Commission of Guangdong Province (A2019520 to J.H.).
Author contributions
Y.D., G.L., and M.X. designed and performed experiments and data analysis. X.Q. and J.T. performed the patch clamp experiment and data analysis. Z.J., Q.Y., M.D., and Z.Lei helped with in vitro cell expansion. Y.H. performed the AFM experiment. Z.Li helped with data analysis and drawing of the proposed model. Z.Liu and Q.W. helped with animal breeding. X.L., G.C., W.K.Z., P.H., L.Z., and R.A.F. contributed to data analysis. Z.Y. and J.H. designed the research and wrote the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Yuanyuan Duan, Guangqiang Li, Miaomiao Xu
Contributor Information
Jianlei Hao, Email: haojianlei@jnu.edu.cn.
Zhinan Yin, Email: zhinan.yin@yale.edu.
Supplementary information
The online version of this article (10.1038/s41423-020-0499-3) contains supplementary material.
References
- 1.Cahalan MD, Chandy KG. The functional network of ion channels in T lymphocytes. Immunol. Rev. 2009;231:59–87. doi: 10.1111/j.1600-065X.2009.00816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cai X, Wang X, Patel S, Clapham DE. Insights into the early evolution of animal calcium signaling machinery: a unicellular point of view. Cell Calcium. 2015;57:166–173. doi: 10.1016/j.ceca.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clapham DE. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 4.Feske S, Concepcion AR, Coetzee WA. Eye on ion channels in immune cells. Sci. Signal. 2019;12:572. doi: 10.1126/scisignal.aaw8014. [DOI] [PubMed] [Google Scholar]
- 5.Feske S. Calcium signalling in lymphocyte activation and disease. Nat. Rev. Immunol. 2007;7:690–702. doi: 10.1038/nri2152. [DOI] [PubMed] [Google Scholar]
- 6.Feske S, Skolnik EY, Prakriya M. Ion channels and transporters in lymphocyte function and immunity. Nat. Rev. Immunol. 2012;12:532–547. doi: 10.1038/nri3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shaw PJ, Qu B, Hoth M, Feske S. Molecular regulation of CRAC channels and their role in lymphocyte function. Cell. Mol. Life Sci. 2013;70:2637–2656. doi: 10.1007/s00018-012-1175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maul-Pavicic A, et al. ORAI1-mediated calcium influx is required for human cytotoxic lymphocyte degranulation and target cell lysis. Proc. Natl Acad. Sci. USA. 2011;108:3324–3329. doi: 10.1073/pnas.1013285108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shaw PJ, Feske S. Regulation of lymphocyte function by ORAI and STIM proteins in infection and autoimmunity. J. Physiol. 2012;590:4157–4167. doi: 10.1113/jphysiol.2012.233221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li FY, et al. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature. 2011;475:471–476. doi: 10.1038/nature10246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chandy KG, DeCoursey TE, Cahalan MD, McLaughlin C, Gupta S. Voltage-gated potassium channels are required for human T lymphocyte activation. J. Exp. Med. 1984;160:369–385. doi: 10.1084/jem.160.2.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeCoursey TE, Chandy KG, Gupta S, Cahalan MD. Voltage-dependent ion channels in T-lymphocytes. J. Neuroimmunol. 1985;10:71–95. doi: 10.1016/0165-5728(85)90035-9. [DOI] [PubMed] [Google Scholar]
- 13.Launay P, et al. TRPM4 regulates calcium oscillations after T cell activation. Science. 2004;306:1374–1377. doi: 10.1126/science.1098845. [DOI] [PubMed] [Google Scholar]
- 14.Launay P, et al. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell. 2002;109:397–407. doi: 10.1016/S0092-8674(02)00719-5. [DOI] [PubMed] [Google Scholar]
- 15.Chimote AA, et al. A defect in KCa3.1 channel activity limits the ability of CD8(+) T cells from cancer patients to infiltrate an adenosine-rich microenvironment. Sci. Signal. 2018;11:527. doi: 10.1126/scisignal.aaq1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crottes D, et al. Immature human dendritic cells enhance their migration through KCa3.1 channel activation. Cell Calcium. 2016;59:198–207. doi: 10.1016/j.ceca.2016.02.008. [DOI] [PubMed] [Google Scholar]
- 17.Kuras Z, Yun YH, Chimote AA, Neumeier L, Conforti L. KCa3.1 and TRPM7 channels at the uropod regulate migration of activated human T cells. PloS ONE. 2012;7:e43859. doi: 10.1371/journal.pone.0043859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riordan JR, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 19.Chen JH, Schulman H, Gardner P. A cAMP-regulated chloride channel in lymphocytes that is affected in cystic fibrosis. Science. 1989;243:657–660. doi: 10.1126/science.2464852. [DOI] [PubMed] [Google Scholar]
- 20.Shah VS, et al. Airway acidification initiates host defense abnormalities in cystic fibrosis mice. Science. 2016;351:503–507. doi: 10.1126/science.aad5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pankow S, et al. F508 CFTR interactome remodelling promotes rescue of cystic fibrosis. Nature. 2015;528:510–516. doi: 10.1038/nature15729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duan Y, et al. Keratin K18 increases cystic fibrosis transmembrane conductance regulator (CFTR) surface expression by binding to its C-terminal hydrophobic patch. J. Biol. Chem. 2012;287:40547–40559. doi: 10.1074/jbc.M112.403584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feske S, Wulff H, Skolnik EY. Ion channels in innate and adaptive immunity. Annu. Rev. Immunol. 2015;33:291–353. doi: 10.1146/annurev-immunol-032414-112212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Puga Molina LC, et al. CFTR/ENaC-dependent regulation of membrane potential during human sperm capacitation is initiated by bicarbonate uptake through NBC. J. Biol. Chem. 2018;293:9924–9936. doi: 10.1074/jbc.RA118.003166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei L, et al. The C-terminal part of the R-domain, but not the PDZ binding motif, of CFTR is involved in interaction with Ca(2+)-activated Cl- channels. Pflug. Arch. 2001;442:280–285. doi: 10.1007/s004240100531. [DOI] [PubMed] [Google Scholar]
- 26.Ogura T, et al. ClC-3B, a novel ClC-3 splicing variant that interacts with EBP50 and facilitates expression of CFTR-regulated ORCC. FASEB J. 2002;16:863–865. doi: 10.1096/fj.01-0845fje. [DOI] [PubMed] [Google Scholar]
- 27.Welling PA, Ho K. A comprehensive guide to the ROMK potassium channel: form and function in health and disease. Am. J. Physiol. Ren. Physiol. 2009;297:F849–F863. doi: 10.1152/ajprenal.00181.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mueller C, et al. Lack of cystic fibrosis transmembrane conductance regulator in CD3+ lymphocytes leads to aberrant cytokine secretion and hyperinflammatory adaptive immune responses. Am. J. Respir. Cell Mol. Biol. 2011;44:922–929. doi: 10.1165/rcmb.2010-0224OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allard JB, et al. Aspergillus fumigatus generates an enhanced Th2-biased immune response in mice with defective cystic fibrosis transmembrane conductance regulator. J. Immunol. 2006;177:5186–5194. doi: 10.4049/jimmunol.177.8.5186. [DOI] [PubMed] [Google Scholar]
- 30.Dorsey J, Gonska T. Bacterial overgrowth, dysbiosis, inflammation, and dysmotility in the Cystic Fibrosis intestine. J. Cyst. Fibros. 2017;16(Suppl 2):S14–S23. doi: 10.1016/j.jcf.2017.07.014. [DOI] [PubMed] [Google Scholar]
- 31.Riquelme SA, et al. Cystic fibrosis transmembrane conductance regulator attaches tumor suppressor PTEN to the membrane and promotes anti pseudomonas aeruginosa immunity. Immunity. 2017;47:1169–81 e7. doi: 10.1016/j.immuni.2017.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holderness J, Hedges JF, Ramstead A, Jutila MA. Comparative biology of gammadelta T cell function in humans, mice, and domestic animals. Annu. Rev. Anim. Biosci. 2013;1:99–124. doi: 10.1146/annurev-animal-031412-103639. [DOI] [PubMed] [Google Scholar]
- 33.Chien YH, Meyer C, Bonneville M. gammadelta T cells: first line of defense and beyond. Annu. Rev. Immunol. 2014;32:121–155. doi: 10.1146/annurev-immunol-032713-120216. [DOI] [PubMed] [Google Scholar]
- 34.Serre K, Silva-Santos B. Molecular mechanisms of differentiation of murine pro-inflammatory gammadelta T cell subsets. Front. Immunol. 2013;4:431. doi: 10.3389/fimmu.2013.00431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sutton CE, et al. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 36.Ferrick DA, et al. Differential production of interferon-gamma and interleukin-4 in response to Th1- and Th2-stimulating pathogens by gamma delta T cells in vivo. Nature. 1995;373:255–257. doi: 10.1038/373255a0. [DOI] [PubMed] [Google Scholar]
- 37.Born WK, Yin Z, Hahn YS, Sun D, O’Brien RL. Analysis of gamma delta T cell functions in the mouse. J. Immunol. 2010;184:4055–4061. doi: 10.4049/jimmunol.0903679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao Y, et al. Gamma delta T cells provide an early source of interferon gamma in tumor immunity. J. Exp. Med. 2003;198:433–442. doi: 10.1084/jem.20030584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He W, et al. Naturally activated V gamma 4 gamma delta T cells play a protective role in tumor immunity through expression of eomesodermin. J. Immunol. 2010;185:126–133. doi: 10.4049/jimmunol.0903767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen L, et al. Epigenetic and transcriptional programs lead to default IFN-gamma production by gammadelta T cells. J. Immunol. 2007;178:2730–2736. doi: 10.4049/jimmunol.178.5.2730. [DOI] [PubMed] [Google Scholar]
- 41.Lo Presti E, et al. Current advances in gammadelta T cell-based tumor immunotherapy. Front. Immunol. 2017;8:1401. doi: 10.3389/fimmu.2017.01401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alnaggar M, et al. Allogenic Vgamma9Vdelta2 T cell as new potential immunotherapy drug for solid tumor: a case study for cholangiocarcinoma. J. Immunother. Cancer. 2019;7:36. doi: 10.1186/s40425-019-0501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hayes SM, Shores EW, Love PE. An architectural perspective on signaling by the pre-, alphabeta and gammadelta T cell receptors. Immunol. Rev. 2003;191:28–37. doi: 10.1034/j.1600-065X.2003.00011.x. [DOI] [PubMed] [Google Scholar]
- 44.Rigau M, et al. Butyrophilin 2A1 is essential for phosphoantigen reactivity by gammadelta T cells. Science. 2020;367:eaay5516. doi: 10.1126/science.aay5516. [DOI] [PubMed] [Google Scholar]
- 45.Bertin S, et al. The ion channel TRPV1 regulates the activation and proinflammatory properties of CD4(+) T cells. Nat. Immunol. 2014;15:1055–1063. doi: 10.1038/ni.3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guggino WB, Stanton BA. New insights into cystic fibrosis: molecular switches that regulate CFTR. Nat. Rev. Mol. Cell Biol. 2006;7:426–436. doi: 10.1038/nrm1949. [DOI] [PubMed] [Google Scholar]
- 47.Roumier A, et al. The membrane-microfilament linker ezrin is involved in the formation of the immunological synapse and in T cell activation. Immunity. 2001;15:715–728. doi: 10.1016/S1074-7613(01)00225-4. [DOI] [PubMed] [Google Scholar]
- 48.Calabia-Linares C, et al. Endosomal clathrin drives actin accumulation at the immunological synapse. J. Cell Sci. 2011;124(Pt 5):820–830. doi: 10.1242/jcs.078832. [DOI] [PubMed] [Google Scholar]
- 49.Roncagalli R, et al. Quantitative proteomics analysis of signalosome dynamics in primary T cells identifies the surface receptor CD6 as a Lat adaptor-independent TCR signaling hub. Nat. Immunol. 2014;15:384–392. doi: 10.1038/ni.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chakraborty AK, Weiss A. Insights into the initiation of TCR signaling. Nat. Immunol. 2014;15:798–807. doi: 10.1038/ni.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kouakanou L, et al. Vitamin C promotes the proliferation and effector functions of human gammadelta T cells. Cell Mol. Immunol. 2019;17:462–473. doi: 10.1038/s41423-019-0247-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carding SR, Egan PJ. Gammadelta T cells: functional plasticity and heterogeneity. Nat. Rev. Immunol. 2002;2:336–345. doi: 10.1038/nri797. [DOI] [PubMed] [Google Scholar]
- 53.Yin Z, et al. Dominance of IL-12 over IL-4 in gamma delta T cell differentiation leads to default production of IFN-gamma: failure to down-regulate IL-12 receptor beta 2-chain expression. J. Immunol. 2000;164:3056–3064. doi: 10.4049/jimmunol.164.6.3056. [DOI] [PubMed] [Google Scholar]
- 54.Yin Z, et al. T-Bet expression and failure of GATA-3 cross-regulation lead to default production of IFN-gamma by gammadelta T cells. J. Immunol. 2002;168:1566–1571. doi: 10.4049/jimmunol.168.4.1566. [DOI] [PubMed] [Google Scholar]
- 55.Sun G, et al. gammadelta T cells provide the early source of IFN-gamma to aggravate lesions in spinal cord injury. J. Exp. Med. 2018;215:521–535. doi: 10.1084/jem.20170686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang Q, et al. Roles of mTORC1 and mTORC2 in controlling gammadelta T1 and gammadelta T17 differentiation and function. Cell Death Differ. 2020;27:2248–2262. doi: 10.1038/s41418-020-0500-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ponzetto A, Holton J, Lucia U. Cancer risk in patients with cystic fibrosis. Gastroenterology. 2018;154:2282–2283. doi: 10.1053/j.gastro.2018.02.040. [DOI] [PubMed] [Google Scholar]
- 58.Liu M, et al. Treatment of human T-cell acute lymphoblastic leukemia cells with CFTR inhibitor CFTRinh-172. Leuk. Res. 2019;86:106225. doi: 10.1016/j.leukres.2019.106225. [DOI] [PubMed] [Google Scholar]
- 59.Yamada A, et al. Risk of gastrointestinal cancers in patients with cystic fibrosis: a systematic review and meta-analysis. Lancet Oncol. 2018;19:758–767. doi: 10.1016/S1470-2045(18)30188-8. [DOI] [PubMed] [Google Scholar]
- 60.Abraham JM, Taylor CJ. Cystic fibrosis & disorders of the large intestine: DIOS, constipation, and colorectal cancer. J. Cyst. Fibros. 2017;16(Suppl 2):S40–S49. doi: 10.1016/j.jcf.2017.06.013. [DOI] [PubMed] [Google Scholar]
- 61.Cao G, et al. mTOR inhibition potentiates cytotoxicity of Vgamma4 gammadelta T cells via up-regulating NKG2D and TNF-alpha. J. Leukoc. Biol. 2016;100:1181–1189. doi: 10.1189/jlb.5A0116-053RR. [DOI] [PubMed] [Google Scholar]
- 62.McEwen GD, et al. Subcellular spectroscopic markers, topography and nanomechanics of human lung cancer and breast cancer cells examined by combined confocal Raman microspectroscopy and atomic force microscopy. Analyst. 2013;138:787–797. doi: 10.1039/C2AN36359C. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.