

Keywords: neurodegenerative disease, Parkinson's disease, insulin resistance, inflammation, GLP-1/GIP receptor unimolecular co-agonist
Abstract
Patients with Parkinson’s disease (PD) have impaired insulin signaling in the brain. Incretin hormones, including glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP), can re-sensitize insulin signaling. In a recent phase II clinical trial, the first GLP-1 mimic, exendin-4, has shown reliable curative effect in patients with PD. DA-CH5 is a novel GLP-1/GIP receptor unimolecular co-agonist with a novel peptide sequence added to cross the blood-brain barrier. Here we showed that both exendin-4 and DA-CH5 protected against 6-hydroxydopamine (6-OHDA) cytotoxicity, inhibited apoptosis, improved mitogenesis and induced autophagy flux in SH-SY5Y cells via activation of the insulin receptor substrate-1 (IRS-1)/alpha serine/threonine-protein kinase (Akt)/cAMP response element-binding protein (CREB) pathway. We also found that DA-CH5 (10 nmol/kg) daily intraperitoneal administration for 30 days post-lesion alleviated motor dysfunction in rats and prevented stereotactic unilateral administration of 6-OHDA induced dopaminergic neurons loss in the substantia nigra pars compacta. However, DA-CH5 showed curative effects in reducing the levels of α-synuclein and the levels of pro-inflammatory cytokines (tumor necrosis factor-α, interleukin-1β). It was also more effective than exendin-4 in inhibiting apoptotic process and protecting mitochondrial functions. In addition, insulin resistance was largely alleviated and the expression of autophagy-related proteins was up-regulated in PD model rats after DA-CH5 treatment. These results in this study indicate DA-CH5 plays a therapeutic role in the 6-OHDA-unilaterally lesioned PD rat model and is superior to GLP-1 analogue exendin-4. The study was approved by the Animal Ethics Committee of Shanxi Medical University of China.
Chinese Library Classification No. R453; R741; Q421
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease in older individuals (after Alzheimer’s disease); there are currently no preventative or disease-modifying therapies available for this chronic, progressive disease (Habibi et al., 2011). Typical pathological characteristics of PD include impaired motor functions, such as bradykinesia, resting tremor, and rigidity; a progressive loss of dopaminergic neurons in the substantia nigra (SN); and the deposition of intraneuronal Lewy bodies (Poewe et al., 2017). The pathogenic mechanisms underlying PD have not yet been fully elucidated, but evidence suggests that various factors bring about the deterioration of vulnerable dopaminergic neurons in the SN, such as chronic inflammation, α-synuclein (α-syn) aggregation, mitochondrial dysfunction, and disruption of the homeostasis between autophagy and apoptosis (Michel et al., 2016). No available treatment can stop dopaminergic neuron degeneration or PD progression. Currently, the most common treatment is the dopamine precursor levodopa, which does not halt or reverse disease progression (De Deurwaerdère et al., 2017). A large number of potentially promising drug candidates have failed to show convincing effects in recent clinical trials (Athauda and Foltynie, 2016a; Athauda et al., 2017; Chatterjee and Kordower, 2019). Thus, the development of effective drugs remains a vital goal in PD research. Recently, it has been observed that people with type 2 diabetes mellitus (T2DM) have an increased risk of developing PD (Sandyk, 1993; Hu et al., 2007; Cereda et al., 2011; Santiago and Potashkin, 2013). Moreover, PD symptoms are enhanced by diabetes, and patients require higher doses of levodopa to reduce their symptoms (Cereda et al., 2012). In addition, non-diabetic PD patients show impaired glucose tolerance and have insulin levels within the pre-diabetes range (Santiago and Potashkin, 2015). In post-mortem brain tissue from PD patients, insulin signaling is impaired, and the phosphorylation levels of insulin receptors in the basal ganglia and SN are greatly increased (Moroo et al., 1994). These observations have prompted research into insulin re-sensitizing drugs, which may be of benefit to PD patients (Athauda and Foltynie, 2016b; Hölscher, 2018).
Glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) are members of the incretin hormone family, and are both growth factors. Incretin-based therapies, including GIP receptor agonists and GLP-1 receptor agonists, show promising curative effects on insulinotropic secretion and the amelioration of insulin resistance. GLP-1 receptor agonists are novel anti-diabetic drugs for the treatment of T2DM (Baggio and Drucker, 2007; Doyle and Egan, 2007). Furthermore, exendin-4 (Exenatide, Byetta, Bydureon), which was the first synthetic GLP-1 receptor agonist to be developed and licensed for the treatment of T2DM, showed therapeutic effects in preclinical tests in PD patients (Harkavyi et al., 2008; Li et al., 2009; Kim et al., 2017). Other GLP-1 receptor agonists currently on the market for the treatment of T2DM (e.g., liraglutide, lixisenatide, and dulaglutide) also show reliable neuroprotective effects in PD animal models (Liu et al., 2015; Li et al., 2016a; Zhang et al., 2019). An open-label pilot clinical trial testing exendin-4 in PD patients (NCT01174810) demonstrated impressive protective effects and reported that this treatment was able to arrest disease progression (Aviles-Olmos et al., 2013, 2014). These findings were confirmed by a subsequent phase II placebo-controlled clinical trial (Athauda et al., 2017). Furthermore, we have tested the ‘sister’ incretin hormone, GIP, in PD animal models. Protease-resistant analogues of GIP have promising neuroprotective effects that are comparable with those of GLP-1 analogues (Li et al., 2016b, 2017). Recently, novel unimolecular dual GLP-1/GIP receptor co-agonists have been developed (Finan et al., 2013). Based on these peptides, we have previously tested four new unimolecular dual GLP-1/GIP receptor co-agonists (DA1-JC, DA3-CH, DA4-JC, and DA-CH5) as novel treatments for neurodegenerative disorders. In our previous studies, we tested these peptides in animal models of PD induced by 1-methyl-4-phenyl-1,2,3,6-tetrahy-1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Kinemuchi et al., 1987; Gerlach et al., 1991) and 6-hydroxydopamine (6-OHDA) (Ma et al., 2014). It has been reported that the older dual agonist (DA1-JC) has neuroprotective properties in the 6-OHDA-lesioned rat model (Jalewa et al., 2017). We also revealed that two novel dual agonists (DA4-JC and DA-CH5) have improved neuroprotective effects compared with a single GLP-1 mimetic (liraglutide) or DA-JC1 in MPTP mouse models (Feng et al., 2018; Zhang et al., 2020). These dual agonist peptides contain motifs that enhance their uptake into the brain (Zhang et al., 2020). However, the MPTP model has limitations; for example, it does not develop neuronal loss in the substantial nigra (SN). It remains unknown whether DA-CH5 has similar protective properties against lesions induced by 6-OHDA in rats.
To answer this question, we evaluated the neuroprotective properties of exendin-4 and DA-CH5 in 6-OHDA-induced cell and rat PD models.
Materials and Methods
Drug preparation
Synthetic exendin-4 and DA-CH5 peptides were provided by GL Biochem (Shanghai) Ltd., Shanghai, China. The amino acid sequences of the exendin-4 and DA-CH5 were as follows: HGEGTFTSDLSKQMEEEAVRLFIEWLKNGGPSSGAPPPS and YXEGTFTSDYSIYLDKQAAXEFVNWLLAGGPSSGAPPPSKRRQRRKKRGY-NH2; X is aminoisobutyric acid. Exendin-4 and DA-CH5 were dissolved in ultrapure water to a concentration of 1 mg/mL and stored at –20°C.
Cell culture and intervention
The SH-SY5Y cell line was purchased from the Cell Bank of Chinese Academy of Sciences. Cells were maintained in Dulbecco’s modified Eagle’s medium/F12 (1:1; Boster Biotechnology, Wuhan, China) supplemented with 10% heat-inactivated fetal bovine serum (Cell Max, Beijing, China) and 1% penicillin/streptomycin (Solarbio, Beijing, China) in a 5% CO2humidified atmosphere at 37°C. The 6-OHDA (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in 0.02% ascorbic acid and was freshly prepared for each experiment. After 6 hours of serum starvation in serum-free Dulbecco’s modified Eagle’s medium/F12 (1:1), the cells were seeded in laminin-coated 96-well plates (Corning, Shanghai, China) at a density of 5 × 104cells/well for 24 hours. The cells were co-treated with different concentrations of 6-OHDA (0, 150, 250, 350, and 450 µM) for 24 hours in the presence or absence of 100 nM exendin-4 or DA-CH5.
Cell viability analysis
Cell viability was measured using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT; Solarbio) assay according to the manufacturer’s instructions. Briefly, after the intervention, cells were incubated with the MTT solution (0.5 mg/mL) for 3 hours, and were then washed with dimethyl sulfoxide to dissolve the formazan crystals. Cell viability was then measured using a plate reader (Spectra Max 190; Molecular Devices, San Jose, CA, USA) at 570 nm absorbance.
Intracellular reactive oxygen species measurement
Superoxide formation was determined using a reactive oxygen species (ROS) Detection Kit (KeyGEN Bio TECH, Nanjing, China) according to the manufacturer’s protocol. Briefly, after the intervention, cells were treated with the fluorescent probe 2′,7′-dichlorofluorescin diacetate (10 mM) for 30 minutes at 37°C. Cells were then collected and washed three times with phosphate-buffered saline (PBS). Next, intracellular ROS levels were detected by 2′,7′-dichlorofluorescein fluorescence (485 nm excitation, 520 nm emission) using a flow cytometer (BD FACSCalibur; BD Biosciences, Franklin Lakes, NJ, USA). The data were analyzed using CellQuestPro (BD Biosciences).
Animals and drug administration
Forty adult male Sprague-Dawley rats (aged 8 weeks, weighing 220–250 g) were purchased from Shanxi Medical University Experimental Animal Center. In a 12/12-hour light/dark cycle, four to six rats were housed per cage with free access to water and a standard rat diet. All studies were carried out in accordance with the Guidelines for the Care and Use of Laboratory Animals from the National Institutes of Health. The study was approved by the Animal Ethics Committee of Shanxi Medical University. The rats were randomly divided into four groups: sham, 6-OHDA, 6-OHDA + exendin-4, and 6-OHDA + DA-CH5 (n = 10).
Animals were anesthetized with ethyl carbamate solution (1000 mg/kg; Aladdin, Shanghai, China) by intraperitoneal injection, and received unilateral stereotactic injections of 6-OHDA (5 µL, 2 mg/mL) using a stereotaxic apparatus (RWD, Shenzhen, China). Thirty minutes prior to the stereotactic surgery, rats received pargyline (5 mg/kg; MedChemExpress, Monmouth Junction, NJ, USA) and desipramine (10 mg/kg; MedChemExpress) to prevent 6-OHDA-induced damage in noradrenergic terminals. Lesions were made in the right medial forebrain bundle (lateral = −1.5 mm, anterior = −2.2 mm, ventral = –8.0 mm) (Paxinos et al., 1980). The syringe was left in place for 10 minutes after 6-OHDA administration. The wound was cleaned and sutured, and the rats were carefully monitored post-surgery.
Rats in the 6-OHDA, 6-OHDA + exendin-4, and 6-OHDA + DA-CH5 groups received daily intraperitoneal injections of saline, exendin-4 (10 nmol/kg), or DA-CH5 (10 nmol/kg), respectively, for 30 days. The overview of the experimental design is shown in Figure 1A.
Figure 1.

Effects of exendin-4 and DA-CH5 on locomotor activity in the open-field test in 6-OHDA-induced PD rats.
(A) Schematic diagram of the animal experimental design. (B–D) Open-field test. Activity was recorded for 10 minutes. (B) Video tracking of locomotor activity patterns. (C) Heat maps of location distributions. (D) The overall distance covered was reduced by 6-OHDA lesions and normalized by exendin-4 or DA-CH5 treatment. Data are expressed as the mean ± SD (n = 8). ‡P < 0.05, ‡‡‡P < 0.001, vs. sham group; $P < 0.05, $$$P < 0.001, vs. 6-OHDA group (one-way analysis of variance followed by Tukey’s multiple comparison test). 6-OHDA: 6-Hydroxydopamine; exendin-4 and DA-CH5: glucagon-like peptide-1/glucose-dependent insulinotropic polypeptide dual receptor agonist; PD: Parkinson’s disease.
Open-field test
The open-field test is often used to measure locomotor activity (Peng et al., 2019). The test apparatus (Noldus Information Technology, Wageningen, Netherlands) consisted of a 75 cm × 75 cm square area surrounded by a wall that was 35 cm high. Each rat was placed into the arena and its locomotor behavior was observed for 10 minutes. The distance traveled and the tracking pattern were measured using a behavior tracking system (Noldus Information Technology, Wageningen, the Netherlands). After each assessment, the apparatus was thoroughly cleaned with 70% ethanol to remove any olfactory interference.
Apomorphine rotation test
The rotational response to apomorphine can be used to evaluate the extent of unilateral nigrostriatal denervation after a unilateral 6-OHDA lesion (Ma et al., 2014). Prior to the behavioral test, animals were habituated to the test room overnight. Rats were then placed into a square open field (as in the open-field test) and allowed to habituate for 10 minutes before the subcutaneous injection of apomorphine (0.05 mg/kg; Absin Bioscience Inc., Shanghai, China). Rats were monitored using a behavior tracking system for 30 minutes, and the numbers of completed ipsilateral rotations were recorded.
Tissue preparation
Rats were sacrificed and transcardially perfused with 4% paraformaldehyde. The brains were removed and fixed in 4% paraformaldehyde for 48 hours, and then transferred to 30% sucrose in PBS for cryoprotection. Next, the brains were snap frozen and cut into coronal sections (40 µm) using a freezing microtome (Leica, Wetzlar, Germany). The ipsilateral SN and striatum were quickly extracted from the brain and stored at −80°C.
Estimation of dopamine and tumor necrosis factor-α in the striatum
Striatal dopamine was estimated using a rat dopamine enzyme-linked immunosorbent assay (ELISA) kit (Cloud-Clone Corp., Wuhan, China). Similarly, tumor necrosis factor-α (TNF-α) was assayed using a commercial ELISA kit (Cloud-Clone Corp.). Both the procedures were performed as per the manufacturer’s instructions. In brief, striatal samples were added to the microtiter plate wells, coated with antibodies specific to dopamine or TNF-α, and incubated at 37°C for 2 hours. After removing the unbound substances from each well, biotin-conjugated antibodies were added to the wells and the plates were incubated at 37°C for 1 hour. After washing, the wells were incubated with avidin-conjugated horseradish peroxidase for 30 minutes at 37°C. Finally, the reaction was terminated by adding a stop solution. The results were quantified at 450 nm using an ELISA reader (ELx808; BioTek, Winooski, VT, USA).
Immunofluorescence staining
After washing with PBS, the SN samples were treated with 5% goat bovine serum albumin (Boster Biotechnology) for 30 minutes. Next, the sections were incubated with rabbit anti-tyrosine hydroxylase (TH) antibody (1:100; Cat# ab75875; Abcam, Cambridge, UK) or rabbit anti-glial fibrillary acidic protein (GFAP) antibody (1:100; Cat# PB0046; Boster Biotechnology) at 4°C overnight. After washing three times with PBS, the sections were treated with fluorescein isothiocyanate-conjugated goat anti-rabbit IgG (1:200; Cat# BA1105; Boster Biotechnology) for 2 hours in the dark at 37°C. Images were taken with a fluorescence microscopy (BX51; Olympus, Tokyo, Japan) and subsequently analyzed using Image J software (National Institutes of Health, Bethesda, MD, USA).
Western blot analysis
For protein extraction, the SN and striatal samples and the cells were lysed in radio-immunoprecipitation assay Lysis Buffer (Beyotime Biotechnology, Shanghai, China) with 1% phenylmethylsulfonyl fluoride (Beyotime Biotechnology) and 1% phosphatase inhibitor (Boster Biotechnology) for 1 hour. The rat SN was lysed using a homogenizer in the radio-immunoprecipitation assay lysis buffer. Before removing insoluble substances, the lysates were centrifuged at 12,000 r/min at 4°C for 20 minutes. A bicinchoninic acid (BCA) Protein Assay Kit (Boster Biotechnology) was then used to determine protein concentrations. Samples were separated on a sodium dodecyl sulphate-polyacrylamide gel (12%) and transferred onto polyethylene difluoride membranes (Boster Biotechnology). The membranes were then blocked with a bovine serum albumin/Tris-buffered saline blocking solution (Boster Biotechnology) at room temperature for 2 hours and incubated with primary antibody in a Tris-buffered saline/Tween solution at 4°C overnight. The primary antibodies were as follows: rabbit anti-TH antibody (1:500; Cat# ab75875; Abcam), rabbit anti-α-syn antibody (1:500; Cat# 2642; Cell Signaling Technology, Danvers, MA, USA), rabbit anti-interleukin-1β (IL-1β) antibody (1:500; Cat# BS6067; Bioworld Technology, St. Louis, MO, USA), rabbit anti-TNF-α antibody (1:500; Cat# BS6000; Bioworld Technology), rabbit anti-insulin receptor substrate 1 (IRS-1) antibody (1:500; Cat# BS3590; Bioworld Technology), rabbit anti-phospho-IRS-1ser312 antibody (1:500; Cat# BS4633; Bioworld Technology), rabbit anti-phospho-protein kinase B (Akt)ser473 antibody (1:500; Cat# 4060; Cell Signaling Technology), rabbit anti-pan-Akt antibody (1:500; Cat# ab8805; Abcam), rabbit anti-phospho-cAMP response element-binding protein (CREB)ser129 antibody (1:500; Cat# BS4781; Bioworld Technology), rabbit anti-pan-CREB antibody (1:500; Cat# BS9811M; Bioworld Technology), rabbit anti-proliferator-activated receptor-gamma coactivator 1α (PGC-1α) antibody (1:500; Cat# BS91062; Bioworld Technology), rabbit anti-nuclear respiratory factor 1 (NRF-1) antibody (1:500; Cat# BS7179; Bioworld Technology), rabbit anti-BCL-2 antibody (1:500; Cat# BS70205; Bioworld Technology), rabbit anti-BAX antibody (1:500; Cat# BS2538; Bioworld Technology), rabbit anti-phospho-BADser136 antibody (1:250; Cat# BS4021; Bioworld Technology), rabbit anti-phospho-BAD antibody (1:250; Cat# BS9832M; Bioworld Technology), rabbit anti-active caspase-3 antibody (1:500; Cat# ab2302; Abcam), rabbit anti-caspase-3 antibody (1:500; Cat# ab4051; Abcam), rabbit anti-Beclin-1 antibody (1:500; Cat# AP0768; Bioworld Technology), rabbit anti-sequestosome (SQSTM) 1 antibody (1:500; Cat# AP6006; Bioworld Technology), rabbit anti-β-actin antibody (1:2000; Cat# AP0714; Bioworld Technology), and rabbit anti-glyceraldehyde-3-phosphate dehydrogenase antibody (1:2000; Cat# AP0063; Bioworld Technology). After the membranes were incubated for 2 hours at room temperature with horseradish peroxidase-labeled goat anti-rabbit IgG (1:5000; Cat# BA1054; Boster Biotechnology), the band intensities were analyzed using a chemiluminescent imaging system (Sage Creation, Beijing, China). The protein levels were expressed as intensity relative to the control, and were normalized using β-actin or glyceraldehyde-3-phosphate dehydrogenase expression.
Statistical analysis
The data are presented as the mean ± standard deviation (SD). Data were analyzed using one-way analysis of variance followed by Tukey’s multiple comparison test, using GraphPad Prism 5.0 software (GraphPad Software, San Diego, CA, USA). A probability of P < 0.05 was considered statistically significant.
Results
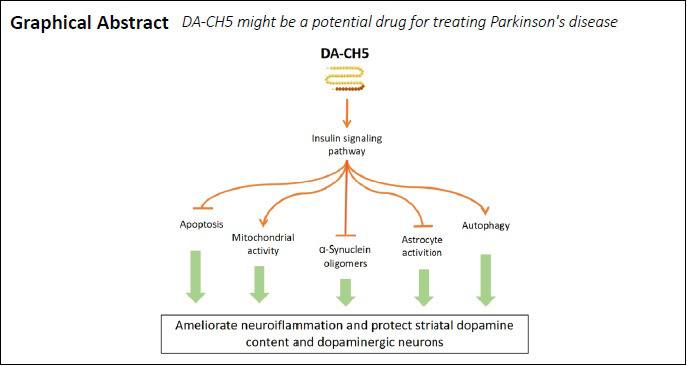
Incretin analogues increase the viability of 6-OHDA-treated SH-SY5Y cells
To examine the neuroprotective effects of exendin-4 and DA-CH5 on SH-SY5Y cells, we used the MTT assay to detect cell viability. Cell viability decreased in a dose-dependent manner with different doses of 6-OHDA for 24 hours. As shown in Figure 2A, 350 µM 6-OHDA induced neuronal death at a rate of approximately 50%. This concentration was used for subsequent experiments. Both exendin-4 and DA-CH5 at 100 nM markedly prevented 6-OHDA-induced neuronal loss (P < 0.05 or P < 0.001). Additionally, cell viability in the 6-OHDA + DA-CH5 group was higher than that in the 6-OHDA + exendin-4 group (P < 0.01; Figure 2B).
Figure 2.
Effects of exendin-4 and DA-CH5 on the viability of 6-OHDA-treated SH-SY5Y cells as detected by the MTT assay.
(A) 6-OHDA treatment induced neuronal death in a dose-dependent manner. (B) Cells were exposed to 350 µM 6-OHDA with or without 100 nM of exendin-4 or DA-CH5 for 24 hours. Exendin-4 and DA-CH5 prevented 6-OHDA-induced neuronal death. Data are expressed as the mean ± SD. The experiments were repeated four (A) or five (B) times. **P < 0.01, ***P < 0.001, vs. control group (CON); ###P < 0.001, vs. 150 µM 6-OHDA group; &&&P < 0.001, vs. 250 µM 6-OHDA group; †P < 0.05, †††P < 0.001, vs. 350 µM 6-OHDA group; §§P < 0.01, vs. 350 6-OHDA + exendin-4 group (one-way analysis of variance followed by Tukey’s multiple comparison test). 6-OHDA: 6-Hydroxydopamine; exendin-4 and DA-CH5: glucagon-like peptide-1/glucose-dependent insulinotropic polypeptide dual receptor agonist; MTT: 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide; PD: Parkinson’s disease.
Incretin analogues reduce intracellular ROS accumulation in 6-OHDA-treated SH-SY5Y cells
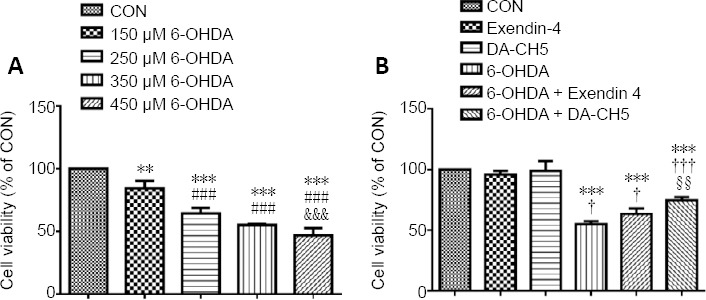
To determine the antioxidative properties of exendin-4 and DA-CH5 in 6-OHDA-mediated neurotoxicity in a neuronal cell line, we used flow cytometry to analyze intracellular ROS levels. 6-OHDA treatment significantly increased ROS levels in SH-SY5Y cells (P < 0.001, vs. control group), while co-treatment with exendin-4 or DA-CH5 significantly suppressed these changes (both P < 0.001, Figure 3). Furthermore, intracellular ROS levels in the 6-OHDA + DA-CH5 group were lower than those in the 6-OHDA + exendin-4 group (P < 0.05).
Figure 3.
Effects of exendin-4 and DA-CH5 on 6-OHDA-induced ROS production in SH-SY5Y cells.
Cells were exposed to 350 µM 6-OHDA with or without 100 nM of exendin-4 or DA-CH5 for 24 hours. (A) ROS generation in SH-SY5Y cells was detected using flow cytometry. (B) Quantitative results of intracellular ROS levels (fluorescence intensity of 2′,7′-dichlorofluorescin diacetate). Data are expressed as the mean ± SD of quadruplicate independent experiments. *P < 0.001, vs. control group (CON); †††P < 0.001, vs. 6-OHDA group; §P < 0.05, vs. 6-OHDA + exendin-4 group (one-way analysis of variance followed by Tukey’s multiple comparison test). 6-OHDA: 6-Hydroxydopamine; DCFH-DA: 2′,7′-dichlorofluorescin diacetate, a fluorescent probe; exendin-4 and DA-CH5: glucagon-like peptide-1/glucose-dependent insulinotropic polypeptide dual receptor agonist; PD: Parkinson’s disease; ROS: reactive oxygen species.
Incretin analogues reverse 6-OHDA-induced apoptosis-related protein expression
Compared with the control group, 6-OHDA markedly increased the expression of BAX (P < 0.001, vs. control group) and decreased the expression of BCL-2 (P < 0.01, vs. control group). Both exendin-4 (BAX, P < 0.01; BCL-2, P < 0.05; vs. 6-OHDA group) and DA-CH5 (BAX, P < 0.001; BCL-2, P < 0.01; vs. 6-OHDA group) reversed the abnormal expression of BAX and BCL-2; DA-CH5 also altered the expression of p-BADser136 (P < 0.01, vs. 6-OHDA group). Moreover, DA-CH5 treatment partially upregulated p-BADser136 phosphorylation compared with exendin-4 treatment (P < 0.05; Figure 4). These results indicate that both exendin-4 and DA-CH5 alleviate 6-OHDA-induced SH-SY5Y cell injury by inhibiting the expression of apoptosis-related proteins.
Figure 4.

Effects of exendin-4 and DA-CH5 on apoptosis-related protein expression in 6-OHDA-treated SH-SY5Y cells.
Cells were treated with 350 µM of 6-OHDA with or without 100 nM of exendin-4 or DA-CH5 for 24 hours. (A) Bands of apoptosis-related proteins. β-Actin was used as the loading control. (B–D) Quantitative results of Bax (B), Bcl-2 (C), and p-Badser136expression. All data are represented as the mean ± SD of quadruplicate independent experiments. **P < 0.01, ***P < 0.001, vs. control group (CON); †P < 0.05, ††P < 0.01, †††P < 0.001, vs. 6-OHDA group; §P < 0.05, vs. 6-OHDA + exendin-4 group (one-way analysis of variance followed by Tukey’s multiple comparison test). 6-OHDA: 6-Hydroxydopamine; exendin-4 and DA-CH5: glucagon-like peptide-1/glucose-dependent insulinotropic polypeptide dual receptor agonist; PD: Parkinson’s disease.
Incretin analogues ameliorate insulin signaling desensitization in 6-OHDA-treated SH-SY5Y cells
Incretin analogues target their receptors, which activate insulin signaling pathways, including IRS-1 and its downstream signaling molecules Akt and CREB (Athauda and Foltynie, 2018). There was a significant increase in p-IRS-1ser312 in the 6-OHDA group compared with the control group (P < 0.001). In contrast, p-IRS-1ser312 levels were slightly decreased in the 6-OHDA + exendin-4 and 6-OHDA + DA-CH5 groups compared with the 6-OHDA group (both P < 0.05; Figure 5A and B). Furthermore, p-Aktser473 expression in the 6-OHDA + exendin-4 and 6-OHDA + DA-CH5 groups was significantly increased compared with the 6-OHDA group (both P < 0.001). Moreover, p-Aktser473 phosphorylation in the 6-OHDA + DA-CH5 group was partially upregulated compared with the 6-OHDA + exendin-4 group (P < 0.05; Figure 5A and C). There was also a significant increase in p-CREBser129 expression in the 6-OHDA + exendin-4 and 6-OHDA + DA-CH5 groups compared with the 6-OHDA group (P < 0.01; Figure 5A and D). These results indicate that exendin-4 and DA-CH5 may alleviate 6-OHDA-induced insulin signaling pathway disturbances by regulating IRS-1, Akt, and CREB phosphorylation.
Figure 5.

Effects of exendin-4 and DA-CH5 on insulin desensitization-related protein expression in 6-OHDA-treated SH-SY5Y cells.
Cells were treated with 350 µM of 6-OHDA with or without 100 nM of exendin-4 or DA-CH5 for 24 hours. (A) Bands of insulin desensitization-related proteins. β-Actin was used as the loading control. (B–D) Quantitative results of p-IRS-1ser312 (B), p-Aktser473 (C), and p-CREBser129 (D) expression. All data are represented as the mean ± SD of quadruplicate independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, vs. control group (CON); †P < 0.05, ††P < 0.01, vs. 6-OHDA group; §P < 0.05, vs. 6-OHDA + exendin-4 group (one-way analysis of variance followed by Tukey’s multiple comparison test). 6-OHDA: 6-Hydroxydopamine; Akt: alpha serine/threonine-protein kinase; CREB: cAMP response element-binding protein; exendin-4 and DA-CH5: glucagon-like peptide-1/glucose-dependent insulinotropic polypeptide dual receptor agonist; IRS-1: insulin receptor substrate-1; PD: Parkinson’s disease.
Incretin analogues normalize mitogenesis-related protein expression in 6-OHDA-treated SH-SY5Y cells
As demonstrated in Figure 6, 6-OHDA treatment suppressed the protein expression of PCG-1α (P < 0.001, vs. control group) and NRF-1 (P < 0.001, vs. control group). This effect was partially attenuated by DA-CH5 treatment (PCG-1: P < 0.05; NRF-1: P < 0.001) in SH-SY5Y cells.
Figure 6.

Effects of exendin-4 and DA-CH5 on mitogenesis-related protein expression in 6-OHDA-treated SH-SY5Y cells.
Cells were treated with 350 µM of 6-OHDA with or without 100 nM of exendin-4 or DA-CH5 for 24 hours. (A) Bands of mitogenesis-related proteins. GAPDH was used as the loading control. (B, C) Quantitative results of PGC-1α (B) and NRF-1 (C) expression. All data are represented as the mean ± SD of quadruplicate independent experiments. *P < 0.001, vs. control group (CON); †P < 0.05, †††P < 0.001, vs. 6-OHDA group; §P < 0.05, vs. 6-OHDA + exendin-4 group (one-way analysis of variance followed by Tukey’s multiple comparison test). 6-OHDA: 6-Hydroxydopamine; exendin-4 and DA-CH5: glucagon-like peptide-1/glucose-dependent insulinotropic polypeptide dual receptor agonist; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; NRF-1: nuclear respiratory factor 1; PD: Parkinson’s disease; PGC-1α: proliferator-activated receptor-gamma coactivator 1α.
Incretin analogues promote autophagy-related protein expression in 6-OHDA-treated SH-SY5Y cells
Both exendin-4 and DA-CH5 upregulated the protein expression of SQSTM1 (both P < 0.01, vs. 6-OHDA group; Figure 7A and B). A similar result was observed in another autophagy-related protein; both exendin-4 and DA-CH5 enhanced Beclin-1 (P < 0.05 or P < 0.01, vs. 6-OHDA group; Figure 7A and C).
Figure 7.

Effects of exendin-4 and DA-CH5 on autophagy-related protein expression in 6-OHDA-treated SH-SY5Y cells.
Cells were treated with 350 µM of 6-OHDA with or without 100 nM of exendin-4 or DA-CH5 for 24 hours. (A) Bands of autophagy-related proteins. GAPDH was used as the loading control. (B, C) Quantitative results of SQSTM1 (B) and Beclin-1 (C). All data are represented as the mean ± SD of quadruplicate independent experiments. **P < 0.01, ***P < 0.001, vs. control group (CON); †P < 0.05, ††P < 0.01, vs. 6-OHDA group (one-way analysis of variance followed by Tukey’s multiple comparison test). 6-OHDA: 6-Hydroxydopamine; exendin-4 and DA-CH5: glucagon-like peptide-1/glucose-dependent insulinotropic polypeptide dual receptor agonist; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; PD: Parkinson’s disease; SQSTM1: sequestosome 1.
Incretin analogues improve cognitive function in a PD model rat
Open-field test
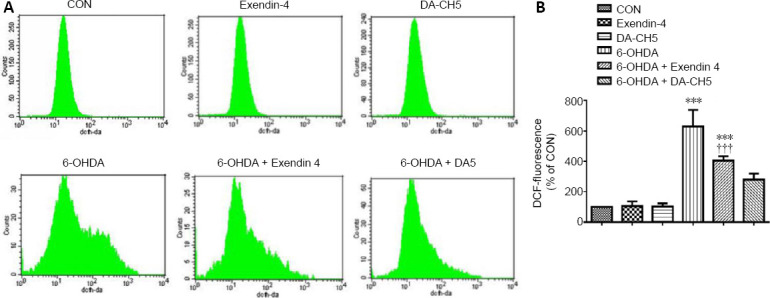
We explored the effects of incretin analogue treatment on locomotor activity in the open-field test in rats with 6-OHDA lesions (Figure 1B–D). The total distance traveled was significantly lower in the 6-OHDA group compared with the sham group (P < 0.001). Furthermore, exendin-4 and DA-CH5 treatment tended to ameliorate this deficit in locomotor behavior (P < 0.05 or P < 0.001, vs. 6-OHDA group).
Apomorphine rotational test
A greater number of ipsilateral rotations were produced by 6-OHDA compared with the sham group (P < 0.001). Both exendin-4 and DA-CH5 treatments were able to prominently reduce the number of rotations induced by 6-OHDA (both P < 0.001). Furthermore, the therapeutic effect of DA-CH5 was superior to that of exendin-4 (P < 0.05; Figure 8).
Figure 8.
Effects of exendin-4 and DA-CH5 on the apomorphine rotation test in 6-OHDA-induced PD rats.
Animals received apomorphine (0.05 mg/kg) via subcutaneous injection and activity was recorded for 30 minutes. (A) Video tracking of the apomorphine rotation test. (B) Quantitative results of contralateral turns in the apomorphine rotation test. Data are expressed as the mean ± SD (n = 10). ‡‡‡P < 0.001, vs. sham group; $$$P < 0.001, vs. 6-OHDA group; &P < 0.05, vs. 6-OHDA + exendin-4 group (one-way analysis of variance followed by Tukey’s multiple comparison test). 6-OHDA: 6-Hydroxydopamine; exendin-4 and DA-CH5: glucagon-like peptide-1/glucose-dependent insulinotropic polypeptide dual receptor agonist; PD: Parkinson’s disease.
Incretin analogues protect striatal dopamine content and dopaminergic neurons in the SN of rats against 6-OHDA induced neurotoxicity
Compared with sham rats, 6-OHDA-treated rats had a marked reduction in striatal dopamine levels (P < 0.01). However, only DA-CH5 treatment was able to partially recover the dopamine depletion compared with the 6-OHDA group (P < 0.05; Figure 9A). To further assess the presence of dopaminergic cell injury caused by 6-OHDA toxicity, immunofluorescence staining was performed to analyze the neuronal presence of TH, as an index of dopamine synthesis (Daubner et al., 2011). There was a reduction in TH immunopositivity in the ipsilateral SN of 6-OHDA-lesioned rats (P < 0.001, vs. sham group). However, there was increased TH immunopositivity in the 6-OHDA + exendin-4 and 6-OHDA + DA-CH5 groups compared with the 6-OHDA group (P < 0.01 or P < 0.001; Figure 9B and C), suggesting that incretin peptides are able to alleviate 6-OHDA-induced toxicity. Similar results were observed in the western blot assay; 6-OHDA-lesioned rats had decreased TH expression in the SN compared with the sham group (P < 0.001). Both exendin-4 and DA-CH5 treatment led to a small increase in TH expression (both P < 0.01, vs. 6-OHDA group; Figure 9D and E).
Figure 9.
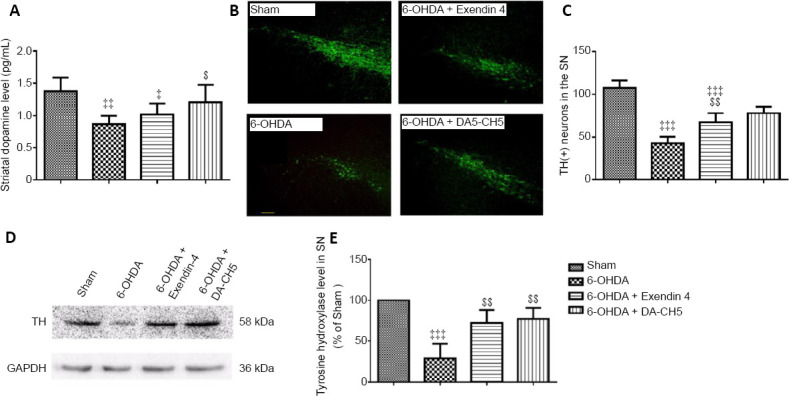
Effects of exendin-4 and DA-CH5 on striatal dopamine levels and TH expression in the SN of 6-OHDA-induced PD rats.
(A) Levels of dopamine in the striatum homogenate as quantified by enzyme-linked immunosorbent assay. (B) Representative fluorescence images of TH (green, stained by fluorescein isothiocyanate) in the SN. Scale bar: 100 µm. (C) Quantification of TH-positive cells in the SN. (D) Bands of TH expression in the SN. (E) Quantification of TH expression in the SN. Data are represented as the mean ± SD of six rats per group (A) or four rats per group (B–E). ‡P < 0.05, ‡‡P < 0.01, ‡‡‡P < 0.001, vs. sham group; $P < 0.05, $$P < 0.01, $$$P < 0.001, vs. 6-OHDA group (one-way analysis of variance followed by Tukey’s multiple comparison test). 6-OHDA: 6-Hydroxydopamine; exendin-4 and DA-CH5: glucagon-like peptide-1/glucose-dependent insulinotropic polypeptide dual receptor agonist; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; PD: Parkinson’s disease; SN: substantia nigra; TH: tyrosine hydroxylase.
Incretin analogues reduce 6-OHDA-induced α-syn expression in the rat SN
Results of western blot assays demonstrated an almost four-fold increase in α-syn expression in 6-OHDA rats compared with sham rats (P < 0.001). After treatment with exendin-4 or DA-CH5, α-syn expression was lower than in the 6-OHDA group (P < 0.05 or P < 0.01; Figure 10).
Figure 10.
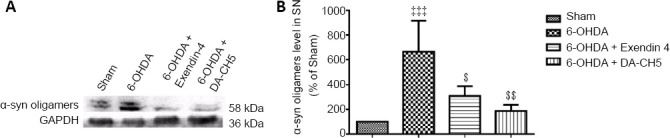
Effects of exendin-4 and DA-CH5 on α-syn expression in the SN of 6-OHDA-induced PD rats.
(A) Bands of α-syn expression in the SN. (B) Quantification of α-syn expression in the SN. Data are represented as the mean ± SD of four rats per group, and analyzed by one-way analysis of variance followed by Tukey’s multiple comparison test. ‡‡‡P < 0.001, vs. sham group; $P < 0.05, $$P < 0.01, vs. 6-OHDA group. 6-OHDA: 6-Hydroxydopamine; α-syn: α-synuclein; exendin-4 and DA-CH5: glucagon-like peptide-1/glucose-dependent insulinotropic polypeptide dual receptor agonist; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; PD: Parkinson’s disease; SN: substantia nigra.
Incretin analogues alleviate astrocyte activation and reduce 6-OHDA-induced increases in pro-inflammatory cytokines in the rat striatum
To investigate whether incretin analogue treatments were able to improve 6-OHDA-induced neuroinflammation, the activation of astrocytes and the expression of IL-1β and TNF-α were evaluated.
GFAP was used as a marker of astrocyte proliferation and activation (Siracusa et al., 2019). Quantitative analysis indicated that 6-OHDA markedly increased the number of GFAP-positive cells compared with the sham group (P < 0.001). Exendin-4 and DA-CH5 attenuated the number of GFAP-positive cells that were induced by 6-OHDA (P < 0.05 or P < 0.01, vs. 6-OHDA group; Figure 11A and B).
Figure 11.
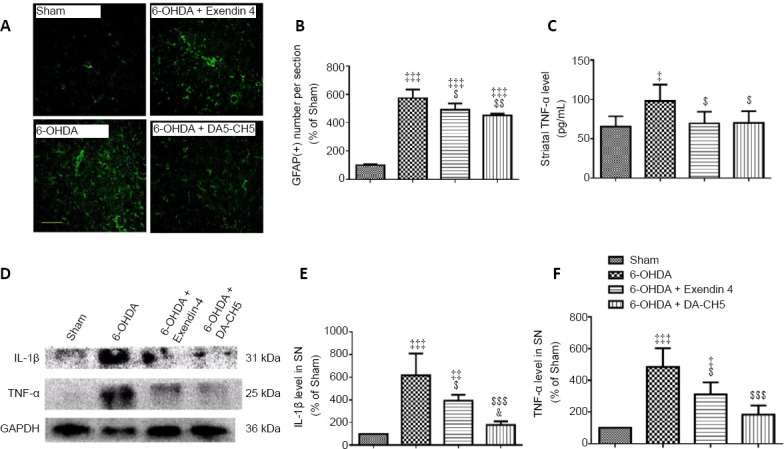
Effects of exendin-4 and DA-CH5 on 6-OHDA-induced alterations in astrocyte activation, striatal TNF-α levels, and pro-inflammatory cytokine levels in the rat striatum.
(A) Representative fluorescence images of GFAP (green, stained by fluorescein isothiocyanate) in the striatum. Scale bar: 100 µm. (B) Quantification of GFAP-positive cells in the striatum. (C) Levels of TNF-α in striatum homogenate as quantified by enzyme-linked immunosorbent assay. (D) Bands of IL-1β and TNF-α in the striatum. (E, F) Quantification of IL-1β (E) and TNF-α (F) expression in the striatum. Data are expressed as the mean ± SD of four rats per group (A–B and D–F) or six rats per group (C). ‡P < 0.05, ‡‡P < 0.01, ‡‡‡P < 0.001, vs. sham group; $P < 0.05, $$P < 0.01, $$$P < 0.001, vs. 6-OHDA group; &P < 0.05, vs. 6-OHDA + exendin-4 group (one-way analysis of variance followed by Tukey’s multiple comparison test). 6-OHDA: 6-Hydroxydopamine; exendin-4 and DA-CH5: glucagon-like peptide-1/glucose-dependent insulinotropic polypeptide dual receptor agonist; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GFAP: glial fibrillary acidic protein; IL-1β: interleukin-1β; PD: Parkinson’s disease; SN: substantia nigra; TNF-α: tumor necrosis factor-α.
ELISA results revealed that TNF-α levels in the striatum of the 6-OHDA group were significantly higher than those in the sham group (P < 0.05). Moreover, exendin-4 and DA-CH5 decreased the TNF-α levels in the striatum compared with the 6-OHDA group (both P < 0.05; Figure 11C).
The results of western blot assays revealed that 6-OHDA toxicity highly upregulated the expression of IL-1β and TNF-α compared with sham rats (both P < 0.001, vs. sham group). However, exendin-4 or DA-CH5 treatment significantly attenuated IL-1β (P < 0.05 or P < 0.001) and TNF-α (P < 0.05 or P < 0.001) expression compared with the 6-OHDA group. Furthermore, IL-1β expression in the 6-OHDA + DA-CH5 group was downregulated compared with the 6-OHDA + exendin-4 group (P < 0.05; Figure 11D–F).
Incretin analogues protect dopaminergic neurons in the rat SN against 6-OHDA-induced insulin resistance
To evaluate the insulin signaling pathway, phosphorylated IRS-1 was analyzed by western blot assay. 6-OHDA significantly elevated the phosphorylation levels of IRS-1 in the SN compared with the sham group (P < 0.001). Moreover, the protein expression of pIRS-1Ser129 was lower in the 6-OHDA + exendin-4 and 6-OHDA + DA-CH5 groups compared with the 6-OHDA group (P < 0.05 or P < 0.01; Figure 12A and B).
Figure 12.

Effects of exendin-4 and DA-CH5 on 6-OHDA-induced desensitization of the insulin signaling pathway in the rat SN.
(A) Bands of proteins in the insulin signaling pathway in SN lysates, as detected by western blot assay. GAPDH was used as the loading control. (B–D) Quantification of p-IRS-1 (B), PGC-1α (C), and NRF-1 (D) expression in the SN. Data are represented as the mean ± SD of four rats per group. ‡‡‡P < 0.001, vs. sham group; $P < 0.05, $$P < 0.01, $$$P < 0.001, vs. 6-OHDA group (one-way analysis of variance followed by Tukey’s multiple comparison test). 6-OHDA: 6-Hydroxydopamine; exendin-4 and DA-CH5: glucagon-like peptide-1/glucose-dependent insulinotropic polypeptide dual receptor agonist; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; IRS-1: insulin receptor substrate-1; NRF-1: nuclear respiratory factor 1; PD: Parkinson’s disease; PGC-1α: proliferator-activated receptor-gamma coactivator 1α; SN: substantia nigra.
Activation of the PCG-1α and NRF-1 pathway is recognized as a key element that confers resistance to mitochondrial stress (Cheng et al., 2010). Compared with the sham group, PCG-1α and NRF-1 expression levels were decreased in the 6-OHDA group (both P < 0.001). However, PCG-1α and NRF-1 expression levels were increased after exendin-4 (PCG-1α, P < 0.05 and NRF-1, P < 0.001; vs. 6-OHDA group) or DA-CH5 (PCG-1α, P < 0.05 and NRF-1, P < 0.01; vs. 6-OHDA group) treatment (Figure 12A, C, and D).
Incretin analogues affect apoptosis-related protein expression in dopaminergic neurons of the SN in 6-OHDA-treated rats
To determine the specificity of the neuroprotective effects against apoptosis, apoptosis-related proteins were investigated using western blot assays. Compared with the sham group, the activated-caspase-3/caspase-3 ratio was elevated under 6-OHDA stress (P < 0.001, vs. sham group), while exendin-4 or DA-CH5 counteracted the effects of 6-OHDA (P < 0.01 or P < 0.001; Figure 13A and B). In the SN of the 6-OHDA group, there was an increase in BAX expression and a reduction in BCL-2 expression (both P < 0.001, vs. sham group), while these abnormal protein expressions were partially improved in the 6-OHDA + exendin-4 (P < 0.01 or P > 0.05, vs. 6-OHDA group) and 6-OHDA + DA-CH5 (both P < 0.001, vs. 6-OHDA group) groups. In addition, the anti-apoptotic effects (as measured by BCL-2 expression) of DA-CH5 were superior to those of exendin-4 (P < 0.05; Figure 13).
Figure 13.

Effects of exendin-4 and DA-CH5 on 6-OHDA-induced apoptosis signaling-related protein expression in the rat SN.
(A) Bands of apoptosis signaling-related proteins in SN lysates as detected by western blot assay. β-Actin was used as the loading control. (B–D) Quantification of activated caspase-3 (a-caspase-3), BCL-2, and BAX expression in SN lysates. Data are represented as the mean ± SD of four rats per group. ‡P < 0.05, ‡‡‡P < 0.001, vs. sham group; $$P < 0.01, $$$P < 0.001, vs. 6-OHDA group; &P < 0.05, vs. 6-OHDA + exendin-4 group (one-way analysis of variance followed by Tukey’s multiple comparison test). 6-OHDA: 6-Hydroxydopamine; ACTIN: β-actin; exendin-4 and DA-CH5: glucagon-like peptide-1/glucose-dependent insulinotropic polypeptide dual receptor agonist; PD: Parkinson’s disease; SN: substantia nigra.
Incretin analogues protect dopaminergic neurons in the SN of rats by normalizing autophagy
Protein markers of autophagy, such as SQSTM1 and Beclin-1, were determined by western blot assay. Treatment with either exendin-4 or DA-CH5 enhanced Beclin-1 expression compared with the 6-OHDA group (P < 0.05 or P < 0.01). However, only DA-CH5 treatment promoted SQSTM1 expression compared with the 6-OHDA group (P < 0.01). Furthermore, SQSTM1 expression was higher in the 6-OHDA + DA-CH5 group than in the 6-OHDA + exendin-4 group (P < 0.01; Figure 14).
Figure 14.

Effects of exendin-4 and DA-CH5 on 6-OHDA-induced reductions in autophagy-related protein expression in the rat SN.
(A) Bands of autophagy-related protein expression in SN lysates as detected by western blot assay. GAPDH was used as the loading control. SQSTM1 (B) and Beclin-1 (C) expression in SN lysates. Data are shown as the mean ± SD of four rats per group, and were analyzed by one-way analysis of variance followed by Tukey’s multiple comparison test. ‡P < 0.05, ‡‡P < 0.01, ‡‡‡P < 0.001, vs. sham group; $P < 0.05, $$P < 0.01, vs. 6-OHDA group; &&P <0.01, vs. 6-OHDA + exendin-4 group (one-way analysis of variance followed by Tukey’s multiple comparison test). 6-OHDA: 6-Hydroxydopamine; exendin-4 and DA-CH5: glucagon-like peptide-1/glucose-dependent insulinotropic polypeptide dual receptor agonist; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; PD: Parkinson’s disease; SN: substantia nigra; SQSTM1: sequestosome 1.
Discussion
T2DM is considered a risk factor for neurodegenerative diseases, and accumulating evidence suggests that the desensitization of insulin signaling is a key underlying pathogenic mechanism of PD (Freiherr et al., 2013; Athauda and Foltynie, 2016b; Hölscher, 2018). Initially, the activity of incretin hormones for the treatment of T2DM was thought to involve the stimulation of pancreatic β-cells to release insulin (Baggio and Drucker, 2007). However, the results of the present and previous studies have demonstrated that these hormones also have potent neuroprotective and neurorestorative properties, making them potential therapies for neurodegenerative disorders (Perry and Greig, 2002; Hölscher, 2018). Importantly, GLP-1 and GIP can re-sensitize insulin signaling in the brain (Bomfim et al., 2012; Long-Smith et al., 2013; Shi et al., 2017). In the current study, both exendin-4 and DA-CH5 had neuroprotective effects in the rat brain, supporting the hypothesis that incretin receptor activation can protect against 6-OHDA neurotoxicity. The unimolecular dual GLP-1/GIP receptor co-agonist DA-CH5 was more effective than the GLP-1 analogue exendin-4. These results support our previous findings, which demonstrated the clear neuroprotective effects of DA-CH5 in an MPTP mouse model of PD (Feng et al., 2018; Zhang et al., 2020).
Various motor symptoms in PD can be attributed to both progressive neuronal loss in specific areas of the SN and dopamine depletion in the striatum (Poewe et al., 2017). The use of 6-OHDA to cause a unilateral lesion in the nigrostriatal pathway of rats, thereby inducing dopaminergic deficiency and unilateral motor deficits, is a commonly used animal model that mimics pathological features of PD (Blandini, 2013; Ma et al., 2014). Consistent with our previous report (Jalewa et al., 2017), we revealed that 4 weeks after 6-OHDA injection into the medial forebrain bundle, animals exhibited ipsilateral rotations after an apomorphine challenge. This finding implies the deterioration of dopamine transmission in the lesioned striatum. Furthermore, rats treated with 6-OHDA had marked reductions in striatal dopamine levels and a loss of dopaminergic neurons in the SN. Thus, the observed neuroprotective effects of exendin-4 and DA-CH5 included the partial alleviation of motor impairments and the reversal of striatal dopamine depletion. Moreover, DA-CH5 was more effective than exendin-4 in restoring dopamine levels in the brain.
Widespread intracellular misfolded α-syn (in Lewy bodies) is another characteristic feature of some forms of PD (Bengoa-Vergniory et al., 2017). The fibrillation process of α-syn includes two stages (Ghosh et al., 2017): (1) the native unfolded monomeric α-syn proteins convert to β-sheet pre-fibrils/oligomers under pathological conditions; and (2) the progressive elongation and aggregation of pre-formed α-syn fibrils can lead to the accumulation of amyloid-like structures, such as Lewy bodies found in fronto-temporal dementia. Some studies appear to show that soluble oligomers and prefibrillar oligomers are the main culprits of neuronal degeneration (Danzer et al., 2007; Winner et al., 2011). Although the underlying pathological mechanism underlying the toxicity of α-syn oligomers has not been demonstrated in PD, it has been proposed that α-syn oligomer-induced neurodegeneration may involve a variety of intracellular pathophysiological activities, such as mitochondrial dysfunction, oxidative stress, neuroinflammation, and autophagy impairment (Bengoa-Vergniory et al., 2017). In the current study, α-syn oligomer levels were significantly reduced after exendin-4 and DA-CH5 treatment.
A wide range of factors can activate glial cells and innate immunity in the central nervous system, which leads to the production of ROS, pro-inflammatory cytokines, and subsequent neuronal damage (Caggiu et al., 2019). Post-mortem studies have found activated microglial and astroglial cells within the SN. In addition, higher concentrations of cytokines, such as IL-1β and TNF-α, have been found in the striatum and cerebrospinal fluid of patients with PD (Hirsch and Hunot, 2009). The aforementioned studies suggest that progressive inflammation is a pathological element, indicating that the inhibition of glial cell overactivation may be a promising treatment strategy for PD. Our data demonstrate that both exendin-4 and DA-CH5 treatment can attenuate 6-OHDA-induced astrogliosis, and can also suppress IL-1β and TNF-α expression in the striatum and SN under 6-OHDA stress; again, DA-CH5 was more effective in the present study. These findings indicate that exendin-4 and DA-CH5 may achieve neuroprotection by suppressing the inflammatory response.
In the brain, insulin signaling significantly impacts on neuronal growth and repair, synaptogenesis, energy utilization, cognitive performance, and memory formation (Ghasemi et al., 2013; Hölscher, 2014). When insulin binds to its receptors, IRS-1 mediates the activation of downstream intracellular secondary messenger cascade pathways to transmit the insulin signal (Adamo et al., 1989). A number of studies have analyzed how insulin resistance might influence neurodegeneration, including in Alzheimer’s disease and PD (Steen et al., 2005; Freiherr et al., 2013; Talbot and Wang, 2014). A key driving force of insulin desensitization is the effect of pro-inflammatory cytokines (Blandini, 2013; Clark and Vissel, 2014). Through activation of the IRS-1/Akt/CREB pathway, insulin can modulate mitochondrial activity by mediating the expression of PCG-1α/NRF-1, which are important promoters that enhance mitogenesis (Patti et al., 2003). Furthermore, reduced PCG-1α and NRF-1 expression promotes mitochondrial dysfunction and plays a critical role in the pathogenesis of PD (Corona and Duchen, 2015). Our previously study demonstrated that another GLP-1 analogue, liraglutide, and the unimolecular GLP-1/GIP receptor co-agonists DA4-JC and DA-CH5 are able to partly reverse insulin desensitization in Alzheimer’s disease animal models (Long-Smith et al., 2013; Shi et al., 2017; Li et al., 2020). In the present study, our results also emphasize the potent neuroprotective actions of exendin-4 and DA-CH5 for restoring insulin sensitization and attenuating mitochondrial dysfunction in this PD rat model.
Mitochondrial homeostasis plays a crucial role for activating both autophagy and apoptosis. Mitochondrial dysfunction is triggered by internal stress, and results in cytochrome c release and apoptotic cell death (Fuchs and Steller, 2011). The mitochondrial outer membrane recruits autophagy-related proteins to initialize the biogenesis of autophagosomes. Mitochondria additionally regulate autophagy by interacting with autophagy initiation complexes and triggering autophagy-mediated cell death by cytochrome c release (Betin and Lane, 2009; Hailey et al., 2010; Takahashi et al., 2011). Furthermore, autophagy and apoptosis share common signaling pathways and exhibit a dynamic balance (Maiuri et al., 2007). For example, Beclin-1 inhibits apoptosis by interacting with BCL-2 family proteins to form Beclin-1–BCL-2/BCLxL complexes (Wei et al., 2008). Furthermore, caspase-mediated cleavage of Beclin-1 generates C- and N-terminal fragments that lose their ability to induce autophagy (Djavaheri-Mergny et al., 2010). The enhancement of autophagy and the inhibition of apoptosis may be novel therapeutic targets for the treatment of PD (Ghavami et al., 2014). Here, we present direct evidence that the protective effects of exendin-4 and DA-CH5 involve the inhibition of apoptosis and the induction of autophagy by reducing the active-Caspase-3/Caspase-3 and BAX/BCL-2 ratios and enhancing autophagic flux. Again, DA-CH5 was superior to exendin-4 in these effects.
In conclusion, we have demonstrated some neuroprotective mechanisms for both exendin-4 and DA-CH5 against 6-OHDA-induced neurotoxicity in vivo and in vitro. The beneficial effects of these two incretin peptides appear to be associated with the inhibition of astrocyte activation and α-syn oligomer formation, the attenuation of desensitized insulin signaling, and the restoration of the imbalance between autophagy and apoptosis. Additionally, our findings suggest that DA-CH5 has advantages in controlling inflammatory cytokine levels, reducing α-syn aggregation, and regulating autophagy and apoptosis homeostasis in the brain. The results of this study suggest that DA-CH5 might be more effective than exendin-4 in treating PD.
Additional files:
Additional file 1: Open peer review report 1 (87KB, pdf) .
Footnotes
P-Reviewer: Goh ELK; C-Editor: Zhao M; S-Editors: Yu J, Li CH; L-Editors: Yu J, Song LP; T-Editor: Jia Y
Funding: The study was supported by the Doctoral Start-Up Foundation of Shanxi Province of China, No. SD1817 (to QQJ).
Conflicts of interest: CH is a named inventor on patent applications that cover the use of dual GLP-1/GIP receptor agonists as treatments of PD.
Financial support: The study was supported by the Doctoral Start-Up Foundation of Shanxi Province of China, No. SD1817 (to QQJ). The funder had no roles in the study design, conduction of experiment, data collection and analysis, decision to publish, or preparation of the manuscript.
Institutional review board statement: The study was approved by the Animal Ethics Committee of Shanxi Medical University.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Eyleen L.K. Goh, Duke-NUS Graduate Medical School, Singapore.
References
- 1.Adamo M, Raizada MK, LeRoith D. Insulin and insulin-like growth factor receptors in the nervous system. Mol Neurobiol. 1989;3:71–100. doi: 10.1007/BF02935589. [DOI] [PubMed] [Google Scholar]
- 2.Athauda D, Foltynie T. The glucagon-like peptide 1 (GLP) receptor as a therapeutic target in Parkinson’s disease: mechanisms of action. Drug Discov Today. 2016a;21:802–818. doi: 10.1016/j.drudis.2016.01.013. [DOI] [PubMed] [Google Scholar]
- 3.Athauda D, Foltynie T. Insulin resistance and Parkinson’s disease: A new target for disease modification. Prog Neurobiol. 2016b;145-146:98–120. doi: 10.1016/j.pneurobio.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Athauda D, Foltynie T. Protective effects of the GLP-1 mimetic exendin-4 in Parkinson’s disease. Neuropharmacology. 2018;136:260–270. doi: 10.1016/j.neuropharm.2017.09.023. [DOI] [PubMed] [Google Scholar]
- 5.Athauda D, Maclagan K, Skene SS, Bajwa-Joseph M, Letchford D, Chowdhury K, Hibbert S, Budnik N, Zampedri L, Dickson J, Li Y, Aviles-Olmos I, Warner TT, Limousin P, Lees AJ, Greig NH, Tebbs S, Foltynie T. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390:1664–1675. doi: 10.1016/S0140-6736(17)31585-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Ell P, Soderlund T, Whitton P, Wyse R, Isaacs T, Lees A, Limousin P, Foltynie T. Exenatide and the treatment of patients with Parkinson’s disease. J Clin Invest. 2013;123:2730–2736. doi: 10.1172/JCI68295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Kahan J, Ell P, Whitton P, Wyse R, Isaacs T, Lees A, Limousin P, Foltynie T. Motor and cognitive advantages persist 12 months after exenatide exposure in Parkinson’s disease. J Parkinsons Dis. 2014;4:337–344. doi: 10.3233/JPD-140364. [DOI] [PubMed] [Google Scholar]
- 8.Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–2157. doi: 10.1053/j.gastro.2007.03.054. [DOI] [PubMed] [Google Scholar]
- 9.Bengoa-Vergniory N, Roberts RF, Wade-Martins R, Alegre-Abarrategui J. Alpha-synuclein oligomers: a new hope. Acta Neuropathol. 2017;134:819–838. doi: 10.1007/s00401-017-1755-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Betin VM, Lane JD. Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J Cell Sci. 2009;122:2554–2566. doi: 10.1242/jcs.046250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blandini F. Neural and immune mechanisms in the pathogenesis of Parkinson’s disease. J Neuroimmune Pharmacol. 2013;8:189–201. doi: 10.1007/s11481-013-9435-y. [DOI] [PubMed] [Google Scholar]
- 12.Bomfim TR, Forny-Germano L, Sathler LB, Brito-Moreira J, Houzel JC, Decker H, Silverman MA, Kazi H, Melo HM, McClean PL, Holscher C, Arnold SE, Talbot K, Klein WL, Munoz DP, Ferreira ST, De Felice FG. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Aβ oligomers. J Clin Invest. 2012;122:1339–1353. doi: 10.1172/JCI57256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caggiu E, Arru G, Hosseini S, Niegowska M, Sechi G, Zarbo IR, Sechi LA. Inflammation, infectious triggers, and Parkinson’s disease. Front Neurol. 2019;10:122. doi: 10.3389/fneur.2019.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cereda E, Barichella M, Cassani E, Caccialanza R, Pezzoli G. Clinical features of Parkinson disease when onset of diabetes came first: A case-control study. Neurology. 2012;78:1507–1511. doi: 10.1212/WNL.0b013e3182553cc9. [DOI] [PubMed] [Google Scholar]
- 15.Cereda E, Barichella M, Pedrolli C, Klersy C, Cassani E, Caccialanza R, Pezzoli G. Diabetes and risk of Parkinson’s disease: a systematic review and meta-analysis. Diabetes Care. 2011;34:2614–2623. doi: 10.2337/dc11-1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chatterjee D, Kordower JH. Immunotherapy in Parkinson’s disease: Current status and future directions. Neurobiol Dis. 2019;132:104587. doi: 10.1016/j.nbd.2019.104587. [DOI] [PubMed] [Google Scholar]
- 17.Cheng Z, Tseng Y, White MF. Insulin signaling meets mitochondria in metabolism. Trends Endocrinol Metab. 2010;21:589–598. doi: 10.1016/j.tem.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clark IA, Vissel B. Inflammation-sleep interface in brain disease: TNF, insulin, orexin. J Neuroinflammation. 2014;11:51. doi: 10.1186/1742-2094-11-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corona JC, Duchen MR. PPARγ and PGC-1α as therapeutic targets in Parkinson’s. Neurochem Res. 2015;40:308–316. doi: 10.1007/s11064-014-1377-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, Kretzschmar H, Hengerer B, Kostka M. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J Neurosci. 2007;27:9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daubner SC, Le T, Wang S. Tyrosine hydroxylase and regulation of dopamine synthesis. Arch Biochem Biophys. 2011;508:1–12. doi: 10.1016/j.abb.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Deurwaerdère P, Di Giovanni G, Millan MJ. Expanding the repertoire of L-DOPA’s actions: A comprehensive review of its functional neurochemistry. Prog Neurobiol. 2017;151:57–100. doi: 10.1016/j.pneurobio.2016.07.002. [DOI] [PubMed] [Google Scholar]
- 23.Djavaheri-Mergny M, Maiuri MC, Kroemer G. Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene. 2010;29:1717–1719. doi: 10.1038/onc.2009.519. [DOI] [PubMed] [Google Scholar]
- 24.Doyle ME, Egan JM. Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacol Ther. 2007;113:546–593. doi: 10.1016/j.pharmthera.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng P, Zhang X, Li D, Ji C, Yuan Z, Wang R, Xue G, Li G, Hölscher C. Two novel dual GLP-1/GIP receptor agonists are neuroprotective in the MPTP mouse model of Parkinson’s disease. Neuropharmacology. 2018;133:385–394. doi: 10.1016/j.neuropharm.2018.02.012. [DOI] [PubMed] [Google Scholar]
- 26.Finan B, Ma T, Ottaway N, Müller TD, Habegger KM, Heppner KM, Kirchner H, Holland J, Hembree J, Raver C, Lockie SH, Smiley DL, Gelfanov V, Yang B, Hofmann S, Bruemmer D, Drucker DJ, Pfluger PT, Perez-Tilve D, Gidda J, et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci Transl Med. 2013;5:209ra151. doi: 10.1126/scitranslmed.3007218. [DOI] [PubMed] [Google Scholar]
- 27.Freiherr J, Hallschmid M, Frey WH, 2nd, Brünner YF, Chapman CD, Hölscher C, Craft S, De Felice FG, Benedict C. Intranasal insulin as a treatment for Alzheimer’s disease: a review of basic research and clinical evidence. CNS Drugs. 2013;27:505–514. doi: 10.1007/s40263-013-0076-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742–758. doi: 10.1016/j.cell.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerlach M, Riederer P, Przuntek H, Youdim MB. MPTP mechanisms of neurotoxicity and their implications for Parkinson’s disease. Eur J Pharmacol. 1991;208:273–286. doi: 10.1016/0922-4106(91)90073-q. [DOI] [PubMed] [Google Scholar]
- 30.Ghasemi R, Dargahi L, Haeri A, Moosavi M, Mohamed Z, Ahmadiani A. Brain insulin dysregulation: implication for neurological and neuropsychiatric disorders. Mol Neurobiol. 2013;47:1045–1065. doi: 10.1007/s12035-013-8404-z. [DOI] [PubMed] [Google Scholar]
- 31.Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, Christoffersson J, Chaabane W, Moghadam AR, Kashani HH, Hashemi M, Owji AA, Łos MJ. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014;112:24–49. doi: 10.1016/j.pneurobio.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 32.Ghosh D, Mehra S, Sahay S, Singh PK, Maji SK. α-synuclein aggregation and its modulation. Int J Biol Macromol. 2017;100:37–54. doi: 10.1016/j.ijbiomac.2016.10.021. [DOI] [PubMed] [Google Scholar]
- 33.Habibi E, Masoudi-Nejad A, Abdolmaleky HM, Haggarty SJ. Emerging roles of epigenetic mechanisms in Parkinson’s disease. Funct Integr Genomics. 2011;11:523–537. doi: 10.1007/s10142-011-0246-z. [DOI] [PubMed] [Google Scholar]
- 34.Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harkavyi A, Abuirmeileh A, Lever R, Kingsbury AE, Biggs CS, Whitton PS. Glucagon-like peptide 1 receptor stimulation reverses key deficits in distinct rodent models of Parkinson’s disease. J Neuroinflammation. 2008;5:19. doi: 10.1186/1742-2094-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection. Lancet Neurol. 2009;8:382–397. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- 37.Hölscher C. Insulin, incretins and other growth factors as potential novel treatments for Alzheimer’s and Parkinson’s diseases. Biochem Soc Trans. 2014;42:593–599. doi: 10.1042/BST20140016. [DOI] [PubMed] [Google Scholar]
- 38.Hölscher C. Novel dual GLP-1/GIP receptor agonists show neuroprotective effects in Alzheimer’s and Parkinson’s disease models. Neuropharmacology. 2018;136:251–259. doi: 10.1016/j.neuropharm.2018.01.040. [DOI] [PubMed] [Google Scholar]
- 39.Hu G, Jousilahti P, Bidel S, Antikainen R, Tuomilehto J. Type 2 diabetes and the risk of Parkinson’s disease. Diabetes Care. 2007;30:842–847. doi: 10.2337/dc06-2011. [DOI] [PubMed] [Google Scholar]
- 40.Jalewa J, Sharma MK, Gengler S, Hölscher C. A novel GLP-1/GIP dual receptor agonist protects from 6-OHDA lesion in a rat model of Parkinson’s disease. Neuropharmacology. 2017;117:238–248. doi: 10.1016/j.neuropharm.2017.02.013. [DOI] [PubMed] [Google Scholar]
- 41.Kim IS, Koppula S, Park SY, Choi DK. Analysis of epidermal growth factor receptor related gene expression changes in a cellular and animal model of Parkinson’s disease. Int J Mol Sci. 2017;18:430. doi: 10.3390/ijms18020430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kinemuchi H, Fowler CJ, Tipton KF. The neurotoxicity of 1-methyl-4-phenyl-1, 2, 3, 6, -tetrahydropyridine (mptp) and its relevance to parkinson’s disease. Neurochem Int. 1987;11:359–373. doi: 10.1016/0197-0186(87)90024-6. [DOI] [PubMed] [Google Scholar]
- 43.Li C, Liu W, Li X, Zhang Z, Qi H, Liu S, Yan N, Xing Y, Hölscher C, Wang Z. The novel GLP-1/GIP analogue DA5-CH reduces tau phosphorylation and normalizes theta rhythm in the icv. STZ rat model of AD. Brain Behav. 2020;10:e01505. doi: 10.1002/brb3.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li L, Liu K, Zhao J, Holscher C, Li GL, Liu YZ. Neuroprotective role of (Val(8))GLP-1-Glu-PAL in an in vitro model of Parkinson’s disease. Neural Regen Res. 2016a;11:326–331. doi: 10.4103/1673-5374.177742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Y, Liu W, Li L, Hölscher C. Neuroprotective effects of a GIP analogue in the MPTP Parkinson’s disease mouse model. Neuropharmacology. 2016b;101:255–263. doi: 10.1016/j.neuropharm.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 46.Li Y, Liu W, Li L, Hölscher C. D-Ala2-GIP-glu-PAL is neuroprotective in a chronic Parkinson’s disease mouse model and increases BNDF expression while reducing neuroinflammation and lipid peroxidation. Eur J Pharmacol. 2017;797:162–172. doi: 10.1016/j.ejphar.2016.11.050. [DOI] [PubMed] [Google Scholar]
- 47.Li Y, Perry T, Kindy MS, Harvey BK, Tweedie D, Holloway HW, Powers K, Shen H, Egan JM, Sambamurti K, Brossi A, Lahiri DK, Mattson MP, Hoffer BJ, Wang Y, Greig NH. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc Natl Acad Sci U S A. 2009;106:1285–1290. doi: 10.1073/pnas.0806720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu W, Jalewa J, Sharma M, Li G, Li L, Hölscher C. Neuroprotective effects of lixisenatide and liraglutide in the 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine mouse model of Parkinson’s disease. Neuroscience. 2015;303:42–50. doi: 10.1016/j.neuroscience.2015.06.054. [DOI] [PubMed] [Google Scholar]
- 49.Long-Smith CM, Manning S, McClean PL, Coakley MF, O’Halloran DJ, Holscher C, O’Neill C. The diabetes drug liraglutide ameliorates aberrant insulin receptor localisation and signalling in parallel with decreasing both amyloid-β plaque and glial pathology in a mouse model of Alzheimer’s disease. Neuromolecular Med. 2013;15:102–114. doi: 10.1007/s12017-012-8199-5. [DOI] [PubMed] [Google Scholar]
- 50.Ma Y, Zhan M, OuYang L, Li Y, Chen S, Wu J, Chen J, Luo C, Lei W. The effects of unilateral 6-OHDA lesion in medial forebrain bundle on the motor, cognitive dysfunctions and vulnerability of different striatal interneuron types in rats. Behav Brain Res. 2014;266:37–45. doi: 10.1016/j.bbr.2014.02.039. [DOI] [PubMed] [Google Scholar]
- 51.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 52.Michel PP, Hirsch EC, Hunot S. Understanding dopaminergic cell death pathways in Parkinson disease. Neuron. 2016;90:675–691. doi: 10.1016/j.neuron.2016.03.038. [DOI] [PubMed] [Google Scholar]
- 53.Moroo I, Yamada T, Makino H, Tooyama I, McGeer PL, McGeer EG, Hirayama K. Loss of insulin receptor immunoreactivity from the substantia nigra pars compacta neurons in Parkinson’s disease. Acta Neuropathol. 1994;87:343–348. doi: 10.1007/BF00313602. [DOI] [PubMed] [Google Scholar]
- 54.Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paxinos G, Watson CR, Emson PC. AChE-stained horizontal sections of the rat brain in stereotaxic coordinates. J Neurosci Methods. 1980;3:129–149. doi: 10.1016/0165-0270(80)90021-7. [DOI] [PubMed] [Google Scholar]
- 56.Peng Q, Zhong S, Tan Y, Zeng W, Wang J, Cheng C, Yang X, Wu Y, Cao X, Xu Y. The rodent models of dyskinesia and their behavioral assessment. Front Neurol. 2019;10:1016. doi: 10.3389/fneur.2019.01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Perry T, Greig NH. The glucagon-like peptides: a new genre in therapeutic targets for intervention in Alzheimer’s disease. J Alzheimers Dis. 2002;4:487–496. doi: 10.3233/jad-2002-4605. [DOI] [PubMed] [Google Scholar]
- 58.Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, Schrag AE, Lang AE. Parkinson disease. Nat Rev Dis Primers. 2017;3:17013. doi: 10.1038/nrdp.2017.13. [DOI] [PubMed] [Google Scholar]
- 59.Sandyk R. The relationship between diabetes mellitus and Parkinson’s disease. Int J Neurosci. 1993;69:125–130. doi: 10.3109/00207459309003322. [DOI] [PubMed] [Google Scholar]
- 60.Santiago JA, Potashkin JA. Shared dysregulated pathways lead to Parkinson’s disease and diabetes. Trends Mol Med. 2013;19:176–186. doi: 10.1016/j.molmed.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 61.Santiago JA, Potashkin JA. Blood biomarkers associated with cognitive decline in early stage and drug-naive Parkinson’s disease patients. PLoS One 10. 2015:e0142582. doi: 10.1371/journal.pone.0142582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shi L, Zhang Z, Li L, Hölscher C. A novel dual GLP-1/GIP receptor agonist alleviates cognitive decline by re-sensitizing insulin signaling in the Alzheimer icv. STZ rat model. Behav Brain Res. 2017;327:65–74. doi: 10.1016/j.bbr.2017.03.032. [DOI] [PubMed] [Google Scholar]
- 63.Siracusa R, Fusco R, Cuzzocrea S. Astrocytes: role and functions in brain pathologies. Front Pharmacol. 2019;10:1114. doi: 10.3389/fphar.2019.01114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes. J Alzheimers Dis. 2005;7:63–80. doi: 10.3233/jad-2005-7107. [DOI] [PubMed] [Google Scholar]
- 65.Takahashi Y, Meyerkord CL, Hori T, Runkle K, Fox TE, Kester M, Loughran TP, Wang HG. Bif-1 regulates Atg9 trafficking by mediating the fission of Golgi membranes during autophagy. Autophagy. 2011;7:61–73. doi: 10.4161/auto.7.1.14015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Talbot K, Wang HY. The nature, significance, and glucagon-like peptide-1 analog treatment of brain insulin resistance in Alzheimer’s disease. Alzheimers Dement. 2014;10:S12–25. doi: 10.1016/j.jalz.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–688. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T, Vilar M, Campioni S, Tzitzilonis C, Soragni A, Jessberger S, Mira H, Consiglio A, Pham E, Masliah E, Gage FH, Riek R. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A. 2011;108:4194–4199. doi: 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang L, Zhang L, Li L, Hölscher C. Semaglutide is Neuroprotective and Reduces α-Synuclein Levels in the Chronic MPTP Mouse Model of Parkinson’s Disease. J Parkinsons Dis. 2019;9:157–171. doi: 10.3233/JPD-181503. [DOI] [PubMed] [Google Scholar]
- 70.Zhang L, Zhang L, Li Y, Li L, Melchiorsen JU, Rosenkilde M, Hölscher C. The novel dual GLP-1/GIP receptor agonist DA-CH5 is superior to single GLP-1 receptor agonists in the MPTP model of Parkinson’s disease. J Parkinsons Dis. 2020;10:523–542. doi: 10.3233/JPD-191768. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.