

Abstract
Streptococcus pneumoniae is an opportunistic respiratory human pathogen that poses a continuing threat to human health. Natural competence for genetic transformation in S. pneumoniae plays an important role in aiding pathogenicity and it is the best-characterized feature to acquire antimicrobial resistance genes by a frequent process of recombination. In S. pneumoniae, competence, along with virulence factor production, is controlled by a cell-density communication mechanism termed the competence regulon. In this review, we present the recent advances in the development of alternative methods to attenuate the pathogenicity of S. pneumoniae by targeting the various stages of the non-essential competence regulon communication system. We mainly focus on new developments related to competitively intercepting the competence regulon signaling through the introduction of promising dominant-negative Competence Stimulating Peptide (dnCSP) scaffolds. We also discuss recent reports on antibiotics that can block CSP export by disturbing the proton motive force (PMF) across the membrane and various ways to control the pneumococcal pathogenicity by activating the counter signaling circuit and targeting the pneumococcal proteome.
Keywords: Streptococcus Pneumoniae, Virulence, Competence, Competence Stimulating Peptide, Cell-cell communication
1. Introduction:
Bacteria live in diverse communities, and the exchange of genetic material and crosstalk among the different species in a community allow the individual species to evolve rapidly, resulting in antibiotic resistance development and robust pathogenic behaviors. Natural competence and virulence are two significant factors that contribute, directly or indirectly, to bacterial pathogenicity and associated infectious diseases in humans.1,2 The healthcare community is continuously working to fight these diseases through the development of antibiotics and vaccines. Bacterial pathogens are extensively treated with antibiotics that act upon several bacterial targets, namely bacterial cell wall biosynthesis and molecular mechanisms responsible for cellular proliferation.3 However, a major concern is that the excessive dose and unprescribed usage of antibiotics causes the rise of resistant bacteria, and most new antibiotics produced nowadays are derivatives of existing antibiotics, to which bacteria can rapidly develop resistance.4–6 As a result of the increasing numbers of antimicrobial resistant pathogens, fighting pathogenic micro-organisms has become increasingly difficult, and the diseases that were once easily treatable are becoming deadly again. This has brought the world to a scenario of a continuing arms race between continued antibiotic development by humans and the emergence of resistance mechanisms by bacteria.7
Streptococcus pneumoniae (pneumococcus) is a commensal organism of the human nasopharyngeal track, and known opportunistic respiratory pathogen associated with several diseases such as bacteremia, meningitis, and pneumonia in immune compromised human hosts.8–10 Over the past four decades, S. pneumoniae has become a severe threat to humans, with annual infections numbering over 1 million people in the United States alone.11 Antibiotic resistance (AMR) in S. pneumoniae is directly associated with pneumococcus genetic plasticity, as it acquires genetic material from the environment through transformation.12–16 The evolutionary lineage of mitis group of streptococci suggests that the cluster of S. pneumoniae/S. mitis/S. pseudopneumoniae evolved together more rapidly because of their co-colonization in the oral cavity and upper respiratory tracts, leaving them exposed to a favorable environment for genetic exchange.17–20 During this parallel evolution, S. pneumoniae acts as a recipient of virulence properties and AMR genes with a high frequency of genetic transformation along with uptake of DNA from cross-species and cell milieu.21,22 This phenomenon has resulted in the development of resistance to several antibiotics including β‐lactams, macrolides, penicillin, and quinolones.15,23,24 As pneumococcal infections developed resistance to several antibiotics, in 2000, a vaccine was developed based on widely observed surface antigens of pneumococci with a seven-valent pneumococcal conjugate vaccine (PCV7).25 Within a decade, non-PCV7 pneumococci serotypes emerged, and these bacteria were found to harbor resistance genes by a frequent process of recombination. In 2010, 6 more serotypes were added to the vaccine, this new combination was administered as PCV13 to protect against pneumococcal infections.26 An additional vaccine is the pneumococcal polysaccharide vaccine (PPSV23), which possesses 23 serotypes and is typically only used on at-risk subjects for pneumococcal disease.26–28 Such rapid accumulation of multi-drug resistance by S. pneumoniae draws more attention to addressing bacterial pathogenicity using alternative approaches. Current rational drug design and development mostly targets essential pathways of pathogenic bacteria, and the constant emergence of resistance is one of the major global health care crises in the 21st century. Thus, there is an urgent need for new strategies to target essential pathways of bacterial survival or targeting non-essential pathways to tackle pneumococcal infections. In S. pneumoniae, quorum sensing (QS), horizontal gene transfer (HGT), and virulence factor production are the major mechanisms for the development of AMR and pathogenicity, and these pathways could act as potential targets to control pneumococcus infections. This review highlights recent reports on alternative methods to inhibit the pathogenicity of S. pneumoniae by targeting the various stages of the non-essential competence regulon communication system.
2. Competence vs Virulence in S. pneumoniae:
Competence is defined as the ability of bacteria to take up genetic information from the environment through HGT. Competent bacteria are then able to integrate this new DNA into their own genome, making modifications to their phenotypes.29,30 Natural genetic transformation in S. pneumoniae was first observed in vivo by Frederick Griffith31 and in vitro by Dawson and Sia.,32 who stated that HGT is the reason for the widespread genomic diversity among S. pneumoniae and for its ability to attain antibiotic resistance. In S. pneumoniae, competence is regulated by a two-component regulatory system (TCS), which is part of the competence regulon QS circuitry.33 Competence and virulence are synchronized phenotypes in pneumococcus; however, it is not clearly understood how these two behaviors are interconnected. The currently acceptable mechanism, proposed by Guiral et al., states that three major cell-wall hydrolases, namely LytA, LytC, and CbpD, along with a two-peptide bacteriocin (CibAB), are involved in the killing of non-competent cells through a process, which they termed as “allolysis”, but is also known as fratricide.34–36 Allolysis favors three significant processes: 1) genetic exchange by DNA release and uptake by competent cells; 2) release of the virulence factor Pneumolysin (Ply) to attack host cell tissue; and 3) benefit from nutrients released by non-competent cells.
Several key players in the development of pneumococcal pathogenicity are surface-exposed components that are also considered virulence factors, including the serotype specific capsule polysaccharide, cell wall polysaccharide, choline-binding proteins, lipoproteins, LPXTG cell wall bound proteins, Ply, autolysin, and Immunoglobulin A1 (IgA1) protease.37,38 A successful pneumococcal invasion begins with a host-pathogen interaction, where pneumococcal virulence factors act by attaching to the host cell surface receptors and allowing for immune evasion by interfering with the host immune system, followed by infection establishment in the host.39 Furthermore, the competence regulon is proposed to be involved in biofilm formation, providing pathogenic reservoirs with reduced susceptibility to antimicrobial agents.40,41 Pneumococcal biofilms are highly resistant to antimicrobials, possessing increased tolerance to environmental changes, and are capable of evolving genetic diversity over short-time scales by upregulating competence genes in co-colonized pneumococcal strains.42 Biofilms play a critical role in pneumococcal survival and adaptation to the host environment during colonization of the nasopharynx.41 Moreover, the extracellular matrix of polysaccharides offers protection and is involved in enhancing S. pneumoniae virulence. In a recent study by Aggarwal et al. the authors identified a novel colonizing factor, BriC (Biofilm regulating peptide induced by Competence), which is regulated by the response regulator of the competence regulon, ComE, and is hypothesized to be a molecular link between pneumococcal competence, biofilm development, and colonization.43,44
3. Components of the Pneumococcal Competence Regulon QS Circuit:
Pneumococcus produces, secretes and detects a peptide pheromone called competence stimulating peptide (CSP) to assess its cell density and turn on the competence regulon QS circuitry.45 The pneumococcal competence regulon QS circuit was shown to be involved in competence as well as in pathogenic behaviors, such as virulence factor production, allolysis, and biofilm formation.34,40,41,46,47 The pro-CSP, encoded by comC, is cleaved into the mature 17-residue CSP45 and exported out of the cell by a proteolytic ATP binding cassette (ABC) transporter, ComAB (Figure 1).48 Upon reaching to a threshold concentration, CSP binds and activates its cognate transmembrane histidine kinase receptor, ComD. Activated ComD phosphorylates the response regulator, ComE, which triggers the autoinduction of comAB and comCDE genes. ComE also initiates the transcription of 24 early genes along with comX.49 The alternative sigma factor (σX) ComX is involved in the expression of 80 late genes, including DNA uptake, transformation and virulence factor production (Figure 1).12,50,51 The majority of S. pneumoniae strains (>90%) produces one of two types of pheromones termed, CSP1 and CSP2, with similar abundance of the two signals across pneumococcal strains. These pheromones share 50% sequence similarity,52 allowing them to confer receptor specificity by activating their respective cognate receptors, ComD1 and ComD2, while requiring a significantly higher signal concentration to effectively activate their non-cognate receptor.53,54 Deletion analysis of comA, comB, comD, comE and comX was found to impair competence induction and reduce the severity of pneumococcal infections.14,19,34,55–57 Thus, this QS circuit can be a prime target for the attenuation of pneumococcal pathogenicity and infections.
Figure 1:
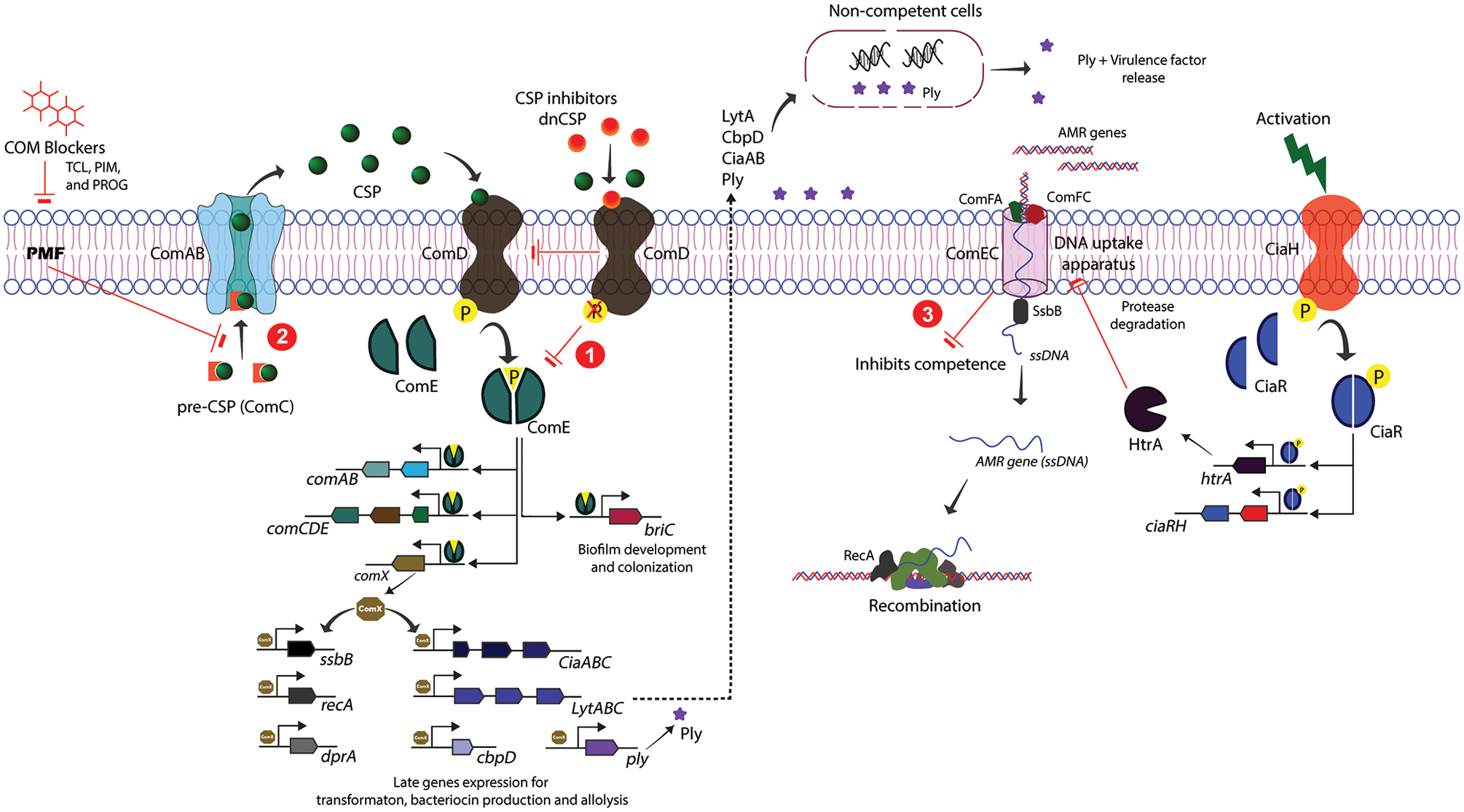
Schematic representation of S. pneumoniae competence regulon QS circuit and inhibitory hot spots. The pro-CSP peptide is processed and exported by the ComAB transporter as a mature CSP to the extracellular environment. Upon reaching threshold concentration, CSP binds and activates the ComD receptor. Activated ComD phosphorylates the response regulator, ComE, which, in turn activates the transcription of comABCDE, comX and other early genes. ComX is then involved in the expression of late genes that facilitate genetic transformation, bacteriocin production, and allolysis. Inhibitory hot spots: (1) CSP-based inhibitors (termed dnCSPs) can competitively inhibit CSP-mediated activation of the ComD receptor. (2) Small molecule-based inhibitors (termed COM blockers) inhibit CSP export by perturbing the PMF, which in turn affects ComD activation and its downstream signaling cascade. (3) The CiaRH TCS attenuates competence indirectly by expressing the protease HtrA, which is involved in degrading the DNA uptake machinery.
3.1. Peptide Based Therapeutic Strategies Targeting the Pneumococcal Competence QS Circuit
3.1.1. Development of QS Modulators Against ComD1
The S. pneumoniae D39 strain regulates the competence QS circuit via CSP1 (group 1).45 The CSP1 sequence can be divided into three regions: N-terminal region (E1-R3), central region (L4-L13), and C-terminal region (Q14-K17) (Figure 2). Initial structure-activity relationship (SAR) studies of CSP1 by Gee Lau and his co-workers demonstrated that one simple modification, Glu1 to Ala (to afford CSP1-E1A), was sufficient to inhibit the competence QS circuitry in S. pneumoniae D39 and its down-stream regulating mechanisms, such as comX induction and HGT in vivo.56,58 CSP1-E1A is even able to attenuate the expression of LytA and CbpD in vitro, which are involved in pneumococcal allolysis to release virulence factors.56 Furthermore, CSP1-E1A attenuates virulence and consequently pneumococcal infections in mouse models, indicated by a significant reduction in the mortality rate.56
Figure 2:
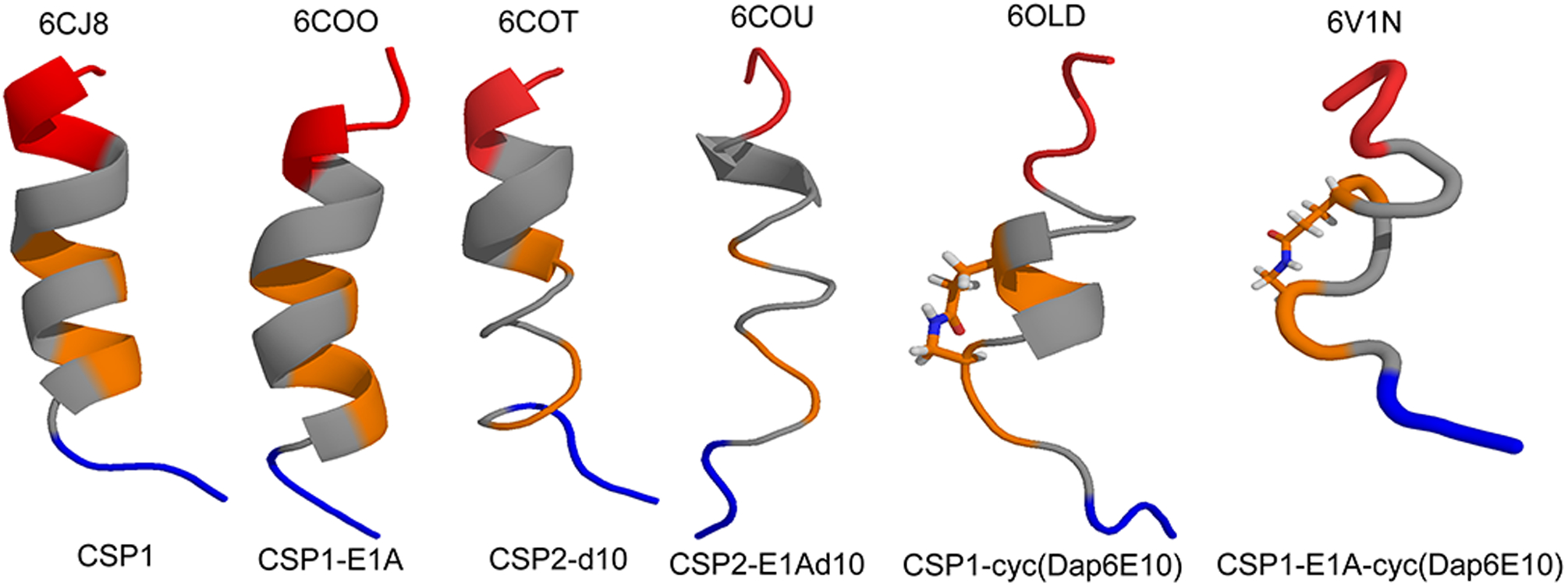
NMR structures of CSP analogs. Backbone ribbon structural representation of CSP1, CSP1-E1A, CSP2-d10, CSP2-E1Ad10, CSP1-cyc(Dap6E10), CSP1-E1A-cyc(Dap6E10). The color code for each structure as follows, blue, N-terminus; grey, hydrophobic patch; orange, hydrophilic patch; and red, C-terminus. All the PDB entries are provided above each structure.
Work from our lab has focused on an in-depth analysis of the structural and functional properties of the S. pneumoniae CSPs through systematic evaluation of each residue in order to determine their importance and uncover potent CSP-based QS-modulators.59 A study from Yang et al. generated a wide range of peptides derived from Ala and D-enantiomer scanning of the CSP1 signal to determine each residue’s contribution to ComD receptor recognition, binding, and activation.60 A follow-up study has focused on the N-terminal role of CSP1 in receptor recognition and activation, and a thorough investigation was carried out at the Glu1 position by introducing amino acids (AAs) with different side-chain length, polarity, and chirality.61 Interestingly, the negatively charged Glu1 side chain and positively charged N-terminal amine were found to be important for receptor activation but not necessarily to initial recognition and binding, providing an explanation as to why a simple E1 to A substitution has led to the formation of a competitive inhibitor (CSP-E1A).60,61 Further competitive cell-based binding assays revealed that CSP1-E1A has an IC50 value of 85.7 nM against the ComD1 receptor (Table 1).60
Table 1:
Summary of structural and functional properties of QS inhibitors
| Peptide Name | Secondary Structure | IC50 (nM) (95% CI) | Metabolic Stability (Half-Life in hrs) | Reference | |
|---|---|---|---|---|---|
| ComD1 | ComD2 | ||||
| CSP1-E1A | α-helix | 85.7 | --- | ~1.5 h | 60 |
| CSP1-E1AK6A | α-helix) | 104 | --- | # | 60 |
| CSP1-E1A-N-Me-K6 | * | 48.0 | --- | ~2 h | 62 |
| CSP1-E1A-N-Me-Q14 | * | 370 | --- | ~6 h | 62 |
| CSP1-E1A-N-Me-K6-N-Me-Q14 | * | 360 | --- | ~6 h | 62 |
| CSP1-E1A-Cyc (Dab6D10) | α-helix | 173 | >1000 | # | 63 |
| CSP1-E1A-Cyc (Dap6D10) | α-helix | 75.8 | 182 | 4 h | 63 |
| CSP1-E1A-DesK16K17Cyc (Dap6D10) | α-helix | 7.57 | 67.2 | # | 63 |
| CSP2-E1A | β-sheet | >1000 | >1000 | # | 60 |
| CSP2-E1Ad10 | α-helix | >1000 | 56.5 | > 3 h | 60 |
| CSP2(15)-E1Ad10 | α-helix | --- | 117 | # | 64 |
| CSP2-E1Ad10-N-Me-F13 | * | >1000 | 64 | ~4 h | 62 |
| CSP2-E1AI4Nvad10L14Q | * | 510 | 18.2 | # | 65 |
| CSP2-E1Ad10L14Q | * | 766 | 49.5 | # | 65 |
| CSP2-E1AI4Ld10L14Q | * | 503 | 87.2 | # | 65 |
| S. mitis-CSP-2-E1AN7II8F | * | 204 | 135 | # | 66 |
| S. mitis-CSP-2-E1AI2MN7FI8F | * | 294 | 418 | # | 66 |
| S. mitis-CSP-2-E1AI2MN7II8F | * | 141 | 32.9 | # | 66 |
Metabolic stability not determined
Secondary structure not evaluated
Evaluating the central region of CSP1 (residues L4–F13) revealed that the hydrophobic residues within this region are playing a key role in receptor binding (residues L4, F7, F8, F11, I12, and L13) while the hydrophilic residues are modifiable (residues S5, K6, R9, and D10), with Ala and D-enantiomer replacements of the hydrophobic residues resulted in a 20–100 fold loss in activity against ComD1 whereas replacements of the hydrophilic residues exhibited only a 3–5 fold decrease in activity, as compared to CSP1 (with an EC50 of 10.5 nM against ComD1).60 Moreover, substitutions of the hydrophilic residues, specifically K6, reduced receptor specificity, allowing for activation of both ComD1 and ComD2, paving the way to the development of pan-group modulators. Indeed, the SAR analysis revealed that CSP1-K6A possesses EC50 values of 51.0 and 24.0 nM against ComD1 and ComD2, respectively. With this knowledge, a double mutant (CSP1-E1AK6A) was designed as a pan-group inhibitor, and this peptide displayed inhibitory activity against ComD1 with an IC50 value of 104 nM, but no inhibitory activity against ComD2 (Table 1).60
To gain information regarding the CSP1 binding pockets within the ComD1 receptor and to enhance the binding interactions between CSP1 and ComD1 by optimizing the degree of occupancy within each binding pocket, Koirala et al. used conservative point mutations at the key hydrophobic residues of CSP1 and substituted each aliphatic AA with Ile Leu, Nle (Norleucine), Val and Nva (Norvaline), and aromatic AA with Phg (Phenylglycine), hPhe (homophenylalanine) and Tyr.67 Aliphatic substitution at positions 4, 12, and 13 were tolerated and the receptor binding pockets for these side chain residues were able to accommodate longer and bulkier groups, suggesting that the degree of occupancy of these binding pockets is not optimized and thus could be improved. In support of that, valine substitutions (smaller side chain residue) at these positions resulted in reduced activity of the modified CSP analog, likely due to reduced receptor binding pocket occupancy. Of these conservative aliphatic analogs, CSP1-L4NV and CSP1-I12L were found to improve activity up to 2-fold compared to CSP1. Aromatic substitutions at positions 7, 8, and 11 (Phe to Phg, nPhe, and Tyr) resulted in an 8 to 80-fold reduction in potency, highlighting the importance of optimally positioning the side chain aromatic ring in the binding pocket.67
Moving to the C-terminal region of CSP1, the last two residues (K16 and K17) were found to be dispensable as modifications or truncations of these positions were well tolerated, with the truncated peptide CSP1-desK16K17 exhibiting EC50 value comparable to that of CSP1 against ComD1.60 These dispensable C-terminal positive-charged residues are presumed to be involved in CSP solubility; however additional studies are required to test this hypothesis.
CSP backbone amide protons are important for stabilizing the peptide bioactive conformation, and thus play a significant role in the CSP activity. Introducing N-methylated AAs was utilized to probe which amide protons are involved in stabilizing the CSP1 bioactive conformation and\or in direct binding to the ComD1 receptor. Additionally, unnatural backbone modifications, including N-methylation, can enhance the peptide proteolytic stability. By successfully balancing between these two parameters (activity and stability), an active CSP analog with improved pharmacological properties could be developed. With this knowledge in mind, the truncated CSP1 sequence, CSP1-desK16K17 (aka CSP1(15)), was used as template in this study.62 A systematic N-methyl scan library was constructed, and the three most potent CSP1(15) N-methylated analogs identified, namely, CSP1(15)-N-Me-K6, CSP1(15)-N-Me-F7 and CSP1(15)-N-Me-Q14, were found to exhibit improved proteolytic stability compared to the native CSP1. Thus, these substitutions were incorporated in the CSP1-E1A template to produce three potential QS inhibitors (also termed dominantly negative CSPs, or dnCSPs), CSP1-E1A-N-Me-K6, CSP1-E1A-N-Me-Q14 and CSP1-E1A-N-Me-K6-N-Me-Q14. These three analogs were found to possess inhibitory activity against the ComD1 receptor, however, expectedly, no activity was observed against the ComD2 receptor. Surprisingly, these CSP1-E1A analogs were also found to be more susceptible to Trypsin/Chymotrypsin digestion as they degraded faster than the truncated CSP1(15) analogs (Table 1).62 The fast degradation was attributed to the presence of the RKK motif at the C-terminus.
3.1.2. Development of QS Modulators against ComD2
The S. pneumoniae TIGR4 strain regulates QS using CSP2 (group2). Previous studies by Lau and co-workers exhibited that CSP2-E1A competitively inhibits competence induction, comX expression, and DNA transformation.56 In a later study, Yang et al. conducted a systematic analysis of the CSP2 sequence by generating Ala and D-enantiomer scan libraries.60 Within the N-terminus region, CSP2-E1A and CSP2-m2 were found to inhibit the ComD2 receptor, albeit only at high peptide concentration (IC50 values of >1,000 nM; Table 1). Truncation of the N-terminal residues (CSP2-desE1 and CSP2-desE1M2) also displayed weak inhibitory activity against the ComD2 receptor reaffirming the role of Glu1 and Met2 in receptor activation.60
The central region of CSP2 is mostly occupied by hydrophobic residues with only 3 hydrophilic residues (S5, R6 and D10). Due to this enrichment of hydrophobic AAs, CSP2 was found to adopt a β-sheet conformation.60,68 One significant finding related to the central region of CSP2 is that a single D-enantiomer modification at the tenth position resulted in an analog, CSP2-d10, that exhibited a significant enhancement in activity compared to CSP2 (EC50 of 2.86 nM for CSP2-d10 compared with EC50 of 50.7 nM for CSP2). Based on the systematic analysis of the N-terminal and central regions of CSP2, a double mutant CSP2-E1Ad10 was generated (Figure 2). This analog was found to be a potent ComD2 inhibitor (IC50 of 56.5 nM) and also possessed weak inhibitory activity against ComD1 (Table 1). Other double and triple mutants (CSP2-m2d10 and CSP2-E1Am2d10) both exhibited weak inhibitory activity against ComD2 with IC50 values of >1,000 nM (Table 1).60
As for the C-terminal region of CSP2, AAs L14-K17 were found to be dispensable, leading to truncated scaffolds such as CSP2-desK16K17 (aka CSP2(15)) and CSP2-desL14-K17 (aka CSP2(13)) retaining activity against the ComD2 receptor.60 Similar to CSP1, the importance of amide protons in receptor binding was studied using N-methyl AA substitutions, utilizing CSP2(13) as the scaffold for this study. N-methylation significantly reduced activity, except in position F13 (CSP2(13)-N-Me-F13) where the N-methyl modification did not alter the activity significantly compared to the parent CSP2(13) analog.62 This mutation was therefore introduced into the CSP2-E1Ad10 scaffold to afford CSP2-E1Ad10-N-Me-F13, which was found to possess similar characteristics in inhibition activity, but also did not offer a better protection against proteolytic degradation (Table 1).62
3.1.3. Development of Pan-Group inhibitors:
As the majority of S. pneumoniae strains produce one of two peptide pheromone variants (CSP1 or CSP2) with similar abundance of the two signals across the pneumococcus population, pan-group dnCSPs capable of blocking the competence regulon in both specificity groups could act as potential therapeutics irrespective of S. pneumoniae serotypes.69 Recently, Koirala et al. reported generating a hybrid sequence of CSP1 and CSP2 that effectively modulates both ComD receptors.65 At first, the authors developed pan-group QS activators by systematically replacing residues within the CSP2 scaffold with their CSP1 counterparts, and then converting these activators into pan-group dnCSPs by replacing the glutamic acid at position 1 (E1) with alanine. Screening was carried out with single, double, triple, quadruple and quintuple substitutions. While this analysis did not lead to the successful construction of a pan-group dnCSP, it allowed the authors to identify the fourth and fourteenth positions as key positions for receptor specificity in both CSP1 and CSP2. Next, the authors utilized the lead CSP2 inhibitor, CSP2-E1Ad10, as template, and by incorporating modifications in the fourth and fourteenth positions were able to identify a pan-group inhibitor, CSP2-E1AI4Nvad10L14Q, with IC50 values at the nanomolar range against both receptors (IC50 of 510 nM against ComD1, and 18.2 nM against ComD2; Table 1). Several other designed CSP2-E1Ad10 analogs were capable of inhibiting the ComD2 receptor with IC50 values in the low nanomolar range (32–90 nM), but exhibited only weak inhibitory activity against the ComD1 receptor.65
In a recent work by Yang et al., the authors reported the development of pan-group dnCSPs through peptide cyclization.63 The authors first evaluated the most suitable positions for peptide cyclization by performing ring position scan and cyclizing the peptides via a lactam bridge between a lysine and aspartic acid that were incorporated in the desired positions. Next, the authors performed a conformational screening mediated by macrocycle ring size alteration on the lead cyclic peptide scaffold selected from the ring position scan (CSP1-cyc(K6D10)). This analysis produced two potent pan-group activators (CSP1-cyc(Dab6E10) and CSP1-cyc(Dap6E10)) with EC50 values at the low nano-molar range against both ComD receptors. Finally, the E1A mutation was introduced to the two lead cyclic activators in an attempt to convert them into pan-group inhibitors. Biological evaluation revealed that CSP1-E1A-cyc(Dap6E10) can inhibit the ComD1 receptor with an IC50 value of 75.8 nM and ComD2 receptor with an IC50 value of 182 nM (Table 1 and Figure 2). Interestingly, the truncated analog, CSP1-E1A-des-K16K17-cyc(Dap6E10), exhibited superior inhibitory activity compared to CSP1-E1A-cyc(Dap6E10), with 10- and 3-fold improvement in IC50 values against the ComD1 and ComD2 receptors, respectively (Table 1). Unfortunately, further in vivo and in vitro analysis of this analog could not be performed due to poor solubility.63
The Mitis group of streptococci is categorized into 13 different species, all of which exhibit commensalism.70 As discussed earlier, the crosstalk between streptococci species results in frequent gene exchange through recombination and leads to the acquirement of antimicrobial resistance.17,18 As a result, coevolved species like S. mitis and S. pneumoniae share up to 80% of gene similarity.20,71,72 Milly et al. recently attempted to uncover potential crosstalk between different species of streptococci. To this end, the native CSPs from the mitis and anginosus groups of streptococci were tested as potential modulators of the S. pneumoniae competence regulon.66 Out of 12 natural CSPs, the CSP2 pheromone from S. mitis was found to activate both the ComD1 and ComD2 receptors and was therefore selected as a scaffold for developing pan-group S. pneumoniae QS modulators. The approach the authors took to afford pan-group modulators was to systematically replace S. mitis-CSP2 residues with the corresponding residues in CSP1 and CSP2 that were found to have a key role in S. pneumoniae ComD receptor binding and activation. Systematic screening of generated peptide libraries ranging from single to triple modifications revealed several S. mitis-CSP2 analogs that exhibit increased potency in activating both pneumococcal ComD receptors with EC50 values at the low nanomolar range. Twelve lead scaffolds were selected to be converted into dnCSPs by introducing the E1A modification. Indeed, three nanomolar range pan-group dnCSPs were identified, namely, S. mitis-CSP-2-E1AN7II8F, S. mitis-CSP-2-E1AI2MN7FI8F, and S. mitis-CSP-2-E1AI2MN7II8F (Table 1).66
3.1.4. Sequence-Structure-Function Correlation:
Traditionally, the primary AA sequence dictates secondary structure characteristics, which inevitably play a key role in peptide and protein function. CSPs follow these well-established sequence-structure-function paradigms. Johnsborg et al.,52 Yang et al.,60,63,68 and Koirala et al.67 extensively studied CSP structural characteristics in aqueous and membrane mimicking environments using circular dichroism (CD) and 2D nuclear magnetic resonance (NMR) spectroscopy. Structural analysis revealed that CSP1 adopts an α-helical conformation in membrane mimicking conditions, and that all the bioactive peptides derived from CSP1 also attain an α-helical conformation (Figure 2). Surprisingly, CSP2 and all derived peptides, with few exceptions, exhibited a β-sheet conformation. Interestingly, unlike CSP2, pan-group modulators designed from the CSP2 scaffold with D-enantiomer substitutions, namely CSP2-d10 and CSP-E1Ad10, shifted the conformation equilibrium from a β-sheet to an α-helix in membrane mimetic environment (Figure 2).68 Together, the structural analysis of CSP1 and CSP2 indicates that the bioactive conformation of pneumococcus CSPs is an α-helix.
3.1.5. Metabolic stability of Pan Group inhibitors:
One of the major characteristics to consider in peptide therapeutics is metabolic stability. Introducing unnatural elements in peptides, such as non-standard AAs, backbone modifications, and cyclization, afford peptide therapeutics a greater chance to overcome protease degradation and become pharmacologically relevant. Yang et al. and Koirala et al. evaluated the proteolytic stability of modified CSP analogs (QS activators and inhibitors) in vitro in the presence of Trypsin/Chymotrypsin and calculated the half-life of these peptides.62–64 This analysis revealed that CSP1, CSP1(15), and CSP1-E1A are highly susceptible to proteolytic degradation with a half-life of approximately 1.5 h (Table 1).64 The CSP1(15)-based truncated analogs with an N-methyl AA substitution, CSP1(15)-N-Me-K6, CSP1(15)-N-Me-F7 and CSP1(15)-N-Me-Q14, exhibited superior proteolytic stability with a half-life that exceeded the 48 h assay duration (Table 1).62 In the case of the CSP1-E1A scaffold where N-methyl modifications at K6 and Q14 were introduced (CSP1-E1A-N-Me-K6-N-Me-Q14 and CSP1-E1A-N-Me-Q14), the half-life was reduced to 6 h (Table 1).62 This reduced stability is likely due to the proteolytic cleavage of the RKK motif at the CSP C-terminus by trypsin. Unlike CSP1, CSP2 and CSP2-E1A form β-sheet structures in aqueous solutions with very low solubility.63 Due to this experimental limitation, the half-life of these analogs could not be determined. Contrary, CSP2-d10 and CSP2-E1Ad10 are soluble in aqueous solutions, and thus their half-life was evaluated and determined to be about 2.5 h and 3 h, respectively.64 The metabolic stability of the N-methyl substituted pan-group QS inhibitor CSP2-E1Ad10-N-Me-F13 was significantly improved with a half-life of approximately 4 h (Table 1).62 As for cyclic CSP analogs, CSP1-E1A-cyc(Dap6E10) has a half-life of about 3 h, which is similar to its linear counterpart, CSP1-E1AK6DapD10E. The half-life of the truncated analog, CSP1-E1A-des-K16K17-cyc(Dap6E10), could not be measured, as this peptide exhibits very low solubility in aqueous solutions (Table 1).63 Overall, the incorporation of unnatural AAs has significantly improved the proteolytic stability of CSPs, with few exceptions, and these studies opened avenues for the development of peptide-based QS modulators with drug-like properties.
3.1.6. QS inhibitors Attenuate Virulence Factor Production:
In S. pneumoniae virulence factor production is proposed to be regulated by the competence regulon.57 The mechanism proposed by Guiral et al. states that competence triggers the release of murein hydrolases, which in turn are involved in the release of the virulence factor Ply.34 Zymogram analysis by Zhu et al. displayed that CSP1-E1A is able to inhibit the expression of LytA and CbpD in S. pneumoniae D39 cells, which are mainly up-regulated by the ComX transcription factor.56 Along with SAR analysis of the native CSPs, Koirala et al. and Yang et al. studied the ability of QS inhibitors to attenuate Ply release.63,64 A pan-group inhibitor, CSP1-E1A-cyc(Dap6E10), was able to attenuate Ply release and Ply-mediated hemolysis in both the D39 and TIGR4 strains.63 Similar results were observed for CSP2-E1Ad10, where Ply release in the TIGR4 strain was attenuated and a consequent reduction in hemolysis was observed.64 These studies highlight the ability of QS inhibitors to attenuate pneumococcus infectivity.
3.1.7. Attenuation of Pneumococcal Infections:
QS modulators with the ability to control the expression of genes involved in virulence factor production can regulate pneumococcal infections. To investigate such dnCSPs mediated attenuation of pneumococcal infections, mice were pre-infected with S. pneumoniae strains and attempts were made to treat the infection with designed QS inhibitors.63 To this end, CD1 mice were intranasally infected with D39 or TIGR4 cells. Then, CSP1-E1A or CSP2-E1Ad10, respectively, were introduced post-infection and the survival rate of mice was monitored for 6 days. The dnCSPs were able to attenuate the infection resulting in a significantly improved survival rate (up to 40%) and delayed mice death kinetics.64 Toxicity of the QS inhibitors was also assessed in major organs (lungs, heart, liver, kidney, and spleen), and the inhibitors appeared to be nontoxic. To evaluate group-specific attenuation, D39-infected mice (group 1) were treated with CSP2-E1Ad10 (group 2 inhibitor) and TIGR4-infected mice (group 2) were treated with CSP1-E1A (group 1 inhibitor). In both cases, the inhibitors were unable to attenuate cross-group infectivity, resulting in 100% mortality rate.63 Importantly, the pan-group inhibitor CSP1-E1A-cyc(Dap6E10) was able to attenuate pneumococcal infections caused by both D39 and TIGR4, leading to a significantly improved survival rates.63 These promising results highlight the potential of peptide-based QS inhibitors to treat pneumococcal infections.
3.2. Targeting the Pneumococcal Competence QS circuit by Proton Motive Force Disruptors:
Bacteria maintain homeostasis in its milieu through electrochemical gradient across the cytoplasmic membrane. The sum of electric potential and the transmembrane proton gradient, called proton motive force (PMF), generated across the cytoplasmic membrane is essential for various processes such as ATP synthesis, flagellar motility, transport (import and export) of various solutes and nutrient import. Recent work from Farha et. al. identified a synergistic combination of compounds that have the potential to disrupt the PMF in methicillin-resistant Staphylococcus aureus (MRSA). These synergistic combinations of compounds exhibited reduced cytotoxicity in mammalian cells, paving the way for novel therapeutic strategies.73 In the case of S. pneumoniae, several antimicrobials activate the competence regulon, leading to acquirement of AMR.14,74–76 A primary step in competence regulation is the export of the mature CSP by the ComAB transporter to the extracellular environment, thus blocking CSP export by disturbing the PMF could hamper competence.77 In a recent work from Domenech et. al. the authors carried out a high-throughput screening of FDA approved drugs to identify compounds capable of attenuating the pneumococcal competence regulon without affecting bacterial growth and termed them COM-blockers.78 Primary screening was carried out by detecting natural competence induction through the use of the ssbB promoter fused to firefly luciferase and screening around 1366 compounds. This high-throughput screening resulted in 46 compounds that were identified as able to inhibit the competence regulon at concentrations that did not inhibit bacterial growth. These COM-blockers belong to the anatomical therapeutic chemical (ATC) library with 7 COM-blockers exhibiting competence inhibitory activity at low concentrations (μg/ml). Three lead compounds: biocide triclosan (TCL), antipsychotic pimozide (PIM), and antimalarial proguanil hydrochloride (PROG), were selected for further evaluation and exhibited superior activity in attenuating pneumococcal competence with potent COM-blocking activity (Figure 1 and 3).
Figure 3:
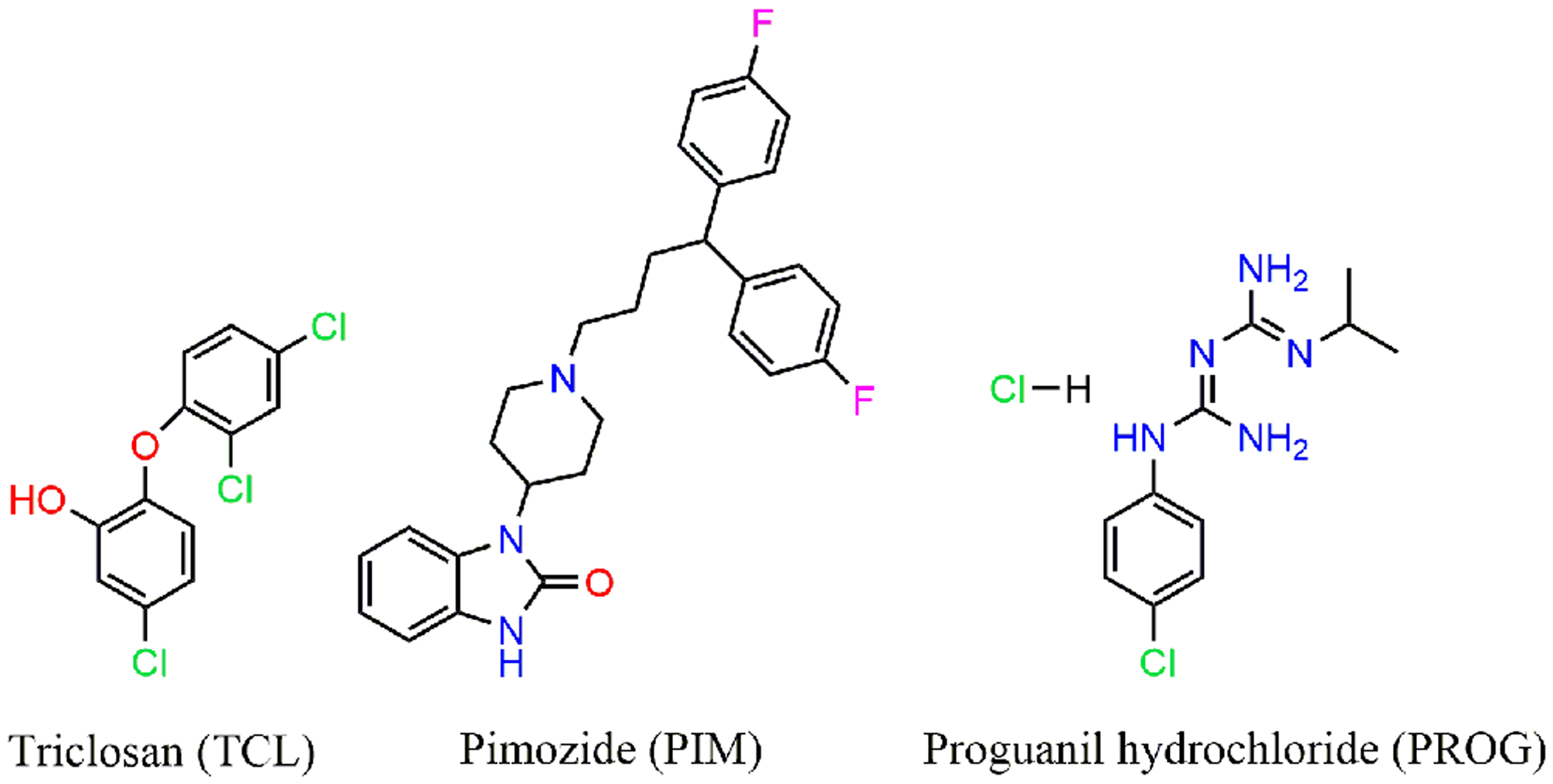
Chemical structures of COM blockers TCL, PIM and PROG.
The COM-blocking ability of these compounds was extensively studied using multiple approaches, starting with IPTG (isopropyl β- d-1-thiogalactopyranoside) induced expression of the comAB and comCDE genes in the presence of COM-blockers, where downregulation of gene expression indicated that competence induction was abolished. Additionally, a wildtype strain and a ΔcomAB mutant were treated with exogenous CSP1 and a rapid competence induction was observed in both strains, with competence being turned off earlier in the ΔcomAB mutant. Upon treating with TCL, faster competence shut down was observed in both strains, suggesting that CSP1 export was abolished, resulting in turning off the CSP-ComD-ComE positive feedback. To understand how COM-blockers perturb the CSP-export, the authors continued to analyze the physiological effects of the lead compounds on pneumococci. Addition of TCL or PROG resulted in a decrease in the internal pH due to PMF disruption-mediated proton assimilation. To maintain cell homeostasis, the cells must rapidly pump out protons using ATP synthase, leading to a decrease in the cellular ATP levels. Under such circumstances, optimal function of ComAB and other transporters is hampered and leads to a reduction in pro-CSP cleavage and export.77 Furthermore, Domenech et. al. exhibited that COM-blockers do not interact directly with F0F1 proton ATPase, which maintains pH homeostasis in S. pneumoniae and only act by perturbing the PMF.78
The transformation ability of the pneumococcal strains D39 and TIGR4, multi-drug resistant pneumococcal strains from the Pneumococcal Molecular Epidemiology Network (PMEN), S. mitis and S. sanguinis was examined by exogenously providing a plasmid with tetracycline-resistance genes. The COM-blocker TCL was able to prevent the strains from acquiring antibiotic resistance through transformation with a minimal transformation inhibition concentration of 1 μg/ml. Moreover, the COM-blockers TCL, PIM, and PROG were also found to inhibit HGT between different pneumococcal strains that were provided with different antibiotic resistance genes in a plasmid. Similar experiments were performed by colonizing the strains on human epithelial lung cells where TCL was found to block both transformation and HGT. Furthermore, in an in vivo mouse model of pneumococcal infection, the antimalarial PROG COM-blocker was able to reduce HGT efficiency by 50% (14 out of 27 animals had no transformants). In a recent work from Westfall et. al. the authors reported that continuous treatment with bacteriostatic concentration of TCL resulted in the development of resistance in E. coli and MRSA. Remarkably, prolonged exposure of the S. pneumoniae D39 strain to TCL (1 μg/ml) prevented competence in all lineages for 30 days with TCL preserving its efficacy as a COM-blocker.78 Controlling the pneumococcal evolution of antibiotic resistance through COM-blockers and caging the pathogenic genome could become a viable treatment strategy since the compounds used are already FDA approved.79
3.3. Potential New Targets to Inhibit Competence
Under stress conditions, pneumococci activate the competence regulon to enhance the acquisition of AMR by assimilating exogenous antibiotic resistant genes through transformation.14,80 Such rapid assimilation of genetic information can be controlled by activating the counter signaling circuit and targeting the pneumococcal proteome at various stages of competence. Recently, the CiaR-H regulatory system was found to activate htrA gene expression, which encodes a membrane bound serine protease, HtrA, that is involved in degrading the components required for DNA uptake in transformation (Figure 1).81–83 The HtrA protease is playing a role in inhibiting pneumococcal competence, and is involved in nasopharyngeal colonization, virulence and bacteriocin production.81,84–87 HtrA homologues are also present in S. mitis and S. gordonii and are proposed to be a key regulator for natural transformation.83 Developing modulators for the CiaR-H TCS could be another promising hotspot to tackle pneumococcal pathogenicity.
Another potential target for attenuating competence in S. pneumoniae is RecA, a protein that is involved in integrating recently acquired genes into S. pneumoniae genome through recombination and repair of DNA damages (Figure 1).88,89 As such, RecA is an essential protein for genetic transformation.88,89 Since RecA is encoded by one of the late genes upregulated by ComX,80 developing molecules that inhibit the RecA association with other proteins or DNA could be a promising downstream approach to control the pathogenicity acquired through HGT.
4. Conclusions and Outlook:
In the past few decades, antibiotic resistance acceleration in pathogenic bacteria has been associated with these pathogens activating their defense mechanisms to acquire AMR, with the misuse and overuse of antibiotics further exasperating the problem. S. pneumoniae remains to be an important cause of invasive diseases, especially in children and the elderly. There is an increase in research focused on exploring novel methodologies to tackle pneumococcal infections to develop better treatment strategies. However, pneumococcal diseases still pose a huge burden on public healthcare, and continuous surveillance of evolving multidrug-resistant serotypes are needed. The foremost objective of the aforementioned studies is to find key checkpoints to prevent the signaling between proteins involved in competence induction, thus caging the genetic information that decodes antibiotic resistance. Successfully designed dnCSPs using a systematic approach were able to inhibit the pneumococcal competence regulon, thus exhibiting the potential to control pneumococcal infections. These studies may open new avenues for future developments of QS-based peptide therapeutics. The synergetic combination of “anti-evolution” drugs or peptide therapeutics with antibiotics could also lead to controlling pathogenic bacteria evolution and treating pneumococcal infections.
Moving forward, the peptide-based approach would likely face challenges associated with the relatively poor pharmacological properties that peptides usually exhibit, while the “anti-evolution” approach would need to overcome the potential off-target effects of the utilized scaffolds (the lead compounds were identified out of a large screen of FDA-approved drugs used to treat other diseases). With the peptide approach, altering the peptide scaffold to construct pharmacologically enhanced peptidomimetic scaffolds could offer a viable pathway to make these lead compounds more druggable. Alternatively, diverging completely from the peptidic scaffolds into small molecule-based scaffolds that still interact with the target histidine kinase receptor, as was successfully implemented in several other systems,90–93 may prove to be the way to convert these anti-virulence compounds into drug leads.
Acknowledgments:
The National Institutes of Health (R35GM128651 and R01HL142626) is acknowledged for the generous support of research in our laboratory.
Biographies

Muralikrishna Lella grew up in the southern part of India and received his B.S. (2008) in Biochemistry from Acharya Nagarjuna University, India, and M.S. (2010) in Biochemistry from Andhra University, India. He received his Ph.D. (2019) in Biological Sciences from the IISER Bhopal, India, working with Dr. R. Mahalakshmi. He is currently working as a Postdoctoral Research Scholar in the Tal-Gan group at University of Nevada, Reno, U.S.A, investigating bacterial quorum sensing in Streptococcus pneumoniae and Enterococcus faecalis.

Yftah Tal-Gan was born in Jerusalem, Israel and received his B.S. (2001), M.S. (2006) and Ph.D. (2011) degrees from The Hebrew University of Jerusalem, working with Chaim Gilon and Alexander Levitzki. He then joined Helen Blackwell’s group at the University of Wisconsin–Madison, U.S.A., as a Postdoctoral Research Associate. Yftah was appointed Assistant Professor in the Department of Chemistry at University of Nevada, Reno, U.S.A. in 2014 and was promoted to Associate Professor with tenure in 2020.
Footnotes
Data Availability:
Data sharing not applicable – no new data generated.
Conflict of Interest:
There are no conflicts to declare.
References:
- 1.Blokesch M Curr Biol 2016, 26, R1126–R1130. [DOI] [PubMed] [Google Scholar]
- 2.Diard M; Hardt WD FEMS Microbiol Rev 2017, 41, 679–697. [DOI] [PubMed] [Google Scholar]
- 3.Kohanski MA; Dwyer DJ; Collins JJ Nat Rev Microbiol 2010, 8, 423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aminov RI Front Microbiol 2010, 1, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Santos-Lopez A; Marshall CW; Scribner MR; Snyder DJ; Cooper VS eLife 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hutchings MI; Truman AW; Wilkinson B Curr Opin Microbiol 2019, 51, 72–80. [DOI] [PubMed] [Google Scholar]
- 7.Karlsson EK; Kwiatkowski DP; Sabeti PC Nat Rev Genet 2014, 15, 379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kadioglu A; Weiser JN; Paton JC; Andrew PW Nat Rev Microbiol 2008, 6, 288–301. [DOI] [PubMed] [Google Scholar]
- 9.Weiser JN J Mol Med (Berl) 2010, 88, 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mehr S; Wood N Paediatr Respir Rev 2012, 13, 258–264. [DOI] [PubMed] [Google Scholar]
- 11.Wahl B; O’Brien KL; Greenbaum A; Majumder A; Liu L; Chu Y; Lukšić I; Nair H; McAllister DA; Campbell H; Rudan I; Black R; Knoll MD Lancet Glob Health 2018, 6, e744–e757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee MS; Morrison DA J Bacteriol 1999, 181, 5004–5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Claverys JP; Prudhomme M; Mortier-Barrière I; Martin B Mol Microbiol 2000, 35, 251–259. [DOI] [PubMed] [Google Scholar]
- 14.Prudhomme M; Attaiech L; Sanchez G; Martin B; Claverys JP Science 2006, 313, 89–92. [DOI] [PubMed] [Google Scholar]
- 15.Croucher NJ; Harris SR; Fraser C; Quail MA; Burton J; van der Linden M; McGee L; von Gottberg A; Song JH; Ko KS; Pichon B; Baker S; Parry CM; Lambertsen LM; Shahinas D; Pillai DR; Mitchell TJ; Dougan G; Tomasz A; Klugman KP; Parkhill J; Hanage WP; Bentley SD Science 2011, 331, 430–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chewapreecha C; Harris SR; Croucher NJ; Turner C; Marttinen P; Cheng L; Pessia A; Aanensen DM; Mather AE; Page AJ; Salter SJ; Harris D; Nosten F; Goldblatt D; Corander J; Parkhill J; Turner P; Bentley SD Nat Genet 2014, 46, 305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janoir C; Podglajen I; Kitzis MD; Poyart C; Gutmann L J Infect Dis 1999, 180, 555–558. [DOI] [PubMed] [Google Scholar]
- 18.Bryskier A Clin Microbiol Infect 2002, 8, 65–69. [DOI] [PubMed] [Google Scholar]
- 19.Kowalko JE; Sebert ME Infect Immun 2008, 76, 3131–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kilian M; Poulsen K; Blomqvist T; Håvarstein LS; Bek-Thomsen M; Tettelin H; Sørensen UB PLoS One 2008, 3, e2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Donkor ES; Bishop CJ; Gould K; Hinds J; Antonio M; Wren B; Hanage WP mBio 2011, 2, e00040–00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marks LR; Reddinger RM; Hakansson AP mBio 2012, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jensen A; Valdórsson O; Frimodt-Møller N; Hollingshead S; Kilian M Antimicrob Agents Chemother 2015, 59, 3529–3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schroeder MR; Stephens DS Front Cell Infect Microbiol 2016, 6, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Practices A. C. o. I. MMWR Recomm Rep 2000, 49, 1–35. [PubMed] [Google Scholar]
- 26.Nuorti JP; Whitney CG; (CDC), C. f. D. C. a. P. MMWR Recomm Rep 2010, 59, 1–18. [PubMed] [Google Scholar]
- 27.Liñares J; Ardanuy C; Pallares R; Fenoll A Clin Microbiol Infect 2010, 16, 402–410. [DOI] [PubMed] [Google Scholar]
- 28.Kim L; McGee L; Tomczyk S; Beall B Clin Microbiol Rev 2016, 29, 525–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Claverys JP; Martin B; Polard P FEMS Microbiol Rev 2009, 33, 643–656. [DOI] [PubMed] [Google Scholar]
- 30.Johnston C; Martin B; Fichant G; Polard P; Claverys JP Nat Rev Microbiol 2014, 12, 181–196. [DOI] [PubMed] [Google Scholar]
- 31.Griffith F J Hyg (Lond) 1928, 27, 113–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dawson MH; Sia RH J Exp Med 1931, 54, 681–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gómez-Mejia A; Gámez G; Hammerschmidt S Int J Med Microbiol 2018, 308, 722–737. [DOI] [PubMed] [Google Scholar]
- 34.Guiral S; Mitchell TJ; Martin B; Claverys JP Proc Natl Acad Sci U S A 2005, 102, 8710–8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Håvarstein LS; Martin B; Johnsborg O; Granadel C; Claverys JP Mol Microbiol 2006, 59, 1297–1307. [DOI] [PubMed] [Google Scholar]
- 36.Claverys JP; Martin B; Håvarstein LS Mol Microbiol 2007, 64, 1423–1433. [DOI] [PubMed] [Google Scholar]
- 37.Jedrzejas MJ Microbiol Mol Biol Rev 2001, 65, 187–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kohler S; Voß F; Gómez Mejia A; Brown JS; Hammerschmidt S FEBS Lett 2016, 590, 3820–3839. [DOI] [PubMed] [Google Scholar]
- 39.Brooks LRK; Mias GI Front Immunol 2018, 9, 1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suntharalingam P; Cvitkovitch DG Trends Microbiol 2005, 13, 3–6. [DOI] [PubMed] [Google Scholar]
- 41.Chao Y; Marks LR; Pettigrew MM; Hakansson AP Front Cell Infect Microbiol 2014, 4, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Domenech M; García E; Moscoso M Microb Biotechnol 2012, 5, 455–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cuevas RA; Eutsey R; Kadam A; West-Roberts JA; Woolford CA; Mitchell AP; Mason KM; Hiller NL Mol Microbiol 2017, 105, 554–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aggarwal SD; Eutsey R; West-Roberts J; Domenech A; Xu W; Abdullah IT; Mitchell AP; Veening JW; Yesilkaya H; Hiller NL PLoS Pathog 2018, 14, e1007328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Håvarstein LS; Coomaraswamy G; Morrison DA Proc Natl Acad Sci U S A 1995, 92, 11140–11144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lau GW; Haataja S; Lonetto M; Kensit SE; Marra A; Bryant AP; McDevitt D; Morrison DA; Holden DW Mol Microbiol 2001, 40, 555–571. [DOI] [PubMed] [Google Scholar]
- 47.Oggioni MR; Trappetti C; Kadioglu A; Cassone M; Iannelli F; Ricci S; Andrew PW; Pozzi G Mol Microbiol 2006, 61, 1196–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hui FM; Zhou L; Morrison DA Gene 1995, 153, 25–31. [DOI] [PubMed] [Google Scholar]
- 49.Ween O; Gaustad P; Håvarstein LS Mol Microbiol 1999, 33, 817–827. [DOI] [PubMed] [Google Scholar]
- 50.Luo P; Li H; Morrison DA Mol Microbiol 2003, 50, 623–633. [DOI] [PubMed] [Google Scholar]
- 51.Peterson SN; Sung CK; Cline R; Desai BV; Snesrud EC; Luo P; Walling J; Li H; Mintz M; Tsegaye G; Burr PC; Do Y; Ahn S; Gilbert J; Fleischmann RD; Morrison DA Mol Microbiol 2004, 51, 1051–1070. [DOI] [PubMed] [Google Scholar]
- 52.Johnsborg O; Kristiansen PE; Blomqvist T; Håvarstein LS J Bacteriol 2006, 188, 1744–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pozzi G; Masala L; Iannelli F; Manganelli R; Havarstein LS; Piccoli L; Simon D; Morrison DA J Bacteriol 1996, 178, 6087–6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Whatmore AM; Barcus VA; Dowson CG J Bacteriol 1999, 181, 3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bartilson M; Marra A; Christine J; Asundi JS; Schneider WP; Hromockyj AE Mol Microbiol 2001, 39, 126–135. [DOI] [PubMed] [Google Scholar]
- 56.Zhu L; Lau GW PLoS Pathog 2011, 7, e1002241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu L; Lin J; Kuang Z; Vidal JE; Lau GW Mol Microbiol 2015, 97, 151–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Duan C; Zhu L; Xu Y; Lau GW PLoS One 2012, 7, e44710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McBrayer DN; Cameron CD; Tal-Gan Y Org Biomol Chem 2020, 18, 7273–7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang Y; Koirala B; Sanchez LA; Phillips NR; Hamry SR; Tal-Gan Y ACS Chem Biol 2017, 12, 1141–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang Y; Tal-Gan Y Bioorg Chem 2019, 89, 102987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Koirala B; Phillips NR; Tal-Gan Y ACS Med Chem Lett 2019, 10, 880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang Y; Lin J; Harrington A; Cornilescu G; Lau GW; Tal-Gan Y Proc Natl Acad Sci U S A 2020, 117, 1689–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koirala B; Lin J; Lau GW; Tal-Gan Y Chembiochem 2018, 19, 2380–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koirala B; Tal-Gan Y Chembiochem 2020, 21, 340–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Milly TA; Tal-Gan Y RSC Chem Biol 2020, 1, 60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Koirala B; Hillman RA; Tiwold EK; Bertucci MA; Tal-Gan Y Beilstein J Org Chem 2018, 14, 1769–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang Y; Cornilescu G; Tal-Gan Y Biochemistry 2018, 57, 5359–5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhu L; Lau GW Expert Rev Anti Infect Ther 2013, 11, 227–229. [DOI] [PubMed] [Google Scholar]
- 70.Håvarstein LS; Hakenbeck R; Gaustad P J Bacteriol 1997, 179, 6589–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shekhar S; Khan R; Ferreira DM; Mitsi E; German E; Rørvik GH; Berild D; Schenck K; Kwon K; Petersen F Front Immunol 2018, 9, 747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Salvadori G; Junges R; Morrison DA; Petersen FC Front Cell Infect Microbiol 2019, 9, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Farha MA; Verschoor CP; Bowdish D; Brown ED Chem Biol 2013, 20, 1168–1178. [DOI] [PubMed] [Google Scholar]
- 74.Slager J; Kjos M; Attaiech L; Veening JW Cell 2014, 157, 395–406. [DOI] [PubMed] [Google Scholar]
- 75.Domenech A; Slager J; Veening JW Cell Rep 2018, 25, 2390–2400.e2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sturød K; Salvadori G; Junges R; Petersen FC Mol Oral Microbiol 2018, 33, 378–387. [DOI] [PubMed] [Google Scholar]
- 77.Moreno-Gámez S; Sorg RA; Domenech A; Kjos M; Weissing FJ; van Doorn GS; Veening JW Nat Commun 2017, 8, 854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Domenech A; Brochado AR; Sender V; Hentrich K; Henriques-Normark B; Typas A; Veening JW Cell Host Microbe 2020, 27, 544–555.e543. [DOI] [PubMed] [Google Scholar]
- 79.Rosch JW; Tuomanen EI Cell Host Microbe 2020, 27, 489–490. [DOI] [PubMed] [Google Scholar]
- 80.Claverys JP; Prudhomme M; Martin B Annu Rev Microbiol 2006, 60, 451–475. [DOI] [PubMed] [Google Scholar]
- 81.Sebert ME; Patel KP; Plotnick M; Weiser JN J Bacteriol 2005, 187, 3969–3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ibrahim YM; Kerr AR; McCluskey J; Mitchell TJ Infect Immun 2004, 72, 3584–3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu Y; Zeng Y; Huang Y; Gu L; Wang S; Li C; Morrison DA; Deng H; Zhang JR Mol Microbiol 2019, 112, 1308–1325. [DOI] [PubMed] [Google Scholar]
- 84.Sebert ME; Palmer LM; Rosenberg M; Weiser JN Infect Immun 2002, 70, 4059–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ibrahim YM; Kerr AR; McCluskey J; Mitchell TJ J Bacteriol 2004, 186, 5258–5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dawid S; Sebert ME; Weiser JN J Bacteriol 2009, 191, 1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kochan TJ; Dawid S J Bacteriol 2013, 195, 1561–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mortier-Barrière I; de Saizieu A; Claverys JP; Martin B Mol Microbiol 1998, 27, 159–170. [DOI] [PubMed] [Google Scholar]
- 89.Mirouze N; Bergé MA; Soulet AL; Mortier-Barrière I; Quentin Y; Fichant G; Granadel C; Noirot-Gros MF; Noirot P; Polard P; Martin B; Claverys JP Proc Natl Acad Sci U S A 2013, 110, E1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wilke KE; Francis S; Carlson EE ACS Chem Biol 2015, 10, 328–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Goswami M; Wilke KE; Carlson EE J Med Chem 2017, 60, 8170–8182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wilke KE; Fihn CA; Carlson EE Bioorg Med Chem 2018, 26, 5322–5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Goswami M; Espinasse A; Carlson EE Chem Sci 2018, 9, 7332–7337. [DOI] [PMC free article] [PubMed] [Google Scholar]