

Abstract
A missense mutation (R620W) of Protein Tyrosine Phosphatase, non-receptor type 22 (PTPN22), which encodes Lymphoid-tyrosine Phosphatase (LYP), confers genetic risk for multiple autoimmune diseases including type 1 diabetes (T1D). LYP has been putatively demonstrated to attenuate proximal T and B cell receptor signaling. However, limited data exist regarding PTPN22 expression within primary T cell subsets and the impact of the T1D risk-variant on human T cell activity. Herein, we demonstrate endogenous PTPN22 is differentially expressed and dynamically controlled following activation. From control subjects homozygous for the non-risk allele, we observed 2.1 (P<0.05) and 3.6-fold (P<0.001) more PTPN22 transcripts in resting CD4+ memory and regulatory T cells (Tregs), respectively, over naïve CD4+ T cells, with expression peaking 24 hours post-activation. When LYP was overexpressed in conventional CD4+ T cells (Tconv), T cell receptor (TCR) signaling and activation were blunted by LYP-620R (P<0.001) but only modestly affected by the LYP-620W risk-variant versus mock-transfected control, with similar results observed in Tregs. LYP overexpression only impacted proliferation following activation by antigen presenting cells, but not anti-CD3 and anti-CD28 coated microbeads, suggesting LYP modulation of pathways other than TCR. Notably, proliferation was signifacanlty lower with LYP-620R than with LYP-620W overexpression in Tconv, but as similar in Treg. These data indicate that the LYP-620W variant is hypomorphic in the context of human CD4+ T cell activation and may have important implications for therapies seeking to restore immunological tolerance in autoimmune disorders.
INTRODUCTION
Genome Wide Association Studies (GWAS) have identified over 60 genetic variants that are thought to confer varying degrees of susceptibility for the T cell-mediated autoimmune disease, Type 1 Diabetes (T1D) (1, 2). Protein tyrosine phosphatase, non-receptor type 22 (PTPN22), which encodes Lymphoid-tyrosine Phosphatase (LYP), is one such candidate gene bearing the strongest T1D risk association after the HLA class II and INS-IGF2 loci (3, 4). A missense C1858T single nucleotide polymorphism (SNP) in the PTPN22 coding sequence results in an LYP variant with an Arginine to Tryptophan substitution at position 620 (R620W) that is associated with increased risk for a number of autoimmune conditions including T1D, rheumatoid arthritis, systemic lupus erythematosus, Graves’ Disease, vitiligo, and myasthenia gravis (3, 5–10). Most early studies of PTPN22-associated risk centered around lymphocytes, given their central role in autoimmune pathogenesis. However to date, no consensus has been reached regarding whether LYP R620W constitutes a gain- or loss-of-function for phosphatase activity (11, 12). Moreover, it remains poorly characterized how the risk variant influences human regulatory T cell (Treg) versus conventional T cell (Tconv) function in a manner that facilitates autoimmune disease development.
Ptpn22 was originally identified in the mouse (13) and later in humans (14) as a cytoplasmic phosphatase predominantly expressed in lymphoid cells. It is composed of a catalytic domain responsible for its phosphatase activity as well as a series of proline rich domains that regulate activity and protein interactions, most notably, with C-terminal Src kinase (CSK) (15). In T cells, LYP dephosphorylates several proximal T cell receptor (TCR) signaling molecules including LCK, CD3ε, CD3ζ, zeta-chain associated protein kinase 70 (ZAP-70), and VAV to down-modulate TCR signaling and is therefore, predicted to have a central role in T cell selection, activation, and differentiation (16). It is known that LYP also functions in B cells, as well as myeloid cells (17–21). As such, LYP has been shown to modulate B cell receptor signaling (22), Toll-like receptor (TLR) signaling (23–25), NLRP3 signaling (19), and outside-in integrin signaling (26, 27). Hence, LYP serves many functions across the cellular immune landscape, and it is possible that modulation of LYP expression levels may play an important role during immune cell development and activation.
In murine models, Ptpn22 deficiency has been associated with increased Treg numbers in both the thymus and the periphery as well as with protection against the induction of experimental autoimmune encephalomyelitis (EAE) (28, 29). Similarly, Ptpn22 deficiency resulted in reduced disease severity and incidence in the SKG mouse model of rheumatoid arthritis as well as enhanced tolerance of allogeneic islet transplants (30, 31). Conversely, while Ptpn22 deficiency does not confer overt autoimmune disease, it has been shown to facilitate autoimmunity when combined with other genetic risk factors, such as hyperactive CD45 E613R mutation, BXSB background, and the KBxN arthritis model (32–34). Similar to its known association with human T1D susceptibility, Ptpn22 has been identified as the candidate gene for the Insulin-dependent diabetes 18.2 (Idd18.2) diabetes susceptibility locus in the non-obese diabetic (NOD) mouse model (35). Interestingly, while silencing Ptpn22 expression in NOD mice resulted in reduced incidence of diabetes (36), expression of the orthologous human risk variant in the endogenous Ptpn22 locus promoted autoimmunity in the NOD strain (37, 38). Altogether, these observations support the notion that LYP likely influences T cell development and selection in the thymus as well as activation in the periphery, suggesting that the autoimmune-associated variant may confer multifaceted effects on host immunity.
T cells isolated from the peripheral blood of human T1D patients carrying the LYP-620W variant exhibited reduced proliferation and calcium flux in response to polyclonal stimulation (39), and heterozygosity for LYP-620W was associated with reduced IL-10 production after stimulation of memory CD4+ T cells (17). There is conflicting evidence as to whether the LYP-620W variant impairs interactions of LYP with CSK, which are important for regulating TCR signal strength (32, 40, 41). Further, it is unclear whether the LYP/CSK interaction strengthens or weakens inactivation of the Src family kinase LCK (40, 41). Thus, there is a need to investigate the complex effects of the LYP-620W variant. In this study, we used a lentiviral gene delivery system to stably overexpress the LYP-620R non-risk and LYP-620W risk variants in human Tconv and Treg. Our objective was to assess the cell-intrinsic effect conferred by LYP-620W in the CD4+ T cell compartment while mitigating epistatic biological variability inherent in studies of genotype-selected donors. The methods herein, may also be applied to other cell types, risk variants, or combinations thereof, to facilitate pathway-directed therapies for autoimmune disorders.
MATERIALS AND METHODS
Genotyping
PTPN22 genotype was determined using a Taqman Genotype Assay (ThermoFisher) for rs2476601 per manufacturer instructions. In this report, we refer to the reverse strand genotype (rs2476601 C>T) to match the PTPN22 gene orientation.
Donors
Healthy control donors (median age 30.9, range 23.6–38.0 y; N=8) that were homozygous for the non-risk (C/C) PTPN22 variant at rs2476601 were selected from the UFDI Biobank. Fresh venous blood was collected in sodium-heparinized Vacutainer tubes (BD Biosciences) after informed consent in accordance with the University of Florida Institutional Review Board approved protocol (IRB201400703). Leukopack blood donations were also obtained from rs2476601 C/C homozygous individuals under IRB-exempt protocols from Life South Blood Centers (N=9) in sodium citrate. All studies involving human samples were conducted in accordance wth the Declaration of Helsinki.
Cell Enrichment and Sorting
For fluorescence-activated cell sorting (FACS) experiments, whole blood or leukopaks were pre-enriched by CD4+ T cell negative selection with RosetteSep (Stemcell Technologies) prior to density gradient centrifugation. CD4+ T cells were labeled with fluorescent antibodies against CD4 (RPA-T4), CD45RA (HI100), CD197 (G043H7), CD127 (A019D5) and CD25 (BC96) as previously described (42). Treg were FACS-purified as CD4+CD25+CD127lo/-, while naïve Tconv were FACS-purified as non-Treg CD4+CD127+CD45RA+CD197+, and memory Tconv were FACS-purified as non-Treg CD4+CD127+CD45RA- on a FACS Aria III (BD Biosciences) as previously described (43).
PTPN22 Transcript Quantification
Leukopak enriched CD4+ T cell subsets were used for endogenous PTPN22 expression kinetic studies. RNA was prepared from 106 naïve Tconv, memory T cells, and Treg either immediately after sorting, or at various time points following activation. Tconv were activated with a 1:1 bead to cell ratio of Dynabeads Human T-Activator CD3/CD28 (ThermoFisher), and Treg were activated with 4:1 ExpAct beads (Miltenyi). Cells were supplemented with growth factor cytokines every 2–3 days: 300U/mL IL-2 for Tregs, 20U/mL IL-2 for Tconv, and after 7 days, 5 ng/μL IL-7 was also added for Tconv. RNA was Qiashredded and extracted using RNeasy Plus Kit (Qiagen) and reverse-transcribed into cDNA using Protoscript First Strand cDNA Synthesis kit (New England Biolabs). The expression level of PTPN22 was measured using an absolute quantification real-time PCR (RT-qPCR) approach on a Lightcycler 480 platform (Roche Diagnostics). Each reaction well contained 1μL PTPN22 TaqMan® Gene Expression Assay solution (ThermoFisher), 2μL LightCycler480 Probe Master Mix (Roche), and 3μL cDNA in a 20μL reaction. To create the DNA standards for the RT-qPCR assay, the pUC57.LYP620R.fuT2A.eGFP (see Lentiviral Vector Design and Construction) plasmid containing PTPN22 coding sequence (CDS) was serially diluted 10 fold. Lambda DNA was used to bring all standards to equivalent concentrations of total DNA (1.2ng/μL). Six concentrations were prepared ranging from 1.2fg/μL to 12ng/μL of plasmid, in addition to an NTC consistiong of 1.2ng/μL Lambda DNA only. 3μL of standard DNA was used in reactions as above. Standards were converted to transcript copy number by dividing the mass of plasmid per reaction in grams by the plasmid formula weight, where 650g/mol was used as the average nucleotide pair formula weight times the plasmid length. Copy number of each standard per well was log-transformed and plotted against the corresponding Ct value to form a standard curve. The standard curve R2>0.998 for each batch, and all samples were within range of the standard curves. PTPN22 expression level of each sample was extrapolated from the standard curve as previously done (44).
Lentiviral Vector Design and Construction
Design of the LYP620R.fuT2A.eGFP construct began with the human PTPN22 full length isoform coding DNA sequence (CDS) containing the non-risk variant (C at position 1858 of the CDS). Two endogenous EcoRI restriction enzyme sites (GAATTC) within the CDS were synonymously mutated, and the stop codon was removed (Supplemental Figure 1A–B). This was followed by a furin cleavage site, a T2A self-cleaving peptide sequence (45), and an enhanced green fluorescent protein (eGFP) coding sequence (stop codon included). The LYP620R.fuT2A.eGFP was synthesized in a pUC57 cloning vector by GenScript (Piscataway, NJ). Site-directed mutagenesis by PCR with pfu DNA polymerase followed by DpnI digest was performed to generate the LYP620W.fuT2A.eGFP construct, which contains the PTPN22 C1858T variant that encodes the LYP R620W amino acid change. These constructs were then subcloned into the pFUGW 3rd generation lentiviral vector (46). pFUGW expressing only eGFP served as the control vector. Lentiviral preparations were conducted as previously described (47). All constructs were validated by restriction enzyme digest and sequence analysis.
Cell Transduction
FACS-isolated naïve Tconv were activated with a 1:1 bead to cell ratio of Dynabeads Human T-Activator CD3/CD28 (ThermoFisher), and Treg were activated with 4:1 ExpAct beads (Miltenyi). On day 2, the cultures were infected with 0.5 titered units of lentivirus per cell in the presence of 8μg/mL protamine sulfate (Sigma), and spinnoculated as previously described (47). The cultures were then allowed to expand and rest for at least 21 days after activation, during which they were supplemented with growth factor cytokines every 2–3 days at dosages of 300U/mL IL-2 for Tregs and 20U/mL IL-2 and for Tconv. After 7 days, 5ng/μL IL-7 was also supplemented every 2–3 days with IL-2 for Tconv. Portions of the cultures were FACS-isolated into stably transduced (eGFP+) and mock-transduced (eGFP-) for cytokine and suppression assays. All other assays used the unsorted fractions of the cultures so that the results from stably transduced cells could be normalized to mock-transduced cells.
Western Blotting
FACS-isolated transduced (eGFP+) and mock-transduced (eGFP-) fractions were lysed with cold lysis buffer (Cell Signaling Technologies) containing protease inhibitors (Sigma). Protein was quantified via BCA assay (Thermo Scientific). 5μg of protein was denatured with Laemmli sample buffer at 95°C for 5 minutes and run on mini protean TGX gels (BioRad). Transfers were performed using the Transblot Turbo Transfer System (BioRad) and Transblot Turbo Transfer Packs (BioRad). Membranes were washed with 1X TBST, blocked with 5% milk in TBST for 1 hour, and incubated with primary antibody (rabbit anti-human LYP (clone D6D1H), β-actin (polyclonal) or GFP (clone D5.1), Cell Signaling Technologies) overnight at 4°C. Membranes were then washed with TBS-T, and incubated with anti-rabbit IgG-HRP secondary antibody (1:1000, Cell Signaling Technologies) for 1 hour at room temperature. Membranes were subsequently washed, developed with Western DuraSignal Substrate (Thermo Scientific), and imaged on the GeneGnome XRQ (Syngene). Western blot band quantification was performed in ImageJ (48). Serial blots were stripped with Restore Western Blot Stripping Buffer (ThermoFisher Scientific) between probes for 15 minutes at room temperature per manufacturer protocol.
Calcium Flux
Stably transduced Tconv or Treg, and their mock-transduced internal control cells, were labeled with 2.5μg/mL FuraRed calcium responsive dye (ThermoFisher) in 1% BSA/HBSS at 37°C for 10 minutes, followed by labeling with Live/Dead Near IR viability dye (ThermoFisher). Cells were then resuspended in HBSS (with Ca2+) containing 10μg/mL goat anti-mouse IgG F(ab’)2 crosslinking antibody (Jackson ImmunoResearch). Next, cells were warmed to 37°C, and fluorescence at 605 nm by a 405 nm laser and at 660 nm by a 532 nm laser was collected on a LSRFortessa (BD Biosciences) to indicate high and low calcium, respectively (Supplemental Figure 3A–C). After recording 30 seconds of basal fluorescence, anti-human CD3 (OKT3) was added at 1μg/mL, and fluorescence was acquired for two minutes. FlowJo software (FlowJo LLC) was used to analyze the mean fluorescence intensity (MFI) kinetics of FuraRed for the high and low [Ca2+] channels after first gating on live cells by forward and side scatter morphology and viability dye exclusion. The percent change from mock in fluxed calcium was calculated as the percent difference between curves of the mock and transduced cells (Supplemental Figure 3D–G).
pERK Signaling
Lentiviral-transduced Tconv or Treg and their mock-transduced internal control cells were labeled with viability dye, then washed into assay buffer (RPMI base medium) and plated at 106 cells per well in 96-well V-bottom plates in the presence of 10μg of anti-mouse IgG crosslinking antibody (Jackson ImmunoResearch). Anti-CD3 (2μg of OKT3) was added at times −10, −5, and −2 minutes. At time 0, wells were fixed with 4% formalin for 10 minutes. Cells were then washed, permeabilized with methanol, and stained with PE-conjugated anti-phospho-ERK (anti-pERK; clone 197G2, Cell Signaling Technology) for 60 minutes. Data was acquired by flow cytometry on an LSRFortessa (BD Biosciences), and pERK MFI of live cells was assessed using FlowJo software (Flowjo LLC).
Proliferation and Suppression Assays
Lentiviral-transduced (eGFP+) and internal control mock-transduced (eGFP-) cells were labeled with Cell Trace Violet (CTV, ThermoFisher) cell tracking dye and activated with either beads, autologous antigen presenting cells (APCs), 10ng/mL phorbol 12-myristate 13-acetate (PMA) plus 1 μM ionomycin, or K562 cell line artificial APCs (aAPCs) which do not express MHC class I or class II molecules but do express murine Fc’ receptor (CD64) and human CD86 (49). In brief, beads were used at a 1:1 bead to cell ratio as described in Cell Transduction above. Autologous APCs, generated from PBMCs that were CD3-depleted (RosetteSep) and exposed to 3000 Rad of gamma irradiation, were cultered 1:1 with T cells in the presence of 2μg/mL anti-CD3 and 1μg/mL anti-CD28. To generate aAPCs, K562 cells (human chronic myeloid leukemia (CML) line) expressing murine CD64 and human CD86 were incubated with various ratios of mouse anti-human CD3 (OKT3) and isotype antibody, and then irradiated with 3000 Rad. The ratios of isotype to OKT3 used were 1:0, 9:1, 1:1, and 0:1, such that the resulting aAPCs were loaded with 0, 10, 50, and 100% anti-CD3, respectively. aAPC were similarly labelled to include mouse anti-human CD4 (OKT4) in ratios of 1:0:0, 1:0.5:0.5, and 0:0.5:0.5 (isotype:OKT3:OKT4), such that the resulting aAPCs were loaded with 0, 5, 25, and 50% each activating antibody. These were cultured at a 1:4 aAPC to T cell ratio.
Suppression assays were performed as previously described (50). Briefly, titering proportions of Tregs labelled with Cell Proliferation Dye eFluor670 (CPD670, ThermoFisher) were co-cultured with autologous CTV-labelled responder Tconv (Tresp) and unlabelled APCs ranging from 1:1:1 to 0:1:1 Treg:Tresp:APC ratios. Suppression assay cultures were set up in triplicate in round-bottom 96-well plates and activated with 2μg/mL anti-CD3 and 1μg/mL anti-CD28. Two versions of the suppression assays were performed. The first, which was designed to assess the response of LYP-modulated Tconv to Treg suppression, consisted of a combination of stably-transduced and mock-transduced Tconv (not eGFP-sorted) being suppressed by unmodulated Treg (mock-transduced sorted eGFP-). The second, which was designed to assess suppression by LYP-modulated Treg, consisted of unmodulated Tconv (mock-transduced sorted eGFP-) being suppressed by stably-transduced Treg (sorted GFP+).
Proliferation and suppression assay data were acquired on an LSRFortessa (BD Biosciences), and analyze using FlowJo software (Flowjo LLC). Live T cells were gated by forward and side scatter morphology, followed by viability dye exclusion. Proliferation of live-gated cell-tracking dye+ cells was assessed by expansion index. Suppression was calculated as the percent decrease in Tresp proliferation when co-cultured with Tregs as compared to when cultured without Tregs.
Surface Marker Activation Kinetics
Stably transduced (eGFP+) and internal control mock-transduced (eGFP-) cells were activated with irradiated autologous APCs at 1:1 in the presence of 2μg/mL anti-CD3 and 1μg/mL anti-CD28. The cultures were harvested at 0h, 4h, 24h, 48h, 72h, and 144h and stained with Live/Dead Near-IR viability dye (ThermoFisher). Next, the cells were washed into 2% FCS in PBS and labelled with CD278 (C398.4A, BioLegend), CD69 (FN50, BioLegend), CD4 (RPA-T4, BioLegend), CD226 (11A8, BioLegend), CD25 (BC96, BioLegend), CD279 (EH12.2H7, eBiosciences), CTLA4 (L3D10, BioLegend). The cells then were fixed in 1% formaldehyde in PBS. Data was acquired on an LSRFortessa (BD Biosciences) and was analyzed using FlowJo software (Flowjo LLC). Live lymphocytes from CD4+ T cell cultures were gated by forward and side scatter morphology, followed by viability dye exclusion before extracting surface marker expression. Fluorescence minus-one (FMO) controls were used to set gates for marker positivity.
Cytokine Detection
Stably transduced (eGFP+) T cells were activated with irradiated autologous APCs at 1:1 in the presence of 2μg/mL anti-CD3 and 1μg/mL anti-CD28. Supernatants were collected at the specified time points, and cytokines were detected via multiplexed bead-based ELISA (42).
Statistics
Data were analyzed by one-way ANOVA with Bonferroni’s post-hoc analysis or by two-way ANOVA with Bonferroni’s posttest, and graphs were prepared using GraphPad Prism software v6. P < 0.05 was considered significant. Subject matching was used to compare donors’ cells across experimental conditions (biological replicates).
RESULTS
PTPN22 expression varies by lymphocyte subset and activation state.
We examined endogenous PTPN22 expression in human naïve, memory, and regulatory CD4+ T cell subsets at rest and following in vitro activation. CD4+ T cells were sorted from leukopacks determined to be homozygous for the T1D non-risk allele, which encodes LYP-620R. Since the transcriptional profile of activated cells is dramatically altered from that of the quiescent state, such that the standard array of housekeeping genes is broadly upregulated, we employed absolute quantification to assess PTPN22 expression. At rest, naïve Tconv expressed significantly less PTPN22 (387±83 transcripts/ng total RNA) than memory Tconv cells (741±169 transcripts/ng total RNA, P < 0.05) and Tregs (1014±120 transcripts/ng total RNA, P < 0.001) (Figure 1A). Following polyclonal activation, all three subsets rapidly increased PTPN22 expression, peaking at 24 hours (Figure 1B). Naïve CD4+ Tconv increased 12.00±5.45 fold from the quiescent state whereas memory Tconv increased 4.01±0.89 fold (P < 0.001 vs. naïve) and Treg increased 2.85±1.31 fold (P < 0.01 vs. naïve). Following the initial increase, PTPN22 expression decreased and plateaued around day 7. Taken together, we found PTPN22 expression to differ across T cell subsets at rest and in response to activation.
Figure 1. Endogenous PTPN22 is differentially expressed in CD4 T cell subsets.
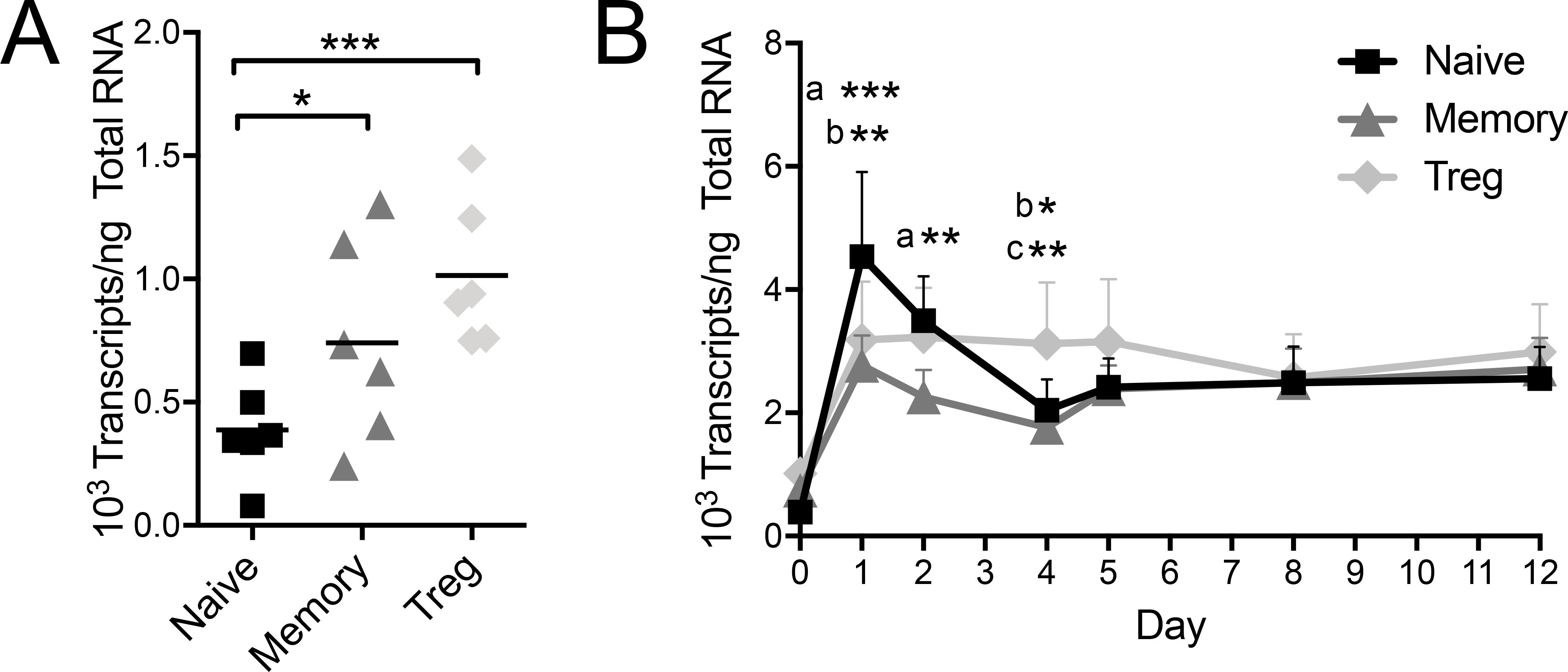
Primary naïve Tconv, memory Tconv and Treg subsets were separated by FACS from leukopack donations and PTPN22 transcripts were quantified by absolute real time PCR. (A) In the resting state (freshly isolated on day 0), naïve Tconv have lower PTPN22 gene expression compared to memory Tconv or Tregs (One-way ANOVA using subject matching with Bonferroni’s posttest). (B) Following in vitro activation with surface-fixed anti-CD3/CD28 beads, naïve Tconv, memory Tconv, and Treg PTPN22 expression was quantified at d1, d2, d4, d5, d8, and d12. (n=6, Two-way ANOVA using subject and timepoint matching with Bonferroni’s posttest, *p<0.05, **p<0.01, ***p<0.001, a: Naïve vs Memory, b: Naïve vs Treg, c: Memory vs Treg)
Overexpression of PTPN22 decreases TCR signaling.
Next, we examined whether LYP-620R and −620W may differentially modulate T cell function in Tconv and Treg populations. Naïve CD4+ Tconv and Treg were sorted from PBMCs from normal healthy control donors that were homozygous for the T1D non-risk PTPN22 allele (C/C at rs2476601 encoding LYP-620R). Cells were then activated and transduced with a lentivirus to express bicistronic constructs of the non-risk LYP-620R or the risk LYP-620W variant with an eGFP reporter following a 2A element, or with eGFP alone for a vector control condition (Supplemental Figure 1A–B). The cultures were expanded, followed by an extended rest period for a total of 21–28 days to allow for reversion of activation programs. We confirmed stable overexpression of PTPN22 transcripts and LYP protien for each variant (Supplemental Figure 1C–E and Supplemental Figure 2).
The proximal signaling events resulting from TCR ligation lead to assembly and activation of the linker for activation of T cells (LAT) signalosome, followed by diverging downstream signaling pathways. These include calcium signaling and MAPK/ERK pathways (51). We sought to determine the functional impact of the non-risk and risk LYP variants on these downstream signaling pathways in Tconv and Treg. We first assessed Ca2+ flux by comparing stably transduced cells to non-transduced (eGFP-) internal culture control cells (Supplemental Figure 3). As expected, LYP-620R expressing Tconv exhibited 25.8±3.7% less Ca2+ flux than non-transduced Tconv, whereas transduction with eGFP alone had no effect (Figure 2A). A similar effect was observed for LYP-620R Tregs in which Ca2+ flux was diminished by 40.3±13.3% compared to non-transduced Tregs (Figure 2B). Conversely, LYP-620W expression only resulted in 4.9±1.6% and 7.9±2.7% less Ca2+ flux in Tconv and Treg, respectively (Figure 2A–B). These differences were not different from T cells transduced with eGFP alone. Thus, the risk LYP-620W variant less effectively downregulated TCR-induced Ca2+ flux.
Figure 2. The risk variant of PTPN22 has a weaker impact on distal TCR signaling than the non-risk variant.
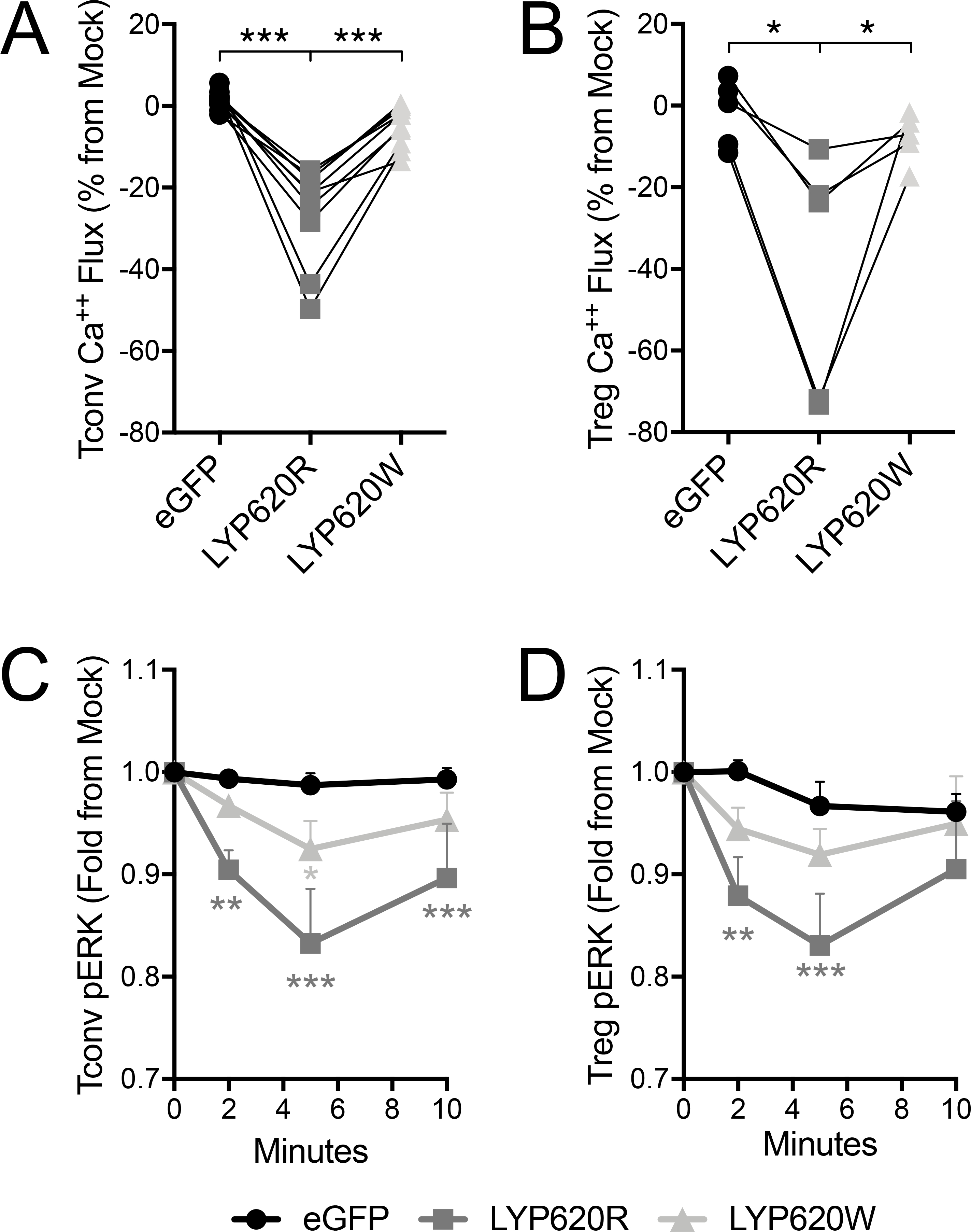
Primary human CD4 Tconv and Treg were transfected to express LYP-620R, LYP-620W risk variant, or eGFP as a control. Calcium flux by (A) Tconv and (B) Treg in response to soluble anti-CD3 and cross-linker was assessed by ratiometric fluorescent dye. The area under the curve during TCR activation was quantified (see Supplemental Figure 3) and then normalized to internal mock control cells (eGFP-). Individual donors are shown with lines connecting their matched T cell transductants (Tconv donors n=10, Treg donors n=5, One-way ANOVA using subject matching with Bonferroni’s posttest). Phospho-ERK (pERK) signaling by Tconv (C) and Treg (D) in response to soluble anti-CD3 and cross-linker was assessed by intracellular phospho-ERK staining. The mean fluorescence intensity (MFI) for each time point was quantified and then normalized to internal mock control cells (eGFP-). Normalized pERK means±SEM are shown with lines connecting the T cell transductant group over the timecourse (Tconv donors n=8, Treg donors n=5, Two-way ANOVA using subject and timepoint matching with Bonferroni’s posttest, *p<0.05, **p<0.01, ***p<0.001, shaded to indicate significance for that group versus eGFP).
We next assessed the impact of LYP modulation on the MAPK/ERK pathway via pERK signaling following TCR ligation. As with Ca2+ signaling, we observed decreased pERK relative to mock transfected cells in both Tconv and Tregs when LYP-620R was overexpressed (Figure 2C–D). Once again, the impact of LYP-620W on TCR signaling was diminished in both subsets when compared to LYP-620R. Taken together, these results indicate that the LYP risk variant has a diminished capacity to regulate TCR signaling activity.
The risk variant of PTPN22 permits increased T cell activation.
In response to Ca2+ flux and pERK signaling, T cells express receptors on their surface to direct activation. We further explored the impact of LYP variants by examining the experssion of a set of these activation markers. Stably transduced T cells were activated with autologous APCs, and surface expression of activation markers was assessed at 0, 4, 24, 48, 72, and 144 hours by flow cytometry. As expected, CD69, ICOS, CD25, CD226, and PD-1 were observed to be dynamically expressed following Tconv activation (Supplemental Figure 4A–D) (52–56). Likewise, Tregs also exhibited activation-induced expression kinetics for CD69, ICOS, CD226, CTLA-4 (Supplemental Figure 4E–F) (57). Interestingly, while most activation marker kinetics involved an intensification followed by a reduction of surface expression, the kinetics of CD226 on Tregs waned before increasing to a level higher than baseline (Supplemental Figure 4H). When normalized to internal mock-transfected controls, the activation marker kinetics on both Tconv (Figure 3A–E) and Tregs (Figure 3F–J) were blunted by overexpression of LYP-620R, with the exception of CD226 on Tconv (Figure 3D). Similar to what was observed with Ca2+ and pERK signaling, cells expressing the LYP-620W variant were less impacted, supporting the notion that the R620W variant confers a loss-of-function for LYP in terms of modulating of T cell activation.
Figure 3. The risk variant of PTPN22 less effectively inhibits activation.
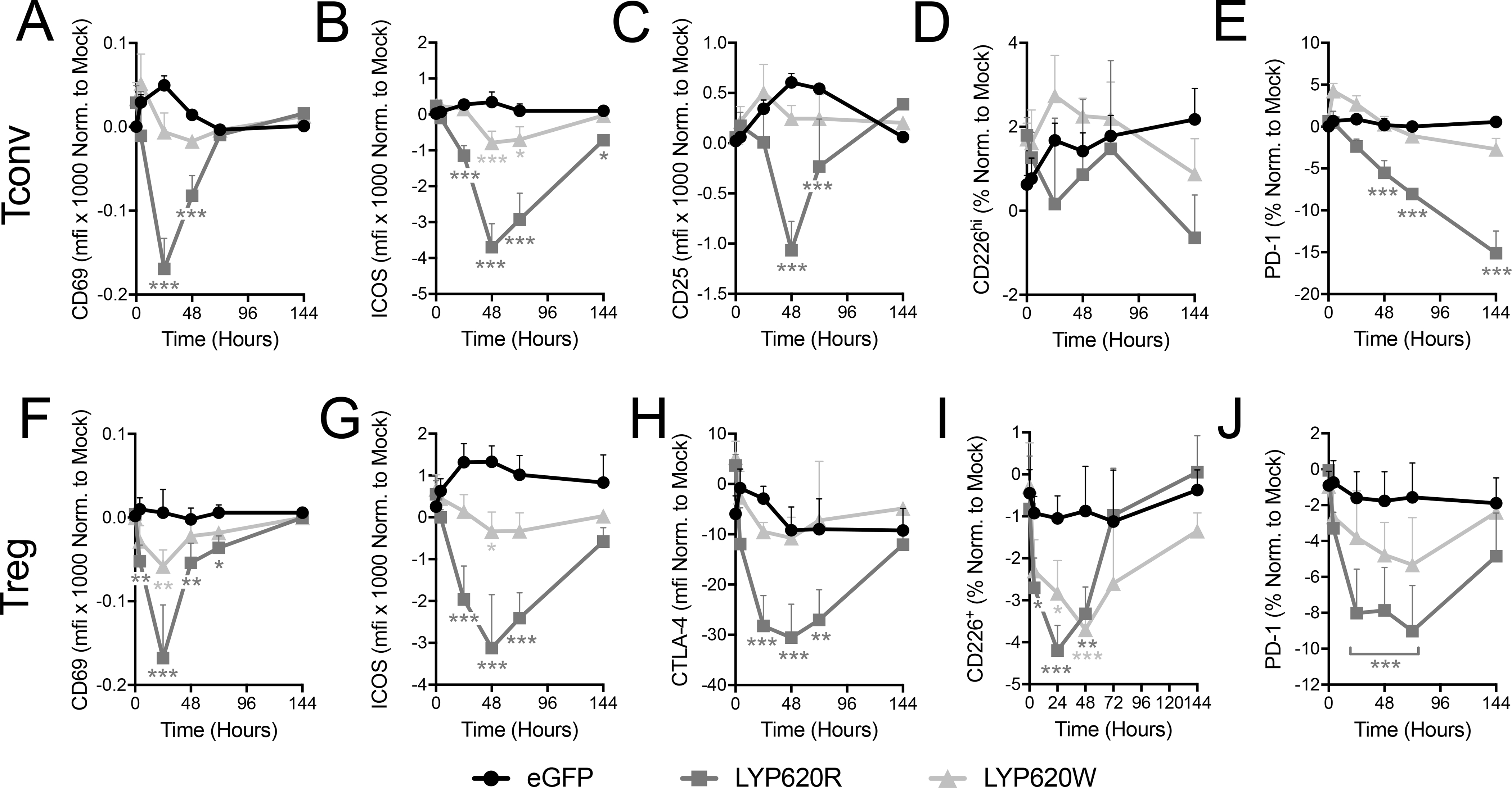
Activation marker expression on Tconv (A-E) and Tregs (F-G) in response to co-culture with antigen presenting cells (APC) and soluble anti-CD3 and anti-CD28 was assessed by surface staining and flow cytometry at 0, 4, 24, 48, 72, and 144 hours post activation. Tconv overexpressing LYP-620R, LYP-620W, or eGFP were assessed for CD69 (A), ICOS (B), CD25 (C), CD226 (D), and PD-1 (E). Treg overexpressing LYP-620R, LYP-620W, or eGFP were assessed for CD69 (F), ICOS (G), CTLA-4 (H), CD226 (I), and PD-1 (J). The mean fluorescent intensity (MFI) or percent positive was quantified and then normalized to internal mock control cells (eGFP-). (Tconv donors n=5, Treg donors n=5, Two-way ANOVA using subject and timepoint matching with Bonferroni posttest, *p<0.05, **p<0.01, ***p<0.001, shaded to indicate significance for that group versus eGFP at that time point).
The PTPN22 risk variant permits proliferation in Tconv, but not in Treg.
We demonstrated that the two LYP variants differentially modulate TCR signaling and expression of T cell activation markers. We next asked whether overexpression of the LYP variants would differentially modulate T cell proliferation. Activation-induced proliferation of Tconv and Treg cells was assessed by dilution of cell tracking dye following in vitro activation. Transduced cells (eGFP+) were cultured with mock-transduced (eGFP-) internal culture control cells. The cultures were activated with either anti-CD3 and anti-CD28 with irradiated APCs, anti-CD3 and anti-CD28 bound to microbeads, or with PMA and ionomycin. While expression of the eGFP reporter did not affect proliferation, overexpression of LYP-620R reduced APC-mediated proliferation of Tconv and Treg by 35.32±15.30% and 19.80±17.26%, respectively (Figure 4A–B). In Tconv, the LYP-620W variant was once again deficient in its ability to downregulate activation, as it did not impede proliferation to the same degree that was observed for the LYP-620R variant (Figure 4A). However, the LYP-620W variant was capable of blocking Treg proliferation to a similar degree as the LYP-620R variant (Figure 4B), indicating a differential impact of PTPN22 on Tconv and Tregs.
Figure 4. PTPN22–mediated abrogation of proliferation requires APC activation.
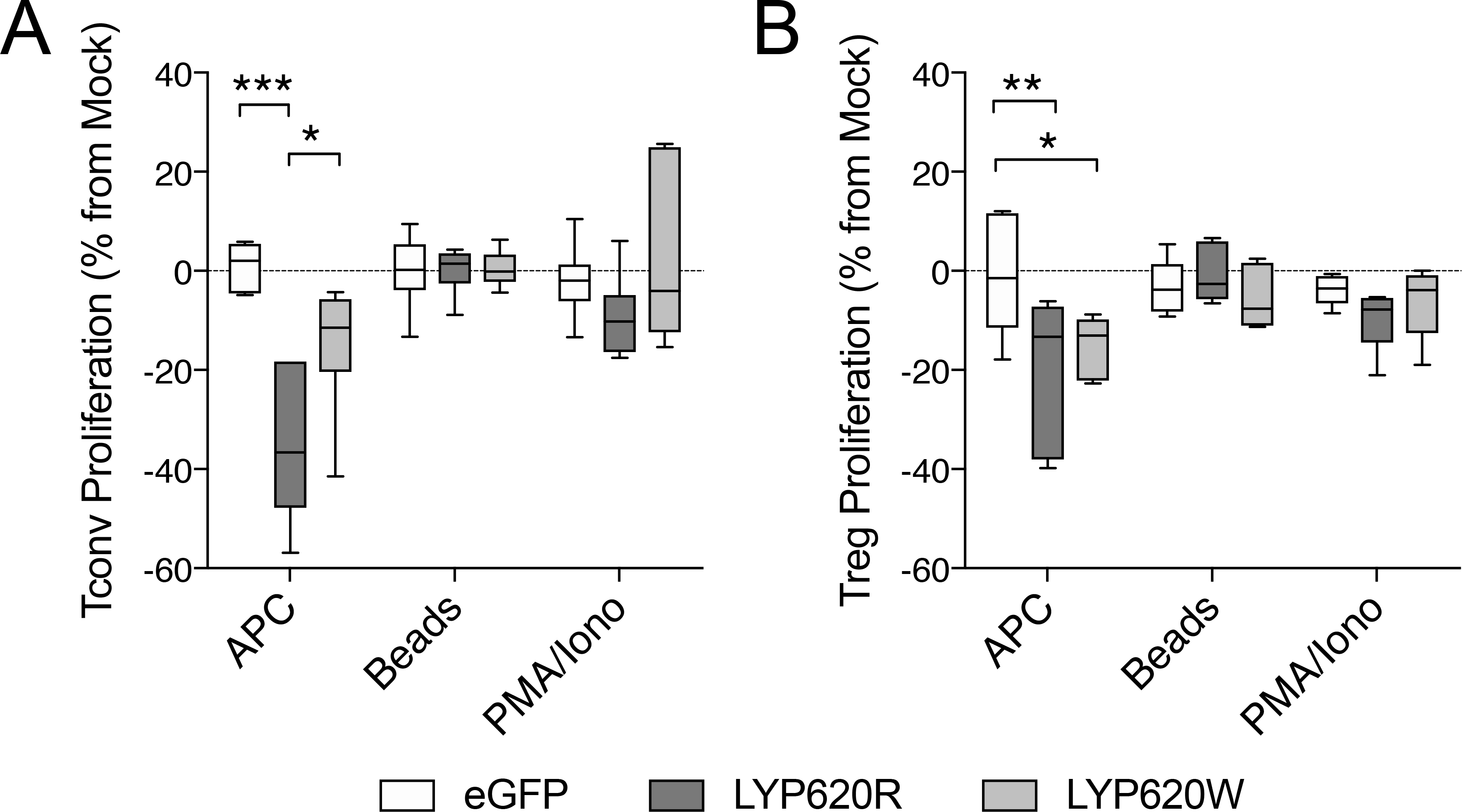
Proliferation was assessed for Tconv (A) and Tregs (B) overexpressing the LYP-620R variant, the LYP-620W risk variant, or eGFP transfected controls. Proliferation assessed by cell tracking dye dilution and was compared to internal mock control cells (GFP-). Three activation stimuli were compared: autologous antigen presenting cells (APC) along with soluble anti-CD3 and anti-CD28, surface-fixed anti-CD3/CD28 beads, and mitogenic compounds that bypass TCR signaling (PMA/Ionomycin). (Tconv donors n=6, Treg donors n=5, Two-way ANOVA using subject and treatment matching with Bonferroni posttest, *p<0.05, **p<0.01, ***p<0.001).
As expected, mitogenic activation by PMA and ionomycin was not altered by LYP overexpression, as all transduced Tconv and Treg proliferated to a comparable degree as their internal mock control cells (Figure 4A–B). Unexpectedly however, proliferation was also unaltered in transduced cells that were activated by microbeads (Figure 4A–B). To assess whether this was due to TCR signal strength, we repeated the experiment with K562 aAPCs loaded with titrated ratios of activating and isotype antibodies. The aAPCs did not induce proliferation when loaded with 0% anti-CD3 (100% isotype), whereas 10% and 50% anti-CD3 induced incrementally more proliferation, with a plateau from 50% to 100% anti-CD3 (Supplemental Figure 5A). In all cases, the proliferation of transduced cells was similar to their internal controls (Supplemental Figure 5B). Similar results were found when CD4 was included in the activation signal to allow for LYP-LCK interactions (Supplemental Figure 5C–D) (30, 58). This suggests that optimized signal strength, as well as fluid membrane interface are not sufficient to recapitulate the differential inhibition of proliferation by the two LYP variants, and that other factors supplied by natural APCs (e.g., adhesion molecules, soluble factors, and/or co-stimulatory and co-inhibitory ligand and downstream signaling interactions) may be required (30, 58).
Overexpression of PTPN22 influences T cell cytokine production.
CD4+ effector T cells modulate host immune responses after encountering cognate antigens, in part, through the production of an array of cytokines. Thus, we sought to assess the impact of LYP variant expression on the secretion of cytokines important in driving Th1 and Th2-associated immunity. Sorted transduced Tconv cells were activated with APCs, anti-CD3, and anti-CD28, and culture supernatants were assayed for cytokines over a 72-hour time period. The net accumulation of IL-2 in this system is a function of activation-induced secretion and of consumption via the IL-2 receptor (IL-2R) because the high-affinity component, CD25, is dramatically upregulated in this time period (Supplemental Figure 4) (59). We found that the net accumulation of IL-2 was not altered by overexpression of the LYP-620R variant, but was increased at the 24-hour time point by the LYP-620W variant (Figure 5A). The secretion of IL-4, IL-5, IL-9, IL-10, and IL-13 was robustly inhibited for the first 48 hours by expression of either LYP variant. At the72-hour time point, inhibition occurred to a somewhat lesser degree by LYP-620W, thought this difference was not significant between variants (Figure 5B–F). Finally, and somewhat surprisingly given the documented Th1 signature implicated in several of the autoimmune disorders that are associated with the rs2476601 risk variant (60, 61), IFNγ secretion was not altered by overexpression of either variant of LYP (Figure 5G). This result is in agreement with studies of Ptpn22 knockout mice (28). Taken together, these data demonstrate that both LYP variants result in similar levels of effector cytokine secretion.
Figure 5. Cytokine production by PTPN22 modulated CD4 T cells.
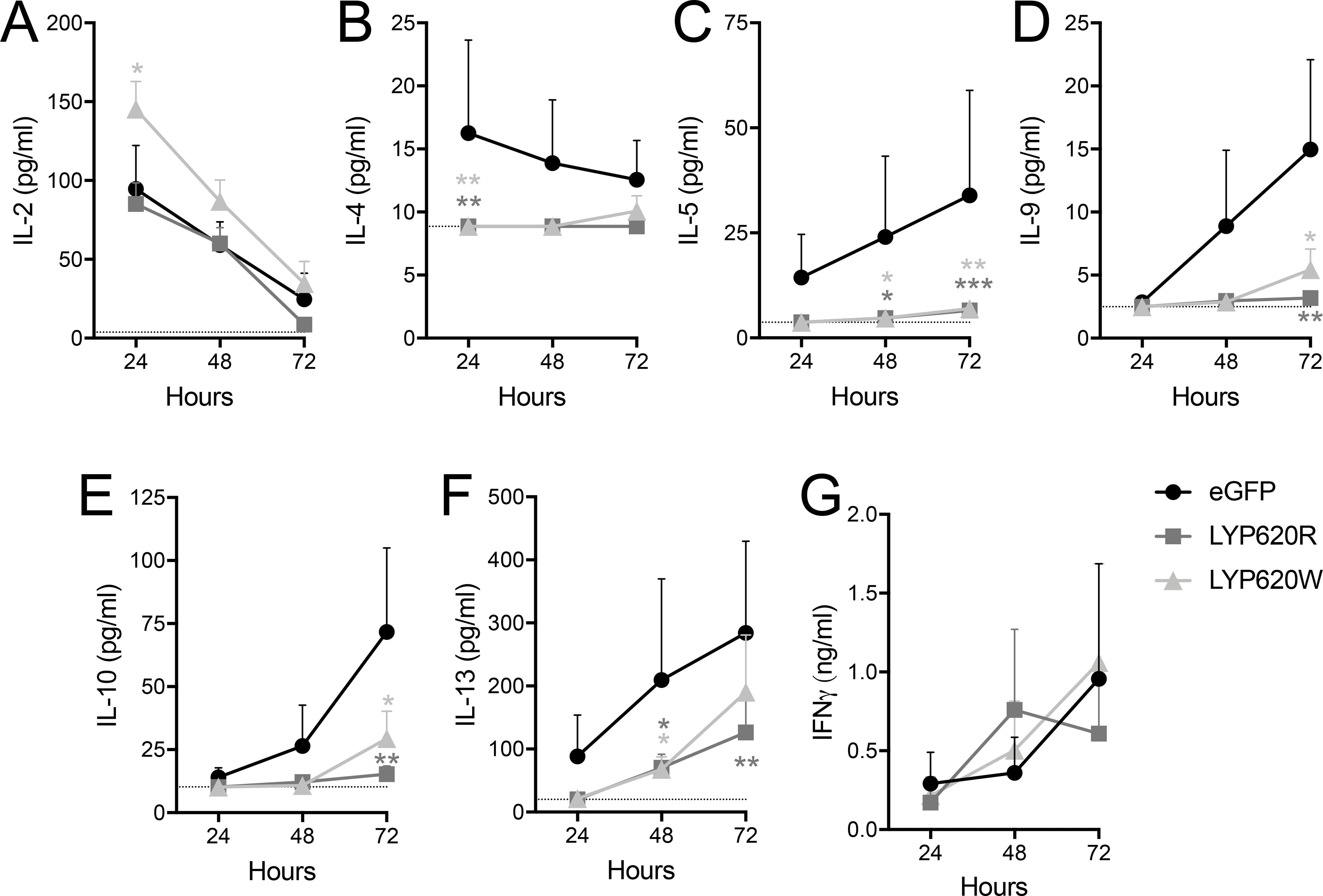
Tconv were sorted for stable transductants (eGFP+) and activated by antigen presenting cells (APCs) with soluble anti-CD3 and anti-CD28. (A) IL-2, (B) IL-4, (C) IL-5, (D) IL-9, (E) IL-10, (F) IL-13, and (G) IFNγ production were assessed by multiplexed immunoassay of culture supernatants at 24, 48, and 72 hours. The limit of detection for each assay is indicated as a dotted line. (Donor n=5, Two-way ANOVA using subject and timepoint matching with Bonferroni’s posttest, *p<0.05, **p<0.01, ***p<0.001, shaded to indicate significance for that group versus eGFP at that time point).
Treg-mediated suppression is altered by overexpressed PTPN22.
Our data demonstrated that PTPN22 is more highly expressed in Treg as compared to Tconv (Figure 1A). We therefore hypothesized that the PTPN22 risk variant may influence disease risk by altering Treg-mediated suppression. Hence, we assessed whether transduced Treg were deficient in their suppressive capacity and whether transduced Tconv were refractory to Treg suppression. In order to determine the effect of overexpressed LYP variants on Tresp, irradiated autologous APCs and titered amounts of Treg were co-cultured with transduced Tconv (eGFP+) and mock-transduced (eGFP-) internal control Tconv. We found that, similar to our previous results (Figure 4A), overexpressed LYP-620R significantly repressed Tresp proliferation while repression by LYP-620W was dramatically reduced across the Treg:Tresp range (Figure 6A). However, the relative suppression of transduced Tresp did not differ significantly from their internal mock controls (Figure 6B). This suggests that the Tconv-intrinsic susceptibility conferred by LYP-620W is due to reduced control over activation, proliferation, and effector mechanisms, rather than a defect in the ability of effector cells to be suppressed by Treg.
Figure 6. Suppression of modulated CD4 T cells and suppression by modulated Tregs is not affected by PTPN22 modulation.
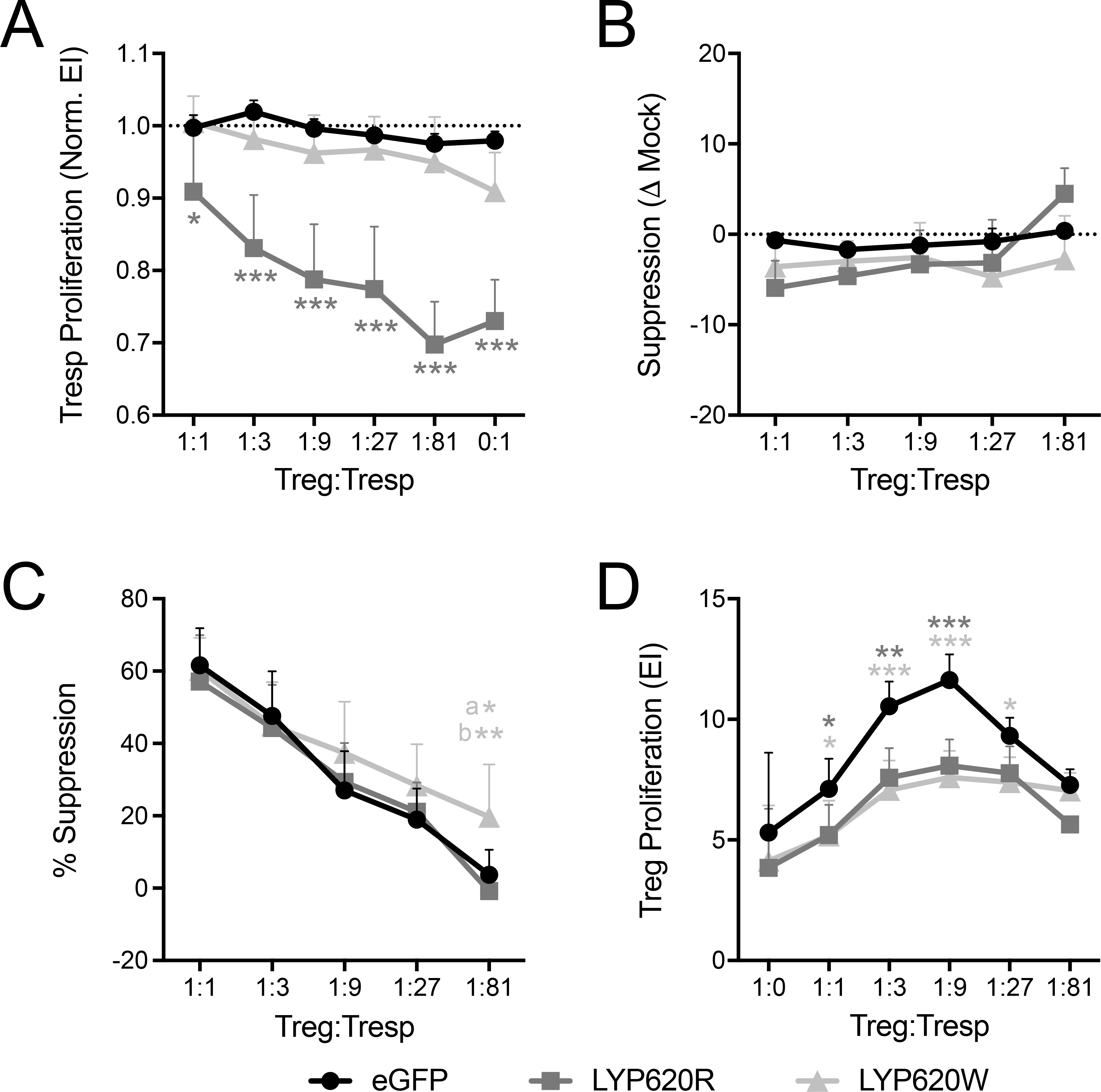
(A, B) Un-modulated regulatory T cells (Tregs, sorted eGFP-) were titrated and co-cultured with Tconv overexpressing LYP-620R, LYP-620W, or eGFP and their respective mock-transduced (eGFP-) internal control Tconv. APCs, anti-CD3, and anti-CD28 were used for activation for 4 days (Donor n=5). (A) Proliferation was assessed as Expansion Index (EI) by cell tracking dye dilution for transduced responder T cells (Tresp) cells and then normalized to the proliferation of internal control mock cells. (B) Percent suppression was calculated for the transduced cells and then normalized to the percent suppression of the mock-transduced cells. (C, D) Tregs overexpressing LYP-620R, LYP-620W, or eGFP (sorted eGFP+) were titrated and co-cultured with un-modulated Tconv (sorted eGFP-), followed by activation with APCs, anti-CD3, and anti-CD28 for 4 days (Donor n=4). (C) Tresp proliferation was assessed and used to calculate percent suppression. (D) Treg proliferation was assessed by cell tracking dye dilution. (Two-way ANOVA using subject and Treg:Tresp matching with Bonferroni posttest *p<0.05, **p<0.01, ***p<0.001, shaded to indicate significance for that group versus eGFP at that time point. In (A) we limited the statistical annotation to the comparisons to eGFP. The following were also significant for LYP-620R vs LYP-620W: 1:1 p<0.05, 1:3 p<0.001, 1:9 p<0.001, 1:27 p<0.001, 1:81 p<0.001, 0:1 p<0.001. In (C) a: LYP-620W vs eGFP, b: LYP-620W vs LYP-620R).
Finally, we assessed the effect of the LYP variants on Treg suppressive capacity by co-culturing titered amounts of sorted transduced (eGFP+) Treg with irradiated autologous APCs and untransduced Tresp. We found suppression by Tregs overexpressing LYP-620R to be similar to eGFP reporter Tregs, while suppression by Tregs expressing the risk LYP-620W variant was slightly increased (Figure 6C). Consistent with our previous result (Figure 4B), Treg proliferation in the suppression assay co-culture was similar for LYP-620R and LYP-620W (Figure 6D). This indicates that the difference in suppressive capacity was not due to differences in Treg proliferation, and suggests a Treg intrinsic impact of LYP-620W to enhance suppressive function.
DISCUSSION
To date, much of the literature has attempted to categorize rs2476601, the PTPN22 C1858T autoimmune-associated missense mutation, as a loss- or gain-of-function variant (11, 12). Because the LYP-620W variant confers risk for T cell-mediated autoimmune diseases (3, 5–8), we were interested in its allotypic effects on TCR signaling and function in the Tconv and Treg subsets. We first evaluated endogenous PTPN22 expression in primary human T cells homozygous for the T1D non-risk allele encoding LYP-620R. We then employed a lentiviral gene delivery system to induce constitutive overexpression of the T1D non-risk variant, LYP-620R, or T1D risk variant, LYP-620W, in primary human CD4+ Tconv and Tregs. There are a few advantages to this approach. First, it enables expansion of primary cell material that stably expresses either the non-risk or risk variant for long-term assays. Second, by isolating LYP variant overexpression to CD4+ T cells, the complicating effects of the LYP variant from other immune subsets are removed so that CD4+ T cell-intrisic effects can be studied. Finally, it controls for epistasis and the associated biologic variability by assessing the functional differences of the LYP variants within the same subjects.
Our observation that endogenous expression of PTPN22 varies by T cell subset and is dynamic throughout activation suggests that SNP-related functional effects are multifaceted and may differ across cell types in a manner that is subject to temporal control and activation state in the periphery. Importantly, our assay measured both splice variants which encode the 85 kD and 105 kD LYP isoforms that are differentially expressed in T cells at rest and after activation, respectively (14), thereby measuring total endogenous PTPN22 expression. Due to the MAF of rs2476601, we were unable to assess PTPN22 expression kinetics of the 620W variant using the methods herein. We will persue this question using scRNA-Seq technology, which requires many fewer cells, thus enabling time course examination of genotype-selected, cryopreserved PBMCs. We next demonstrated that, as expected, overexpression of LYP-620R decreases T cell Ca2+ flux, pERK signaling, surface expression of activation markers, and proliferation in both Tconv and Tregs. However, with the exception of Treg proliferation, overexpression of LYP-620W had little effect on TCR activation-induced responses. We therefore conclude that LYP-620W is hypomorphic in terms of CD4+ T cell responses to TCR activation.
Interestingly, proliferation was only affected by PTPN22 modulation in the context of APC activation, indicating a requirement for a natural immune synapse or specific costimulatory signaling. Burn et al. showed that primary human T cells with the endogenous LYP-620W variant had increased pERK1/2 induced by LFA-1/ICAM-1 outside-in signaling as compared to T cells expressing the LYP-620R variant (27). This resulted in stronger integrin-mediated adhesion that was resistant to shear forces, which may also apply to cell-cell interactions. Thus, whereas interactions between autoreactive T cells and APCs may normally be transient (62), expression of the LYP-620W variant may stabilize the immune synapse allowing for more efficient activation and initiation of autoreactivity. Similar experiments comparing LYP variants in the APC are necessitated to determine if the reported effect on synapse stability occurs bidirectionally.
Treg proliferation was blunted by LYP overexpression, but contrary to Tconv, Treg proliferation was not differentially impacted by the two LYP variants. This was the case when cultured alone with APCs as well as with APCs and Tconv in the context of suppression assays. Overall suppressive capacity was not affected for LYP-620R Tregs and was enhanced for LYP-620W Tregs. The data presented here further indicate that certain mechanisms of suppression, perhaps IL-2 consumption or regulatory cytokine production, may not be affected by LYP expression, while other mechanisms, such as contact-dependent co-inhibitory receptor interactions, may only be affected by the LYP-620W variant. It was previously shown that Tregs from Ptpn22 deficient mice had improved suppressive function due to increased and prolonged LFA-1 interactions between Tresp and Tregs (26). This is in line with the finding that LYP-620W is a loss-of-function variant with respect to outside-in integrin signaling, which results in sustained LFA-1 interactions (27). We previously showed that Tregs expressing a higher affinity TCR are more potent suppresors (63). Sustained Treg interactions conferred by LYP-620W may be functionally similar to higher affinity TCR engagement, resulting in enhanced suppressive capacity per cell (Figure 6C). Nevertheless, enhanced Treg suppressive capacity does not support a mechanism for autoimmune pathogenesis. In fact, suppression assays from rs2476601 T/T individuals were recently shown to exhibit lower suppression than those from C/C subjects (64). Thus, we posit that progression to autoimmunity may occur when the LYP-620W deficit in restraint of Tconv proliferation overcomes the LYP-620W enhancement of Treg suppression.
In terms of cytokine production, IL-2 accumulation was not altered in T cells overexpressing LYP-620R, but was increased by the LYP-620W variant. This finding is consistent with observations of the murine Pep-R619W ortholog knock-in (37). While further studies are required to determine if this is attributable to increased production or reduced consumption, this result may corroborate the observed differential effects of LYP variants on Tconv proliferation. The secretion of effector cytokines (IL-4, IL-5, IL-9, IL-10, and IL-13) was inhibited by overexpression of either variant, but this effect appeared to wane in LYP-620W Tconv by the 72-hour time point. IFNγ production by Tconv was not altered by overexperession of either LYP variant. This is in contrast to observations in a murine model of Ptpn22 overexpression, which exhibited decreased IFNγ production in effector T cells (65). We also did not observe increased IFNγ production in in LYP-620W expressing cells relative to LYP-620R expressing cells. Conversely, Anderson et al. recently reported increased IFNγ production and activation marker expression in PTPTN22-deficient human CD4+ T cells after 48 hours of reactivation (66). Those results suggests that a loss-of-function variant may also result in enhanced IFNγ secretion. While, the reason for these observed differences between experimental model systems is not clear at this point, we posit that constitutive overexpression and CRISPR knock-out systems impact LYP stoichiometry with TCR signaling molecules in an opposing manner. As such, cytokine regulation may not be affected in the same manner as activation and proliferation.
Faced with inconsistent reports in the current literature (12, 17, 39), our data nonetheless support the notion that the risk LYP-620W variant is generally a loss-of-function variant in Tconv, as TCR signaling, activation, and proliferation are less affected by its overexpression compared to LYP-620R. Still, the fundamental question of whether the LYP-620W functional decrease results from abrogated phosphatase activity or altered localization remains outstanding. While altered localization may also expose a distinct set of targets to dephosphorylation, it likely also disengages LYP-620W from the TCR signalosome, where its modulatory capacity is specifically tied to weaker TCR signaling (67, 68). In this regard, differences in activation threshold, signal strength, and costimulation between Tconv and Treg may make them differentially amenable to LYP modulation (69, 70). In the context of recurrent autoreactive T cell responses, which tend confer weaker low-affinity TCR signaling in memory T cells (62, 71), the net effect may be the outgrowth of autoreactive Tconv relative to Treg. Future studies will examine the nature of our observed loss of function.
Despite the advantages of the lentiviral gene delivery methods utilized herein, there are caveats to consider when interpretating our results. First, while constitutive overexpression induced by our lentiviral constructs is useful for unmasking differences between variants, it does not provide equivalent kinetics to those observed for endogenous PTPN22 expression. Hence, more careful control of expression may be required for mechanistic studies of temporal LYP activity. Second, in this experimental system we are examining the the effect of LYP variant expression in the context of endogenous LYP-620R. In that regard, these data are analogous to comparing homozygous non-risk to heterozygous risk LYP variants, with the caveat of continual and mild overexpression (3–6 fold higher LYP protein expression, Supplemental Figure 1E) from the LV construct. However, a qPCR assay could have been designed for a more thorough analysis of exogenous vs endogenous expression. This is an important consideration because amount of LYP relative to its binding parters, most notably CSK, influences the degree of dephosphorylation of TCR signaling (32, 72). Further studies are required to determine how this modest LYP overexpression relative to CSK impacts the biological outcome. Third, for efficient integration to occur, we transduce actively dividing cells. We are therefore unable to examine LYP functional aspects during primary activation, for example to study naïve vs memory or to determine the impact on polarization. Finally, we have only examined the 620W effect in the full length isoform. Several isoforms of LYP are expressed in human T cells, and variation in phosphatase activity, cellular localization, and association with autoimmune disease have been reported (73, 74).
Considering the various roles for LYP in modulating immune responsiveness of T cells, B cells, APCs, neutrophils and more (11, 17, 23, 24, 29, 75), it is not surprising that the effects of the LYP-R620W are complex, even within the CD4+ T cell compartment. Further, there is a need for understanding the role of LYP in B and T cell central tolerance. Indeed, it is likely that the phenotypes observed here from T cell LYP variant expression would have additional implications in vivo and that cell-specific downstream effects of the PTPN22 SNP (rs2476601) may work in concert for altered effector function. Hence, there is a need moving forward for additional investigation of SNP-mediated in vitro versus in vivo effects. We plan to address these questions through ongoing efforts to build isogenic cellular systems to model multiple cell-cell interactions utilizing gene editing technologies (76). It is clear that rs2476601 contributes susceptibility for a number of autoimmune diseases including T1D (3, 4), likely related to functional consequences within the T cell compartment. We demonstrated, herein, that the risk variant imparts a hypomorphic phenotype marked by impaired ability to down-regulate T cell activation and effector function, supporting the notion that PTPN22 represents an important target for therapies aimed at preventing or reversing autoimmune disorders, including T1D.
Supplementary Material
KEY POINTS.
The PTPN22 autoimmune risk allele confers loss-of-function in human T cells
Natural APC activation is required for PTPN22 to inhibit CD4+ T cell proliferation
Inhibition of proliferation by the PTPN22 risk variant is not defective in Treg
ACKNOWLEDGEMENTS
This work was supported by grants from the JDRF (2-2012-280 and 2-PDF-2016-207-A-N), the National Institutes of Health (P01 AI042288, R01 DK106191, and T32 DK108736), and the NIH Human Islet Research Network (HIRN) (UC4 DK104194 and UG3 DK122638). The authors declare that no conflicts of interest exist pertaining to the contents of this manuscript.
Guarantor Statement
Todd M. Brusko is the guarantor of this work and as such, assumes full responsibility for the ethical acquisition and reporting of data.
REFERENCES
- 1.Bergholdt R, Brorsson C, Palleja A, Berchtold LA, Floyel T, Bang-Berthelsen CH, Frederiksen KS, Jensen LJ, Storling J, and Pociot F. 2012. Identification of novel type 1 diabetes candidate genes by integrating genome-wide association data, protein-protein interactions, and human pancreatic islet gene expression. Diabetes 61: 954–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roizen JD, Bradfield JP, and Hakonarson H. 2015. Progress in understanding type 1 diabetes through its genetic overlap with other autoimmune diseases. Curr Diab Rep 15: 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, MacMurray J, Meloni GF, Lucarelli P, Pellecchia M, Eisenbarth GS, Comings D, and Mustelin T. 2004. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet 36: 337–338. [DOI] [PubMed] [Google Scholar]
- 4.Xuan C, Lun LM, Zhao JX, Wang HW, Zhu BZ, Yu S, Liu Z, and He GW. 2013. PTPN22 gene polymorphism (C1858T) is associated with susceptibility to type 1 diabetes: a meta-analysis of 19,495 cases and 25,341 controls. Ann Hum Genet 77: 191–203. [DOI] [PubMed] [Google Scholar]
- 5.Smyth D, Cooper JD, Collins JE, Heward JM, Franklyn JA, Howson JM, Vella A, Nutland S, Rance HE, Maier L, Barratt BJ, Guja C, Ionescu-Tirgoviste C, Savage DA, Dunger DB, Widmer B, Strachan DP, Ring SM, Walker N, Clayton DG, Twells RC, Gough SC, and Todd JA. 2004. Replication of an association between the lymphoid tyrosine phosphatase locus (LYP/PTPN22) with type 1 diabetes, and evidence for its role as a general autoimmunity locus. Diabetes 53: 3020–3023. [DOI] [PubMed] [Google Scholar]
- 6.Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, Ardlie KG, Huang Q, Smith AM, Spoerke JM, Conn MT, Chang M, Chang SY, Saiki RK, Catanese JJ, Leong DU, Garcia VE, McAllister LB, Jeffery DA, Lee AT, Batliwalla F, Remmers E, Criswell LA, Seldin MF, Kastner DL, Amos CI, Sninsky JJ, and Gregersen PK. 2004. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet 75: 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kyogoku C, Langefeld CD, Ortmann WA, Lee A, Selby S, Carlton VE, Chang M, Ramos P, Baechler EC, Batliwalla FM, Novitzke J, Williams AH, Gillett C, Rodine P, Graham RR, Ardlie KG, Gaffney PM, Moser KL, Petri M, Begovich AB, Gregersen PK, and Behrens TW. 2004. Genetic association of the R620W polymorphism of protein tyrosine phosphatase PTPN22 with human SLE. Am J Hum Genet 75: 504–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Velaga MR, Wilson V, Jennings CE, Owen CJ, Herington S, Donaldson PT, Ball SG, James RA, Quinton R, Perros P, and Pearce SH. 2004. The codon 620 tryptophan allele of the lymphoid tyrosine phosphatase (LYP) gene is a major determinant of Graves’ disease. J Clin Endocrinol Metab 89: 5862–5865. [DOI] [PubMed] [Google Scholar]
- 9.Canton I, Akhtar S, Gavalas NG, Gawkrodger DJ, Blomhoff A, Watson PF, Weetman AP, and Kemp EH. 2005. A single-nucleotide polymorphism in the gene encoding lymphoid protein tyrosine phosphatase (PTPN22) confers susceptibility to generalised vitiligo. Genes Immun 6: 584–587. [DOI] [PubMed] [Google Scholar]
- 10.Vandiedonck C, Capdevielle C, Giraud M, Krumeich S, Jais JP, Eymard B, Tranchant C, Gajdos P, and Garchon HJ. 2006. Association of the PTPN22*R620W polymorphism with autoimmune myasthenia gravis. Ann Neurol 59: 404–407. [DOI] [PubMed] [Google Scholar]
- 11.Gianchecchi E, Palombi M, and Fierabracci A. 2013. The putative role of the C1858T polymorphism of protein tyrosine phosphatase PTPN22 gene in autoimmunity. Autoimmun Rev 12: 717–725. [DOI] [PubMed] [Google Scholar]
- 12.Vang T, Congia M, Macis MD, Musumeci L, Orru V, Zavattari P, Nika K, Tautz L, Tasken K, Cucca F, Mustelin T, and Bottini N. 2005. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet 37: 1317–1319. [DOI] [PubMed] [Google Scholar]
- 13.Matthews RJ, Bowne DB, Flores E, and Thomas ML. 1992. Characterization of hematopoietic intracellular protein tyrosine phosphatases: description of a phosphatase containing an SH2 domain and another enriched in proline-, glutamic acid-, serine-, and threonine-rich sequences. Mol Cell Biol 12: 2396–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohen S, Dadi H, Shaoul E, Sharfe N, and Roifman CM. 1999. Cloning and characterization of a lymphoid-specific, inducible human protein tyrosine phosphatase, Lyp. Blood 93: 2013–2024. [PubMed] [Google Scholar]
- 15.Cloutier JF, and Veillette A. 1996. Association of inhibitory tyrosine protein kinase p50csk with protein tyrosine phosphatase PEP in T cells and other hemopoietic cells. Embo j 15: 4909–4918. [PMC free article] [PubMed] [Google Scholar]
- 16.Wu J, Katrekar A, Honigberg LA, Smith AM, Conn MT, Tang J, Jeffery D, Mortara K, Sampang J, Williams SR, Buggy J, and Clark JM. 2006. Identification of substrates of human protein-tyrosine phosphatase PTPN22. J Biol Chem 281: 11002–11010. [DOI] [PubMed] [Google Scholar]
- 17.Rieck M, Arechiga A, Onengut-Gumuscu S, Greenbaum C, Concannon P, and Buckner JH. 2007. Genetic variation in PTPN22 corresponds to altered function of T and B lymphocytes. J Immunol 179: 4704–4710. [DOI] [PubMed] [Google Scholar]
- 18.Schickel JN, Kuhny M, Baldo A, Bannock JM, Massad C, Wang H, Katz N, Oe T, Menard L, Soulas-Sprauel P, Strowig T, Flavell R, and Meffre E. 2016. PTPN22 inhibition resets defective human central B cell tolerance. Sci Immunol 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spalinger MR, Kasper S, Gottier C, Lang S, Atrott K, Vavricka SR, Scharl S, Raselli T, Frey-Wagner I, Gutte PM, Grutter MG, Beer HD, Contassot E, Chan AC, Dai X, Rawlings DJ, Mair F, Becher B, Falk W, Fried M, Rogler G, and Scharl M. 2016. NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J Clin Invest 126: 4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vermeren S, Miles K, Chu JY, Salter D, Zamoyska R, and Gray M. 2016. PTPN22 Is a Critical Regulator of Fcγ Receptor-Mediated Neutrophil Activation. J Immunol 197: 4771–4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bayley R, Kite KA, McGettrick HM, Smith JP, Kitas GD, Buckley CD, and Young SP. 2015. The autoimmune-associated genetic variant PTPN22 R620W enhances neutrophil activation and function in patients with rheumatoid arthritis and healthy individuals. Ann Rheum Dis 74: 1588–1595. [DOI] [PubMed] [Google Scholar]
- 22.Arechiga AF, Habib T, He Y, Zhang X, Zhang ZY, Funk A, and Buckner JH. 2009. Cutting edge: the PTPN22 allelic variant associated with autoimmunity impairs B cell signaling. J Immunol 182: 3343–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Shaked I, Stanford SM, Zhou W, Curtsinger JM, Mikulski Z, Shaheen ZR, Cheng G, Sawatzke K, Campbell AM, Auger JL, Bilgic H, Shoyama FM, Schmeling DO, Balfour HH Jr., Hasegawa K, Chan AC, Corbett JA, Binstadt BA, Mescher MF, Ley K, Bottini N, and Peterson EJ. 2013. The autoimmunity-associated gene PTPN22 potentiates toll-like receptor-driven, type 1 interferon-dependent immunity. Immunity 39: 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gianchecchi E, Crino A, Giorda E, Luciano R, Perri V, Russo AL, Cappa M, Rosado MM, and Fierabracci A. 2014. Altered B cell homeostasis and toll-like receptor 9-driven response in type 1 diabetes carriers of the C1858T PTPN22 allelic variant: implications in the disease pathogenesis. PLoS One 9: e110755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Ewart D, Crabtree JN, Yamamoto A, Baechler EC, Fazeli P, and Peterson EJ. 2015. PTPN22 Variant R620W Is Associated With Reduced Toll-like Receptor 7-Induced Type I Interferon in Systemic Lupus Erythematosus. Arthritis Rheumatol 67: 2403–2414. [DOI] [PubMed] [Google Scholar]
- 26.Brownlie RJ, Miosge LA, Vassilakos D, Svensson LM, Cope A, and Zamoyska R. 2012. Lack of the phosphatase PTPN22 increases adhesion of murine regulatory T cells to improve their immunosuppressive function. Sci Signal 5: ra87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burn GL, Cornish GH, Potrzebowska K, Samuelsson M, Griffié J, Minoughan S, Yates M, Ashdown G, Pernodet N, Morrison VL, Sanchez-Blanco C, Purvis H, Clarke F, Brownlie RJ, Vyse TJ, Zamoyska R, Owen DM, Svensson LM, and Cope AP. 2016. Superresolution imaging of the cytoplasmic phosphatase PTPN22 links integrin-mediated T cell adhesion with autoimmunity. Sci Signal 9: ra99. [DOI] [PubMed] [Google Scholar]
- 28.Fousteri G, Jofra T, Debernardis I, Stanford SM, Laurenzi A, Bottini N, and Battaglia M. 2014. The protein tyrosine phosphatase PTPN22 controls forkhead box protein 3 T regulatory cell induction but is dispensable for T helper type 1 cell polarization. Clin Exp Immunol 178: 178–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maine CJ, Hamilton-Williams EE, Cheung J, Stanford SM, Bottini N, Wicker LS, and Sherman LA. 2012. PTPN22 alters the development of regulatory T cells in the thymus. J Immunol 188: 5267–5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sood S, Brownlie RJ, Garcia C, Cowan G, Salmond RJ, Sakaguchi S, and Zamoyska R. 2016. Loss of the Protein Tyrosine Phosphatase PTPN22 Reduces Mannan-Induced Autoimmune Arthritis in SKG Mice. J Immunol 197: 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fousteri G, Jofra T, Di Fonte R, Gagliani N, Morsiani C, Stabilini A, and Battaglia M. 2015. Lack of the protein tyrosine phosphatase PTPN22 strengthens transplant tolerance to pancreatic islets in mice. Diabetologia 58: 1319–1328. [DOI] [PubMed] [Google Scholar]
- 32.Zikherman J, Hermiston M, Steiner D, Hasegawa K, Chan A, and Weiss A. 2009. PTPN22 deficiency cooperates with the CD45 E613R allele to break tolerance on a non-autoimmune background. J Immunol 182: 4093–4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maine CJ, Marquardt K, Scatizzi JC, Pollard KM, Kono DH, and Sherman LA. 2015. The effect of the autoimmunity-associated gene, PTPN22, on a BXSB-derived model of lupus. Clin Immunol 156: 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maine CJ, Marquardt K, Cheung J, and Sherman LA. 2014. PTPN22 controls the germinal center by influencing the numbers and activity of T follicular helper cells. J Immunol 192: 1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fraser HI, Howlett S, Clark J, Rainbow DB, Stanford SM, Wu DJ, Hsieh YW, Maine CJ, Christensen M, Kuchroo V, Sherman LA, Podolin PL, Todd JA, Steward CA, Peterson LB, Bottini N, and Wicker LS. 2015. Ptpn22 and Cd2 Variations Are Associated with Altered Protein Expression and Susceptibility to Type 1 Diabetes in Nonobese Diabetic Mice. J Immunol 195: 4841–4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng P, and Kissler S. 2013. PTPN22 silencing in the NOD model indicates the type 1 diabetes-associated allele is not a loss-of-function variant. Diabetes 62: 896–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dai X, James RG, Habib T, Singh S, Jackson S, Khim S, Moon RT, Liggitt D, Wolf-Yadlin A, Buckner JH, and Rawlings DJ. 2013. A disease-associated PTPN22 variant promotes systemic autoimmunity in murine models. J Clin Invest 123: 2024–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin X, Pelletier S, Gingras S, Rigaud S, Maine CJ, Marquardt K, Dai YD, Sauer K, Rodriguez AR, Martin G, Kupriyanov S, Jiang L, Yu L, Green DR, and Sherman LA. 2016. CRISPR-Cas9-Mediated Modification of the NOD Mouse Genome With Ptpn22R619W Mutation Increases Autoimmune Diabetes. Diabetes 65: 2134–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aarnisalo J, Treszl A, Svec P, Marttila J, Oling V, Simell O, Knip M, Korner A, Madacsy L, Vasarhelyi B, Ilonen J, and Hermann R. 2008. Reduced CD4+T cell activation in children with type 1 diabetes carrying the PTPN22/Lyp 620Trp variant. J Autoimmun 31: 13–21. [DOI] [PubMed] [Google Scholar]
- 40.Vang T, Liu WH, Delacroix L, Wu S, Vasile S, Dahl R, Yang L, Musumeci L, Francis D, Landskron J, Tasken K, Tremblay ML, Lie BA, Page R, Mustelin T, Rahmouni S, Rickert RC, and Tautz L. 2012. LYP inhibits T-cell activation when dissociated from CSK. Nat Chem Biol 8: 437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de la Puerta ML, Trinidad AG, Rodriguez Mdel C, de Pereda JM, Sanchez Crespo M, Bayon Y, and Alonso A. 2013. The autoimmunity risk variant LYP-W620 cooperates with CSK in the regulation of TCR signaling. PLoS One 8: e54569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fuhrman CA, Yeh WI, Seay HR, Saikumar Lakshmi P, Chopra G, Zhang L, Perry DJ, McClymont SA, Yadav M, Lopez MC, Baker HV, Zhang Y, Li Y, Whitley M, von Schack D, Atkinson MA, Bluestone JA, and Brusko TM. 2015. Divergent Phenotypes of Human Regulatory T Cells Expressing the Receptors TIGIT and CD226. J Immunol 195: 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Putnam AL, Brusko TM, Lee MR, Liu W, Szot GL, Ghosh T, Atkinson MA, and Bluestone JA. 2009. Expansion of human regulatory T-cells from patients with type 1 diabetes. Diabetes 58: 652–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whelan JA, Russell NB, and Whelan MA. 2003. A method for the absolute quantification of cDNA using real-time PCR. J Immunol Methods 278: 261–269. [DOI] [PubMed] [Google Scholar]
- 45.Szymczak AL, and Vignali DA. 2005. Development of 2A peptide-based strategies in the design of multicistronic vectors. Expert Opin Biol Ther 5: 627–638. [DOI] [PubMed] [Google Scholar]
- 46.Lois C, Hong EJ, Pease S, Brown EJ, and Baltimore D. 2002. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295: 868–872. [DOI] [PubMed] [Google Scholar]
- 47.Brusko TM, Koya RC, Zhu S, Lee MR, Putnam AL, McClymont SA, Nishimura MI, Han S, Chang LJ, Atkinson MA, Ribas A, and Bluestone JA. 2010. Human antigen-specific regulatory T cells generated by T cell receptor gene transfer. PLoS One 5: e11726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneider CA, Rasband WS, and Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Butler MO, and Hirano N. 2014. Human cell-based artificial antigen-presenting cells for cancer immunotherapy. Immunol Rev 257: 191–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brusko TM, Hulme MA, Myhr CB, Haller MJ, and Atkinson MA. 2007. Assessing the in vitro suppressive capacity of regulatory T cells. Immunol Invest 36: 607–628. [DOI] [PubMed] [Google Scholar]
- 51.Brownlie RJ, and Zamoyska R. 2013. T cell receptor signalling networks: branched, diversified and bounded. Nat Rev Immunol 13: 257–269. [DOI] [PubMed] [Google Scholar]
- 52.Simms PE, and Ellis TM. 1996. Utility of flow cytometric detection of CD69 expression as a rapid method for determining poly- and oligoclonal lymphocyte activation. Clin Diagn Lab Immunol 3: 301–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, and Kroczek RA. 1999. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature 397: 263–266. [DOI] [PubMed] [Google Scholar]
- 54.Adler SH, Chiffoleau E, Xu L, Dalton NM, Burg JM, Wells AD, Wolfe MS, Turka LA, and Pear WS. 2003. Notch signaling augments T cell responsiveness by enhancing CD25 expression. J Immunol 171: 2896–2903. [DOI] [PubMed] [Google Scholar]
- 55.Lozano E, Dominguez-Villar M, Kuchroo V, and Hafler DA. 2012. The TIGIT/CD226 axis regulates human T cell function. J Immunol 188: 3869–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Keir ME, Francisco LM, and Sharpe AH. 2007. PD-1 and its ligands in T-cell immunity. Curr Opin Immunol 19: 309–314. [DOI] [PubMed] [Google Scholar]
- 57.Jago CB, Yates J, Camara NO, Lechler RI, and Lombardi G. 2004. Differential expression of CTLA-4 among T cell subsets. Clin Exp Immunol 136: 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nyakeriga AM, Fichtenbaum CJ, Goebel J, Nicolaou SA, Conforti L, and Chougnet CA. 2009. Engagement of the CD4 Receptor Affects the Redistribution of Lck to the Immunological Synapse in Primary T Cells: Implications for T-Cell Activation during Human Immunodeficiency Virus Type 1 Infection ▿. J Virol 83: 1193–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brenchley JM, Douek DC, Ambrozak DR, Chatterji M, Betts MR, Davis LS, and Koup RA. 2002. Expansion of activated human naïve T-cells precedes effector function. Clin Exp Immunol 130: 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spalinger MR, Zeitz J, Biedermann L, Rossel JB, Sulz MC, Frei P, Scharl S, Vavricka SR, Fried M, Rogler G, and Scharl M. 2016. Genotype-Phenotype Associations of the CD-Associated Single Nucleotide Polymorphism within the Gene Locus Encoding Protein Tyrosine Phosphatase Non-Receptor Type 22 in Patients of the Swiss IBD Cohort. PLoS One 11: e0160215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vang T, Landskron J, Viken MK, Oberprieler N, Torgersen KM, Mustelin T, Tasken K, Tautz L, Rickert RC, and Lie BA. 2013. The autoimmune-predisposing variant of lymphoid tyrosine phosphatase favors T helper 1 responses. Hum Immunol 74: 574–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schubert DA, Gordo S, Sabatino JJ, Vardhana S, Gagnon E, Sethi DK, Seth NP, Choudhuri K, Reijonen H, Nepom GT, Evavold BD, Dustin ML, and Wucherpfennig KW. 2012. Self-reactive human CD4 T cell clones form unusual immunological synapses. J Exp Med 209: 335–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yeh WI, Seay HR, Newby B, Posgai AL, Moniz FB, Michels A, Mathews CE, Bluestone JA, and Brusko TM. 2017. Avidity and Bystander Suppressive Capacity of Human Regulatory T Cells Expressing De Novo Autoreactive T-Cell Receptors in Type 1 Diabetes. Front Immunol 8: 1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ferreira RC, Castro Dopico X, Oliveira JJ, Rainbow DB, Yang JH, Trzupek D, Todd SA, McNeill M, Steri M, Orrù V, Fiorillo E, Crouch DJM, Pekalski ML, Cucca F, Tree TI, Vyse TJ, Wicker LS, and Todd JA. 2019. Chronic Immune Activation in Systemic Lupus Erythematosus and the Autoimmune PTPN22 Trp. Front Immunol 10: 2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yeh LT, Miaw SC, Lin MH, Chou FC, Shieh SJ, Chuang YP, Lin SH, Chang DM, and Sytwu HK. 2013. Different modulation of Ptpn22 in effector and regulatory T cells leads to attenuation of autoimmune diabetes in transgenic nonobese diabetic mice. J Immunol 191: 594–607. [DOI] [PubMed] [Google Scholar]
- 66.Anderson W, Thorpe J, Long SA, and Rawlings DJ. 2019. Efficient CRISPR/Cas9 Disruption of Autoimmune-Associated Genes Reveals Key Signaling Programs in Primary Human T Cells. J Immunol 203: 3166–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Salmond RJ, Brownlie RJ, Morrison VL, and Zamoyska R. 2014. The tyrosine phosphatase PTPN22 discriminates weak self peptides from strong agonist TCR signals. Nat Immunol 15: 875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bray C, Wright D, Haupt S, Thomas S, Stauss H, and Zamoyska R. 2018. Crispr/Cas Mediated Deletion of PTPN22 in Jurkat T Cells Enhances TCR Signaling and Production of IL-2. Front Immunol 9: 2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yan D, Farache J, Mingueneau M, Mathis D, and Benoist C. 2015. Imbalanced signal transduction in regulatory T cells expressing the transcription factor FoxP3. Proc Natl Acad Sci U S A 112: 14942–14947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sprouse ML, Shevchenko I, Scavuzzo MA, Joseph F, Lee T, Blum S, Borowiak M, Bettini ML, and Bettini M. 2018. Cutting Edge: Low-Affinity TCRs Support Regulatory T Cell Function in Autoimmunity. J Immunol 200: 909–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Koehli S, Naeher D, Galati-Fournier V, Zehn D, and Palmer E. 2014. Optimal T-cell receptor affinity for inducing autoimmunity. Proc Natl Acad Sci U S A 111: 17248–17253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Davidson D, Zhong MC, Pandolfi PP, Bolland S, Xavier RJ, Seed B, Li X, Gu H, and Veillette A. 2016. The Csk-Associated Adaptor PAG Inhibits Effector T Cell Activation in Cooperation with Phosphatase PTPN22 and Dok Adaptors. Cell Rep 17: 2776–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chang HH, Tai TS, Lu B, Iannaccone C, Cernadas M, Weinblatt M, Shadick N, Miaw SC, and Ho IC. 2012. PTPN22.6, a dominant negative isoform of PTPN22 and potential biomarker of rheumatoid arthritis. PLoS One 7: e33067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chang HH, Tseng W, Cui J, Costenbader K, and Ho IC. 2014. Altered expression of protein tyrosine phosphatase, non-receptor type 22 isoforms in systemic lupus erythematosus. Arthritis Res Ther 16: R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Habib T, Funk A, Rieck M, Brahmandam A, Dai X, Panigrahi AK, Luning Prak ET, Meyer-Bahlburg A, Sanda S, Greenbaum C, Rawlings DJ, and Buckner JH. 2012. Altered B cell homeostasis is associated with type I diabetes and carriers of the PTPN22 allelic variant. J Immunol 188: 487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wallet MA, Santostefano KE, Terada N, and Brusko TM. 2017. Isogenic Cellular Systems Model the Impact of Genetic Risk Variants in the Pathogenesis of Type 1 Diabetes. Front Endocrinol (Lausanne) 8: 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.