

Abstract
Asthma is a common inflammatory lung disease with no known cure. Previously, we uncovered a lung TNFR2+ conventional DC2 subset (cDC2s) that induces regulatory T cells (Tregs) maintaining lung tolerance at steady state but promotes TH2 response during house dust mite (HDM)–induced asthma. Lung IFNβ is essential for TNFR2+ cDC2s–mediated lung tolerance. Here, we showed that exogenous IFNβ reprogrammed TH2-promoting pathogenic TNFR2+ cDC2s back to tolerogenic DCs, alleviating eosinophilic asthma and preventing asthma exacerbation. Mechanistically, inhaled IFNβ, not IFNα, activated ERK2 signaling in pathogenic lung TNFR2+ cDC2s, leading to enhanced fatty acid oxidation (FAO) and lung Treg induction. Last, human IFNβ reprogrammed pathogenic human lung TNFR2+ cDC2s from patients with emphysema ex vivo. Thus, we identified an IFNβ-specific ERK2-FAO pathway that might be harnessed for DC therapy.
INTRODUCTION
Dendritic cells (DCs) consist of functionally distinct subsets that generate either immunogenic, pathogenic, or tolerogenic immune responses in vivo. Some DC subsets can induce both immunogenic and tolerogenic immune responses. PD-L2+CD11b+ dermal DCs can elicit T helper cell 2 (TH2) responses or prime regulatory T cells (Tregs) (1). CD103+ DCs in the gut have a dual role in tolerogenic and immunogenic T cell responses (2). We previously identified a tumor necrosis factor receptor 2–positive (TNFR2+) conventional DC2 subset (cDC2s) in the lung that generates TH2 responses or Tregs depending on the stimuli (3, 4). DCs in the peripheral tissue sense local environmental cues that can serve dual purposes: maintaining peripheral tolerance during homeostasis and mounting immunogenic responses upon infections or tissue injury. The underlying mechanisms for this dual function of peripheral DCs are unclear.
Growing evidence suggests that metabolic programming acts as a master regulatory switch in determining immunogenic or tolerogenic DC cell fate (5). Immunogenic DCs adopt a glycolytic metabolic state, whereas tolerogenic DCs favor oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO) (5). Lowering glucose concentrations in culture medium from 10 to 0 mM diminishes CD40 and CD86 expression, the production of interleukin-12 (IL-12) and IL-23, and T cell priming capability in lipopolysaccharide-stimulated murine bone marrow–derived DCs (6). In contrast, in vitro differentiated tolerogenic DCs have increased expression of OXPHOS genes including electron transport chain complexes II and IV, high mitochondrial activity, and increased reactive oxygen species production (7). The mitochondrial activity is associated with an increase in FAO (7). Inhibition of the mitochondrial outer membrane protein carnitine palmitoyltransferase I (CPT1), the rate-limiting enzyme of FAO, with etomoxir (ETO) increases responder T cell activation (7). On the other hand, the activation of adenosine monophosphate (AMP)–activated protein kinase (AMPK) inhibits acetyl–coenzyme A (CoA) carboxylase (ACC), the rate-limiting enzyme in fatty acid synthesis, and enhances FAO (8, 9). Last, metabolic enzymes indolamine-2,3-dioxygenase (IDO-1) and arginase 1 (Arg-1) control tolerogenic DC function and maintain peripheral tolerance (10, 11). Steady-state lung TNFR2+ cDC2s cells constitutively express IDO-1 and Arg-1, which are reduced upon house dust mite (HDM) stimulation (3), suggesting that a metabolic shift may control TNFR2+ cDC2s immunogenic, tolerogenic fates.
Tolerogenic lung TNFR2+ cDC2s cells are induced and maintained by tissue interferon-β (IFNβ) (3). IFNβ belongs to the type I IFN family, which increases glycolysis in splenic DCs ex vivo (12). However, type I IFNs up-regulate OXPHOS and FAO in plasmacytoid DCs (pDCs) and cDCs (13). Type I IFN mediates induction of glycolysis by canonical pathway activation through Tyk2 and STAT1 (signal transducer and activator of transcription 1), whereas OXPHOS and FAO are regulated in a cell type–specific manner, which might involve a noncanonical type I IFN pathway (14). IFNβ, not IFNα, is effective in the treatment of multiple sclerosis, which correlates to increased IL-10 production (15) and markedly improves the frequency and suppressive function of Tregs in these patients (16). However, the potential IFNβ-specific tolerogenic signaling pathway connected to TNRF2+ cDC2s is unknown.
In this study, we identified an IFNβ-specific extracellular signal–regulated kinase 2 (ERK2)–FAO pathway in lung TNFR2+ cDC2s and successfully reprogrammed TH2-promoting pathogenic TNFR2+ cDC2s back to tolerogenic DCs in vivo. Thus, in vivo reprogramming of pathogenic DCs is possible via IFNβ, presenting potential therapeutic implications for inflammatory diseases such as asthma.
RESULTS
Intranasal administration of IFNβ alleviated HDM-induced acute and chronic asthma
Steady-state lung IFNβ maintains lung tolerance (3). We hypothesized that exogenous IFNβ might be therapeutic for inflammatory lung diseases by restoring lung tolerance. We used two HDM mouse models: a 2-week, HDM-induced acute asthma model (Fig. 1) (17) and a 5-week HDM-induced chronic asthma model (fig. S1) (18). We administered IFNβ intranasally after asthma was established in these mice (Fig. 1A and fig. S1A). In the acute asthma mouse model, IFNβ treatment reduced eosinophils in the bronchoalveolar fluid (BALF) (Fig. 1B). Serum HDM-specific immunoglobulin G1 (IgG1) and lung IL-4+ CD4 T cells were decreased in IFNβ-treated HDM mice (Fig. 1, C and D). Hematoxylin and eosin (H&E) staining showed reduced lung inflammation in IFNβ-treated HDM mice (Fig. 1E). Furthermore, examining HDM-specific memory TH2 cells (IL-5+) by ex vivo recall studies showed a reduction in lungs and lung draining lymph nodes from IFNβ-treated HDM mice (Fig. 1F). Last, lung function analysis showed that IFNβ treatment improved lung function with decreased airway resistance and pulmonary artery pressure, as well as increased pulmonary compliance in HDM mice (Fig. 1G).
Fig. 1. Inhaled IFNβ alleviated HDM-induced asthma in mice.
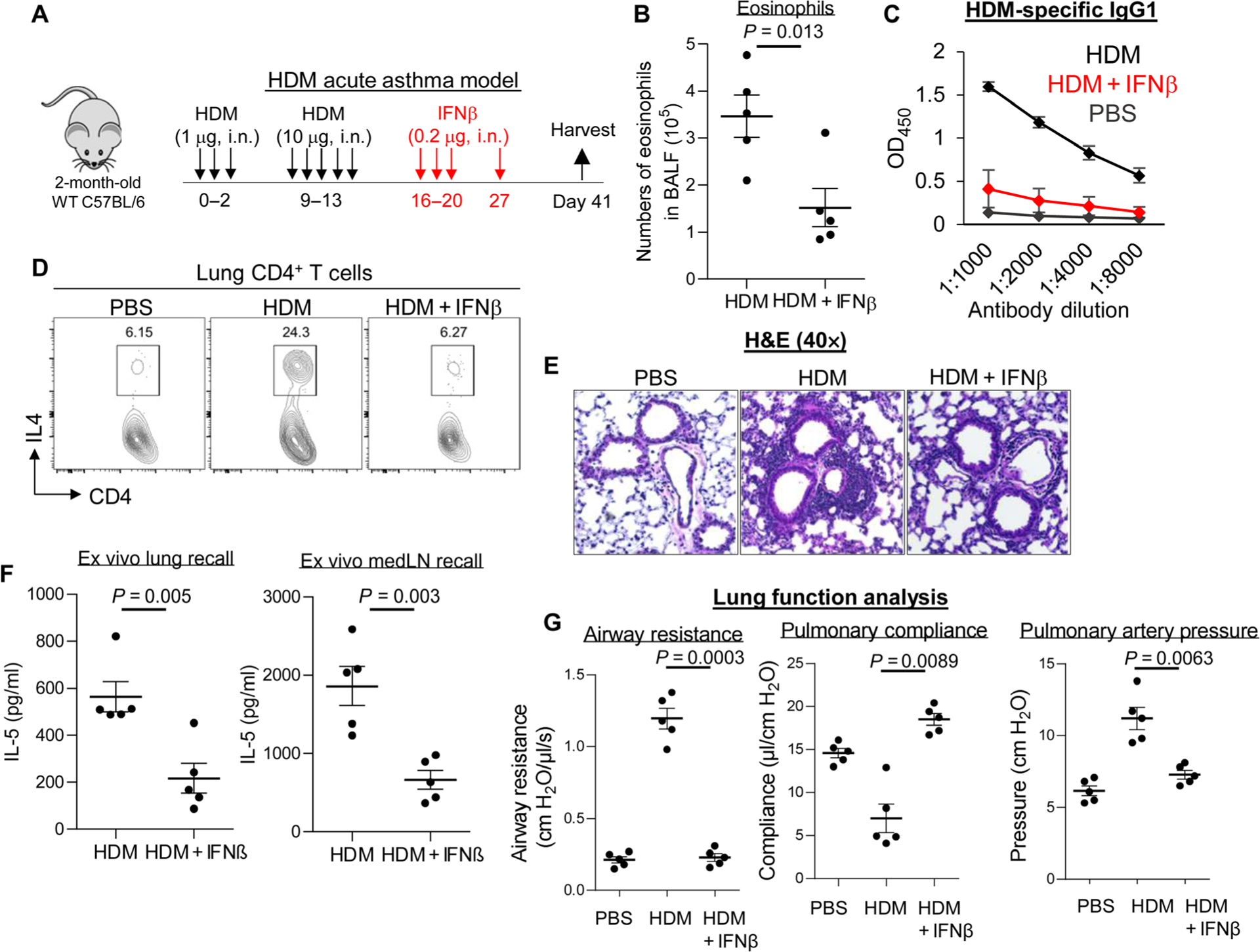
(A) Experimental protocol for HDM-induced acute asthma. Mice were intranasally (i.n.) administered PBS or IFNβ (0.2 μg). (B) Absolute numbers of BALF eosinophils of asthmatic mice treated with IFNβ (n = 3 to 7 mice per group). Data are representative of three independent experiments. (C) Serum levels of HDM-specific IgG1 from asthmatic mice treated with IFNβ in (B). Data are representative of three independent experiments. (D) Flow cytometry plots of IL-4–producing lung CD4+ T cells from asthmatic mice treated with IFNβ in (B). Data are representative of three independent experiments. (E) Representative H&E staining of lung sections from asthmatic mice treated with PBS or IFNβ from (B). Data are representative of three independent experiments. (F) Cytokine production by lung and lung draining lymph nodes (mLNs) from asthmatic mice from (B) restimulated ex vivo for 4 days with HDM (25 μg/ml). Data are representative of three independent experiments. (G) Airway resistance, pulmonary compliance, and pulmonary artery pressure were determined in asthmatic mice as described in Materials and Methods (n = 3 to 5 mice per group). Data are representative of three independent experiments. Graphs represent the mean, with error bars indicating SEM. P values were determined by unpaired Student’s t test (B) and (F) or one-way ANOVA with Tukey’s multiple comparison test (G). OD450, optical density at 450 nm.
We asked whether IFNβ treatment also alleviated chronic asthma. Intranasal administration of IFNβ inhibited IL-4–producing CD4 T cells, eosinophil infiltration, and HDM-specific IgE and IgG1 in chronic asthma mice (fig. S1, B to F). Intranasal administration of IFNβ inhibited neutrophil infiltration (fig. S1G) and TH17 responses (fig. S1H) in chronic asthma mice. Thus, intranasal exogenous IFNβ was therapeutic in HDM-induced asthma in mice.
Intranasal IFNβ administration prevented HDM-induced asthma exacerbation
According to the U.S. Centers for Disease Control and Prevention, ~50% of asthmatic patients have asthma exacerbation each year. We asked whether IFNβ treatment might prevent asthma exacerbation. We challenged IFNβ-treated acute asthma mice with HDM (Fig. 2A). IFNβ-treated asthma mice were completely protected from a second HDM challenge, correlating to reduced HDM-specific IgG1, IgE, eosinophil infiltration, and TH2 cell induction (Fig. 2, B to F). Lung function analysis showed that IFNβ treatment improved lung function with decreased airway resistance and pulmonary artery pressure, as well as increased pulmonary compliance during asthma exacerbation (Fig. 2G). Thus, intranasal IFNβ administration alleviated asthma exacerbation.
Fig. 2. Inhaled IFNβ prevented HDM-induced asthma exacerbation.
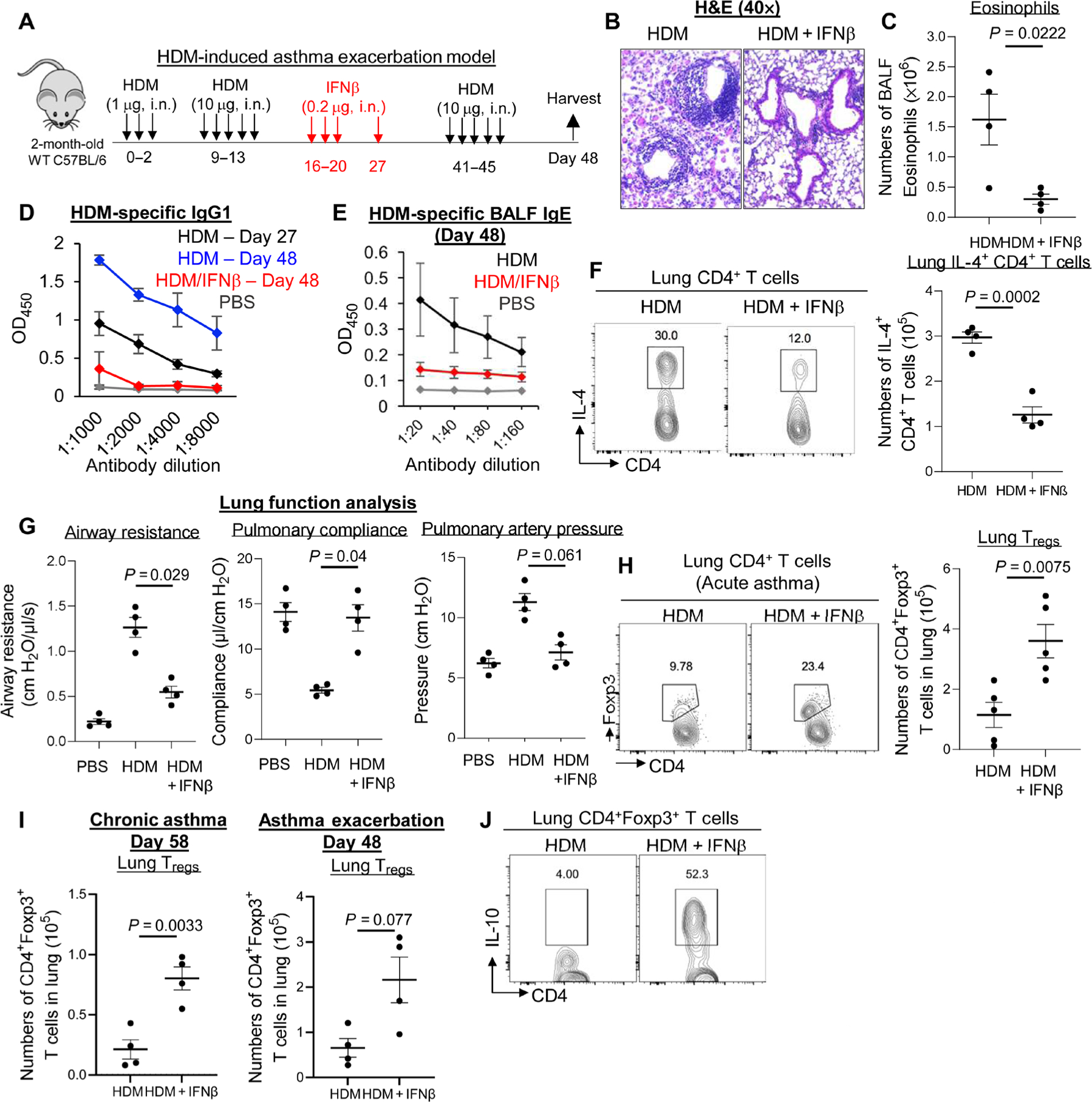
(A) Experimental protocol for treating HDM-induced asthma exacerbation. Mice were intranasally administered PBS or IFNβ (0.2 μg). (B) Representative H&E staining of lung sections from asthmatic mice treated with PBS or IFNβ (n = 3 to 4 mice per group). Data are representative of three independent experiments. (C) Absolute numbers of BALF eosinophils in asthmatic mice treated with IFNβ (n = 3 to 4 mice per group). Data are representative of three independent experiments. (D and E) Serum levels of HDM-specific IgG1 and IgE from asthmatic mice treated with IFNβ (n = 3 to 4 mice per group). Data are representative of three independent experiments. (F) Flow cytometry plots of IL-4–producing lung CD4+ T cells from asthmatic mice treated with IFNβ (n = 3 to 4 mice per group). Data are representative of three independent experiments. (G) Airway resistance, pulmonary compliance, and pulmonary artery pressure were determined in asthmatic mice as described in Materials and Methods (n = 3 to 4 mice per group). Data are representative of three independent experiments. (H) Flow cytometry analysis (left) and absolute number (right) of lung CD4+Foxp3+ Tregs in acute asthmatic mice treated with IFNβ (n = 3 to 5 mice per group). Data are representative of three independent experiments. (I) Absolute number of lung CD4+Foxp3+ Tregs in asthmatic mice treated with IFNβ (n = 3 to 4 mice per group). Data are representative of three independent experiments. (J) Flow cytometry analysis of IL-10–producing lung Tregs from asthmatic mice in (H). Data are representative of three independent experiments. Graphs represent the mean, with error bars indicating SEM. P values were determined by one-way ANOVA with Tukey’s multiple comparison test (G) or unpaired Student’s t test (C, F, H, and I).
Intranasal IFNβ administration generated lung Tregs in asthmatic mice
Lung IFNβ generated lung Tregs at steady state (3). We hypothesized that exogenous IFNβ might induce Tregs in the lung of asthmatic mice. Intranasal administration of IFNβ increased lung Tregs in acute, chronic asthma and asthma exacerbation (Fig. 2, H and I). Moreover, IFNβ-induced lung Tregs produce IL-10 (Fig. 2J).
The induction of Tregs in the lungs is therapeutic for inflammatory lung diseases (19, 20). To confirm that, we administered (intranasally) anti-CD25 monoclonal antibody (mAb) (PC-61.5.3) (21) to deplete lung Tregs after IFNβ treatment. Asthma exacerbation was induced afterward, and asthma phenotypes were determined 3 days later (fig. S2A). Anti-CD25 mAb reduced IFNβ-induced lung Tregs and eliminated IFNβ-induced protection against asthma exacerbation, including the generation of anti-HDM–specific IgG1 (fig. S2, B to D). Thus, we proposed that exogenous IFNβ induced lung Treg production in HDM-treated mice, alleviating asthmatic symptoms.
Depleting lung TNFR2+ cDC2s in vivo ablated the therapeutic effect of IFNβ in asthmatic mice
Lung IFNβ acts on lung TNFR2+ cDC2s to generate Tregs at steady state (3). Lung TNFR2+ cDC2s rely on the constitutive TNFR2 signaling for survival in vivo (3). Intranasal administration of TNFR2 blocking antibody eliminates the lung TNFR2+ cDC2s population (3). Here, we found that depleting TNFR2+ cDC2s before, during, and after IFNβ treatment in asthmatic mice by anti-TNFR2 blocking mAb (Fig. 3, A to C) resulted in the loss of lung Tregs and the suppression of anti-HDM IgG1 by IFNβ (Fig. 3, D and E). Anti-TNFR2 antibody did not significantly affect lung cDC1 numbers, and the numbers of monocyte DCs (moDCs) decreased slightly but were not significant (Fig. 3C).
Fig. 3. Depleting lung TNFR2+ cDC2s rendered the IFNβ therapy ineffective.
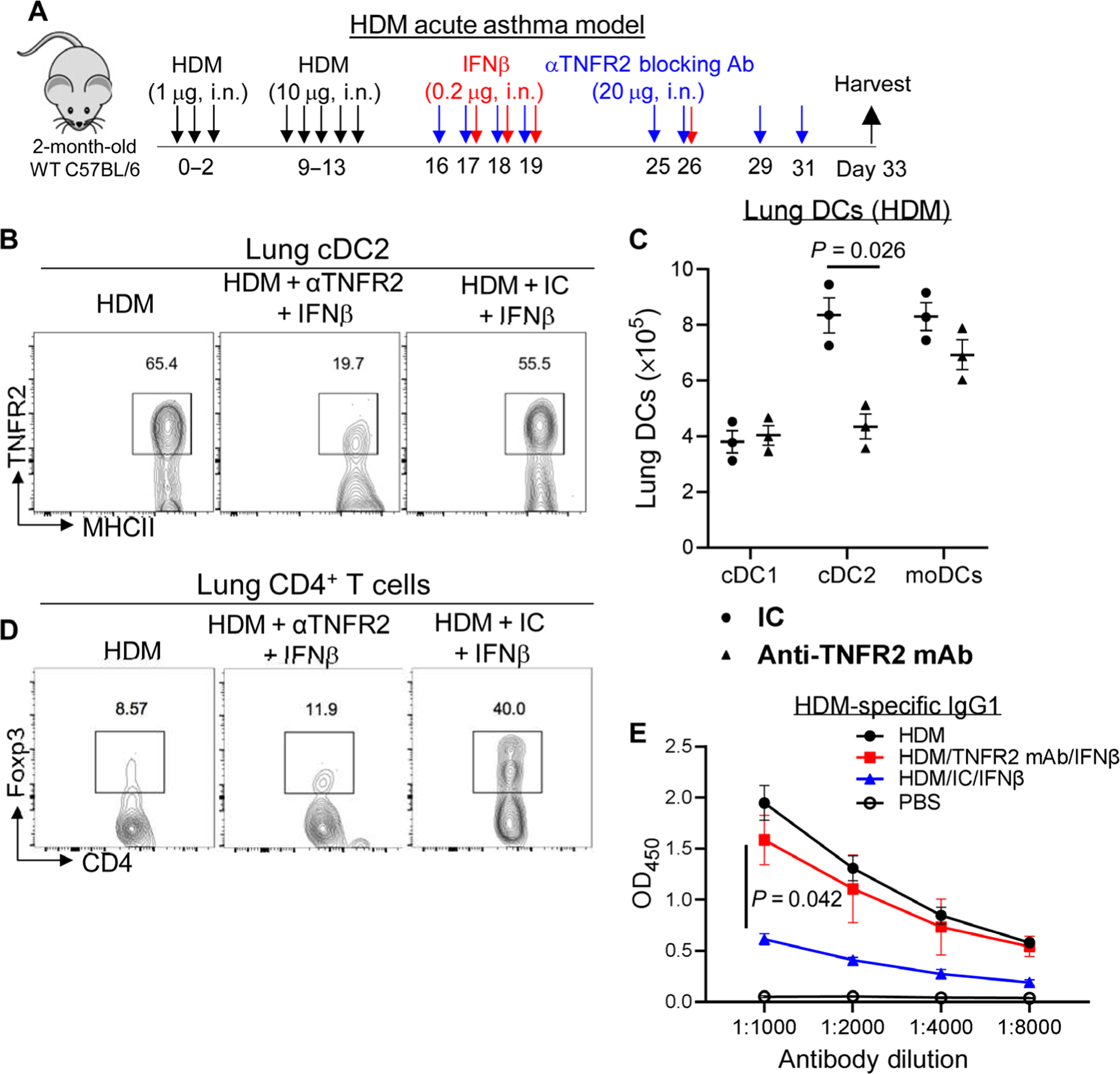
(A) Experimental protocol for the depletion of TNFR2-expressing cells in asthmatic mice. (B) Flow cytometry analysis of TNFR2+ cDC2s in asthmatic mice treated with IFNβ/isotype control or IFNβ/anti-TNFR2 antibody (20 μg) (n = 3 mice per group). Data are representative of two independent experiments. (C) Total numbers of lung DC subsets in (B) (n = 3 mice per group). Data are representative of two independent experiments. (D) Flow cytometry analysis of Tregs in asthmatic mice treated with IFNβ (0.2 μg)/isotype control or IFNβ/anti-TNFR2 antibody (20 μg) (n = 3 mice per group). Data are representative of two independent experiments. (E) Serum levels of HDM-specific IgG1 in asthmatic mice treated with IFNβ/isotype control or IFNβ/anti-TNFR2 antibody (20 μg) (n = 3 mice per group). Data are representative of two independent experiments. Graphs represent the mean, with error bars indicating SEM. P values were determined by two-way ANOVA with Sidak’s multiple comparison test (C) or two-way ANOVA with Tukey’s multiple comparison test (E).
cDC2s are a heterogeneous population consisting of functionally distinct subsets (22, 23). The pathogenic TNFR2+ cDC2s are gated on TNFR2+CD24+CD11B+CD64−CD172a+CD26+FMO− lung DCs (fig. S3, A to F), which distinguished them from recently characterized CD64+ inflammatory cDC2s (23). Compared with the steady state, HDM treatment increased lung cDC1, TNFR2+ cDC2s, and moDC numbers (fig. S3B). However, IFNβ treatment did not alter total lung TNFR2+ cDC2s numbers in asthmatic mice (fig. S3, C to F).
IFNβ reprogrammed pathogenic lung TNFR2+ cDC2s in vivo to generate lung Tregs
Lung TNFR2+ cDC2s are plastic and promote TH2 responses upon HDM treatment (3). We hypothesized that exogenous IFNβ might reprogram pathogenic TNFR2+ cDC2s back to tolerogenic DCs and generate lung Tregs. Tolerogenic TNFR2+ cDC2s cells express programmed cell death protein–1 (PD-L1), Arg-1, and transforming growth factor–β1 (TGFβ1), all required for lung Treg induction (24). Intranasal IFNβ administration enhanced PD-L1, Arg-1, and TGFβ1 expression in TNFR2+ cDC2s in asthmatic mice (Fig. 4, A to C). Meanwhile, intranasal IFNβ administration reduced inflammatory molecules T1ST2 (IL-33R), OX40L, and ICOSL on pathogenic TNFR2+ cDC2s in asthmatic mice (Fig. 4, D to F) (3).
Fig. 4. IFNβ reprogrammed pathogenic TNFR2+ CDC2S in vivo to generate lung Tregs.
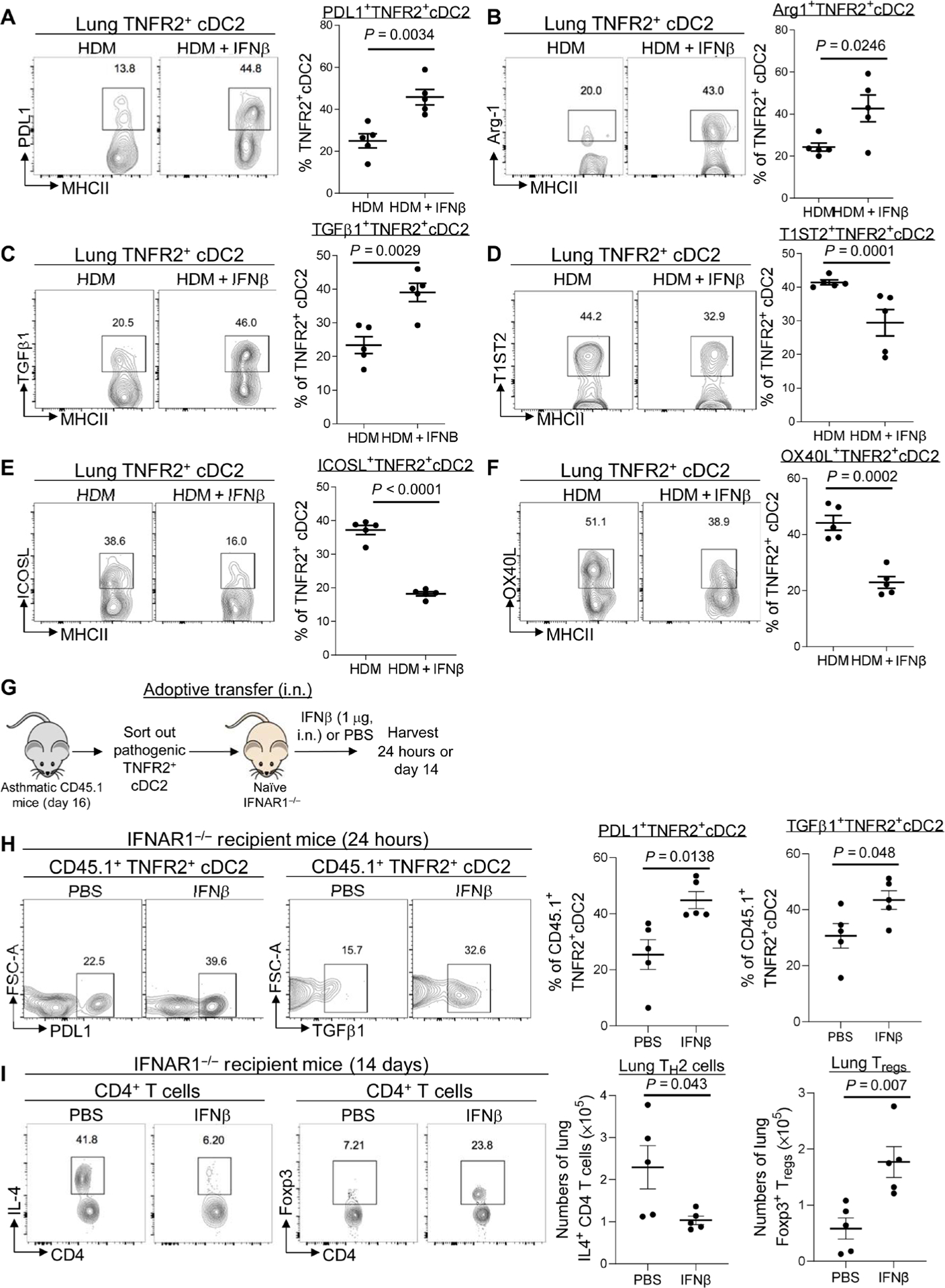
(A to C) Flow cytometry analysis and frequency of PD-L1, Arg-1, and TGFβ1 expression on TNFR2+ cDC2s in asthmatic mice treated with IFNβ (0.2 μg). Lung cDC2s are MHCIIhiCD11c+CD11b+CD64−CD24+ (n = 3 to 5 mice per group). Data are representative of two to three independent experiments. (D to F) Flow cytometry analysis and frequency of T1ST2, OX40L, and ICOSL expression on lung TNFR2+ cDC2s in asthmatic mice treated with IFNβ (0.2 μg) (n = 3 to 5 mice per group). Data are representative of two to three independent experiments. (G) Experimental design for adoptive transfer. Lung TNFR2+ cDC2s cells were sorted out of asthmatic CD45.1 mice on day 16. About 60,000 TNFR2+ cDC2s cells were transferred intranasally into IFNAR1−/− recipient mice. Recipient mice were treated with IFNβ (1 μg) or PBS. (H) Flow cytometry analysis and frequency of CD45.1+TNFR2+ cDC2s in the recipient mice after IFNβ treatment (n = 3 to 5 mice per group). Data are representative of two independent experiments. (I) Flow cytometry analysis and numbers of IL-4–producing CD4+ T cells (left) and Tregs (right) in recipient mice treated with IFNβ on day 14 after IFNβ treatment (n = 3 to 5 mice per group). Data are representative of two independent experiments. Graphs represent the mean, with error bars indicating SEM. P values were determined by unpaired Student’s t test.
To demonstrate that intranasal administration of IFNβ directly reprogrammed TNFR2+ cDC2s, we performed adoptive cell transfer experiments (Fig. 4G). We sorted out TNFR2+ cDC2s from asthmatic CD45.1 mice on day 16 (fig. S4A) and transferred (intranasally) them into naïve IFNAR1−/− mice. The recipient IFNAR1−/− mice were then treated (intranasally) with IFNβ or phosphate-buffered serum (PBS). CD45.1+ lung cells were examined after 24 hours (fig. S4, B and D to F). The numbers of recovered CD45.1 wild-type (WT) TNFR2+ cDC2s from PBS- or IFNβ-treated IFNAR1−/− mice were similar (fig. S4D). Moreover, the recovered CD45.1 cells were TNFR2+, CD24+, TNFR2+, CD26+, CD172a+ but CD64− and Mar-1−, confirming that they were TNFR2+ cDC2 (fig. S4, B, E, and F).
IFNβ administration enhanced tolerogenic molecules PD-L1 and TGFβ1 in CD45.1 cells in the IFNAR1−/− recipient mice, indicating that IFNβ directly acted on the adoptively transferred CD45.1 TNFR2+ cDC2s cells to enhance their tolerogenicity (Fig. 4H). TNFR2+ cDC2s primed TH2 cells or Tregs in the lungs (3). We further examined lung IL-4+ TH2 cells and Tregs in the recipient IFNAR1−/− mice 14 days after adoptive cell transfer. Adoptively transferred pathogenic CD45.1 TNFR2+ cDC2s generated lung IL-4+ TH2, not Treg cells, in IFNAR1−/− mice (Fig. 4I). In contrast, IFNβ treatment generated Tregs in IFNAR1−/− mice receiving the pathogenic TNFR2+ cDC2s (Fig. 4I). We concluded that IFNβ directly reprogrammed pathogenic lung TNFR2+ cDC2s in vivo, leading to the reduction in lung TH2 cells and the enhancement of lung Tregs in asthmatic mice.
IFNβ-reprogrammed CD45.1 TNFR2+ cDC2s primed bystander IFNAR1−/− TNFR2+ cDC2s to generate lung Tregs
Unexpectedly, in IFNβ-treated IFNAR1−/− recipient mice with CD45.1 WT TNFR2 cDC2s, the endogenous IFNAR1−/− CD45.2 TNFR2+ cDC2s had increased TGFβ1 and PD-L1 expression (fig. S5, A to C). We proposed that the donor CD45.1 WT TNFR2+ cDC2s activated the recipient CD45.2 IFNAR1−/− TNFR2+ cDC2s in vivo upon intranasal IFNβ administration. TGFβ1 promotes the conversion of peripheral naïve T cells to Tregs and likely plays a central role in DC-induced long-term peripheral tolerance (25–27). Intranasally administered IFNβ induces TGFβ1 production in TNFR2+ cDC2s, which is essential for lung Treg induction (3).
To determine whether these primed TGFβ1-producing bystander endogenous TNFR2+ cDC2s could induce Tregs, we sorted out the CD45.2 endogenous TNFR2+ cDC2s from the IFNAR1−/− recipient mice and adoptively transferred them into naïve IFNAR1−/− mice (fig. S5D). Fourteen days later, lung Tregs were examined. Compared with the CD45.2 TNFR2+ cDC2s from PBS-treated IFNAR1−/− recipient mice, the CD45.2 TNFR2+ cDC2s from IFNβ-treated IFNAR1−/− recipient mice increased lung Tregs when transferred into naïve IFNAR1−/− mice (fig. S5E). Naïve IFNAR1−/− mice had reduced lung Tregs compared with their WT littermates due to the disruption of lung IFNβ-TNFR2+cDC2-Tregs tolerogenic axis (3). In conclusion, the Treg-inducing IFNβ therapy could be amplified via the secondary activation of bystander TNFR2+ cDC2s.
IFNβ enhanced FAO in pathogenic lung TNFR2+ cDC2s in vivo
We asked how exogenous IFNβ reprogrammed pathogenic TNFR2+ cDC2s in vivo. We first found that intranasal administration of IFNβ reduced the activation of mammalian target of rapamycin complex 1 (mTORC1) indicated by reduced kinase S6K1 phosphorylation (Fig. 5A) and decreased glucose transporter 1 (GLUT1) expression (Fig. 5B) in TNFR2+ cDC2s in the asthmatic mice. In contrast, IFNβ treatment enhanced CD36 expression and fatty acid uptake in pathogenic TNFR2+ cDC2s in asthmatic mice (Fig. 5, C and D), indicating an enhanced fatty acid metabolism. We hypothesized that inhaled IFNβ might have altered the cellular metabolism in pathogenic TNFR2+ cDC2s in vivo.
Fig. 5. IFNβ enhanced mitochondrial FAO in pathogenic lung TNFR2+ cDC2s.
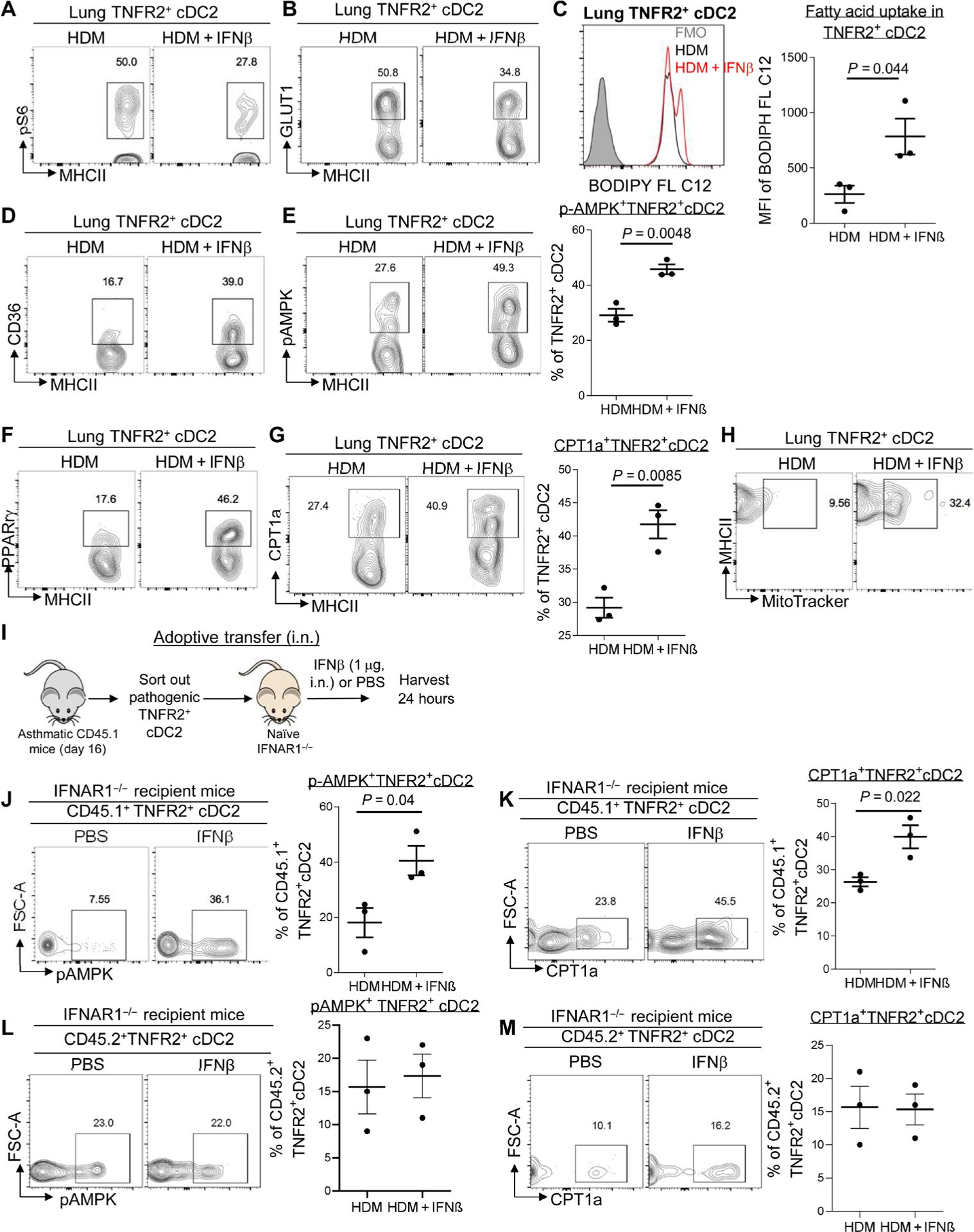
(A and B) Flow cytometry analysis of pS6 and GLUT1 expression in TNFR2+ cDC2s of asthmatic mice treated with IFNβ as in Fig. 1 (n = 3 mice per group). Data are representative of three independent experiments. (C) Representative histogram (left) and quantitative results of the geometric mean fluorescence intensity (right) of BODIPY C12 in TNFR2+ cDC2s from (A). Data are representative of three independent experiments. (D to G) Flow cytometry analysis of CD36 (D), pAMPK (E), PPARγ (F), and CPT1a (G) expression in TNFR2+ cDC2s of C57BL/6J mice from (A). Data are representative of three independent experiments. (H) Flow cytometry analysis of MitoTracker Green in TNFR2+ cDC2s of the lung from C57BL/6J mice from (A). Data are representative of three independent experiments. (I) Experimental design for adoptive transfer. Lung TNFR2+ cDC2s cells were sorted out of asthmatic CD45.1 mice on day 16. About 60,000 TNFR2+ cDC2s cells were transferred intranasally into IFNAR1−/− recipient mice. Recipient mice were treated with IFNβ (1 μg) or PBS. (J and K) Flow cytometry analysis of pAMPK (J) and CPT1a (K) expression in CD45.1+TNFR2+cDC2s from the recipient mice after 24 hours (n = 3 mice per group). Data are representative of two independent experiments. (L and M) Flow cytometry analysis of pAMPK and CPT1a expression in CD45.2+ TNFR2+cDC2s from the recipient mice after 24 hours (n = 3 mice per group). Data are representative of two independent experiments. Graphs represent the mean, with error bars indicating SEM. P values were determined by unpaired Student’s t test. Rabbit mAb isotype control staining for pAMPK and pS6 can be found in fig. S6.
Enhanced FAO is associated with immune tolerance (5). FAO is mediated by AMPK activation and enhanced peroxisome proliferator–activated receptor γ (PPARγ), CPT1a expression that transports acyl-CoA into mitochondria for oxidation (7–9). IFNβ treatment increased AMPK activation and PPARγ, CPT1a expression in TNFR2+ cDC2s in the asthmatic mice (Fig. 5, E to G). Last, mitotracker staining was increased in lung TNFR2+ cDC2s from IFNβ-treated asthmatic mice, indicating an increase of mitochondria mass and activity (Fig. 5H).
To confirm that IFNβ treatment directly acted on TNFR2+ cDC2s in asthmatic mice to enhance FAO, we did the CD45.1+ cell adoptive transfer in the IFNAR1−/− mice (Fig. 5I). Lung TNFR2+ cDC2s from asthmatic CD45.1 mice were transferred (intranasally) into IFNAR1−/− recipient mice. The recipient mice subsequently received (intranasally) IFNβ (Fig. 5I). After 24 hours, CD45.1 cells were examined in the IFNAR1−/− mice. IFNβ treatment enhanced AMPK activation and CPT1a expression in CD45.1+ TNFR2+ cDC2s in IFNAR1−/− mice (Fig. 5, J and K), indicating that IFNβ treatment directly enhanced FAO in adoptively transferred pathogenic TNFR2+ cDC2s in asthmatic mice. The endogenous IFNAR1−/− CD45.2+ cells did not have elevated AMPK activation or CPT1a expression (Fig. 5, L and M), indicating that FAO induction in TNFR2+cDC2s required direct IFNβ-IFNAR1 signaling.
Inhibiting FAO in TNFR2+ cDC2s suppressed IFNβ therapy in asthmatic mice
To demonstrate that FAO in TNFR2+ cDC2s was essential for the therapeutic effect of IFNβ, we did the TNFR2+ cDC2s adoptive transfer experiments with ETO treatment (Fig. 6A). ETO is a widely used small-molecule inhibitor of FAO via the irreversible inhibitory effects on CPT1a preventing the mitochondria transportation of acyl-CoA. Adoptively transferred pathogenic TNFR2+ cDC2s could generate TH2 cells in naïve mice (Fig. 4I). We sorted out TNFR2+ cDC2s from asthmatic mice, treated them with IFNβ or IFNβ plus ETO ex vivo, and adoptively transferred (intranasally) them into a naïve C57BL/6J mouse (Fig. 6A). Ex vivo ETO treatment of TNFR2+ cDC2s for 30 min did not affect their viability (fig. S4C). The recipient mice were treated with 20 μg of HDM daily for three consecutive days. Mice were harvested 5 days later to measure asthma symptoms (Fig. 6A).
Fig. 6. Inhibiting FAO in TNFR2+ cDC2s reduced IFNβ efficacy in HDM-induced asthma.
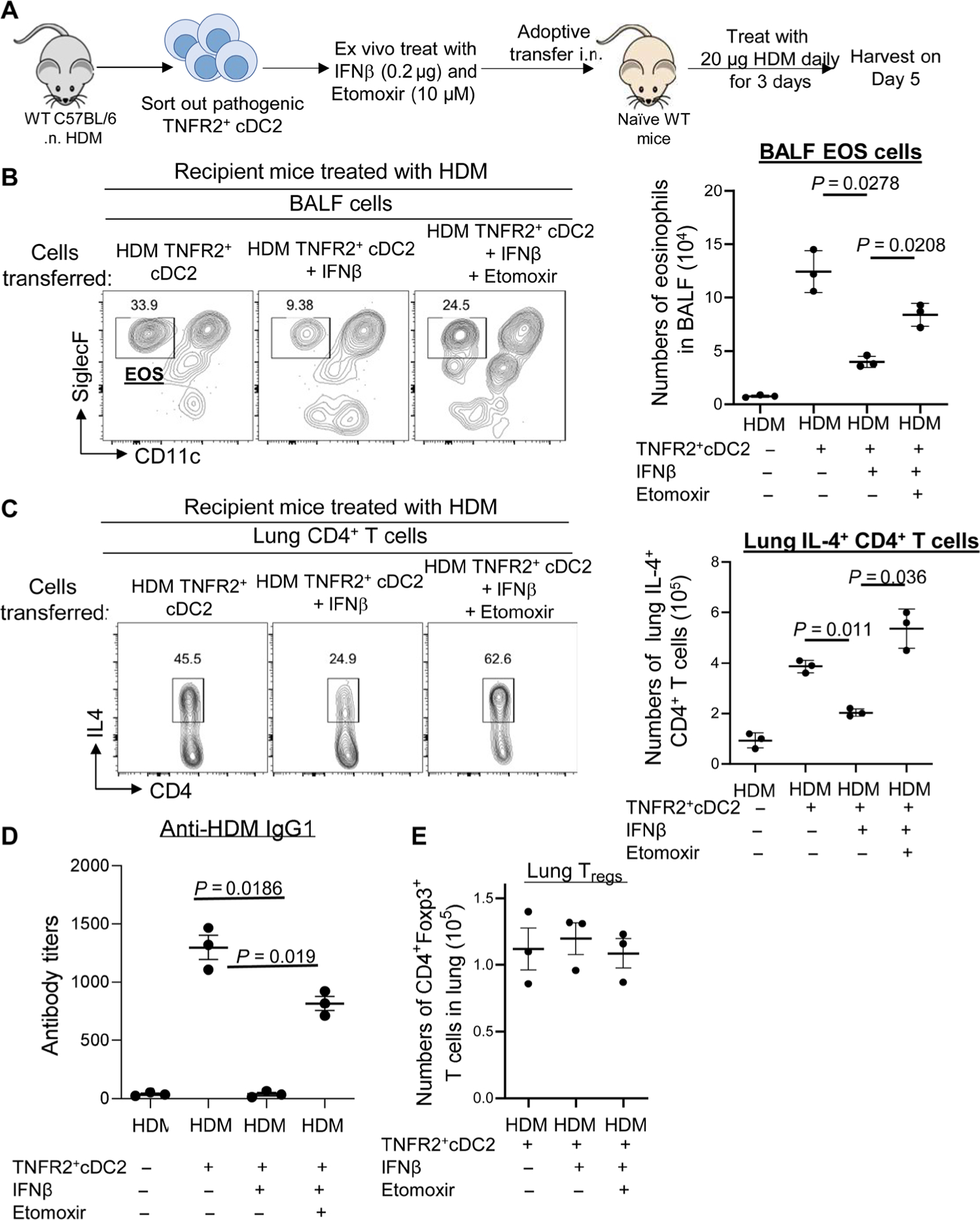
(A) Experimental design for adoptive transfer. TNFR2+ cDC2s cells were sorted out of WT mice 24 hours after treatment with HDM (100 μg). Sorted TNFR2+ cDC2s cells were treated with PBS, IFNβ (0.2 μg), and ETO (10 μM) for 30 min at 37°C. Sixty thousand treated TNFR2+ cDC2s cells were transferred intranasally into naïve C57BL/6J mice. Recipient mice were treated with 20 μg of HDM for three consecutive days. Mice were harvested 5 days after the last HDM treatment. (B and C) Flow cytometry analysis and total numbers of eosinophils (B) and IL-4 production by lung CD4+ T cells (C) in recipient mice (n = 3 mice per group). Data are representative of two independent experiments. (D) Serum levels of HDM-specific IgG1 in recipient mice (n = 3 mice per group). Data are representative of two independent experiments. (E) Total numbers of lung Tregs in recipient mice from (B) (n = 3 mice per group). Data are representative of two independent experiments. Graphs represent the mean, with error bars indicating SEM. P values were determined by one-way ANOVA with Tukey’s multiple comparison test.
Consistent with a previous report (28), three doses of 20 μg of HDM alone did not induce asthma in the naïve C57BL/6J mice because of the lack of previous sensitization (Fig. 6, B to D). However, 3 × 20 μg HDM induced asthma in naïve C57BL/6J mice that received TNFR2+ cDC2s from asthmatic mice (Fig. 6, B to D). These mice had IL-4+ TH2 cells, eosinophil infiltration, and anti-HDM IgG1 (Fig. 6, B to D), suggesting that TNFR2+ cDC2s from asthmatic mice sensitized naïve mice to develop asthma. As expected, pathogenic TNFR2+ cDC2s treated with IFNβ did not sensitize the naïve mice to develop asthma (Fig. 6, B to D). Critically, pathogenic TNFR2+ cDC2s treated with IFNβ plus ETO sensitized the naïve mice to develop asthma (Fig. 6, B to D). Thus, IFNβ-induced FAO in pathogenic TNFR2+ cDC2s is likely required for the therapeutic efficacy of IFNβ in asthmatic mice.
We also examined lung Tregs in the recipient mice. We harvested the recipient mice on day 5 after HDM treatment, which was too early for the generation of lung Tregs. Accordingly, lung Tregs numbers were similar in mice that received IFNβ- or IFNβ + ETO–treated pathogenic TNFR2+ cDC2s on day 5 (Fig. 6E).
IFNα did not generate lung Tregs or reprogram pathogenic lung TNFR2+ cDC2s in vivo
IFNα and IFNβ belong to type I IFNs and signal via a common IFNAR1/2–Tyk2/Janus kinase 1 (JAK1)–STAT1 pathway. However, replacing IFNβ with IFNα in HDM asthmatic mice did not generate lung Tregs (fig. S6, A and B). Both IFNβ (intranasally) and IFNα (intranasally) activated STAT1 in lung TNFR2+ cDC2s (fig. S6C). However, only IFNβ enhanced PD-L1 and TGFβ1 expression in TNFR2+ cDC2s in asthmatic mice (fig. S6, D and E). IFNα treatment also did not induce AMPK activation or inhibit S6 activation (mTORC1 pathway) (fig. S6, F and G) in the TNFR2+ cDC2s in the asthmatic mice. Instead, compared with IFNβ treatment, intranasal IFNα enhanced GLUT1 expression and STAT3 activation in TNFR2+ cDC2s (fig. S6, H and I). Thus, the lung Treg-inducing function is unique to IFNβ.
ERK2 expression in CD11C+ cells mediated the efficacy of IFNβ therapy in asthmatic mice
To investigate IFNβ-specific tolerogenic pathway in TNFR2+ cDC2s, we first focused on STAT3, which mediates alternative type I IFN signaling (29). We examined IFNβ-induced Treg induction in STAT3fl/flCD11cCre mice using a modified intranasal ovalbumin (OVA) model (3). Mice were treated (intranasally) with one dose of OVA (2 μg) or OVA (2 μg)/IFNβ (0.2 μg). Lungs were harvested on day 14 to examine lung Treg induction. The IFNβ-induced lung Tregs in STAT3fl/fl CD11cCre mice were similar to STAT3fl/fl mice (fig. S7A). Moreover, IFNβ treatment reduced BALF eosinophil infiltration in HDM-induced asthmatic STAT3fl/fl CD11cCre mice (Fig. 7A). Thus, STAT3 is dispensable for the tolerogenic IFNβ signaling in lung TNFR2+ cDC2s leading to Treg induction.
Fig. 7. IFNβ activated Erk2 in lung TNFR2+ cDC2s to promote FAO.
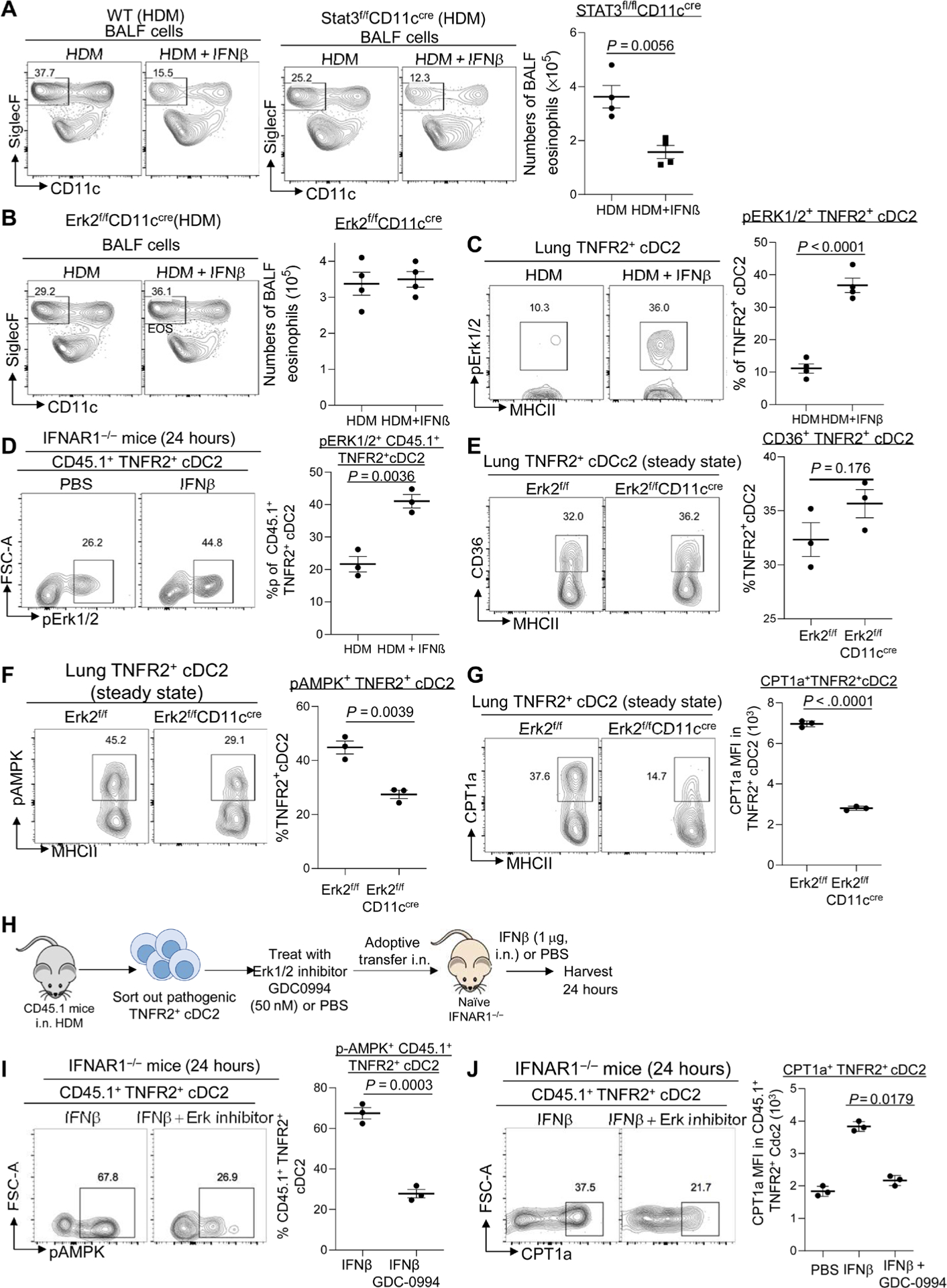
(A and B) Flow cytometry analysis of eosinophils in the BALF of WT, Erk2f/fCD11ccre, and Stat3f/fCD11ccre mice treated with HDM or HDM/IFNβ (0.2 μg) (n = 4 mice per group). Data are representative of three independent experiments. (C) Flow cytometry analysis of pErk1/2 in HDM mice treated with IFNβ (0.2 μg) (n = 3 to 4 mice per group). Data are representative of three independent experiments. (D) Lung CD45.1+TNFR2+cDC2s from HDM-induced asthmatic mice were adoptively transferred into IFNAR1−/− CD45.2 recipient mice. Recipient mice were treated with IFNβ (1 μg). Flow cytometry analysis (left) and frequency (right) of pErk1/2 in CD45.1+ TNFR2+cDC2s after IFNβ treatment (n = 3 mice per group). Data are representative of two independent experiments. (E to G) Flow cytometry analysis of CD36 (E), pAMPK (F), and CPT1a (G) expression in TNFR2+ cDC2s from Erk2f/f and Erk2f/fCD11ccre mice at steady state (n = 3 mice per group). Data are representative of three independent experiments. (H) Experimental design for adoptive transfer. Lung TNFR2+ cDC2s cells were sorted out of asthmatic CD45.1 mice on day 16. Sorted TNFR2+ cDC2s cells were treated with PBS or GDC-0994 (50 nM) for 30 min at 37°C. About 60,000 treated TNFR2+ cDC2s cells were transferred intranasally into IFNAR1−/− recipient mice. Recipient mice were treated with IFNβ (1 μg) or PBS. (I and J) Flow cytometry analysis of pAMPK (I) and CPT1a (J) expression in CD45.1+TNFR2+cDC2s after IFNβ or IFNβ plus Erk1/2 inhibitor (GDC-0994) treatment (n = 3 mice per group). Data are representative of two independent experiments. Graphs represent the mean, with error bars indicating SEM. P values were determined by one-way ANOVA with Tukey’s multiple comparison test (J) or by unpaired Student’s t test (A to G and I). Rabbit mAb isotype control staining for pAMPK and pErk1/2 can be found in fig. S6.
IFNβ can activate ERK2 (30). We found that ERK2fl/flCD11cCre mice did not have IFNβ-induced lung Tregs (fig. S7B). Furthermore, IFNβ treatment failed to suppress HDM-induced eosinophil infiltration in the ERK2fl/flCD11cCre mice (Fig. 7B). We propose that ERK2 mediates the tolerogenic IFNβ signaling in TNFR2+ cDC2s and promotes the anti-inflammatory effect of IFNβ in asthmatic mice.
IFNβ activated ERK1/2 in TNFR2+ cDC2s in vivo
We asked whether IFNβ activated ERK1/2 in TNFR2+ cDC2s. Lung TNFR2+ cDC2s at steady state are constitutively primed by endogenous lung IFNβ and are tolerogenic (3). Steady-state TNFR2+ cDC2s had constitutive ERK1/2 activation (fig. S7C). Furthermore, exogenous IFNβ induced ERK1/2 activation in the TNFR2+ cDC2s in asthmatic mice (Fig. 7C). To establish that IFNβ directly activated ERK1/2 in TNFR2+ cDC2s, we did adoptive cell transfers as in Fig. 4. Pathogenic TNFR2+ cDC2s from asthmatic CD45.1 mice were transferred into IFNAR1−/− mice. The IFNAR1−/− mice were subsequently treated (intranasally) with IFNβ. IFNβ activated ERK1/2 in CD45.1+ TNFR2+ cDC2s in the IFNAR1−/− mice (Fig. 7D). In contrast, CD45.2+ TNFR2+ cDC2s in the IFNAR1−/− mice did not have ERK1/2 activation (fig. S7D). IFNα does not activate phosphorylated ERK1/2 (pERK1/2) in TNFR2+ cDC2s (fig. S6J). We concluded that IFNβ specifically stimulated ERK1/2 in TNFR2+ cDC2s, whereas IFNα activates STAT3 in TNFR2+ cDC2s.
IFNβ activated ERK2 to promote FAO in TNFR2+ cDC2s in vivo
The therapeutic effect of IFNβ depended on the induction of FAO in TNFR2+ cDC2s (Fig. 6). We hypothesized that ERK2 activation in TNFR2+ cDC2s promotes FAO. Efficient cellular FAO depends on enhanced fatty acid uptake via CD36, the activation of AMPK (8, 9), transcription factor PPARγ, and CPT1, the rate-limiting enzyme of FAO (7).
CD36 expression (Fig. 7E) and fatty acid uptake indicated by BODIPY-C12 staining (fig. S6E) in TNFR2+ cDC2s were not altered in ERK2fl/flCD11Ccre mice. AMPK activation and CPT1a expression were reduced in ERK2fl/flCD11Ccre mice (Fig. 7, F and G), indicating that ERK2 expression in CD11C+ cells was required for FAO in steady-state TNFR2+ cDC2s.
To evaluate the role of ERK1/2 activation in IFNβ-induced FAO in pathogenic TNFR2+ cDC2s, we used the ERK1/2 inhibitor ravoxertinib (GDC-0994). GDC-0994 is a potent and highly selective ERK1/2 inhibitor with an IC50 (median inhibitory concentration) of 1.1 nM (ERK1) and 0.3 nM (ERK2), respectively (31). We sorted out pathogenic TNFR2+ cDC2s from asthmatic CD45.1 mice, treated them with GDC-0994 or PBS, and transferred them into IFNAR1−/− mice. The IFNAR1−/− mice were subsequently treated (intranasally) with IFNβ (Fig. 7H). GDC-0994–treated TNFR2+ cDC2s failed to up-regulate CPT1a or activate AMPK upon IFNβ administration in the IFNAR1−/− mice (Fig. 7, I and J), indicating that IFNβ-induced ERK1/2 activation in TNFR2+ cDC2s was required for FAO induction. GDC-0994 treatment did not inhibit fatty acid uptake (BODIPY C12 stain) in adoptively transferred TNFR2+ cDC2s (fig. S7F).
ERK1/2 activation in TNFR2+ cDC2s was required for the therapeutic effect of IFNβ in vivo
To further establish the IFNβ-ERK2-FAO pathway in TNFR2+ cDC2s, we sorted out pathogenic TNFR2+ cDC2s from HDM-induced asthmatic C57BL/6J mice and treated sorted cells with IFNβ or IFNβ plus GDC-0994 ex vivo. We transferred the treated cells into naïve C57BL/6J mice and followed with 20 μg of HDM (intranasally) daily for 3 days (Fig. 8A). Asthma phenotypes were examined 5 days later. Ex vivo GDC-0994 treatment of TNFR2+ cDC2s for 30 min did not affect their viability (fig. S4, C and D). Naïve mice did not have an asthmatic phenotype upon 3 × 20 μg HDM treatment because of the lack of previous sensitization (Fig. 8, B to D). In contrast, naïve mice that received pathogenic TNFR2+ cDC2s developed asthmatic symptoms indicated by the induction of IL-4+ TH2 cells (Fig. 8B), BALF eosinophil infiltration (Fig. 8C), and HDM-specific antibodies (Fig. 8D). As expected, naïve mice that received IFNβ-treated pathogenic TNFR2+ cDC2s did not develop asthma (Fig. 8, B to D). Critically, naïve mice that received IFNβ plus GDC-0994–treated pathogenic TNFR2+ cDC2s developed asthma similar to the naïve mice that received untreated pathogenic TNFR2+ cDC2s (Fig. 8, B to D). Lung Treg numbers were similar in mice that received IFNβ- or IFNβ + GDC-0994–treated pathogenic TNFR2+ cDC2s on day 5 after HDM challenge (Fig. 8E). We propose that ERK2 activation in TNFR2+ cDC2s mediated IFNβ therapy in asthmatic mice.
Fig. 8. Inhibiting Erk1/2 activation in TNFR2+ cDC2s reduced IFNβ efficacy in HDM-induced asthma.
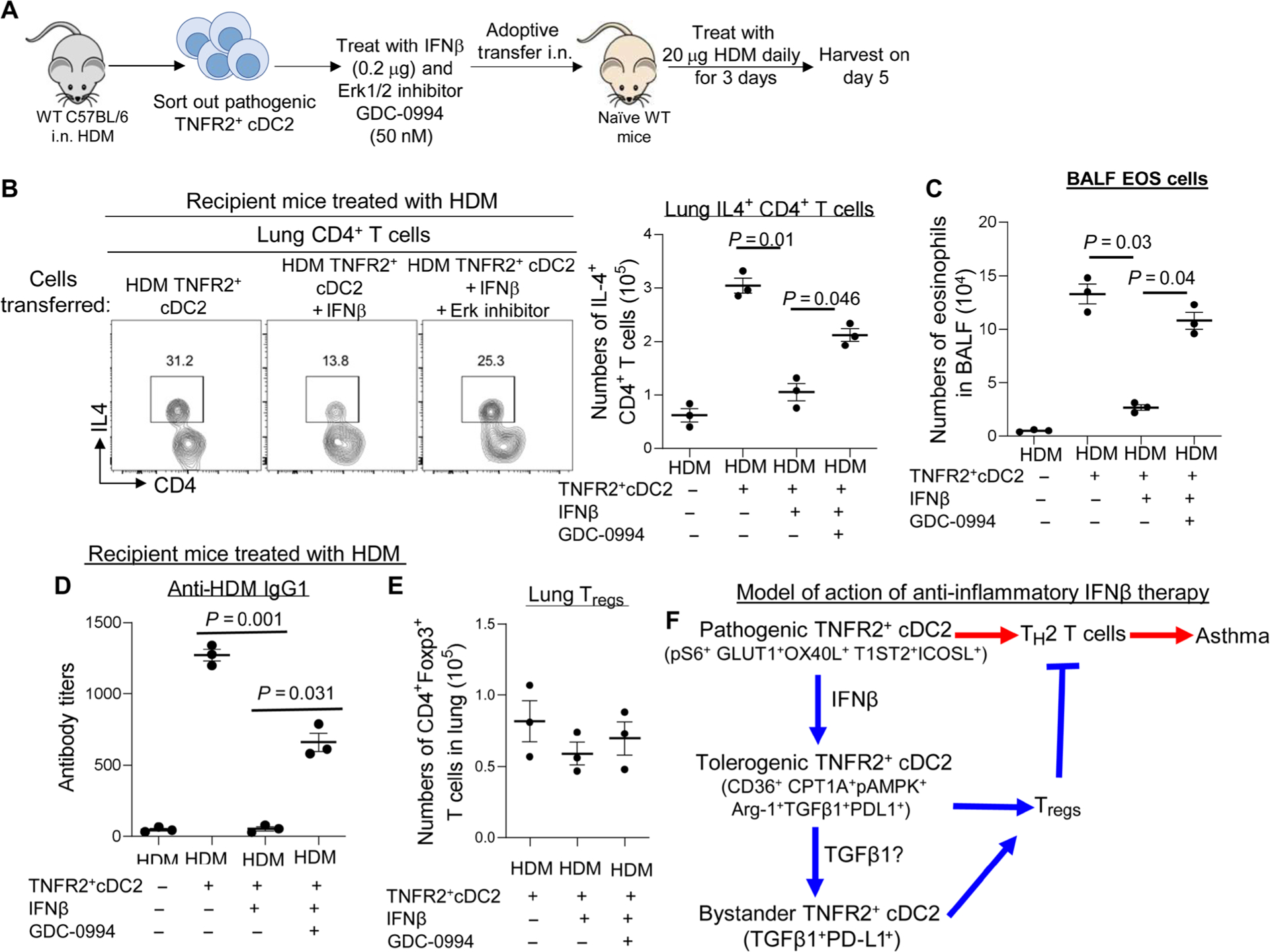
(A) Experimental design for adoptive transfer. TNFR2+ cDC2s cells were sorted out of WT mice 24 hours after treatment with HDM (100 μg). TNFR2+ cDC2s cells were treated with PBS, IFNβ (0.2 μg), or IFNβ plus Erk1/2 inhibitor GDC-0994 (50 nM) for 30 min at 37°C. Sixty thousand TNFR2+ cDC2s cells were transferred intranasally into naïve C57BL/6J mice. Recipient mice were treated with 20 μg of HDM for three consecutive days. Mice were harvested 5 days after the last HDM treatment. (B) Flow cytometry plots of IL-4–producing lung CD4+ T cells from recipient mice (n = 3 mice per group). Data are representative of two independent experiments. (C) Numbers of BALF eosinophils in recipient mice (n = 3 mice per group). Data are representative of two independent experiments. (D) Serum levels of HDM-specific IgG1 in recipient mice (n = 3 mice per group). Data are representative of two independent experiments. (E) Total numbers of lung Tregs in recipient mice in (B) (n = 3 mice per group). Data are representative of two independent experiments. (F) A model illustrates the proposed anti-inflammatory role of IFNβ in asthma. Red lines indicate inflammatory responses driven by pathogenic TNFR2+ cDC2 in asthma. The blue lines represent the anti-inflammatory responses induced by IFNβ therapy during the treatment. Graphs represent the mean, with error bars indicating SEM. P values were determined by one-way ANOVA with Tukey’s multiple comparison test.
IFNβ enhanced tolerogenic marker expression in human lung TNFR2+ cDC2s from patients with emphysema ex vivo
Healthy human lungs have a similar TNFR2+ cDC2s population that constitutively expresses TGFβ1, IDO-1, and Arg-1 (3). Furthermore, TNFR2+ cDC2s from patients with emphysema had decreased TGFβ1, Arg-1, and PD-L1 but increased IL-4 expression (3). We asked whether human IFNβ could reprogram pathogenic human lung TNFR2+ cDC2s.
Total lung cells were isolated from lung explants of patients with emphysema. Human IFNβ was added to the human lung cell culture. Lung TNFR2 cDC2s was analyzed after 24 hours. Emphysema lung cells treated with IFNβ had increased TGFβ1 and Arg1 expression and decreased IL-4 production in TNFR2 cDC2s (fig. S8). Thus, similar to mice, IFNβ could reprogram pathogenic human lung TNFR2+ cDC2s.
DISCUSSION
Here, we show that IFNβ treatment reprograms pathogenic TNFR2+ cDC2s, subsequently inhibiting asthma through multiple steps. First, by reprogramming pathogenic TNFR2+ cDC2s, IFNβ therapy reduces the number of TH2-promoting pathogenic TNFR2+ cDC2s. Second, the generation of tolerogenic TNFR2+ cDC2s induces lung Tregs that suppress lung inflammation. Third, IFNβ-reprogrammed TNFR2+cDC2s likely primes bystander TNFR2+ cDC2s in vivo that may amplify the anti-inflammatory effect. Thus, multiple in vivo mechanisms underlie IFNβ therapy through TNFR2+ cDC2s–dependent anti-inflammatory effects (Fig. 8F).
Because of its central role in controlling immune responses, the ability to manipulate DCs in vivo is highly desired. A phase 1 clinical trial has started using tolerogenic DCs to reduce islet-specific inflammation in type 1 diabetes (32). Another DC trial is underway for rheumatoid arthritis using autologous DC metabolically modified with dexamethasone and vitamin D3 and loaded with synovial fluid (33). Current DC therapies mainly use ex vivo modulated DCs that require differentiating and expanding patients’ DCs ex vivo, loading specific antigens, and subsequently injecting back to patients. These pharmacological agents differentiated ex vivo DCs that likely do not reflect the tolerogenic DCs in vivo. Furthermore, they may become immunogenic once in vivo influenced by the inflammatory microenvironment. Last, loading them with specific antigens poses a challenge for autoimmune diseases with unknown or many autoantigens, such as systemic lupus erythematosus. Directly reprograming pathogenic DCs in vivo may be superior to the current ex vivo DC therapy, bypassing the needs for ex vivo culture, autoantigens, and suppressive pharmacological agents, thus reducing the cost and time and increasing efficacy for DC therapy.
IFNβ is a U.S. Food and Drug Administration–approved medicine for multiple sclerosis (34). Its safety profile in humans is well established. Inhaled IFNβ is also in a phase 2 clinical trial for virus-induced asthma exacerbation (clinicaltrials.gov, identifier NCT01126177), although the trial is based on the antiviral function of IFNβ (35, 36). We proposed that inhaled IFNβ therapy has the potential to treat asthma caused by allergens other than HDM by reprogramming pathogenic lung TNFR2+ cDC2s and generating Tregs. The inhaled IFNβ therapy could prevent asthma exacerbation in mice. Under current asthma treatments, ~50% of patients have asthma exacerbation every year. It is crucial to determine the efficacy of inhaled IFNβ therapy in patients treated with or compared with traditional asthma treatments, such as steroids.
IFNα treatment could not enhance FAO in TNFR2+ cDC2s, indicating an IFNβ-specific signaling pathway. The IFNβ-specific anti-inflammatory activity has been well documented (37, 38). IFNβ binds IFNAR1 much tighter (100 nM affinity) than IFNα (1 to 5 μM) (39). The binding difference for IFNAR1 affects the antiviral and anti-proliferative function of type I IFNs. The EC50 (median effective concentration) for eliciting antiproliferative activity is much higher than that for the antiviral activity (40). Consequently, a 50-fold lower concentration of IFNβ than that of IFNα is required to elicit antiproliferative activity (40). We speculated that the high binding affinity of IFNβ to IFNAR1 may aid in its unique ability to activate ERK2 and FAO in TNFR2+ cDC2s. Larner’s group reports that IFNAR1 directly binds ERK2 in vitro and in vivo (30). Whether ERK2 binds to IFNAR1 in TNFR2+ cDC2s needs further investigation.
In summary, inhaled IFNβ therapy reprogrammed pathogenic lung TNFR2+ cDC2s in vivo to become tolerogenic TNFR2+ cDC2s and alleviated asthma in mice. The limitations of the study include the usage of mouse models for human asthma, the lack of safety evaluation for inhaled IFNβ, and the off-target effects of inhibitors ETO and GDC-0996. Further studies are needed to advance TNFR2+ cDC2s–dependent treatments for inflammatory diseases, especially for asthma.
MATERIALS AND METHODS
Study design
The study was designed to (i) examine inhaled IFNβ in treating and preventing asthma in mice and (ii) identify the cellular and molecular mechanisms of the anti-inflammatory IFNβ therapy in mice. The impact of inhaled IFNβ therapy on asthmatic mice was based on (i) anti-HDM antibody, (ii) lung H&E stain, (iii) lung TH2 cells, (iv) BALF eosinophils and neutrophils, and (v) lung functions. Flow cytometry and adoptive cell transfer were used to establish the in vivo cellular and molecular mechanism of IFNβ therapy. All the repeats were biological replications that involve the same experimental procedures on different mice. Where possible, treatments were assigned blindly to the experimenter by another individual in the laboratory. When comparing samples from different groups, samples from each group were analyzed in concert, thereby preventing any biases that might arise from analyzing individual treatments on different days. All experiments were repeated at least twice.
Mice
Age- and sex-matched mice (8 to 18 weeks old) were used for all experiments. C57BL/6, B6.CD45.1, Erk2fl/f, Stat3fl/f, IFNAR1−/−, and CD11ccre mice on a C57BL/6 background were purchased from The Jackson Laboratory. Mice were housed and bred under pathogen-free conditions in the Animal Research Facility at the University of Florida. All mouse experiments were performed by the regulations and approval of the Institutional Animal Care and Use Committee at the University of Florida, IACUC number 201909362.
Reagents
Recombinant mouse IFNβ (R&D Systems, catalog no. 8234-MB) or recombinant mouse IFNα1 (R&D Systems, catalog no.10148-IF) was administered intranasally in 40 μl of PBS. Endotoxin-free OVA from InvivoGen (catalog no. vac-pova) was administered intranasally in 40 μl of PBS. Anti-TNFR2 mAb (TR75-54.7, BioXcell) and isotype control (BE0091, BioXcell) were administered intranasally in 40 μl of PBS. ETO (Sigma-Aldrich, catalog no. E1905) and an ERK1/2 inhibitor (GDC-0994, Selleckchem, catalog no. S7910) were used to treat ex vivo TNFR2+ cDC2s cells. All reagents are summarized in table S1.
HDM-induced asthma
HDMs, Dermatophagoides pteronyssinus (HDM-Der p1, Greer Laboratories, catalog no. XPB82D3A2.5), were suspended in endotoxin-free PBS at a concentration of 5 mg/ml. HDM was freshly prepared by mixing equal parts of HDM-Der p1 in PBS. To induce asthma, two protocols were used: acute (low dose) and chronic asthma (high dose). Acute asthma mice were sensitized intranasally with three daily doses of 1 μg of HDM on days 0 to 2 and were later challenged with 10 μg of HDM intranasally on days 9 to 13. Chronic asthma mice were established with 25 μg of HDM (intranasally), three times a week for 5 weeks. Bronchoalveolar lavage (BAL) fluid, blood, lungs, and mediastinal lymph nodes (medLNs) were collected 3 days after the last HDM administration (17).
IFNβ therapy for asthmatic mice
Acute asthma was established in mice as above. IFNβ was administered (intranasally) (200 ng, ~240,000 IU) on days 16, 18, 20, and 27. Chronic asthma was established in mice as above. IFNβ was administered (intranasally) (400 ng, ~480,000 IU) starting on day 35 and every other day for 2 weeks. BAL fluid, blood, lungs, and medLNs were collected 2 weeks after the last HDM administration. To study the preventative effects of IFNβ, acute asthma mice were treated with IFNβ as mentioned above. Subsequently, treated mice were challenged again with five doses of 10 μg of HDM on days 41 to 45. BAL fluid, blood, lungs, and medLNs were collected on day 48.
Induction of lung Tregs by IFNβ in naïve mice
Mice were treated (intranasally) with one dose of OVA (2 μg) or OVA/IFNβ (0.2 μg) in 40 μl of PBS as previously described (3). Fourteen days later, lungs were harvested and lung Tregs were analyzed by flow cytometry.
Lung histology
Lungs were fixed in 10% formalin, paraffin-embedded, and cut into 4-μm sections. Lung sections were then stained for H&E. All staining procedures were performed by the histology core at the University of Florida. Briefly, tissue sections were immersed in Harris hematoxylin for 10 s and then washed with tap water. Cleared sections were reimmersed in eosin stain for ~30 s. The sections were washed with tap water until clear and then dehydrated in ascending alcohol solutions (50%, 70%, 80%, 95% × 2, 100% × 2). Afterward, the sections were cleared with xylene (3× to 4×). The sections were mounted on glass slides with Permount organic mounting medium for visualization.
Lung function
Pulmonary function was evaluated using an isolated, buffer-perfused mouse lung apparatus (Hugo Sachs Elektronik, March-Hugstetten, Germany), as previously described (41). Briefly, mice were anesthetized with ketamine and xylazine and a tracheostomy was performed, and animals were ventilated with room air at 100 breaths/min at a tidal volume of 7 μl/g body weight with a positive end-expiratory pressure of 2 cm H2O using a pressure-controlled ventilator (Hugo Sachs Elektronik, March-Hugstetten, Germany).
Isolation of lung cells
Cells were isolated from the lung as previously described (3). The lungs were perfused with ice-cold PBS and removed. Lungs were digested in Dulbecco’s modified Eagle’s medium containing deoxy-ribonuclease I (200 μg/ml) (Roche, 10104159001) and Liberase TM (25 μg/ml) (Roche, 05401119001) at 37°C for 2 hours. Red blood cells were then lysed, and a single cell suspension was prepared by filtering through a 70-μm cell strainer.
HDM ELISA
HDM-specific IgG1 and IgE were measured by enzyme-linked immunosorbent assay (ELISA) in the serum of HDM-treated mice. Secondary antibodies used were anti-mouse IgG1-HRP (horseradish peroxidase) (Southern Biotech, catalog no.1070-05) and anti-mouse IgE-HRP (Southern Biotech, catalog no.1110-05). To measure HDM-specific T cell responses, lung cells were restimulated with HDM (25 μg/ml) for 4 days. IL-5 cytokines were measured in the supernatant by ELISA.
Flow cytometry
Single-cell suspensions were stained with fluorescent dye–conjugated antibodies in PBS containing 2% fetal bovine serum and 1 mM EDTA. Surface stains were performed at 4°C for 20 min. For intracellular cytokine or transcription factor staining of murine and human cells, cells were fixed and permeabilized with the Foxp3 staining buffer set (eBioscience, catalog no 00-5523-00). Cells were washed and stained with surface markers. Cells were then fixed and permeabilized (eBioscience, catalog no. 00-5523-00) for intracellular cytokine stain. Data were acquired on a BD LSRFortessa and analyzed using the FlowJo software package (FlowJo LLC). Cell sorting was performed on the BD FACSAria III flow cytometer and cell sorter. More information on the antibodies used can be found in table S1.
Adoptive transfer
cDC2s cells were sorted from the lungs of CD45.1 HDM-treated mice (two to three mice) with a FACSAria III flow cytometer. cDC2s cells were identified as MHCII+CD11c+CD11b+CD24+CD64−. Sixty thousand cells in 40 μl of PBS were administered intranasally into recipient IFNAR1−/− mice as before (3). Briefly, mice were anesthetized by inhalation of isoflurane (1 to 5%) using a rodent anesthesia machine with a vaporizer. Cells, in 40 μl of PBS, were administered into the nostrils of an anesthetized mouse. Donor cells in the lungs were analyzed 24 hours later. T cells in the lung were analyzed 14 days later.
Mitochondrion and fatty acid uptake assay
To measure mitochondrial membrane potential, cells were washed and incubated with RPMI containing MitoTracker Green FM (Thermo Fisher Scientific, catalog no. M7514) (100 nM) for 15 min at 37°C. Cells were washed and resuspended in fluorescence-activated cell sorting (FACS) buffer. To measure fatty acid uptake, cells were incubated in RPMI medium containing C1-BODIPY 500/510 C12 (Life Technologies, catalog no. D-382) at a final concentration of 1 μM for 15 min at 37°C. Cells were washed with FACS buffer.
DC transfer model of asthma
cDC2s cells were sorted from the lungs of B6.CD45.1 mice and treated with HDM (100 μg) for 24 hours. Sorted cells (~60,000) were treated with IFNβ (0.2 μg), IFNβ/ETO (10 μM), or IFNβ/Erk1/2 inhibitor (GDC-0994) (50 nM) for 30 min at 37°C. Cells were then transferred intranasally into naïve recipient C57BL/6J mice as described above. Recipient mice were then treated with 20 μg of HDM daily for 3 days. Recipient mice were harvested on day 5.
Human lung explants
Human lung explants were procured at the Lung Transplant Center, Division of Pulmonary, Critical Care and Sleep Medicine, Department of Medicine, University of Florida. Donor and patient consent was obtained for a research protocol (UF IRB201902955—Treatment with IFNβ induces tolerogenic lung dendritic cells in human advanced lung disease). Healthy donor lungs were surgically removed postmortem and perfused, and small pieces were cut from the right middle and lower lobes for research purposes and stored in cold Perfadex at 4°C for no more than 12 hours before processing. Explanted lungs from emphysema lung transplant patients were stored in cold Perfadex at 4°C for no more than 12 hours before the process. No lung explants were procured from prisoners.
Statistical analysis
To gain statistical power, we use three to four mice per group to characterize lung immunity. The statistical justification for group size was calculated using the SAS program to calculate the animal numbers. The analysis was carried out using a standard error of 0.5 for immunological assays and a power of 0.9. All data are expressed as means ± SEM. Statistical significance was evaluated using Prism 7.0 software. Comparisons between two groups were analyzed by performing an unpaired Student’s t test. Comparisons between more than two groups were analyzed by performing a one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test.
Supplementary Material
Acknowledgments:
We thank the Center for Immunology and Transplantation at the University of Florida for the assistance. We thank past and present members of the Jin laboratory for helpful discussion and technical support.
Funding:
This work was supported by NIH grants AI110606, AI132865, and HL152163 (to L.J.) and Spevak Memorial Fund for Asthma Research (to L.J.). S.M. was supported through The American Association of Immunologists Careers in Immunology Fellowship Program.
Footnotes
SUPPLEMENTARY MATERIALS
immunology.sciencemag.org/cgi/content/full/6/61/eabi8472/DC1
Figs. S1 to S8
Tables S1 and S2
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Tordesillas L, Lozano-Ojalvo D, Dunkin D, Mondoulet L, Agudo J, Merad M, Sampson HA, Berin MC, PDL2+ CD11b+ dermal dendritic cells capture topical antigen through hair follicles to prime LAP+ Tregs. Nat. Commun 9, 5238 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Semmrich M, Plantinga M, Svensson-Frej M, Uronen-Hansson H, Gustafsson T, Mowat AM, Yrlid U, Lambrecht BN, Agace WW, Directed antigen targeting in vivo identifies a role for CD103+ dendritic cells in both tolerogenic and immunogenic T cell responses. Mucosal Immunol. 5, 150–160 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mansouri S, Katikaneni DS, Gogoi H, Pipkin M, Machuca TN, Emtiazjoo AM, Jin L, Lung IFNAR1hi TNFR2+ cDC2 promotes lung regulatory T cells induction and maintains lung mucosal tolerance at steady state. Mucosal Immunol. 13, 595–608 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mansouri S, Patel S, Katikaneni DS, Blaauboer SM, Wang W, Schattgen S, Fitzgerald K, Jin L, Immature lung TNFR2(−) conventional DC 2 subpopulation activates moDCs to promote cyclic di-GMP mucosal adjuvant responses in vivo. Mucosal Immunol. 12, 277–289 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pearce EJ, Everts B, Dendritic cell metabolism. Nat. Rev. Immunol 15, 18–29 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, Pearce EJ, Toll-like receptor–induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115, 4742–4749 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malinarich F, Duan K, Hamid RA, Bijin A, Lin WX, Poidinger M, Fairhurst AM, Connolly JE, High mitochondrial respiration and glycolytic capacity represent a metabolic phenotype of human tolerogenic dendritic cells. J. Immunol 194, 5174–5186 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Everts B, Pearce EJ, Metabolic control of dendritic cell activation and function: Recent advances and clinical implications. Front. Immunol 5, 203 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Munday MR, Campbell DG, Carling D, Hardie DG, Identification by amino acid sequencing of three major regulatory phosphorylation sites on rat acetyl-CoA carboxylase. Eur. J. Biochem 175, 331–338 (1988). [DOI] [PubMed] [Google Scholar]
- 10.Mondanelli G, Bianchi R, Pallotta MT, Orabona C, Albini E, Iacono A, Belladonna ML, Vacca C, Fallarino F, Macchiarulo A, Ugel S, Bronte V, Gevi F, Zolla L, Verhaar A, Peppelenbosch M, Mazza EMC, Bicciato S, Laouar Y, Santambrogio L, Puccetti P, Volpi C, Grohmann U, A relay pathway between arginine and tryptophan metabolism confers immunosuppressive properties on dendritic cells. Immunity 46, 233–244 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, Servillo G, Brunacci C, Calvitti M, Bicciato S, Mazza EMC, Boon L, Grassi F, Fioretti MC, Fallarino F, Puccetti P, Grohmann U, Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat. Immunol 12, 870–878 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Pantel A, Teixeira A, Haddad E, Wood EG, Steinman RM, Longhi MP, Direct type I IFN but not MDA5/TLR3 activation of dendritic cells is required for maturation and metabolic shift to glycolysis after poly IC stimulation. PLOS Biol. 12, e1001759 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu D, Sanin DE, Everts B, Chen Q, Qiu J, Buck MD, Patterson A, Smith AM, Chang CH, Liu Z, Artyomov MN, Pearce EL, Cella M, Pearce EJ, Type 1 interferons induce changes in core metabolism that are critical for immune function. Immunity 44, 1325–1336 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fritsch SD, Weichhart T, Effects of interferons and viruses on metabolism. Front. Immunol 7, 630 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rudick RA, Ransohoff RM, Peppler R, Medendorp SV, Lehmann P, Alam J, Interferon β induces interleukin-10 expression: Relevance to multiple sclerosis. Ann. Neurol 40, 618–627 (1996). [DOI] [PubMed] [Google Scholar]
- 16.Vandenbark AA, Huan J, Agotsch M, la Tocha D, Goelz S, Offner H, Lanker S, Bourdette D, Interferon-β1a treatment increases CD56bright natural killer cells and CD4+CD25+ Foxp3 expression in subjects with multiple sclerosis. J. Neuroimmunol 215, 125–128 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Haspeslagh E, Debeuf N, Hammad H, Lambrecht BN, Murine models of allergic asthma. Methods Mol. Biol 1559, 121–136 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Debeuf N, Haspeslagh E, van Helden M, Hammad H, Lambrecht BN, Mouse models of asthma. Curr. Protoc. Mouse Biol 6, 169–184 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Strickland DH, Stumbles PA, Zosky GR, Subrata LS, Thomas JA, Turner DJ, Sly PD, Holt PG, Reversal of airway hyperresponsiveness by induction of airway mucosal CD4+CD25+ regulatory T cells. J. Exp. Med 203, 2649–2660 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Josefowicz SZ, Niec RE, Kim HY, Treuting P, Chinen T, Zheng Y, Umetsu DT, Rudensky AY, Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 482, 395–399 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.D’Alessio FR, Tsushima K, Aggarwal NR, West EE, Willett MH, Britos MF, Pipeling MR, Brower RG, Tuder RM, McDyer JF, King LS, CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J. Clin. Invest 119, 2898–2913 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cabeza-Cabrerizo M, Cardoso A, Minutti CM, Pereira da Costa M, Reis ESC, Dendritic cells revisited. Annu. Rev. Immunol 39, 131–166 (2021). [DOI] [PubMed] [Google Scholar]
- 23.Bosteels C, Neyt K, Vanheerswynghels M, van Helden MJ, Sichien D, Debeuf N, De Prijck S, Bosteels V, Vandamme N, Martens L, Saeys Y, Louagie E, Lesage M, Williams DL, Tang S-C, Mayer JU, Ronchese F, Scott CL, Hammad H, Guilliams M, Lambrecht BN, Inflammatory Type 2 cDCs acquire features of cDC1s and macrophages to orchestrate immunity to respiratory virus infection. Immunity 52, 1039–1056.e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hasegawa H, Matsumoto T, Mechanisms of tolerance induction by dendritic cells in vivo. Front. Immunol 9, 350 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM, Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J. Exp. Med 198, 1875–1886 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamazaki S, Bonito AJ, Spisek R, Dhodapkar M, Inaba K, Steinman RM, Dendritic cells are specialized accessory cells along with TGF- for the differentiation of Foxp3+ CD4+ regulatory T cells from peripheral Foxp3 precursors. Blood 110, 4293–4302 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coombes JL, Siddiqui KRR, Arancibia-Cárcamo CV, Hall J, Sun CM, Belkaid Y, Powrie F, A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β and retinoic acid–dependent mechanism. J. Exp. Med 204, 1757–1764 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Plantinga M, Guilliams M, Vanheerswynghels M, Deswarte K, Branco-Madeira F, Toussaint W, Vanhoutte L, Neyt K, Killeen N, Malissen B, Hammad H, Lambrecht BN, Conventional and monocyte-derived CD11b+ dendritic cells initiate and maintain T helper 2 cell–mediated immunity to house dust mite allergen. Immunity 38, 322–335 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Ivashkiv LB, Donlin LT, Regulation of type I interferon responses. Nat. Rev. Immunol 14, 36–49 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.David M, Petricoin E III, Benjamin C, Pine R, Weber M, Larner A, Requirement for MAP kinase (ERK2) activity in interferon α– and interferon β–stimulated gene expression through STAT proteins. Science 269, 1721–1723 (1995). [DOI] [PubMed] [Google Scholar]
- 31.Blake JF, Burkard M, Chan J, Chen H, Chou KJ, Diaz D, Dudley DA, Gaudino JJ, Gould SE, Grina J, Hunsaker T, Liu L, Martinson M, Moreno D, Mueller L, Orr C, Pacheco P, Qin A, Rasor K, Ren L, Robarge K, Shahidi-Latham S, Stults J, Sullivan F, Wang W, Yin J, Zhou A, Belvin M, Merchant M, Moffat J, Schwarz JB, Discovery of (S)-1-(1-(4-chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino) pyrimidin-4-yl)pyridin-2(1H)-one (GDC-0994), an extracellular signal–regulated kinase 1/2 (ERK1/2) inhibitor in early clinical development. J. Med. Chem 59, 5650–5660 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Creusot RJ, Giannoukakis N, Trucco M, Clare-Salzler MJ, Fathman CG, It is time to bring dendritic cell therapy to type 1 diabetes. Diabetes 63, 20–30 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hilkens CM, Isaacs JD, Tolerogenic dendritic cell therapy for rheumatoid arthritis: Where are we now? Clin. Exp. Immunol 172, 148–157 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacobs L, Brownscheidle CM, Appropriate use of interferon β−1a in multiple sclerosis. BioDrugs 11, 155–163 (1999). [DOI] [PubMed] [Google Scholar]
- 35.Djukanovic R, Harrison T, Johnston SL, Gabbay F, Wark P, Thomson NC, Niven R, Singh D, Reddel HK, Davies DE, Marsden R, Boxall C, Dudley S, Plagnol V, Holgate ST, Monk P; INTERCIA Study Group, The effect of inhaled IFN-β on worsening of asthma symptoms caused by viral infections. A randomized trial. Am. J. Respir. Crit. Care Med 190, 145–154 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jackson DJ, Inhaled interferon: A novel treatment for virus-induced asthma? Am. J. Respir. Crit. Care Med 190, 123–124 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kasper LH, Reder AT, Immunomodulatory activity of interferon-β. Ann. Clin. Transl. Neurol 1, 622–631 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gonzalez-Navajas JM, Lee J, David M, Raz E, Immunomodulatory functions of type I interferons. Nat. Rev. Immunol 12, 125–135 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lavoie TB, Kalie E, Crisafulli-Cabatu S, Abramovich R, DiGioia G, Moolchan K, Pestka S, Schreiber G, Binding and activity of all human alpha interferon subtypes. Cytokine 56, 282–289 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Schreiber G, The molecular basis for differential type I interferon signaling. J. Biol. Chem 292, 7285–7294 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cai J, Gehrau R, Tu Z, Leroy V, Su G, Shang J, Mas VR, Emtiazjoo A, Pelaez A, Atkinson C, Machuca T, Upchurch GR Jr., Sharma AK, MicroRNA-206 antagomiR–enriched extracellular vesicles attenuate lung ischemia–reperfusion injury through CXCL1 regulation in alveolar epithelial cells. J. Heart Lung Transplant 39, 1476–1490 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.