

Abstract
Mitochondria can be released by astrocytes as part of a help-me signaling process in stroke. In this study, we investigated the molecular mechanisms that underlie mitochondria secretion, redox status and functional regulation in the extracellular environment. Exposure of rat primary astrocytes to NAD or cADPR elicited an increase in mitochondrial calcium through ryanodine receptor (RyR) in the endoplasmic reticulum (ER). Importantly, CD38 stimulation with NAD accelerated ATP production along with increasing glutathione reductase (GR) and dipicolinic acid (DPA) in intracellular mitochondria. When RyR was blocked by Dantrolene, all effects were clearly diminished. Mitochondrial functional assay showed that these activated mitochondria appeared to be resistant to H2O2 exposure and sustained mitochondrial membrane potential, while inhibition of RyR resulted in disrupted membrane potential under oxidative stress. Finally, a gain- or loss-of-function assay demonstrated that treatment with DPA in control mitochondria preserved GR contents and increased mitochondrial membrane potential, whereas inhibiting GR with carmustine decreased membrane potentials in extracellular mitochondria released from astrocytes. Collectively, these data suggest that ER-mitochondrial interaction mediated by CD38 stimulation may support mitochondrial energy production and redox homeostasis during the mode of mitochondrial transfer from astrocytes.
Keywords: CD38, astrocytes, dipicolinic acid, glutathione reductase, mitochondria, endoplasmic reticulum, ryanodine receptor
Introduction
Astrocytes have multicomplex roles in the regulation of neurodevelopment, neurotransmission, cerebral metabolism and blood flow in the CNS physiology and pathophysiology [1–6]. Normal astrocytes protect neurons against oxidative stress and excitotoxicity [7, 8]. In contrast, astrocytes under inflammatory and oxidative stress conditions may release deleterious factors that damage neurons [9–13]. It has been reported that activating astrocytic mitochondrial metabolism reverses ischemic neuronal damage [14–16], while inhibition of astrocytic mitochondria retards neurovascular remodeling after stroke [17]. Therefore, astrocytic metabolic health may be essential for these neuroglial protective mechanisms.
Mitochondria are essential for maintaining intracellular function in the central nervous system (CNS) [18, 19]. Perturbations in mitochondrial function has been implicated in almost all forms of CNS injury and disease, e.g. stroke [20], intracerebral hemorrhage [21], spinal cord injury [22], Alzheimer’s disease [23], Parkinson’s disease [24], and amyotrophic lateral sclerosis [25]. Recently, it has been demonstrated that mitochondria can also be secreted, and extracellular mitochondria play key roles in CNS disease, including stroke [26]. Within the conceptual framework of help-me signaling [27], mitochondria may be transferred from astrocytes into adjacent neurons in a protective manner [28], and mitochondria transplantation has been pursued as a candidate therapeutic approach for CNS injury and disease [29–32]. However, many gaps in knowledge remain, and the underlying molecular mechanisms are not fully understood, including those that mediate mitochondrial secretion and uptake, maintenance of functionality in the extracellular compartment, and pathways that lead to neuroprotection, neuroplasticity, and inflammatory modulation [33–36].
Although the mechanisms of mitochondrial release remain to be fully elucidated, several pathways are being actively investigated. A recent study demonstrated that the interaction between mitochondria and the ER may facilitate mitochondrial transfer within the osteocyte dendritic network [37]. In the context of cerebral ischemia, we previously found that CD38 may be a critical mechanism involved in the release of extracellular mitochondria from astrocytes and their transfer into vulnerable neurons [28]. CD38 is multifunctional ecto-enzyme that participates in NAD+ metabolism in order to generate second messengers and regulate intracellular calcium levels [38]. Activation of NAD+-dependent pathways is intimately involved in the regulation of mitochondrial metabolism and redox homeostasis [39]. Therefore, we hypothesize that the ability of CD38 to induce mitochondria secretion from astrocytes may be related to (i) NAD+ and calcium signaling between mitochondria and endoplasmic reticulum, and (ii) regulation of redox status that contributes to mitochondria functionality in the extracellular compartment.
Methods and Materials
Reagents:
β-Nicotinamide adenine dinucleotide sodium salt (Sigma Aldrich, N0632–1G), cyclic ADP ribose (Sigma Aldrich, C7344–5MG), Dantrolene sodium salt (Tocris, 0507), CellLight Mitochondria-GFP, BacMam 2.0 (Thermo Fisher Scientific, C10600), CellLight Golgi-RFP, BacMam 2.0 (Thermo Fisher Scientific, C10593), ER-Tracker Blue-White DPX (Thermo Fisher Scientific, E12353), Dantrolene (Tocris, 507). DPA/Terbium for membrane fusion assay kit (Biotium, 80104), 2,6-Pyridinedecarboxilic acid (Sigma Aldrich, P63808), Carmustine (Millipore Sigma, C0400).
Primary astrocyte cultures:
Primary astrocyte cultures were prepared from cerebral cortices of 2-day-old neonatal Sprague-Dawley rats. Briefly, dissociated cortical cells were suspended in Dulbecco’s modified Eagle medium (DMEM, Life Technology, 11965–084) containing 25 mM glucose, 4 mM glutamine, 1 mM sodium pyruvate, and 10% fetal bovine serum and plated on uncoated 25 cm2 flasks at a density of 6×105 cells/cm2. Monolayers of type 1 astrocytes were obtained 12–14 days after plating. Non-astrocytic cells such as microglia and neurons were detached from the flasks by shaking and removed by changing the medium. In this system, more than 95% of the cells were identified as type 1 astrocytes by GFAP staining and their flattened, polygonal morphology [40, 41]. To detect extracellular mitochondria, these rat cortical astrocytes were incubated with CellLight Mitochondria-GFP overnight, resulting in expressing GFP in mitochondrial E1 alpha pyruvate dehydrogenase. Following treatments, astrocyte-conditioned media (ACM) were collected for FACS analysis.
Reagent treatment in astrocytes:
Fig 1A: Nicotinamide adenine dinucleotide (NAD: 5 mM) was treated for 1 hour and astrocytes were fixed with 4% PFA for further staining processes. Fig 1B–C: NAD (5 mM) or cADPR (100 μM) was co-treated with a ryanodine receptor blocker Dantrolene (10 μM) for 30 minutes. Fig 2–6: NAD (5 mM) or NAD + Dantrolene (10 μM) was incubated in astrocytes for 24 hrs. Dantrolene was co-incubated with NAD in astrocytes. For aconitase inhibition with fluorocitrate (FC: 0.5 mM), astrocytes were first incubated with FC for 24 hrs followed by NAD treatment for another 24 hrs to assess mitochondrial DPA contents. Fig.6: NAD (5 mM) or NAD + Carumustine (250 μM) was treated for 24 hrs.
Fig. 1. CD38 - cADPR signals increase mitochondrial calcium via ryanodine receptor:
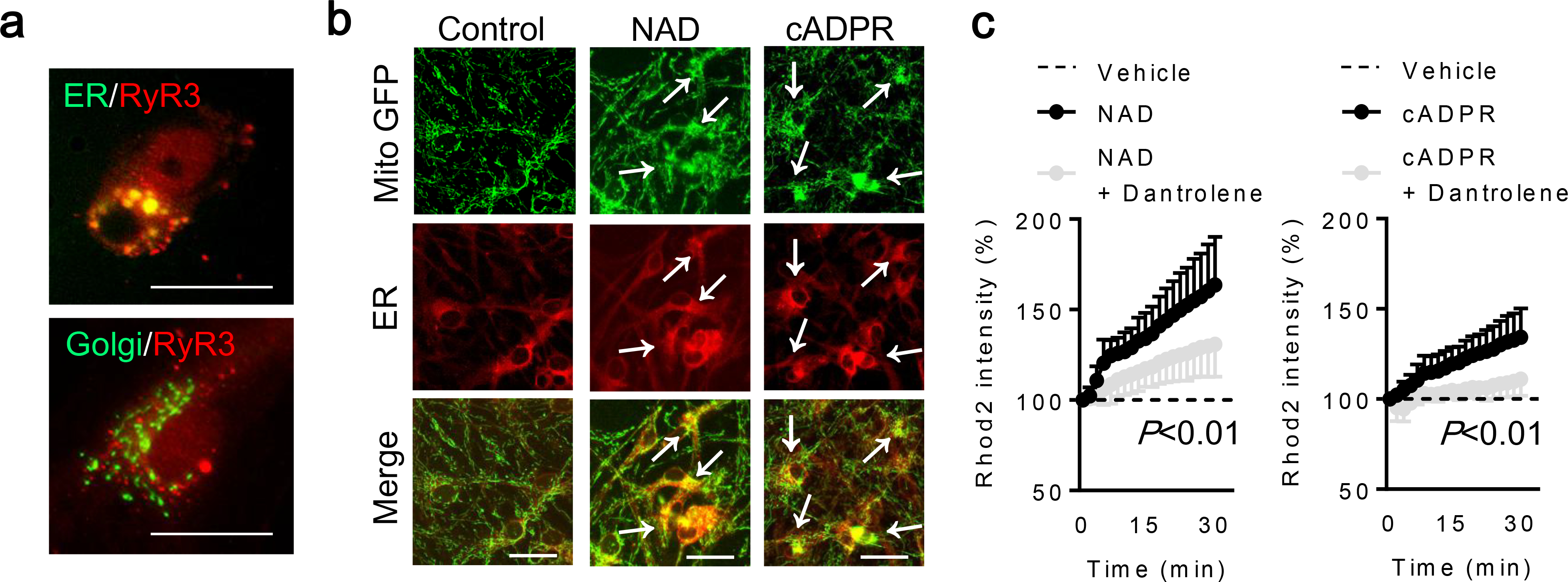
a. NAD (5 mM) was added to rat cortical astrocytes for an hour. Immunostaining demonstrated that brain-type ryanodine receptor RyR3 was expressed in the endoplasmic reticulum (ER) in astrocytes. Scale: 20 μm. b. Fluorescent staining demonstrated that mitochondria were close proximity to the endoplasmic reticulum (ER) at 30 minutes after treatment with either NAD (5 mM) or cADPR (100 μM). c. Rhod2 intensity was increased after NAD or cADPR treatment, and a ryanodine receptor blocker Dantrolene (10 μM) blocked these effect (NAD; n= 11, NAD + Dantrolene; n=15, cADPR; n= 7, cADPR + Dantrolene; n=7). Note: Each data point was normalized by vehicle-treated astrocytes (dotted line). RM two-way ANOVA. All values are mean +/− SD.
Fig. 2. CD38 increases mitochondrial ATP production via ryanodine receptor:
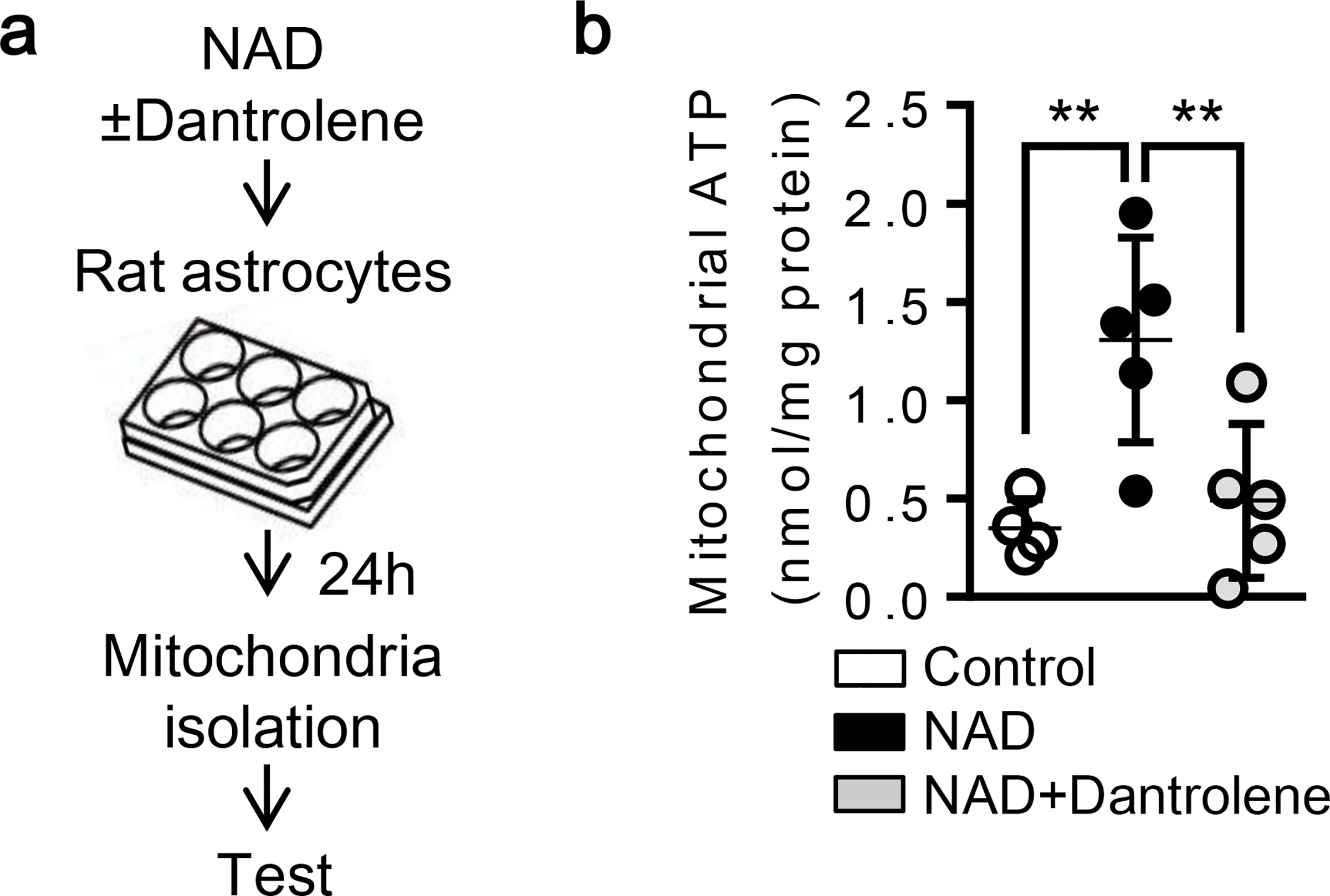
a. Rat cortical astrocytes were treated with NAD (5 mM) with or without Dantrolene (10 μM) for 24 hrs. b. Luminescence assay showed that NAD treatment increased ATP and Dantrolene diminished the effect (Control; n= 4, NAD; n= 5, NAD + Dantrolene; n=5). *P<0.05, one-way ANOVA followed by Fisher’s LSD test. All values are mean +/− SD.
Fig. 6. Effect of glutathione reductase in maintaining membrane potential in mitochondria released from astrocytes:
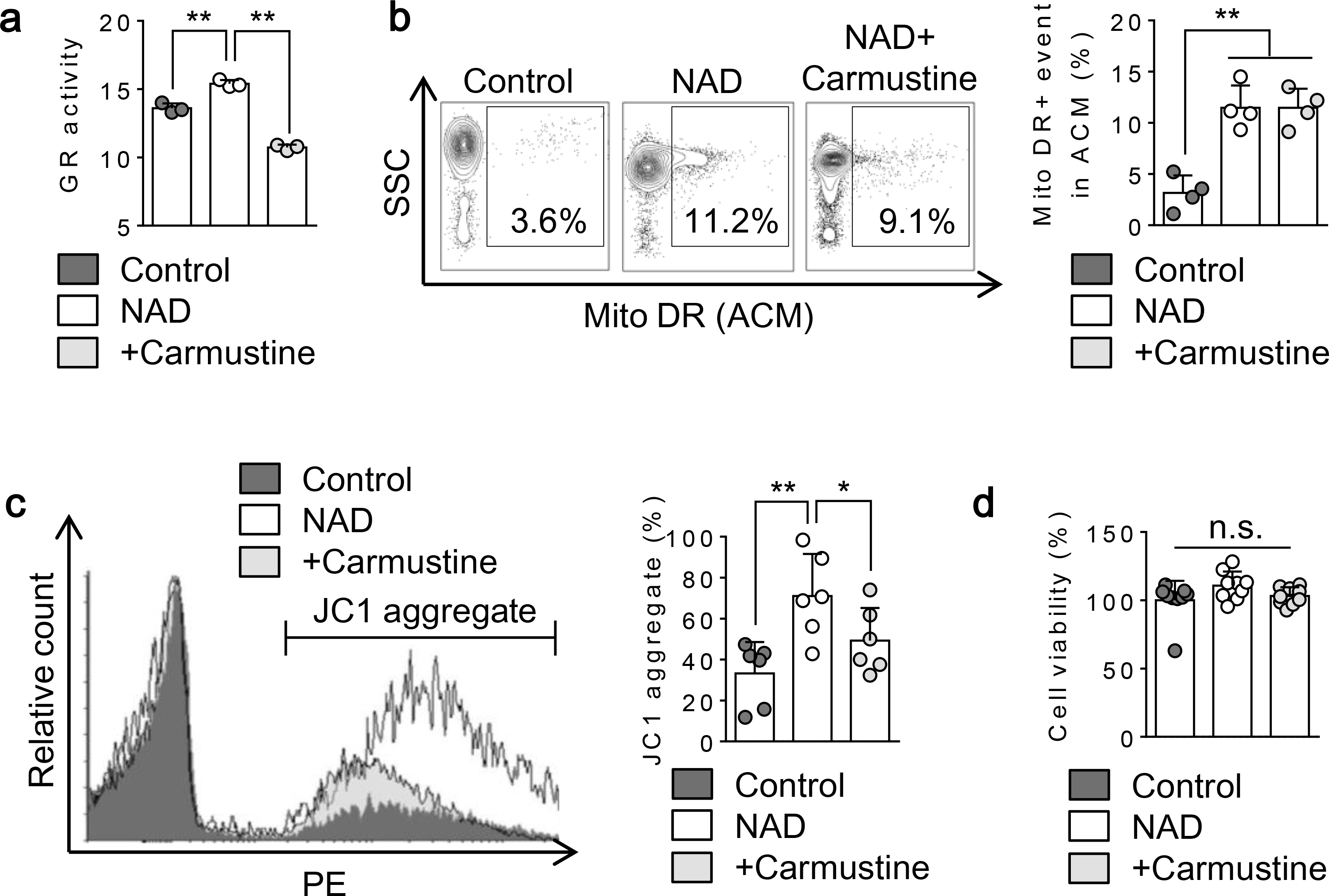
Extracellular mitochondria were identified by Mitotracker DR. a. NAD (5 mM) treatment for 24 hrs significantly increased glutathione reductase activity in astrocytes while inhibition of glutathione reductase with Carmustine (250 μM) significantly decreased the activity (n=3). **P<0.01, one-way ANOVA followed by Fisher’s LSD test. b. FACS analysis showed that NAD treatment for 24 hrs increased extracellular mitochondrial events (Mitotracker DR) and co-treatment with carmustine did not affect mitochondrial secretion induced by NAD (n= 4). **P<0.01, one-way ANOVA followed by Fisher’s LSD test. c. Further FACS analysis using JC1 demonstrated that NAD-treated astrocytes released mitochondria with higher membrane potential compared to control, whereas co-treatment with carmustine lowered mitochondrial membrane potential in extracellular mitochondria (n= 6). *P<0.05, **P<0.01, one-way ANOVA followed by Fisher’s LSD test. d. All conditions did not influence cell viability (n=9). All values are mean +/− SD.
Glutathione reductase activity assay:
Glutathione reductase activity was measured by Glutathione reductase assay kit (ab83461, Abcam). Briefly, mitochondria were isolated from astrocytes stimulated with NAD (5 mM) or NAD plus carmustine (250 μM) for 24 hrs. Each 5 μg of mitochondria was processed in the kit according to the manufacture instruction. In the assay, glutathione reductase reduced GSSG to GSH, which reacts with 5, 5’-Dithiobis (2-nitrobenzoic acid) (DTNB), followed by the generated TNB2 (yellow color, λmax = 405 nm) was analyzed by a microplate reader.
Cell Viability Assays:
Neuronal injury was measured by Cell Counting Kit 8 cytotoxicity assay (DOJINDO, CK04–13). The relative assessments of neuronal injury were normalized by comparison with control cell as 100% cell survival.
ATP measurement:
Mitochondrial ATP was determined by CellTiter-Glo luminescence (Promega, G7570). Briefly, opaque-walled 96-well plates with mitochondria suspension (2 μg per 50 μL) were prepared. CellTiter-Glo luminescence test solution (50 μL) was added and incubated for 10 min at room temperature. Luminescent signal was determined by luminescence plate reader.
Mitochondria membrane potential measurement:
To monitor mitochondrial health, JC-1 dye (invitrogen, T-3168) was used to assess mitochondrial membrane potential. Astrocyte conditioned media were incubated with JC1 (0.8 μM) for 30 min at 37 °C. JC1 dye exhibits potential-dependent accumulation in mitochondria, indicated by fluorescence emission shift from green (Ex 485 nm/Em 516 nm) to red (Ex 579 nm/Em 599 nm). Mitochondria membrane potential was determined by the fluorescent ratio with a fluorescent microplate reader.
Terbium - Dipicolinic acid (DPA) measurement:
DPA was measured by the Terbium fluorescence. Briefly, mitochondria samples were prepared in PRO-PREP solution (Bulldog Bio, 17081) before analysis and the protein concentration was adjusted to 10 μg/mL. Each sample (20 μL) was mixed with 100 μL of terbium chloride solution (2.5 mM) in wells of a 96-well flat-bottomed black plates (Fisher, 14–245-197A). After incubating for 10 min at room temperature, fluorescence was detected by fluorescent microplate reader (Ex/Em, 276 nm/490 nm).
FACS analysis:
Standard FACS analysis was performed by BD Fortessa. Extracellular mitochondria were identified by mitochondrial protein E1 alpha pyruvate dehydrogenase-GFP or Mitotracker Deep Red. Astrocyte-conditioned medium (ACM) was collected from rat cortical astrocytes. Briefly, ACM were centrifuged by 700g for 5 minutes in order to exclude cellular debris. It is important to note that astrocytic viability was not affected by various stimuli in this study. Therefore, passively released mitochondria from dying/dead cells during ACM collection should not impact on changes in the number of actively secreted extracellular mitochondria. When ACM were collected from astrocytes labeled by mitochondria GFP, samples were directly analyzed by BD Fortessa following the centrifugation. Other than that, ACM were incubated with Mitotracker Deep Red (50 nM) for 30 min at 37°C. For FACS analysis, control beads were utilized to gate population ranging in size less than 900 nm as previously reported [28].
Western blot analysis:
Each sample was loaded onto 4–20% Tris-glycine gels. After electrophoresis and transferring to nitrocellulose membranes, the membranes were blocked in Tris-buffered saline containing 0.1% Tween 20 and 0.2% I-block (Tropix, T2015) for 90 min at room temperature. Membranes were then incubated overnight at 4°C with following primary antibodies, anti-TOM40 (1:200, Santacruz, sc-11414), anti-glutathione reductase (1:500, Abcam, ab137513), anti-SOD2 (1:2000, Enzo Life Sciences, ADI-SOD-111-F). After incubation with peroxidase-conjugated secondary antibodies, visualization was enhanced by chemiluminescence (GE Healthcare, NA931- anti-mouse, or NA934- anti-rabbit, or NA935- anti-rat). Optical density was assessed using the NIH Image analysis software.
Immunocytochemistry:.
After staining with primary antibody (anti-ryanodine receptor 3 antibody, 1:100, MilliporeSigma, AB9082), fluorescent-tagged secondary antibody, nuclei were counterstained with or without 4,6-diamidino-2-phenylindole (DAPI), and coverslips were placed. Immunostaining images or time lapse images were obtained with a fluorescence microscope (Nikon ECLIPSE Ti-S) interfaced with a digital charge-coupled device camera and an image analysis system.
Statistical analysis:
Results were expressed as mean ± SD. All of experiments were performed with full blinding, allocation concealment and randomization. When only two groups were compared, unpaired t-test (two-tailed) was used. Multiple comparisons were evaluated by one-way or two-way ANOVA followed by Fisher’s LSD test or repeated two-way ANOVA in the Graph Pad prism version 6. P<0.05 was considered to be statistically significant.
Results
Astrocytic CD38 signaling through the endoplasmic reticulum ryanodine receptor increases mitochondrial calcium and ATP, and increases extracellular mitochondrial secretion.
CD38 catalyzes the synthesis of a calcium messenger, cyclic ADP ribose (cADPR) from nicotinamide adenine dinucleotide (NAD+) [42]. cADPR then activates ryanodine receptors (RyRs) in the endoplasmic reticulum (ER) and triggers Ca2+ release into cytosol [43]. Therefore, we asked whether CD38-cADPR signaling stimulated ryanodine receptors in our astrocyte mitochondrial release model. Based on previous studies [44–47], nicotinamide adenine dinucleotide (NAD: 5 mM) was added to rat cortical astrocytes to stimulate CD38 pathway. Immunocytochemistry showed that an hour after NAD treatment in astrocytes, the brain type ryanodine receptor 3 (RyR3) appeared to be co-expressed with the ER; no colocalization was detected with the Golgi (Fig. 1a). At 30 minutes following treatment with NAD or cADPR, mitochondria were localized closely to the ER (Fig. 1b) in accompanied by increasing mitochondrial calcium uptake (Fig. 1c). When the ryanodine receptor was blocked by co-treatment with Dantrolene, this effect was significantly diminished (Fig. 1c). Concomitantly, CD38 stimulation with NAD significantly increased ATP production in mitochondria and blocking RyR by Dantrolene treatment canceled the effect, consistent with a mitochondrial calcium response (Fig. 2a–b). Taken together, these data suggest that ER-mitochondrial interaction through calcium signals may amplify mitochondrial energy production after CD38 activation. Flow cytometry showed that NAD increased the secretion of astrocytic mitochondria into extracellular space (Fig. 3a–b). Blocking the ryanodine receptor with Dantrolene significantly decreased mitochondria secretion without affecting astrocyte viability (Fig. 3a–c). Collectively, mitochondrial secretion from astrocytes may be powered by increased mitochondrial ATP levels via a CD38-endoplasmic reticulum signaling pathway.
Fig. 3. CD38 - cADPR signals increase extracellular mitochondria via ryanodine receptor:
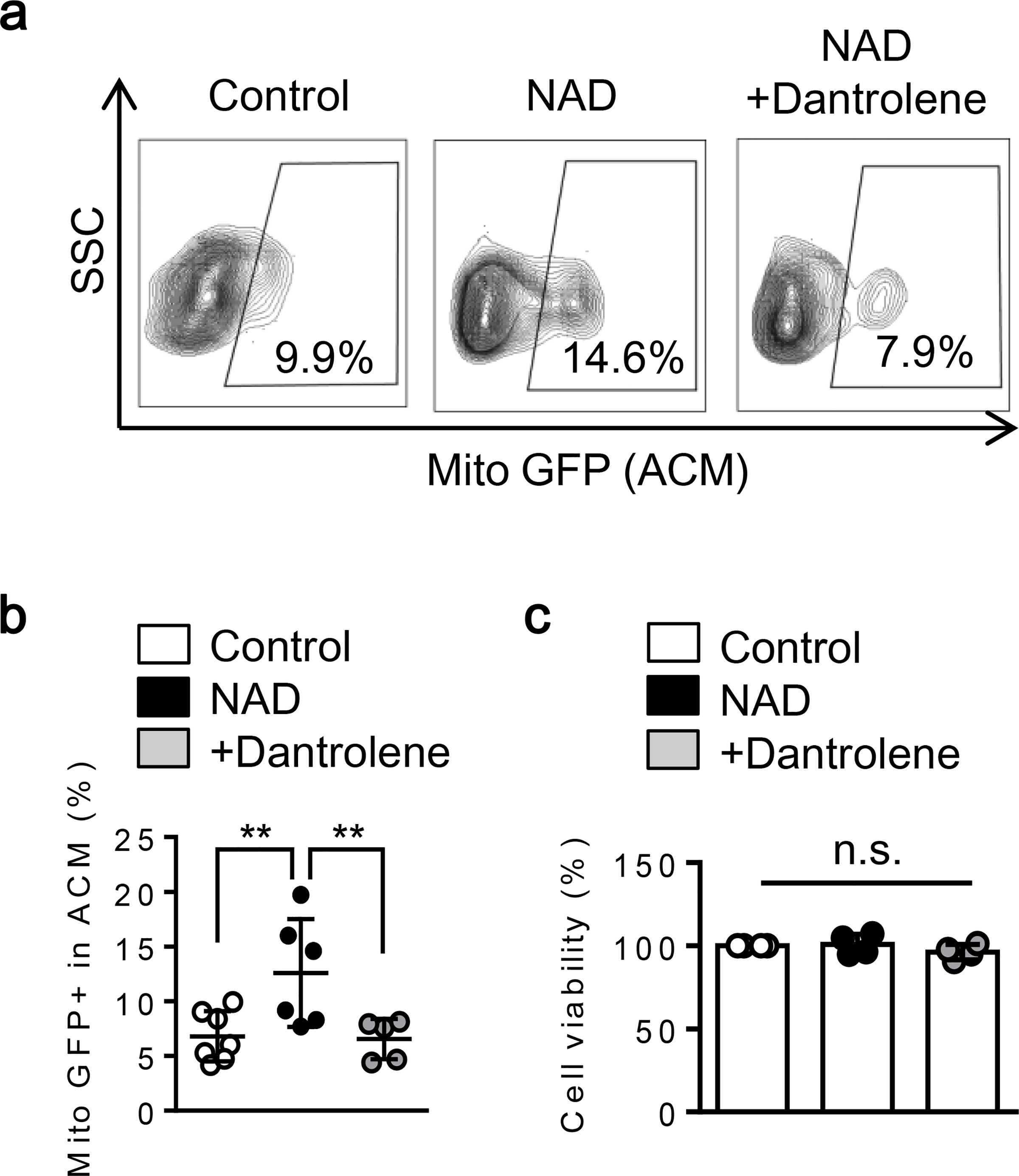
Extracellular mitochondria were identified by mitochondrial protein E1 alpha pyruvate dehydrogenase labeled by GFP. a-b. FACS analysis showed that NAD treatment for 24 hrs increased extracellular mitochondrial GFP events and co-treatment with Dantrolene diminished the effects (Control; n= 7, NAD; n= 6, NAD + Dantrolene; n=5). c. All conditions did not influence cell viability (n=4). *P<0.05, one-way ANOVA followed by Fisher’s LSD test. All values are mean +/− SD.
CD38-ryanodine receptor signaling increases antioxidant enzymes in astrocytic mitochondria.
Mitochondria are important for regulating redox homeostasis [48]. The mitochondria-associated antioxidant enzyme MnSOD converts O2 •− to hydrogen peroxide (H2O2). H2O2 is then catalyzed by glutathione to become H2O and O2. With another antioxidant enzyme glutathione reductase, oxidized glutathione is then reduced back to glutathione [49]. Therefore, we assessed the two key antioxidant enzymes MnSOD and glutathione reductase to ask whether CD38-mediated ER signals were also involved in the regulation of the mitochondria redox system. Rat cortical astrocytes were stimulated by NAD and intracellular mitochondria were isolated and analyzed over time. Western blots showed that mitochondrial glutathione reductase was increased by NAD treatment, whereas the change in MnSOD was moderate (Fig. 4a–b). As a control, the mitochondrial outer membrane protein TOM40 was not changed by NAD treatment (Fig. 4a–b). At 24 hrs, when RyR in the ER was inhibited by Dantrolene, the increase of glutathione reductase was prevented (Fig. 4c). Recently, dipicolinic acid (pyridine-2,6-dicarboxylic acid, DPA) has been identified as a novel antioxidant in mammalian cells that can also interact with glutathione reductase enzyme activity [50]. Therefore, we also assessed mitochondrial dipicolinic acid in our system. Upon NAD treatment, mitochondrial dipicolinic acid steadily increased in a time-dependent manner (Fig. 4d). Blocking the ryanodine receptor (co-treatment with NAD for 24 hrs) or inhibiting astrocytic aconitase to interfere with the TCA cycle (pre-inhibition for 24 hrs followed by NAD treatment for another 24 hrs) abolished this dipicolinic acid response (Fig. 4e–f). Taken together, these glutathione reductase and dipicolinic acid data suggest mitochondrial redox status is regulated by endoplasmic reticulum ryanodine receptor-mediated calcium signals after CD38 stimulation.
Fig. 4. CD38-stimulated astrocytes increased glutathione reductase (GR) and dipicolinic acid via RyR:
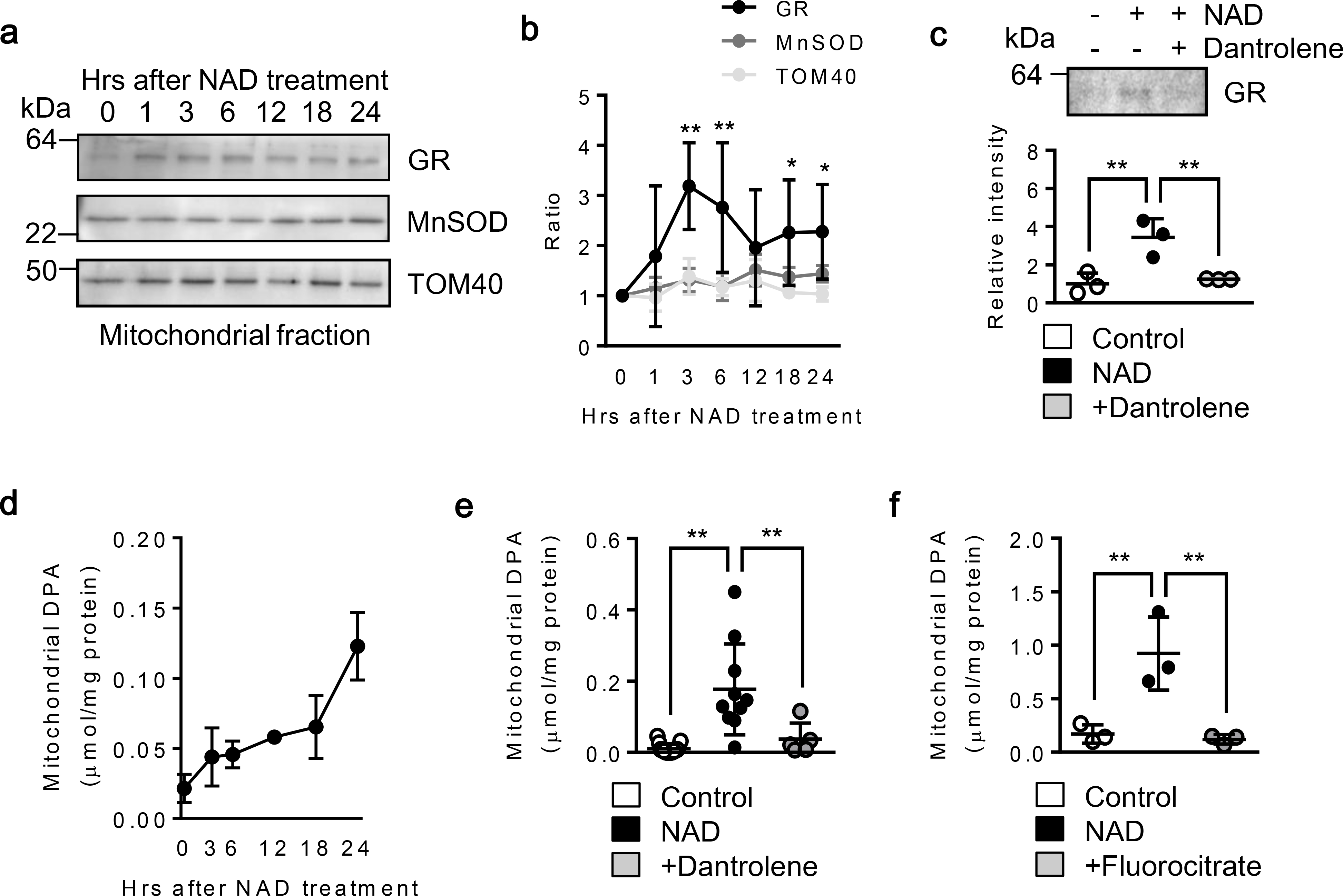
Rat cortical astrocytes were treated with NAD (5 mM). a-b. Western blot analysis revealed that antioxidant enzyme glutathione reductase (GR) was increased in astrocytic mitochondria over time after NAD treatment, whereas superoxide dismutase 2 (MnSOD) was moderately changed (n= 3). *P<0.05, **P<0.01 vs TOM40. Two-way ANOVA followed by Fisher’s LSD test. c. Blocking ryanodine receptor with Dantrolene (10 μM) significantly decreased GR expression in astrocytic mitochondria (n=3). *P<0.05, **P<0.01, one-way ANOVA followed by Fisher’s LSD test. d. Mitochondrial dipicolinic acid (DPA) was measured by reading fluorescence emitted by terbium (Tb3)-DPA complex (Ex:Em=276 nm:490 nm). DPA was increased in astrocytic mitochondria over time after NAD (5 mm) treatment (n= 4). e. NAD (5 mM) or NAD + Dantrolene (10 μM) was incubated in astrocytes for 24 hrs. NAD significantly increased DPA, but co-treatment with Dantrolene diminished the effect (Control; n= 15, NAD; n= 10, NAD + Dantrolene; n= 5). f. Astrocytes were first incubated with fluorocitrate (FC: 0.5 mM) for 24 hrs to disrupt astrocyte TCA cycle followed by NAD (5 mM) treatment for another 24 hrs. FC significantly reduced mitochondrial DPA content (n= 3). **P<0.01, one-way ANOVA followed by Fisher’s LSD test. All values are mean +/− SD.
CD38-endoplasmic reticulum-mediated signaling supports mitochondrial redox homeostasis after oxidative stress.
So far, our data show that a CD38 - ryanodine receptor pathway may increase energy production and antioxidants in mitochondria. If these antioxidants are keys for mitochondrial redox mechanism, then one should expect that interfering with this signaling pathways with Dantrolene should correlate with a decrease in mitochondrial function under oxidative stress. To address the question, we isolated “antioxidants-enriched” mitochondria and those where antioxidants were diminished with Dantrolene. Then these two groups of mitochondria were exposed to H2O2 followed by JC1 measurements. Strikingly, antioxidants-enriched mitochondria were able to maintain mitochondrial membrane potential following H2O2 exposure (Fig. 5a–b). In contrast, antioxidant-diminished mitochondria significantly lost membrane potential after oxidative stress (Fig. 5a–b). Next, a gain-of-function experiment was performed by adding dipicolinic acid to control mitochondria isolated from normal rat astrocytes (Fig. 5c). Compared to freshly isolated mitochondria, a 24 hr incubation at room temperature decreased glutathione reductase expression along with a reduction of mitochondrial membrane potential (Fig. 5d–e). Addition of dipicolinic acid to isolated mitochondria restored glutathione reductase and mitochondrial membrane potential in a concentration-dependent manner (Fig. 5d–e). Collectively, these data suggest that CD38-endoplasmic reticulum signaling boosts mitochondrial redox status and protects mitochondria against oxidative stress.
Fig. 5. Effect of CD38-ER-mediated signals on mitochondrial antioxidant redox potential in oxidative stress environment:
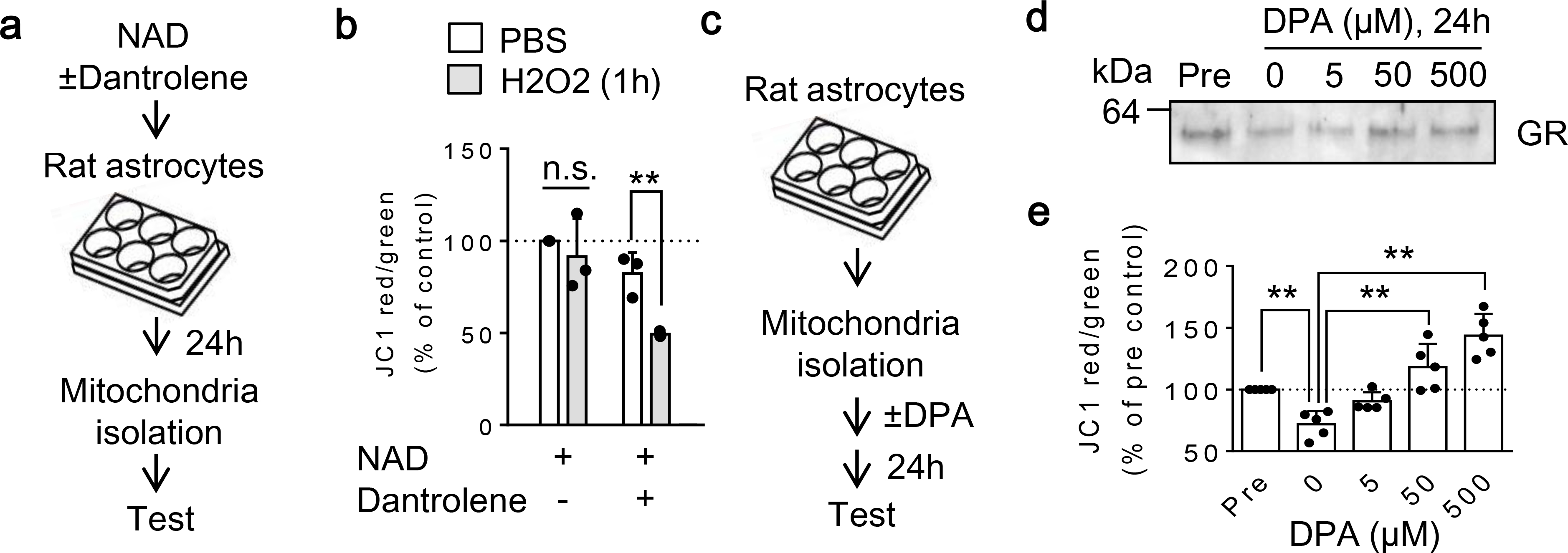
a. Rat astrocytes were treated with NAD (5 mM) with or without Dantrolene (10 μM) for 24 hours. Then mitochondria were extracted from each group for JC1 analysis after H2O2 exposure. b. Under normal condition with PBS solution, JC1 values was slightly lower in Dantrolene-treated group, but it was not statistically significance. Importantly, antioxidants-enriched mitochondria (NAD-treated) were able to maintain JC1 values following oxidative stress induced by H2O2, whereas antioxidants-declined mitochondria by Dantrolene significantly decreased mitochondrial membrane potential after H2O2 treatment (n=3). c. Mitochondria were isolated from normal rat astrocytes and applied DPA (0–500 μM) for 24 hours at room temperature. d-e. Without DPA supplement, GR expression was decreased (d) in accompanied by significant reduction of mitochondrial membrane potential (e) compared to freshly isolated mitochondria (Pre). Importantly, addition of DPA to isolated mitochondria restored GR content (d) and mitochondrial membrane potential (e) in a concentration-dependent manner (n=5). b: **P<0.01, unpaired t-test (two-tailed). e: *P<0.05, **P<0.01, one-way ANOVA followed by Fisher’s LSD test. All values are mean +/− SD.
Blockade of glutathione reductase with carmustine decreased membrane potential in extracellular mitochondria released by astrocytes.
To obtain further evidence of the involvement of mitochondrial redox signaling in supporting extracellular mitochondria, we attempted to inhibit glutathione reductase by carmustine in astrocytes. Enzyme activity assay demonstrated that NAD treatment significantly increased glutathione reductase activity in astrocytes while co-treatment with carmustine successfully inhibited the enzyme activity in astrocytes (Fig. 6a). FACS analysis revealed that co-treatment with carmustine did not change the number of total extracellular mitochondria released by CD38-stimulated astrocytes (Fig. 6b). But inhibition of glutathione reductase with carmustine significantly reduced membrane potential in extracellular mitochondria (Fig. 6c). Either treatment with NAD or NAD plus carmustine did not influence astrocytic viability (Fig. 6d).
Discussion
Taken together, our data demonstrate that (i) CD38 to RyR in the ER pathway increases mitochondrial calcium and ATP production in mitochondria, (ii) the same signaling pathway enhances mitochondrial antioxidant potential in accompanied by increases of glutathione reductase and dipicolinic acid, and (iii) these alternation in intracellular mitochondria may allow them to be ready for the extracellular environment after secretion from astrocytes (Fig. 7).
Fig. 7.
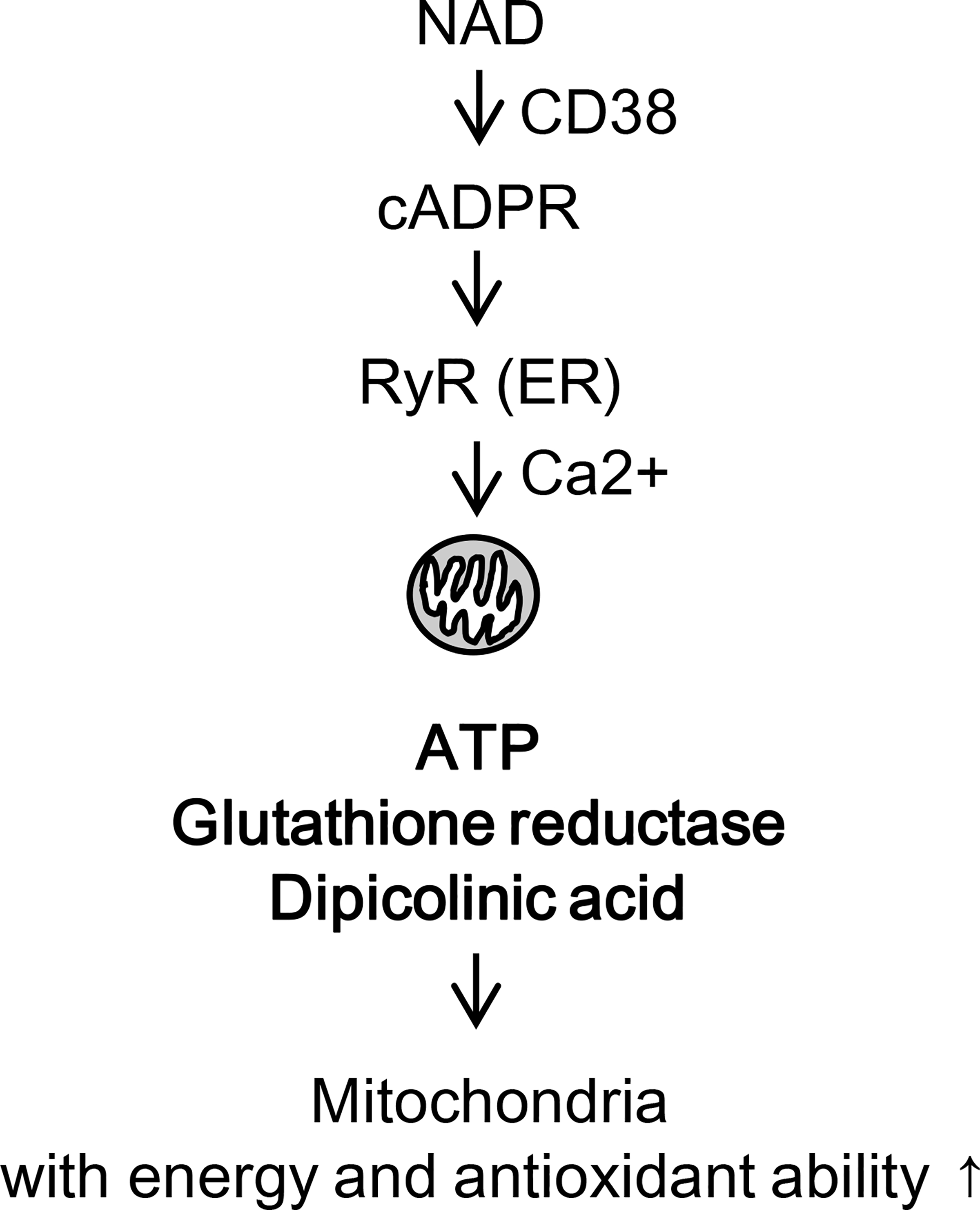
Proposed pathway for mitochondrial acquirement of ATP and antioxidants through ER-mitochondria interaction.
Mitochondria are the energy center of cells, and a key regulator of the balance between cell function and death [51–53]. Emerging findings suggest that endoplasmic reticulum-mitochondria contact sites control calcium and redox homeostasis, mitochondria metabolism and intracellular signaling pathways that are essential for normal cellular physiology [54, 55]. In contrast, dysfunctional communication between endoplasmic reticulum and mitochondria may result in neurodegenerative diseases such as Alzheimer’s disease and amyotrophic lateral sclerosis [25, 56–58]. It is now recognized that interaction between ER and mitochondria play a crucial role in energy metabolism through mitochondria-associated endoplasmic reticulum membranes (MAM) [59, 60]. The interface enables close contacts between ER and mitochondria and regulates calcium signaling, lipid transport, mitochondrial functions and cell survival [59, 60]. This ER-mitochondria interaction may also facilitate mitochondrial transfer between cells [37]. In this study, CD38-stimulated astrocytes induced intracellular mitochondria to produce ATP and antioxidants through RyR in the endoplasmic reticulum, suggesting that astrocytic CD38 may be a new target to amplify mitochondrial energetic and redox metabolism through the endoplasmic reticulum-mitochondria signaling.
Dipicolinic acid was originally found in bacterial spores and is responsible for bacterial survival in times of extreme environmental stress [61]. Moreover, the synthesis of dipicolinic acid is correlated with an increased incorporation of calcium into the endospores, and calcium starvation results in spores deficient in dipicolinic acid [62]. Mitochondria have been postulated to be originated from the integration of an endosymbiotic alpha-proteobacterium into the eukaryotic ancestor [63]. Moreover, alpha-proteobacterium may have the ability to produce spores and dipicolinic acid [64]. Therefore, it is interesting to ask whether the self-protection machinery evolved in the prokaryotic cells takes place in the powerhouse of eukaryotes during the process of the extracellular secretion. In this proof-of-concept study, we found that ER-mitochondria pathways regulated dipicolinic acid accumulation in astrocytic mitochondria, suggesting that stimulated mitochondria may potentially acquire dipicolinic acid-mediated resilience to various environmental stresses.
Nevertheless, there are a few caveats that warrant further investigation. First, mechanisms of the interaction between ER and mitochondria remain to be fully dissected. ER-mitochondria interface contains multiplex tethering complexes including MFN2 and MFN1/2, BAP31 and Fis1 and IP3R and VDAC via Grp75 etc [65]. Whether RyR in the endoplasmic reticulum may also establish functional bridge with mitochondrial protein should be further addressed. Second, physiological concentrations of NAD+ and NADH in vivo may be in the micromolar range, but our study used 5 mM of NAD to stimulate cultured astrocytes in vitro. It has been reported that the millimolar range of NAD was sufficient for signaling experiments in vitro [44, 45]. Moreover, NAD has a short half-life of about 1 hour [46]. Therefore, a relatively higher concentration of NAD compared to physiological levels was needed to ensure CD38 stimulation in vitro. Our previous study demonstrated that 5 mM of NAD successfully promoted CD38-mediated mitochondrial secretion in rat astrocytes in vitro [47]. From the collective literature, we acknowledge that NAD concentrations are typically higher for in vitro experiments compared to in vivo models. Third, we are fully aware that mitochondria may be passively released from dying cells [66] besides CD38-mediated active secretion pathway. In this context, all drug treatments did not affect astrocytic viability. Therefore, the non-specific mitochondrial extrusion or leakage from dead or dying cells should not be the concern in our current study. Nonetheless, it would be of interest to assess differences in functional properties between actively secreted mitochondria and ones released by dying cells in future studies. Fourth, the mechanism of DPA production in astrocytic mitochondria remains unknown. In the prokaryotes, dipicolinic acid synthesis pathway utilizes multiple enzymes including dapG, asd, and dapA. Then dipicolinic acid synthesis is completed with another enzymes including dpaAB, etfA, and isf [67]. Do mitochondria have these enzymes? Future studies are warranted to identify the mechanisms in mammalian cells. Fifth, it is known that dipicolinic acid is found in substances such as Natto that may lower risks of cardiovascular disease and death [68]. Whether dipicolinic acid supplement is useful to preserve functionality of extracellular mitochondria presenting in biological fluid and ameliorate inflammation and aging pathology in the CNS should be addressed in future studies. Finally, our study demonstrates that the endoplasmic reticulum - mitochondria interaction may regulate mitochondrial secretion. However, the underlying mechanisms remain to be fully understood. Vacuole formation is a known pathway for mitochondrial extrusion in reticulocytes [69], and the ER may be the main membrane source for vacuole formation [70]. How the endoplasmic reticulum-mediated vacuole formation regulates mitochondrial transfer should be explored. Assessing causality in a multifactorial phenomenon is always difficult. Our glutathione reductase blocking experiment confirms this specific step, but future studies are required to truly establish causal mechanisms in all steps of the signaling cascade.
Emerging findings suggest that mitochondria interact with the endoplasmic reticulum through the close contact sites in order to maintain metabolic homeostasis. Within the emerging paradigm of targeting mitochondria after CNS injury [71–73], our study suggests that CD38 signaling may activate RyR in the endoplasmic reticulum, boost mitochondria ATP and amplify redox homeostasis mechanisms in order to defend extracellular mitochondria under oxidative stress conditions. These proof-of-concept molecular and cellular data may provide a potential basis for understanding how secreted mitochondria maintain functionality in extracellular compartments within normal or diseased tissue. Further studies are warranted to dissect and validate the in vivo consequences of this phenomenon in animal models of stroke, brain injury and neurodegeneration.
Acknowledgments
This work was supported by grants from the National Institute of Health (NIH), National Institute of Neurological Disorders and Stroke (NINDS). Cytometric findings reported here were performed in the MGH Department of Pathology Flow and Image Cytometry Research Core.
Funding:
This work was supported by grants from NIH (K.H.) R01NS094756.
Footnotes
Conflicts of interest: There is no conflict of interest
Declarations: Not applicable
References
- [1].Mazucanti CH, Kawamoto EM, Mattson MP, Scavone C, Camandola S. Activity-dependent neuronal Klotho enhances astrocytic aerobic glycolysis. J Cereb Blood Flow Metab. 2019;39:1544–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Blochet C, Buscemi L, Clement T, Gehri S, Badaut J, Hirt L. Involvement of caveolin-1 in neurovascular unit remodeling after stroke: Effects on neovascularization and astrogliosis. J Cereb Blood Flow Metab. 2020;40:163–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tefera TW, Borges K. Neuronal glucose metabolism is impaired while astrocytic TCA cycling is unaffected at symptomatic stages in the hSOD1(G93A) mouse model of amyotrophic lateral sclerosis. J Cereb Blood Flow Metab. 2019;39:1710–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wang F, Han J, Higashimori H, Wang J, Liu J, Tong L, et al. Long-term depression induced by endogenous cannabinoids produces neuroprotection via astroglial CB1R after stroke in rodents. J Cereb Blood Flow Metab. 2019;39:1122–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chen Y, Swanson RA. Astrocytes and brain injury. J Cereb Blood Flow Metab. 2003;23:137–49. [DOI] [PubMed] [Google Scholar]
- [6].Loftspring MC, Johnson HL, Johnson AJ, Clark JF. Depletion of GR-1-Positive Cells Is Associated with Reduced Neutrophil Inflammation and Astrocyte Reactivity after Experimental Intracerebral Hemorrhage. Transl Stroke Res. 2012;3:147–54. [DOI] [PubMed] [Google Scholar]
- [7].Boussicault L, Herard AS, Calingasan N, Petit F, Malgorn C, Merienne N, et al. Impaired brain energy metabolism in the BACHD mouse model of Huntington’s disease: critical role of astrocyte-neuron interactions. J Cereb Blood Flow Metab. 2014;34:1500–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shields J, Kimbler DE, Radwan W, Yanasak N, Sukumari-Ramesh S, Dhandapani KM. Therapeutic targeting of astrocytes after traumatic brain injury. Transl Stroke Res. 2011;2:633–42. [DOI] [PubMed] [Google Scholar]
- [9].Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;29:824–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Al-Senani FM, Zhao X, Grotta JC, Shirzadi A, Strong R, Aronowski J. Proteasome Inhibitor Reduces Astrocytic iNOS Expression and Functional Deficit after Experimental Intracerebral Hemorrhage in Rats. Transl Stroke Res. 2012;3:146–53. [DOI] [PubMed] [Google Scholar]
- [12].Wu X, Luo J, Liu H, Cui W, Guo K, Zhao L, et al. Recombinant Adiponectin Peptide Ameliorates Brain Injury Following Intracerebral Hemorrhage by Suppressing Astrocyte-Derived Inflammation via the Inhibition of Drp1-Mediated Mitochondrial Fission. Transl Stroke Res. 2020. [DOI] [PubMed] [Google Scholar]
- [13].Bao Y, Qin L, Kim E, Bhosle S, Guo H, Febbraio M, et al. CD36 is involved in astrocyte activation and astroglial scar formation. J Cereb Blood Flow Metab. 2012;32:1567–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zheng W, Talley Watts L, Holstein DM, Wewer J, Lechleiter JD. P2Y1R-initiated, IP3R-dependent stimulation of astrocyte mitochondrial metabolism reduces and partially reverses ischemic neuronal damage in mouse. J Cereb Blood Flow Metab. 2013;33:600–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zong X, Dong Y, Li Y, Yang L, Yang B, Tucker L, et al. Beneficial Effects of Theta-Burst Transcranial Magnetic Stimulation on Stroke Injury via Improving Neuronal Microenvironment and Mitochondrial Integrity. Transl Stroke Res. 2020;11:450–67. [DOI] [PubMed] [Google Scholar]
- [16].Voloboueva LA, Duan M, Ouyang Y, Emery JF, Stoy C, Giffard RG. Overexpression of mitochondrial Hsp70/Hsp75 protects astrocytes against ischemic injury in vitro. J Cereb Blood Flow Metab. 2008;28:1009–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hayakawa K, Nakano T, Irie K, Higuchi S, Fujioka M, Orito K, et al. Inhibition of reactive astrocytes with fluorocitrate retards neurovascular remodeling and recovery after focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2010;30:871–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Devine MJ, Kittler JT. Mitochondria at the neuronal presynapse in health and disease. Nat Rev Neurosci. 2018;19:63–80. [DOI] [PubMed] [Google Scholar]
- [19].Bas-Orth C, Schneider J, Lewen A, McQueen J, Hasenpusch-Theil K, Theil T, et al. The mitochondrial calcium uniporter is crucial for the generation of fast cortical network rhythms. J Cereb Blood Flow Metab. 2019:271678X19887777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rutkai I, Merdzo I, Wunnava SV, Curtin GT, Katakam PV, Busija DW. Cerebrovascular function and mitochondrial bioenergetics after ischemia-reperfusion in male rats. J Cereb Blood Flow Metab. 2019;39:1056–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang Z, Zhou F, Dou Y, Tian X, Liu C, Li H, et al. Melatonin Alleviates Intracerebral Hemorrhage-Induced Secondary Brain Injury in Rats via Suppressing Apoptosis, Inflammation, Oxidative Stress, DNA Damage, and Mitochondria Injury. Transl Stroke Res. 2018;9:74–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gollihue JL, Patel SP, Eldahan KC, Cox DH, Donahue RR, Taylor BK, et al. Effects of Mitochondrial Transplantation on Bioenergetics, Cellular Incorporation, and Functional Recovery after Spinal Cord Injury. J Neurotrauma. 2018;35:1800–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wang W, Zhao F, Ma X, Perry G, Zhu X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol Neurodegener. 2020;15:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Requejo-Aguilar R, Bolanos JP. Mitochondrial control of cell bioenergetics in Parkinson’s disease. Free Radic Biol Med. 2016;100:123–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tadic V, Prell T, Lautenschlaeger J, Grosskreutz J. The ER mitochondria calcium cycle and ER stress response as therapeutic targets in amyotrophic lateral sclerosis. Front Cell Neurosci. 2014;8:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hayakawa K, Bruzzese M, Chou SH, Ning M, Ji X, Lo EH. Extracellular Mitochondria for Therapy and Diagnosis in Acute Central Nervous System Injury. JAMA Neurol. 2018;75:119–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Xing C, Lo EH. Help-me signaling: Non-cell autonomous mechanisms of neuroprotection and neurorecovery. Prog Neurobiol. 2017;152:181–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, et al. Transfer of mitochondria from astrocytes to neurons after stroke. Nature. 2016;535:551–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Borlongan CV, Nguyen H, Lippert T, Russo E, Tuazon J, Xu K, et al. May the force be with you: Transfer of healthy mitochondria from stem cells to stroke cells. J Cereb Blood Flow Metab. 2019;39:367–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chen W, Huang J, Hu Y, Khoshnam SE, Sarkaki A. Mitochondrial Transfer as a Therapeutic Strategy Against Ischemic Stroke. Transl Stroke Res. 2020. [DOI] [PubMed] [Google Scholar]
- [31].Nakamura Y, Park JH, Hayakawa K. Therapeutic use of extracellular mitochondria in CNS injury and disease. Exp Neurol. 2019;324:113114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chou SH, Lan J, Esposito E, Ning M, Balaj L, Ji X, et al. Extracellular Mitochondria in Cerebrospinal Fluid and Neurological Recovery After Subarachnoid Hemorrhage. Stroke. 2017;48:2231–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rocca CJ, Goodman SM, Dulin JN, Haquang JH, Gertsman I, Blondelle J, et al. Transplantation of wild-type mouse hematopoietic stem and progenitor cells ameliorates deficits in a mouse model of Friedreich’s ataxia. Sci Transl Med. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Davis CH, Kim KY, Bushong EA, Mills EA, Boassa D, Shih T, et al. Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci U S A. 2014;111:9633–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Eun Jung J, Sun G, Bautista Garrido J, Obertas L, Mobley AS, Ting SM, et al. The mitochondria-derived peptide humanin improves recovery from intracerebral hemorrhage: implication of mitochondria transfer and microglia phenotype change. J Neurosci. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Nakamura Y, Lo EH, Hayakawa K. Placental mitochondria therapy for cerebral ischemia-reperfusion injury in mice. Stroke. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gao J, Qin A, Liu D, Ruan R, Wang Q, Yuan J, et al. Endoplasmic reticulum mediates mitochondrial transfer within the osteocyte dendritic network. Sci Adv. 2019;5:eaaw7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zocchi E, Daga A, Usai C, Franco L, Guida L, Bruzzone S, et al. Expression of CD38 increases intracellular calcium concentration and reduces doubling time in HeLa and 3T3 cells. J Biol Chem. 1998;273:8017–24. [DOI] [PubMed] [Google Scholar]
- [39].Pehar M, Harlan BA, Killoy KM, Vargas MR. Nicotinamide Adenine Dinucleotide Metabolism and Neurodegeneration. Antioxid Redox Signal. 2018;28:1652–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hayakawa K, Arai K, Lo EH. Role of ERK map kinase and CRM1 in IL-1beta-stimulated release of HMGB1 from cortical astrocytes. Glia. 2010;58:1007–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hayakawa K, Pham LD, Katusic ZS, Arai K, Lo EH. Astrocytic high-mobility group box 1 promotes endothelial progenitor cell-mediated neurovascular remodeling during stroke recovery. Proc Natl Acad Sci U S A. 2012;109:7505–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Aksoy P, White TA, Thompson M, Chini EN. Regulation of intracellular levels of NAD: a novel role for CD38. Biochem Biophys Res Commun. 2006;345:1386–92. [DOI] [PubMed] [Google Scholar]
- [43].Ogunbayo OA, Zhu Y, Rossi D, Sorrentino V, Ma J, Zhu MX, et al. Cyclic adenosine diphosphate ribose activates ryanodine receptors, whereas NAADP activates two-pore domain channels. J Biol Chem. 2011;286:9136–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Harkcom WT, Ghosh AK, Sung MS, Matov A, Brown KD, Giannakakou P, et al. NAD+ and SIRT3 control microtubule dynamics and reduce susceptibility to antimicrotubule agents. Proc Natl Acad Sci U S A. 2014;111:E2443–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sasaki Y, Araki T, Milbrandt J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J Neurosci. 2006;26:8484–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Rechsteiner M, Hillyard D, Olivera BM. Turnover at nicotinamide adenine dinucleotide in cultures of human cells. J Cell Physiol. 1976;88:207–17. [DOI] [PubMed] [Google Scholar]
- [47].Park J, Nakamura Y, Li W, Hamanaka G, Arai K, Lo E, et al. Effects of O-GlcNAcylation on functional mitochondrial transfer from astrocytes. Journal of Cerebra Blood Flow and Metabolism. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Willems PH, Rossignol R, Dieteren CE, Murphy MP, Koopman WJ. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab. 2015;22:207–18. [DOI] [PubMed] [Google Scholar]
- [49].Ribas V, Garcia-Ruiz C, Fernandez-Checa JC. Glutathione and mitochondria. Front Pharmacol. 2014;5:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Murakami K, Yoshino M. Dipocolinic acid as an Antioxidant: Protection of Glutathione Reductase from the Inactivation by Copper. Biomedical Research. 1999;20:321–6. [Google Scholar]
- [51].Baxter P, Chen Y, Xu Y, Swanson RA. Mitochondrial dysfunction induced by nuclear poly(ADP-ribose) polymerase-1: a treatable cause of cell death in stroke. Transl Stroke Res. 2014;5:136–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Owens K, Park JH, Schuh R, Kristian T. Mitochondrial dysfunction and NAD(+) metabolism alterations in the pathophysiology of acute brain injury. Transl Stroke Res. 2013;4:618–34. [DOI] [PubMed] [Google Scholar]
- [53].Liu Y, Hu XB, Zhang LZ, Wang Z, Fu R. Knockdown of Arginyl-tRNA Synthetase Attenuates Ischemia-Induced Cerebral Cortex Injury in Rats After Middle Cerebral Artery Occlusion. Transl Stroke Res. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Rieusset J The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: an update. Cell Death Dis. 2018;9:388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ouyang YB, Giffard RG. ER-Mitochondria Crosstalk during Cerebral Ischemia: Molecular Chaperones and ER-Mitochondrial Calcium Transfer. Int J Cell Biol. 2012;2012:493934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Area-Gomez E, de Groof AJ, Boldogh I, Bird TD, Gibson GE, Koehler CM, et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am J Pathol. 2009;175:1810–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Lee KS, Huh S, Lee S, Wu Z, Kim AK, Kang HY, et al. Altered ER-mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc Natl Acad Sci U S A. 2018;115:E8844–E53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Moltedo O, Remondelli P, Amodio G. The Mitochondria-Endoplasmic Reticulum Contacts and Their Critical Role in Aging and Age-Associated Diseases. Front Cell Dev Biol. 2019;7:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].van Vliet AR, Verfaillie T, Agostinis P. New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta. 2014;1843:2253–62. [DOI] [PubMed] [Google Scholar]
- [60].Giorgi C, Missiroli S, Patergnani S, Duszynski J, Wieckowski MR, Pinton P. Mitochondria-associated membranes: composition, molecular mechanisms, and physiopathological implications. Antioxid Redox Signal. 2015;22:995–1019. [DOI] [PubMed] [Google Scholar]
- [61].Forman M, Aronson A. Regulation of dipicolinic acid biosynthesis in sporulating Bacillus cereus. Characterization of enzymic changes and analysis of mutants. Biochem J. 1972;126:503–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Black SH, Hashimoto T, Gerhardt P. Calcium reversal of the heat susceptibility and dipicolinate deficiency of spores formed “endotrophically” in water. Can J Microbiol. 1960;6:213–24. [DOI] [PubMed] [Google Scholar]
- [63].Roger AJ, Munoz-Gomez SA, Kamikawa R. The Origin and Diversification of Mitochondria. Curr Biol. 2017;27:R1177–R92. [DOI] [PubMed] [Google Scholar]
- [64].Kirsebom LA, Dasgupta S, Pettersson BM. Pleiomorphism in Mycobacterium. Advances in Applied Microbiology. 2012;80:81–112. [DOI] [PubMed] [Google Scholar]
- [65].Liu Y, Zhu X. Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases. Transl Neurodegener. 2017;6:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Maeda A, Fadeel B. Mitochondria released by cells undergoing TNF-alpha-induced necroptosis act as danger signals. Cell Death Dis. 2014;5:e1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Gordon B, Duellman P, Salvucci A, De Lorme M. Detecting dipicolinic acid production and biosynthesis pathways in Bacilli and Clostridia. biorxiv. 2019;[preprint]. [Google Scholar]
- [68].Katagiri R, Sawada N, Goto A, Yamaji T, Iwasaki M, Noda M, et al. Association of soy and fermented soy product intake with total and cause specific mortality: prospective cohort study. BMJ. 2020;368:m34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Simpson CF, Kling JM. The mechanism of mitochondrial extrusion from phenylhydrazine-induced reticulocytes in the circulating blood. J Cell Biol. 1968;36:103–9. [PubMed] [Google Scholar]
- [70].Viotti C ER and vacuoles: never been closer. Front Plant Sci. 2014;5:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Simon R, Meller R, Yang T, Pearson A, Wilson G. Enhancing Base Excision Repair of Mitochondrial DNA to Reduce Ischemic Injury Following Reperfusion. Transl Stroke Res. 2019;10:664–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Vekaria HJ, Talley Watts L, Lin AL, Sullivan PG. Targeting mitochondrial dysfunction in CNS injury using Methylene Blue; still a magic bullet? Neurochem Int. 2017;109:117–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].She DT, Jo DG, Arumugam TV. Emerging Roles of Sirtuins in Ischemic Stroke. Transl Stroke Res. 2017. [DOI] [PubMed] [Google Scholar]