

Summary
Regenerative medicine approaches utilizing stem cells offer a promising strategy to address tendinopathy, a class of common tendon disorders associated with pain and impaired function. Tendon progenitor cells (TPCs) are important in healing and maintaining tendon tissues. Here we provide a comprehensive single cell transcriptomic profiling of TPCs from three normal and three clinically classified tendinopathy samples in response to mechanical stimuli. Analysis reveals seven distinct TPC subpopulations including subsets that are responsive to the mechanical stress, highly clonogenic, and specialized in cytokine or growth factor expression. The single cell transcriptomic profiling of TPCs and their subsets serves as a foundation for further investigation into the pathology and molecular hallmarks of tendinopathy in mechanical stimulation conditions.
Keywords: Single Cell RNAseq, Tendon, Tendon progenitor cell, Mechanical stress, Tendinopathy, Single cell analysis, Stem cells, Tendon disorders, Tendinopathy markers
Graphical abstract
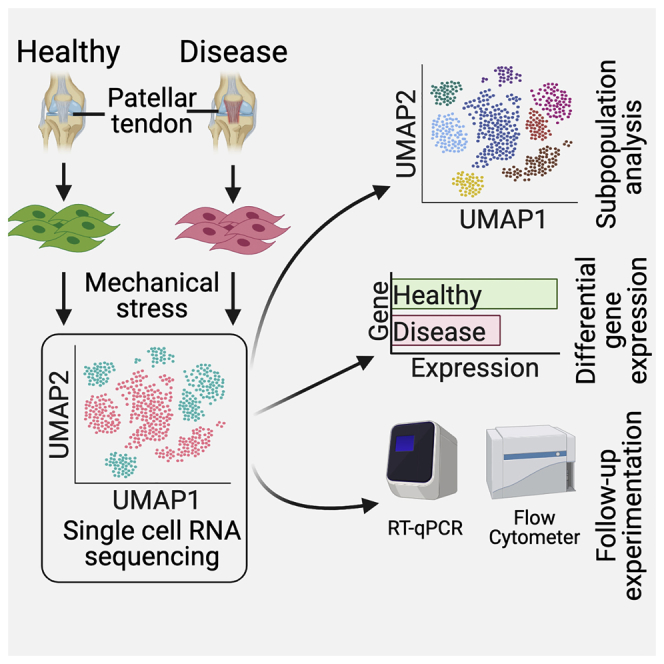
Highlights
Single cell profiling of tendon progenitor cells under mechanical stimulus
Seven distinct tendon progenitor subpopulations are identified
A subpopulation shows different mechanical response at the single cell level
Mitochondria factors TUFM and TFAM downregulated in diseased state under stimulus
Still et al. use single-cell RNA sequencing to profile subpopulations of tendon progenitor cells (TPCs) from normal and diseased patient tendon tissues under mechanical stretching. They identify seven TPC subpopulations and observe different mechanical responses of TPCs at the single-cell level.
Introduction
Tendon injuries comprise 30% of musculoskeletal injuries.1 Tendinopathy, a common disorder of the tendon that results in pain and impaired function, has two primary forms: acute and chronic.2 Although acute tendinopathy is often caused by extrinsic conditions (i.e., trauma) or inflammation, chronic tendinopathy has a mixed etiology including both extrinsic, intrinsic, and genetic factors.2 Current options to treat chronic tendinopathy are very limited.1 The use of tendon-derived adult stem cells and/or tendon progenitor cells (TPCs) provides a potential pathway for tendon regeneration. However, this is limited by our poor understanding of the disease pathogenesis and TPCs.3
Discovered originally from the mouse patellar tendon and the human hamstring tendon, TPCs are described as a highly heterogeneous cell population by their morphology and doubling time.4 Subpopulations of TPCs are suggested to come from different sources within the tendon, including fascicle, epitenon, or perivascular compartments.3, 4, 5, 6 In addition, it is likely that different TPC subpopulations display different behaviors in response to stress and in disease states, which have a crucial role in the clinically observed TPC functions and in future diagnostics and treatment.
Recent advances in single-cell gene expression analysis have improved our understanding of individual subpopulations of tenocytes and TPCs. However, researchers did not introduce physical stimulation to the cells they profiled.5,6 Tendon cells require physical stimulation to mimic their natural state, which is an important consideration in the context of studying TPCs.7 Simply extracting and enriching TPCs from patients may not provide a complete picture of their functional differences in normal and disease contexts.4,7, 8, 9 Providing physical stimulation could potentially better identify functional differences related to TPC subpopulations and their transcriptomic profiles in normal and tendinopathic contexts.
To improve our understanding of TPC populations for tendinopathy treatment, we combined single-cell RNA sequencing (scRNA-seq) with physical stimulation. We hypothesized that we could distinguish unique subpopulations among the patient-derived TPCs and that these would vary between the normal and diseased cohorts. We further hypothesized that using mechanical stimulation in vitro, we could better understand tendon pathology.
Results
Patient-derived normal or tendinopathy tendon samples show distinct characteristics
All six subjects were males with a mean age of 26.9 ± 5.4 years (Table S1). There was no significant difference in age between the normal (27.0 ± 7.9 years) and diseased cohorts (26.7 ± 5.0) (p = 0.944) (Table S1). The mean duration of symptoms was 1.7 ± 0.6 years (Table S1). The diseased tendons (10.8 ± 1.8 mm) were significantly thicker compared with the healthy tendons (5.7 ± 0.2 mm) (p = 0.002) (Figure S1A). Two of the diseased subjects (D1 and D3) had (Popkin-Golman [PG]) grade 3 tendinopathy and one subject (D2) had grade 4 (Table S1).10 Cells extracted from patients were cultured to passage 2 and subjected to flow cytometry to measure expression of common TPC markers. TPC markers of both healthy and diseased cohorts showed expression of the expected antigen profile CD31− CD34− CD45− CD73+ CD90+ CD105+ (Figure S1B; Table S2), as reported previously.4
Single-cell transcriptomic profiling of the non-stimulated TPC samples reveals heterogeneous subpopulations
We first performed scRNA-seq experiments on human patellar-tendon-derived TPCs from one normal (N1) and one tendinopathy (D1) sample (Figure S2A; Table S1).11 We profiled the transcriptomes of 5,870 cells for each sample with an average of 2,056 genes per cell for a combined dataset of 11,740 cells (Figure S2B). Cells within the datasets expressed some genes expected of TPCs such as COL1A1 and THY1 as well as genes less commonly associated with TPCs such as CXCL1 and IL8 (Figures S2C and S2D). The normal and tendinopathy TPCs showed high similarity, with a Pearson correlation coefficient of 0.965 (Figure S2B). This is consistent with our earlier study that suggested that TPCs mainly exhibited functional differences upon physical stimulation.7 Within the dataset we additionally noted subpopulations with potential functional importance to modulate inflammation, specifically a subset of TPCs expressing cytokines IL8 and CXCL1 (Figure S2E). The high similarity between healthy and tendinopathy samples prompted us to investigate their difference under mechanical stimuli.
Single-cell transcriptomic profiling of mechanically stimulated TPCs identifies major subpopulation differences between healthy and tendinopathic tendons
We next studied TPCs derived from three normal (N1–N3) and three tendinopathy (D1–D3) samples under mechanical stress (Figure 1A). Tendons are constantly stretched in vivo, which has been shown to be critical for TPC development and function.7, 8, 9 We recently found that stretching TPCs can significantly accentuate functional differences between healthy and tendinopathy samples.7 A total of 11,358 cells from all six subjects were profiled, with an average of 3,811 genes analyzed per cell (Figure 1B). Normal and tendinopathy cohorts each contained 5,679 cells (Figure 1C). We found greater intra-correlation within the normal and tendinopathy cohorts compared with inter-correlation between the cohorts (p < 0.05; Figures 1D and S3). This likely suggests a category of pathological transcriptomic signatures of tendinopathy TPCs under the mechanical-stress condition that is greater than human genetic variations. Furthermore, we found one tendinopathy subject (D2) who correlated less with the other two tendinopathy samples (Figure 1D). That lower correlation is consistent with a difference in tendinopathy-grade classification of the samples (Table S1).
Figure 1.

Single-cell transcriptomic profiling of TPCs from normal and diseased samples under mechanical stretching reveals heterogeneous subpopulations
(A) Overview of stretching and scRNA-seq experiment.
(B) UMAP plot of 11,298 cells from healthy and disease patients. Chart to the right displays association of TPC subsets with either the normal or the diseased cohort. Data represent three biological replicates per cohort, and both normal and diseased cohorts are shown.
(C) UMAP plots of cells from the normal cohort and diseased cohort. (Bottom) Cohort association for each cluster. Data represent 3 biological replicates per cohort, and both normal and diseased cohorts are shown separately.
(D) Pearson correlation matrix examining patient to patient similarity. Scale bar indicates pearson correlation.
Patient variability in TPC subtype presence
We examined variability in the presence of TPC subtypes between individual patient samples (Figure S4). All clusters are represented by each patient, except for sample N2, which did not contain SLC40A1+ TPCs (Figure S4). We were also able to observe different organization (position in the Uniform Manifold Approximation and Projection [UMAP] plot) of TPCs within a cell type depending on patient. This may suggest some biological variance in the TPC subset. The TPC population levels for each patient were quantified in Table S3.
Identification and characterization of subpopulations with high mitochondrially expressed genes in healthy, but not tendinopathy, TPCs
To elucidate differences between the normal and the tendinopathy cohorts, we ran differential gene expression (DGE) analysis using MAST.12 Many mitochondrially expressed genes (MEGs) were found to be enriched in the normal cohort (Figure 2A). Genes related to cell motility and insulin-like growth factor receptor signaling were also expressed more among healthy TPCs (adjusted p = 1.25e−2 and 1.28e−2, respectively) (Figure 2A). We observed a subset of cells with high percentage (15%–50%) of MEG expression, which was primarily seen within the normal cohort (Figure 2B). Mapping the distribution of cells as a function of MEG showed a bimodal distribution of cells in both cohorts (Figure 2C). This pattern was consistent in individual members of each cohort (Figure 2D). Recent work showed higher MEG percentage detected by the 10× Chromium V3 method compared with the Drop-seq approach.13 Taking this inherent technology difference into account, we compared the MEG percentage distribution between non-mechanically stimulated TPCs and stimulated TPCs for two samples (Figure 2E; see Method details). Both non-stimulated samples displayed a very low MEG percentage (<15%) with a unimodal distribution (Figure 2E). However, upon mechanical stimulus, the unimodal distribution switched to a bimodal distribution in both samples (Figure 2E).
Figure 2.

Mechanically responsive TPCs (mrTPCs) are a subset of TPCs with high numbers of mitochondrially expressed genes (MEGs) that are enriched among normal TPCs
(A) GO term analysis of more highly expressed genes in the normal donor-derived TPC cohort. Darker red colors correspond to higher p values (shown). Genes were automatically chosen by the Enrichr program.
(B) UMAP plots showing MEG levels within the total TPC populations for normal and disease cohorts. Data represent three biological replicates per cohort, and both normal and diseased cohorts are shown separately.
(C) Histogram of MEG percentages for normal and disease cohorts. Data represent three biological replicates per cohort, and both normal and diseased cohorts are shown separately.
(D) Violin plot showing individual sample MEG percentage. Black dashed lines represent sample averages. ∗p < 0.05. Data represent individual biological replicates per sample.
(E) Histogram of MEG percentages for TPCs in either resting or post-stretching condition. Grey bars of the histogram are resting TPC MEG percentages, and yellow bars are after stretching TPCs. Data represent one biological replicate and one technical replicate of the single-cell profiling.
(F) Ridge plot showing MEG percentage for TPCs within each TPC subset. Data represent all six biological replicates from both cohorts.
(G) Individual sample association with mrTPC subsets. Each dot represents an individual biological replicate, and three biological replicates from each cohort are shown. Error bars represent standard deviation.
(H) Percentage of cells in healthy and disease cohorts that are apoptotic, dead, or both in the resting and stretched conditions. Blue bars represent the normal cohort, and red bars represent diseased cohort, with three biological replicates per cohort, and one technical replicate per cohort member. ∗p < 0.05. Error bars represent min/max values.
(I) Mitotracker red CMXRos staining. Dotted line represents 0. Counts are log10 of the ratio of the median stretched compared with its resting counterpart. Three biological replicates per cohort, and one technical replicate per biological sample.
(J) qRT-PCR results for TFAM, TUFM, and BAX. Three biological replicates per cohort, and three technical replicates per cohort. ∗p < 0.05.
A distinct cluster of TPCs showed higher MEG levels (15%–50%) in the mechanically stimulated dataset (Figure 2F), and we named them mechanically responsive TPCs (mrTPCs). mrTPC abundance was enriched among two of the healthy TPC samples compared with that of the tendinopathy samples (Figure 2G).
MrTPC populations represented stressed TPCs in response to the mechanical stretching
The use of mechanical stretching to model strain on the TPCs is an important aspect of our study. Mechanical stretching is also associated with inducing strain on cells, potentially harming or stressing the cells.13,14 Furthermore, mechanical strain has been noted to affect mitochondria behavior in cells.15 Thus, we wanted to further examine why cells in our dataset showed exceptionally high MEG levels, especially because, in scRNA-seq, this can be a sign of dead or dying cells. We hypothesized three possible reasons for the high MEG levels: the cells are dying or undergoing apoptosis because of the increased mechanical stress, the cells may have higher mitochondria and energetic demands, or the cells were undergoing increased stress but not necessarily dying because of the stress.
We initially tested cell death using a combination of propidium iodide (PI) staining and annexin V surface staining. With similar survival in resting condition as the baseline for healthy (94.63% ± 1.51%) and diseased samples (95.37% ± 0.90%), we saw no significant difference in cell death between normal and diseased cohorts under stretch with survival rates at 95.33% ± 0.80% and 94.90% ± 2.25%, respectively (Figures 2H and S5A). These results further suggest no significant changes in cell-survival transitioning from resting to stretching in both cohorts. The UVC-exposed normal TPCs were included in Figure S5B to validate the cell death assay with a significant decrease of 28.20% in survival at the highest UV-dosage tested at 24 h. In addition, UVC exposure was shown to cause mitochondria-dependent caspase-9 activation and apoptotic cell death in human cells.16
We next wanted to understand whether normal-cohort TPCs are more energetically active, which could explain the heightened MEG levels. Cells were stained with MitoTracker red CMXRos to measure mitochondria membrane potential and, in effect, mitochondria energy production (Figure 2I). We note that this marks mitochondria health and, in effect, cellular health. It was interesting to see the increase in MitoTracker red staining after stretching, compared with resting, suggesting a general increase in TPC energy production in response to the mechanical stimulus. When examining changes to MitoTracker red staining, comparing stretched samples to their resting counterparts, we saw a small decrease of the diseased cohorts compared with the normal cohort, despite the difference not being statistically significant.
We also measured expression of two mitochondria biogenesis factors, TFAM and TUFM, using quantitative reverse transcriptase PCR (qRT-PCR) (Figure 2J). TFAM and TUFM both showed significantly higher expression levels in the normal cohort compared with the diseased counterpart before stretching (p = 0.0190 and 0.0061, respectively). This suggests normal TPCs might have higher resting levels of mitochondria biogenesis. There was no significant difference between stretched normal and disease cohort TFAM and TUFM expression. We did see significant increases of TFAM when comparing the diseased stretched cells versus their resting counterpart (p = 0.0453). Similarly, TUFM was shown to be significantly upregulated in the normal stretched cells compared with their resting counterparts (p = 0.0459).
Finally, we also looked at BAX expression level by qRT-PCR as an assessment of cellular stress (Figure 2J). BAX is active in inducing cell apoptosis, and its expression is linked to the cell stress response.17,18 We noted that both normal and diseased cohorts showed a significant increase in BAX levels upon mechanical stretching of the cells (p = 0.0483 and 0.0297, respectively). Furthermore, we found that, even among non-stretched cells, there was a significant difference in BAX levels between the normal and diseased cohorts (p = 0.0201).
Cell cycle scoring reveals highly clonogenic TPC subsets
Previous literature noted a highly clonogenic subset of TPCs with tenogenic potential marked by high NESTIN (NES) expression.5 We sought to identify whether this clonogenic population was also present in our study and understand differences between healthy and tendinopathy samples. We characterized the cell cycle states of cells for each TPC subset (see Method details).19,20 We calculated a fraction for each TPC subset of cells associated with either the G2M or S phase of the cell cycle. This became the fraction of cells cycling for the TPC subset (Figure 3A). As expected, the subset of TPCs with the highest levels of NES also had the most cells cycling (100%) (Figures 3A and 3B). Additionally, we observed another subset of TPCs with a high level of cycling cells: the TPC state C cells (69%) (Figure 3A). NES+ clonogenic TPCs and TPC state C cells showed varying population sizes among subjects with the NES+ clonogenic TPCs enriched among the diseased cohort, whereas TPC state C cells were enriched among healthy populations (Figures 1B and 1C). Considering that TPC state C cells clustered close to the NES+ clonogenic TPCs (Figure 1B), TPC state C cells showed higher MEG levels (Figure 2F), and TPC state C cells showed a high number of cells cycling (Figure 3A), we suspect these cells may be stressed versions of NES+ clonogenic TPCs.
Figure 3.

scRNA-seq reveals clonogenic TPC subpopulations
(A) Fraction of cells identified as cycling (G2M or S phase) for each TPC subset. The right panel shows a UMAP plot with color-matched TPC subsets. Data represent all six biological replicates from both cohorts.
(B) Dot plot showing distinct expression markers for the NES+ clonogenic TPCs, SLC40A1+ TPCs, and piTPCs. Data represent all six biological replicates from both cohorts.
(C) Enriched Bioplanet 2019 pathways for NES+ clonogenic TPCs, SLC40A1+ TPCs, and piTPCs. Data represent all six biological replicates from both cohorts.
Identification of pro-inflammatory TPCs and SLC40A1+ TPC subpopulations related to tendinopathy
We also identified a pro-inflammatory TPCs (piTPCs) population and a SLC40A1+ TPC population (Figure 1B). The piTPCs expressed several cytokines, including IL8, IL6, and CXCL1 (Figure 3B). SLC40A1+ TPCs expressed high levels of TGFB2 (2.44-fold higher, p < 0.05) and TGFB3 (1.81-fold higher, p < 0.05), important factors for tendon development and general wound repair (Figure 3B).21
We examined BioPlanet 2019 pathway enrichment for these two TPC subsets. For the SLC40A1+ TPCs, we observed transforming growth factor (TGF-β) regulation of the extracellular matrix (ECM) as the top pathway (Figure 3C). This is consistent with the heightened levels of TGFB2 and TGFB3 observed for this subpopulation (Figure 3B). We also saw TGFB1 enriched in the SLC40A1+ TPCs (see Data S1). SLC40A1+ TPCs also had exceptionally high expression of MAFB (8.31-fold increase over other TPC populations), a transcription factor traditionally noted to have a role in lineage-specific hematopoiesis (Figures S6A and S6B; see Data S1).22 The top enriched pathway of piTPCs was interleukin-1 regulation of extracellular matrix and, in general, showed higher expression of pro-inflammatory genes compared with other TPC populations (Figures 3B and 3C).
Identification of TPC-, tenogenesis-, and tendinopathy-related genes among TPC subsets
We assessed the expression of a broad array of genes related to tenogenesis and tendinopathy among the TPC subsets (Figure 4). From our analysis, although many of the genes were heterogeneously expressed by TPC subsets, with some showing low or almost no expression, other TPC subsets have most of their cells expressing the gene (Figure 4). Additionally, some of the genes showed differential expression among normal and disease cohorts for a specific TPC subset (Figure 4).
Figure 4.

Expression of tendon-related genes among TPC subpopulations
A dot plot of genes known to be expressed in tendon development or expressed in the mature tendon. Size of each dot correlates with the percentage of cells for the TPC subset that express the gene. Color shows the magnitude of expression. The dot plot shows gene expression for all six biological samples from both cohorts. Normal cohort is in blue, and diseases cohort is in red.
Discussion
Despite the overall importance of TPCs to the tendon tissue, an understanding of their heterogeneity has, thus far, been limited. 3 We were able to confirm our hypothesis that, using scRNA-seq, we could uncover multiple TPC subpopulations including mrTPCs, piTPCs, and SLC40A1+ TPCs. We were also able to map known tenogenesis and tendon markers sourced from the literature among our TPC subpopulations.5
DGE between normal and disease cohorts showed that healthy TPCs were enriched for MEGs. This change in MEG levels led us to speculate that there may be alterations in mitochondrial function or numbers in the TPCs, high amounts of dead or apoptotic cells, or cells under stress because of the mechanical stretching. Follow-up on the scRNA-seq data, qPCR, and MitoTracker staining data illustrated changes in mitochondria behavior after stretching and key differences between the normal and disease cohorts in expression of mitochondria biogenesis factors TFAM and TUFM. There have been limited links between TPCs and mitochondria regulation. Recently, it has been shown that, under hypoxic conditions, similar to an environment experienced by tenocytes after rotator cuff injury (RCI), tenocytes will increase mitochondria biogenesis.23 However, in this context, individual mitochondria were less healthy and effective at energy production.23 Although we are not assessing TPC behavior under hypoxic conditions, we are introducing mechanical strain on these cells, and this does seem to lead to higher MEG levels. Furthermore, fluoroquinolones, a class of antibiotics that are suspected to have negative effects on mitochondria health, have been linked to tendinopathy and increased chance of tendon rupture, particularly in the Achilles tendon.24, 25, 26, 27, 28 Despite this evidence linking together fluoroquinolones and mitochondria health and the correlation between fluoroquinolones and tendinopathy, the mechanism by which fluoroquinolones may induce tendinopathy is still debated.24 Our results may help better clarify why fluoroquinolones are strongly correlated with tendon rupture and highlights the lack of TPC control over mitochondria function as a potential cause of the pathology in tendinopathy.
Another major question posed by the high levels of MEGs was whether the cells in our study were apoptotic or dead after stretching. We did not find many cells in either cohort that were apoptotic or dead. BAX, a pro-apoptotic factor, is linked to stress.17,18 We found that stretching induced higher levels of BAX in both cohort, suggesting increased stress, which is well in line with other studies that introduced mechanical perturbation to cells.13,14 This agrees with the scRNA-seq data showing a high proportion of cells with high levels of MEGs suggestive of stress.29 The higher BAX level that we observed in resting normal-cohort cells may indicate they have more issues adapting to cell culture compared with their diseased counterparts. This is likely due to TPCs being heavily dependent on the ECM, and although healthy TPCs experience a highly structured ECM, diseased TPCs have adapted to a much less-structured one, making them more amenable to cell culture conditions. These results, along with the higher MEG levels seen in stretched TPCs compared with their resting counterparts, suggest we are observing cellular stress in the scRNA-seq data.
Since the initial discovery of TPCs, it has been suggested that there are different subsets of TPCs with varying clonogenic rates.3,4 More recently, Yin et al.5 observed and characterized a NES+ clonogenic TPC subset using single-cell qRT-PCR. We also observed this NESTIN+, highly clonogenic TPC subset and provided a full transcriptome measurement. In addition, we found a second population of TPCs with high amounts of cycling cells, the TPC state C cells. Given that these TPC state C cells have higher levels of MEGs, we think these may be the stressed version of the NES+ clonogenic TPCs.
Recently, inflammation was re-examined as a potential factor leading to the development of tendinopathy.30, 31, 32, 33 Consistent with that, cytokines and their potential to function as biomarkers for tendonitis and its progression have been considered.33 In addition, the interaction of TPCs and tenocytes with immune cells has been studied.32 piTPCs, given their unique gene expression, may have a role in the inflammation-mediated pathology of tendinitis. As a next step, it will be interesting to investigate the effect of piTPC-expressed cytokines on surrounding TPCs as well as mature tenocytes. Similar investigation has been done specifically looking at the effects of TNF, IL6, and IL10 in human tenocytes.34 These studies suggested cytokines have a role in tenocyte function and, therefore, also, likely, in TPC function.34 Moreover, cytokine signaling is not a ubiquitous function among all TPCs but primarily the role of a specialized subset (Figure 3B). This subset of TPCs, therefore, may be particularly relevant to specific tendinopathy treatments focused on stimulating inflammation for repair, such as prolotherapy.35
SLC40A1+ TPCs are a population with higher expression of TGFB2 and TGFB3. TGFB2 and TGFB3 have been shown to be important in tendon development, a double-mutant results in the loss of most tendons in the developing mouse embryo.21 In addition, TGFB family members have been implicated in the tendon repair process.36, 37, 38 Tendinopathy has been characterized as a prolonged aberrant healing response of the tendon, which could explain why a TPC subset with higher TGFB2 and TGFB3 levels could be relevant to tendinopathy.39,40 Further examination of the SLC40A1+ TPCs will be necessary to fully understand their role in the tendon.
Our dataset provides many opportunities for follow-up. First, the work provides a more-extensive understanding of the stressed mrTPC population and how they tolerate increased mechanical stress. Second, contextual understanding needs to be further explored for piTPCs and SLC40A1+ TPCs because they were found only in a small subset of samples. Third, many genes were found, consistently, to be differentially expressed between healthy and diseased TPCs, even though they weren’t necessarily tendon associated. Although acquiring more samples may show some of these to be patient variation only, further examination of their potential role in tendinopathy could lead to more therapeutic options.
Limitations of the study
Our study expands the understanding of TPCs as a heterogenous group with specialized functionalities. Unfortunately, a difficulty in our study was finding unique surface marker for each subset of TPC we observed. Given that our study only observed the transcriptome, there may be differences in the protein level of certain surface markers. Expansion of our study to cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) may provide further information on unique surface markers.6,41 Acquiring TPC samples from healthy and diseased patients provided many barriers, thus limiting cohort sizes. These barriers usually came in the form of patient recruitment as for healthy tendon samples, patients had to require an anterior cruciate ligament (ACL) reconstruction surgery. In terms of tendinopathy patient recruitment, it is rare that patients with tendinopathy require tissue to be excised, thus limiting the number of diseased samples that can be collected. Additionally, another potential limitation of our study is the lack of female-derived tendon samples. With that said, according to a recent study, there were no significant differences found between female and male samples6. In the future, building a broad consortium to uniformly collect samples can provide a larger sample size.
Conclusion
In conclusion, extending on our previous study, we profiled TPCs with and without mechanical stimulus.7 We were able to identify seven subpopulations with unique characteristics; in doing so, we have been able to establish a foundational dataset upon which to better understand TPCs. Further examination to better clarify some of the subpopulations and their functions will be vital toward gaining a better understanding of TPCs and the tendon tissue. In addition, identification of markers of tendinopathy and their association with TPC subsets can help to better identify the etiology of tendinopathy.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TotalSeq-A0251 anti-human Hashtag 1 Antibody | BioLegend | 394601; RRID:AB_2750015 |
| TotalSeq-A0252 anti-human Hashtag 2 Antibody | BioLegend | 394603; RRID:AB_2750016 |
| TotalSeq-A0254 anti-human Hashtag 4 Antibody | BioLegend | 394607; RRID:AB_2750018 |
| APC/Cyanine7 anti-human CD45 Antibody | BioLegend | 368515; RRID:AB_2566375 |
| PerCP/Cyanine5.5 anti-human CD31 Antibody | BioLegend | 303131; RRID:AB_2566174 |
| FITC Mouse Anti-Human CD34 | BD PharMingen | 560942; RRID:AB_10562559 |
| APC Mouse anti-Human CD73 | BD PharMingen | 560847; RRID:AB_10612019 |
| PE-Cy7 Mouse Anti-Human CD90 | BD PharMingen | 561558; RRID:AB_10714644 |
| PE Mouse anti-Human CD105 | BD PharMingen | 560839; RRID:AB_2033932 |
| FITC Annexin V Apoptosis Detection Kit with PI | BioLegend | 640914 |
| APC/Cyanine7 Mouse IgG1, κ Isotype Ctrl Antibody | BioLegend | 400128; RRID:AB_2892538 |
| PerCP/Cyanine5.5 Mouse IgG1, κ Isotype Ctrl Antibody | BioLegend | 400149; RRID:AB_893680 |
| PE-Cy7 Mouse IgG1 κ Isotype Control | BD PharMingen | 557872; RRID:AB_396914 |
| APC Mouse IgG1 κ Isotype Control | BD PharMingen | 554681; RRID:AB_398576 |
| PE Mouse IgG1, κ Isotype Control | BD PharMingen | 555749; RRID:AB_396091 |
| FITC Mouse IgG1, κ Isotype Control | BD PharMingen | 555748; RRID:AB_396090 |
| Biological samples | ||
| Human tendon progenitor cells | Patellar tendon samples attained by Dr. Jason Dragoo | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Fixable Viability Stain 450 | BD PharMingen | 562247 |
| Deposited data | ||
| Single cell RNA sequencing datasets | This paper | GEO accession GEO: GSE150482 |
| Oligonucleotides | ||
| TUFM qPCR primers | IDT | Hs.PT.58.4007037 |
| TFAM qPCR primers | IDT | Hs.PT.58.4391001 |
| BAX qPCR primers | IDT | Hs.PT.56a.19141193.g |
| GAPDH qPCR F primer | IDT | CAATGACCCCTTCATTGACC |
| GAPDH qPCR R primer | IDT | TTGATTTTGGAGGGATCTCG |
| Software and algorithms | ||
| R (4.0.1) | CRAN | N/A |
| Seurat | Stuart∗,Butler∗,et al., Cell 2019 | N/A |
| Excel | Microsoft | N/A |
| GraphPad Prism | GraphPad | N/A |
| CITE-seq-Count | https://github.com/Hoohm/CITE-seq-Count | N/A |
| MAST | Finak et al., Genome Biol 2015 | N/A |
| FlowJo | Tree Star | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Lei S. Qi (slqi@stanford.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The datasets generated during this study are available at the NCBI GEO accession code GEO: GSE150482. Examples of code are available in Data S1.
Experimental model and subject details
Ethical approval
The study was reviewed and approved by the Institutional Review Board of Research Compliance Office at Stanford University (approval numbers 48131 & 56522) and was conducted according to the guidelines of the Declaration of Helsinki. All patient information was confidentially maintained, and written informed consent was obtained.
Tendon tissue collection
All donors were provided informed consent prior to tissue collection. The patient data on each subject was limited to age, gender, and medical description related to tendon disease diagnosis. Tendon samples were collected surgically from either patellar tendinopathy patients diagnosed at class 3 or 4 by magnetic resonance imaging (N = 3) or healthy patellar tendons in patients undergoing bone-patellar tendon-bone anterior cruciate ligament reconstruction (N = 3). All donors were male with age ranging from 22–33 years for tendinopathy donors (26.67 5.03 years) and 18–33 years for normal donors (27.00 7.94 years). For the normal cohort, three donors were aged 30 (thickness of tendon tissue = 5.75 mm), 19 (thickness of tendon tissue = 5.47 mm), and 33 (thickness of tendon tissue = 5.77 mm). For the patellar tendinopathy patient cohort, three donors were aged 33 (insidious, classification grade = 3, duration of symptoms ~2 years, thickness of tendon tissue = 8.98 mm), 26 (traumatic, classification grade = 4, duration of symptoms ~1 year, thickness of tendon tissue = 10.9 mm), aged 22 (insidious, classification grade = 3, duration of symptoms ~2 years, thickness of tendon tissue = 12.5mm). Information on donor samples including age, gender, and health status can also be found in Table S1.
Tendon progenitor cells (TPCs) isolation and maintenance
The peritendinous connective tissues were completely removed from the harvested tendons before processing. Tissues were digested in 0.1% type IV collagenase (Sigma-Aldrich, St. Louis, MO) in low-glucose Dulbecco’s modified Eagle medium (DMEM/Invitrogen Corporation, Carlsbad, CA) at 37C with gentle agitation for overnight. After wash with the medium, cell suspension was passed through a cell strainer (70 um; Becton Dickinson, Franklin Lakes, NJ) to obtain a single-cell suspension. The number of viable cells was determined by NucBlue Live Ready Probes Reagent (Invitrogen Corporation) staining, according to the manufacturer’s procedure. Cells were seeded at 500 viable cells/cm2 in culture dishes with DMEM/10%FBS and incubated at 37C /5% CO2 for 14 days. Once the plate reached 90% confluency, cells were designated as passage 0, and split to two new plates for expansion. Passage 2 cells were used for all experiments. For the purposes of this study, all progenitor cells harvested from the surgical tissues will be referred to as TPCs and validated by fluorescence activated cell sorting with specific cell surface markers for mesenchymal stem cells.
Methods details
Cell stretch device and experiments
We used a cell stretch device identical to the one shown in Figure 1 of Chang et al.7The mold used to make the silicone stretchable wells was fabricated using Delrin Acetal Resin and stainless steel screws from McMaster-Carr (Santa Fe Springs, CA). Polydimethylsiloxane (PDMS) solution (Sylgard 184 Silicone Elastomer from Dow Corning; Sigma-Aldrich) was prepared at 1:10 mixture with solution A and B for the silicone block and the thin layer, and cured at 70C for 120 minutes. For surface coating, a spin coater from Specialty Coating Systems (Indianapolis, IN) was used. To create the thin layer for cell attachment, a microscope slide (75 × 38 × 1mm; Fisher Scientific, Pittsburgh, PA) was coated with a sacrifice layer of 10% polyvinyl alcohol (Sigma-Aldrich) solution at 1000 rpm/60 s. Once dried, a thin layer of PDMS solution was coated on the top of it at 500 rpm/60 s. The final stretchable culture wells were generated by bonding the well block to the thin layer on glass slide with PDMS solution. Once cured, the assembly was soaked in water to dissolve the sacrifice layer and release the final product. The stretchable culture wells were then cleaned thoroughly and autoclaved. To provide the physical force for the mechanical loading, a stretch platform on a syringe pump (Model NE-1600-U; New Era Pump Systems, Inc.) was designed from a pair of 90 angle aluminum elbows (McMaster-Carr, Santa Fe Springs, CA) attached with parallel pins as well anchors. More details are shown in a close-up. For cell attachment, sterile silicone wells were coated with 0.4% gelatin (Sigma-Aldrich) and incubated overnight at 37C. After drying, cells were seeded at 50 × 103 per cm2 in triplicates and cultured overnight in DMEM/10%FBS before subjecting to cyclic stretch at moderate strength, 6% clamp-to-clamp, for 12 hours per day for 3 days with 1 day rest before harvest.
Drop-seq test of resting cells
Drop-seq was carried out as previously described.11 Cells were dissociated using TrypLE Express (ThermoFisher Scientific Cat#: 12604013) and spun down at 1000 rpm for 3 mins. Two washes were done using PBS-BSA (0.01% BSA, ThermoFisher Scientific Cat#: 15260037). Cells were loaded in PBS-BSA at a concentration of 280 Cells/ul. Drop-seq beads (Chemgenes Cat#: Macosko-2011-10) were loaded into another syringe at 280 beads/ul in the Drop-seq lysis buffer.11 Cells, beads, and oil were ran through a Dolomite scRNaseq chip (Dolomite Cat#: 3200583) with cells and beads at 30ul/min and the oil at 200ul/min as recommended in the product literature. Following the microfluidics step, library generation was conducted consistent with the standard Drop-seq protocol.11 Briefly, following the microfluidic step beads underwent a reverse transcription reaction using the Maxima H Minus Reverse transcriptase (Thermofisher scientific Cat#: EP0751). cDNA was then amplified from beads for 13 cycles and purified using 0.6X and 1X AMPure XP bead cleanup (Beckman Coulter Cat#: A63881). 0.6 ng of cDNA was used for the tagmentation reaction using the Nextera XT DNA library preparation kit (Illumina Cat#: FC-131-1096). Following this, libraries were cleaned up again using AMPure XP beads, with a 0.45X high cleanup (remove excessively long library strands) followed by two 0.6X low cleanups (remove smaller fragments and primers) (Beckman Coulter Cat#: A63881).
Cell hashing
Cell hashing was carried out as previously described.41 Briefly, cells were washed and filtered into a single cell suspension. After counting cells were resuspended in 100ul of cell staining buffer (BioLegend Cat#: 420201). Cells were then blocked using the Human TruStain FcX Fc Blocking reagent (BioLegend Cat#: 422301). Finally, cells had their appropriate cell hashing antibody administered and incubated at 4°C for 30 minutes. Wash cells three times with 1ml of cell staining buffer before mixing cells to the appropriate concentration for the single cell capture. See Table S1 for cell hashing antibodies used for each sample.
10X Chromium test of stretched cells
Cells were subjected to 10X Chromium Controller 3′ counting at the Stanford Functional Genomics facility (SFGF) consistent with standard 10X Chromium protocols. Cells were hashed and submitted to the facility at a concentration of 1000 cells/ml with a target of 10,000 cell capture. Cell viability checks were also done during cell counting. Cell hashing and scRNaseq transcriptome libraries were produced at the SFGF.
Sequencing
Each Drop-seq sample was subject to sequencing on the HiSeq 4000 using a custom read 1 primer specified in the Drop-seq protocol.11 Reads were trimmed to 25bp on read 1 and 50 bp on read 2. 10X Chromium libraries were multiplexed onto a NovaSeq 6000 sequencer for sequencing. Reads were trimmed to 28bp on read 1 and 98 bp on read 2. Hashing libraries were separately sequenced in Mi-seq runs.
Mitotracker staining
Mitotracker Red CMXRos (Cell Signaling cat#: 9082S) aliquots were resuspended in DMSO to make a 1mM stock. The stain was further diluted in DMEM (ThermoFisher Scientific cat#: 10569-044) + 10% FBS (Alstem cat#: FB500) to a working concentration of 100nM. Cells were dissociated, spun down at 1000 rpm for 3 minutes and then resuspended in 1mL of PBS (Fisher Scientific cat#: 14190250). Cells were counted and 100,000 cells were apportioned for staining. Cells were spun down again as before, and resuspended in 1ml of MitoTracker Red CMXRos stain. Cells were stained for 30 minutes and then washed twice with PBS and staining buffer (BioLegend cat#:420201). Flow cytometry was then done.
Propidium iodide and Annexin 5 staining
The FITC Annexin V Apoptosis Detection kit with Propidium Iodide (PI) was used (Biolegend cat#: 640914). Cells were washed with cell staining buffer before being resuspended in Annexin V binding buffer at a concentration of 1e7 cells/mL. 100ul of the cells were transferred to a 5ml test tube. 5ul of FITC Annexin V antibody and 10ul of propidium iodide solution were added to the 100ul cell suspension. The solution was then gently vortexed and incubated in the dark at room temperature (25C) for 15 minutes. 400ul of Annexin V binding buffer was then added to the tube and analyzed by flow cytometry.
qRT-PCR
qRT-PCR was conducted using the Kapa sybr fast qPCR master mix (2X) (Roche Cat#: KK4602) and iScript (Bio-Rad Cat#: 7959397001). 1000ng of RNA was used as input for each sample cDNA synthesis. Each gene was tested with 3 technical replicates per sample of 15ul reactions, 37.5ng cDNA per reaction. Reactions were run on a CFX384 Touch Real-Time PCR Detection System (Bio-Rad Cat#:1855485). Gene expression was normalized to GAPDH. Microsoft excel was used to calculate standard deviations for the error bars. Significance of difference between cohorts and conditions was determined by an unpaired t test in GraphPad Prism (GraphPad). For TUFM, the forward primer is 5′ - CCCACTTCATGCCTGTCAT-3′ and reverse primer is 5′- AACGCTGGCCTTTCTCTAAG-3′, this corresponds to IDT primetime assay ID: Hs.PT.58.4007037. For TFAM, the forward primer is 5′ -TGGGAAGGTCTGGAGCA-3′ and reverse primer is 5′-GCCAAGACAGATGAAAACCAC-3′, this corresponds to IDT primetime assay ID: Hs.PT.58.4391001. For BAX, the forward primer is 5′ - TCTGAGCAGATCATGAAGACAG −3′ and reverse primer is 5′-CCACTCGGAAAAAGACCTCT-3′, this corresponds to IDT primetime assay ID: Hs.PT.56a.19141193.g. For GAPDH, the forward primer is 5′ - CAATGACCCCTTCATTGACC-3′ and reverse primer is 5′- TTGATTTTGGAGGGATCTCG-3′.
Quantification and statistical analysis
Data analysis, drop-seq
Drop-seq libraries were converted to a matrix using a combination of the Drop-seq core computational protocol (referred to as the Drop-seq Alignment Cookbook) and hisat2.42 After generation of an expression matrix, datasets were analyzed primarily using Seurat.19,20 Cells were selected by a minimum gene threshold of 500 genes. See Data S2 attached coding examples for further details.
Data analysis, 10x chromium
10X Chromium controller generated datasets were converted to expression matrices using Cellranger v3.1 (https://support.10xgenomics.com/permalink/3IQFKIvEuskMoEWkWUis2s). Cellranger uses STAR for alignment of reads to the genome. Data was primarily analyzed using Seurat,19,20 Cells were selected by a minimum gene threshold of 1400 genes, excluding cells in the dataset with a relatively low diversity of genes (Figure S7). CellCycleScoring function from the Seurat package was used to assign cell cycle states. CellCycleScoring was done using the RNA assay after log normalization and data scaling was applied. Enrichr was used to analyze some of the DGE analysis.43,44 Heatmaps were generated using the Pheatmap package.45 See Data S2 examples for further details.
Statistics
Subject demographic data was recorded including age at time of surgery, gender, and duration of symptoms. Continuous variable data were reported as mean ± standard deviation from the mean. For qRT-PCR comparison between samples, an unpaired t test in GraphPad Prism (GraphPad) was used to determine significance. A p value cutoff of 0.05 was used to determine significance. For DGE, a Bonferroni correction was applied to generate adjusted p values. DGE analysis was done using the MAST package.12
Acknowledgments
The authors would like to thank the Stanford functional genomics facility for their guidance and help on single-cell experiments. We acknowledge the Sherlock cluster, operated by the Stanford Research Computing Center. We acknowledge help from Qi laboratory members for technical support and comments on the manuscript. We thank Fulgent for their sequencing services. Some figures were created with BioRender.com. C.S. acknowledges support from the NSF graduate research fellowship program (GRFP) and the Stanford graduate fellowship in science and engineering (SGF). L.S.Q. acknowledges support from Pew Charitable Trusts and the Li Ka Shing Foundation. The work is supported by a gift fund from the Li Ka Shing Foundation and from Stanford University School of Medicine startup funds (to L.S.Q.).
Author contributions
C.S., J.L.D., and L.S.Q. conceived the idea. C.S., W.-T.C., J.L.D., and L.S.Q. designed the experiments. J.L.D. harvested tendon tissues from patient subjects. W.-T.C. processed tendon tissues and enriched TPCs. W.-T.C. performed, cyclical stretching. K.R.S. performed clinical-grade classification and analysis of patient-derived samples. C.S. performed all single-cell experiments, including Drop-seq,sample prep for 10× chromium controller 3′ counting, managed/set-up sequencing runs, and performed qRT-PCR experiments. C.S. performed all computational analysis. W.-T.C. conducted PI and Annexin V staining. C.S. and L.S.Q. wrote the manuscript with inputs from all authors. All authors read and revised the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: July 20, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100343.
Supplemental information
References
- 1.Andarawis-Puri N., Flatow E.L., Soslowsky Tendon basic science: development, repair, regeneration, and healing. J. Orthop. Res. 2015;33:780–784. doi: 10.1002/jor.22869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharma P., Maffulli N. Biology of tendon injury: healing, modeling and remodeling. J. Musculoskelet. Neuronal Interact. 2006;6:181–190. [PubMed] [Google Scholar]
- 3.Walia B., Huang A.H. Tendon stem progenitor cells: understanding the biology to inform therapeutic strategies for tendon repair. J. Orthop. Res. 2019;37:1270–1280. doi: 10.1002/jor.24156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bi Y., Ehirchiou D., Kilts T.M., Inkson C.A., Embree M.C., Sonoyama W., Li L., Leet A.I., Seo B.M., Zhang L. Identification of tendon stem/progenitor cells and the role of the extracellular matrix in their niche. Nat. Med. 2007;13:1219–1227. doi: 10.1038/nm1630. [DOI] [PubMed] [Google Scholar]
- 5.Yin Z., Hu J.J., Yang L., Zheng Z.F., An C.R., Wu B.B., Zhang C., Shen W.L., Liu H.H., Chen J.L. Single-cell analysis reveals a nestin+ tendon stem/progenitor cell population with strong tenogenic potentiality. Sci. Adv. 2016;2:e1600874. doi: 10.1126/sciadv.1600874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kendall A., Layton T., Al-Mossawi H., Brown R., Loizou C., Rogers M., Sharp M., Dakin S., Appleton L., Carr A. Identification of human tendon cell populations in healthy and diseased tissue using combined single cell transcriptomics and proteomics. bioRxiv. 2019 doi: 10.1101/2019.12.09.869933. [DOI] [Google Scholar]
- 7.Chang W., Callan K.T., Dragoo J.L. The behavior of tendon progenitor cells from tendinopathic tendons: implications for treatment. Tissue Eng. Part A. 2020;26:38–46. doi: 10.1089/ten.TEA.2019.0042. [DOI] [PubMed] [Google Scholar]
- 8.Galloway M.T., Lalley A.L., Shearn J.T. The role of mechanical loading in tendon development, maintenance, injury, and repair. J. Bone Joint Surg. Am. 2013;95:1620–1628. doi: 10.2106/JBJS.L.01004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang C., Zhu J., Zhou Y., Thampatty B.P., Wang J.H.C. Tendon stem/progenitor cells and their interactions with extracellular matrix and mechanical loading. Stem Cells Int. 2019;2019:3674647. doi: 10.1155/2019/3674647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Golman M., Wright M.L., Wong T.T., Lynch T.S., Ahmad C.S., Thomopoulos S., Popkin C.A. Rethinking patellar tendinopathy and partial patellar tendon tears: a novel classification system. Am. J. Sports Med. 2020;48:359–369. doi: 10.1177/0363546519894333. [DOI] [PubMed] [Google Scholar]
- 11.Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finak G., McDavid A., Yajima M., Deng J., Gersuk V., Shalek A.K., Slichter C.K., Miller H.W., McElrath M.J., Prlic M. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278. doi: 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liang X., Wang Z., Gao M., Wu S., Zhang J., Liu Q., Yu Y., Wang J., Liu W. Cyclic stretch induced oxidative stress by mitochondrial and NADPH oxidase in retinal pigment epithelial cells. BMC Ophthalmol. 2019;19:79. doi: 10.1186/s12886-019-1087-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen H., Chen L., Cheng B., Jiang C. Cyclic mechanical stretching induces autophagic cell death in tenofibroblasts through activation of prostaglandin E2 production. Cell. Physiol. Biochem. 2015;36:24–33. doi: 10.1159/000374050. [DOI] [PubMed] [Google Scholar]
- 15.Helle S.C.J., Feng Q., Aebersold M.J., Hirt L., Grüter R.R., Vahid A., Sirianni A., Mostowy S., Snedeker J.G., Šarić A. Mechanical force induces mitochondrial fission. eLife. 2017;6:1–26. doi: 10.7554/eLife.30292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takasawa R., Nakamura H., Mori T., Tanuma S. Differential apoptotic pathways in human keratinocyte HaCaT cells exposed to UVB and UVC. Apoptosis. 2005;10:1121–1130. doi: 10.1007/s10495-005-0901-8. [DOI] [PubMed] [Google Scholar]
- 17.Lindenboim L., Grozki D., Amsalem-Zafran A.R., Peña-Blanco A., Gundersen G.G., Borner C., Hodzic D., Garcia-Sáez A.J., Worman H.J., Stein R. Apoptotic stress induces Bax-dependent, caspase-independent redistribution of LINC complex nesprins. Cell Death Discov. 2020;6:90. doi: 10.1038/s41420-020-00327-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pihán P., Carreras-Sureda A., Hetz C. BCL-2 family: integrating stress responses at the ER to control cell demise. Cell Death Differ. 2017;24:1478–1487. doi: 10.1038/cdd.2017.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W. Comprehensive integration of single cell data. bioRxiv. 2018:460147. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pryce B.A., Watson S.S., Murchison N.D., Staverosky J.A., Dünker N., Schweitzer R. Recruitment and maintenance of tendon progenitors by TGFβ signaling are essential for tendon formation. Development. 2009;136:1351–1361. doi: 10.1242/dev.027342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelly L.M., Englmeier U., Lafon I., Sieweke M.H., Graf T. MafB is an inducer of monocytic differentiation. EMBO J. 2000;19:1987–1997. doi: 10.1093/emboj/19.9.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thankam F.G., Chandra I.S., Kovilam A.N., Diaz C.G., Volberding B.T., Dilisio M.F., Radwan M.M., Gross R.M., Agrawal D.K. Amplification of mitochondrial activity in the healing response following rotator cuff tendon injury. Sci. Rep. 2018;8:17027. doi: 10.1038/s41598-018-35391-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sakaan S.A., Self T.H. Minimizing the risk of tendon injury associated with fluoroquinolone use. Infect. Dis. Consultant. 2017;57:541–542. [Google Scholar]
- 25.Wise B.L., Peloquin C., Choi H., Lane N.E., Zhang Y. Impact of age, sex, obesity, and steroid use on quinolone-associated tendon disorders. Am. J. Med. 2012;125:1228.e23–1228.e28. doi: 10.1016/j.amjmed.2012.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Owens R.C., Jr., Ambrose P.G. Antimicrobial safety: focus on fluoroquinolones. Clin. Infect. Dis. 2005;41:S144–S157. doi: 10.1086/428055. [DOI] [PubMed] [Google Scholar]
- 27.Khaliq Y., Zhanel G.G. Fluoroquinolone-associated tendinopathy: a critical review of the literature. Clin. Infect. Dis. 2003;36:1404–1410. doi: 10.1086/375078. [DOI] [PubMed] [Google Scholar]
- 28.Lipsky B.A., Baker C.A. Fluoroquinolone toxicity profiles : a review focusing on newer agents. Clin. Infect. Dis. 1999;28:352–364. doi: 10.1086/515104. [DOI] [PubMed] [Google Scholar]
- 29.Luecken M.D., Theis F.J. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol. 2019;15:e8746. doi: 10.15252/msb.20188746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rees J.D., Stride M., Scott A. Tendons--time to revisit inflammation. Br. J. Sports Med. 2014;48:1553–1557. doi: 10.1136/bjsports-2012-091957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Charnoff J., Naqvi U. StatPearls: Content Is King. StatPearls Publishing; 2020. Tendinosis. [Google Scholar]
- 32.Stolk M., Klatte-Schulz F., Schmock A., Minkwitz S., Wildemann B., Seifert M. New insights into tenocyte-immune cell interplay in an in vitro model of inflammation. Sci. Rep. 2017;7:9801. doi: 10.1038/s41598-017-09875-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morita W., Dakin S.G., Snelling S.J.B., Carr A.J. Cytokines in tendon disease: a systematic review. Bone Joint Res. 2017;6:656–664. doi: 10.1302/2046-3758.612.BJR-2017-0112.R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.John T., Lodka D., Kohl B., Ertel W., Jammrath J., Conrad C., Stoll C., Busch C., Schulze-Tanzil G. Effect of pro-inflammatory and immunoregulatory cytokines on human tenocytes. J. Orthop. Res. 2010;28:1071–1077. doi: 10.1002/jor.21079. [DOI] [PubMed] [Google Scholar]
- 35.Goerl K. Getting tendinopathy treatment (and terminology) right. J. Fam. Pract. 2020;69:127–134. [PubMed] [Google Scholar]
- 36.Tsubone T., Moran S.L., Subramaniam M., Amadio P.C., Spelsberg T.C., An K.N. Effect of TGF-beta inducible early gene deficiency on flexor tendon healing. J. Orthop. Res. 2006;24:569–575. doi: 10.1002/jor.20101. [DOI] [PubMed] [Google Scholar]
- 37.Branford O.A., Klass B.R., Grobbelaar A.O., Rolfe K.J. The growth factors involved in flexor tendon repair and adhesion formation. J. Hand Surg. Eur. Vol. 2014;39:60–70. doi: 10.1177/1753193413509231. [DOI] [PubMed] [Google Scholar]
- 38.Jiang K., Chun G., Wang Z., Du Q., Wang A., Xiong Y. Effect of transforming growth factor-β3 on the expression of Smad3 and Smad7 in tenocytes. Mol. Med. Rep. 2016;13:3567–3573. doi: 10.3892/mmr.2016.4944. [DOI] [PubMed] [Google Scholar]
- 39.Fu S.C., Rolf C., Cheuk Y.C., Lui P.P.Y., Chan K.M. Deciphering the pathogenesis of tendinopathy: a three-stages process. Sport Med. Arthrosc. Rehabil. Ther. Technol. 2010;2:30. doi: 10.1186/1758-2555-2-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ackermann P.W., Renström P. Tendinopathy in sport. Sports Health. 2012;4:193–201. doi: 10.1177/1941738112440957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stoeckius M., Hafemeister C., Stephenson W., Houck-Loomis B., Chattopadhyay P.K., Swerdlow H., Satija R., Smibert P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods. 2017;14:865–868. doi: 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kolde R. R Foundation for Statistical Computing; 2012. Pheatmap: Pretty Heatmaps. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study are available at the NCBI GEO accession code GEO: GSE150482. Examples of code are available in Data S1.