

Summary
We have established experimental systems to assess the effects of early-life exposures to antibiotics on the intestinal microbiota and gene expression in the brain. This model system is highly relevant to human exposure and may be developed into a preclinical model of neurodevelopmental disorders in which the gut–brain axis is perturbed, leading to organizational effects that permanently alter the structure and function of the brain. Exposing newborn mice to low-dose penicillin led to substantial changes in intestinal microbiota population structure and composition. Transcriptomic alterations implicate pathways perturbed in neurodevelopmental and neuropsychiatric disorders. There also were substantial effects on frontal cortex and amygdala gene expression by bioinformatic interrogation, affecting multiple pathways underlying neurodevelopment. Informatic analyses established linkages between specific intestinal microbial populations and the early-life expression of particular affected genes. These studies provide translational models to explore intestinal microbiome roles in the normal and abnormal maturation of the vulnerable central nervous system.
Subject areas: developmental neuroscience, microbiome
Graphical abstract
Highlights
-
•
Low-dose antibiotic exposure perturbs the infant gut mouse microbiome to PND10
-
•
Frontal cortex and amygdala gene expression were substantially affected
-
•
Multiple pathways underlying neurodevelopment were affected
-
•
Specific gut microbial populations were linked with expression of particular genes
Developmental neuroscience; Microbiome
Introduction
Early life is a critical period for neurodevelopment (Krebs et al., 2017; Perry et al., 2017; Robinson-Drummer et al., 2019). In recent decades, there has been a rise in the incidence of childhood neurodevelopmental disorders including autism spectrum disorder (ASD), attention deficit/hyperactivity disorder (ADHD), and learning disabilities worldwide (Antshel and Barkley, 2020; Baio et al., 2018; Davidovitch et al., 2020; DuPaul et al., 2013; Hertz-Picciotto et al., 2018). Although increased awareness and diagnosis are likely contributing factors, environmental perturbations early in development are posited as critical in initiating pathology. The rapid increases in case numbers are consistent with changes in environmental exposures, but few exposures are sufficiently global or of significant scale to explain the development of these disorders in multiple countries around the world. The challenge is identifying the environmental perturbations, understanding the neurobehavioral targets, and identifying interventions for public health benefit.
One class of exposures to nearly all children of the world, which essentially did not exist before 1945, is antibiotics. In the USA, an average child receives nearly 3 courses of antibiotics before the age of 2 years, and similar or greater exposure rates have been found across the globe including both developed and low- and middle-income nations (Rogawski et al., 2017). Antibiotics could affect the brain directly, or could have effects via their actions on the human microbiome (Cox et al., 2014, 2019; Cox and Weiner, 2018). In recent years, there has been a focus on the human microbiome as an important environmental factor to which all children are exposed (Blaser, 2006). The human microbiome is a vast consortium of microorganisms that live in and on us, persisting throughout the life span. For every human cell, there is more than one microbial cell, and for each of the 23,000 human genes, there are hundreds of microbial genes (Sender et al., 2016; Tierney et al., 2019). The particular microbiome taxa living with us are ancient, dating to our earliest origins (Ochman et al., 2010). The microbiome performs many salutary functions for the host, including producing needed vitamins, aiding in digestion, and defending against invaders (Cho and Blaser, 2012; Spor et al., 2011; Tremaroli and Bäckhed, 2012).
However, when the microbiome is altered, such as with antibiotic treatment, the host may have long-term effects. For example, for the past 70 years, farmers have administered sub-therapeutic antibiotic treatment (STAT) to livestock for growth promotion. This is an effective and widely used practice (Ozawa, 1955), indicating that antibiotic exposure changes early-life development with lifelong consequences; that growth promotion is ineffective in germ-free animals indicates that the antibiotic effects are transduced via the microbiome (Coates et al., 1963), which also was shown in experimental murine models (Cox et al., 2014).
A growing body of evidence links phenomena in the intestinal tract with signaling to the brain, a field of inquiry known as the ‘gut–brain axis’ (Cryan et al., 2019; Margolis et al., 2021; Mayer et al., 2014). There is heightened interest in this fast-moving field to help explain whether or not modulating the gut–brain axis, especially early in development, can alter risk in developing neuropsychiatric (Fetissov and Déchelotte, 2011; Malan-Muller et al., 2018; Warner, 2019) and/or neurodegenerative disorders (Breen et al., 2019; Cox et al., 2014, 2019; Cox and Weiner, 2018; Parashar and Udayabanu, 2017) in adulthood. Such investigations may open new avenues of therapeutic interventions for severe central nervous system (CNS) diseases that currently are incurable.
To study mechanisms underlying early antibiotic exposure, we have established models of STAT in developing mice (Cho et al., 2012; Lieber et al., 2019; Mahana et al., 2016; Schulfer et al., 2018). These mice display permanently altered metabolism and increased adiposity among other externalities (Cho and Blaser, 2012; Cox et al., 2014; Lieber et al., 2019; Mahana et al., 2016; Schulfer et al., 2018). For example, in mouse pups, even transient effects of STAT on community composition of the maturing gut microbiome may have permanent developmental metabolic consequences (Cox et al., 2014; Schulfer et al., 2018). Moreover, multiple independent research groups studying rodents have shown that early antibiotic exposure likely has a profound, organizational impact on gene expression, brain structure and function, and behavior (Diaz Heijtz et al., 2011; Guida et al., 2018; Leclercq et al., 2017; Minter et al., 2016; Ogawa et al., 2020; Schmidtner et al., 2019). These findings have direct translational relevance since antibiotic exposure at birth in humans has been shown to alter recognition memory in one-month-old infants, as assessed by event-related potentials (Hickey et al., 2021). Taken together, existing research is provocative but incomplete, clearly indicating that additional preclinical research is necessary to determine the overall impact of early antibiotic exposure on neurodevelopment.
Although early-life antibiotic treatment has long been considered to have minimal adverse effects, much less than the benefit for their use, one hypothesis we are testing herein is that antibiotic exposure may have a potentially deleterious organizational effect by perturbing the developing microbiome in early life and subsequently altering signaling within the developing brain. This hypothesis could help explain the rising incidence in neuropsychiatric diseases of childhood, but must be tested rigorously to assess causation. Although epidemiologic and observational studies have associated early-life antibiotic exposures with an increased risk of health-related outcomes including obesity, asthma, and celiac disease (Aversa et al., 2021; Hviid et al., 2011; Manco, 2012; Patrick et al., 2020; Trasande et al., 2013; Wickens et al., 1999) and importantly neurodevelopmental disorders including ASD and ADHD (Aversa et al., 2021; Hamad et al., 2019; Lee et al., 2019; Slob et al., 2021; Wimberley et al., 2018), the results of these correlational analyses often conflict (Hamad et al., 2018, 2019; Łukasik et al., 2019; Slob et al., 2021) and do not infer causality. To initiate mechanistic inquiry, we began studies exposing newborn mice to low-dose (sub-therapeutic) levels of penicillin (LDP) to determine whether there might be effects in vulnerable portions in the developing brain. We chose penicillin because beta-lactams, such as penicillin, are the most widely used antibiotics in early childhood (Chai et al., 2012) and because we have extensive experience in mouse models using LDP, showing that these have substantial effects on the development of metabolism (Cho et al., 2012; Cox et al., 2014; Lieber et al., 2019; Mahana et al., 2016; Schulfer et al., 2018) and inflammation (Cho et al., 2012; Cox et al., 2014; Lieber et al., 2019; Mahana et al., 2016; Schulfer et al., 2018). We now ask (Figure 1) whether exposure of newborn mice to LDP will again alter the early-life intestinal microbiome and whether it will affect gene expression in the frontal cortex and amygdala, two critical areas underlying emotional and cognitive functions that are vulnerable to early-life perturbations (Raineki et al., 2019; Robinson-Drummer et al., 2019; Tottenham and Sheridan, 2010). The infant amygdala and frontal cortex circuit is also recognized as acutely sensitive to maternal rearing (Sullivan et al., 2000). Moreover, triggers that impact maternal caregiving can readily influence the social behavioral switch from attraction to fear learning in pups that contribute to lifelong emotional and learning deficiencies (Moriceau and Sullivan, 2006; Sullivan et al., 2000). Accordingly, using the established murine LDP model and interrogating the frontal cortex and amygdala, we tested the hypothesis that selective gut microbiome changes in specific taxa linked to brain level gene expression differences can be identified during the early postnatal period within mouse brain regions relevant to dysfunction observed in human neurodevelopmental disorders.
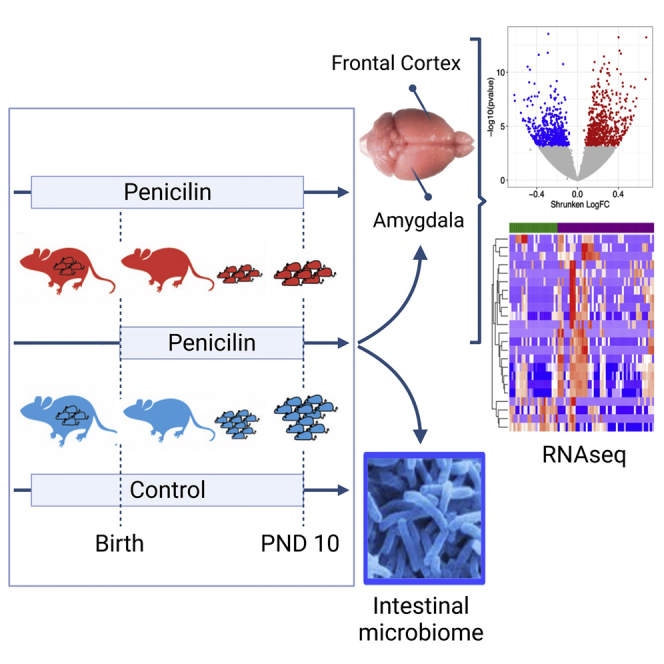
Figure 1.

Study design: assessing effects of early-life antibiotics on gene expression in the brain
Pregnant C57BL/6 mice were either exposed to low-dose penicillin G, as described [Cox], during the last week of pregnancy and until sacrifice of their pups on PND10 (STAT group; n = 8 pups), or exposed starting on the day of birth (STAT-Birth; n = 7 pups), or were unexposed (Control; n = 8 pups). The pups were sacrificed at PND10 for examination of microbiome and CNS transcription. Pups were born on P0, and all dams were sacrificed by P22.
Results
Microbiome analyses
The composition and population structure of the intestinal microbiomes of the pups and their dams and sires were significantly different, as expected (Figures S1 and S2). There were no significant differences in the alpha-diversity in the small intestinal and cecal contents of the pups according to their treatment groups (antibiotic-exposed groups [STAT, STAT-Birth] and the Control group) (Figure S1). However, the groups differed significantly in their population structure; in the cecum, all three groups differed from one another and in the small intestine, the Control and STAT groups were significantly different (Figure 2).
Figure 2.

Beta-diversity of pup intestinal contents, based on a Bray–Curtis distance matrix
The significance of differences was tested by Permanova-pairwise test with 999 permutations (∗p < 0.05; ∗∗p < 0.01).
The antibiotic exposures selected for particular taxa and against others (Figure 3). The strongest and most consistent were in the small intestinal microbiota; the antibiotic treatments strongly selected against the dominant Lactobacillus species, with replacement by Corynebacterium, Lachnospiraceae, Sphingomonaceae, Enterococcus, Pseudomonas, and Stenotrophomonas, among others. The most numerous significant effects were in the cecum, with selection against Staphylococcus, Lactobacillus and Roseburia species, and Oscillospiraceae, among others, with replacement by Enterobacteriales, Enterococcus, and Pseudomonales, among others. Taken together, these results indicate that exposure to even low (sub-therapeutic) doses of penicillin substantially changes the early-life intestinal microbiota in developing mice.
Figure 3.

Heatmap representing taxa (at genus level) in the pup samples that are significantly different between STAT vs. Control and STAT-Birth vs. Control
Each row represents a different taxon and each column a different treatment. Log10 taxon relative abundance is shown in the scales for each intestinal sampling site at right. Significantly different taxa were inferred by MaAsLin2 analysis.
(A) Cecal contents.
(B) Colonic contents.
(C) Small intestinal contents.
Differential regulation of key genes within the frontal cortex and amygdala by early-life antibiotic exposure
We examined transcriptional profiles in the frontal cortex and amygdala of the pups at a global level across all measured transcripts. The major comparison was between each of the antibiotic-exposed groups (STAT, STAT-Birth) versus the Control group. By applying principal component analysis (PCA), we found that patterns of gene expression in both the frontal cortex and amygdala differed significantly across the three groups along the PC1 axis (Figure 4). We then examined differential expression across individual genes to identify statistically significant differences between the exposed and control groups in each brain region. In each of these comparisons between the controls and antibiotic-exposed groups, there were a large number (>500) of significantly differentially expressed genes (DEGs) in both the frontal cortex and amygdala (Figure 5). Since these analyses identified hundreds of DEGs in each comparison (Tables S1–S4), we increased the stringency of the comparisons. Genes were first filtered based on having a robust fold change in at least one condition (>1.5 absolute log2-fold change) and an adjusted p value <0.01, with false discovery rate (FDR) under 5% (Tables S5–S8). We then highlighted genes found to be differentially expressed in either of the comparisons between the control group and the antibiotic-exposed (STAT and STAT-Birth) groups.
Figure 4.

Gene expression in the frontal cortex and amygdala in the three study groups
In the groups (STAT, STAT-Birth, and Control), there are three samples per pup. Global expression data for each sample in each site are represented by PCA. The significance of differences was tested by pairwise Mann–Whitney tests on PC1 (∗p < 0.05; ∗∗p < 0.01; not significant: p > 0.05). See also Figure S3.
Figure 5.

Volcano plots of differentially expressed genes in the frontal cortex (left) and amygdala (right) between antibiotic-exposed and control pups
Genes differentially expressed with p < 0.01 are represented by colored points (e.g., no change in gray, upregulation in red, and downregulation in blue).
(A) Comparison of STAT vs. Control.
(B) Comparison of STAT-Birth vs. Control.
Using these criteria, we identified 209 genes in the frontal cortex and 41 genes in the amygdala, some of which overlap between the two antibiotic groups (Figure 6; Tables S9 and S10). Comparing across treatment conditions, there were 17 genes in the frontal cortex and 3 genes in the amygdala that were significantly differentially regulated in relation to the Control group (Table 1). Significant frontal cortex genes include adenylate kinase 7 (Ak7) (2.01-fold upregulated in STAT and 1.58-fold upregulated in STAT-Birth); coiled-coil domain containing 121 (Ccdc121) (1.78-fold upregulated in STAT and 1.57-fold upregulated in STAT-Birth); canopy FGF signaling regulator 1 (Cnpy1) (2.52-fold upregulated in STAT and 2.87-fold upregulated in STAT-Birth); dynein axonemal light intermediate chain 1 (Dnali1) (2.24-fold upregulated in STAT and 1.81-fold upregulated in STAT-Birth); ETS homologous factor (Ehf) (1.96-fold upregulated in STAT and 1.91-fold upregulated in STAT-Birth); neurotensin (Nts) (1.64-fold upregulated in STAT and 1.97-fold upregulated in STAT-Birth); serpin family A member 9 (Serpina9) (2.91-fold upregulated in STAT and 3.69-fold upregulated in STAT-Birth); SIX homeobox 3 (Six3) (3.14-fold upregulated in STAT and 4.51-fold upregulated in STAT-Birth); and solute carrier family 6 member 4 (Slc6a4; also known as serotonin transporter SERT [2.00-fold upregulated in STAT and 2.46-fold upregulated in STAT-Birth]). The amygdala genes included diacylglycerol kinase kappa (Dgkk) (1.57-fold upregulated in STAT and 1.66-fold upregulated in STAT-Birth) and gastrulation brain homeobox 1 (Gbx1) (2.18-fold upregulated in Control vs STAT and 2.11-fold upregulated in Control vs STAT-Birth].
Figure 6.

Heatmap of genes in the frontal cortex and amygdala that are differentially expressed in the Control vs. STAT and/or the Control vs. STAT-Birth mice comparisons
Genes that are significantly significant (p < 0.01 with log fold change > 1.5) are depicted. See also Figure S4.
Table 1.
Overlap in differential gene expression in the two antibiotic groups vs. controls
| A. Frontal cortex, n = 17 genes | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Control vs. STAT-Birth |
Control vs. STAT |
||||||||||
| Base mean | log2FC | log2FC-SE | Z-stats | p value | p-Adj | Base mean | log2FC | log2FC-SE | Z-stats | p value | p-Adj | |
| 2610528A11Rik | 5.62 | 1.63 | 0.45 | 3.67 | 2.48e-4 | 1.75e-3 | 6.25 | 1.78 | 0.48 | 3.73 | 1.8e-4 | 3.33e-3 |
| Ak7 | 3.35 | 1.58 | 0.39 | 4.05 | 5.15e-5 | 4.90e-4 | 4.35 | 2.01 | 0.38 | 5.34 | 9.05e-8 | 1.62e-5 |
| Ccdc121 | 1.29 | 1.56 | 0.45 | 3.48 | 4.00e-4 | 3.00e-3 | 1.55 | 1.78 | 0.50 | 5.60 | 3.20e-4 | 4.91e-3 |
| Cnpy1 | 8.49 | 2.86 | 0.52 | 5.53 | 3.23e-8 | 1.37e-6 | 7.12 | 2.52 | 0.52 | 3.40 | 1.12e-6 | 8.81e-5 |
| Col10a1 | 2.86 | 1.63 | 0.39 | 4.22 | 2.42e-5 | 2.70e-4 | 2.87 | 1.57 | 0.45 | 4.87 | 4.40e-4 | 6.15e-3 |
| Dnali1 | 1.31 | 1.81 | 0.55 | 3.33 | 8.80e-4 | 4.74e-3 | 1.77 | 2.23 | 0.55 | 3.51 | 4.13e-5 | 1.17e-3 |
| Ehf | 2.96 | 1.90 | 0.49 | 3.90 | 9.72e-5 | 8.00e-4 | 3.18 | 1.95 | 0.50 | 4.10 | 8.19e-5 | 1.89e-3 |
| Fam216b | 1.76 | 1.83 | 0.55 | 3.34 | 8.40e-4 | 4.61e-3 | 2.40 | 2.26 | 0.50 | 3.94 | 5.44e-6 | 2.80e-4 |
| Lhx1os | 3.25 | 2.39 | 0.56 | 4.28 | 1.84e-5 | 2.20e-4 | 2.39 | 1.74 | 0.47 | 4.55 | 2.00e-4 | 3.50e-3 |
| Nts | 14.67 | 1.97 | 0.26 | 7.64 | 2.04e-14 | 1.12e-11 | 12.49 | 1.64 | 0.35 | 3.72 | 3.54e-6 | 2.10e-4 |
| Pcp2 | 2.22 | 3.29 | 0.99 | 3.33 | 8.50e-4 | 4.62e-3 | 2.91 | 3.66 | 0.97 | 3.78 | 1.50e-4 | 2.92e-3 |
| Serpina9 | 2.03 | 3.69 | 0.88 | 4.17 | 2.99e-5 | 3.20e-4 | 1.21 | 2.91 | 0.84 | 3.48 | 5.00e-4 | 6.76e-3 |
| Six3 | 3.82 | 4.51 | 1.13 | 3.99 | 6.72e-5 | 6.10e-4 | 1.55 | 3.14 | 0.78 | 4.00 | 6.28e-5 | 1.57e-3 |
| Slc6a4 | 4.48 | 2.46 | 0.38 | 6.47 | 9.90e-11 | 1.23e-8 | 3.17 | 1.99 | 0.39 | 5.06 | 4.16e-7 | 4.64e-5 |
| Tjp3 | 2.00 | 1.96 | 0.39 | 5.07 | 4.08e-7 | 1.07e-5 | 1.73 | 1.66 | 0.45 | 3.69 | 2.20e-4 | 3.72e-3 |
| Tmem212 | 1.80 | 3.64 | 0.88 | 4.18 | 3.36e-5 | 3.10e-4 | 1.40 | 3.21 | 0.82 | 3.93 | 8.43e-5 | 1.95e-3 |
| Tnnc1 | 54.70 | -1.69 | 0.33 | -5.29 | 1.18e-7 | 3.97e-6 | 57.26 | -1.54 | 0.30 | -5.17 | 2.31e-7 | 3.01e-5 |
| B. Amygdala, n=3 genes | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Control vs. STAT-Birth |
Control vs. STAT |
||||||||||
| Base mean | log2FC | log2FC-SE | Z-stats | p value | p-Adj | Base mean | log2FC | log2FC-SE | Z-stats | p- value | p-Adj | |
| Dgkk | 61.48 | 1.65 | 0.36 | 4.52 | 6.15e-6 | 1.20e-4 | 48.00 | 1.57 | 0.41 | 3.82 | 1.30e-4 | 3.08e-3 |
| Gbx1 | 2.67 | 2.11 | 0.56 | 3.73 | 1.80e-4 | 1.89e-3 | 2.19 | 2.18 | 0.61 | 3.58 | 3.40e-4 | 5.90e-3 |
| LOC102641351 | 4.45 | -1.66 | 0.29 | -5.69 | 1.24e-8 | 8.99e-7 | 3.63 | -1.78 | 0.34 | -5.25 | 1.49e-7 | 2.19e-5 |
We also performed an analysis combining the STAT and STAT-Birth group into a single “Antibiotics” group compared with the Control group (Tables S11 and S12), which revealed differential regulation of 74 frontal cortex genes and 23 amygdala genes (Figure S3; Tables S13 and S14). These included overlapping genes from the aforementioned analyses in the frontal cortex (Ak7, Ccdc121, Cnpy1, Dnali1, Ehf, Nts, Serpina9, Six3, and Slc6a4) along with notable genes including: Draxin (1.74-fold upregulated in Antibiotics); gamma-aminobutyric acid type A receptor subunit rho1 (Gabrr1) (1.44-fold upregulated in Antibiotics); interleukin 12B (Il12b) (1.52-fold downregulated in Antibiotics); interleukin-2 receptor subunit beta 2 (Il12rb2) (1.65-fold upregulated in Antibiotics); troponin C1 (Tnnc1) (1.61-fold downregulated in Antibiotics); T-Box transcription factor 22 (Tbx22) (1.51-fold downregulated in Antibiotics); and transthyretin (Ttr) (1.44-fold upregulated in Antibiotics). Similarly, overlap was observed with amygdala genes Dgkk and Gbx1 and additional notable genes including: gamma-aminobutyric acid type A receptor subunit alpha6 (Gabra6) (2.84-fold upregulated in Antibiotics); potassium voltage-gated channel subfamily E regulatory subunit 1 (Kcne1) (1.54-fold downregulated in Antibiotics); LIM homeobox 8 (Lhx8) (2.09-fold upregulated in Antibiotics); resistin-like alpha (Retnla) (2.06-fold upregulated in Antibiotics); serpin family clade A, member 3G (Serpina3g) (1.65-fold upregulated in Antibiotics); somatostatin receptor 5 (Sstr5) (1.63-fold upregulated in Antibiotics); and Ttr (3.48-fold upregulated in Antibiotics).
Differential regulation of key pathways within the frontal cortex and amygdala by early-life antibiotic exposure
To assess differences at the pathway level based on DEGs, we used the DAVID database to complete pathway enrichment, focusing on genes that were significantly differential by p value, fold change, and FDR value (Table 2). Multiple pathways related to neuronal development were identified as significantly upregulated in STAT-Birth vs. Control, including neuron differentiation (GO:0030182) which included key genes such as Six3 and Slc6a4. Similarly, within the amygdala, a key neuronal development pathway, neuron fate commitment (GO:0048663), was significantly upregulated in the STAT-Birth vs. Control group. We also performed pathway analysis combining the STAT and STAT-Birth group into a single “Antibiotics” group and compared with the control mice (Figure S4). This analysis resulted in identification of the multicellular organism development GO pathway (GO:0007275, %FDR 3.11) in the amygdala which was differentially upregulated (LHX1, NTRK1, DRAXIN, OTX2, SIX3, EOMES, ISL1, and MAB21L1 genes upregulated and BMP8A gene downregulated) in the Antibiotic group compared with the Control group.
Table 2.
Significantly differential pathways of the DEGs, by site and comparison (DAVID database: log FC > 1.5, p<0.01, and FDR<5%)∗
| A. Frontal cortex: STAT-Birth vs. Control | |||
|---|---|---|---|
| GO Term | Genes | Directionality of the genes | FDR% |
| GO:0007275~multicellular organism development | IRX3, TSHZ1, GDF6, DRAXIN, BARHL2, OTX2, SIX3, EOMES, LMX1A, EN2, ISL1, MEIS1, SFRP5, LHX1, SHISA3, NTRK1, KRT8, WNT9B, NHLH2, MAB21L1, UNCX | Upregulated | 0.01 |
| GO:0045944~positive regulation of transcription from RNA polymerase II promoter | DRD3, DRD2, BARHL1, BARHL2, OTX2, SIX3, EOMES, EHF, EN2, LMX1A, GAL, ISL1, MEIS1, GRHL2, GALR1, SP7, TFAP2D, TFAP2C, PBX3 | Upregulated | 0.09 |
| GO:0045665~negative regulation of neuron differentiation | IRX3, SLC6A4, SIX3, LMX1A, ISL1, MEIS1 | Upregulated | 0.19 |
| GO:0021516~dorsal spinal cord development | DRAXIN, PBX3, UNCX | Upregulated | 0.92 |
| GO:0006355~regulation of transcription, DNA-templated | IRX3, TSHZ1, TBX22, TBX21, IRX2, ZFP663, EHF, MEIS1, LHX1, LHX9, BARHL1, OTX2, BARHL2, SIX3, EOMES, EN2, LMX1A, ISL1, GRHL2, NHLH2, TFAP2D, SP7, ST18, TFAP2C, SP8, SP9, PBX3, UNCX | Upregulated except for TBX22 | 1.41 |
| GO:0030182~neuron differentiation | OTX2, BARHL2, WNT9B, LMX1A, EN2, ISL1 | Upregulated | 2.02 |
| GO:0030901~midbrain development | BARHL1, OTX2, LMX1A, EN2 | Upregulated | 3.20 |
| B. Amygdala: STAT-Birth vs. Control | |||
|---|---|---|---|
| GO Term | Genes | Directionality of the genes | FDR% |
| GO:0048663~neuron fate commitment | OTX2, GBX1, NKX2-1 | Upregulated | 0.96 |
| GO:0007411~axon guidance | DRAXIN, OTX2, GBX2, NKX2-1 | Upregulated | 1.10 |
| GO:0002016~regulation of blood volume by renin-angiotensin | DRD3, NKX2-1 | Upregulated | 4.77 |
All directionality is in relation to the control.
Linking CNS transcriptional alterations to changes in microbiota after early-life antibiotic exposure
We linked observed changes in CNS gene expression with differences in the abundances of specific taxa observed in the intestine (Figure 7). Normalized mean expression for each DEG (DEseq analysis) was divided into tertiles, which were used to group the relative abundance of each taxon in each sample. We identified 39 significant gene–microbiota relationships in the frontal cortex and 19 significant gene–microbiota relationships in the amygdala (Figure 7 and Tables S15–S19). Significant relationships were found with taxa in the small intestine, cecum, and colon. Both Enterococcus and Lactobacillus had multiple significant positive or negative associations with specific gene expression in the frontal cortex. Specifically, there was an increased relative abundance of colonic Lactobacillus in subjects with the highest tertile of Gm30875 and 1700019L22Rik and reduced relative abundance of colonic Lactobacillus in subjects with the highest tertile expression of Tbx22 and Gm42359 in the frontal cortex. Increased relative abundance of colonic Enterococcus was found in subjects with the highest levels of Lhx9, Lhx1, and Chst9 in the frontal cortex (Figure 7A). In the amygdala, those with the highest expression of Sstr5 also had increased relative abundance of both cecal and colonic Lactobacillus (Figures 7C and 7D). The highest tertile of Gabra6 and Cbln3 expression was associated with increased colonic Erysipelotrichaceae and Enterococcus, respectively (Figure 7D). Subjects with the highest Rnase2a and Retnla expression in the amygdala had increased Enterococcus relative abundance in the small intestine (Figure 7E).
Figure 7.

Comparison of significantly expressed genes in the frontal cortex and amygdala from both STAT and STAT-Birth conditions with microbiota identified from MaAsLin2 analysis
Normalized mean expression for each differentially expressed gene (determined by DEseq analysis) was divided into tertiles, which were used to group the relative abundance of each taxon in each sample. A pairwise Kruskal–Wallis rank-sum test was performed to compare the gene tertiles with relative abundance of each taxon identified by the MaAsLin2 analysis. Taxa with p value <0.05 were selected for the post-hoc Mann–Whitney test between the tertiles with Benjamini–Hochberg p value adjustment. Each of the taxa shown had at least one comparison with p value <0.05.
(A) Frontal cortex vs colon.
(B) Frontal cortex vs. small intestine (No significant differences were found in the comparison between frontal cortex vs. ceca).
(C) Amygdala vs. ceca.
(D) Amygdala vs. colon.
(E) Amygdala vs. small intestine.
Discussion
We and others have found changes in the microbiome as a result of the low-dose STAT (Cho et al., 2012; Cox et al., 2014; Lieber et al., 2019; Mahana et al., 2016; Schulfer et al., 2018). However, since we did not want to assume that there would be effects, we sought experimental validation. This also was the earliest time point (PND10) we have examined in our studies. Moreover, most studies of antibiotics in early life use super-pharmacological regimens, which are not particularly relevant physiologically to childhood exposures, but which cause massive changes to the microbiome.
As posited, early-life low-dose penicillin exposure altered the composition and community structure of the intestinal microbiome of PND10 mice. Specific changes in microbiota are consistent with our previous findings in the STAT model (Cho et al., 2012; Cox et al., 2014; Lieber et al., 2019; Mahana et al., 2016; Schulfer et al., 2018), but in this analysis, we focused on PND10, a key time point during the period of obligate lactation of the pups (Pantoja-Feliciano et al., 2013). Not surprisingly, we saw a diminishment in the dominant Lactobacillus species colonizing normal mice (Pantoja-Feliciano et al., 2013). Also as expected, the changes in the microbiota were more extensive in the STAT mice in which the exposure began during the gestation of the dams, ensuring that the first transferred microbiota from dam to pup was already affected, before the exposure in the STAT-Birth mice. Although differing in extent, many of the taxonomic changes were similar, which facilitated combining the groups, in addition to the individual comparisons with Control mice.
We observed significant changes in gene expression in both the frontal cortex and amygdala in mice exposed in their early life to LDP (Figures 5 and 6). Other groups have focused on select gene expression changes and/or behavioral endpoints after early antibiotic exposure in rodents (Diaz Heijtz et al., 2011; Guida et al., 2018; Leclercq et al., 2017; Minter et al., 2016; Ogawa et al., 2020; Schmidtner et al., 2019). However, before this study, little information was available on the extent of expression level changes within these brain regions which are vulnerable to perinatal insults that lead to long-lasting organizational changes in how individuals behave and react to stressors as they develop into adults.
Limitations of the study
A limitation of this study is that we were not able to assess whether the changes were due to LDP exposure directly or were due to the effects on the intestinal microbiota. That we used low-dose (sub-therapeutic) levels of penicillin, approximately 1/50th of therapeutic doses makes a direct effect less likely, but this was not confirmed experimentally. Another limitation is that the mice received the penicillin through the milk of their mother, and the effects observed in CNS expression could also be due to the penicillin effects on the mother herself, with secondary effects on the development of the pups. Although we cannot rule out that possibility, the low doses used in this study suggest this is not likely. Because of this route of differential exposure to penicillin or not, we could not randomize the pups within a litter, which is another limitation, since litter effects could bias interpretation of the findings. Finally, studying the pups at only a single time point (PND10) did not permit an assessment of the durability of the findings; this question must be examined in future studies.
In any event, the changes in gene expression involved specific DEGs, often the same gene in the independent analyses of the STAT group vs Control, and the STAT-Birth group vs. control (e.g., Slc6a4 and Dgkk), a conservation increasing confidence that the findings were robust. Genes that play significant role(s) in early-life neurodevelopment and have important signaling relevance in adulthood include Slc6a4 observed in the frontal cortex and Dgkk observed in the amygdala. Upregulation of Slc6a4 (SERT) after STAT and STAT-Birth may indicate microbiota perturbations impacting serotonergic signaling in the developing brain that could have organizational effects persisting into adulthood. Early-life serotonergic signaling system alterations also link antibiotic treatment and neurobehavioral deficits. Gut enterochromaffin cells are a major source of serotonin (Liu et al., 2008), and the blood–brain barrier (BBB) is open to peripherally produced serotonin early in life, but closes later. Increases in peripherally derived serotonin early in life alters brain development (Bonnin et al., 2011; Bonnin and Levitt, 2011) and have been associated with mood disorders and limbic system dysfunction (Gross and Hen, 2004). Transient increases in serotonin levels, induced by antibiotic effects in the microbiome, may have long-lasting effects in the brain. Our observed alteration of the expression of SERT, a key regulator of serotonin signaling in the synaptic cleft, supports this contention.
In addition to specific genes in the frontal cortex and amygdala that were altered by LDP, we identified key pathways through DAVID with specific early-life neural development functions that were significantly altered by the antibiotic exposure. GO (Gene Ontology) pathways regulating neuron differentiation, fate development, and axon guidance were altered by LDP (Table 2). The preponderance of GO pathways related to neurodevelopment that were altered by early antibiotic exposure suggests high potential for organizational change in the developing brain, especially in vulnerable regions with cognitive and emotional functions.
Importantly, our analyses linked LDP-induced changes in specific microbial taxa and differences in CNS gene expression, with key translational implications. Multiple linkages involved relative abundances of Lactobacillus species, which dominate during the obligate lactation period in mouse development (Pantoja-Feliciano et al., 2013). That differences in abundances of these critical species in early life might be directly associated and/or mechanistic drivers of a mosaic CNS including select gene expression and/or key developmental pathways is a foundational finding and requires further investigation. Conversely, Enterococcus species, enriched by LDP, emerged as an important potential actor in the gut–brain axis interaction. Whether these changes are important per se for neurodevelopment within critical brain regions or merely reflective of broader taxonomic changes remains to be determined.
In summary, these initial proof-of-concept studies establish the STAT model for investigating the effects of early-life antibiotic perturbation of the intestinal microbiome in the context of CNS development. Epidemiologic and observational studies have shown associations of early-life antibiotic exposure with neurodevelopmental disorders including ASD and ADHD (Aversa et al., 2021; Hamad et al., 2019; Lee et al., 2019; Slob et al., 2021; Wimberley et al., 2018), but these findings are only correlative, and the literature is conflicting (Hamad et al., 2018, 2019; Łukasik et al., 2019; Slob et al., 2021). Therefore, future experimental studies should be carried out in suitable antibiotic-exposure models, with extensive assessment of the microbiome, expression profiling of key brain circuits, and physiological and/or behavioral readouts to allow testing for causal inferences. Further experiments to validate these findings are needed to generalize from these data.
By analogy, these results provide evidence that early-life antibiotic exposure in humans may have effects not only on the infant microbiome but also on gene expression within critical brain structures, including the frontal cortex and amygdala, which are vulnerable to perinatal insults. These initial findings require further validation but suggest a paradigm shift in early-life antibiotic use should be considered. Although not considered a potentially severe CNS teratogen such as alcohol, cocaine, or toxoplasmosis, other (such as syphilis, varicella, mumps, parvovirus, and HIV), rubella, cytomegalovirus, herpes simplex (TORCH) pathogens (Al-Haddad et al., 2019; Bennett, 1999), early-life antibiotic use may have unexpected consequences. Our present findings should trigger re-examining widespread antibiotic prescriptions when their use is not directly indicated.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Brain tissue | This study | N/A |
| Fecal samples | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Penicillin G | Sigma, St. Louis MO | P3032 |
| Critical commercial assays | ||
| PowerSoil-htp 96-Well DNA Isolation Kit | MoBio, Carlsbad CA | Cat # 12855-50 |
| Quant-iT PicoGreen dsDNA assay kit | Life Technologies, Eugene OR | Cat # P11496 |
| Qiaquick PCR purification kit | Qiagen, Valencia CA | Cat # 28104 |
| high-sensitivity dsDNA assay kit | Life Technologies, Eugene OR | Cat # Q33120 |
| mRNAeasy Micro Kit | Qiagen, Valencia CA | Cat # 74004 |
| TruSeq RNA Sample Preparation Kit v2 | Illumina, San Diego CA | Cat # RS-122-2001 |
| Deposited data | ||
| Raw 16S rRNA and RNA-Seq data | This paper | SRA Accession #’s PRJNA731131(16S); PRJNA730313 (RNA -Seq) |
| Mouse genome | https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.20/ | GRCm38 (mm10) |
| Silva 138 | [Pruesse et al., 2007 #15858] | Release of December 2019 |
| Experimental models: Organisms | ||
| Mice | Jackson Laboratories, Bar Harbor ME | C57BL/6 mice |
| Oligonucleotides | ||
| Primers 515F/806R | Walters et al. (2015) | IDT |
| Software and algorithms | ||
| TrimGalore | bioinformatics.babraham.ac.uk | v. 0.5.0 |
| Bbmap | jgi.doe.gov/data-and-tools/bbtools/bb-tools-user- guide/bbmap-guide/ | v. 38.25 |
| Samtools | http://www.htslib.org/doc/ | v. 1.9 |
| featureCounts | [Liao et al., 2014 #15514] | Subread package v.1.6.3 |
| DESeq2 package | https://bioconductor.org/packages/release/bioc/html/DESeq2.html | v 1.22.2 |
| QIIME2 | https://qiime2.org/ | v. 2020.8 |
| DADA2 | [Callahan et al., 2016 #15857] | v. 1.16 |
| MaAsLin2 | https://huttenhower.sph.harvard.edu/maaslin/ | R package version 1.4.0 |
| GraphPadPrism® software | Graph Pad Software, San Diego CA | v. 6 |
Resource availability
Lead contact
Information and requests for resources should be directed to and will be fulfilled by the lead contact, Martin J. Blaser martin.blaser@cabm.rutgers.edu.
Materials availability
No new materials were generated in this study.
Data and code availability
The 16S rRNA and RNA-Seq FASTQ files generated during this study are available at SRA with accession numbers: SRA: PRJNA730313 (RNA-Seq) and SRA: PRJNA731131 (16S).
Animals and protocols. Mice were housed in a specific pathogen-free environment with a 12-hr light/dark cycle. C57BL/6 mice (Jackson Laboratories, Bar Harbor ME) were obtained at 6 weeks of age and acclimated to the animal facility for 1 week before initiating breeding. After a 5-day period, breeder pairs were separated, and pregnant dams were randomized into 3 treatment groups. One-third of the dams were given low-dose penicillin G (Sigma, St. Louis, MO) in their drinking water (STAT) starting during the last week of pregnancy and continuing to postnatal day 10 (PND10) as described (Cox et al., 2014; Schulfer et al., 2018). One-third of the dams were given STAT in their drinking water starting at birth and continuing to PND10 (STAT-Birth). One-third of the dams did not receive antibiotics in the drinking water and served as controls (CTR). The STAT dams received penicillin G at 1 mg/kg body weight daily, effectively the mid-range of the Food and Drug Administration (FDA)–approved dosing for use of penicillin for growth promotion in farm animals, approximately 2% of the levels used therapeutically (Cho et al., 2012; Cox et al., 2014); pups received the penicillin via their mother's milk. We used this approach to avoid handling the pups directly, which could create trauma and bias. For this preliminary study, for the pups to receive the antibiotic through their mother's milk seemed optimal under the circumstances to minimize excessive handing. Animal protocols for this study were compliant with NIH guidelines and approved by the Institutional Animal Care and Use Committee (IACUC) of the NYU Grossman School of Medicine.
Analysis of CNS transcription. Brains were removed from offspring at PND10 after cervical dislocation. For RNA-seq studies, the third male pup of each litter was used. A regional dissection containing the frontal cortex (FC) and the amygdala (A) were performed on frozen brain slabs using microforceps and a dissecting microscope using landmarks from the Allen Developing Mouse Brain Atlas (2017) and the procedure followed by Chen et al. (Chen et al., 2017). RNA was isolated from frozen FC and A samples using the mRNAeasy Micro Kit (Qiagen, Valencia, CA) according to manufacturer specifications. The Agilent 2100 Bioanalyzer (Agilent Biotechnologies, Santa Clara, CA) was used to determine RNA concentration and quality. RNA integrity number (RIN) values ranged from 9.6 to 10. Construction of the mRNA sequencing libraries and subsequent RNAseq was performed at the NYU Grossman School of Medicine Genome Technology Center (NYUGSOM GTC) Core Facility. Approximately 300 ng of total RNA was applied to the TruSeq RNA Sample Preparation Kit v2 (Illumina, San Diego, CA) per manufacturer recommendations. The quality of the libraries was assessed using Agilent bioanalysis. Libraries were applied to runs of paired-end 50-base pair sequencing on a HiSeq 4000 platform (Illumina), using the rapid run mode. A total of 23 samples were successfully used for RNA-seq (12 FC and 11 A) from four mice per condition (STAT, STAT-Birth, and CTR), with one exception. For the RNA-seq examination of the A samples, three STAT-Birth mice were studied.
FASTQ files were trimmed to a quality score of 30 with TrimGalore (v 0.5.0) (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) and then aligned to the GRCm38 (mm10) mouse genome (https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.20/) from Genome Reference Consortium with bbmap (v 38.25) [jgi.doe.gov/data-and-tools/bbtools/bb-tools-user-guide/bbmap-guide/] using the first best site and minimum alignment identity of 0.9. Samtools (v 1.9) () were used to sort and convert aligned SAM files to BAM format. Reads were quantified with the featureCounts (Liao et al., 2014) command line tool by counting only primary alignments and ignoring duplicate reads. The DESeq2 package (v 1.22.2) (Love et al., 2014) in R (v 3.5.2) was used to identify differentially expressed genes. Genes with p value <0.01 and log2-fold change of 1.5 were selected for GO enrichment analysis by utilizing DAVID (Huang et al., 2009a, 2009b) database. PCA was performed on genes separately from the amygdala and frontal cortex to find differences between groups (STAT, STAT-Birth, and Control). Mann-Whitney test was applied to principal component 1 to calculate the significance for the comparisons between the groups.
Microbiome sample collection and processing. At sacrifice, the contents of the small intestine, cecum, and colon were collected in cryopreservative tubes, then snap frozen in liquid nitrogen, and stored at - 80°C. Fecal samples were obtained from dams and sires and used to compare with the pup samples. The microbiota DNA was extracted using the PowerSoil-htp 96-Well DNA Isolation Kit (MoBio, Carlsbad, CA). The V4 region of bacterial 16S rRNA genes was amplified in triplicate reactions using barcoded fusion primers 515F/806R, which amplifies bacterial and archaeal 16S genes (Walters, 2016 #15856). The DNA concentration of the V4 amplicons for each sample was measured using the Quant-iT PicoGreen dsDNA assay kit (Life Technologies, Eugene, OR), and samples were pooled in equal quantities. These pools were treated with the Qiaquick PCR purification kit (Qiagen) to remove primers, quantified using the high-sensitivity dsDNA assay kit and the Qubit 2.0 Fluorometer (Life Technologies), and then combined at equal concentrations to form the sequencing library. The ∼254-bp V4 region was sequenced using the Ilumina MiSeq 2 × 150 bp platform at the NYUGSOM GTC.
16S rRNA sequence analysis. Quantitative insights for microbial ecology (QIIME, version Qiime 2–2020.8) was used for quality filtering and downstream analysis for α-diversity, β-diversity, and compositional analysis (https://view.qiime2.org). Sequences were filtered for quality, trimmed, de-noised, merged, and then the chimeric sequences were removed using DADA2 (Callahan et al., 2016) to generate the feature table. The Shannon index was used as a measure of the intra-individual (α-) diversity [Figure S1]. The inter-individual (β-) diversity was computed as Bray–Curtis distance matrix (Bray and Curtis, 1957), and differences in β-diversity were visualized with principal coordinates analysis (PCoA) plots [Figure S2]. Taxonomy was assigned using Silva 138 (Pruesse et al., 2007) (release of December 2019). Differential features of relative abundance of bacterial taxa were identified using the MaAsLin algorithm (https://huttenhower.sph.harvard.edu/maaslin/).
Statistical analysis for microbial data. Graphical representation of the data was obtained using GraphPadPrism software (version 6; GraphPad Software, San Diego, CA) and R. For the microbiota analysis, significant differences in α-diversity between experimental groups were determined using the Kruskal–Wallis method, while differences in β-diversity were tested by Pairwise PERMANOVA with 999 permutations. Significant differences in relative abundance were assessed using MaAsLin 2 (http://huttenhower.sph.harvard.edu/maaslin2) R package, version 1.4.0, and thresholds for significance were performed at the default setting.
Comparison between microbiome and CNS transcription. DEGs from both STAT and STAT-Birth conditions from the frontal cortex and amygdala generated with DESeq2 package were compared with microbiota from three different sites resulted from MaAsLin2 analysis in the following manner. Normalized mean expression for each differentially expressed gene was divided into tertiles, which were used to group relative abundance of each bacterial species in each sample. Pairwise Kruskal–Wallis rank-sum test was performed to compare the gene tertiles with relative abundance of each bacteria identified by previous analysis. In the correlation analysis, each tertile had an equal number of samples in it. Each RNA-seq sample represented an average of the three normalized replicates. Bacteria with a p value <0.05 were selected for the post-hoc Mann–Whitney test between the tertiles with Benjamini–Hochberg p value adjustment. The reported bacteria had at least one comparison with a p value <0.05.
Acknowledgments
The authors thank the Emch Foundation and an anonymous donor for their support. Supported by NIH grants: U01-AI22285 and P30CA016087 supporting the NYU Genome Technology Center, and AG014449, AG043375, and AG017617 (SDG).
Author contributions
Angelina Volkova conducted analyses of transcriptome and microbiome data. Kelly Ruggles conducted analyses of transcriptome and microbiome data. Anjelique Schulfer conceived the project and conducted the mouse challenge experiments, secured the specimens for analysis, and prepared the microbiome libraries for sequence analysis. Zhan Gao analyzed the microbiome findings. Stephen D. Ginsberg conceived the project and dissected the brains, prepared the libraries for sequencing, and performed the initial informatic analyses. Martin J. Blaser conceived the project and supervised all aspects of its conduct and analysis. All authors contributed to the writing of the manuscript, and reviewed and approved the final manuscript.
Declaration of interest
The authors declare no competing interests.
Published: July 14, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102797.
Contributor Information
Stephen D. Ginsberg, Email: ginsberg@nki.rfmh.org.
Martin J. Blaser, Email: martin.blaser@cabm.rutgers.edu.
Supplemental information
References
- Al-Haddad B.J.S., Oler E., Armistead B., Elsayed N.A., Weinberger D.R., Bernier R., Burd I., Kapur R., Jacobsson B., Wang C. The fetal origins of mental illness. Am. J. Obstet. Gynecol. 2019;221:549–562. doi: 10.1016/j.ajog.2019.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antshel K.M., Barkley R. Attention deficit hyperactivity disorder. Handb. Clin. Neurol. 2020;174:37–45. doi: 10.1016/B978-0-444-64148-9.00003-X. [DOI] [PubMed] [Google Scholar]
- Aversa Z., Atkinson E.J., Schafer M.J., Theiler R.N., Rocca W.A., Blaser M.J., Lebrasseur N.K. Association of infant antibiotic exposure with childhood health outcomes. Mayo Clin. Proc. 2021;96:66–77. doi: 10.1016/j.mayocp.2020.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baio J., Wiggins L., Christensen D.L., Maenner M.J., Daniels J., Warren Z., Kurzius-Spencer M., Zahorodny W., Robinson Rosenberg C., White T. Prevalence of autism spectrum disorder among children aged 8 Years - autism and developmental disabilities monitoring network, 11 sites, United States, 2014. MMWR Surveill. Summ. 2018;67:1–23. doi: 10.15585/mmwr.ss6706a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett A.D. Perinatal substance abuse and the drug-exposed neonate. Adv. Nurse Pract. 1999;7:32–36. [PubMed] [Google Scholar]
- Blaser M.J. Who are we? Indigenous microbes and the ecology of human diseases. EMBO Rep. 2006;7:956–960. doi: 10.1038/sj.embor.7400812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnin A., Levitt P. Fetal, maternal, and placental sources of serotonin and new implications for developmental programming of the brain. Neuroscience. 2011;197:1–7. doi: 10.1016/j.neuroscience.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnin A., Goeden N., Chen K., Wilson M.L., King J., Shih J.C., Blakely R.D., Deneris E.S., Levitt P. A transient placental source of serotonin for the fetal forebrain. Nature. 2011;472:347–350. doi: 10.1038/nature09972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray J.R., Curtis J.T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 1957;27:325–349. [Google Scholar]
- Breen D.P., Halliday G.M., Lang A.E. Gut-brain axis and the spread of α-synuclein pathology: vagal highway or dead end? Mov Disord. 2019;34:307–316. doi: 10.1002/mds.27556. [DOI] [PubMed] [Google Scholar]
- Callahan B.J., Mcmurdie P.J., Rosen M.J., Han A.W., Johnson A.J.A., Holmes S.P. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods. 2016;13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai G., Governale L., Mcmahon A.W., Trinidad J.P., Staffa J., Murphy D. Trends of outpatient prescription drug utilization in US children, 2002-2010. Pediatrics. 2012;130:23–31. doi: 10.1542/peds.2011-2879. [DOI] [PubMed] [Google Scholar]
- Chen V.S., Morrison J.P., Southwell M.F., Foley J.F., Bolon B., Elmore S.A. Histology Atlas of the developing prenatal and postnatal mouse central nervous system, with emphasis on prenatal days E7.5 to E18.5. Toxicol. Pathol. 2017;45:705–744. doi: 10.1177/0192623317728134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho I., Blaser M.J. The human microbiome: at the interface of health and disease. Nat. Rev. Genet. 2012;13:260–270. doi: 10.1038/nrg3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho I., Yamanishi S., Cox L., Methé B.A., Zavadil J., Li K., Gao Z., Mahana D., Raju K., Teitler I. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature. 2012;488:621–626. doi: 10.1038/nature11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coates M.E., Fuller R., Harrison G.F., Lev M., Suffolk S.F. A comparison of the growth of chicks in the Gustafsson germ-free apparatus and in a conventional environment, with and without dietary supplements of penicillin. Br. J. Nutr. 1963;17:141–150. doi: 10.1079/bjn19630015. [DOI] [PubMed] [Google Scholar]
- Cox L.M., Weiner H.L. Microbiota signaling pathways that influence neurologic disease. Neurotherapeutics. 2018;15:135–145. doi: 10.1007/s13311-017-0598-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox L.M., Yamanishi S., Sohn J., Alekseyenko A.V., Leung J.M., Cho I., Kim S.G., Li H., Gao Z., Mahana D. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell. 2014;158:705–721. doi: 10.1016/j.cell.2014.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox L.M., Abou-El-Hassan H., Maghzi A.H., Vincentini J., Weiner H.L. The sex-specific interaction of the microbiome in neurodegenerative diseases. Brain Res. 2019;1724:146385. doi: 10.1016/j.brainres.2019.146385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryan J.F., O'riordan K.J., Cowan C.S.M., Sandhu K.V., Bastiaanssen T.F.S., Boehme M., Codagnone M.G., Cussotto S., Fulling C., Golubeva A.V. The microbiota-gut-brain Axis. Physiol. Rev. 2019;99:1877–2013. doi: 10.1152/physrev.00018.2018. [DOI] [PubMed] [Google Scholar]
- Davidovitch M., Slobodin O., Weisskopf M.G., Rotem R.S. Age-specific time trends in incidence rates of autism spectrum disorder following adaptation of DSM-5 and other ASD-related regulatory changes in Israel. Autism Res. 2020;13:1893–1901. doi: 10.1002/aur.2420. [DOI] [PubMed] [Google Scholar]
- Diaz Heijtz R., Wang S., Anuar F., Qian Y., Björkholm B., Samuelsson A., Hibberd M.L., Forssberg H., Pettersson S. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. U S A. 2011;108:3047–3052. doi: 10.1073/pnas.1010529108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPaul G.J., Gormley M.J., Laracy S.D. Comorbidity of LD and ADHD: implications of DSM-5 for assessment and treatment. J. Learn. Disabil. 2013;46:43–51. doi: 10.1177/0022219412464351. [DOI] [PubMed] [Google Scholar]
- Fetissov S.O., Déchelotte P. The new link between gut-brain axis and neuropsychiatric disorders. Curr. Opin. Clin. Nutr. Metab. Care. 2011;14:477–482. doi: 10.1097/MCO.0b013e32834936e7. [DOI] [PubMed] [Google Scholar]
- Gross C., Hen R. The developmental origins of anxiety. Nat. Rev. Neurosci. 2004;5:545–552. doi: 10.1038/nrn1429. [DOI] [PubMed] [Google Scholar]
- Guida F., Turco F., Iannotta M., De Gregorio D., Palumbo I., Sarnelli G., Furiano A., Napolitano F., Boccella S., Luongo L. Antibiotic-induced microbiota perturbation causes gut endocannabinoidome changes, hippocampal neuroglial reorganization and depression in mice. Brain Behav. Immun. 2018;67:230–245. doi: 10.1016/j.bbi.2017.09.001. [DOI] [PubMed] [Google Scholar]
- Hamad A.F., Alessi-Severini S., Mahmud S.M., Brownell M., Kuo I.F. Early childhood antibiotics use and autism spectrum disorders: a population-based cohort study. Int. J. Epidemiol. 2018;47:1497–1506. doi: 10.1093/ije/dyy162. [DOI] [PubMed] [Google Scholar]
- Hamad A.F., Alessi-Severini S., Mahmud S.M., Brownell M., Kuo I.F. Prenatal antibiotics exposure and the risk of autism spectrum disorders: a population-based cohort study. PLoS One. 2019;14:e0221921. doi: 10.1371/journal.pone.0221921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz-Picciotto I., Schmidt R.J., Krakowiak P. Understanding environmental contributions to autism: causal concepts and the state of science. Autism Res. 2018;11:554–586. doi: 10.1002/aur.1938. [DOI] [PubMed] [Google Scholar]
- Hickey M.K., Miller N.C., Haapala J., Demerath E.W., Pfister K.M., Georgieff M.K., Gale C.A. Infants exposed to antibiotics after birth have altered recognition memory responses at one month of age. Pediatr. Res. 2021;89:1500–1507. doi: 10.1038/s41390-020-01117-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D.W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D.W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Hviid A., Svanström H., Frisch M. Antibiotic use and inflammatory bowel diseases in childhood. Gut. 2011;60:49–54. doi: 10.1136/gut.2010.219683. [DOI] [PubMed] [Google Scholar]
- Krebs N.F., Lozoff B., Georgieff M.K. Neurodevelopment: the impact of nutrition and inflammation during infancy in low-resource settings. Pediatrics. 2017;139:S50–S58. doi: 10.1542/peds.2016-2828G. [DOI] [PubMed] [Google Scholar]
- Leclercq S., Mian F.M., Stanisz A.M., Bindels L.B., Cambier E., Ben-Amram H., Koren O., Forsythe P., Bienenstock J. Low-dose penicillin in early life induces long-term changes in murine gut microbiota, brain cytokines and behavior. Nat. Commun. 2017;8:15062. doi: 10.1038/ncomms15062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E., Cho J., Kim K.Y. The association between autism spectrum disorder and pre- and postnatal antibiotic exposure in childhood-A systematic review with meta-analysis. Int. J. Environ. Res. Public Health. 2019;16:4042. doi: 10.3390/ijerph16204042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Lieber A.D., Beier U.H., Xiao H., Wilkins B.J., Jiao J., Li X.S., Schugar R.C., Strauch C.M., Wang Z., Brown J.M. Loss of HDAC6 alters gut microbiota and worsens obesity. FASEB J. 2019;33:1098–1109. doi: 10.1096/fj.201701586R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Yang Q., Sun W., Vogel P., Heydorn W., Yu X.Q., Hu Z., Yu W., Jonas B., Pineda R. Discovery and characterization of novel tryptophan hydroxylase inhibitors that selectively inhibit serotonin synthesis in the gastrointestinal tract. J. Pharmacol. Exp. Ther. 2008;325:47–55. doi: 10.1124/jpet.107.132670. [DOI] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahana D., Trent C.M., Kurtz Z.D., Bokulich N.A., Battaglia T., Chung J., Müller C.L., Li H., Bonneau R.A., Blaser M.J. Antibiotic perturbation of the murine gut microbiome enhances the adiposity, insulin resistance, and liver disease associated with high-fat diet. Genome Med. 2016;8:48. doi: 10.1186/s13073-016-0297-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malan-Muller S., Valles-Colomer M., Raes J., Lowry C.A., Seedat S., Hemmings S.M.J. The gut microbiome and mental health: implications for anxiety- and trauma-related disorders. Omics. 2018;22:90–107. doi: 10.1089/omi.2017.0077. [DOI] [PubMed] [Google Scholar]
- Manco M. Gut microbiota and developmental programming of the brain: from evidence in behavioral endophenotypes to novel perspective in obesity. Front. Cell. Infect. Microbiol. 2012;2:109. doi: 10.3389/fcimb.2012.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis K.G., Cryan J.F., Mayer E.A. The microbiota-gut-brain Axis: from motility to mood. Gastroenterology. 2021;160:1486–1501. doi: 10.1053/j.gastro.2020.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer E.A., Knight R., Mazmanian S.K., Cryan J.F., Tillisch K. Gut microbes and the brain: paradigm shift in neuroscience. J. Neurosci. 2014;34:15490–15496. doi: 10.1523/JNEUROSCI.3299-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minter M.R., Zhang C., Leone V., Ringus D.L., Zhang X., Oyler-Castrillo P., Musch M.W., Liao F., Ward J.F., Holtzman D.M. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 2016;6:30028. doi: 10.1038/srep30028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriceau S., Sullivan R.M. Maternal presence serves as a switch between learning fear and attraction in infancy. Nat. Neurosci. 2006;9:1004–1006. doi: 10.1038/nn1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H., Worobey M., Kuo C.-H., Ndjango J.-B.N., Peeters M., Hahn B.H., Hugenholtz P. Evolutionary relationships of wild hominids recapitulated by gut microbial communities. PLoS Biol. 2010;8:e1000546. doi: 10.1371/journal.pbio.1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa Y., Miyoshi C., Obana N., Yajima K., Hotta-Hirashima N., Ikkyu A., Kanno S., Soga T., Fukuda S., Yanagisawa M. Gut microbiota depletion by chronic antibiotic treatment alters the sleep/wake architecture and sleep EEG power spectra in mice. Sci. Rep. 2020;10:19554. doi: 10.1038/s41598-020-76562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa E. Studies on growth promotion by antibiotics. I. Effects of chlortetracycline on growth. J. Antibiot. 1955;8:205–211. [PubMed] [Google Scholar]
- Pantoja-Feliciano I.G., Clemente J.C., Costello E.K., Perez M.E., Blaser M.J., Knight R., Dominguez-Bello M.G. Biphasic assembly of the murine intestinal microbiota during early development. ISME J. 2013;7:1112–1115. doi: 10.1038/ismej.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parashar A., Udayabanu M. Gut microbiota: implications in Parkinson's disease. Parkinsonism Relat. Disord. 2017;38:1–7. doi: 10.1016/j.parkreldis.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick D.M., Sbihi H., Dai D.L.Y., Al Mamun A., Rasali D., Rose C., Marra F., Boutin R.C.T., Petersen C., Stiemsma L.T. Decreasing antibiotic use, the gut microbiota, and asthma incidence in children: evidence from population-based and prospective cohort studies. Lancet Respir. Med. 2020;8:1094–1105. doi: 10.1016/S2213-2600(20)30052-7. [DOI] [PubMed] [Google Scholar]
- Perry R.E., Blair C., Sullivan R.M. Neurobiology of infant attachment: attachment despite adversity and parental programming of emotionality. Curr. Opin. Psychol. 2017;17:1–6. doi: 10.1016/j.copsyc.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse E., Quast C., Knittel K., Fuchs B.M., Ludwig W., Peplies J., Glöckner F.O. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raineki C., Opendak M., Sarro E., Showler A., Bui K., Mcewen B.S., Wilson D.A., Sullivan R.M. During infant maltreatment, stress targets hippocampus, but stress with mother present targets amygdala and social behavior. Proc. Natl. Acad. Sci. U S A. 2019;116:22821. doi: 10.1073/pnas.1907170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson-Drummer P.A., Opendak M., Blomkvist A., Chan S., Tan S., Delmer C., Wood K., Sloan A., Jacobs L., Fine E. Infant trauma alters social buffering of threat learning: emerging role of prefrontal cortex in preadolescence. Front. Behav. Neurosci. 2019;13:132. doi: 10.3389/fnbeh.2019.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogawski E.T., Platts-Mills J.A., Seidman J.C., John S., Mahfuz M., Ulak M., Shrestha S., Soofi S.B., Yori P.P., Mduma E. Early antibiotic exposure in low-resource settings is associated with increased weight in the first two years of life. J. Pediatr. Gastroenterol. Nutr. 2017;65:350–356. doi: 10.1097/MPG.0000000000001640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtner A.K., Slattery D.A., Gläsner J., Hiergeist A., Gryksa K., Malik V.A., Hellmann-Regen J., Heuser I., Baghai T.C., Gessner A. Minocycline alters behavior, microglia and the gut microbiome in a trait-anxiety-dependent manner. Transl. Psychiatry. 2019;9:223. doi: 10.1038/s41398-019-0556-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulfer A.F., Battaglia T., Alvarez Y., Bijnens L., Ruiz V.E., Ho M., Robinson S., Ward T., Cox L.M., Rogers A.B. Intergenerational Transfer of antibiotic-perturbed microbiota enhances colitis in susceptible mice. Nat. Microbiol. 2018;3:234–242. doi: 10.1038/s41564-017-0075-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sender R., Fuchs S., Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14:e1002533. doi: 10.1371/journal.pbio.1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slob E.M., Brew B.K., Vijverberg S.J., Dijs T., Van Beijsterveldt C.E., Koppelman G.H., Bartels M., Dolan C.V., Larsson H., Lundström S. Early-life antibiotic use and risk of attention-deficit hyperactivity disorder and autism spectrum disorder: results of a discordant twin study. Int. J. Epidemiol. 2021;50:475–484. doi: 10.1093/ije/dyaa168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spor A., Koren O., Ley R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 2011;9:279–290. doi: 10.1038/nrmicro2540. [DOI] [PubMed] [Google Scholar]
- Sullivan R.M., Landers M., Yeaman B., Wilson D.A. Good memories of bad events in infancy. Nature. 2000;407:38–39. doi: 10.1038/35024156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tierney B.T., Yang Z., Luber J.M., Beaudin M., Wibowo M.C., Baek C., Mehlenbacher E., Patel C.J., Kostic A.D. The landscape of genetic content in the gut and oral human microbiome. Cell Host Microbe. 2019;26:283–295.e8. doi: 10.1016/j.chom.2019.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tottenham N., Sheridan M.A. A review of adversity, the amygdala and the hippocampus: a consideration of developmental timing. Front. Hum. Neurosci. 2010;3:68. doi: 10.3389/neuro.09.068.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trasande L., Blustein J., Liu M., Corwin E., Cox L.M., Blaser M.J. Infant antibiotic exposures and early-life body mass. Int. J. Obes. 2013;37:16–23. doi: 10.1038/ijo.2012.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremaroli V., Bäckhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489:242–249. doi: 10.1038/nature11552. [DOI] [PubMed] [Google Scholar]
- Walters W., Hyde E.R., Berg-Lyons D., Ackermann G., Humphrey G., Parada A., Gilbert J.A., Jansson J.K., Caporaso J.G., Fuhrman J.A. Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems. 2015;1 doi: 10.1128/mSystems.00009-15. e00009–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner B.B. The contribution of the gut microbiome to neurodevelopment and neuropsychiatric disorders. Pediatr. Res. 2019;85:216–224. doi: 10.1038/s41390-018-0191-9. [DOI] [PubMed] [Google Scholar]
- Wickens K., Pearce N., Crane J., Beasley R. Antibiotic use in early childhood and the development of asthma. Clin. Exp. Allergy. 1999;29:766–771. doi: 10.1046/j.1365-2222.1999.00536.x. [DOI] [PubMed] [Google Scholar]
- Wimberley T., Agerbo E., Pedersen C.B., Dalsgaard S., Horsdal H.T., Mortensen P.B., Thompson W.K., Köhler-Forsberg O., Yolken R.H. Otitis media, antibiotics, and risk of autism spectrum disorder. Autism Res. 2018;11:1432–1440. doi: 10.1002/aur.2015. [DOI] [PubMed] [Google Scholar]
- Łukasik J., Patro-Gołąb B., Horvath A., Baron R., Szajewska H. Early life exposure to antibiotics and autism spectrum disorders: a systematic review. J. Autism Dev. Disord. 2019;49:3866–3876. doi: 10.1007/s10803-019-04093-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The 16S rRNA and RNA-Seq FASTQ files generated during this study are available at SRA with accession numbers: SRA: PRJNA730313 (RNA-Seq) and SRA: PRJNA731131 (16S).
Animals and protocols. Mice were housed in a specific pathogen-free environment with a 12-hr light/dark cycle. C57BL/6 mice (Jackson Laboratories, Bar Harbor ME) were obtained at 6 weeks of age and acclimated to the animal facility for 1 week before initiating breeding. After a 5-day period, breeder pairs were separated, and pregnant dams were randomized into 3 treatment groups. One-third of the dams were given low-dose penicillin G (Sigma, St. Louis, MO) in their drinking water (STAT) starting during the last week of pregnancy and continuing to postnatal day 10 (PND10) as described (Cox et al., 2014; Schulfer et al., 2018). One-third of the dams were given STAT in their drinking water starting at birth and continuing to PND10 (STAT-Birth). One-third of the dams did not receive antibiotics in the drinking water and served as controls (CTR). The STAT dams received penicillin G at 1 mg/kg body weight daily, effectively the mid-range of the Food and Drug Administration (FDA)–approved dosing for use of penicillin for growth promotion in farm animals, approximately 2% of the levels used therapeutically (Cho et al., 2012; Cox et al., 2014); pups received the penicillin via their mother's milk. We used this approach to avoid handling the pups directly, which could create trauma and bias. For this preliminary study, for the pups to receive the antibiotic through their mother's milk seemed optimal under the circumstances to minimize excessive handing. Animal protocols for this study were compliant with NIH guidelines and approved by the Institutional Animal Care and Use Committee (IACUC) of the NYU Grossman School of Medicine.
Analysis of CNS transcription. Brains were removed from offspring at PND10 after cervical dislocation. For RNA-seq studies, the third male pup of each litter was used. A regional dissection containing the frontal cortex (FC) and the amygdala (A) were performed on frozen brain slabs using microforceps and a dissecting microscope using landmarks from the Allen Developing Mouse Brain Atlas (2017) and the procedure followed by Chen et al. (Chen et al., 2017). RNA was isolated from frozen FC and A samples using the mRNAeasy Micro Kit (Qiagen, Valencia, CA) according to manufacturer specifications. The Agilent 2100 Bioanalyzer (Agilent Biotechnologies, Santa Clara, CA) was used to determine RNA concentration and quality. RNA integrity number (RIN) values ranged from 9.6 to 10. Construction of the mRNA sequencing libraries and subsequent RNAseq was performed at the NYU Grossman School of Medicine Genome Technology Center (NYUGSOM GTC) Core Facility. Approximately 300 ng of total RNA was applied to the TruSeq RNA Sample Preparation Kit v2 (Illumina, San Diego, CA) per manufacturer recommendations. The quality of the libraries was assessed using Agilent bioanalysis. Libraries were applied to runs of paired-end 50-base pair sequencing on a HiSeq 4000 platform (Illumina), using the rapid run mode. A total of 23 samples were successfully used for RNA-seq (12 FC and 11 A) from four mice per condition (STAT, STAT-Birth, and CTR), with one exception. For the RNA-seq examination of the A samples, three STAT-Birth mice were studied.
FASTQ files were trimmed to a quality score of 30 with TrimGalore (v 0.5.0) (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) and then aligned to the GRCm38 (mm10) mouse genome (https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.20/) from Genome Reference Consortium with bbmap (v 38.25) [jgi.doe.gov/data-and-tools/bbtools/bb-tools-user-guide/bbmap-guide/] using the first best site and minimum alignment identity of 0.9. Samtools (v 1.9) () were used to sort and convert aligned SAM files to BAM format. Reads were quantified with the featureCounts (Liao et al., 2014) command line tool by counting only primary alignments and ignoring duplicate reads. The DESeq2 package (v 1.22.2) (Love et al., 2014) in R (v 3.5.2) was used to identify differentially expressed genes. Genes with p value <0.01 and log2-fold change of 1.5 were selected for GO enrichment analysis by utilizing DAVID (Huang et al., 2009a, 2009b) database. PCA was performed on genes separately from the amygdala and frontal cortex to find differences between groups (STAT, STAT-Birth, and Control). Mann-Whitney test was applied to principal component 1 to calculate the significance for the comparisons between the groups.
Microbiome sample collection and processing. At sacrifice, the contents of the small intestine, cecum, and colon were collected in cryopreservative tubes, then snap frozen in liquid nitrogen, and stored at - 80°C. Fecal samples were obtained from dams and sires and used to compare with the pup samples. The microbiota DNA was extracted using the PowerSoil-htp 96-Well DNA Isolation Kit (MoBio, Carlsbad, CA). The V4 region of bacterial 16S rRNA genes was amplified in triplicate reactions using barcoded fusion primers 515F/806R, which amplifies bacterial and archaeal 16S genes (Walters, 2016 #15856). The DNA concentration of the V4 amplicons for each sample was measured using the Quant-iT PicoGreen dsDNA assay kit (Life Technologies, Eugene, OR), and samples were pooled in equal quantities. These pools were treated with the Qiaquick PCR purification kit (Qiagen) to remove primers, quantified using the high-sensitivity dsDNA assay kit and the Qubit 2.0 Fluorometer (Life Technologies), and then combined at equal concentrations to form the sequencing library. The ∼254-bp V4 region was sequenced using the Ilumina MiSeq 2 × 150 bp platform at the NYUGSOM GTC.
16S rRNA sequence analysis. Quantitative insights for microbial ecology (QIIME, version Qiime 2–2020.8) was used for quality filtering and downstream analysis for α-diversity, β-diversity, and compositional analysis (https://view.qiime2.org). Sequences were filtered for quality, trimmed, de-noised, merged, and then the chimeric sequences were removed using DADA2 (Callahan et al., 2016) to generate the feature table. The Shannon index was used as a measure of the intra-individual (α-) diversity [Figure S1]. The inter-individual (β-) diversity was computed as Bray–Curtis distance matrix (Bray and Curtis, 1957), and differences in β-diversity were visualized with principal coordinates analysis (PCoA) plots [Figure S2]. Taxonomy was assigned using Silva 138 (Pruesse et al., 2007) (release of December 2019). Differential features of relative abundance of bacterial taxa were identified using the MaAsLin algorithm (https://huttenhower.sph.harvard.edu/maaslin/).
Statistical analysis for microbial data. Graphical representation of the data was obtained using GraphPadPrism software (version 6; GraphPad Software, San Diego, CA) and R. For the microbiota analysis, significant differences in α-diversity between experimental groups were determined using the Kruskal–Wallis method, while differences in β-diversity were tested by Pairwise PERMANOVA with 999 permutations. Significant differences in relative abundance were assessed using MaAsLin 2 (http://huttenhower.sph.harvard.edu/maaslin2) R package, version 1.4.0, and thresholds for significance were performed at the default setting.
Comparison between microbiome and CNS transcription. DEGs from both STAT and STAT-Birth conditions from the frontal cortex and amygdala generated with DESeq2 package were compared with microbiota from three different sites resulted from MaAsLin2 analysis in the following manner. Normalized mean expression for each differentially expressed gene was divided into tertiles, which were used to group relative abundance of each bacterial species in each sample. Pairwise Kruskal–Wallis rank-sum test was performed to compare the gene tertiles with relative abundance of each bacteria identified by previous analysis. In the correlation analysis, each tertile had an equal number of samples in it. Each RNA-seq sample represented an average of the three normalized replicates. Bacteria with a p value <0.05 were selected for the post-hoc Mann–Whitney test between the tertiles with Benjamini–Hochberg p value adjustment. The reported bacteria had at least one comparison with a p value <0.05.