

Abstract
Development of catalytic amide bond-forming methods is important because they could potentially address the existing limitations of classical methods using superstoichiometric activating reagents. In this paper, we disclose an Umpolung amidation reaction of carboxylic acids with nitroarenes and nitroalkanes enabled by the triplet synergistic catalysis of FeI2, P(V)/P(III) and photoredox catalysis, which avoids the production of byproducts from stoichiometric coupling reagents. A wide range of carboxylic acids, including aliphatic, aromatic and alkenyl acids participate smoothly in such reactions, generating structurally diverse amides in good yields (86 examples, up to 97% yield). This Umpolung amidation strategy opens a method to address challenging regioselectivity issues between nucleophilic functional groups, and complements the functional group compatibility of the classical amidation protocols. The synthetic robustness of the reaction is demonstrated by late-stage modification of complex molecules and gram-scale applications.
Subject terms: Synthetic chemistry methodology, Photocatalysis
Catalytic amide bond-forming methods is important because they could potentially address the existing limitations of classical methods using superstoichiometric activating reagents. Here the authors show an Umpolung amidation reaction of carboxylic acids with nitroarenes and nitroalkanes enabled by FeI2, P(V)/P(III) and photoredox catalysis that avoids the production of byproducts.
Introduction
The amide functional group is an important moiety present in a broad spectrum of biologically active compounds, synthetic materials and building blocks1,2. About 25% of natural and synthetic drugs on the market contain at least one amide3 and consequently, the development of new synthetic strategy for the construction of amide bonds is pivotal in both organic synthesis and pharmaceutical production4–6. In addition to the biosynthetic routes7, classical synthetic routes from readily available carboxylic acids generally require either reagents which can activate carboxylic acids or preparation of reactive intermediates, such as esters, anhydrides or acyl chlorides for subsequent amidation with nucleophilic amines (Fig. 1a)8–19. Nitroarenes are readily available and cheap feedstock reagents20–27 and have recently been applied with reduction in situ as amination reagents in amidation28–30. For example, Ma et al. recently achieved an elegant one-pot stoichiometric amidation protocol of carboxylic acids with nitroarenes30. Although these typical amide bond formation strategies are powerful, the development of a catalytic amidation protocol of carboxylic acids is still being actively pursued in the context of sustainable synthetic chemistry1,31 as the carboxylic acid activating reagents not only result in some harmful byproducts but also would compromise the functional group tolerance.
Fig. 1. The state-of-the-art of amidation of carboxylic acids.

a Well-established amidation protocols of carboxylic acids. b Iron/P(III)/Photoredox catalysis for Umpolung amidation. PC photocatalyst.
Recently, our group developed a photoredox, Ph3P radical cation-mediated deoxygenation of aromatic acids, generating the corresponding acyl radical which participates in a series of novel organic transformations32–34. At the same time, Doyle and others also disclosed a similar activation mode of carboxylic acids35–37. However the use of stoichiometric amounts of Ph3P will diminish the total reaction economy, leading to issues in purification, and the aliphatic carboxylic acids remain challenging substrates with Ph3P as deoxygenation reagent under photoredox conditions due to the possibility of their decarboxylation38–41. Inspired by Radosevich’s recent seminal work on the P(III)/P(V) catalytic cycle25,42–46, we questioned if Umpolung amidation of carboxylic acids is possible with electrophilic amination reagents of nitroarenes. Such a reaction could proceed by means of synergistic photoredox and R3P/R3P = O catalysis, where the generated nucleophilic acyl radical undergoes radical addition to electrophilic amination reagents as shown in Fig. 1b. This kind of amide bond formation strategy would avoid the use of nucleophilic amine reagents and address the regioselectivity issue between different amine motifs, thus improving the functional group compatibility. A significant difficulty for the Umpolung amidation originates from the fact that the reaction rates between carboxylic C-O homolysis, R3P = O and nitroarenes reduction should be matched to support the concerted catalytic cycles. Herein we report an Umpolung amidation strategy of various carboxylic acids with commercially abundant nitroarenes and nitroalkanes by means of iron/R3P/photoredox multi-cooperative catalysis.
Results
Reaction optimization
Initially, we selected n-heptanoic acid (1a) and nitrobenzene (2a) as model reactants with which to investigate the Umpolung amidation conditions. As shown in Table 1, the standard conditions include synergistic catalysis by 15 mol% FeI2, 30 mol% organophosphine (P-A) and 1 mol% PC-I in the presence of PhSiH3 as the reductant (entry 1, also see Supplementary Information for details). Under the standard conditions, the desired amide (3a) is obtained in 95% isolated yield. The use of other organophosphine precatalysts ranging from P-B to P-F significantly decreased the reaction efficiency (entries 2-6). We presumed that P-A (R3P = O) undergoes rapid reduction rates in the presence of FeI2 and silanes to generate R3P at room temperature47–49. Replacement of FeI2 with other iron-based catalysts also led to sharply decreased yields (entries 7 and 8). It was speculated that iron-based catalysts would not only accelerate the reduction of R3P = O to R3P but would also favor the reduction of nitrobenzene to nitrosobenzene under mild conditions26,50–52. Notably, the use of other silanes in place of PhSiH3 did not improve the reaction yields (entries 9 and 10, and also see Supplementary Table 3 for details). Control experiments suggested that all the factors, FeI2, organophosphine precatalyst, photocatalyst and light irradiation were important for the successful Umpolung amidation (entries 11-14). The triplet catalytic systems should work in concert to meet the total reaction rate demand. A single faster or slower catalytic cycle will mismatch the synergistic effect, thus negatively influencing the reaction.
Table 1.
Optimization of catalytic Umpolung amidation conditionsa.
| Entry | Variation of standard conditions | Yield(%)b |
|---|---|---|
| 1 | None | 95 |
| 2 | P-B instead of P-A | 40 |
| 3 | P-C instead of P-A | 30 |
| 4 | P-D instead of P-A | Trace |
| 5 | P-E instead of P-A | 8 |
| 6 | P-F instead of P-A | Trace |
| 7 | FeCl2 instead of FeI2 | 38 |
| 8 | Fe(acac)3 instead of FeI2 | Trace |
| 9 | Ph2SiH2 instead of PhSiH3 | 18 |
| 10 | Et3SiH instead of PhSiH3 | Trace |
| 11 | Without photocatalyst PC-I | ND |
| 12 | Without organocatalyst P-A | ND |
| 13 | Without FeI2 | Trace |
| 14 | Without light irradiation | ND |
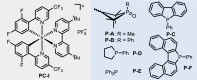 | ||
ND not detected.
aStandard conditions: 1a (0.1 mmol), 2a (0.12 mmol), PC-I (1 mol%), P-A (30 mol%), FeI2 (15 mol%), K2HPO4 (0.5 equiv), PhSiH3 (0.5 mmol), MeCN (2 ml), blue LEDs, ambient temperature, 24 h.
bIsolated yield.
Substrate scope
With the optimal conditions in hand, we explored the substrate scope of carboxylic acids. As can be seen in Fig. 2, this Umpolung amidation has a satisfactory functional group compatibility and a broad carboxylic acid substrate scope. A series of structurally diverse carboxylic acids (1a-1rr) are competent starting materials, affording the desired amides (3a-3rr) in moderate to good yields (44 examples, up to 95% yield). Primary, secondary and tertiary aliphatic acids (1a-1o, 1q-1s, 1u-1ee) all tolerate the conditions well and the protocol has excellent functional group tolerance. A great number of versatile functional groups, such as ketone (1h, 1m, 1s), iodo (1y), bromo (1gg), cyclopropanyl (1l), alkene (1p, 1r, 1u), alkyne (1w, 1ii), ester (1z), heterocycle (1o, 1dd-1ff, 1hh, 1jj-1nn) remain intact. Besides aliphatic carboxylic acids, α,β-unsaturated acids (1p and 1t) and aromatic acids (1gg-1ii, 1oo-1rr) as well as heteroaromatic acids (1ff, 1jj-1nn) are also efficient coupling partners and can be directly employed for the construction of the corresponding amides. Interestingly, when the chiral amino acids (1cc and 1dd) were subjected to this protocol, the chiral amides were obtained without racemization as determined by HPLC analysis. The use of organophosphine precatalyst (P-A) as the deoxygenation catalyst appears to complement Ph3P-mediated deoxygenative coupling, significantly expanding the carboxylic acid scope32–34.
Fig. 2. The scope of carboxylic acids.

Standard conditions: carboxylic acids 1 (0.2 mmol), 2a (1.2 equiv), PC-I (1 mol%), P-A (30 mol%), FeI2 (15 mol%), K2HPO4 (0.5 equiv), PhSiH3 (1 mmol), MeCN (4 ml), blue LEDs, ambient temperature, 24-40 h.
Subsequently, we studied the scope of the organonitro compounds (Fig. 3). A number of different nitroarenes can be successfully employed for selective formation of desired amides (3ss-3ax). Both electron-donating and withdrawing groups on the phenyl rings barely influence the reaction efficiency, delivering the desired products in yields of up to 97%. Importantly, when the nitroarenes bearing nucleophilic functional groups, such free amino (3uu), hydroxy (3al), NH-free indole (3ao), the amide bond formation between carboxylic acid and nitro groups is still predictable. These are cases which are challenging for classical amidation strategies with coupling reagents or transition metal-catalyzed aminocarbonylation53–55. There is also good functional group tolerance for nitroarenes. The aldehyde (3af), ketone (3ae), halogen (3tt, 3ac, 3ad, 3ah, 3ap), heteroarene (3an, 3ao) and Bpin (3ab) are quite compatible. The use of nitroalkanes, such as nitromethane, nitroethane, nitropropane and nitrocyclopentane can successfully result in isolation of the desired amides (3aq-3ax) in acceptable yields of 47–72%.
Fig. 3. The scope of the organonitro compounds.

Standard conditions: 1a (0.2 mmol), R-NO2 2 (0.24 mmol), PC-I (1 mol%), P-A (30 mol%), FeI2 (15 mol%), K2HPO4 (0.5 equiv), PhSiH3 (1 mmol), MeCN (4 ml), blue LEDs, ambient temperature, 24-36 h. a 2.0 equivalent nitromethane was used.
Synthetic application
To further demonstrate the synthetic robustness of Umpolung amidation, we used this amide bond formation strategy with complex molecules (Fig. 4a). The excellent functional group tolerance and high selectivity of the reaction supports a predictable amide bond formation method. Complex carboxylic acids derived from AD-Acid (3ay), ambrisentan (3az), acolen (3ba), actiprofen (3bc), acemetacin (3bd), etodolac (3be), gluconorm (3bf) and aminocyclo-propanecarboxylic acid (3bg) are produced smoothly. Complex nitroarenes can also be successfully subjected to this Umpolung amidation reaction, affording for example, the desired product (3bh) in 61% yield. Importantly, it was found that this protocol can be compatible for the late-stage modification of some peptides. Under the standard conditions, the carboxylic acid group in dipeptides of aspartame and L-alanylglycine can be employed for construction of amide bond in moderate isolated yields (3bi and 3bj). The success of these complex molecules suggests the potential application of the Umpolung amidation method in the late-stage modification of complex molecules. Furthermore, with modified standard conditions using only 0.1 mol% photocatalyst, a scaled-up experiment of 10 mmol can be conducted smoothly to afford the desired product in 72% yield (Fig. 4b).
Fig. 4. Synthetic application.

a Late-stage Umpolung amidation of complex molecules. Standard conditions for late-stage modification: carboxylic acids 1 (0.2 mmol), nitroarenes 2 (0.24 mmol), PC-I (1 mol%), P-A (30 mol%), FeI2 (15 mol%), K2HPO4 (0.5 equiv), PhSiH3 (1 mmol), MeCN (4 ml), blue LEDs, 24-60 h. b Gram-scale test with 0.1 mol% photocatalyst. Standard conditions for gram-scale experiment: n-heptanoic acid 1a (10 mmol), nitrobenzene 2a (12 mmol), PC-I (0.1 mol%), P-A (30 mol%), FeI2 (10 mol%), K2HPO4 (0.5 equiv), PhSiH3 (5 equiv), MeCN (100 ml), blue LEDs, 48 h.
Mechanistic studies
The following control experiments were performed to gain insight the mechanism of the reaction (Fig. 5). Under the standard conditions, upon addition of 2 equiv of TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl radical) into the reaction mixture, the reaction was completely inhibited and the corresponding acyl radical was trapped by TEMPO (Fig. 5a), giving a product which was identified by high resolution mass spectroscopy (HRMS). In general, with the use of nitroarenes as electrophilic reagents, there are several kinds of potential N-based intermediates27,50,52,56,57. Among the potential intermediates that were screened, we found that the use of nitrosobenzene under experimental conditions could afford the desired product (3a) in 45% yield, whereas the other electrophilic N-based intermediates such as 1,2-diphenyldiazene-1-oxide gave only a little product (Fig. 5b). This suggests that nitrosobenzene potentially is the intermediate which reacts with the nucleophilic acyl radical. The use of aniline failed to generate the product (3a) but N-phenylhydroxylamine gave 4% of the desired product (Fig. 5c). We envisioned that the photoredox conditions with [*Ir(dF(CF3)ppy)2(dtbbpy)]PF6 [1/2Ered (*IrIII/IrII) = + 1.21 V]58 would slowly oxidize N-phenylhydroxylamine to the corresponding nitrosobenzene. In addition, under FeI2 catalysis together with PhSiH3, the prepared intermediate (9), which can also be detected by HRMS in the reaction mixture, can directly be reduced to the final amide (3a). In the light of previous work32–37, a plausible mechanism was proposed and is shown in Fig. 5d. After irradiation with blue LEDs, the excited photocatalyst [*Ir(dF(CF3)ppy)2(dtbbpy)]PF6 [1/2Ered (*IrIII/IrII) = + 1.21 V]58 causes a single electron oxidation of electron-rich R3P (4), which can be formed by reduction in situ from the precatalyst R3P = O (P-A) in the presence of PhSiH3 and FeI2, generating the corresponding phosphine radical cation species (5). The Stern-Volmer quenching experiments demonstrated that the photoexcited [*Ir(dF(CF3)ppy)2(dtbbpy)]PF6 could be quenched by R3P rather than R3P = O (P-A), n-heptanoic acid (1a) or nitrobenzene (2a). Subsequently, this species (5) can recombine with the carboxylate anion to furnish the radical intermediate (6). Owing to the high affinity of P- and O-atoms, intermediate (6) prefers to undergo β-scission to generate nucleophilic acyl radical (7) and complete the organophosphine catalytic cycle. Under the reductive reaction conditions with FeI2, the nitrobenzene (2a) tends to form nitrosobenzene. Once the concentration of nitrosobenzene is relatively high, rapid nucleophilic acyl radical addition to nitrosobenzene occurs readily to give rise to N-centered radical intermediate (8). Donation of one electron to the Ir(II)-species would generate intermediate (9), completing the photoredox cycle. Finally, our control experiment showed that reduction of intermediate (9) with FeI2/PhSiH3 to amide (3a) at ambient temperature was almost quantitative. Less than 10% of amide (3a) was obtained under the identical conditions without the addition of catalytic amount of FeI2.
Fig. 5. Mechanistic studies and proposed mechanism.

a Radical inhibition experiment. b Potential N-based intermediate. c Reduction of 9 to product 3a. d Proposed mechanism.
Discussion
We have developed an unprecedented synergistic catalysis system of iron/P(V-III)/photoredox catalysis for Umpolung amidation of carboxylic acids and nitroarenes or nitroalkanes. A wide range of commercially abundant and inexpensive carboxylic acids and nitroarenes are competent coupling partners in this direct amidation, affording a rich library of structurally diverse amides in yields of up to 97%. This amidation strategy surpasses the classical amide bond-forming method via carboxylic acid activation and subsequent amidation with nucleophilic amines, thus creating promising synthetic robustness especially when the substrates already have several sensitive functional groups or competing nucleophilic substituents. This protocol is readily scaled-up with 0.1 mol% photocatalyst for preparative systems. The excellent functional group compatibility and reaction selectivity render it useful in future peptide modification and drug discovery.
Methods
General procedure for amidation
To an 8 mL transparent vial equipped with a stirring bar, P-A (10.4 mg, 30 mol%), PC-I (2.2 mg, 1 mol%), FeI2 (9.3 mg, 15 mol%), K2HPO4 (17.4 mg, 0.1 mmol) were added successively. Then the vial was carried into glovebox which was equipped with nitrogen. Then MeCN (4.0 ml), PhSiH3 (1 mmol), carboxylic acids 1 (0.2 mmol) and nitroarenes 2 (0.24 mmol) were added in sequence under N2 atmosphere. The reaction mixture was stirred under the irradiation of 45 W blue LEDs (distance app. 10.0 cm from the bulb) at ambient temperature for 24–60 h. When the reaction finished, the mixture was quenched with water and extracted with ethyl acetate (3 × 10 mL). The organic layers were combined and concentrated under vacuo. The product was purified by flash column chromatography on silica gel (eluent: n-hexane: ethyl acetate).
Supplementary information
Acknowledgements
We thank the National Natural Science Foundation of China (21971108, 21971111, 21732003), the Natural Science Foundation of Jiangsu Province (Grant No. BK20190006), Fundamental Research Funds for the Central Universities (0205/14380252), “Innovation & Entrepreneurship Talents Plan” of Jiangsu Province, and Foundation of Advanced Catalytic Engineering Research Center of the Ministry of Education of Hunan University. Shan Gao, Yubo Pang, Jian Han, Xiaopeng Wu are warmly acknowledged to reproduce experimental procedures for products (3a, 3nn, 3oo and 3hh). Dedicated to Prof. Christian Bruneau for his outstanding contribution to catalysis.
Author contributions
J.X. and Y.N. conceived and designed the project. Y.N. and S.W. performed and analyzed the experimental data. J.X. wrote the manuscript with input from all authors and discussed the manuscript with M.L., J.H. and C.Z.
Data availability
We declare that all other data supporting the findings of this study are available within the article and Supplementary Information files, and also are available from the corresponding author upon reasonable request. The X-ray crystallographic data of product 3tt in this study has been deposited in the Cambridge Crystallographic Data Centre under accession code CCDC 2055473.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-021-24908-w.
References
- 1.Sabatini MT, Boulton LT, Sneddon HF, Sheppard TD. A green chemistry perspective on catalytic amide bond formation. Nat. Catal. 2019;2:10–17. doi: 10.1038/s41929-018-0211-5. [DOI] [Google Scholar]
- 2.Wang X. Challenges and outlook for catalytic direct amidation reactions. Nat. Catal. 2019;2:98–102. doi: 10.1038/s41929-018-0215-1. [DOI] [Google Scholar]
- 3.Roughley SD, Jordan AM. The medicinal chemist’s toolbox: An analysis of reactions used in the pursuit of drug candidates. J. Med. Chem. 2011;54:3451–3479. doi: 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]
- 4.Carey JS, et al. Analysis of the reactions used for the preparation of drug candidate molecules. Org. Biomol. Chem. 2006;4:2337–2347. doi: 10.1039/b602413k. [DOI] [PubMed] [Google Scholar]
- 5.Narendar Reddy T, Beatriz A, Jayathirtha Rao V, de Lima DP. Carbonyl compounds’ journey to amide bond formation. Chem. Asian J. 2019;14:344–388. doi: 10.1002/asia.201801560. [DOI] [PubMed] [Google Scholar]
- 6.Massolo, E., Pirola, M. & Benaglia, M. Amide bond formation strategies: latest advances on a dateless transformation. Eur. J. Org. Chem. 30, 4641–4651 (2020).
- 7.Dorr BM, Fuerst DE. Enzymatic amidation for industrial applications. Curr. Opin. Chem. Bio. 2018;43:127–133. doi: 10.1016/j.cbpa.2018.01.008. [DOI] [PubMed] [Google Scholar]
- 8.Valeur E, Bradley M. Amide bond formation: beyond the myth of coupling reagents. Chem. Soc. Rev. 2009;38:606–631. doi: 10.1039/B701677H. [DOI] [PubMed] [Google Scholar]
- 9.Hu L, et al. Ynamides as racemization-free coupling reagents for amide and peptide synthesis. J. Am. Chem. Soc. 2016;138:13135–13138. doi: 10.1021/jacs.6b07230. [DOI] [PubMed] [Google Scholar]
- 10.Dunetz JR, Magano J, Weisenburger GA. Large-scale applications of amide coupling reagents for the synthesis of pharmaceuticals. Org. Process Res. Dev. 2016;20:140–177. doi: 10.1021/op500305s. [DOI] [Google Scholar]
- 11.Braddock DC, et al. Tetramethyl orthosilicate (TMOS) as a reagent for direct amidation of carboxylic acids. Org. Lett. 2018;20:950–953. doi: 10.1021/acs.orglett.7b03841. [DOI] [PubMed] [Google Scholar]
- 12.Sayes M, Charette AB. Diphenylsilane as a coupling reagent for amide bond formation. Green. Chem. 2017;19:5060–5064. doi: 10.1039/C7GC02643A. [DOI] [Google Scholar]
- 13.Allen CL, Williams JMJ. Metal-catalysed approaches to amide bond formation. Chem. Soc. Rev. 2011;40:3405–3415. doi: 10.1039/c0cs00196a. [DOI] [PubMed] [Google Scholar]
- 14.Li G, Ji C, Hong X, Szostak M. Highly chemoselective, transition-metal-free transamidation of unactivated amides and direct amidation of alkyl esters by N-C/O-C cleavage. J. Am. Chem. Soc. 2019;141:11161–11172. doi: 10.1021/jacs.9b04136. [DOI] [PubMed] [Google Scholar]
- 15.Ishihara K, Ohara S, Yamamoto H. 3,4,5-Trifluorobenzeneboronic acid as an extremely active amidation catalyst. J. Org. Chem. 1996;61:4196–4197. doi: 10.1021/jo9606564. [DOI] [PubMed] [Google Scholar]
- 16.Sawant DN, et al. Diboron-catalyzed dehydrative amidation of aromatic carboxylic acids with amines. Org. Lett. 2018;20:4397–4400. doi: 10.1021/acs.orglett.8b01480. [DOI] [PubMed] [Google Scholar]
- 17.Du Y, et al. A solid-supported arylboronic acid catalyst for direct amidation. Chem. Commun. 2019;55:2916–2919. doi: 10.1039/C8CC09913H. [DOI] [PubMed] [Google Scholar]
- 18.Movahed FS, et al. Tris(o-phenylenedioxy)cyclotriphosphazene as a promoter for the formation of amide bonds between aromatic acids and amines. Synthesis. 2020;52:3253–3262. doi: 10.1055/s-0040-1707174. [DOI] [Google Scholar]
- 19.Ramachandran PV, Hamann HJ. Ammonia-borane as a catalyst for the direct amidation of carboxylic acids. Org. Lett. 2021;23:2938–2942. doi: 10.1021/acs.orglett.1c00591. [DOI] [PubMed] [Google Scholar]
- 20.Gao Y, Yang S, Huo Y, Hu X‐Q. Recent progress on reductive coupling of nitroarenes by using organosilanes as convenient reductants. Adv. Synth. Catal. 2020;362:3971–3986. doi: 10.1002/adsc.202000370. [DOI] [Google Scholar]
- 21.Li, G. et al. Light-promoted C-N coupling of aryl halides with nitroarenes. Angew. Chem. Int. Ed.60, 5230–5234 (2021). [DOI] [PubMed]
- 22.Cheung CW, Ploeger ML, Hu X. Direct amidation of esters with nitroarenes. Nat. Commun. 2017;8:14878. doi: 10.1038/ncomms14878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gui J, et al. Practical olefin hydroamination with nitroarenes. Science. 2015;348:886–891. doi: 10.1126/science.aab0245. [DOI] [PubMed] [Google Scholar]
- 24.Rauser M, Ascheberg C, Niggemann M. Electrophilic amination with nitroarenes. Angew. Chem. Int. Ed. 2017;56:11570–11574. doi: 10.1002/anie.201705356. [DOI] [PubMed] [Google Scholar]
- 25.Li G, et al. An improved P(III)/P(V)=O catalyzed reductive C-N coupling of nitroaromatics and boronic acids by mechanistic differentiation of rate- and product-determining steps. J. Am. Chem. Soc. 2020;142:6786–6799. doi: 10.1021/jacs.0c01666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song H, Yang Z, Tung C-H, Wang W. Iron-catalyzed reductive coupling of nitroarenes with olefins: intermediate of iron–nitroso complex. ACS Catal. 2019;10:276–281. doi: 10.1021/acscatal.9b03604. [DOI] [Google Scholar]
- 27.Xiao J, He Y, Ye F, Zhu S. Remote sp3 C–H amination of alkenes with nitroarenes. Chem. 2018;4:1645–1657. doi: 10.1016/j.chempr.2018.04.008. [DOI] [Google Scholar]
- 28.Lee K, Kim J, Kim J. One pot conversion of nitroarenes into N-arylamides. Bull. Korean Chem. Soc. 2002;23:1359–1360. doi: 10.5012/bkcs.2002.23.10.1359. [DOI] [Google Scholar]
- 29.Kumar V, Kumar M, Sharma S, Kumar N. Highly selective direct reductive amidation of nitroarenes with carboxylic acids using cobalt(II) phthalocyanine/PMHS. RSC Adv. 2014;4:11826–11830. doi: 10.1039/c3ra46619a. [DOI] [Google Scholar]
- 30.Wang S, Cheung CW, Ma J. Direct amidation of carboxylic acids with nitroarenes. J. Org. Chem. 2019;84:13922–13934. doi: 10.1021/acs.joc.9b02068. [DOI] [PubMed] [Google Scholar]
- 31.Todorovic M, Perrin DM. Recent developments in catalytic amide bond formation. Pept. Sci. 2020;112:e24210. doi: 10.1002/pep2.24210. [DOI] [Google Scholar]
- 32.Zhang M, Xie J, Zhu C. A general deoxygenation approach for synthesis of ketones from aromatic carboxylic acids and alkenes. Nat. Commun. 2018;9:3517. doi: 10.1038/s41467-018-06019-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruzi R, Liu K, Zhu C, Xie J. Upgrading ketone synthesis direct from carboxylic acids and organohalides. Nat. Commun. 2020;11:3312. doi: 10.1038/s41467-020-17224-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang M, Yuan X-A, Zhu C, Xie J. Deoxygenative deuteration of carboxylic acids with D2O. Angew. Chem. Int. Ed. 2019;58:312–316. doi: 10.1002/anie.201811522. [DOI] [PubMed] [Google Scholar]
- 35.Martinez Alvarado JI, Ertel AB, Stegner A, Stache EE, Doyle AG. Direct use of carboxylic acids in the photocatalytic hydroacylation of styrenes to generate dialkyl ketones. Org. Lett. 2019;21:9940–9944. doi: 10.1021/acs.orglett.9b03871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stache EE, Ertel AB, Tomislav R, Doyle AG. Generation of phosphoranyl radicals via photoredox catalysis enables voltage-independent activation of strong C-O bonds. ACS Catal. 2018;8:11134–11139. doi: 10.1021/acscatal.8b03592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang L, et al. Reductive C–C coupling by desulfurizing gold-catalyzed photoreactions. ACS Catal. 2019;9:6118–6123. doi: 10.1021/acscatal.9b01368. [DOI] [Google Scholar]
- 38.Zhou Q, et al. Decarboxylative alkynylation and carbonylative alkynylation of carboxylic acids enabled by visible-light photoredox catalysis. Angew. Chem. Int. Ed. 2015;54:11196–11199. doi: 10.1002/anie.201504559. [DOI] [PubMed] [Google Scholar]
- 39.Patra T, Mukherjee S, Ma J, Strieth-Kalthoff F, Glorius F. Visible-light-photosensitized aryl and alkyl decarboxylative functionalization reactions. Angew. Chem. Int. Ed. 2019;58:10514–10520. doi: 10.1002/anie.201904671. [DOI] [PubMed] [Google Scholar]
- 40.Shibatomi K, et al. Enantioselective decarboxylative chlorination of beta-ketocarboxylic acids. Nat. Commun. 2017;8:15600. doi: 10.1038/ncomms15600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liang Y, Zhang X, MacMillan DWC. Decarboxylative sp3 C–N coupling via dual copper and photoredox catalysis. Nature. 2018;559:83–88. doi: 10.1038/s41586-018-0234-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li G, Grotenhuis C, Radosevich AT. Reductive Csp2–N coupling by PIII/PV=O catalysis. Trends Chem. 2021;3:72–73. doi: 10.1016/j.trechm.2020.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lecomte M, Lipshultz JM, Kim-Lee SH, Li G, Radosevich AT. Driving recursive dehydration by P(III)/P(V) catalysis: Annulation of amines and carboxylic acids by sequential C-N and C-C bond formation. J. Am. Chem. Soc. 2019;141:12507–12512. doi: 10.1021/jacs.9b06277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nykaza TV, et al. Intermolecular reductive C-N cross coupling of nitroarenes and boronic acids by PIII/PV=O catalysis. J. Am. Chem. Soc. 2018;140:15200–15205. doi: 10.1021/jacs.8b10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li G, Qin Z, Radosevich AT. P(III)/P(V)-catalyzed methylamination of arylboronic acids and esters: reductive C–N coupling with nitromethane as a methylamine surrogate. J. Am. Chem. Soc. 2020;142:16205–16210. doi: 10.1021/jacs.0c08035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nykaza TV, Li G, Yang J, Luzung MR, Radosevich AT. PIII/PV=O catalyzed cascade synthesis of N‐functionalized azaheterocycles. Angew. Chem. Int. Ed. 2020;59:4505–4510. doi: 10.1002/anie.201914851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O’Brien CJ, et al. Recycling the waste: the development of a catalytic Wittig reaction. Angew. Chem. Int. Ed. 2009;48:6836–6839. doi: 10.1002/anie.200902525. [DOI] [PubMed] [Google Scholar]
- 48.Marsi KL. Phenylsilane reduction of phosphine oxides with complete stereospecificity. J. Org. Chem. 1974;39:265–267. doi: 10.1021/jo00916a041. [DOI] [Google Scholar]
- 49.Kirk AM, O’Brien CJ, Krenske EH. Why do silanes reduce electron-rich phosphine oxides faster than electron-poor phosphine oxides? Chem. Commun. 2020;56:1227–1230. doi: 10.1039/C9CC08718D. [DOI] [PubMed] [Google Scholar]
- 50.Junge K, Wendt B, Shaikh N, Beller M. Iron-catalyzed selective reduction of nitroarenes to anilines using organosilanes. Chem. Commun. 2010;46:1769–1771. doi: 10.1039/b924228g. [DOI] [PubMed] [Google Scholar]
- 51.Lo JC, Yabe Y, Baran PS. A practical and catalytic reductive olefin coupling. J. Am. Chem. Soc. 2014;136:1304–1307. doi: 10.1021/ja4117632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu K, Shaver MP, Thomas SP. Chemoselective nitro reduction and hydroamination using a single iron catalyst. Chem. Sci. 2016;7:3031–3035. doi: 10.1039/C5SC04471E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang L, Neumann H, Beller M. Palladium-catalyzed methylation of nitroarenes with methanol. Angew. Chem. Int. Ed. 2019;58:5417–5421. doi: 10.1002/anie.201814146. [DOI] [PubMed] [Google Scholar]
- 54.Zheng Y-L, Newman SG. Methyl esters as cross-coupling electrophiles: Direct synthesis of amide bonds. ACS Catal. 2019;9:4426–4433. doi: 10.1021/acscatal.9b00884. [DOI] [Google Scholar]
- 55.Yuan Y, et al. Copper-catalyzed carbonylative hydroamidation of styrenes to branched amides. Angew. Chem. Int. Ed. 2020;59:22441–22445. doi: 10.1002/anie.202010509. [DOI] [PubMed] [Google Scholar]
- 56.Cheung CW, Leendert Ploeger M, Hu X. Amide synthesis via nickel-catalysed reductive aminocarbonylation of aryl halides with nitroarenes. Chem. Sci. 2018;9:655–659. doi: 10.1039/C7SC03950F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang X-J, Chen B, Zheng L-Q, Wu L-Z, Tung C-H. Highly efficient and selective photocatalytic hydrogenation of functionalized nitrobenzenes. Green. Chem. 2014;16:1082–1086. doi: 10.1039/C3GC42042F. [DOI] [Google Scholar]
- 58.Prier CK, Rankic DA, MacMillan DWC. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We declare that all other data supporting the findings of this study are available within the article and Supplementary Information files, and also are available from the corresponding author upon reasonable request. The X-ray crystallographic data of product 3tt in this study has been deposited in the Cambridge Crystallographic Data Centre under accession code CCDC 2055473.