

Abstract
Interferons (IFNs) are a broad class of cytokines that have multifaceted roles. Type I IFNs have variable effects when it comes to host susceptibility to bacterial infections, that is, the resulting outcomes can either be protective or deleterious. The mechanisms identified to-date have been wide and varied between pathogens. In this review, we discuss recent literature that provides new insights into the mechanisms of how type I IFN signaling exerts its effects on the outcome to infection from the host’s point of view.
Keywords: bacteria, infection, type I IFN, type I interferons, interferon, IFN
Introduction
Interferons (IFNs) are a broad class of pleiotropic cytokines elicited upon encounter of the innate immune system with pathogens. These innate immune mediators owe their name to the initial observation that they could “interfere” with viral replication [1]. Although they were originally identified for their antiviral properties, it is now recognized that they also play a multitude of roles in cancer, autoimmunity and can modulate infection with a range of other microorganisms including parasites, fungi and bacteria [2–7]. This review will focus on the role of type I IFNs in bacterial infection, with a focus on recent studies that have defined their impact on infection resolution.
There are three classes of interferons type I, type II and type III. In humans, the type I interferon (IFN) family includes IFN-α (13 subtypes), IFN-β, IFN-ε, IFN-κ and IFN-ω subtypes. In mice, 14 IFN-α subtypes have been identified along with individual IFN-β, IFN-ε, IFN-κ and IFN-ζ subtypes [8]. IFN-α and IFN-β are the best characterized and most broadly expressed genes of this family. All type I IFNs interact with a single heterodimeric receptor composed of two subunits, IFNAR1 and IFNAR2. This receptor is ubiquitously expressed and binding of its ligand signals through the JAK-STAT pathway and interferon regulatory factors (IRFs) that induces expression of hundreds of interferon stimulated genes (ISGs), as well as autocrine and paracrine signaling [9] (Figure 1).
Figure 1: Type I IFN signaling in the context of bacterial infection.

Type I IFNs are induced when bacteria are recognized by PRR, including nucleotide binding leucine rich repeat proteins such as NOD-1 and NOD-2 (NLR), RIG-I like receptors (RLR) and toll-like receptors (TLR) and cyclic GMP-AMP Synthase (cGAS). PRR sensing activates the transcription factors of the interferon regulatory factors (IRF) family, which, with NF-κB, stimulate the expression of type I IFNs, depicted here with IFN-β. IFN-β is then secreted and binds to the IFNAR receptor which signals through the JAK-STAT pathway. The phosphorylated forms of STAT-1 and STAT-2 and the interferon regulatory factors form a transcription factor complex that translocates to the nucleus where it induces the expression of hundreds of interferon stimulated genes (ISGs). IFN-β is also produced, allowing a positive feedback loop and paracrine signaling.
Type I IFNs are induced through extracellular and intracellular pattern recognition receptors (PRR) of the innate immune system. These PRR mediate the recognition of specific motifs found on pathogens, called pathogen-associated molecular patterns (PAMPs). PAMPs are comprised of structural components of the bacterial cell wall such as lipopolysaccharides (LPS), lipoproteins, peptidoglycan fragments and flagellin subunits. Other microbial components such as nucleic acids (DNA and RNA) can also be sensed by these receptors [10]. Several families of PRR have been identified in mammals. They include Toll-like receptors (TLRs) which are the primary sensors of extracellular bacteria, nucleotide binding leucine rich repeat (NLRs) proteins that detect cytosolic bacteria, RIG-I like receptors (RLR) that detect short RNA sequences in the cytosol and the DNA sensors also found in this subcellular compartment [10]. Although activation of most of these PRR leads to the expression of proinflammatory and antibacterial genes, only a subset of them have been linked with type I IFN production during bacterial infection. In the TLR family, TLR2, TLR4, TLR7/8, TLR9 and TLR13 have been shown to stimulate a type I IFN response after binding of their respective ligands [11–16]. TLR3 has been shown to stimulate the production of IFN-β after sensing of commensal bacteria, but not in the context of bacterial infections [17]. In the NLR family, recognition of bacterial peptides by the nucleotide-binding and oligomerizing domain (NOD) like receptors, NOD1 and NOD2 also elicits type I IFN production [18, 19]. cGAS-dependent and independent stimulation of STING has also been linked with type I IFN induction via sensing of intracellular DNA [12, 20–22]. Finally, the cytosolic sensor RIG-I has been linked to the induction of type I IFN production via sensing of bacterial RNA [23, 24]. The transcription factors IRFs, in particular IRF1, IRF3, IRF5 and IRF7, together with NF-κB are subsequently activated and lead to expression of type I IFNs [14, 19, 25].
The roles of type I IFNs in bacterial infections
Because of the multifaceted roles of these cytokines, the effect of type I IFN signaling on host susceptibility to bacterial infections are diverse. Many factors can influence this response and the infection outcome. These can be intrinsic to the bacterium and its capacity to activate different PRR, its replication programs, virulence factor expression and immune evasion strategies. The types of cells that encounter the pathogen, the target organs, and its cellular lifestyle all can influence this response. In many cases (Table. 1) a differential effect is seen based on the route of infection. As there are several reviews on the ability of bacteria to activate type I IFN signaling [26, 27], here, we summarize existing data (Table. 1) and discuss below, recent literature that provides new insights into the mechanisms of how type I IFN signaling exerts its effects on the outcome to infection from the host point of view (Figure. 2).
Table 1:
Impact of type I IFN signaling on the outcome of bacterial infections
| Bacterium | Known IFN signaling receptors | Mechanism | Outcome | Impact of type I IFN signaling1 | Reference |
|---|---|---|---|---|---|
| Acinetobacter baumannii | TRIF |
|
|
Host protection | [34] |
| Brucella abortus | cGAS, STING, IRF5 |
|
|
Differential effects between studies | [21, 32] |
| Chlamydia muridarum | STING, IRF3 |
|
|
Detrimental | [76–78] |
| Coxiella burnetii | - |
|
Differential effects | [69] | |
| Escherichia coli | - |
|
Host protection | [30] | |
| Escherichia coli- viral |
|
|
Detrimental effect | [79] | |
| Francisella novicida | cGAS, STING, IRF3/7 |
|
|
Detrimental | [37] |
| Francisella tularensis | IRF3 |
|
|
Detrimental | [80] |
| Haemophilus influenza (nontypeable) | cGAS, STING, TBK1 |
|
Detrimental | [57, 81] | |
| Helicobacter pylori | NOD1, IRF7 |
|
Host protection | [18] | |
| Legionella pneumophila | STING, IRF3 |
|
Host protection | [82, 83] | |
| Listeria monocytogenes | RIG-I, STING, TLR2, TRIF |
|
|
Dual effects | [23, 24, 40–43, 70, 80, 84–89] |
| Mycobacterium abscessus | TLR2, TLR4, MyD88, TRIF, IRF3 |
|
|
In vitro: Host protection | [11] |
| Mycobacterium bovis | - |
|
|
Detrimental | [55] |
| Mycobacterium smegmatis | cGAS, STING, TBK1, IRF3/7 |
|
Detrimental | [20] | |
| Mycobacterium tuberculosis | Early phase: cGAS, STING, TBK1, IRF3 Late phase: RIG-I, MAVS, TBK1, IRF7 |
|
|
In vivo: Detrimental In vitro: Host protection | [53, 54, 90–93] |
| Neisseria gonorrhoeae | cGAS, STING, TLR4, TRIF, IRF3 |
|
In vitro: detrimental | [12] | |
| Pseudomonas aeruginosa | TLR4, TRIF, MD2, TBK1 |
|
|
Dual effects | [73, 74, 94] |
| Pseudomonas aeruginosa-viral |
|
|
Detrimental | [64, 79] | |
| Rickettsia parkeri | cGAS, IRF5 |
|
|
Host protection | [31] |
| Salmonella enterica serovar Typhimurium | TLR3, TLR4, TRIF |
|
|
Dual effects | [70, 71, 95, 96] |
| Salmonella Typhimurium-viral |
|
|
Detrimental | [97] | |
| Staphylococcus aureus | TLR9, NOD2, MyD88, IRF1, IRF5, cGAS, STING |
|
|
Dual effects | [14, 33, 43, 45, 46, 51, 98] |
| Staphylococcus aureus-viral |
|
|
Detrimental | [47, 79] | |
| Streptococcus agalactiae | cGAS, STING |
|
|
Host protection | [30, 99] |
| Streptococcus pneumoniae | STING, TBK1, IRF3 |
|
|
Host protection | [29, 58–60] |
| S. pneumoniae-viral |
|
|
Detrimental | [43, 61–68] | |
| Streptococcus pyogenes | STING, TBK1, MyD88, IRF3, IRF5 |
|
|
Host protection | [15, 28, 36] |
| Yersinia enterocolitica | TLR4, TRIF |
|
|
Host protection | [100] |
| Yersinia pestis | TLR7 |
|
Dual effects | [101, 102] |
Abbreviations: ARDS: acute respiratory distress syndrome, c-di-GMP: cyclic dimeric guanosine monophosphate, STING: stimulator of interferon genes, NTHi: nontypeable Hemophilus influenzae, sRNAs: noncoding small RNAs. RIG-I: retinoic acid inducible gene I, RBP: RNA-Binding protein, ROS: reactive oxygen species, iNOS-inducible nitric oxide species
Impact is based on in vivo data unless otherwise stated
Figure 2: Recently described effects of type I IFN signaling on clearance during bacterial infections.
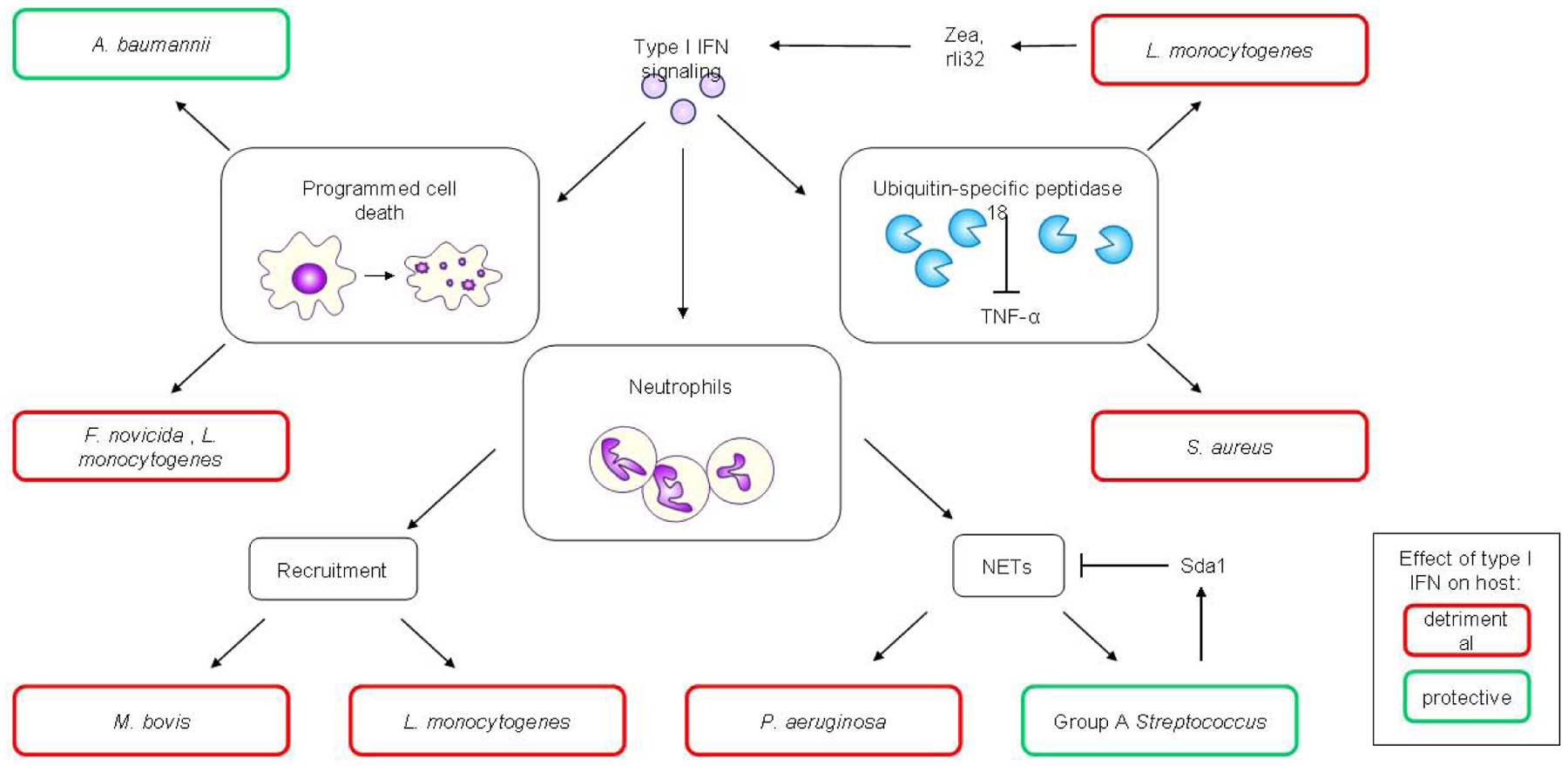
Type I IFN signaling induces different cell death programs, which in the context of A.baumannii infection is beneficial for the host as it aided bacterial clearance. However, in the context of F. novicida and L. monocytogenes infections, type I IFN-mediated cell death is detrimental to the host. Type I IFN signaling exerts different effects on neutrophils: in the context of M. bovis infection, increased neutrophil recruitment is observed and is detrimental to the host. In the context of L.monocytogenes infection, neutrophil recruitment is increased and is detrimental to the host, while during P .aeruginosa infection, type I IFN-mediated production of neutrophil extracellular traps (NETs) facilitiates biofilm production and bacterial persistence, making this process detrimental to the host. However, in the context of Group A Streptococcus (GAS) infection, type I IFN-mediated NET production aids in bacterial clearance, making type I IFN a protective host factor. GAS produces the bacterial DNase, SdaI that degrades NETs, allowing the bacteria to evade this immune response. Finally, type I IFN signaling induces the production of USP18, an ISG able to inhibit the antibacterial effects of TNF-α. This contributes to the detrimental effects of infections with S. aureus and L. monocytogenes. The latter is also able to produces factors such as small noncoding RNAs and RNA binding proteins that stimulate type I IFN production, reinforcing its detrimental effects.
1-. Protective effects of type I IFNs
Any aspect of the immune system is typically viewed as serving a critical role against invading organisms. However, sometimes this activation disrupts the balance of controlling infection and maintaining a harmonious state. Until recently, most studies exploring the mechanism of type I IFN-mediated protection have largely converged on neutrophils and the ability of type I IFN to positively regulate their antimicrobial production, such as reactive nitrogen species [20, 28–31] [32, 33]. In recent studies, two unique mechanisms whereby type I IFN can be beneficial to the host are discussed.
In a pulmonary infection model of Acinetobacter baumannii infection, Ifnar−/− and Irf3−/− /Irf7−/− mice, in which IFN-β signaling and production is impaired, exhibited significantly higher bacterial burdens in their lung and spleen compared to WT mice [34]. Type I IFN signaling also initiated cell death, via activation of apoptosis, necroptosis and pyroptosis. This was evident by activation of the NLRP3 inflammasome and caspase-11. A. baumanni-induced type I IFN was shown to generate epigenetic modifications (H3K27ac marks) at the promoters of these key programmed cell death mediators. While it was recently shown that interferon exposure can induce an immunological innate memory [35], this study is the first to implicate bacteria in inducing a type I IFN mediated epigenetic change to influence host outcome.
Two recent studies were able to demonstrate that bacterial proteins can directly suppress the type I IFN response to reduce the beneficial effects afforded to the host. Group A Streptococcus (GAS) produces a DNase, Sda1, involved in protection against neutrophil extracellular traps, which can also impair TLR9-mediated type I IFN production. Mice infected with a GAS sda1 mutant, produced higher type I IFN levels, which reduced bacterial numbers and lesion sizes [36]. While a phenotype was not observed in IFNAR knockout mice, this is presumably due to the active suppression mediated by Sda1. The obligate cytosolic human pathogen Rickettsia parkeri is also sensitive to type-I IFN-mediated killing. Increased mortality and bacterial burdens are observed when both type I and II interferon receptors are inactivated. While a specific factor is yet to be identified, it has been demonstrated to reduce type I IFN production in macrophages by antagonizing the inflammasome [31].
2-. Detrimental effects of type I IFN on the host
There have been several examples in the literature whereby type I IFNs sensitize cells to apoptosis and recently this was shown to be evident with Francisella novicida [37]. Inactivation of either component of the IFNAR receptor led to increased survival after infection with F. novicida infection. Likewise, inactivation of the downstream IRF, Irf3 (as well Irf3/Irf7 double knockout mice) also had a protective phenotype. While previous reports have suggested that type I IFN can regulate the AIM2 inflammasome [38], which helps in infection protection, it was still functional in the mice lacking IFNAR. Type I IFN was shown to exert its negative effects through enhancing apoptosis, as shown in liver tissue with increased active caspases 3, 7 and 8. The TNF-related apoptosis inducing ligand (TRAIL) is upregulated by type I IFNs [37] and binding to its receptor, DR5, triggers apoptosis [39]. This was shown to be the likely mechanism in this case, as neutralization of TRAIL aided in mouse survival [37].
One of the first bacterial pathogens to be identified as activating a detrimental type I IFN host immune response was Listeria monocytogenes [40, 41]. Several different mechanisms have been documented to explain this including: suppression of the Th17 response, sensitizing cells to apoptosis, T cell death, enhanced IL-10 production, decreased neutrophil recruitment and promotion of actin-based motility (Table. 1). Most of these studies have been conducted in systemic models of infection. The exception to this rule to-date has been a study investigating oral infection (intragastric inoculation) as a model for foodborne contamination with L. monocytogenes that didn’t see a change in outcome in WT versus Ifnar−/− mice [42]. Focusing back on sepsis, recent studies have been able to elucidate molecular mechanisms behind this response as well as bacterial products to manipulate this response to their advantage.
Two different bacterial products of L. monocytogenes have been shown to help facilitate infection by activating type I IFN signaling. Frantz et al [24] identified several small RNAs (sRNAs) that could induce type I IFNs. One of these sRNAs that induced the highest levels of IFN-β was rli32. It induced IFN-β via RIG-I and indicative of this strong type I IFN response, was able to inhibit influenza virus replication. rli32 was shown to promote intracellular survival of L. monocytogenes in a type-I IFN-dependent manner and aided in resistance to hydrogen peroxide [24]. A second L. monocytogenes product, this time the RNA binding protein, Zea, also leads to enhanced type I IFN signaling, mediated through RIG-I. Zea is able to bind to several RNAs that accumulate in the extracellular medium, potentiating type I IFN production. Inactivation of Zea attenuates virulence [23].
Ubiquitin-specific peptidase 18 (USP18) was identified as an interferon stimulated gene (ISG) increased in macrophages and dendritic cells after L. monocytogenes stimulation. USP18 was primarily responsible for the deleterious effects of type IFN signaling during mice infection with L. monocytogenes [43]. Likewise, in the context of superinfection with acute lymphocytic choriomeningitis virus (LCMV), LCMV enhanced L. monocytogenes persistence in a type I IFN- dependent manner via CD11c+ cells. CD11c+ cells were identified as the cause, as inactivation of Ifnar or Usp18 in CD11c+ cells lead to reduced bacterial titers in multiple organs as well as increased survival rates. USP18 is known to prevent TNF-α signaling by targeting TAK1 and NEMO for deubiquitination [44]. This proved to be the mechanism behind the phenotype with USP18, by inhibiting TNF-α production it promoted bacterial replication. This observation did not prove to be unique to L. monocytogenes. Respiratory infection with Staphylococcus aureus, yielded similar observations that were dependent upon signaling through CD11c+ cells and USP18 [43].
S. aureus also benefits from the activation of type I IFN signaling [14, 45, 46]. This detrimental impact of type I IFN on the host is further exacerbated with antecedent viral infection. Influenza decreases IL-17, IL-22 and IL-23, which are important for S. aureus clearance [47]. It was also observed that mice lacking STAT2 (downstream of IFNAR) exhibited increased susceptibility to influenza infection but decreased lethality and improved bacterial clearance upon super-infection with methicillin resistant S. aureus [48]. This mechanism could be explained by a compensatory effect of type II IFN driving the induction of M1-polarized macrophages. In the study mentioned above for L. monocytogenes [43], it was also shown that CD11c+ DCs appear to be integral mediators in the negative response to S. aureus, as inactivation of Ifnar in these cells confers an improved outcome. Specific deletion of the ISG USP18, which can regulate type I IFN signaling, in CD11c+ dendritic cells also led to significant reductions in bacteria. It remains to be determined how this protein negatively impacts bacterial clearance. Several studies have shown in the context pneumonia that type I IFN signaling benefits S. aureus infection [14, 43, 45, 46]. As an example that mice can vary their phenotype between suppliers, facilities and housing conditions, a recent study using a neutralizing antibody observed a protective role for type I IFN with S. aureus [33]. In this case, type I IFN was observed to enhance granzyme production in neutrophils and thus facilitate bacterial killing. This study would concur with Kaplan et al [49], which observed direct antibacterial killing by IFN-β. However, this study also saw direct killing against L. monocytogenes that, as described above, benefits from type I IFN signaling. The discrepancy between these studies maybe true when examining differences between in vitro and in vivo but could be due to the specific strains studied as well.
We were able to recently demonstrate significant diversity within S. aureus in its ability to activate the production of type I IFNs. It had been assumed that within a given species activation was somewhat conserved. We identified two strains with divergent activation [14, 46] before screening dozens of S. aureus isolates. We identified a broad range of IFN-β activation potential with vancomycin intermediate strains generating reduced amounts of IFN-β. These low levels of type I IFN induction correlated with increased resistance to autolysis and lysostaphin in vitro. This is probably as a result of the thickened cell wall seen in vancomycin intermediate strains [50], protecting the bacterial cells from endosomal processing and release of PAMPS to receptors to signal. In an in vivo model of acute pneumonia, we observed that an S. aureus strain with reduced type I IFN induction ability to be more readily cleared than a strain with higher IFN-β induction propensity, however whether this was solely due to their differences in type I IFN induction needs to be further investigated [51].
In the context of mycobacterial infections, type I IFN response has been associated with pathogenesis [52, 53]. The detrimental phenotype of type I IFN to Mycobacterium does not necessarily hold true in vitro. Type I IFN signaling enhanced the intrinsic capability of macrophages to effectively clear the M. tuberculosis and M. abscessus by inducing nitric oxide production [11, 54]. M. tuberculosis was also shown to inhibit autocrine type I IFN signaling (by 50–60%) via reduced phosphorylation of the IFNAR-associated protein kinases JAK1 and TYK2, leading to reduced phosphorylation of STAT1 and STAT2 [54]. Suggesting that the type I IFN response could be detrimental to the pathogen but a good example of how in vitro does not always correlate to in vivo. Murine models with the bovine turbercule bacilli, Mycobacterium bovis are protected against infection when IFNAR is neutralized [55]. Both cellular and immune signaling differences were noted. A reduction in neutrophil recruitment was observed in vivo along with reduced IL-10, IL-6 and increased in IFN-γ and IL-1β. In vitro, macrophages treated with αIFNAR1 induced decreased levels of M2 markers such as Arg1, Ym1 and Mrc1 and increased expression of M1 markers such as Nos2, and Ifng suggesting that type I IFN signaling mediates macrophage polarization toward an anti-inflammatory profile during M. bovis infection [55]. Another study found that macrophages deficient in either IFNAR or STAT exhibited increased viability compared to WT cells after infection with M.tuberculosis [56]. The authors also observed that IFNAR antibody blockade increased the protective effects of rifampin, a first-line tuberculosis drug.
Type I IFN-mediated effects on macrophage function were also observed with nontypeable Haemophilus influenzae (NTHi). WT macrophages pre-treated with IFN-β showed impaired phagocytosis and bacterial killing, while Ifnar−/− macrophages had increased phagocytic and killing abilities compared to WT cells. In vivo infection corroborated these results, Ifnar−/− mice showing reduced susceptibility to NTHi infection and reduced weight loss. Likewise, in a COPD model, animals treated with IFN-β and NTHi fared worse compared to controls. Type I IFNs also induced enhanced proinflammatory signaling through MAP kinase activation [57].
3-. Dual effects of type I IFN signaling on infection outcomes
We have summarized (Table. 1) and discussed so far, several examples where contrary phenotypes exist. This tends to occur when different routes of infection are studied, further highlighting what is beneficial for one organ can be detrimental to another. A good example of this binary phenotype is Streptococcus pneumoniae. Type I IFN signaling has been shown to be important for protection against pneumococcal infection however, with preceding influenza infection, this creates a more susceptible environment that is propagated by type I IFN signaling [29, 43, 58–68] (Table. 1). Where mechanisms are known, this further the illuminates the pleiotropic effects type I IFN signaling can exert on the host. In the context of respiratory tract infection with Coxiella burnetii, the dual effect of type I IFN signaling has also been documented [69]. Inactivating IFNAR led to reduced bacterial burdens and better weight retention. When WT mice received an injection of recombinant IFN-α, disease-induced weight loss was exacerbated, suggesting that type I IFN signaling is deleterious. However, when mice received recombinant IFN- α intratracheally, bacterial replication was decreased in all tissues. A reduction in IL-1β expression was observed in the lung of mice that received recombinant IFN-α intraperitoneally, thus inflammatory cytokine dampening could be responsible for this, tissue specific, dual phenotype [69].
A reduction in cell death improved the outcome in Ifnar−/− mice infected with Salmonella enterica Serovar Typhimurium [70, 71] and recent work showed that the absence of STAT2- dependent type I IFN signaling led to decreased reactive oxygen species (ROS) production by neutrophils and disruption of hypoxia in the intestinal epithelium, resulting in respiration inhibition of S. Typhimurium and impaired luminal expansion [72]. Suggesting that type I IFN signaling is beneficial for the bacterium, in this context. However, a unique study recently examined the influence of pregnancy on the outcome to infection with L. monocytogenes and S. Typhimurium in the presence and absence of type I IFN. While pregnancy did not influence the detrimental outcome conferred by type I IFNs in L. monocytogenes infection, the protection afforded in Ifnar−/− mice to S. Typhimurium infection was lost in pregnant mice. The compromised outcome in pregnant mice to Salmonella was attributed to decreased production of several cytokines including IFN-γ, TNF, MCP-1 and IL-12 [70].
A further recent example in which type I IFN signaling appears to have dual effects was with Pseudomonas aeruginosa. In a murine two-hit infection model to reproduce sepsis-related acute respiratory distress syndrome (ARDS) consisting of cecal ligation and puncture (CLP)- mediated peritoneal sepsis followed by respiratory tract infection with Pseudomonas aeruginosa, IFN-β production was beneficial to the host [73]. IFN-β administration reversed the suppressive effects of prior sepsis on the functions of alveolar macrophages, improving their phagocytosis and increasing CXCL1-mediated neutrophil recruitment. Lung bacterial burdens were reduced, mouse survival was improved and sepsis-related ARDS reduced [73]. IFN-β administration after CLP but before pneumonia also reduced mortality, lung bacterial burden and lung injury score [73]. This contrasts to a mono-infection acute lung injury model of P. aeruginosa infection. In this model, type I IFN led to activation of neutrophils which mediated tissue damage and also supported biofilm formation and tissue persistence by P. aeruginosa [74]. Mouse knockouts in both Ifnar1 and Ifnb1 exhibited lower lung colonization of P. aeruginosa and reduced tissue damage compared to WT controls. Type I IFN-deficient neutrophils were found to be impaired in their ability to produce and release long neutrophil extracellular traps (NETs) and ROS. Upon infection with P. aeruginosa, NETs were found to support bacterial biofilm formation and thereby to promote persistence of the pathogen in the lung by protecting it from the immune system. The direct effect of IFN-β on NETosis and biofilm formation was also demonstrated [74]. These examples of dual effects highlight how the model and infection site as well as immune status can alter the outcome, while antimicrobial products, such as reactive oxygen species can both support and repress bacterial clearance depending upon the pathogen.
Concluding remarks
The studies summarized here illustrate the complex interactions of type I IFN signaling with the immune system in the context of bacterial infections. These cytokines can have drastically different effects on the host, ranging from deleterious to beneficial. The specific reasons behind these phenotypes are still poorly understood (see Outstanding Questions). The type of bacterial pathogen and their mode of infection can account for some of these differences. However, other examples show that the context of infection (different tissues, cell types and many other host factors such as pregnancy and prior exposure to heterologous pathogens) can be as crucial as the bacterial species in determining the outcome of infection. Some progress has been made with the discovery of specific bacterial factors (noncoding RNA and RNA-binding proteins) that can directly modulate IFN expression. The identification of ISGs that contribute to the deleterious effects of type I IFN signaling in bacterial infections is a further step towards characterizing these responses, but many questions remain (see Outstanding Questions). Furthermore, the entirety of the studies discussed in this review focused on the effect of IFN-α and β on host response to bacterial infections. With the exception of one publication demonstrating the protective role of IFN-ε against Chlamydia muridarum-induced sexually transmitted infection [75], very little is known about the effects of other type I IFNs on bacterial infection outcomes. Future research that aims to define the conditions in which type I IFN signaling leads to enhanced susceptibility or protection to bacterial infections would be most informative.
Outstanding Questions Box.
Why can type I IFN activation in the same site be beneficial to one microbe but detrimental to another?
How does the route of infection impact type I IFN induction and infection outcome?
How does the magnitude and duration of type I IFN induction impact infection outcome?
What controls the magnitude of type I IFN induction between strains?
How similar is the signaling for type I IFN to the related type III IFN pathway?
Identification of bacterial factors that can manipulate this pathway.
Identification of specific ISGs that influence bacterial clearance.
What are the cells influenced by type I IFN from species-to-species?
What specific factors determine the protective and detrimental effects of type I IFN signaling on the host during bacterial infections?
What impact does epigenetics play in the type I IFN response and does it impact subsequent infections?
Due to the diverse roles of type I interferons in bacterial infections, we are left without any unifying theme that could, based on niche, infection site, genus or species, predict pathogen susceptibility to type I IFN. This is exemplified by the observations that even with the same pathogen, there are examples of dual effects of type I IFN on the outcome to infection. We have observed that within the same species, a diversity of induction can occur strain-to-strain. The level of induction evoked by each specific strain, the duration of this induction and the location of infection may all dictate the outcome to infection. This leaves us with the question of what the true role of type I IFN is. It might have evolved as an antiviral pathway and has adapted additional innate sensors to respond to bacterial products; however, its role is truly variable from pathogen-to-pathogen. Based on the data to-date, it would be presumptive to assume any given species or strain would behave the same way as a standard laboratory strain analyzed. It is very clear that there are significant differences in the ability to induce type I IFN and the infection outcome between different species, strains and sites of infection. Likewise, the ability to activate this pathway is not a one size fits all system that again varies considerably between species and within species. It is unlikely in the short term that we will come to a unified conclusion on what specific factors and events lead to a positive or negative outcome in regards to type I IFN activation. It will not be until we have a significant body of work that investigates a single infection site with different species that might activate type I IFN through the same receptors we will get closer to this point. But given the complexity already observed amongst different pathogens and strains, this would require some very large labor intensive experiments. What is clear, is that type I IFN signaling can have a major impact on the outcome to bacterial infections, the outcome of which can be both positive and detrimental to the host.
Highlights box.
Type I IFN signaling can be detrimental or beneficial to the host during bacterial infections and this varies between species and by infection site
Bacterial factors can directly modulate type I IFN signaling and its downstream effects
Significant diversity is seen between strains of the same species to activate this response
Type I IFN can cause epigenetic changes that aid in cell death for bacterial clearance but can also sensitize cells to apoptosis
Expression of USP18 in CD11c+ cells suppresses antibacterial production of TNF
Type I IFNs can manipulate neutrophil recruitment for the benefit and detriment of the host, while their products can aid in bacterial persistence
Acknowledgements
We apologize to the authors whose works were not included in this review as we have focused on protection mechanisms and discussion of recent works. Work in the laboratory is funded by NIH grant R01HL134870 to DP.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Isaacs A and Lindenmann J (2015) Pillars Article: Virus Interference. I. The Interferon.Proc R Soc Lond B Biol Sci. 1957. 147: 258–267. The Journal of Immunology 195 (5), 1911. [PubMed] [Google Scholar]
- 2.Yu X et al. (2016) Cross-Regulation of Two Type I Interferon Signaling Pathways in Plasmacytoid Dendritic Cells Controls Anti-malaria Immunity and Host Mortality. Immunity 45 (5), 1093–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parker D (2014) Bacterial activation of type I interferons, Springer. [Google Scholar]
- 4.Zitvogel L et al. (2015) Type I interferons in anticancer immunity. Nat Rev Immunol 15 (7), 405–14. [DOI] [PubMed] [Google Scholar]
- 5.Chessler AD et al. (2011) Type I interferons increase host susceptibility to Trypanosoma cruzi infection. Infect Immun 79 (5), 2112–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biondo C et al. (2008) IFN-alpha/beta signaling is required for polarization of cytokine responses toward a protective type 1 pattern during experimental cryptococcosis. J Immunol 181 (1), 566–73. [DOI] [PubMed] [Google Scholar]
- 7.Crow MK and Ronnblom L (2019) Type I interferons in host defence and inflammatory diseases. Lupus Sci Med 6 (1), e000336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krause CD and Pestka S (2015) Cut, copy, move, delete: The study of human interferon genes reveal multiple mechanisms underlying their evolution in amniotes. Cytokine 76 (2), 480–495. [DOI] [PubMed] [Google Scholar]
- 9.Schreiber G (2017) The molecular basis for differential type I interferon signaling. J Biol Chem 292 (18), 7285–7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Odendall C and Kagan JC (2019) Host-Encoded Sensors of Bacteria: Our Windows into the Microbial World. Microbiol Spectr 7 (3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruangkiattikul N et al. (2019) Type I interferon induced by TLR2-TLR4-MyD88-TRIF-IRF3 controls Mycobacterium abscessus subsp. abscessus persistence in murine macrophages via nitric oxide. Int J Med Microbiol 309 (5), 307–318. [DOI] [PubMed] [Google Scholar]
- 12.Andrade WA et al. (2016) Type I Interferon Induction by Neisseria gonorrhoeae: Dual Requirement of Cyclic GMP-AMP Synthase and Toll-like Receptor 4. Cell Rep 15 (11), 2438–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mancuso G et al. (2009) Bacterial recognition by TLR7 in the lysosomes of conventional dendritic cells. Nat Immunol 10 (6), 587–94. [DOI] [PubMed] [Google Scholar]
- 14.Parker D and Prince A (2012) Staphylococcus aureus induces type I IFN signaling in dendritic cells via TLR9. J Immunol 189 (8), 4040–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castiglia V et al. (2016) Type I Interferon Signaling Prevents IL-1beta-Driven Lethal Systemic Hyperinflammation during Invasive Bacterial Infection of Soft Tissue. Cell Host Microbe 19 (3), 375–87. [DOI] [PubMed] [Google Scholar]
- 16.Moen SH et al. (2019) Human Toll-like Receptor 8 (TLR8) Is an Important Sensor of Pyogenic Bacteria, and Is Attenuated by Cell Surface TLR Signaling. Front Immunol 10, 1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawashima T et al. (2013) Double-stranded RNA of intestinal commensal but not pathogenic bacteria triggers production of protective interferon-beta. Immunity 38 (6), 1187–97. [DOI] [PubMed] [Google Scholar]
- 18.Watanabe T et al. (2010) NOD1 contributes to mouse host defense against Helicobacter pylori via induction of type I IFN and activation of the ISGF3 signaling pathway. J Clin Invest 120 (5), 1645–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pandey AK et al. (2009) NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS Pathog 5 (7), e1000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruangkiattikul N et al. (2017) cGAS-STING-TBK1-IRF3/7 induced interferon-beta contributes to the clearing of non tuberculous mycobacterial infection in mice. Virulence 8 (7), 1303–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Costa Franco MM et al. (2018) Brucella abortus Triggers a cGAS-Independent STING Pathway To Induce Host Protection That Involves Guanylate-Binding Proteins and Inflammasome Activation. J Immunol 200 (2), 607–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lienard J et al. (2020) The Mycobacterium marinum ESX-1 system mediates phagosomal permeabilization and type I interferon production via separable mechanisms. Proc Natl Acad Sci U S A 117 (2), 1160–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pagliuso A et al. (2019) An RNA-Binding Protein Secreted by a Bacterial Pathogen Modulates RIG-I Signaling. Cell Host Microbe 26 (6), 823–835 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frantz R et al. (2019) The secRNome of Listeria monocytogenes Harbors Small Noncoding RNAs That Are Potent Inducers of Beta Interferon. mBio 10 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Honda K et al. (2006) Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25 (3), 349–60. [DOI] [PubMed] [Google Scholar]
- 26.Kovarik P et al. (2016) Type I Interferons in Bacterial Infections: A Balancing Act. Front Immunol 7, 652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boxx GM and Cheng G (2016) The Roles of Type I Interferon in Bacterial Infection. Cell Host Microbe 19 (6), 760–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gratz N et al. (2011) Type I interferon production induced by Streptococcus pyogenes-derived nucleic acids is required for host protection. PLoS Pathog 7 (5), e1001345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Damjanovic D et al. (2014) Type 1 interferon gene transfer enhances host defense against pulmonary Streptococcus pneumoniae infection via activating innate leukocytes. Mol Ther Methods Clin Dev 1, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mancuso G et al. (2007) Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol 178 (5), 3126–33. [DOI] [PubMed] [Google Scholar]
- 31.Burke TP et al. (2020) Inflammasome-mediated antagonism of type I interferon enhances Rickettsia pathogenesis. Nature Microbiology 5 (5), 688–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Almeida LA et al. (2011) MyD88 and STING signaling pathways are required for IRF3-mediated IFN-beta induction in response to Brucella abortus infection. PLoS One 6 (8), e23135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spolski R et al. (2019) IL-21/type I interferon interplay regulates neutrophil-dependent innate immune responses to Staphylococcus aureus. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Y et al. (2018) Type I IFN operates pyroptosis and necroptosis during multidrug-resistant A. baumannii infection. Cell Death Differ 25 (7), 1304–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kamada R et al. (2018) Interferon stimulation creates chromatin marks and establishes transcriptional memory. Proc Natl Acad Sci U S A 115 (39), E9162–e9171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keller N et al. (2019) Group A Streptococcal DNase Sda1 Impairs Plasmacytoid Dendritic Cells’ Type 1 Interferon Response. J Invest Dermatol 139 (6), 1284–1293. [DOI] [PubMed] [Google Scholar]
- 37.Zhu Q et al. (2018) Detrimental Type I Interferon Signaling Dominates Protective AIM2 Inflammasome Responses during Francisella novicida Infection. Cell Rep 22 (12), 3168–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fernandes-Alnemri T et al. (2010) The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol 11 (5), 385–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shlyakhtina Y et al. (2017) Dual role of DR5 in death and survival signaling leads to TRAIL resistance in cancer cells. Cell Death Dis 8 (8), e3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Auerbuch V et al. (2004) Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med 200 (4), 527–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carrero JA et al. (2004) Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J Exp Med 200 (4), 535–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pitts MG et al. (2016) Type I IFN Does Not Promote Susceptibility to Foodborne Listeria monocytogenes. J Immunol 196 (7), 3109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shaabani N et al. (2018) The probacterial effect of type I interferon signaling requires its own negative regulator USP18. Science immunology 3 (27), eaau2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang Z et al. (2015) USP18 negatively regulates NF-kappaB signaling by targeting TAK1 and NEMO for deubiquitination through distinct mechanisms. Sci Rep 5, 12738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martin FJ et al. (2009) Staphylococcus aureus activates type I IFN signaling in mice and humans through the Xr repeated sequences of protein A. J Clin Invest 119 (7), 1931–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parker D et al. (2014) Induction of type I interferon signaling determines the relative pathogenicity of Staphylococcus aureus strains. PLoS Pathog 10 (2), e1003951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robinson KM et al. (2015) The role of IL-27 in susceptibility to post-influenza Staphylococcus aureus pneumonia. Respir Res 16, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gopal R et al. (2018) STAT2 Signaling Regulates Macrophage Phenotype During Influenza and Bacterial Super-Infection. Front Immunol 9, 2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaplan A et al. (2017) Direct Antimicrobial Activity of IFN-β. Journal of immunology (Baltimore, Md. : 1950) 198 (10), 4036–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Howden BP et al. (2014) The evolution of vancomycin intermediate Staphylococcus aureus (VISA) and heterogenous-VISA. Infect Genet Evol 21, 575–82. [DOI] [PubMed] [Google Scholar]
- 51.Peignier A et al. (2020) Differential Induction of Type I and III Interferons by Staphylococcus aureus. Infect Immun 88 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Manca C et al. (2001) Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha /beta. Proc Natl Acad Sci U S A 98 (10), 5752–5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stanley SA et al. (2007) The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J Immunol 178 (5), 3143–52. [DOI] [PubMed] [Google Scholar]
- 54.Banks DA et al. (2019) Mycobacterium tuberculosis Inhibits Autocrine Type I IFN Signaling to Increase Intracellular Survival. J Immunol 202 (8), 2348–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang J et al. (2019) Inhibition of type I interferon signaling abrogates early Mycobacterium bovis infection. BMC Infect Dis 19 (1), 1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang L et al. (2021) Type I interferon signaling mediates Mycobacterium tuberculosis-induced macrophage death. J Exp Med 218 (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang S et al. (2019) Type I interferon induced by DNA of nontypeable Haemophilus influenza modulates inflammatory cytokine profile to promote susceptibility to this bacterium. Int Immunopharmacol 74, 105710. [DOI] [PubMed] [Google Scholar]
- 58.Parker D et al. (2011) Streptococcus pneumoniae DNA initiates type I interferon signaling in the respiratory tract. mBio 2 (3), e00016–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maier BB et al. (2016) Type I interferon promotes alveolar epithelial type II cell survival during pulmonary Streptococcus pneumoniae infection and sterile lung injury in mice. Eur J Immunol 46 (9), 2175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.LeMessurier KS et al. (2013) Type I interferon protects against pneumococcal invasive disease by inhibiting bacterial transmigration across the lung. PLoS Pathog 9 (11), e1003727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shahangian A et al. (2009) Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest 119 (7), 1910–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shirey KA et al. (2019) Influenza “Trains” the Host for Enhanced Susceptibility to Secondary Bacterial Infection. mBio 10 (3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cao J et al. (2014) Activation of IL-27 signalling promotes development of postinfluenza pneumococcal pneumonia. EMBO Mol Med 6 (1), 120–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Merches K et al. (2015) Virus-Induced Type I Interferon Deteriorates Control of Systemic Pseudomonas aeruginosa Infection. Cell Physiol Biochem 36 (6), 2379–92. [DOI] [PubMed] [Google Scholar]
- 65.Li W et al. (2012) Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of gammadelta T cells. J Virol 86 (22), 12304–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nakamura S et al. (2011) Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. J Clin Invest 121 (9), 3657–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shepardson KM et al. (2016) Differential Type I Interferon Signaling Is a Master Regulator of Susceptibility to Postinfluenza Bacterial Superinfection. mBio 7 (3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Berg J et al. (2017) Tyk2 as a target for immune regulation in human viral/bacterial pneumonia. Eur Respir J 50 (1). [DOI] [PubMed] [Google Scholar]
- 69.Hedges JF et al. (2016) Type I Interferon Counters or Promotes Coxiella burnetii Replication Dependent on Tissue. Infect Immun 84 (6), 1815–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Agbayani G et al. (2019) Type I interferons differentially modulate maternal host immunity to infection by Listeria monocytogenes and Salmonella enterica serovar Typhimuriumduring pregnancy. Am J Reprod Immunol 81 (1), e13068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Robinson N et al. (2012) Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol 13 (10), 954–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wilson RP et al. (2019) STAT2 dependent Type I Interferon response promotes dysbiosis and luminal expansion of the enteric pathogen Salmonella Typhimurium. PLoS Pathog 15 (4), e1007745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hiruma T et al. (2018) IFN-beta Improves Sepsis-related Alveolar Macrophage Dysfunction and Postseptic Acute Respiratory Distress Syndrome-related Mortality. Am J Respir Cell Mol Biol 59 (1), 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pylaeva E et al. (2019) Detrimental Effect of Type I IFNs During Acute Lung Infection With Pseudomonas aeruginosa Is Mediated Through the Stimulation of Neutrophil NETosis. Front Immunol 10, 2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fung KY et al. (2013) Interferon-epsilon protects the female reproductive tract from viral and bacterial infection. Science 339 (6123), 1088–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nagarajan UM et al. (2008) Type I interferon signaling exacerbates Chlamydia muridarum genital infection in a murine model. Infect Immun 76 (10), 4642–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Qiu H et al. (2008) Type I IFNs enhance susceptibility to Chlamydia muridarum lung infection by enhancing apoptosis of local macrophages. J Immunol 181 (3), 2092–102. [DOI] [PubMed] [Google Scholar]
- 78.Prantner D et al. (2010) Stimulator of IFN gene is critical for induction of IFN-beta duringChlamydia muridarum infection. J Immunol 184 (5), 2551–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee B et al. (2015) Influenza-induced type I interferon enhances susceptibility to gram-negative and gram-positive bacterial pneumonia in mice. Am J Physiol Lung Cell Mol Physiol 309 (2), L158–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Henry T et al. (2010) Type I IFN signaling constrains IL-17A/F secretion by gammadelta T cells during bacterial infections. J Immunol 184 (7), 3755–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lu C et al. (2018) Nontypeable Haemophilus influenzae DNA stimulates type I interferon expression via STING signaling pathway. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1865 (4), 665–673. [DOI] [PubMed] [Google Scholar]
- 82.Schiavoni G et al. (2004) Type I IFN protects permissive macrophages from Legionella pneumophila infection through an IFN-gamma-independent pathway. J Immunol 173 (2),1266–75. [DOI] [PubMed] [Google Scholar]
- 83.Lippmann J et al. (2011) Dissection of a type I interferon pathway in controlling bacterial intracellular infection in mice. Cell Microbiol 13 (11), 1668–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Valderrama C et al. (2017) Listeria monocytogenes induces an interferon-enhanced activation of the integrated stress response that is detrimental for resolution of infection in mice. Eur J Immunol 47 (5), 830–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Osborne SE et al. (2017) Type I interferon promotes cell-to-cell spread of Listeria monocytogenes. Cell Microbiol 19 (3). [DOI] [PubMed] [Google Scholar]
- 86.Kernbauer E et al. (2013) Route of Infection Determines the Impact of Type I Interferons on Innate Immunity to Listeria monocytogenes. PLoS One 8 (6), e65007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.O’Connell RM et al. (2004) Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med 200 (4), 437–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rayamajhi M et al. (2010) Induction of IFN-alphabeta enables Listeria monocytogenes to suppress macrophage activation by IFN-gamma. J Exp Med 207 (2), 327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aubry C et al. (2012) Both TLR2 and TRIF contribute to interferon-beta production during Listeria infection. PLoS One 7 (3), e33299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cheng Y and Schorey JS (2018) Mycobacterium tuberculosis-induced IFN-β production requires cytosolic DNA and RNA sensing pathways. Journal of Experimental Medicine 215 (11), 2919–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Novikov A et al. (2011) Mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL-1beta production in human macrophages. J Immunol 187 (5), 2540–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wiens KE and Ernst JD (2016) The Mechanism for Type I Interferon Induction by Mycobacterium tuberculosis is Bacterial Strain-Dependent. PLoS Pathog 12 (8), e1005809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Berry MP et al. (2010) An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466 (7309), 973–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Parker D et al. (2012) Induction of type I interferon signaling by Pseudomonas aeruginosais diminished in cystic fibrosis epithelial cells. Am J Respir Cell Mol Biol 46 (1), 6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Perkins DJ et al. (2015) Salmonella Typhimurium Co-Opts the Host Type I IFN System To Restrict Macrophage Innate Immune Transcriptional Responses Selectively. J Immunol 195 (5),2461–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Owen KA et al. (2016) Salmonella Suppresses the TRIF-Dependent Type I Interferon Response in Macrophages. mBio 7 (1), e02051–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Deriu E et al. (2016) Influenza Virus Affects Intestinal Microbiota and Secondary Salmonella Infection in the Gut through Type I Interferons. PLoS Pathog 12 (5), e1005572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Scumpia PO et al. (2017) Opposing roles of Toll-like receptor and cytosolic DNA-STING signaling pathways for Staphylococcus aureus cutaneous host defense. PLoS Pathog 13 (7), e1006496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Andrade WA et al. (2016) Group B Streptococcus Degrades Cyclic-di-AMP to Modulate STING-Dependent Type I Interferon Production. Cell Host Microbe 20 (1), 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sotolongo J et al. (2011) Host innate recognition of an intestinal bacterial pathogen induces TRIF-dependent protective immunity. J Exp Med 208 (13), 2705–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Patel AA et al. (2012) Opposing roles for interferon regulatory factor-3 (IRF-3) and type I interferon signaling during plague. PLoS Pathog 8 (7), e1002817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dhariwala MO et al. (2017) Induction of Type I Interferon through a Noncanonical Toll-Like Receptor 7 Pathway during Yersinia pestis Infection. Infect Immun 85 (11). [DOI] [PMC free article] [PubMed] [Google Scholar]