

Abstract
Despite the value of mass spectrometry in modern natural products discovery workflows, it remains very difficult to compare datasets between laboratories. In this study we compared mass spectrometry data for the same sample set from two different laboratories (quadrupole time-of-flight and quadrupole-Orbitrap) and evaluated the similarity between these two datasets in terms of both mass spectrometry features and their ability to describe the chemical composition of the sample set. Somewhat surprisingly, the two datasets, collected with appropriate controls and replication, had very low feature overlap (25.7% of Laboratory A features overlapping 21.8% of Laboratory B features). Our data clearly demonstrate that differences in fragmentation, charge state, and adduct formation in the ionization source are a major underlying cause for these differences. Consistent with other recent literature, these findings challenge the conventional wisdom that electrospray ionization mass spectrometry (ESI-MS) yields a simple one-to-one correspondence between analytes in solution and features in the dataset. Importantly, despite low overlap in feature lists, principal component analysis (PCA) generated qualitatively similar PCA plots. Overall, our findings demonstrate that comparing untargeted metabolomics data between laboratories is challenging, but that datasets with low feature overlap can yield the same qualitative description of a sample set using PCA.
TOC Graphic
Mass spectrometry-based metabolomics is emerging as a central tool for determining the chemical composition of natural product samples, including botanical extracts.1,2 Botanical natural products are often evaluated using targeted methods, where the sample is analyzed to determine the presence of ‘marker compounds’ from a defined target list.3,4 The inherent disadvantage of this approach is that metabolites outside this target list are not considered. This can result in contaminants being overlooked or lead to incorrect taxonomic or origin assignments. These issues can be addressed using untargeted metabolomics approaches. With untargeted metabolomics, all detectable features (m/z-retention time pairs) are reported in ‘feature lists’, which can subsequently be annotated for known molecules using reference compounds or in silico methods. However, there is currently lack of consensus within the natural products community on the optimal methods for acquiring or processing untargeted metabolomics datasets.
Untargeted mass spectrometry workflow design includes a large number of instrumental and analytical parameters, each of which can influence data quality and content. In this study, we analyzed a set of botanical extracts in two independent laboratories on two different mass spectrometry platforms using similar acquisition and processing methods. From these data, we first evaluated the importance of replicate type selection on data quality. We assessed the impact on feature list composition of performing replicate injections in addition to replicate extractions, to provide guidance on replicate type selection. Secondly, we assessed how closely the results aligned between the two laboratories when acquired under optimized conditions to better understand the opportunities and challenges associated with sharing untargeted metabolomics datasets between research sites.
Results and Discussion
Botanical Selection.
We selected Camellia sinensis (green tea) as the test species for this study. Camellia sinensis has been studied extensively for chemical composition.5–7 Consequently, many of the common constituents are known, and authentic reference samples of both the complex botanical and many of its components are commercially available. These studies were conducted using a set of 37 different sources of green tea, including cut-leaves, powders, and supplements (green tea capsules) from 31 commercial vendors (Supporting Information Table S1). These same green tea samples have been used in previous studies in our laboratory, which provided detailed data about their chemical composition.8 This sample set included the NIST (National Institute of Standards and Technology) green tea leaf standard (SRM 3254), which was used as an authentic green tea (positive) control and a mixture of Curcuma longa and Zingiber officinale (turmeric and ginger) tea, which was employed as a non-green tea (negative) control.
Experimental design.
The key considerations for data collection were: the number and type of replicates; instrument type; data acquisition method; chromatographic conditions; and data analysis strategy (Figure 1). To assess the relative influence of performing replicate extractions versus replicate injections, we prepared three separate extractions of each sample, and analyzed each extraction replicate by mass spectrometry as injection replicates. Replicate extractions were all prepared by the same researcher using the same source material and equipment. Extracts were concentrated to dryness under nitrogen gas and stored at −20 °C prior to analysis.
Figure 1.
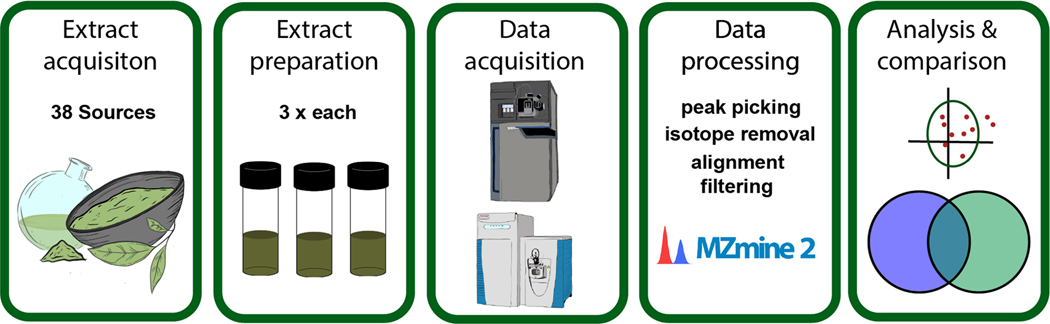
Project workflow.
For chemical analysis, we employed two different ‘high-resolution’ instruments; a Waters SYNAPT G2-Si qTOF (Laboratory A) and a Thermo Fisher Q-Exactive Plus Orbitrap (Laboratory B). Both instruments were equipped with electrospray ionization (ESI) sources. In addition, the SYNAPT is equipped with a traveling wave ion mobility cell, which was disabled for this study. Both types of instruments (qTOF and Orbitrap) are commonly encountered in academic and industrial natural products research groups and are instruments of choice for many natural product-based metabolomics studies. Although they function in fundamentally different ways, both instruments generate similar types of data (MS1 and optionally MS2 data, typically coupled to a liquid chromatography inlet), making it practical to compare the results of analyses. While this study does compare the features detected between two different MS platforms, the goal was not to directly compare the capabilities of these instruments. This is because performance with a single sample type and under a single set of conditions is not representative of the overall performance of any system. For this study the data collected on the Waters SYNAPT G2-Si instrument are labeled Laboratory A, while the data collected in the Thermo Fisher Scientific Q-Exactive Orbitrap are labeled Laboratory B.
Following initial method development (Supporting Information), a short linear gradient method was selected, and the same column and chromatographic conditions were used on both instruments (Experimental Section). This method balances chromatographic separation against analysis acquisition time. The method was selected as representative of those used for untargeted metabolomics studies of natural products mixtures, rather than being extensively tailored to achieve the best possible separation for C. sinensis.
The two mass spectrometers used in the study differ in several important ways with respect to data acquisition parameters. To obtain datasets that were directly comparable, we selected acquisition parameters that could easily be performed on both instruments. Data were obtained in positive ionization mode, using data-dependent acquisition (DDA) for MS2 data with a maximum of five target masses per MS1 scan. The same inclusion list of precursor masses (Table S2) was used for all analyses.
As has been clearly demonstrated, data processing software and parameter selection can have a dramatic impact on feature list composition.9,10 A recent study from Hohrenk et al. showed that processing the same experimental dataset using four different software platforms afforded feature lists with a maximum common overlap of 10%.10 Differences in instrument design and output data formats often complicate data analysis tool selection. For example, the SYNAPT instrument includes a ‘lockspray’ calibrant that is infused into the instrument from a separate nebulizer at a defined interval during the acquisition. These calibration scans complicate processing of the MS1 data and are not well tolerated by all open-source tools. Similarly, the Orbitrap instrument can be run in fast polarity switching mode, but not all processing packages can correctly identify and segregate these two data types from a single output file. To generate feature lists that were directly comparable between instruments, we selected the popular open source software platform MZmine 2, which could handle. raw data from both mass spectrometry platforms.11
Influence of Replicate Analyses on Data Quality.
A number of previous studies have highlighted the importance of performing replicate analyses for untargeted metabolomics analysis of natural products mixtures.1,12–14 However, analysts often have difficulty determining which type of replication is appropriate. Replication in chemical analysis fits into two broad categories, biological replicates, in which samples are collected from different individuals or pooled samples from a given population (i.e. multiple samples of different plants of the same species or multiple batches of bacterial cells) or analytical replicates, also called technical replicates, where the analysis is repeated multiple times on the same sample.15 The type of replicate that is appropriate depends entirely on the scientific question being asked. In the test case used for this study, we were not asking a biological question, but rather sought to compare the chemical composition of a series of botanical natural product samples (green tea) all obtained from different suppliers. Thus, we chose to create analytical replicates that captured the variability in the entire process of extraction and analysis of the different samples. We first extracted each sample three times, creating a series of extraction replicates. The question arose in these studies as to whether it would be helpful to conduct replicate analyses of each of these extraction replicates with the mass spectrometer (replicate injections). We addressed this question using the three separate replicate extractions prepared from the same sample of the NIST standard of C. sinensis (Experimental Section). Aliquots of each replicate extraction were delivered to Laboratories A and B, resuspended in MeOH, and each extract analyzed with three replicate injections using the standard column, gradient, mass spectrometer acquisition parameters, and schedule of blank injections (Experimental Section).
Data processing was performed with MZmine 2. The nine replicates of the NIST standard (three replicate injections of three replicate extractions) were peak picked using the ADAP workflow (chromatogram builder and deconvolution). The same key ADAP settings were used for data from both instruments (e.g. 5 ppm for feature matching and 0.01 min for retention time tolerance).16 Intensity filter values were selected individually for each laboratory as the instruments have different sensitivities and dynamic ranges, and samples were therefore injected at different concentrations. Final feature lists were then aligned and filtered (160–1300 m/z and 1–9 min) to remove features from the column wash (9 – 11 min).
The output from this analysis was a set of nine feature lists from each laboratory, containing the features identified from each replicate analysis. These feature lists were internally consistent in terms of scale, with average feature counts of 2056 ± 49 (mean ± standard deviation) for Laboratory A and 6229 ± 156 for Laboratory B. Using these lists, we examined the variability in feature list composition as a function of replicate count for both replicate extractions and replicate injections (Figure 2). In one case, we grouped data from all three injection replicates for a single extraction replicate (Figure 2 panels Ai and Bi), while in the other case we grouped data from all three extraction replicates for a single injection replicate (Figure 2 panels Aii and Bii). In both cases, we assessed the number of features present in at least one, two or three replicates. Interestingly, in all cases feature counts were similar, with variation typically < 10%. This was true both within replicate types (i.e. between the three extracts in panel i) and between replicate types (i.e. between panels i and ii). In all cases, a sharp decrease in feature counts was observed between those present in at least one replicate and those present in at least two replicates. On average, 60% of the features in each list were observed only once, highlighting the variability of low intensity features between replicates.
Figure 2.
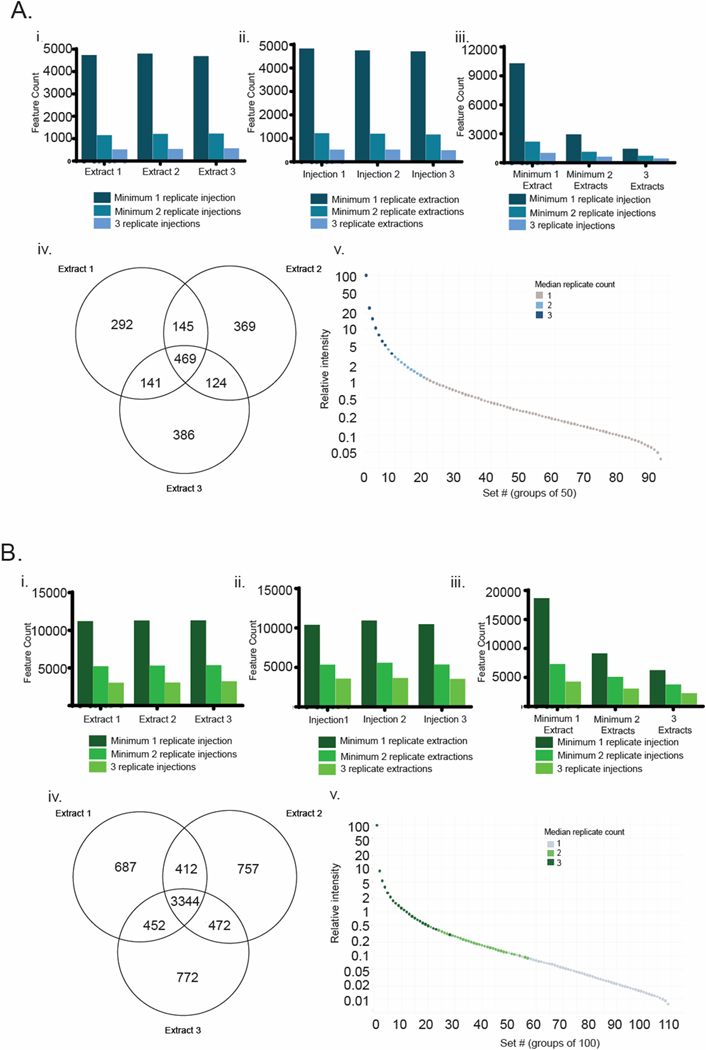
Replicate distributions of features for Laboratory A (blue) and B (green). i) distribution of features between replicate injections ii) distribution of features between replicate extractions iii) distribution of features between replicate extractions and replicate injections iv) Venn diagram of feature distributions between replicate extractions for features that are present in at least two replicate injections and v) relationship between feature intensity and replicate count for data from panel iv.
Interestingly, the absolute numbers of features and the relative changes in feature count were closely aligned between both type of replicate for both laboratories. If variations in extraction protocol had a dramatic impact on the constitution or concentration of individual extracts, then we would have expected to observe a higher rate of decrease in feature counts in replicate extractions versus replicate injections. Instead, we see low variation between the feature counts in the two replicate methods as we increase the requirement to be in at least one, two or three replicates, suggesting that there is low variation in chemical composition between extraction replicates. Finally, we combined these two replicate methods in panel iii which provides the feature counts for all possible combinations of replicate counts. The largest bar (far left) is the count of all features present in at least one injection replicate and one extraction replicate. The smallest bar (far right) provides a count of features present in all three injection replicates of all three extraction replicates (the most stringent criterion). These results illustrate the impact of setting different requirements for feature presence in replicates and the dramatic effect that this can have on feature counts for downstream analysis.
However, feature counts alone are not sufficient to fully assess overlap between analyses. To examine the value of performing injection replicates in addition to extraction replicates, we used the data from panel iii to filter the feature lists for each extraction replicate to retain only those features present in at least two injection replicates. Using these filtered lists, we then determined the distribution of features between extraction replicates (Venn diagrams, Figure 2 panels Aiv and Biv). Overall, Laboratory A detected a lower number of features (1926) compared to the feature list from Laboratory B (6896). Furthermore, the distribution of features within each set was quite different, with 46% of features from Laboratory A appearing at least twice, and 68% of features appearing at least twice in the dataset from Laboratory B. Intrigued by the source of this discrepancy, we ordered the features by intensity and placed them in groups of 50 (Laboratory A) or 100 (Laboratory B) and then plotted median group intensity, color coding the data points by median extraction replicate count (Figure 2 panels Av and Bv). This analysis demonstrates, perhaps unsurprisingly, the relationship between replicate extraction count and intensity. Intense features are consistently present in all three analyses, while weaker features are often present just once or twice in the dataset.
Overall, our study shows that a significant portion of the features detectable in a single replicate are not observed in 2/3 replicates (Figure 2); therefore, inclusion of injection replicates will significantly reduce feature lists if features appearing only once are removed. The question of the relative value of extraction replicates as compared to injection replicates is best addressed by considering how the data will be interpreted. If the goal of the study is to employ untargeted metabolomics to compare chemical composition between samples (as in the case presented here), rigorous experimental design requires that the replicates reproduce the entire method, from extraction through to analysis by the mass spectrometer. Failure to do so could introduce systematic errors into the dataset, whereby variations in extraction efficiency could be interpreted as variability in chemical composition of samples. The importance of conducting replicate extractions for rigorous metabolomics studies appears to be generally accepted by the metabolomics community. For example, in a recent survey, 73% of metabolomics practitioners reported the use of replicate extractions in their analyses.17 Although there has been no similar survey specifically directed at the natural products community employing metabolomics, it is likely that the use of replicate extractions is not particularly common among natural products scientists. This is at least in part because the goal of many natural products studies is not to compare different sources of the same material, but to profile, as comprehensively as possible, the chemical content of a single natural product extract. For such applications, replicates are advisable to improve dataset quality and ensure a robust feature list. However, triplicate injections of a single extract are sufficient if the analyst does not seek to make quantitative comparisons between different extracts. Finally, for the study conducted herein, the data suggest that there is little value to conducing injection replicates of replicate extracts, particularly given the increased instrument time required to analyze these samples (nine analyses per sample rather than three).
Comparison of Feature Lists Across Laboratories.
An overarching objective of our research programs is the development of methods for the accurate characterization of natural products mixtures using MS-based untargeted metabolomics methods. In contrast to targeted metabolomics, where the goal is to determine the presence of compounds from a fixed target list, untargeted metabolomics aims to generate a complete description of the small molecules (metabolites) present in any mixture. Targeted methods are tolerant of feature lists with very high false positive rates, because these erroneous features can be ignored provided that they do not accidentally align with the target list. By contrast, untargeted methods aim to create feature lists that contain exclusively features that derive from real molecules in the test mixture. The extent to which this goal is met in a given MS metabolomics study is difficult to evaluate, given the large size of metabolomics datasets and that the samples being analyzed are typically of unknown composition. Here we sought to compare datasets between laboratories to evaluate the extent of overlap as a strategy to address dataset quality.
An additional observation from Figure 2 is the discrepancy between the absolute number of features identified in the two laboratories (10,047 from Laboratory A vs 18,247 from Laboratory B). It is tempting to conclude that the data from Laboratory B is in some way ‘better’ because more features were detected. However, if these additional features did not derive from the actual sample, then the results from Laboratory A would be a more accurate representation of the chemical constitution of the test mixture. Alternatively, if the two instruments produce different numbers of adducts and in-source fragments, then the two datasets could be equally valuable for describing composition, even though the absolute numbers of features are very different. We were interested to examine this discrepancy in more depth, to try to understand the source of differences in number of features present in the datasets from the different laboratories.
We asked three questions about the data from these replicated feature lists: 1) How many of a set of known reference compounds were observable in each dataset? 2) How many features overlapped between the two datasets? and 3) What was the origin of the unique features from each feature list? Question 1 was designed to determine whether the difference in feature list sizes was due solely to detection limit, and therefore to reduced coverage of low abundance molecules in Laboratory A. Question 2 assessed the issue of coverage from a different perspective, by determining whether the feature list from Laboratory A was a subset of the list from Laboratory B, or whether the two lists contained fundamentally different features. Finally, question 3 was designed to evaluate whether we could propose an origin for these unique features, based on differences in background signals or ionization and in-source fragmentation behavior between these two mass spectrometers.
To define a reference set of known chemical constituents of C. sinensis, we purchased fifteen reference compounds (Table S2) and analyzed them in both laboratories to generate a full suite of UHPLC-MS/MS data on both MS platforms. Manual inspection of the raw MS data for the extract prepared from the NIST standard of C. sinensis by Laboratories A and B demonstrated that all 15 reference compounds were detectable, based on retention time and MS feature matching (MS1 and MS2 data, Supporting Information Figure S1). The reference compounds covered a broad range of retention times and intensities, from caffeine, which was the strongest signal in the sample, to caffeic acid and gallic acid, both of which were present at intensities close to the intensity cutoff assigned to each instrument.
Feature lists were created from a single injection of each replicate extraction on each instrument, with the requirement that features be present in at least two of three extraction replicates. These feature lists were then compared against the reference table for the commercial reference compounds (Table S2) and presence or absence determined by the detection of the required [M+H]+ adduct within 5 ppm and 0.05 min of the reference table. In addition, we compared the results with (Table 1A) and without (Table 1B) the inclusion of the ‘isotope filtering’ option in MZmine 2, which excludes features from the feature list that do not have at least one 13C isotope feature.
Table 1.
A) Reference compounds identification from feature lists generated without isotope filtering and B) reference compounds identification from feature lists generated with isotope filtering in the NIST leaf extract (SRM 3254)
| A. | |||
|---|---|---|---|
| Standard | Laboratory A | Both | Laboratory B |
| Caffeic acid | + | ||
| Caffeine | + | ||
| Chlorogenic acid | + | ||
| Coumaric acid | - | ||
| Epicatechin | + | ||
| Epicatechin gallate | + | ||
| Epigallocatechin | + | ||
| Gallic acid | + | ||
| Gallocatechin | + | ||
| Gallocatechin gallate | + | ||
| Kaempferol | + | ||
| Myricetin | + | ||
| Quercetin | + | ||
| Rutin | + | ||
| Theanine | + | ||
| B. | |||
| Standard | Laboratory A | Both | Laboratory B |
| Caffeic acid | - | ||
| Caffeine | + | ||
| Chlorogenic acid | - | ||
| Coumaric acid | - | ||
| Epicatechin | + | ||
| Epicatechin gallate | + | ||
| Epigallocatechin | + | ||
| Gallic acid | - | ||
| Gallocatechin | + | ||
| Gallocatechin gallate | + | ||
| Kaempferol | + | ||
| Myricetin | + | ||
| Quercetin | + | ||
| Rutin | + | ||
| Theanine | + | ||
Interestingly, the number of identified reference compounds was similar for the feature lists from the two laboratories, notwithstanding the differences in feature counts. Without isotope filtering, 10 of the compounds represented in the list of reference compounds were identified by both laboratories, with Laboratory A identifying one additional reference compound, and Laboratory B identifying three additional reference compounds. With the inclusion of isotope filtering, the number of identified reference compounds reduced, with seven reference compounds identified by both laboratories, Laboratory B identifying three additional reference compounds, and Laboratory A identifying one additional reference compound. The inclusion of isotope filtering selects against low abundance features because of the requirement to observe at least one additional isotopologue peak, which is typically at an intensity of just a few percent compared to the base all 12C peak for small molecules. Because it negatively impacted limit of detection for the known reference compounds, the isotope filtering feature was excluded from subsequent analyses.
Manual inspection of the data revealed that gallic acid, chlorogenic acid and caffeic acid were detected with very low signal intensity in the analysis of the NIST green tea standard extract, and were not detected by MZmine 2 in the dataset from either laboratory even without the isotope filter. Similarly, coumaric acid was present with such low signal intensity that it was not identified in either dataset. By contrast, caffeine was present with a very high signal intensity. This interfered with the mass accuracy in Laboratory A, causing a mass error > 5 ppm and preventing identification of the [M + H]+ signal.
We repeated this analysis on a mixture of all 15 reference compounds at a fixed concentration (6.25 μg/mL Laboratory A; 0.78 μg/mL Laboratory B) using the same chromatographic conditions. Results from this analysis mirrored those from the individual reference compounds, with both laboratories identifying 14 reference compounds, indicating that combining the reference compounds into a single mixture did not impact their detection under these chromatographic conditions.
We next sought to assess what percentage of the features in each list were common between the two datasets. In theory, data acquired on two identical systems should produce two identical feature lists. In reality, however, differences such as background contamination, clustering and fragmentation within the source, environmental conditions, and column performance will all contribute to differences in feature lists. In this study, differences in feature lists were further increased by the use of two different types of mass spectrometers. A major objective of this study was to assess the degree of similarity between datasets acquired on these two instruments, to evaluate the feasibility of data comparison between the two laboratories.
To relate features in the datasets acquired on the two different instruments, we first considered using a match factor or cosine score between corresponding MS2 spectra.18 However, this approach suffers from the major drawback that many of these features are of low intensity, and were not selected for DDA MS2 analysis.
An alternative strategy is to match features between lists based on retention time and mass to charge (m/z) ratio. To be viable as a matching strategy, there must be a defined relationship between the retention times of compounds on the two instruments. Plotting retention time in Laboratory A vs retention time in Laboratory B for the reference compounds demonstrated a strong linear relationship, with an R2 value of 0.99 (Figure 3A). Using the line of best fit to correct for a small deviation in the void volumes of the two systems (Experimental Section), we compared the corrected retention times and mass to charge ratios between the two feature lists and identified all features that were within 0.1 min retention time and 5 ppm m/z of one another (Figure 3B).
Figure 3.
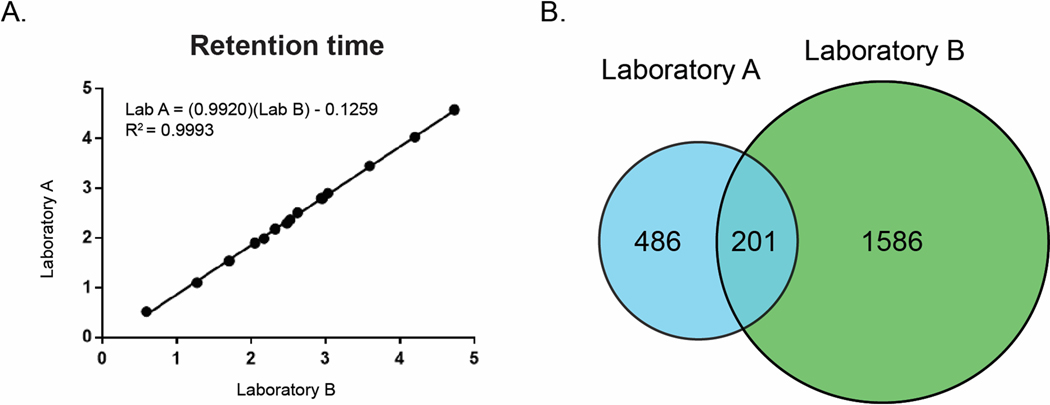
A) Relationship between retention times for reference compounds detected in the NIST Camillia sinensis standard from Laboratory A (y-axis) and Laboratory B (x-axis). B) Venn diagram of feature list overlap for NIST standard between Laboratory A (blue) and Laboratory B (green).
To our surprise, these feature lists from the two laboratories showed a maximum 29% overlap. Given that both instruments used the same column make and model, elution conditions, and ionization source type (ESI), we expected a higher degree of overlap between feature lists at the MS1 level. Use of the same column and gradient conditions ensures that the mixture of analytes eluting at a given point in the chromatogram should be similar on each instrument. Under identical ionization conditions, one would expect similar MS1 signals, and therefore feature lists of similar magnitude and composition. Several possible justifications for this discrepancy present themselves. Firstly, it is possible that one or both of the feature lists contain a large number of interference features that are not related to true analytes in the mixture. Secondly, it is possible that different molecules are ionized between the two instruments. Thirdly, it is possible that the two instruments generate significantly different MS1 features (e.g., charge states, fragments, adducts etc.) for the same set of molecules.
Each of these possibilities raises different concerns for researchers wishing to compare metabolomics datasets between laboratories. Central among these is the question of whether differences in feature lists are due to differing degrees of signal interference, detection of different components of complex mixtures, or different signals from the same set of components. The detection of 11 of 15 reference compounds in both feature lists (Table 1) suggests that in this case differences in compound ionization is not a principal driver of feature list variation. All feature lists were subject to blank subtraction to remove chemical interference signals deriving from background chemical contamination (The procedure for blank subtraction is available in the Experimental Section). The requirement that features be present in at least two of three replicates decreases the likelihood that the features outside the combined region of the Venn diagram (Figure 3B) are largely due to electronic interference signals (Figure 1). Instead, we hypothesized that these differences were due in large part to differences in MS1 signal generation between the two instruments. To test this hypothesis, we analyzed the mixture of the 15 C. sinensis reference compounds (Table 1) in triplicate on both instruments. Following the standard processing workflow, we manually identified the [M+H]+ signal for each observable reference compound and, using an in-house Python script that matched features based on alignment of chromatographic peak shapes, assembled all of the MS features associated with each compound. A comparison of the features for each reference compound from each laboratory is presented in Figure 4.
Figure 4.
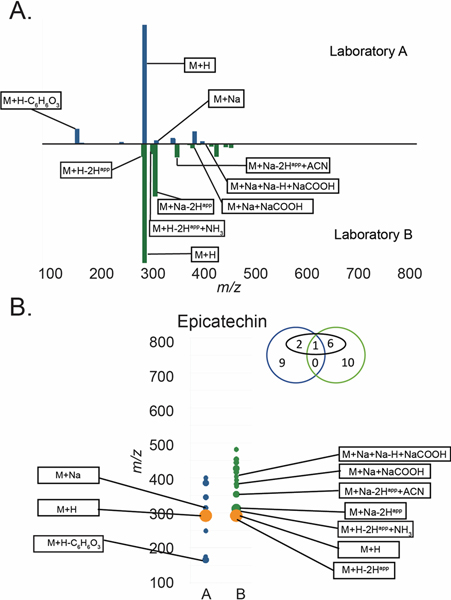
A) Butterfly plot of MS features for epicatechin in both laboratories and B) a simplified view of the butterfly plot in panel A. For panel B orange = features present in both datasets, blue = features only present in Laboratory A, green = features only present in Laboratory B. Diameter is proportional to the relative intensity of each datapoint. The Venn diagram above the trace indicates the number of features from Laboratory A (blue) or Laboratory B (green). Features in black boxes were annotatable adducts or fragments (e.g. [M+H]+, [M+Na]+, [M+H-H2O]+ etc.). Interpretation of the Venn Diagram for epicatechin is as follows: 12 and 17 features were grouped with the molecule for Laboratories A and B respectively. Four of the epicatechin-associated features in the dataset for Laboratory A and seven of the epicatechin-associated features in the dataset for Laboratory B were annotatable, as adducts or clusters (numbers in black circle). Only one of the epicatechin-associated features was detected by both Laboratory A and Laboratory B, and none (zero) of the unidentified peaks were present in both datasets.
The results from Figure 5 support the hypothesis that differences between the feature lists from the two instruments are driven to a large extent by differential formation of adducts, clusters and in-source fragments. Interestingly, although the [M+H]+ signal was present in both datasets in almost all cases, the overall alignment between features was very low, with most compounds having <10% alignment. Also surprising was the observation that very few of the features for each compound could be assigned to common adducts or fragments (black ovals in Venn diagrams above each plot) in either laboratory. This is congruent with several recent reports that have demonstrated that typical untargeted metabolomics feature lists contain vastly more features than compounds, and that these features derive at least in part from the generation of many unique features for each analyte during the ionization process.19,20,21
Figure 5.
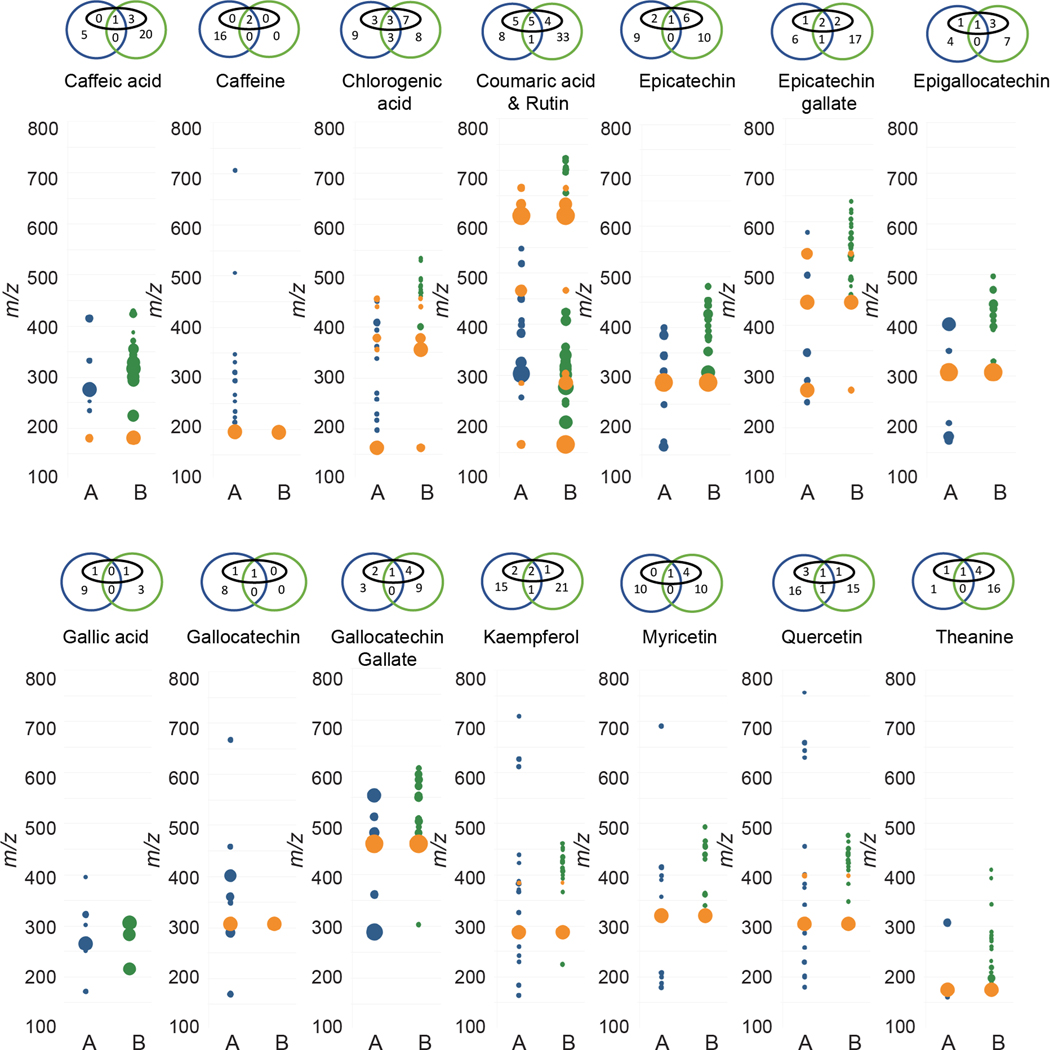
Comparison of MS features for reference compounds analyzed in Laboratories A and B. Orange = features present in both datasets, blue = features only present in Laboratory A, green = features only present in Laboratory B. Diameter proportional to relative intensity. Venn diagram above each trace indicates number of features from Laboratory A (blue) or Laboratory B (green). Features in black ovals were annotatable adducts or fragments (e.g. [M+H]+, [M+Na]+, [M+Na+NaCOOH]+, [M+H-H2O]+, [2M+Na]+, etc.).
To test the hypothesis that variations in feature list composition between instruments were driven at least in part by differences in MS feature formation for the same set of molecules, we extended our study to a large set of samples containing a higher degree of chemical complexity. If this hypothesis were sound, then independent grouping of related samples based on metabolomic profiles should provide comparable results from both laboratories, regardless of the precise distribution of MS features generated by each analyte. By contrast, if differences in feature list composition were due in large part to chemical interference within the datasets, created by background noise from solvents, atmosphere surrounding the source, or contamination within the liquid chromatograph or mass spectrometer, then either one or both datasets should perform poorly at grouping chemically similar samples.
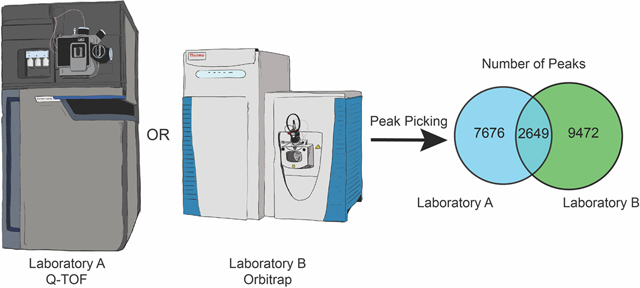
We prepared three replicate extractions for each of the 37 sources of green tea (Table S1),8 and the control sample mixture of C. longa and Z. officinale. This included three NIST standards; NIST 3254 (green tea leaf) and NIST 3255 and 3256 (green tea supplements). We analyzed this sample set independently in Laboratories A and B using the same workflow employed throughout this study, and created replicated feature lists for each sample with the requirement that features be present in a minimum of two replicates. A total of 10,325 features were detected in the dataset obtained by Laboratory A, and 12,121 features in the dataset from Laboratory B, of which 2,649 were detected in both datasets (Figure 6A). These results were broadly aligned with the previous evaluation of the reference compounds mixture, with ~20% of the features being present in both datasets.
Figure 6.
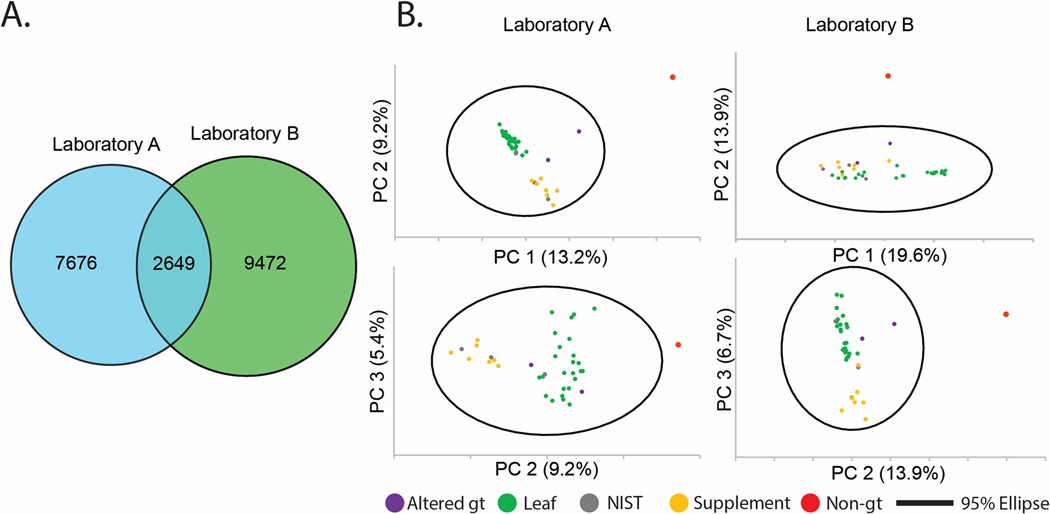
A) Venn diagram of unique feature list counts and shared features between Laboratories A (blue) and B (green). B) Principal component analysis (PCA) scores plots of green tea samples: 27 “leaf” products, either whole-leaf teas (21) or powders (6), seven supplements, a single non-green tea (“non-gt”, turmeric-ginger tea) negative control, and three Camellia sinensis standard reference materials from NIST, drawn with Hotelling’s 95% confidence ellipse from Laboratory A (left) and B (right). Top plots are PC1 vs. PC2. Bottom plots are PC2 vs. PC3.
Next, the two feature lists were subjected to principal component analysis (PCA) to assess their ability to discriminate between sample types, a common and practical application of untargeted metabolomics datasets. Inspection of the PCA scores plot using combinations of the first three components (Figure 6B) yielded similar sample distributions. The non-green tea sample (containing C. longa and Z. officinale) was located beyond the Hotelling’s 95% confidence ellipse in all cases (red dot in Figure 6B), and two green teas that contained additional botanical components (shown in orange in Figure 6B) were spatially located away from the main cluster of green tea samples. The scores plot from the data from Laboratory A evidenced greater separation between the tea and supplement samples when PC1 versus PC2 was plotted (as compared to the plot of PC2 versus PC3), while the scores plot from Laboratory B showed better discrimination between tea leaves and supplements in the PC2 versus PC3 plot than in the PC1 versus PC2 plot. Overall, the qualitative appearance of the PCA plots, with green tea leaves and supplements grouped separately and NIST standards located within the relevant data clusters, is consistent with the previously published data on this same sample set.8 The ability to effectively distinguish the various types of samples in the PCA scores plots served to strengthen the conclusion that the differences between feature lists represent ‘real’ features (i.e., those arising from green tea metabolites) and not noise or contamination.
One question that arises when interpreting the data in Figures 5 and 6 is whether the larger number of features observed for Laboratory B compared with Laboratory A could be due to differences in the ability to detect low abundance ions. Under the conditions used in these analyses, the limits of detection for Laboratory A varied depending on the analyte but were typically 5 to 10 times higher than those for Laboratory B (Table 6S). To adjust for this difference, the C. sinensis samples were analyzed by Laboratory A at 10-fold higher concentration (1 mg/mL) than for Laboratory B (0.1 mg/mL). Even with the concentrations adjusted to address differences in limit of detection, the feature lists for Laboratory B were still quite different in both identity and number of features as compared to Laboratory A. Thus, we conclude that the differences in feature lists are due at least in part to differences in ionization behavior beyond what could be expected due to differences in limits of detection.
When comparing the differences in datasets across the two laboratories, it is also important to consider the question of whether interpretation might have been confounded by differences in the acquisition methodologies. For the design of this study, we intentionally attempted to harmonize the data acquisition parameters between the two laboratories to facilitate meaningful comparisons between the datasets acquired on the qTOF (Laboratory A) and the Q-Exactive Orbitrap (Laboratory B). Despite these efforts, there were some minor methodological differences across laboratories that were overlooked in the design step and did not become apparent until the data analysis stage. As described in the Experimental section, these included the type of solvent used as diluent prior to HPLC injection, the mass spectrometer scan rate, the column temperature, and the number of precursors chosen in the data dependent analysis. Even with these methodological differences, we contend that the conclusion that different platforms generated different numbers of features and types of adducts detected remains sound. The diluent used prior to injection to LC-MS should have negligible impact on ionization efficiency (given that it is diluted many-fold in the LC solvent) and the differences in MS scan rate, while impacting duty cycle and sampling across peaks, should not change the identity of the clusters and fragments detected. Any minor differences in retention time that may have arisen from the slightly different column temperatures used were addressed in the process of retention time correction demonstrated in Figure 3.
Importantly, even if all acquisition parameters were identical, it would be impossible to make a head-to-head comparison in the two mass spectrometry platforms due to dramatic differences in the instrument hardware. Although both instruments were equipped with an electrospray ionization source, the source design and ion optics differs between vendors. Furthermore, the two instruments are equipped with different types of mass analyzers, a hybrid quadrupole-time-of-flight for Laboratory A and a hybrid quadrupole-Orbitrap for Laboratory B. Some compromise was necessary to collect datasets with similar parameters on these two instruments, and, consequently, the parameters were not fully optimized for either instrument. Therefore, it is not possible based on this single study to draw a concrete conclusion as to which platform is “better.” We can, however, say based on the data presented here that both instruments effectively characterized the chemical differences in the C. sinensis samples.
Conclusion
The majority of previous interlaboratory comparisons in MS have focused on targeted analysis; comparison of the abundance of known analytes in the datasets from different laboratories.22–24 This study attempted an interlaboratory comparison using an untargeted approach, comparing feature lists generated independently by each laboratory on the same sample set. We showed that retention times were strongly correlated between the datasets and could be used to facilitate feature-by-feature comparisons. In comparing the datasets, we observed dramatic differences in feature identity, which appear to result from differences in the adducts, fragments, charge states, and clusters generated by different mass spectrometers. Importantly, datasets with very different feature composition collected in two independent laboratories still effectively described qualitative differences in samples, which is typically the stated objective of metabolomics studies.
Our studies have some interesting implications for the community of scientists seeking to employ untargeted metabolomics to characterize natural products samples. Firstly, we demonstrated that the requirement that a feature be detected in at least two of three replicates results in a dramatic reduction in the number of features in an untargeted metabolomics dataset. Thus, the inclusion of replicates is important for improving dataset quality and preventing erroneous data interpretation, but will also result in rejection of some low-abundance features that may represent real analytes. Secondly, these data suggest that unique feature lists are to be expected when analyses are conducted on different instruments, an observation that makes the typical inter-laboratory comparisons for validating analytical approaches very difficult. Our findings emphasize the important conclusion that untargeted metabolomics feature lists are not a description of the chemical constitution of the sample, but rather an instrument-specific snapshot of how the chemical entities in the sample respond to analysis by the particular mass spectrometer. As such, feature lists are useful for comparing samples all analyzed on the same MS platform, but such comparisons should not be extended to samples analyzed on different platforms or in different laboratories. Results from the evaluation of standards in both laboratories highlights the large number of adducts, fragments, and charge states that can derive from a single compound. As has previously been noted, these inflate feature list counts and artificially increase the assumed sample complexity in many cases. An effective and accurate deconvolution tool to reduce a complex feature list down to its component list of individual analytes (chemical entities) would greatly improve the ability to compare MS datasets across laboratories.
Experimental Section
Chemicals.
Unless otherwise noted, all chemicals were of reagent or spectroscopic grade and obtained from Fisher Scientific
Green Tea Product Selection and Extraction.
Commercially available green tea products were selected using consumer sales25 and product quality reports.26,27 The 34 products included 21 whole-leaf teas, six powders, and seven supplements (Table S1). A single non-green tea (turmeric-ginger tea) was included as a negative control (GT23), and Camellia sinensis standard reference materials from NIST for loose leaf tea (GT26), supplement (GT27), and oral dosage form (GT37) (nos. 3254, 3255, and 3256, respectively) served as positive controls (Table S1). Two green teas that contained other botanical additives were also selected for comparison (GT24 and GT38). A retention sample of each product, containing several grams of material, was maintained in Laboratory B for future reference. Data about the chemical composition of the green tea samples used in this study is publicly available on the Center of Excellence for Natural Product Drug Interaction Research Data Repository, https://repo.napdi.org/.
Green tea products were extracted in triplicate. For each sample, to 200 mg tea sample was added 20 mL of reagent-grade MeOH, and the mixtures shaken overnight at room temperature, filtered, concentrated to dryness under nitrogen gas and stored at −20 °C prior to analysis.
Sample Preparation.
Weighed aliquots of dried samples prepared in Laboratory B (~10 mg each) were transferred to new 1-dram vials and shipped to Laboratory A over dry ice. Laboratory A prepared the sample in Optima grade MeOH to a concentration of 5 mg/mL. Vials were then sonicated and vortexed before being dilution to a final concentration of 1 mg/mL in a mixture of 50:50 MeOH:H2O. Laboratory B reconstituted the dried samples to 4 or 6 mg/mL in Optima grade MeOH depending on the volume limits of the vial. Vials were then sonicated and vortexed before being diluted to a final concentration of 0.1 mg/mL with Optima grade MeOH.
A set of mixes was also created using 15 reference compounds purchased from ChromaDex. For consistency, the mixtures were prepared in Laboratory B, divided into two and one aliquot was shipped to Laboratory A over dry ice. Mixtures were made in equivalent amounts of each reference compound beginning with 100 μg/mL and following half dilutions ending with 0.195 μg/mL.
LCMS Conditions.
A total of nine samples (3 replicate extractions × 3 replicate injections) were injected for green tea sample 26 (GT-26; NIST sample SRM 3254). All other green tea samples were collected as single injections of replicate extractions. Additionally, each of the reference compound mixes were injected in triplicate. Approximately 20% of the sample lists were blank samples dispersed throughout, three were selected from each lab to be used for blank subtraction
All measurements were performed with an Acquity I-Class UPLC (Waters) for Laboratory A, or Acquity H-class UPLC (Waters) for Laboratory B, with both laboratories using an HSS T3 C18 column (100 mm × 2.1 mm, 1.8 μm, Waters). Separation of 5 μL of sample was achieved by a gradient of (A) H2O + 0.1% formic acid(FA) to (B) MeCN + 0.1% FA at a flow rate of 500 μL/min and for 12.8 min (5% MeCN, 0–0.3 min; 5–90% MeCN, 0.3–9.1 min; 90–98% MeCN, 9.1–10.7 min; 98% MeCN, 10.7–11 min; 5% MeCN, 11.01–12.8 min).
For Laboratory A, the LC flow was directly infused into a SYNAPT G2-Si qTOF (Waters) operated in positive ion mode. Analysis was conducted using DDA mode with an inclusion list for the reference compounds and a maximum of three MS2 acquisitions per MS1 survey scan. The instrument was operated in electrospray mode with 200 pg/mL leucine enkephalin lockspray infusion enabled every 10 seconds. Mass spectra were acquired from 50–1500 m/z at a 5 Hz scan rate in centroid mode with lockmass correction.
For Laboratory B the LC flow was directly infused into a Q-Exactive Plus quadrupole-Orbitrap mass spectrometer (Thermo Scientific) operated in positive ion mode. Analysis was conducted using DDA mode with an inclusion list for the reference compounds and a maximum of five MS2 acquisitions per MS1 survey scan. Mass spectra were acquired from 120–1200 m/z at a 11 Hz scan rate in profile mode.
Data Analysis.
All Samples were processed using MZmine 2 (http://mzmine.github.io/) version 2.51.11 Peak detection was performed using mass detection and the ADAP chromatogram builder module with using MS level 1, an m/z tolerance of 0.001 Da or 5 ppm, and a minimum group size of four scans for both labs. For peak detection Laboratory A used a mass detection (noise level) and group intensity threshold of zero, and a minimum highest intensity of 600, Laboratory B used a mass detection (noise level) of 1 × 104, a group intensity threshold of 5 × 104, and a minimum highest intensity of 1 × 105. Deconvolution was then performed using ADAP deconvolution with a signal to noise threshold of 10, peak duration range of 0–3, and a retention time wavelet range of 0–0.1 for both laboratories. Laboratory A used a coefficient area threshold of 50 and a minimum feature height of 600, while Laboratory B uses a coefficient area threshold of 120 and a minimum feature height of 1 × 105. Feature lists were then deisotoped using an m/z tolerance of 0.001 Da or 5 ppm and a retention time tolerance of 0.05 min. Feature lists for each sample (in triplicate) were then join aligned using a weight of 10 for both m/z and retention time, and a retention time tolerance of 0.05 min. The join aligned list was then row filtered for mass range 160–1300 m/z, time range of 0–9 min, and requiring occurrence in a minimum of two out of three samples. The new aligned and filtered lists for each sample were then join aligned together along with a list of blank samples processed identically to the samples, followed by deletion of features that showed up at least two of three times in the blank samples.
Comparison of Feature Lists.
Feature lists were compared using an in-house Python script (https://github.com/liningtonlab/green_tea_ms). The script first performs a correction to the retention times for each feature from laboratory A using the line of best fit from Figure 3A and the following equation:
Secondly, the script identifies features between the two lists that have matching m/z and corrected retention times within a 0.05 Da (m/z window) and 0.1 min (retention time window).
Feature Grouping and Annotation of Reference Compound Mixes.
To group features from each feature list, first the scan-by-scan data for each feature was exported from MZmine 2.0 as a csv file. An in-house Python script (https://github.com/liningtonlab/green_tea_ms) was then used on the scan-by-scan data to compare intensities of pairs of features as a function of scan number. Plotting intensity vs intensity as a function of scan number provides a measure of the change in relative intensity for each feature pair. Features that displayed an r2 > 0.9 from the linear regression of these plots were grouped together and defined as a single analyte. Features associated with each analyte were retained if they were associated with that analyte in at least two out of three replicate injections.
In MZmine 2, MS1 annotations were performed through the identification modules (custom database and adduct searches). In addition to the calculated [M+H]+ of each reference compound, the platform MZedDB was employed to generate lists of potential adducts specific to each reference compound.28 For additional adducts and neutral losses, a list was compiled using reference data from several different sources. 29–34 The annotation modules were employed with a m/z tolerance of 0.002 Da or 5 ppm, and a retention time tolerance of 0.05 min. A maximum relative adducts peak height of 10,000% were set for adducts and neutral masses. The annotation results are reported as [M+CC+NM]+ according to the model proposed by Kachman et al., 31 where CC denotes the Charge Carrier, and NM is the proposed Neutral Mass (gain or loss). The lists compiling the adducts and neutral losses are presented in Table S5.
Supplementary Material
ACKNOWLEDGMENT
We thank E. D. Wallace for technical assistance in preparing and analyzing green tea extracts.
Funding Sources
These studies were funded in part by the Center of Excellence for Natural Product Drug Interaction Research (NaPDI) and the Center for High Content Functional Annotation of Natural Products (HiFAN). These centers are supported under grant numbers U54AT008909 (NBC) and U41AT008718 (NBC and RGL), respectively, from the National Center for Complementary and Integrative Health (NCCIH) and the Office of Dietary Supplements (ODS), components of the US National Institutes of Health. Funding for this research was also provided by an NSERC Discovery grant (RGL).
Dedicated to Dr. A. Douglas Kinghorn, The Ohio State University, for his pioneering work on bioactive natural products.
Footnotes
Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website at DOI: Additional methods (Note S1), green tea sample sourcing details (Table S1), green tea reference compound identity and details (Table S2), presence of reference compounds in green tea samples (Table S3), MS2 butterfly plots for each standard (Figure S1) annotation details for both Laboratory A (Table S4A) and Laboratory B (Table S4B), adduct/fragment distribution per standard analyte (Figure S2), detailed source material for possible adduct and fragment annotation (Table S5a–c), Blank injection chromatograms (Figure S3), EIC chromatograms of the reference compounds (Figure S4), and LOD comparison between the two instruments (Table S6) (PDF).
References
- (1).Salem MA; Perez de Souza L; Serag A; Fernie AR; Farag MA; Ezzat SM; Alseekh S. Metabolomics in the Context of Plant Natural Products Research: From Sample Preparation to Metabolite Analysis. Metabolites. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Rahman S; Ul Haq F; Ali A; Khan MN; Shah SMZ; Adhikhari A; El-Seedi HR; Musharraf SG Combining Untargeted and Targeted Metabolomics Approaches for the Standardization of Polyherbal Formulations through UPLC–MS/MS. Metabolomics 2019, 15 (9), 116. [DOI] [PubMed] [Google Scholar]
- (3).Millán L; Sampedro MC; Sánchez A; Delporte C; Van Antwerpen P; Goicolea MA; Barrio RJ Liquid Chromatography–Quadrupole Time of Flight Tandem Mass Spectrometry–Based Targeted Metabolomic Study for Varietal Discrimination of Grapes According to Plant Sterols Content. J. Chromatogr. A 2016, 1454, 67–77. [DOI] [PubMed] [Google Scholar]
- (4).Alvarenga R. F. Ramos; Friesen J. Brent; Nikolić D; Simmler C; Napolitano JG; Breemen R. van; Lankin DC; McAlpine JB; Pauli GF; Chen S-N. K-Targeted Metabolomic Analysis Extends Chemical Subtraction to DESIGNER Extracts: Selective Depletion of Extracts of Hops (Humulus Lupulus). J. Nat. Prod 2014, 77 (12), 2595–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lambert JD; Elias RJ The Antioxidant and Pro-Oxidant Activities of Green Tea Polyphenols: A Role in Cancer Prevention. Arch. Biochem. Biophys 2010, 501 (1), 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Friedman M. Overview of Antibacterial, Antitoxin, Antiviral, and Antifungal Activities of Tea Flavonoids and Teas. Mol. Nutr. Food Res 2007, 51 (1), 116–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Moore RJ; Jackson KG; Minihane AM Green Tea (Camellia Sinensis) Catechins and Vascular Function. Br. J. Nutr 2009, 102 (12), 1790–1802. [DOI] [PubMed] [Google Scholar]
- (8).Kellogg JJ; Graf TN; Paine MF; McCune JS; Kvalheim OM; Oberlies NH; Cech NB Comparison of Metabolomics Approaches for Evaluating the Variability of Complex Botanical Preparations: Green Tea (Camellia Sinensis) as a Case Study. J. Nat. Prod 2017, 80 (5), 1457–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Schiffman C; Petrick L; Perttula K; Yano Y; Carlsson H; Whitehead T; Metayer C; Hayes J; Rappaport S; Dudoit S. Filtering Procedures for Untargeted LC-MS Metabolomics Data. BMC Bioinformatics 2019, 20 (1), 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hohrenk LL Itzel F; Baetz N; Tuerk J; Vosough M; Schmidt C, Comparison T. of Software Tools for Liquid Chromatography–High-Resolution Mass Spectrometry Data Processing in Nontarget Screening of Environmental Samples. Anal. Chem 2019, 92 (2), 1898–1907. [DOI] [PubMed] [Google Scholar]
- (11).Pluskal T; Castillo S; Villar-Briones A; Orešič M. MZmine 2: Modular Framework for Processing, Visualizing, and Analyzing Mass Spectrometry-Based Molecular Profile Data. BMC Bioinformatics 2010, 11 (1), 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Caesar LK; Kvalheim OM; Cech NB Hierarchical Cluster Analysis of Technical Replicates to Identify Interferents in Untargeted Mass Spectrometry Metabolomics. Anal. Chim. Acta 2018, 1021, 69–77.29681286 [Google Scholar]
- (13).Sumner LW; Amberg A; Barrett D; Beale MH; Beger R; Daykin CA; Fan TW-M; Fiehn O; Goodacre R; Griffin JL; Hankemeier T; Hardy N; Harnly J; Higashi R; Kopka J; Lane AN; Lindon JC; Marriott P; Nicholls AW; Reily MD; Thaden JJ; Viant MR Proposed Minimum Reporting Standards for Chemical Analysis. Metabolomics 2007, 3 (3), 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hoffmann T; Krug D; Hüttel S; Müller R. Improving Natural Products Identification through Targeted LC-MS/MS in an Untargeted Secondary Metabolomics Workflow. Anal. Chem 2014, 86 (21), 10780–10788. [DOI] [PubMed] [Google Scholar]
- (15).Sumner LW; Amberg A; Barrett D; Beale MH; Beger R; Daykin CA; Fan TW-M; Fiehn O; Goodacre R; Griffin JL; Hankemeier T; Hardy N; Harnly J; Higashi R; Kopka J; Lane AN; Lindon JC; Marriott P; Nicholls AW; Reily MD; Thaden JJ; Viant MR Proposed Minimum Reporting Standards for Chemical Analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3 (3), 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Myers OD; Sumner JS; Li S; Barnes S; Du X. Detailed Investigation and Comparison of the XCMS and MZmine 2 Chromatogram Construction and Chromatographic Peak Detection Methods for Preprocessing Mass Spectrometry Metabolomics Data. Anal. Chem 2017, 89 (17), 8689–8695. [DOI] [PubMed] [Google Scholar]
- (17).Dunn WB; Broadhurst DI; Edison A; Guillou C; Viant MR; Bearden DW; Beger RD Quality Assurance and Quality Control Processes: Summary of a Metabolomics Community Questionnaire. Metabolomics 2017, 13 (5), 50. [Google Scholar]
- (18).Wang M; Carver JJ; Phelan VV; Sanchez LM; Garg N; Peng Y; Nguyen DD; Watrous J; Kapono CA; Luzzatto-Knaan T; Porto C; Bouslimani A; Melnik AV; Meehan MJ; Liu W-T; Crüsemann M; Boudreau PD; Esquenazi E; Sandoval-Calderón M; Kersten RD; Pace LA; Quinn RA; Duncan KR; Hsu C-C; Floros DJ; Gavilan RG; Kleigrewe K; Northen T; Dutton RJ; Parrot D; Carlson EE; Aigle B; Michelsen CF; Jelsbak L; Sohlenkamp C; Pevzner P; Edlund A; McLean J; Piel J; Murphy BT; Gerwick L; Liaw C-C; Yang Y-L; Humpf H-U; Maansson M; Keyzers RA; Sims AC; Johnson AR; Sidebottom AM; Sedio BE; Klitgaard A; Larson CB; Boya P CA; Torres-Mendoza D; Gonzalez DJ; Silva DB; Marques LM; Demarque DP; Pociute E; O’Neill EC; Briand E; Helfrich EJN; Granatosky EA; Glukhov E; Ryffel F; Houson H; Mohimani H; Kharbush JJ; Zeng Y; Vorholt JA; Kurita KL; Charusanti P; McPhail KL; Nielsen KF; Vuong L; Elfeki M; Traxler MF; Engene N; Koyama N; Vining OB; Baric R; Silva RR; Mascuch SJ; Tomasi S; Jenkins S; Macherla V; Hoffman T; Agarwal V; Williams PG; Dai J; Neupane R; Gurr J; Rodríguez AMC; Lamsa A; Zhang C; Dorrestein K; Duggan BM; Almaliti J; Allard P-M; Phapale P; Nothias L-F; Alexandrov T; Litaudon M; Wolfender J-L; Kyle JE; Metz TO; Peryea T; Nguyen D-T; VanLeer D; Shinn P; Jadhav A; Müller R; Waters KM; Shi W; Liu X; Zhang L; Knight R; Jensen PR; Palsson BØ; Pogliano K; Linington RG; Gutiérrez M; Lopes NP; Gerwick WH; Moore BS; Dorrestein PC; Bandeira N. Sharing and Community Curation of Mass Spectrometry Data with Global Natural Products Social Molecular Networking. Nat. Biotechnol 2016, 34 (8), 828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sindelar M; Patti J, Chemical G. Discovery in the Era of Metabolomics. J. Am. Chem. Soc 2020, 142 (20), 9097–9105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Mahieu NG; Patti GJ Systems-Level Annotation of a Metabolomics Data Set Reduces 25 000 Features to Fewer than 1000 Unique Metabolites. Anal. Chem 2017, 89 (19), 10397–10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Broeckling CD; Afsar FA; Neumann S; Ben-Hur A; Prenni JE RAMClust: A Novel Feature Clustering Method Enables Spectral-Matching-Based Annotation for Metabolomics Data. Anal. Chem 2014, 86 (14), 6812–6817. [DOI] [PubMed] [Google Scholar]
- (22).Martin J-C; Maillot M; Mazerolles G; Verdu A; Lyan B; Migné C; Defoort C; Canlet C; Junot C; Guillou C; Manach C; Jabob D; Bouveresse DJ-R; Paris E; Pujos-Guillot E; Jourdan F; Giacomoni F; Courant F; Favé G; Le Gall G; Chassaigne H; Tabet J-C; Martin J-F; Antignac J-P; Shintu L; Defernez M; Philo M; Alexandre-Gouaubau M-C; Amiot-Carlin M-J; Bossis M; Triba MN; Stojilkovic N; Banzet N; Molinié R; Bott R; Goulitquer S; Caldarelli S; Rutledge DN Can We Trust Untargeted Metabolomics? Results of the Metabo-Ring Initiative, a Large-Scale, Multi-Instrument Inter-Laboratory Study. Metabolomics 2015, 11 (4), 807–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lin Y; Caldwell GW; Li Y; Lang W; Masucci J. Inter-Laboratory Reproducibility of an Untargeted Metabolomics GC–MS Assay for Analysis of Human Plasma. Sci. Rep 2020, 10 (1), 10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Benton HP; Want E; Keun HC; Amberg A; Plumb RS; Goldfain-Blanc F; Walther B; Reily MD; Lindon JC; Holmes E; Nicholson JK; Ebbels TMD Intra- and Interlaboratory Reproducibility of Ultra Performance Liquid Chromatography-Time-of-Flight Mass Spectrometry for Urinary Metabolic Profiling. Anal. Chem 2012, 84 (5), 2424–2432. [DOI] [PubMed] [Google Scholar]
- (25).Amazon.com, AmazonBest Sellers http://www.amazon.com/Best-Sellers-Grocery-Gourmet-Food-Green-Tea-Beverages/zgbs/grocery/16318471 (accessed Nov 11, 2015). [Google Scholar]
- (26).Health J. Green Tea Supplement Reviews. [Google Scholar]
- (27).Anonymous. Consum. Rep; 2003. [Google Scholar]
- (28).Draper J; Enot DP; Parker D; Beckmann M; Snowdon S; Lin W; Zubair H. Metabolite Signal Identification in Accurate Mass Metabolomics Data with MZedDB, an Interactive m/z Annotation Tool Utilising Predicted Ionisation Behaviour “Rules.” BMC Bioinformatics 2009, 10 (1), 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Damont A; Olivier M-F; Warnet A; Lyan B; Pujos-Guillot E; Jamin EL; Debrauwer L; Bernillon S; Junot C; Tabet J-C; Fenaille F. Proposal for a Chemically Consistent Way to Annotate Ions Arising from the Analysis of Reference Compounds under ESI Conditions: A Prerequisite to Proper Mass Spectral Database Constitution in Metabolomics. J. Mass Spectrom 2019, 54 (6), 567–582. [DOI] [PubMed] [Google Scholar]
- (30).Kuhl C; Tautenhahn R; Böttcher C; Larson TR; Neumann S. CAMERA: An Integrated Strategy for Compound Spectra Extraction and Annotation of Liquid Chromatography/Mass Spectrometry Data Sets. Anal. Chem 2011, 84 (1), 283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kachman M; Habra H; Duren W; Wigginton J; Sajjakulnukit P; Michailidis G; Burant C; Karnovsky A. Deep Annotation of Untargeted LC-MS Metabolomics Data with Binner. Bioinformatics 2020, 36 (6), 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Li H-J; Deinzer ML Tandem Mass Spectrometry for Sequencing Proanthocyanidins. Anal. Chem 2007, 79 (4), 1739–1748. [DOI] [PubMed] [Google Scholar]
- (33).Frebault F; Luparia M; Oliveira MT; Goddard R; Maulide N. A Versatile and Stereoselective Synthesis of Functionalized Cyclobutenes. Angew. Chem. Int. Ed. Engl 2010, 49 (33), 5672–5676. [DOI] [PubMed] [Google Scholar]
- (34).Feihn Lab. Mass Spectrometry Adduct Calculator 2016. https://fiehnlab.ucdavis.edu/staff/kind/metabolomics/ms-adduct-calculator/ (accessed Jul 8, 2020). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.