

Abstract
Hereditary hemorrhagic telangiectasia (HHT, Osler-Weber-Rendu disease) is a rare multisystem vascular disorder that causes chronic gastrointestinal bleeding, epistaxis, and severe anemia. Bevacizumab, an anti-vascular endothelial growth factor antibody, may be effective to treat bleeding in HHT. This international, multicenter, retrospective study evaluated the use of systemic bevacizumab to treat HHTassociated bleeding and anemia at 12 HHT treatment centers. Hemoglobin, Epistaxis Severity Score (ESS), red cell units transfused, and intravenous iron infusions before and after treatment were evaluated using paired means testing and mixed-effects linear models. Bevacizumab was given to 238 HHT patients for a median of 12 (range, 1-96) months. Compared with pretreatment, bevacizumab increased mean hemoglobin by 3.2 g/dL (95% confidence interval: 2.9-3.5 g/dL); i.e., from a mean hemoglobin of 8.6 (8.5-8.8) g/dL to 11.8 (11.5-12.1) g/dL; P<0.0001) and decreased the ESS by 3.4 (3.2-3.7) points (mean ESS 6.8 [6.6-7.1] versus 3.4 [3.2-3.7]; P<0.0001) during the first year of treatment. Compared with 6 months before treatment, the number of red blood cell units transfused decreased by 82% (median of 6.0 [interquartile range, 0.0-13.0] units versus 0 [0.0-1.0] units; P<0.0001) and iron infusions decreased by 70% (median of 6.0 [1.0-18.0] infusions versus 1.0 [0.0-4.0] infusions, P<0.0001) during the first 6 months of bevacizumab treatment. Outcomes were similar regardless of the underlying pathogenic mutation. Following initial induction infusions, continuous/scheduled bevacizumab maintenance achieved higher hemoglobin and lower ESS than intermittent/as-needed maintenance but with more drug exposure. Bevacizumab was well tolerated: hypertension, fatigue, and proteinuria were the most common adverse events. Venous thromboembolism occurred in 2% of patients. In conclusion, systemic bevacizumab was safe and effective for managing chronic bleeding and anemia in HHT.
Introduction
Hereditary hemorrhagic telangiectasia (HHT, Osler- Weber-Rendu disease) is an autosomal dominant multisystem disease of disordered angiogenesis.1 It occurs due to mutations in genes encoding proteins that mediate signaling via the transforming growth factor-b superfamily.2 The vast majority of patients with HHT have mutations in endoglin (ENG) or activin receptor-like kinase 1 (ACVRL1/ALK1), resulting in angiogenic dysregulation, formation of telangiectasias on mucocutaneous surfaces, local hyperfibrinolysis within telangiectasias, and development of arteriovenous malformations in visceral organs.3-5 Fragile mucocutaneous telangiectasias in the nasal mucosa (>95% of patients) and throughout the gastrointestinal tract (75% of patients) lead to severe, recurrent epistaxis and chronic gastrointestinal bleeding, with consequent severe iron deficiency anemia that is often transfusion-dependent.6-8 Severe recurrent epistaxis, which may last for hours per day, also results in psychosocial morbidity, social isolation, and challenges with employment, travel, and routine daily activities.9 Visceral arteriovenous malformations may involve the liver, lung, and central nervous system and can result in severe complications including high-output cardiac failure, liver failure, pulmonary hemorrhage, stroke and intracerebral hemorrhage.10 Thus, chronic bleeding and visceral arteriovenous malformations in HHT are associated with considerable morbidity and mortality.10,11
With a prevalence of one case in 5,000 people, HHT is classified as a rare bleeding disorder by the Centers for Disease Control and Prevention, but is actually the second most common hereditary bleeding disorder in the USA and worldwide (following only von Willebrand disease in prevalence). Despite this, there are no United States Food and Drug Administration or European Medicines Agency-approved treatments for HHT-associated bleeding. The current standard of care for bleeding in HHT includes supportive red blood cell (RBC) transfusion and intravenous iron infusion to treat anemia and local nasal and endoscopic hemostatic procedures to reduce bleeding symptoms. Systemic non-specific hemostatic therapies, such as antifibrinolytic agents, are of limited benefit.12 Most importantly, none of these modalities addresses the underlying pro-angiogenic pathophysiology, and the natural history of HHT in many patients is of unremitting telangiectasia formation and progressively worsening bleeding over the lifespan.9,13-15
Although several angiogenic proteins are dysregulated in HHT,5,16 the raised level of vascular endothelial growth factor (VEGF)17 is of particular clinical interest due to the availability of targeted anti-VEGF agents. Bevacizumab (Avastin®), a recombinant, humanized monoclonal IgG1 antibody that binds to and neutralizes circulating VEGF, is one such agent and is widely used to treat malignancies and age-related macular degeneration.18,19 After promising case reports were published describing successful use of systemic bevacizumab to manage bleeding and anemia in HHT,20-22 several HHT Centers of Excellence worldwide began using this agent off-label to treat HHT patients, given the profound unmet clinical need. Currently, data describing effectiveness and safety of systemic bevacizumab to treat chronic bleeding and anemia in HHT is limited to case reports and small, retrospective, singlecenter case series.23-28 The International HHT Intravenous Bevacizumab Investigative Team study of Bleeding (InHIBIT-Bleed) was therefore designed as an international collaboration of 12 HHT centers seeking to better define the safety and effectiveness of systemic bevacizumab in the treatment of moderate-to-severe HHTassociated bleeding.
Methods
Patients and data collection
This study was approved by the Institutional Review Board of Partners Healthcare (approval 2016P002753/PHS). Nine centers in the USA and one center each in Argentina, Israel, and France participated in the study. All patients aged >18 years treated with systemic (intravenous) bevacizumab for HHT-associated bleeding (epistaxis, gastrointestinal bleeding, or both) from January 1, 2011 until May 1, 2019 were identified at each participating institution. General criteria used by centers to offer systemic bevacizumab for HHT-associated bleeding are detailed in Online Supplementary Table S1. Additional data collection information is given in the Online Supplementary Methods.
Effectiveness measures
Hemoglobin
Anemia was defined as a baseline hemoglobin (Hb) <11 g/dL regardless of gender. Baseline hemoglobin was calculated as the average of all measured hemoglobin values in the 6 months prior to bevacizumab initiation (at least 2 values required) and compared with mean on-treatment hemoglobin (mean of values collected at 3, 6, 9, and 12 months depending on treatment duration).
Epistaxis Severity Score
Background information regarding the Epistaxis Severity Score (ESS) is provided in the Online Supplementary Methods. The baseline ESS at initiation of bevacizumab was compared with the mean on-treatment ESS (mean of values collected at 3, 6, 9, and 12 months depending on treatment duration).
Red blood cell transfusion and iron infusion
The number of red blood cell (RBC) units transfused in the first 6 months on treatment was compared with the number of units transfused in the 6 months prior to treatment. Additionally, events in the second 6 months on treatment were compared with those in the first 6 months on treatment to evaluate ongoing response. The same analysis was performed for the number of intravenous iron infusions administered.
To be included in effectiveness analyses, patients needed to receive ≥3 months of treatment (Hb and ESS) or ≥6 months of treatment (RBC transfusion and iron infusion).
Subgroup analyses
Subgroup analyses for each effectiveness outcome were performed by genotype (ENG vs. ACVRL1 mutation) as well as by maintenance dosing method (continuous vs. intermittent bevacizumab maintenance following an initial series of induction infusions).
Receipt of concurrent bleeding and anemia-directed therapies
Rates of freedom from hemostatic procedures and medical treatments (antifibrinolytic or erythropoiesis-stimulating agents) in the year following bevacizumab initiation were calculated, and effectiveness outcomes in patients who received these concurrent HHT-directed therapies were compared to those in patients who did not receive concurrent HHT-directed therapies.
Safety analysis
Treatment-emergent adverse events (TEAE) were defined according to Common Terminology Criteria for Adverse Events v. 5.0 (National Cancer Institute) and recorded. To evaluate any impact of longer-term treatment on TEAE incidence, the number of TEAE per patient in those treated for 2 years or longer was compared with the number of TEAE per patient in those treated for less than 2 years.
Statistical analysis
Baseline hemoglobin concentration and ESS were compared with on-treatment mean values using the paired t-test. Pretreatment RBC transfusion and iron infusion requirements were compared with on-treatment values using the Wilcoxon signed-rank test. To estimate the change in each outcome measure over time on treatment utilizing a method robust in the setting of missing data, a mixed effects linear model with a random intercept was also used for each outcome measure. Additional details regarding statistics, including analysis of subgroups, are detailed in the Online Supplementary Methods.
Results
Patients’ characteristics
Two hundred fifty-seven HHT patients were treated with systemic bevacizumab for epistaxis, gastrointestinal bleeding, or both for the purpose of alleviating bleeding and consequent iron-deficiency anemia at the 12 participating centers during the study period. After 19 patients had been excluded because of inadequate chart data (missing baseline data or incomplete bevacizumab dosing information), 238 patients were included in the data analysis. Table 1 lists the patients’ baseline characteristics. Patients were treated for a median of 12 (range, 1-96) months, receiving a median of 11 (range, 1-74) infusions of intravenous bevacizumab. This totaled 343.9 patientyears of systemic bevacizumab treatment of HHT-associated bleeding in the cohort.
Bevacizumab induction and maintenance dosing
Specific dosing protocols were institution-dependent. Two hundred twenty-one patients (93%) began treatment by receiving four to six induction infusions of bevacizumab 5 mg/kg administered every 2 weeks; 46 (19%) also received an additional four induction infusions administered every 4 weeks. Induction courses typically lasted 2-4 months but never extended beyond 6 months. A total of 181 patients received maintenance treatment, which was defined as additional bevacizumab doses administered to prevent or treat recurrent bleeding after completion of the initial induction infusion course. These maintenance doses were administered using one of two different protocols. The majority of patients (n=136) received continuous maintenance, defined as bevacizumab administered on a regular schedule every 4-12 weeks. The remainder (n=45) received intermittent maintenance, defined as retreatment only following recurrence or worsening of bleeding symptoms or anemia, with one to four doses administered every 2 weeks once the need for maintenance was triggered. Ninety-two percent of patients were given maintenance doses of 5 mg/kg per infusion (regardless of maintenance schedule) but doses of 1, 2.5, 3, and 7.5 mg/kg were also used occasionally. Compared with the relatively universal induction dose intensity of 10.0 mg/kg/month, mean maintenance dose intensity was 4.0 mg/kg/month in patients receiving continuous maintenance and 3.0 mg/kg/month in patients receiving intermittent maintenance.
Table 1.
Baseline characteristics of 238 patients with hereditary hemorrhagic telangiectasia treated with bevacizumab for chronic bleeding and iron deficiency anemia.

Effect of bevacizumab on hemoglobin concentration
Of the 197 anemic (Hb <11 g/dL) patients treated, 185 patients had been treated for ≥3 months and were included in the hemoglobin analysis. Mean hemoglobin increased by 3.2 g/dL (95% confidence interval [95% CI]: 2.9-3.4, P<0.0001) from baseline following bevacizumab treatment (Table 2). In the evaluation of on-treatment mean hemoglobin, freedom from anemia (Hb ≥11 g/dL) was observed in 67% (n=123/185) of patients. In the evaluation of hemoglobin values at each time point, freedom from anemia was observed in 64% of patients at 3 months, 73% at 6 months, 76% at 9 months, and 71% at 12 months (Figure 1A). The change in hemoglobin concentration from baseline to on-treatment ranged from a 1.0 g/dL reduction to a 7.9 g/dL increase. A mixed linear model estimated the improvement in mean hemoglobin at between 3.0 g/dL (95% CI: 2.7-3.3 g/dL; P<0.0001) and 3.4 g/dL (95% CI: 3.1-3.7 g/dL; P<0.0001) at each time point, stable over the course of treatment (Online Supplementary Table S2, Online Supplementary Figure S1A).
Effect of bevacizumab on Epistaxis Severity Score
Of the 175 patients treated for epistaxis with complete ESS data available, 146 patients had been treated for ≥3 months and were included in the ESS analysis. Mean ESS decreased by 3.37 points (95% CI: -3.69 to -3.05; P<0.0001) from baseline following bevacizumab treatment (Table 2). Change in ESS from baseline to on-treatment ranged from a 1.92-point increase to a 10-point reduction. A clinically meaningful reduction in epistaxis (ESS decrease of ≥0.71 post-treatment) was seen in 92% (n=132/146) of patients. The ESS reduction was evident by 3 months (Figure 1B). A mixed linear model estimated the decrease in mean ESS to be between -2.96 (95% CI: - 3.26 to -2.66; P<0.0001) and -3.73 (95% CI: -4.05 to -3.43; P<0.0001) at each time point, stable over the course of treatment (Online Supplementary Table S2, Online Supplementary Figure S1B).
Effect of bevacizumab on red blood cell transfusion
All patients
A total of 191 patients who had been treated for ≥6 months had complete RBC transfusion data available and were included in the RBC transfusion analysis. In analysis of all included patients (those who required RBC transfusion in the 6 months pretreatment and those who did not), the median number of RBC units transfused decreased from 6.0 (interquartile range [IQR], 0.0-13.0) in the 6 months pretreatment to 0.0 (IQR, 0.0-1.0) in the first 6 months after treatment (P<0.0001) (Table 2). This improvement was maintained into the second 6 months of bevacizumab treatment (median of 0.0 units, IQR 0.0-0.0; P=0.0005). The findings of a mixed linear model were similar (Online Supplementary Table S2, Online Supplementary Figure S1C).
Table 2.
Impact of bevacizumab on hemoglobin, Epistaxis Severity Score, red blood cell transfusions, and iron infusions over the first year of treatment.

Patients requiring prior red blood cell transfusion
In an analysis of only those patients who required RBC transfusion in the 6 months prior to treatment (n=137), the median number of RBC units transfused decreased from 9.0 (IQR, 5.0-16.0) in the 6 months before treatment to 0.0 (IQR, 0.0-2.0) in the first 6 months after treatment (P<0.0001); 80/137 patients (58%), were RBC transfusion- free. There was maintained and continued reduction into the second 6 months of bevacizumab treatment (median of 0.0 units, IQR 0.0-0.0; P=0.0005), with 97/121 (80%) being RBC transfusion-free (Figure 1C). The decline in RBC transfusion requirements was observed regardless of baseline disease severity (Figure 2A).
Effect of bevacizumab on iron infusion
All patients
A total of 183 patients who had been treated for ≥6 months had complete iron infusion data available and were included in the iron infusion analysis. In the analysis of all included patients (those who required iron infusion in the 6 months prior to treatment and those who did not), the median number of iron infusions decreased from 6.0 (IQR, 1.0-18.0) in the 6 months before treatment to 1.0 (IQR, 0.0-4.0) in the first 6 months after treatment (P<0.0001) (Table 2). This improvement continued into the second 6 months of bevacizumab treatment (median of 0.0 infusions, IQR 0.0-2.0; P<0.0001). The findings of a mixed linear model were similar (Online Supplementary Table S2, Online Supplementary Figure S1D).
Patients requiring prior iron infusion
In the analysis of only those patients who received iron infusion in the 6 months prior to treatment (n=155), median number of iron infusions decreased from 8.0 (IQR, 3.0-20.0) in the 6 months before treatment to 2.0 (IQR, 0.0-5.0) in the first 6 months after treatment (P<0.0001); 48/155 (31%) patients were iron infusion-free. There was a maintained and continued reduction into the second 6 months of bevacizumab treatment (median of 0.0 units, IQR 0.0-3.0), with 84/138 (61%) patients being iron infusion-free (Figure 1D). The decline in iron infusion requirements was observed regardless of baseline disease severity (Figure 2B).
Figure 1.

Box-and-whisker plots showing the effect of intravenous bevacizumab on hematologic parameters and epistaxis severity. A box represents the median and interquartile range and tails represent minimum and maximum values. Numbers at each time-point reflect the number of hereditary hemorrhagic telangiectasia (HHT) patients in a given analysis treated for at least that duration with complete data. (A) Effect on hemoglobin in HHT patients with baseline anemia (n=185). (B) Effect on Epistaxis Severity Score in patients treated for epistaxis (n=146). (C) Effect on red blood cell (RBC) transfusions in patients receiving transfusion in the 6 months prior to bevacizumab initiation (n=137). (D) Effect on iron infusion events in patients receiving intravenous iron in the 6 months prior to bevacizumab initiation (n=155). RBC: red blood cell; Mo: months; Tx: treatment.
Subgroup analysis by genotype
There were no significant differences between baseline hematologic parameters or effect of bevacizumab treatment on these parameters between patients with ENG and ACVRL1 mutations (Online Supplementary Table S3).
Subgroup analysis by maintenance dosing strategy
As compared with patients receiving continuous scheduled bevacizumab maintenance, those receiving intermittent (as-needed) bevacizumab maintenance had a lower mean hemoglobin (10.8 g/dL vs. 12.3 g/dL; P=0.0002) and higher ESS (4.96 vs. 2.88; P<0.0001) during the second 6 months of bevacizumab treatment (Online Supplementary Table S4). There were no significant differences in iron infusions or RBC transfusions between the two groups.
Use of concurrent therapies to treat hereditary hemorrhagic telengiectasia-associated bleeding
Fifty-three patients (22%) received one (39 patients) or more (14 patients) local hemostatic procedures to treat epistaxis (41 patients, 17%), gastrointestinal bleeding (12 patients, 5%), or both (1 patient) in the first year after initiating bevacizumab. Thirty-three patients (14%) received antifibrinolytic therapy and one patient received an erythropoiesis- stimulating agent during the first year after initiating bevacizumab. In total, 73 patients (31%) did receive and 165 patients (69%) did not receive any of these therapies.
Patients receiving concurrent therapies were similar at baseline to those not receiving them, except for a slightly higher mean ESS (Online Supplementary Table S5). On bevacizumab treatment, patients receiving concurrent treatments for HHT-associated bleeding had slightly lower ontreatment mean hemoglobin and slightly higher on-treatment mean ESS, median number of RBC transfusions, and median number of iron infusions than those not receiving concurrent treatments (Online Supplementary Table S5).
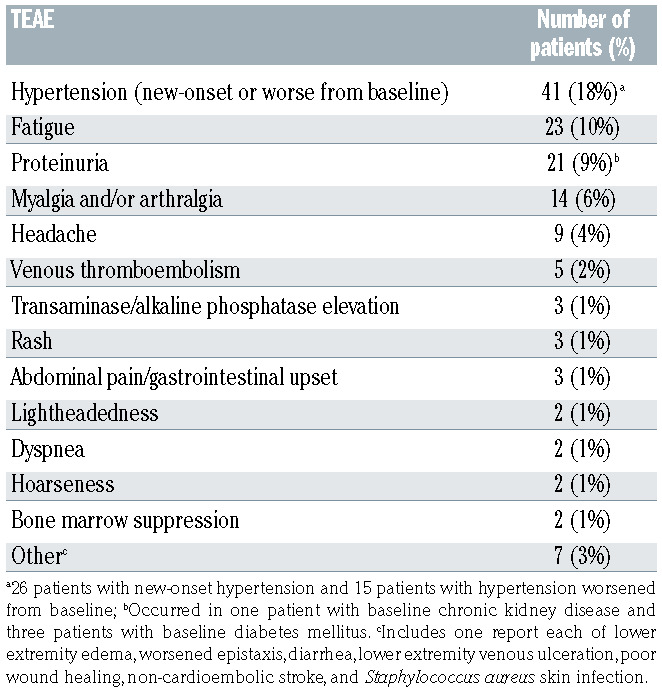
Safety analysis
The safety analysis included 232 patients with complete and continuous chart data available over the duration of bevacizumab treatment, totaling 340.1 patient-years at risk of bevacizumab administration. TEAE possibly or likely related to bevacizumab treatment are summarized in Table 3. The only TEAE reported in >1% of patients were hypertension (18%), fatigue (10%), proteinuria (9%), myalgia and/or arthralgia (6%), headache (4%), and venous thromboembolism (VTE, 2%). Overall, 88 patients (38%) experienced at least one TEAE possibly or likely related to bevacizumab. There were no fatal TEAE. The TEAE rate was slightly higher in patients receiving intermittent maintenance (51%) than in those receiving continuous maintenance (32%). There was no significant difference in number of reported TEAE in patients with long-term exposure to bevacizumab (≥2 years) and those with shorter-term exposure (<2 years) with a mean of 0.57 versus 0.53 TEAE per patient, respectively (P=0.80, Mann-Whitney U test).
Figure 2.

Red blood cell transfusions and iron infusions pretreatment and on-bevacizumab treatment by hematologic support requirements in the 6 months prior to bevacizumab initiation. (A) All patients requiring red blood cell (RBC) transfusion before treatment (n=137) were divided into quartiles (Q1-Q4) according to their pretreatment RBC transfusion requirements. Reductions in RBC transfusions were observed following bevacizumab treatment regardless of the pretreatment disease severity. (B) All patients requiring iron infusion pretreatment (n=155) were divided into quartiles according to their pretreatment iron infusion requirements. Reductions in iron infusions were observed following bevacizumab treatment regardless of the pretreatment disease severity. Mo: months; PreTx, pretreatment; OnTx, on treatment.
The rate of VTE in HHT patients receiving bevacizumab was 1.5 events per 100 patient-years at risk. Online Supplementary Table S6 shows the venous thromboembolic events occurring in the cohort of patients.
Bevacizumab discontinuation
Twelve patients (5%) discontinued bevacizumab treatment because of adverse events. Online Supplementary Table S7 describes the adverse events prompting treatment discontinuation. Eleven patients (5%) discontinued bevacizumab because of inadequate treatment effect. Discontinuation due to excessive financial cost for patients occurred in two cases (1%).
Discussion
We present a large, international, multicenter study of systemic bevacizumab treatment of HHT-associated bleeding, observing striking improvements in bleeding and anemia with bevacizumab treatment (Figure 1, Online Supplementary Figure S1). Historically, reports on bevacizumab treatment for HHT-related bleeding have been limited to single case reports and small cohorts from individual treatment centers. Before the present study, the largest study of systemic bevacizumab for bleeding and anemia in HHT, evaluating critical hematologic outcomes such as hemoglobin, blood transfusion and iron infusion, was a 13-patient case series.23
Table 3.
Treatment-emergent adverse events (TEAE) identified by treating clinicians as likely or possibly due to bevacizumab. TEAE were evaluable in 232 patients.
We found that bevacizumab was effective at reducing HHT-associated bleeding regardless of the patients’ genotype, with an improvement in mean hemoglobin of 3.2 g/dL and resolution of anemia in two-thirds of patients. The severity of epistaxis also declined sharply, with a drop in mean ESS of 3.37 points, nearly five times the minimal clinically- important difference of 0.71 for this well-validated clinical bleeding score.29 Concurrent with the improvements in hemoglobin, RBC transfusions and iron infusions dropped precipitously: 80% and 61% of patients previously requiring RBC transfusions and iron infusions, respectively, were liberated entirely from these hematologic support modalities after 6 months of treatment. Improvement was observed irrespective of baseline bleeding or anemia severity and included the most severely afflicted patients (Figure 2). Similar improvement was observed regardless of the underlying pathogenic mutation (Online Supplementary Table S3). Most patients did not receive any additional concurrent treatments to manage HHT-associated bleeding once on bevacizumab, and outcomes of the patients who did were actually slightly worse than those who did not (Online Supplementary Table S5). This is likely reflective of less improvement with bevacizumab in these patients prompting the need for additional therapies. The fact that patients not receiving any other bleeding-directed treatments had such significant improvement in hematologic parameters with bevacizumab initiation increases our confidence in the relationship between bevacizumab treatment and the dramatic improvement observed in these parameters.
The effectiveness observed with systemic bevacizumab treatment in this study is considerably better than that found for other systemic agents evaluated for HHT-associated bleeding.30 Small randomized studies of tranexamic acid versus placebo suggest mild to moderate improvements in epistaxis severity but no improvement in hemoglobin with the active treatment.12,31 A small randomized study of oral estrogen compared with placebo found no significant improvement in epistaxis.32 Topical nasal pharmacotherapy has been similarly disappointing, with randomized studies of topical timolol, estrogen, and tranexamic acid all showing no difference in the comparison with placebo.33,34 Bevacizumab via topical nasal spray has also been evaluated, with no improvement noted in epistaxis severity in the comparison with placebo.33,35 Therefore, given the lack of other effective options and the continued lack of any Food and Drug Administration-approved treatment, our findings suggest it may be appropriate to consider systemic bevacizumab in patients with moderate-to-severe HHT-associated bleeding (Online Supplementary Table S1) without contraindications. This may be particularly true for patients with significant gastrointestinal bleeding, who constituted over half the cohort in the present study, for whom there are no studies of other pharmacological interventions. Oral anti-angiogenics such as pomalidomide36 and pazopanib37,38 are additionally under investigation for treatment of HHTassociated bleeding. Should these agents demonstrate efficacy, head-to-head studies comparing them with bevacizumab will be needed.
While initial bevacizumab induction treatment schedules were quite similar between different centers, maintenance strategies varied considerably, with some centers employing continuous scheduled maintenance and others opting for an intermittent, as-needed approach to minimize overall bevacizumab exposure. Both approaches were effective, but intermittent maintenance (which employed 75% of the overall bevacizumab dose intensity used in continuous maintenance) resulted in significantly lower mean hemoglobin and higher mean ESS than continuous maintenance (Online Supplementary Table S4). This is not unexpected given that recurrence of bleeding and/or anemia are the triggers for re-treatment with an intermittent maintenance approach. It is unclear whether the financial savings of administering less drug in the intermittent maintenance approach offsets the potential disadvantages. Additionally, although patients in this study were treated for up to 8 years without any complications specifically attributed to extended-duration treatment, the impact of indefinite bevacizumab exposure in HHT (either positive or negative) is not known. How maintenance strategy and overall dose intensity could affect this impact is therefore also unknown.
Bevacizumab was well-tolerated overall, with hypertension, proteinuria, fatigue, and myalgia/arthralgia being the most common TEAE, consistent with prior reports in HHT patients.23-27,39-42 Only 5% of patients discontinued treatment because of adverse events. An understanding of the adverse event profile of single-agent systemic bevacizumab in HHT patients is critical, given that the vast majority of prior bevacizumab studies added bevacizumab (typically at 2-3 times the common 5 mg/kg dose used in this study) to multiagent cytotoxic chemotherapy regimens and administered this combination to patients with metastatic cancer. Therefore, the adverse event rates described in the prescribing information, such as the relatively high rates of serious events including VTE or intestinal perforation, are not reflective of single-agent bevacizumab use in HHT patients. This point is highlighted by the VTE rate in this study of 2%, with a median duration of bevacizumab exposure considerably longer than that of most cancer studies finding VTE rates in the 5-10% range. Furthermore, two out of five of the VTE observed were provoked events immediately following major joint replacement surgery (Online Supplementary Table S6). We observed no central nervous system bleeding, hemoptysis, pulmonary hemorrhage, or intestinal perforation. Additionally, while anticoagulation in HHT patients can be very challenging and dramatically exacerbate bleeding, patients with VTE in this study receiving anticoagulation did well with concurrent bevacizumab treatment without significantly increased bleeding (Online Supplementary Table S6).
Our study has several limitations consistent with its retrospective nature. These include center variability in the management of HHT and variability in the use and availability of adjunctive measures to control bleeding. The lack of randomization allows for the presence of confounding factors that could have resulted in clinical improvement independent of bevacizumab use. However, the fact that significant improvements in all outcome measures occurred following initiation of bevacizumab treatment increases our confidence that bevacizumab was the cause of the clinical improvements. Additionally, we analyzed known confounders such as concurrent use of antifibrinolytics and receipt of local hemostatic procedures and found no significant difference in outcomes between the two groups. To minimize the impact of patient heterogeneity in this disease, we employed a pre/post-treatment analytical design with paired analyses such that each patient served as his or her own internal control in our effectiveness analyses. Adverse event reporting was limited by the retrospective design, but we expect it is unlikely that any serious adverse events would not have been appropriately documented. Finally, our study evaluated number of iron infusion events, not exact milligrams of elemental iron infused. Although less precise, we believe that the observed reduction in iron infusions to be valid for two reasons: the median number of iron infusions went from six before treatment to zero after treatment (and the amount of elemental iron in 0 iron infusions is none) and more importantly, the reduction in iron infusions paralleled the dramatic reduction in RBC transfusions.
In conclusion, we observed that systemic bevacizumab was effective in the management of severe HHT-related epistaxis and gastrointestinal bleeding in a cohort of 238 HHT patients. While data from large, randomized prospective studies are needed to confirm these findings, in this large observational study, bevacizumab was associated with significant improvements in hemoglobin and ESS, along with significant reductions in the need for RBC transfusion and intravenous iron infusion. Improvement was similar regardless of the underlying pathogenic mutation. Following an initial sequence of induction treatments, continuous scheduled maintenance therapy and intermittent as-needed maintenance therapy were both reasonable to maintain treatment effect. Hypertension, fatigue, proteinuria, and myalgia/arthralgia were the most common TEAE; VTE and treatment discontinuation for adverse events were rare, occurring in 2% and 5% of patients, respectively.
Supplementary Material
Acknowledgments
HA-S is the recipient of the National Hemophilia Foundation- Shire Clinical Fellowship Award, the Harvard KL2/Catalyst Medical Research Investigator Training Award, and the American Society of Hematology Scholar Award.
References
- 1.Fuchizaki U, Miyamori H, Kitagawa S, Kaneko S, Kobayashi K. Hereditary haemorrhagic telangiectasia (Rendu-Osler-Weber disease). Lancet. 2003;362(9394):1490-1494. [DOI] [PubMed] [Google Scholar]
- 2.Kritharis A, Al-Samkari H, Kuter DJ. Hereditary hemorrhagic telangiectasia: diagnosis and management from the hematologist's perspective. Haematologica. 2018;103(9):1433-1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kwaan HC, Silverman S. Fibrinolytic activity in lesions of hereditary hemorrhagic telangiectasia. Arch Dermatol. 1973;107(4): 571-573. [PubMed] [Google Scholar]
- 4.Watanabe M, Hanawa S, Morishima T. Fibrinolytic activity in cutaneous lesions of hereditary hemorrhagic telangiectasia. Nihon Hifuka Gakkai Zasshi. 1985;95(1):11. [PubMed] [Google Scholar]
- 5.Shovlin CL. Hereditary haemorrhagic telangiectasia: pathophysiology, diagnosis and treatment. Blood Rev. 2010;24(6):203-219. [DOI] [PubMed] [Google Scholar]
- 6.Letteboer TG, Mager HJ, Snijder RJ, et al. Genotype-phenotype relationship for localization and age distribution of telangiectases in hereditary hemorrhagic telangiectasia. Am J Med Genet A. 2008;146A(21):2733-2739. [DOI] [PubMed] [Google Scholar]
- 7.Abdalla SA, Geisthoff UW, Bonneau D, et al. Visceral manifestations in hereditary haemorrhagic telangiectasia type 2. J Med Genet. 2003;40(7):494-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kasthuri RS, Montifar M, Nelson J, et al. Prevalence and predictors of anemia in hereditary hemorrhagic telangiectasia. Am J Hematol. 2017. Jun 22. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.OS AA, Friedman CM, White RI. The natural history of epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope. 1991; 101(9):977-980. [DOI] [PubMed] [Google Scholar]
- 10.Donaldson JW, McKeever TM, Hall IP, Hubbard RB, Fogarty AW. Complications and mortality in hereditary hemorrhagic telangiectasia: a population-based study. Neurology. 2015;84(18):1886-1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kjeldsen AD, Vase P, Green A. [Hereditary hemorrhagic telangiectasia. A populationbased study on prevalence and mortality among Danish HHT patients]. Ugeskr Laeger. 2000;162(25):3597-3601. [PubMed] [Google Scholar]
- 12.Gaillard S, Dupuis-Girod S, Boutitie F, et al. Tranexamic acid for epistaxis in hereditary hemorrhagic telangiectasia patients: a European cross-over controlled trial in a rare disease. J Thromb Haemost. 2014;12(9): 1494-1502. [DOI] [PubMed] [Google Scholar]
- 13.Ingrosso M, Sabba C, Pisani A, et al. Evidence of small-bowel involvement in hereditary hemorrhagic telangiectasia: a capsule-endoscopic study. Endoscopy. 2004;36(12):1074-1079. [DOI] [PubMed] [Google Scholar]
- 14.Iyer VN, Brinjikji W, Apala D, et al. Impact of age on outcomes in hospitalized patients with hereditary hemorrhagic telangiectasia. Adv Hematol. 2018;2018:4798425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brinjikji W, Wood CP, Lanzino G, et al. High rates of bleeding complications among hospitalized patients with hereditary hemorrhagic telangiectasia in the United States. Ann Am Thorac Soc. 2016;13(9):1505-1511. [DOI] [PubMed] [Google Scholar]
- 16.Letarte M, McDonald ML, Li C, et al. Reduced endothelial secretion and plasma levels of transforming growth factor-beta1 in patients with hereditary hemorrhagic telangiectasia type 1. Cardiovasc Res. 2005;68(1):155-164. [DOI] [PubMed] [Google Scholar]
- 17.Cirulli A, Liso A, D'Ovidio F, et al. Vascular endothelial growth factor serum levels are elevated in patients with hereditary hemorrhagic telangiectasia. Acta Haematol. 2003;110(1):29-32. [DOI] [PubMed] [Google Scholar]
- 18.Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335-2342. [DOI] [PubMed] [Google Scholar]
- 19.Michels S, Rosenfeld PJ, Puliafito CA, Marcus EN, Venkatraman AS. Systemic bevacizumab (Avastin) therapy for neovascular age-related macular degeneration twelve-week results of an uncontrolled open-label clinical study. Ophthalmology. 2005;112(6):1035-1047. [DOI] [PubMed] [Google Scholar]
- 20.Bose P, Holter JL, Selby GB. Bevacizumab in hereditary hemorrhagic telangiectasia. N Engl J Med. 2009;360(20):2143-2144. [DOI] [PubMed] [Google Scholar]
- 21.Oosting S, Nagengast W, de Vries E. More on bevacizumab in hereditary hemorrhagic telangiectasia. N Engl J Med. 2009;361(9):931; author reply 931-932. [DOI] [PubMed] [Google Scholar]
- 22.Brinkerhoff BT, Poetker DM, Choong NW. Long-term therapy with bevacizumab in hereditary hemorrhagic telangiectasia. N Engl J Med. 2011;364(7):688-689. [DOI] [PubMed] [Google Scholar]
- 23.Al-Samkari H, Kritharis A, Rodriguez-Lopez JM, Kuter DJ. Systemic bevacizumab for the treatment of chronic bleeding in hereditary haemorrhagic telangiectasia. J Intern Med. 2019;285(2):223-231. [DOI] [PubMed] [Google Scholar]
- 24.Al-Samkari H. Systemic bevacizumab for hereditary hemorrhagic telangiectasia: considerations from observational studies. Otolaryngol Head Neck Surg. 2019;160(2): 368. [DOI] [PubMed] [Google Scholar]
- 25.Iyer VN, Apala DR, Pannu BS, et al. Intravenous bevacizumab for refractory hereditary hemorrhagic telangiectasia-related epistaxis and gastrointestinal bleeding. Mayo Clin Proc. 2018;93(2):155-166. [DOI] [PubMed] [Google Scholar]
- 26.Guilhem A, Fargeton AE, Simon AC, et al. Intra-venous bevacizumab in hereditary hemorrhagic telangiectasia (HHT): a retrospective study of 46 patients. PLoS One. 2017;12(11):e0188943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Epperla N, Kapke JT, Karafin M, Friedman KD, Foy P. Effect of systemic bevacizumab in severe hereditary hemorrhagic telangiectasia associated with bleeding. Am J Hematol. 2016;91(6):E313-314. [DOI] [PubMed] [Google Scholar]
- 28.Epperla N, Kleman A, Karafin M, Foy P. Retreatment versus extended treatment strategy of systemic bevacizumab in hereditary hemorrhagic telangiectasia: which is better? Ann Hematol. 2018;97(9):1727-1729. [DOI] [PubMed] [Google Scholar]
- 29.Yin LX, Reh DD, Hoag JB, et al. The minimal important difference of the epistaxis severity score in hereditary hemorrhagic telangiectasia. Laryngoscope. 2016;126(5): 1029-1032. [DOI] [PubMed] [Google Scholar]
- 30.Hsu YP, Hsu CW, Bai CH, Cheng SW, Chen C. Medical treatment for epistaxis in hereditary hemorrhagic telangiectasia: a metaanalysis. Otolaryngol Head Neck Surg. 2018;160(1):22-35. [DOI] [PubMed] [Google Scholar]
- 31.Geisthoff UW, Seyfert UT, Kubler M, Bieg B, Plinkert PK, Konig J. Treatment of epistaxis in hereditary hemorrhagic telangiectasia with tranexamic acid - a double-blind placebo-controlled cross-over phase IIIB study. Thromb Res. 2014;134(3):565-571. [DOI] [PubMed] [Google Scholar]
- 32.Vase P. Estrogen treatment of hereditary hemorrhagic telangiectasia. A double-blind controlled clinical trial. Acta Med Scand. 1981;209(5):393-396. [DOI] [PubMed] [Google Scholar]
- 33.Whitehead KJ, Sautter NB, McWilliams JP, et al. Effect of topical intranasal therapy on epistaxis frequency in patients with hereditary hemorrhagic telangiectasia: a randomized clinical trial. JAMA. 2016;316(9):943-951. [DOI] [PubMed] [Google Scholar]
- 34.Dupuis-Girod S, Pitiot V, Bergerot C, et al. Efficacy of TIMOLOL nasal spray as a treatment for epistaxis in hereditary hemorrhagic telangiectasia. A double-blind, randomized, placebo-controlled trial. Sci Rep. 2019;9(1):11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dupuis-Girod S, Ambrun A, Decullier E, et al. Effect of bevacizumab nasal spray on epistaxis duration in hereditary hemorrhagic telangectasia: a randomized clinical trial. JAMA. 2016;316(9):934-942. [DOI] [PubMed] [Google Scholar]
- 36.Samour M, Saygin C, Abdallah R, Kundu S, McCrae KR. Pomalidomide in hereditary hemorrhagic telangiectasia: interim results of a phase I study. Blood. 2016;128(22):210. [Google Scholar]
- 37.Faughnan ME, Gossage JR, Chakinala MM, et al. Pazopanib may reduce bleeding in hereditary hemorrhagic telangiectasia. Angiogenesis. 2019;22(1):145-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parambil JG, Woodard TD, Koc ON. Pazopanib effective for bevacizumab-unresponsive epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope. 2018;128(10):2234-2236. [DOI] [PubMed] [Google Scholar]
- 39.Buscarini E, Botella LM, Geisthoff U, et al. Safety of thalidomide and bevacizumab in patients with hereditary hemorrhagic telangiectasia. Orphanet J Rare Dis. 2019;14(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dupuis-Girod S, Ginon I, Saurin JC, et al. Bevacizumab in patients with hereditary hemorrhagic telangiectasia and severe hepatic vascular malformations and high cardiac output. JAMA. 2012;307(9):948-955. [DOI] [PubMed] [Google Scholar]
- 41.Al-Samkari H, Albitar HA, Olitsky SE, Clancy MS, Iyer VN. An international survey to evaluate systemic bevacizumab for chronic bleeding in hereditary haemorrhagic telangiectasia. Haemophilia. 2020;26(6): 1038-1045. [DOI] [PubMed] [Google Scholar]
- 42.Al-Samkari H, Albitar HA, Olitsky SE, Clancy MS, Iyer VN. Systemic bevacizumab for high-output cardiac failure in hereditary hemorrhagic telangiectasia: an international survey of HHT centers. Orphanet J Rare Dis. 2019;14(1):256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.