

Abstract
R-loops are stable chromatin structures comprising a DNA:RNA hybrid and a displaced single-stranded DNA. R-loops have been implicated in gene expression and chromatin structure, as well as in replication blocks and genome instability. Here, we conducted a genome-wide identification of R-loops and identified more than 700,000 R-loop peaks in the maize (Zea mays) genome. We found that sense R-loops were mainly enriched in promoters and transcription termination sites and relatively less enriched in gene bodies, which is different from the main gene-body localization of sense R-loops in Arabidopsis and Oryza sativa. At the chromosome scale, maize R-loops were enriched in pericentromeric heterochromatin regions, and a significant portion of R-loops were derived from transposable elements. In centromeres, R-loops preferentially formed within the binding regions of the centromere-specific histone CENH3, and centromeric retrotransposons were strongly associated with R-loop formation. Furthermore, centromeric retrotransposon R-loops were observed by applying the single-molecule imaging technique of atomic force microscopy. These findings elucidate the fundamental character of R-loops in the maize genome and reveal the potential role of R-loops in centromeres.
During transcription, the nascent RNA sometimes threads back to hybridize with the transiently accessible template strand to form an R-loop, which includes the DNA:RNA duplex and the displaced nontemplate DNA strand (Belotserkovskii et al. 2018). Structurally, the hybrid adopts an intermediate A/B conformation, carrying more stability than either double-stranded DNA (dsDNA, B form) or dsRNA (A form) (Shaw and Arya 2008). R-loop structures have long been considered to be mere “by-products” of transcription that occur exclusively in cis, at the site of transcription (Aguilera and García-Muse 2012). However, the development of DRIP-seq (DNA:RNA immunoprecipitation by S9.6, followed by sequencing) provided an overall picture of R-loop distribution in human cells, which showed that these structures are far more prevalent than expected (Ginno et al. 2012).
Growing evidence suggests that R-loops play important roles in cellular processes. For instance, R-loops (1) are implicated in numerous stimulatory and inhibitory effects upon transcription by regulating chromatin structure, DNA modification, and recruiting transcriptional regulators (Niehrs and Luke 2020), (2) promote the establishment of centromere function to ensure accurate chromosome segregation (Kabeche et al. 2018; Liu et al. 2020), (3) drive recombination at short telomeres to maintain them to achieve immortality (Graf et al. 2017; Feretzaki et al. 2020), and (4) initiate replication of bacterial plasmids, mitochondrial DNA, and phage genomes (Kreuzer and Brister 2010; Pohjoismäki et al. 2010). However, R-loops can also be detrimental to genome instability because the single-stranded DNA (ssDNA) in the R-loop structure is more prone to nucleotide changes and strand breakage (Aguilera and García-Muse 2012). This damage is strongly associated with blocks to replication fork progression and transcription-replication conflicts (Gan et al. 2011; Helmrich et al. 2011; Stork et al. 2016). To minimize the harmful effects, cells encode a number of helicases to resolve R-loops once they form. Among these helicases, Ribonuclease H (RNase H) enzymes play a key role in all cells in specifically degrading RNA within DNA:RNA hybrids (Wahba et al. 2011; Nguyen et al. 2017; Yang et al. 2017).
Heterochromatin is a compacted state of chromatin that plays an essential role in chromatin condensation, epigenetic regulation, and sister chromatid cohesion at centromeres (Allshire and Madhani 2018). Heterochromatin is categorized into two major types, constitutive and facultative. Both types are transcriptionally repressed and exhibit high nucleosome density (Grewal and Jia 2007). Constitutive heterochromatin, which consists of repetitive tandem repeats, is mainly formed at the gene-poor regions of pericentromeres (Saksouk et al. 2015). As the hallmarks of heterochromatin, DNA methylation and H3K9me2 combinatorically maintain the epigenetic silencing of transposons (Underwood et al. 2017). Recently, R-loops were proposed to play an important role in the regulation of heterochromatin formation (Nakama et al. 2012). Heterochromatic noncoding RNAs (ncRNAs) are retained on chromatin via the formation of DNA:RNA hybrids, which provide a platform for the RNAi-mediated heterochromatin assembly in yeast (Nakama et al. 2012). A recent study also showed R-loops are tightly connected with the repressive chromatin compaction mark, histone H3 serine 10 phosphorylation, in both yeast and human cells at centromeres and pericentromeric regions (Castellano-Pozo et al. 2013). Most of these studies were conducted in yeast and animals. However, the association of R-loops and heterochromatin in plants is largely unknown.
Centromeres are the fundamental chromosomal structure where kinetochores form and microtubules attach to segregate eukaryotic chromosomes during cell division. Centromeres are defined by the presence of CENH3, a variant of histone H3, which is also known as CENPA in animals (Henikoff et al. 2001; Dhatchinamoorthy et al. 2018). In animals, centromeric DNA is comprised of megabases of tandemly repeated “satellite” sequences (Manuelidis 1978), whereas in plants, multiple retrotransposons are interspersed with tandem repeats in the centromeric regions. For example, in maize, tandemly arranged CentC repeats and interspersed centromeric retrotransposons (CRM) are the major DNA components of maize centromeres (Birchler and Han 2009). One of the common features of centromere sequences is their transcription into noncoding centromere RNAs (Talbert and Henikoff 2018). These ncRNAs that are transcribed from centromeric repeats can form R-loops, which are regulated in a cell cycle-dependent manner (Kabeche et al. 2018; Liu et al. 2020). In human cells, the displaced ssDNA at the centromere R-loop is bound by replication protein A (RPA) and recruits ATR serine/threonine kinase (ATR), leading to the activation of aurora kinase B (AURKB), which promotes faithful chromosome segregation (Kabeche et al. 2018). In maize, circular RNA derived from centromeric CRM retroelements binds to the centromere through R-loops, thereby formatting chromatin loops to regulate CENH3 loading (Liu et al. 2020). These studies imply that R-loops might be involved in centromere organization and centromere maintenance.
Growing evidence suggests that R-loops are associated with functional roles in plants. They have been found to play a role in gene expression and plant development (Sun et al. 2013; Xu et al. 2017, 2020; Yang et al. 2017; Fang et al. 2019; Yuan et al. 2019). High-throughput genome-wide R-loop identification in plants was only studied in Arabidopsis thaliana and Oryza sativa that possess small genome sizes and a relative paucity of repeats (Xu et al. 2017; Fang et al. 2019). Maize has a moderately large genome (2.3 Gbp) with a high concentration of repetitive sequences located in heterochromatic blocks and extensively interspersed in the euchromatic portion of the genome (Schnable et al. 2009; Jiao et al. 2017), which makes maize an ideal species for investigating the association between repetitive elements and R-loop formation.
In this study, we present genome-wide R-loop maps of maize leaf generated by ssDRIP-seq (single-strand DNA ligation-based library construction from DNA:RNA hybrid immunoprecipitation, followed by sequencing) and determine the general characteristics of the R-loop pattern in the maize genome. We studied the relationships between R-loops and repetitive sequences in maize, such as tandem repeats and TEs. We also provide the first R-loop observation by atomic force microscopy (AFM) in plants.
Results
Genome-wide identification of R-loops in maize
To investigate the genome-wide R-loop distribution in maize, we used the reported ssDRIP-seq approach (Xu et al. 2017) on genomic DNA from two biological replicates of young leaf of inbred B73, as well as the same samples but treated with RNase H (negative control) (Supplemental Fig. S1A). R-loop distribution was highly replicable, with the Pearson correlation coefficient between two replicates reaching 0.86, whereas the R-loops were almost undetectable in the RNase H-treatment sample (Fig. 1A; Supplemental Fig. S1B–D).
Figure 1.

Genome-wide detection of R-loops in maize by ssDRIP-seq. (A) A representative region from Chromosome 6 showing the ssDRIP-seq data. Line 1, gene annotations; line 2, unstranded (unstrd) R-loop signal, normalized to the genome-wide mean; line 3, unstrd R-loop peaks; line 4, normalized wR-loops signal (green); line 5, wR-loop peaks; line 6, normalized cR-loop signal (purple); line 7, cR-loops peaks; line 8, DRIP signal of RNase H treatment (gray), normalized to the genome-wide mean of unstrd R-loops; line 9, input, normalized to the genome-wide mean. (B) Location analysis of unstranded R-loop peaks (upper) compared with the expected genomic distribution (lower) on the chromosome. The maize genome was characterized into seven classes that included six classes of genic regions (promoter, 5′ UTR, 3′ UTR, coding exon, intron, and terminator) and intergenic regions. Promoter, −1 kb to 100 bp of TSS; terminator, −100 bp to 1 kb of TTS. (C) Relative enrichment of unstranded R-loop peaks in different genomic elements. The dashed line represents enrichment fold = 1.0. Permutation test; asterisks denote the observed value was >90% of the permutation value. (D) DNA motif in the peak regions of unstranded R-loops, wR-loops, and cR-loops that were identified by MEME-ChIP. E-values are provided at the top. (E) Metaplots of sense (orange) and antisense (cyan) R-loop peaks centered on non-TE genes (B73_RefGen_v4), 95% mean.
In total, ssDRIP-seq identified 504,205 Watson DNA strand-related R-loops (wR-loops) and 510,342 Crick DNA strand-related R-loops (cR-loops) (Supplemental Fig. S2A), which constituted ∼10% of the maize genome (Supplemental Fig. S2B). Only 65,084 wR-loops and cR-loops peaks were mapped in the same genomic regions (dR-loops) (Supplemental Fig. S2A,C). Most of the peaks were 100 to 500 base pairs (bp) long (Supplemental Fig. S2D), which is consistent with the peak size distribution of R-loops in the Arabidopsis genome (Xu et al. 2017). These peaks were further validated by DRIP-qPCR assay (DRIP followed by quantitative PCR) (Supplemental Fig. S3). Association analysis of R-loops with various genomic features indicated that R-loops were enriched in the promoter and 5′ untranslated regions (UTRs) of the genome (Fig. 1B,C). De novo motif analysis of R-loop peaks using the MEME software programs identified a GA-rich motif (Fig. 1D), which was consistent with the R-loops strong correlation with both GC (DNA strand bias for GC composition) and AT skews (DNA strand bias for AT composition) (Supplemental Fig. S4). Altogether, maize R-loops showed conserved sequence characters compared with those in humans and Arabidopsis (Ginno et al. 2012; Xu et al. 2017).
In our data, we identified numerous antisense R-loops in the maize genome, even more than sense R-loops (Supplemental Fig. S5A,B). We classified the non-TE genes into four categories: (1) genes with sense only R-loops (SO-R-loops); (2) genes with antisense only R-loops (ASO-R-loops); (3) genes with both sense and antisense R-loops (S-AS-R-loops); and (4) genes without R-loops (No-R-loops). We found that more than 24,000 non-TE genes exhibited S-AS-R-loops formation (Supplemental Fig. S5C). About 3002 and 8093 non-TE genes had SO-R-loops and ASO-R-loops, respectively (Supplemental Fig. S5C). After examining the expression level of non-TE genes with different types of R-loops, we found that the average expression level of non-TE genes with ASO-R-loops or S-AS-R-loops was higher than that of non-TE genes with SO-R-loops or without R-loops (Supplemental Fig. S5D). To assess the enrichment of R-loops relative to non-TE genes, we plotted the sense and antisense R-loop read densities across non-TE genes and their 1-kb surrounding regions. Different from the gene-body localization of sense R-loops in Arabidopsis and Oryza sativa, maize sense R-loops mainly formed in both promoters and transcription termination sites (TTSs), and relatively less formed in gene bodies, indicating a species-specific R-loop pattern (Fig. 1E). The antisense R-loop localization pattern was similar to that in Arabidopsis and Oryza sativa, which occurred around the transcription start sites (TSSs) compared to flanking regions (Fig. 1E).
R-loops are enriched in pericentromeric regions
We next analyzed the distribution pattern of R-loop peaks along each chromosome. Although R-loops are thought to be localized to euchromatin and are implicated in transcription (Xu et al. 2020), we found a gentle, chromosome-level trend of R-loop increasing toward pericentromeres and centromeres (Fig. 2A; Supplemental Fig. S6). Plant centromeres are surrounded by retrotransposon-dense pericentromeric heterochromatin that is epigenetically silenced by H3K9me2 and DNA methylation in CG and CHG sequence contexts (where “H” indicates A, C, or T, respectively) but are depleted of CHH methylation, gene density, and the gene-associated euchromatic modification H3K56ac (Gent et al. 2014; Zhao et al. 2016). Because of pericentromeric R-loop enrichment, we observed positive correlations with retrotransposon density, DNA methylation in CG and CHG sequence contexts, as well as the heterochromatic histone modification H3K9me2 around the centromeric regions (Fig. 2A; Supplemental Fig. S6A).
Figure 2.

R-loops are enriched in pericentromeric regions. (A) Map of R-loop peaks on Chromosome 1. Diagram of Chromosome 1 with the pericentromeres shaded in black (top). The x-axis represents positions on Chromosome 1. The y-axis represents numbers of R-loop peaks. See Supplemental Figure S6, for the other chromosomes. Shown below are patterns of genes expressed in the leaf (yellow, genes/1Mb), TEs (DNA-TE/1Mb, dark yellow; RNA-TEs/1Mb, dark blue), histone modification (H3K9me2 [purple, log2{ChIP/input}], H3K56ac [light blue, log2{ChIP/input}]), DNA methylation (proportion methylated in CG [orange], CHG [gray], and CHH [dark blue] contexts/500-kb). (B) R-loops (green) and CRM1 (red) were costained on pachytene stage chromosomes. RNase H-treatment abolishes the R-loop signals on the pachytene chromosomes. The dashed line indicates the section of chromatin used for quantifying signal intensity. Scale bar = 10 μm. (C) Fluorescent profiles indicates that R-loops mainly localized to both sides of the CRM1 signals. Twelve axis sections of 100 pixels centered on pericentromeric heterochromatin were used to quantify signal intensity.
The meiotic pachytene chromosomes, which are often more than 10 times longer than somatic metaphase chromosomes (Koo and Jiang 2009), provide an efficient way for the observation of R-loop distribution. Thus, we analyzed spread nuclei at the pachytene stage that were immunostained with anti-S9.6 antibody. We found that the R-loops were spread on the chromosomes, but the signals showed strength differences across chromosomes (Fig. 2B). To assess R-loop signals through the centromeres, we performed fluorescence in situ hybridization for CRM1 and co-immunostained for anti-S9.6 antibody. We tracked and quantified axial R-loop, DAPI, and CRM1 signals as they traversed pericentromeric heterochromatin. We centered the analysis on pericentromeric heterochromatin over a distance of 100 pixels in 12 sections from a total of six meiocytes. We observed that mean R-loop and CRM1 signal intensity were anticorrelated over the tracked regions (rs = −0.07) (Fig. 2C). Though derived from different development stages, these cytological data are consistent with ssDRIP-seq enrichment correlating with pericentromeric heterochromatin, except that the ssDRIP-seq data did not reveal the hypo-R-loop level associated with centromere chromatin (Fig. 2A).
Transposable elements are the primary source of R-loops
The pericentromeric heterochromatin is mainly composed of TEs (Schnable et al. 2009), and the results that R-loops are enriched in pericentromeric regions prompted us to investigate whether TEs contributed to R-loop formation. Based on the mechanism of transposition, TEs are categorized into two major classes: class I (retroelement) transposing through reverse transcription of an RNA intermediate (copy and paste mechanism); and class II (DNA element) using a DNA intermediate (cut and paste mechanism) to transpose (Wicker et al. 2007). These two types of TEs can be further classified into various families based on their structure, encoded genes, and phylogeny, and each family of TEs has its own functional properties (Wicker et al. 2007). Therefore, we examined our R-loop data to identify which may be derived from a TE (TE-R-loops). To do so, we intersected the R-loop peaks with the maize TE annotation. Overall, a total of 513,106 TE-R-loops were identified, which account for 66.6% of the 770,889 total R-loops (Supplemental Table S1). Retrotransposons and DNA transposons account for 458,807 (89.4%) and 54,399 (10.6%) of the TE-R-loops, respectively (Supplemental Table S1). Because the copy-number of each element differs in the maize genome, enrichment analysis was performed to examine the significance of contributions of different TEs to R-loops. We estimated the proportion length among all annotated TEs including LTR/Gypsy, LTR/Copia, and other element superfamilies. We then compared these percentages to the proportion length among TE-R-loops. We found that, although LTR/Gypsy and LTR/Copia were the two major contributors to the TE-R-loops, they gave rise to TE-R-loops at roughly the expected proportion (24.49% vs. 26.87% for Copia and 52.49% vs. 51.90% for Gypsy), relative to their representation in the TEs (Supplemental Table S1). Particularly notable is the fact that Helitron and CACTA DNA transposons produced TE-R-loops in our data set at ∼1.23- and 1.75-fold higher rates than expected (Supplemental Table S1). Altogether, our results revealed that most maize R-loops derive from TEs, with Helitron and CACTA DNA transposons being most significantly enriched.
Analysis of tandem repeat sequences in ssDRIP-seq reads
In addition to the TEs, the maize genome contains a large number of tandemly repeated arrays (Plohl et al. 2008). These arrays include the centromere-associated CentC repeat of 156 bp (Ananiev et al. 1998a), a 45S ribosomal DNA (rDNA) repeat of 9349 bp, and a 5S rDNA repeat of 341 bp (Rivin et al. 1986). In addition, maize contains knob repeats that are composed primarily of two classes of tandemly repeated sequences, the major knob180 repeat (180 bp), and the minor TR-1 knob repeat (350 bp) (Peacock et al. 1981; Ananiev et al. 1998b). Tandem repeats cause sequencing and genome assembly challenges. Reads mapping to such regions with commonly used mapping programs for high-throughput sequence analysis, including BWA and Bowtie 2, often lead to false-positive signals. To investigate the R-loop distribution characteristics in these tandem repeats, we used a different strategy—the filtered, trimmed, and adapter-free DNA fragment reads of ssDRIP-seq and input were subjected to BLAST analysis (Altschul et al. 1990) against the knob180, TR-1, 5S, and 45S rDNAs, CentC tandem repeat, as well as another two centromeric-specific retrotransposons CRM1 and CRM2. The resulting read counts for each tandem repeat type and retrotransposons in ssDRIP-seq and input were then normalized to the total number of reads to obtain the percentage of reads that align to each tandem repeat and retrotransposons.
We found that about 11% of the total ssDRIP-seq reads could be mapped onto the 45S rDNA, which is much more abundant than other types of tandem repeats (Fig. 3A). To address this difference, fold enrichment was calculated through comparing the percentage of ssDRIP-seq reads matching each tandem repeat to the percentage of input reads. The knob180, TR-1, and CentC repeats did not show R-loop enrichment, whereas the 5S and 45S rDNA repeats have over three- and sixfold enrichment, respectively, relative to input (Fig. 3B). To further provide cytological support of R-loop enrichment in 5S and 45S repeats, we prepared mitotic chromosome spreads that were immunostained for anti-S9.6 antibody and a fluorescence in situ hybridization probe for the 45S and 5S probe. As expected, we found R-loops were strongly accumulated at the 45S rDNA region (Fig. 3C). However, we did not detect obvious R-loop signals in the 5S region as revealed by the extremely low ssDRIP-seq percentage reads across 5S repeats (Fig. 3A; Supplemental Fig. S7).
Figure 3.
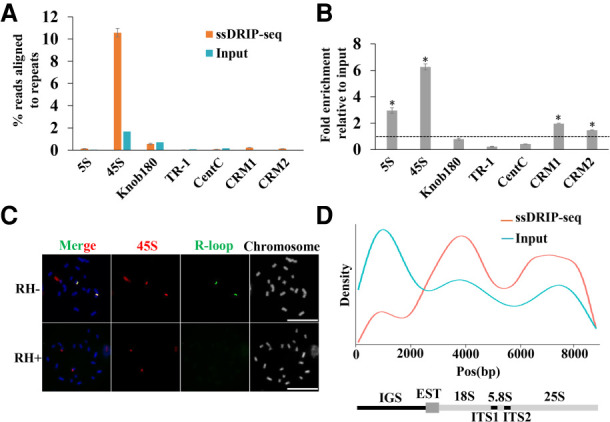
R-loops for tandem repeat sequences. (A,B) Tandem repeat and CRM1/2 consensus sequences were analyzed against the trimmed ssDRIP-seq and input reads using BLAST, independent of mapping to the reference genome, to estimate the abundance of these repetitive sequences in the ssDRIP-seq reads. (A) The percentage of reads corresponding to each tandem repeat sequence and CRM1/2. Data are means ± standard errors (SE) of two biological ssDRIP-seq replicates. (B) The fold enrichment of each tandem repeat and CRM1/2 relative to the amount in input. Data were means ± SE of two biological ssDRIP-seq replicates. Permutation test; asterisks denote the observed value was >90% of the permutation value. (C) R-loops (green) were colocalized to 45S (red) loci on spread chromosomes. RNase H-treatment abolished the R-loop signals on the spread chromosomes. Scale bar = 10 μm. (D) ssDRIP-seq data mapped on the maize 45S rDNA reference sequence shows two enriched regions. Below is the entire 45 rDNA gene locus with intergenic spacer region (IGS).
The 45S rDNA of maize includes the 5.8S, 18S, and 25S rDNA genes, the internal transcribed spacer (ITS1 and ITS2), and the intergenic spacer IGS. To determine which structural elements in 45S are the major contributors for R-loop formation, we used consensus sequences for the 45S rDNA to query trimmed ssDRIP-seq and input reads using BLAST software and a nonstringent E-value to allow for variants of each repeat. We found that the input reads were enriched in the IGS region, possibly due to overrepresentation of IGS sequences in the genome (Fig. 3D). However, R-loops were detected all along the rDNA but peaked at the region of the 18S gene body, dropped over the ITS, increased over the 25S, and declined over the terminator region (Fig. 3D). These results are consistent with the reports in wild-type yeast cells, which represent a high transcription rate.
Contribution of CRM1 and CRM2 to R-loop formation in the maize centromeres
Immunofluorescence assays revealed that there were R-loop signals in the core centromere domains, though not as strong as in pericentromeres (Fig. 2B). We thus investigated what kinds of sequences in the core centromeres contribute to R-loop formation. The maize core centromeres contain highly repetitive DNA sequences, including several centromeric retrotransposons (CRM1 and CRM2) and the CentC tandem repeat (Birchler and Han 2009). The CentC tandem repeats did not show R-loop enrichment, whereas CRM1 and CRM2 were enriched in R-loops (Fig. 3B). CRM1 and CRM2 are members of the Ty3-gypsy family of retrotransposons that consist of two identical long terminal repeats (LTRs) at their 5′ and 3′ UTR and an open reading frame encoding a polyprotein (Sharma et al. 2008). To determine which structural elements in CRM1 and CRM2 are the major contributions for R-loop formation, we analyzed the distribution of R-loops along the full length of the CRM1 and CRM2 retrotransposons. We also mapped maize ESTs to CRM1 and CRM2 elements to identify the TSSs within them. A total of 2025 and 1188 maize ESTs can be mapped to CRM1 and CRM2 elements. We found that both sense and antisense R-loops localized to the TSSs, as defined by the distribution of ESTs (Fig. 4A,B; Supplemental Fig. S8).
Figure 4.

Contribution of CRM1 and CRM2 to R-loop formation in the maize centromere. (A,B) Distribution of R-loops and ESTs on CRM1 (A) and CRM2 (B). The blue line represents the distribution of 10,000 randomly selected 150-bp reads that were used as controls for comparison. The red and green lines represent the distribution of ESTs and R-loops, respectively. The entire CRM1 and CRM2 elements are shown below with domains highlighted. (RT) Reverse transcriptase, (RH) RNase H, (INT) integrase. (C) Diagram of the main steps of the in vitro transcription system. (RE) Restriction enzyme, (RH) RNase H, (RA) RNase A (Pro) promoter, (Ter) terminator, (RNAP) RNA polymerase II. (D,E) The circular plasmids pET-30a-CRM1/CRM2 were transcribed in vitro and treated or not with RNase H or RNase A as indicated. After purification, the DNA was run on agarose gels (D) or spotted on a membrane to perform dot blot analysis with the anti-S9.6 antibody (E). (F) pET-30a-CRM1/CRM2 and pET-30a-Control were processed as in Figure 4C and visualized using AFM. Magnifications of the molecules are identified by arrows. Scale bar = 200 nm.
To gain a better insight into the architecture of R-loops formed at CRM1 and CRM2, we used an in vitro transcription system to characterize the R-loop formation on CRM1 and CRM2 (Fig. 4C). The ∼4000-bp sequences from the CRM1 and CRM2 DNA of B73, containing the presumed R-loop region, were used as the in vitro transcription template driven by T7 RNA polymerase (Fig. 4C). As reported previously (Carrasco-Salas et al. 2019; Pan et al. 2020), R-loop formation on circular templates induced a pronounced mobility shift under electrophoresis in a native gel. Treatment with RNase H, which specifically digests the R-loop structure, completely reverted this upward shift (Fig. 4D). The formation of stable R-loops at CRM1 and CRM2 was also validated by dot blots with the anti-S9.6 antibody (Fig. 4E).
To further characterize the defined region of R-loops at CRM1 and CRM2, we applied AFM on mica surfaces to visualize R-loop formation on linear CRM1 and CRM2 fragments. We also constructed a control CRM1 sequence vector without R-loop formation in our ssDRIP-seq data. The circular DNA that contained or did not contain the R-loop was linearized with KpnI and HindIII after in vitro transcription to generate the CRM1 and CRM2 sequences and the plasmid backbone (Fig. 4C). Both fragments were then purified and imaged together on mica surfaces. The displaced ssDNA in R-loops allowed us to unambiguously identify R-loop structures in AFM images (Fig. 4F). We observed R-loops near one of the ends of the DNA (Fig. 4F), which is consistent with the distribution of ssDRIP-seq reads on CRM1 and CRM2 (Fig. 4A,B). In contrast, we did not observe an R-loop structure in the CRM1 control in AFM (Fig. 4F), indicating that not all transcribed elements can form R-loops in the in vitro transcription system. By applying AFM, Carrasco-Salas and colleagues found three different types of R-loop objects that formed after in vitro transcription. “Blobs” represent R-loop objects aligned on the main axis of the DNA molecule, whereas “spurs” come away from this axis; “Loops” correspond to objects formed when a “blob” sits at the base of a loop of DNA (Carrasco-Salas et al. 2019). In our data, we also found two types of R-loops corresponding to “loops” and “spurs” (Fig. 4F). R-loop architectures impose significant short-range mechanical constraints on the surrounding DNA via the introduction of kinks in the template (Carrasco-Salas et al. 2019). Thus, it is conceivable that these different structure-forming R-loops may represent functionally important differences. Collectively, these results from agarose gels, S9.6 dot blot, and AFM validated the formation of R-loops on the CRM1 and CRM2 templates.
CENH3 binding regions are favorable for R-loop formation
Centromeres are defined by the presence of CENH3, a variant of histone H3. Genome-wide mapping of sequences associated with CENH3 nucleosomes revealed CENH3-enriched and CENH3-depleted subdomains in the centromeres of rice, maize, and other species (Yan et al. 2008; Su et al. 2016, 2019; Zhao et al. 2016). We identified a total of 8.9 Mb mappable regions in maize centromeres, in which CENH3-enriched and CENH3-depleted subdomains account for about 66% and 34% of the centromeres, respectively (Supplemental Table S2). A total of 4422 R-loop peaks localized to the 10 maize centromeres, of which 3265 were located in CENH3-enriched subdomains and 1157 were located in CENH3-depleted regions (Supplemental Table S2). We next analyzed the potential correlation of the distribution of centromeric R-loops with CENH3-containing nucleosomes using two different methods. First, we determined CENH3 nucleosome positions using nucleR software and plotted the R-loop distribution around CENH3 nucleosomes. We obtained 40,867 CENH3 nucleosomes, in which 8994 (22.01%) CENH3 nucleosomes were identified overlapping with R-loops. We found that R-loops were relatively colocalized with CENH3 nucleosomes. However, there was no R-loop enrichment for input nucleosomes (Fig. 5A,D). Second, analysis of the CENH3-ChIP reads distribution profile around the centromeric R-loops indicated that the centromeric R-loop formation regions were highly enriched in CENH3 (Fig. 5B,D). To provide experimental support for the R-loop formation in CENH3 binding regions, we used CENH3-ChIP to capture DNA and then subjected it to dot blot analysis with anti-S9.6 antibody (Supplemental Fig. S9). Dot blots confirmed the R-loop formation in CENH3-ChIP DNA (Supplemental Fig. S9). However, we cannot exclude the background interference of whole genomic DNA immunoprecipitated by the anti-CENH3 antibody. The S9.6 dot blotting signals may represent part of R-loops in association with CENH3 nucleosomes. We further conducted CENH3 ChIP followed by S9.6 pull-down in the same experiment, with the final DNA being used for qPCR assay. We designed four pairs of primers. Primer 1 (P1-CRM1) and primer 2 (P2-CRM2) were designed from CRM1 and CRM2 regions, respectively, which were highly enriched with both CENH3 nucleosomes and R-loops. Primer 3 (P3-CENH3) was designed from a CRM1 region, which was enriched with CENH3 nucleosomes but less enriched with R-loops. Primer 4 (P4-NO) was designed in a centromeric region, which was not enriched with either CENH3 nucleosomes or R-loops. We found that the P1-CRM1 and P2-CRM2 were highly enriched in the CENH3 ChIPed DNA. P1-CRM1 and P2-CRM2 were also enriched in the CENH3-ChIP-S9.6-immunoprecipitated DNA sample and were sensitive to RNase H-treatment. P3-CENH3 showed enrichment in the CENH3 ChIPed DNA but did not show enrichment in R-loops. P4-NO did not show enrichment for either CENH3 ChIPed DNA or R-loops. Taken together, these results indicate the potential copresence of CENH3-nucleosomes with R-loops in maize centromeres (Fig. 5C).
Figure 5.

CENH3 binding regions are favorable for R-loop formation. (A) Metaplots of R-loop peaks along the CENH3 and input nucleosome and the up- and downstream regions. (B) Metaplots of CENH3-ChIP reads along the R-loop peaks and the up- and downstream regions. (C) Procedure of CENH3-ChIP-S9.6-qPCR (top). Data are means ± SE of three independent experiments. The values were compared by Student's t-test. (*) P-value < 0.05. (IgG) Immunoglobulin G. (D) A representative region showing CENH3-ChIP and R-loop data. Line 1, unstranded (unstrd) R-loops signal, normalized to the genome-wide mean; line 2, unstrd R-loop peaks; line 3, normalized wR-loops signal (green); line 4, wR-loop peaks; line 5, normalized cR-loop signal (purple); line 6, cR-loops peaks; line7, CENH3 ChIP-seq reads; line 8, CENH3 nucleosome positions identified by nucleR. The arrowheads indicate the colocalization regions of CENH3 nucleosomes and R-loops.
To assess whether the colocalization pattern was conserved in Arabidopsis, we investigated the CENH3 and R-loop correlation using publicly available CENH3-ChIP and R-loop data in Arabidopsis. Manhattan plots indicated that R-loops were also enriched in Arabidopsis pericentromeric and centromeric regions (Supplemental Fig. S10A). In Arabidopsis, centromeric regions contain large satellite arrays comprised of thousands of copies of 180-bp repeats, and transcripts are found from both strands of centromeric satellite repeats (May et al. 2005). We also found that Arabidopsis centromeric repeat regions contained R-loops in both the sense and antisense orientation (Supplemental Fig. S10C). Metaplot analysis indicated that R-loops also tended to form in CENH3 binding regions in Arabidopsis (Supplemental Fig. S10B,C). Taken together, these results demonstrated that CENH3 nucleosomes may provide a permissible environment for R-loops.
Discussion
Work in a range of eukaryotic organisms, including fungi, plants, and animals, is revealing a widespread regulatory role for R-loops (Niehrs and Luke 2020). In this study, we present genome-wide mapping of R-loops in maize by ssDRIP-seq. We found that the R-loop length distribution and preferential localization in promoter regions is largely conserved in plants (Fig. 1C; Supplemental Fig. S2D). The GA-rich motif identified in maize was also very similar to that in Arabidopsis and rice (Fig. 1D), indicating a conserved pattern of preference and bias for nucleic acid composition across plants, which could have implications for mechanisms of R-loop formation, gene regulation, and other related processes. At the gene level, the antisense R-loops were mainly detected around TSS, which showed a conserved pattern in plants (Fig. 1E). However, the sense R-loop distribution pattern was different from that in Arabidopsis and rice. Arabidopsis and rice sense R-loops are highly enriched in gene bodies, which were thought to be formed in a cotranscription manner (Xu et al. 2017; Fang et al. 2019), whereas in maize, sense R-loops are mainly detected in the promoter and TTS (Fig. 1E). These differences could be partially attributed to the finding that gene structure in large genomes are different from that in small genomes, as larger genomes have longer introns and a higher proportion of mobile elements (Lynch and Conery 2003). Mobile elements also impact gene structure and expression, as they can insert into genes, including introns and exons, and thus contribute to the evolution of genes (Stival Sena et al. 2014).
ncRNAs transcribed from pericentromeric repeats can form R-loops, which mediate the heterochromatin states of the pericentromere (Nakama et al. 2012). In maize, we observed the greatest R-loop enrichment in proximity to the centromeres and within pericentromeric heterochromatin at the chromosome scale, which represents an interphase chromatin state (Fig. 2A). We also observed strong R-loop signals in pericentromeric heterochromatin during meiosis (Fig. 2B). These results indicated that R-loops may act as more structural components to the pericentromeric chromatin rather than merely being by-products of transcription. Indeed, work in Arabidopsis revealed R-loop dynamics were not strongly associated with RNA abundance (Xu et al. 2020). Recently, using the newly elucidated global DNA:RNA interaction sequencing (GRID-seq), Hao and colleagues reported that various active retrotransposons, especially those from the gypsy family of the LTR class, produced a large amount of repeat RNAs with the ability to act in both cis and trans on chromatin to help maintain pericentromeric heterochromatin (Hao et al. 2020). In maize, retrotransposons were major components of pericentromeric heterochromatin, and we found that the majority of R-loops were derived from LTR/Gypsy and LTR/Copia families (Supplemental Table S1). Therefore, it is possible that R-loops derived from retrotransposons play a role in maintaining pericentromeric heterochromatin.
Centromeres are specific regions where kinetochores assemble (Henikoff et al. 2001). Although these regions were long considered to be silent, some experimental studies have demonstrated that transcription occurs in centromeres to generate ncRNAs and these ncRNAs are associated with a broad range of functions such as heterochromatin establishment and maintenance, chromatin structure, kinetochore assembly, centromeric protein loading, and inner centromere signaling (Henikoff et al. 2001). Although centromeres are essential for ensuring accurate chromosome segregation, there are no DNA sequence similarities in this region among different species (Ideue and Tani 2020). Therefore, centromere RNAs (cenRNAs) derived from these DNA sequences also shared no similarities in sequence and structure. Nevertheless, they all functioned at the centromere. This finding raises an interesting question: what mechanisms underlie the maintenance of cenRNAs at the centromere? Studies in Schizosaccharomyces pombe and maize indicate that the heterochromatic ncRNAs and centromere circRNAs are retained on chromatin via the formation of DNA:RNA hybrids, which suggests that DNA:RNA hybrid formation plays a role in ncRNA function (Nakama et al. 2012; Liu et al. 2020). In our data, R-loops have also been observed at maize centromeres (Fig. 2B). Our bioinformatics and experimental analysis suggested that the centromeric-specific retrotransposons CRM1 and CRM2 were strongly associated with R-loop formation (Fig. 4). In addition, both sense and antisense cenRNAs of CRM and CentC were detected in maize (Topp et al. 2004). We also correspondingly observed that maize centromeric repeat regions contained R-loops in both the sense and antisense orientations (Supplemental Fig. S8). Thus, one possible mechanism for how cenRNAs regulate centromere function is via the DNA:RNA hybrid, which may represent a conserved scenario across species.
CENH3-containing nucleosomes are thought to be the landmark for the active centromeric region. Therefore, elucidating how CENH3 nucleosomes seed new centromere assembly and maintain centromere location to guide faithful chromosome inheritance is an important question. In this study, we observed that those CENH3 nucleosomes that are associated with retrotransposons (CRM1 or CRM2) may be able to form R-loops in maize (Fig. 5D). Recent work showed that centromere R-loops, which are generated by mitotic processes in repetitive DNA sequences, promote the localization of the chromosome passenger complex (CPC) to the inner centromere (Moran et al. 2021). It was also recently reported that centromere R-loops are required to activate aurora kinase B, which promotes faithful chromosome segregation (Kabeche et al. 2018). Thus, CENH3 nucleosomes provide a permissible environment for R-loops, which may act as a marker for recruitment of other centromere proteins to direct chromosome segregation. This association between R-loops and centromere proteins may be directed by CENPC, one component of the important constitutive centromere-associated network. CENPC has been shown to be required for CENPA assembly (Carroll et al. 2010). CENPC also has extensive nucleic acid binding activity (Du et al. 2010), and both budding yeast and human CENPC associates with AT-rich DNA (Arunkumar and Melters 2020). Furthermore, maize CENPC has both DNA-binding and RNA-binding capacities, and the centromeric RNA facilitates the binding of maize CENPC to centromeric DNA (Du et al. 2010). Hence, direct and in vivo evidence in the future may demonstrate whether or not maize CENPC binds centromeric R-loops and therefore could facilitate the centromere-specific assembly of CENH3.
Methods
Sample preparation and ssDRIP-seq library construction
Leaf tissues were collected from 10-d-old B73 maize plants grown in a greenhouse under a 15-h light (28°C)/9-h dark (25°C) photoperiod. The collected leaves were ground into a fine powder using liquid nitrogen. ssDRIP-seq library construction, including nuclei isolation, was performed according to published procedures (Xu et al. 2017) with some modifications. Briefly, nuclei were isolated using Honda buffer (0.44 M sucrose, 1.25% Ficoll, 2.5% Dextran T40, 20 mM HEPES, 10 mM MgCl2, and 0.5% Triton X-100 [pH 7.4]), followed by SDS/Proteinase K digestion in a constant temperature shaker for 14 h at 37°C. Genomic DNA was extracted through a classic salt-ethanol precipitation. DNA fragmentation was performed using a cocktail of restriction enzymes (SspI, BamHI, HindIII, DdeI, MspI, and RsaI; New England Biolabs). The negative control was treated with RNase H (Takara 2151) overnight at 37°C. DRIP was performed with the commercial S9.6 antibody (Kerafast ENH001). The DRIPed DNA was sonicated using a S220 focused-ultrasonicator (Covaris) with 10% duty cycle, 175 peak incident power, 200 cycles per burst, and 70-sec treatment time to achieve an average fragment size of ∼200-bp. The ssDRIP-seq libraries were constructed from the sonicated DNA using the Accel-NGS 1S Plus DNA Library kit (Swift Biosciences), following instructions from the manufacturer. The libraries were checked on a fragment analyzer, followed by sequencing on an Illumina NovaSeq system. We constructed a total of five DRIP-seq libraries for sequencing, two replicates for positive and two for RNase H-treatment and one for input.
Data processing, quantification, and statistical analyses
ssDRIP-seq raw data processing and alignment
Raw reads were trimmed with Trimmomatic v.0.36 (Bolger et al. 2014) to remove adaptor, low-quality bases. The quality-controlled reads were then aligned to the B73_RefGen_v4 (Jiao et al. 2017) using Burrows Wheeler Aligner BWA-MEM (Li and Durbin 2009) with default parameters. Reads with mapping quality greater than 20 were extracted, and clonal duplicates were removed using SAMtools v.1.3.1 (Li et al. 2009). The ssDRIP-seq total mapped reads (nonstrand R-loops) were divided into forward (wR-loops, representing an R-loop formation containing ssDNA on the Watson strand and an DNA:RNA hybrid on the Crick strand) and reverse reads (cR-loops, representing an R-loop formation containing ssDNA on the Crick strand and an DNA:RNA hybrid on the Watson strand) (Supplemental Fig. S2). MACS2 (Zhang et al. 2008) was used to call peaks using input as a control with the settings: -f BAMPE -g 2.3e9. Overlaps between features were calculated using BEDTools intersect v2.27.1 (Quinlan and Hall 2010).
ChIP-seq raw data processing and alignment
Histone ChIP-seq data for H3K9me2 and H3K56ac were downloaded from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) database, accession SRR1584364 and SRR7889772, respectively. For the association analysis of R-loops and CENH3, the maize and Arabidopsis anti-CENH3 ChIP-seq data sets were obtained from the GEO database with accession numbers SRR2000635 and SRR4430537, respectively, and the input databases for maize and Arabidopsis were obtained with accession numbers SRR2000646 and SRR4430555, respectively. The reads were quality-trimmed and aligned as described for the ssDRIP-seq reads. The positions of CENH3-containing and canonical nucleosomes were determined using nucleR (Flores and Orozco 2011). To generate the anti-CENH3 antibody, peptides corresponding to ZmCENH3 (RPGTVALREIRKYQKS) were generated by GL Biochem. The core centromeric regions were defined as follows (in bp): Chr 1: 137,002,540–137,116,367; Chr 2: 95,504,395–97,492,600; Chr 3: 85,886,475–86,787,530; Chr 4: 109,089,400–110,364,670; Chr 5: 104,638,000–106,769,930; Chr 6: 52,293,450–52,452,350; Chr 7: 56,622,700–56,663,097; Chr 8: 50,328,200–52,053,600; Chr 9: 53,772,400–55,421,800; Chr 10: 51,522,000–52,773,750.
RNA-seq raw data processing, alignment, and expression quantification
The public RNA-seq data set was downloaded from the GEO database with accession number SRR3329316. The reads were quality-trimmed as described for the ChIP-seq reads. The trimmed reads were aligned to the B73_RefGen_v4 using HISAT2 v.2.0.5 (Kim et al. 2015). Gene expression values were computed using StringTie v.1.3.3b (Pertea et al. 2016) with the maize annotation version AGPv4.36.
Whole-genome bisulfite sequencing (WGBS) raw data processing, alignment, and calculation of methylation level
WGBS data were downloaded from the GEO database with accession number SRR850328. Raw reads were trimmed with Trimmomatic v.0.36 (Bolger et al. 2014). The trimmed reads were mapped to the B73_RefGen_v4 using Bismark v.0.22.1 (Krueger and Andrews 2011), allowing for no mismatch. Methylated cytosines were extracted from aligned reads using the Bismark methylation extractor with parameters ‐‐CX. The proportion of CG, CHG, and CHH methylation was determined as weighted methylation levels in 100-bp windows across the genome.
Normalization of read counts
The regions ±1 kb upstream of and downstream from TSSs and TTSs from non-TE genes with R-loops were divided into 10-bp windows for normalization. The number of reads per sliding window was first divided by the window length (10-bp) and then by the number of all uniquely mappable reads within the genome (Mb).
In vitro transcription of R-loops and AFM imaging
The regions spanning CRM1 (153–3956) and CRM2 (1–4000) were amplified from B73 genomic DNA and cloned into pET-30a (Novagen 70781) using standard protocols. For a control CRM1 sequence without R-loop formation in our ssDRIP-seq data, we amplified a region spanning CRM1(2468–4223) and then cloned it into pET-30a for AFM observation. The circular plasmids pET-30a-CRM1, pET-30a-CRM2, and pET-30a-control-CRM1 were incubated for 30 min at 37°C with T7 RNA Polymerase (New England Biolabs M0251) in a transcription buffer (40 mM Tris-HCl [pH 7.9], 6 mM MgCl2, 2 mM spermidine, 1 mM DTT) containing 0.5 mM of rATP, rCTP, rUTP, and rGTP (Promega P1221). Transcription was terminated by heat inactivation of the T7 RNA polymerase for 10 min at 65°C. The RNase A (Takara 2158) was then added for 30 min at 37°C to digest soluble RNAs. For negative control DNA without the R-loops, RNase A and RNase H were both used. The circular DNA was further linearized with KpnI and BamHI to generate two fragments, followed by purification using phenol/chloroform extraction. For AFM sample preparation, 5 ng of DNA were diluted in TN buffer (10 mM Tris [pH 7.4], 5 mM NiCl2) and incubated on the surface of freshly cleaved aminopropyl silatrane (APS)-mica for 5 min, rinsed with 200 μL of Milli-Q filtered ultrapure water, and dried with a gentle stream of nitrogen gas. Samples were measured using cantilevers (ScanAsyst-Air, Bruker) of nominal force constant 0.4 N/m, resonance frequency of 70 kHz, and tip radius 2 nm. All images were collected under ambient air conditions using a Bruker MultiMode 8 AFM with a nanoscope IIIa controller in ScanAsyst mode. All primers are listed in Supplemental Table S3.
Dot blot analysis
The genomic DNA of B73 was extracted using hexadecyl trimethyl ammonium bromide. DNase I (New England Biolabs M0303S), RNase H, and RNase R (Epicentre RNR07250) treatments were performed for 3 h at 37°C. The treated or untreated DNA was loaded onto a Hybond-N+ membrane (Amersham RPN203B) and cross-linked twice with UV (0.12 J). After air-drying, the membrane was blocked with 5% skimmed milk in Tris-buffered saline Tween-20 (TBST) for 1 h at room temperature (27°C), and then incubated overnight with anti-S9.6 antibody (1:10,000 dilution) dissolved in 5% milk/TBST at 4°C. After washing three times in TBST for 5 min each time, secondary antibody (goat antimouse antibody conjugated to horseradish peroxidase, 1:20,000 dilution in 5% milk/TBST, GE Healthcare NA931) was added to the buffer, followed by incubation for 1.5 h at room temperature. The membrane was washed three times with TBST for 5 min each time, followed by Tris-buffered saline for 5 min. Detection was performed using enhanced chemiluminescence reagent.
Immunostaining and fluorescence in situ hybridization (FISH)
Male inflorescences at the meiotic stage were fixed in ethanol: acetic acid (3:1, v/v) overnight at 4°C and then washed three times in 70% ethanol. Anthers at the pachytene stage were collected and added to a tube of enzyme cocktail (1% pectolyase and 2% cellulase dissolved in citric buffer) and incubated for 20 min at 37°C. The anthers were gently rinsed in 100% ethanol several times and then were broken apart with a dull dissecting probe, and then centrifuged in a microcentrifuge at 1000 rpm for 10 sec to remove the supernatant. The pellet was dissolved in acetic acid and ∼8 µL of cell suspension was dropped onto a glass slide in a humid chamber and allowed to dry. Prepared glass slides showing correct staging and spreading of the meiotic cells were used for incubating overnight with anti-S9.6 antibody (1:100 dilution) dissolved in 3% bovine serum albumin at 4°C. Samples were then washed in phosphate-buffered saline (PBS) (137 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, and 2 mm KH2PO4 [pH 7.4]) three times, each for 10 min. The secondary antibody (goat antimouse antibody labeled by FITC green, 1:500 dilution, Jackson ImmunoResearch 115-096-003) was added and allowed to bind for 2 h at 37°C. After washing the slides in PBS three times, each for 5 min, samples were stained with 4′,6-diamidino-2-phenylindole. FISH was performed as described (Liu et al. 2017) with CRM1, 45S, and 5S probes labeled with Texas-red-5-dUTP. The samples were observed by confocal microscopy (Zeiss Cell Observer SD), and the images were processed with ZEN 2009 Light Edition (Zeiss, http://www.zeiss.com/) and Adobe Photoshop CS 6.0 software.
Data access
The ssDRIP-seq data generated in this study have been submitted to the NGDC Genome Sequence Archive (GSA; https://bigd.big.ac.cn/gsa/) under accession number CRA003770.
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31920103006, 31630049, and 31991212) and a National Science Foundation plant genome grant (IOS-1444514).
Author contributions: F.H., Y.L., Q.L., and H.S. planned and designed the research; Y.L., Q.L., H.S., K.L., X.X., W.L., Q.S., J.A.B., and F.H. performed experiments, conducted field work, and analyzed data; Y.L., F.H., and J.A.B. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.275270.121.
Competing interest statement
The authors declare no competing interests.
References
- Aguilera A, García-Muse T. 2012. R loops: from transcription byproducts to threats to genome stability. Mol Cell 46: 115–124. 10.1016/j.molcel.2012.04.009 [DOI] [PubMed] [Google Scholar]
- Allshire RC, Madhani HD. 2018. Ten principles of heterochromatin formation and function. Nat Rev Mol Cell Biol 19: 229–244. 10.1038/nrm.2017.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215: 403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- Ananiev EV, Phillips RL, Rines HW. 1998a. Chromosome-specific molecular organization of maize (Zea mays L.) centromeric regions. Proc Natl Acad Sci 95: 13073–13078. 10.1073/pnas.95.22.13073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananiev EV, Phillips RL, Rines HW. 1998b. A knob-associated tandem repeat in maize capable of forming fold-back DNA segments: are chromosome knobs megatransposons? Proc Natl Acad Sci 95: 10785–10790. 10.1073/pnas.95.18.10785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunkumar G, Melters DP. 2020. Centromeric transcription: a conserved Swiss-Army knife. Genes (Basel) 11: 911. 10.3390/genes11080911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belotserkovskii BP, Tornaletti S, D'Souza AD, Hanawalt PC. 2018. R-loop generation during transcription: formation, processing and cellular outcomes. DNA Repair (Amst) 71: 69–81. 10.1016/j.dnarep.2018.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA, Han F. 2009. Maize centromeres: structure, function, epigenetics. Annu Rev Genet 43: 287–303. 10.1146/annurev-genet-102108-134834 [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco-Salas Y, Malapert A, Sulthana S, Molcrette B, Chazot-Franguiadakis L, Bernard P, Chédin F, Faivre-Moskalenko C, Vanoosthuyse V. 2019. The extruded non-template strand determines the architecture of R-loops. Nucleic Acids Res 47: 6783–6795. 10.1093/nar/gkz341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CW, Milks KJ, Straight AF. 2010. Dual recognition of CENP-A nucleosomes is required for centromere assembly. J Cell Biol 189: 1143–1155. 10.1083/jcb.201001013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano-Pozo M, Santos-Pereira José M, Rondón Ana G, Barroso S, Andújar E, Pérez-Alegre M, García-Muse T, Aguilera A. 2013. R loops are linked to histone H3 S10 phosphorylation and chromatin condensation. Mol Cell 52: 583–590. 10.1016/j.molcel.2013.10.006 [DOI] [PubMed] [Google Scholar]
- Dhatchinamoorthy K, Mattingly M, Gerton JL. 2018. Regulation of kinetochore configuration during mitosis. Curr Genet 64: 1197–1203. 10.1007/s00294-018-0841-9 [DOI] [PubMed] [Google Scholar]
- Du Y, Topp CN, Dawe RK. 2010. DNA binding of centromere protein C (CENPC) is stabilized by single-stranded RNA. PLoS Genet 6: e1000835. 10.1371/journal.pgen.1000835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Chen L, Lin K, Feng Y, Zhang P, Pan X, Sanders J, Wu Y, Wang X-E, Su Z, et al. 2019. Characterization of functional relationships of R-loops with gene transcription and epigenetic modifications in rice. Genome Res 29: 1287–1297. 10.1101/gr.246009.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feretzaki M, Pospisilova M, Valador Fernandes R, Lunardi T, Krejci L, Lingner J. 2020. RAD51-dependent recruitment of TERRA lncRNA to telomeres through R-loops. Nature 587: 303–308. 10.1038/s41586-020-2815-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores O, Orozco M. 2011. nucleR: a package for non-parametric nucleosome positioning. Bioinformatics 27: 2149–2150. 10.1093/bioinformatics/btr345 [DOI] [PubMed] [Google Scholar]
- Gan W, Guan Z, Liu J, Gui T, Shen K, Manley JL, Li X. 2011. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev 25: 2041–2056. 10.1101/gad.17010011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gent JI, Madzima TF, Bader R, Kent MR, Zhang X, Stam M, McGinnis KM, Dawe RK. 2014. Accessible DNA and relative depletion of H3K9me2 at maize loci undergoing RNA-directed DNA methylation. Plant Cell 26: 4903–4917. 10.1105/tpc.114.130427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginno PA, Lott PL, Christensen HC, Korf I, Chédin F. 2012. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol cell 45: 814–825. 10.1016/j.molcel.2012.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf M, Bonetti D, Lockhart A, Serhal K, Kellner V, Maicher A, Jolivet P, Teixeira MT, Luke B. 2017. Telomere length determines TERRA and R-loop regulation through the cell cycle. Cell 170: 72–85.e14. 10.1016/j.cell.2017.06.006 [DOI] [PubMed] [Google Scholar]
- Grewal SIS, Jia S. 2007. Heterochromatin revisited. Nat Rev Genet 8: 35–46. 10.1038/nrg2008 [DOI] [PubMed] [Google Scholar]
- Hao Y, Wang D, Wu S, Li X, Shao C, Zhang P, Chen J-Y, Lim D-H, Fu X-D, Chen R, et al. 2020. Active retrotransposons help maintain pericentromeric heterochromatin required for faithful cell division. Genome Res 30: 1570–1582. 10.1101/gr.256131.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmrich A, Ballarino M, Tora L. 2011. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol Cell 44: 966–977. 10.1016/j.molcel.2011.10.013 [DOI] [PubMed] [Google Scholar]
- Henikoff S, Ahmad K, Malik HS. 2001. The centromere paradox: stable inheritance with rapidly evolving DNA. Science 293: 1098–1102. 10.1126/science.1062939 [DOI] [PubMed] [Google Scholar]
- Ideue T, Tani T. 2020. Centromeric non-coding RNAs: conservation and diversity in function. Noncoding RNA 6: 4. 10.3390/ncrna6010004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Peluso P, Shi J, Liang T, Stitzer MC, Wang B, Campbell MS, Stein JC, Wei X, Chin C-S, et al. 2017. Improved maize reference genome with single-molecule technologies. Nature 546: 524–527. 10.1038/nature22971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeche L, Nguyen HD, Buisson R, Zou L. 2018. A mitosis-specific and R loop–driven ATR pathway promotes faithful chromosome segregation. Science 359: 108–114. 10.1126/science.aan6490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360. 10.1038/nmeth.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo D-H, Jiang J. 2009. Super-stretched pachytene chromosomes for fluorescence in situ hybridization mapping and immunodetection of DNA methylation. The Plant J 59: 509–516. 10.1111/j.1365-313X.2009.03881.x [DOI] [PubMed] [Google Scholar]
- Kreuzer KN, Brister JR. 2010. Initiation of bacteriophage T4 DNA replication and replication fork dynamics: a review in the Virology Journal series on bacteriophage T4 and its relatives. Virol J 7: 358. 10.1186/1743-422X-7-358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR. 2011. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27: 1571–1572. 10.1093/bioinformatics/btr167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Su H, Liu Y, Zhang J, Dong Q, Birchler JA, Han F. 2017. Cohesion and centromere activity are required for phosphorylation of histone H3 in maize. The Plant J 92: 1121–1131. 10.1111/tpj.13748 [DOI] [PubMed] [Google Scholar]
- Liu Y, Su H, Zhang J, Liu Y, Feng C, Han F. 2020. Back-spliced RNA from retrotransposon binds to centromere and regulates centromeric chromatin loops in maize. PLoS Biol 18: e3000582. 10.1371/journal.pbio.3000582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Conery JS. 2003. The origins of genome complexity. Science 302: 1401–1404. 10.1126/science.1089370 [DOI] [PubMed] [Google Scholar]
- Manuelidis L. 1978. Complex and simple sequences in human repeated DNAs. Chromosoma 66: 1–21. 10.1007/BF00285812 [DOI] [PubMed] [Google Scholar]
- May BP, Lippman ZB, Fang Y, Spector DL, Martienssen RA. 2005. Differential regulation of strand-specific transcripts from Arabidopsis centromeric satellite repeats. PLoS Genet 1: e79. 10.1371/journal.pgen.0010079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran EC, Liu L, Zasadzinska E, Kestner CA, Sarkeshik A, DeHoyos H, Yates JR, Foltz D, Stukenberg PT. 2021. Mitotic R-loops direct Aurora B kinase to maintain centromeric cohesion. bioRxiv 10.1101/2021.01.14.426738 [DOI]
- Nakama M, Kawakami K, Kajitani T, Urano T, Murakami Y. 2012. DNA–RNA hybrid formation mediates RNAi-directed heterochromatin formation. Genes Cells 17: 218–233. 10.1111/j.1365-2443.2012.01583.x [DOI] [PubMed] [Google Scholar]
- Nguyen HD, Yadav T, Giri S, Saez B, Graubert TA, Zou L. 2017. Functions of replication protein A as a sensor of R loops and a regulator of RNaseH1. Mol Cell 65: 832–847.e4. 10.1016/j.molcel.2017.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehrs C, Luke B. 2020. Regulatory R-loops as facilitators of gene expression and genome stability. Nat Rev Mol Cell Biol 21: 167–178. 10.1038/s41580-019-0206-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H, Jin M, Ghadiyaram A, Kaur P, Miller HE, Ta HM, Liu M, Fan Y, Mahn C, Gorthi A, et al. 2020. Cohesin SA1 and SA2 are RNA binding proteins that localize to RNA containing regions on DNA. Nucleic Acids Res 48: 5639–5655. 10.1093/nar/gkaa284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock WJ, Dennis ES, Rhoades MM, Pryor AJ. 1981. Highly repeated DNA sequence limited to knob heterochromatin in maize. Proc Natl Acad Sci 78: 4490–4494. 10.1073/pnas.78.7.4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. 2016. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 11: 1650–1667. 10.1038/nprot.2016.095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plohl M, Luchetti A, Meštrović N, Mantovani B. 2008. Satellite DNAs between selfishness and functionality: structure, genomics and evolution of tandem repeats in centromeric (hetero)chromatin. Gene 409: 72–82. 10.1016/j.gene.2007.11.013 [DOI] [PubMed] [Google Scholar]
- Pohjoismäki JLO, Holmes JB, Wood SR, Yang M-Y, Yasukawa T, Reyes A, Bailey LJ, Cluett TJ, Goffart S, Willcox S, et al. 2010. Mammalian mitochondrial DNA replication intermediates are essentially duplex but contain extensive tracts of RNA/DNA hybrid. J Mol Biol 397: 1144–1155. 10.1016/j.jmb.2010.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivin CJ, Cullis CA, Walbot V. 1986. Evaluating quantitative variation in the genome of Zea mays. Genetics 113: 1009–1019. 10.1093/genetics/113.4.1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saksouk N, Simboeck E, Déjardin J. 2015. Constitutive heterochromatin formation and transcription in mammals. Epigenetics Chromatin 8: 3. 10.1186/1756-8935-8-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, et al. 2009. The B73 maize genome: complexity, diversity, and dynamics. Science 326: 1112–1115. 10.1126/science.1178534 [DOI] [PubMed] [Google Scholar]
- Sharma A, Schneider KL, Presting GG. 2008. Sustained retrotransposition is mediated by nucleotide deletions and interelement recombinations. Proc Natl Acad Sci 105: 15470–15474. 10.1073/pnas.0805694105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw NN, Arya DP. 2008. Recognition of the unique structure of DNA:RNA hybrids. Biochimie 90: 1026–1039. 10.1016/j.biochi.2008.04.011 [DOI] [PubMed] [Google Scholar]
- Stival Sena J, Giguère I, Boyle B, Rigault P, Birol I, Zuccolo A, Ritland K, Ritland C, Bohlmann J, Jones S, et al. 2014. Evolution of gene structure in the conifer Picea glauca: a comparative analysis of the impact of intron size. BMC Plant Biol 14: 95. 10.1186/1471-2229-14-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stork CT, Bocek M, Crossley MP, Sollier J, Sanz LA, Chédin F, Swigut T, Cimprich KA. 2016. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. eLife 5: e17548. 10.7554/eLife.17548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, Liu Y, Liu Y-X, Lv Z, Li H, Xie S, Gao Z, Pang J, Wang X-J, Lai J, et al. 2016. Dynamic chromatin changes associated with de novo centromere formation in maize euchromatin. The Plant J 88: 854–866. 10.1111/tpj.13305 [DOI] [PubMed] [Google Scholar]
- Su H, Liu Y, Liu C, Shi Q, Huang Y, Han F. 2019. Centromere satellite repeats have undergone rapid changes in polyploid wheat subgenomes. Plant Cell 31: 2035–2051. 10.1105/tpc.19.00133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Csorba T, Skourti-Stathaki K, Proudfoot NJ, Dean C. 2013. R-loop stabilization represses antisense transcription at the Arabidopsis FLC locus. Science 340: 619–621. 10.1126/science.1234848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert PB, Henikoff S. 2018. Transcribing centromeres: noncoding RNAs and kinetochore assembly. Trends Genet 34: 587–599. 10.1016/j.tig.2018.05.001 [DOI] [PubMed] [Google Scholar]
- Topp CN, Zhong CX, Dawe RK. 2004. Centromere-encoded RNAs are integral components of the maize kinetochore. Proc Natl Acad Sci 101: 15986–15991. 10.1073/pnas.0407154101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood CJ, Henderson IR, Martienssen RA. 2017. Genetic and epigenetic variation of transposable elements in Arabidopsis. Curr Opin Plant Biol 36: 135–141. 10.1016/j.pbi.2017.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahba L, Amon JD, Koshland D, Vuica-Ross M. 2011. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol Cell 44: 978–988. 10.1016/j.molcel.2011.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, et al. 2007. A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8: 973–982. 10.1038/nrg2165 [DOI] [PubMed] [Google Scholar]
- Xu W, Xu H, Li K, Fan Y, Liu Y, Yang X, Sun Q. 2017. The R-loop is a common chromatin feature of the Arabidopsis genome. Nat Plants 3: 704–714. 10.1038/s41477-017-0004-x [DOI] [PubMed] [Google Scholar]
- Xu W, Li K, Li S, Hou Q, Zhang Y, Liu K, Sun Q. 2020. The R-loop atlas of Arabidopsis development and responses to environmental stimuli. Plant Cell 32: 888–903. 10.1105/tpc.19.00802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Talbert PB, Lee H-R, Jett J, Henikoff S, Chen F, Jiang J. 2008. Intergenic locations of rice centromeric chromatin. PLoS Biol 6: e286–e286. 10.1371/journal.pbio.0060286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Hou Q, Cheng L, Xu W, Hong Y, Li S, Sun Q. 2017. RNase H1 cooperates with DNA gyrases to restrict R-loops and maintain genome integrity in Arabidopsis chloroplasts. Plant Cell 29: 2478–2497. 10.1105/tpc.17.00305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Zhou J, Tong J, Zhuo W, Wang L, Li Y, Sun Q, Qian W. 2019. ALBA protein complex reads genic R-loops to maintain genome stability in Arabidopsis. Sci Adv 5: eaav9040. 10.1126/sciadv.aav9040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based Analysis of ChIP-Seq (MACS). Genome Biol 9: R137. 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Zhu X, Wang K, Gent JI, Zhang W, Dawe RK, Jiang J. 2016. Gene expression and chromatin modifications associated with maize centromeres. G3 (Bethesda) 6: 183–192. 10.1534/g3.115.022764 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.