

Abstract
Dry leaves of kratom (mitragyna speciosa) are anecdotally consumed as pain relievers and antidotes against opioid withdrawal and alcohol use disorders. There are at least 54 alkaloids in kratom; however, investigations to date have focused around mitragynine, 7-hydroxy mitragynine (7OH), and mitragynine pseudoindoxyl (MP). Herein, we probe a few minor indole and oxindole based alkaloids, reporting the receptor affinity, G-protein activity, and βarrestin-2 signaling of corynantheidine, corynoxine, corynoxine B, mitraciliatine, and isopaynantheine at mouse and human opioid receptors. We identify corynantheidine as a mu opioid receptor (MOR) partial agonist, whereas its oxindole derivative corynoxine was an MOR full agonist. Similarly, another alkaloid mitraciliatine was found to be an MOR partial agonist, while isopaynantheine was a KOR agonist which showed reduced βarrestin-2 recruitment. Corynantheidine, corynoxine, and mitraciliatine showed MOR dependent antinociception in mice, but mitraciliatine and corynoxine displayed attenuated respiratory depression and hyperlocomotion compared to the prototypic MOR agonist morphine in vivo when administered supraspinally. Isopaynantheine on the other hand was identified as the first kratom derived KOR agonist in vivo. While these minor alkaloids are unlikely to play the majority role in the biological actions of kratom, they represent excellent starting points for further diversification as well as distinct efficacy and signaling profiles with which to probe opioid actions in vivo.
Keywords: Respiration, kratom, oxindoles, corynoxine, mitraciliatine, partial agonism
INTRODUCTION
Opioid receptor agonists like morphine and fentanyl are clinically used analgesics. However, activation of a mu opioid receptor (MOR) is associated with adverse e ects like respiratory depression, dependence, and abuse potential.1–3 Mismanagement of prolonged use of opioids for treating pain, coupled with the spread of illicit synthetic opioids such as fentanyl, has led to an opioid epidemic estimated to take an average of 130 American lives daily.4–6 Synthesis of a nonaddictive and unexploitable analgesic is a pressing scientific priority in addressing the causes of the current opioid epidemic.
Various approaches to design functionally selective opioids have been proposed in the opioid field. These include ligands displaying biased agonism,7–9 targeting heteromers,10,11 allosteric modulators,12–14 targeting MOR splice variants,15–17 and compounds with polypharmacological actions at multiple opioid targets.18–22 A recent approach aims at revisiting low efficacy partial agonism in newer assays with limited signal amplification.23–25
The kappa26 and delta opioid receptor (DOR)27,28 have been proposed as alternate targets to develop safer analgesics as well. Peripherally restricted kappa agonists26 and kappa antagonists29 have been reported to preclude or ameliorate the negative affect associated with pain. Similar to MOR based approaches, biased agonism7,24,30 and allosterism31 have been proposed as avenues to develop functionally selective opioids at these two receptors. A recent paper from our group32 shows that dual partial agonism at MOR/KOR subtypes may offer some additional advantages over targeting partial agonism at a single subtype.
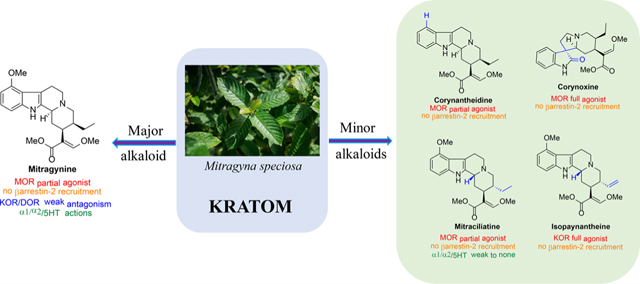
Natural products offer another approach, with unique opportunities to discover probes with druglike properties.33–35 Nearly 30% of all FDA approved drugs are either natural products or their derivatives.36 In search of templates distinct from morphine and fentanyl, our group has been interested in the chemical and pharmacological characterization of kratom based natural products. Mitragyna speciosa is a psychoactive plant from Southeast Asia, which is also known as “kratom”.37,38 In Malaysia it is well-known as “biak biak”. Kratom products are commercially accessible in the U.S. and sold as dried leaves, capsules, and liquid concentrates.38–40 Individuals use it to relieve pain and alcoholism, and it is also used in withdrawal medications.
Kratom leaves mostly consist of alkaloids, which contain indole or oxindole moiety (Figure 1). Roughly 54 alkaloids have already been extracted from this plant, with mitragynine being the major (∼66%) alkaloid component.41–45 About 1– 2% of the total dry leaf material in kratom is mitragynine. The relative concentrations of each alkaloid present in this plant are largely dependent on its origin and geographical factors. Variation in climates, soil types, and environment have been proposed to play a significant role in the overall distribution of alkaloids in kratom.46,47 While we have studied kratom from various sources, we have largely focused our recent studies on the “Red Indonesian Micro Powder” from Moon Kratom (Austin, TX). In our hands, we find approximately 48.6% of mitragynine (1, Figure 1), 5.8% of paynantheine (2), 3.5% of speciogynine (3), and 7.7% of speciociliatine (4) along with a much smaller amount (<0.02% of mitraciliatine, <0.03% of isopaynantheine) from the crude kratom alkaloid extracts (Figure 1). Although we did not find other minor alkaloids like corynantheidine (5), corynoxine (9), corynoxine B (10), and 7-hydroxymitragynine (8) from “Red Indonesian Micro Powder”, other sources of kratom have been reported to have these minor alkaloids.44,48,49
Figure 1.

Chemical structures and configurations at C3 and C20 of select indole and oxindole moiety based kratom alkaloids.
Pharmacological characterization of kratom,39,43,50 mitragynine,51,52 7-hydroxy mitragynine (7OH),51,53 mitragynine pseudoindoxyl (MP)51,54,55 (Figure 1), and other major alkaloids paynantheine,43 speciogynine,43,45,56 and speciocilliatine are known in the literature. However, studies are limited on contributions of the minor alkaloids to biological actions of kratom, and the signaling profile of these individual alkaloids remains unknown to date. This report describes the characterization of low abundance indole alkaloids: corynantheidine57 (5), isopaynantheine (6), mitraciliatine (7), and the oxindole alkaloids48 corynoxine (9) and corynoxine B (10). Alkaloids were characterized pharmacologically in cell lines expressing opioid receptors using binding assays, [35S]GTPγS assays, and BRET based Gi-1 and βarrestin-2 signaling, with selected alkaloids tested in vivo for antinociception and adverse effects like respiratory depression and locomotor activity in mice. Off-target evaluation on three alkaloids was also carried out using PDSP-NIMH screening.58
Presently, we report identification of three novel antinociceptive natural products and compounds with unique opioid receptor mediated signaling profiles including partial agonism at MOR by corynantheidine and mitraciliatine, antinociceptive agents with attenuated adverse effects (corynoxine and mitraciliatine), and reduced off-target receptor labeling by mitraciliatine. Another natural product, isopaynantheine with a C20-vinyl group instead of C20-ethyl in the case of mitraciliatine, was a KOR agonist with reduced recruitment of βarrestin-2 in cell lines and showed KOR dependent–MOR independent antinociception in mice.
RESULTS AND DISCUSSION
Chemistry.
Corynantheidine (5), one of the minor indole alkaloids from kratom is also known as 9-desmethoxymitragynine (Figure 1).49,59,60 Corynantheidine has the same stereochemistry at C3 (3S) and C20 (20S) as mitragynine. About 1% of alkaloid extract is believed to be corynantheidine in some extracts according to literature reports.61 We were not able to find this alkaloid in “Red Indonesian Micro Powder”, so we chemically synthesized corynantheidine from mitragynine as shown in Scheme 1.
Scheme 1.

Chemical Synthesis of Corynantheidine.
Extraction of mitragynine (1) from kratom powder was performed using a literature reported protocol by our group.51 9-Hydroxymitragynine (12, Scheme 1) was synthesized from 1 using AlCl3 as the Lewis acid in the presence of ethanethiol.57 Hydroxy compound 12 was treated with N-phenyl-bis(trifluoromethanesulfonimide) in the presence of a base to furnish corresponding triflate 13. Synthesis of corynantheidine (5) was achieved by palladium-catalyzed removal of the triflate ester (13) in the presence of formic acid.
Mitraciliatine (7), a diastereoisomer of mitragynine, has also been isolated as a minor alkaloid from mitragyna speciosa leaves.62–65 This tetracyclic indole alkaloid represents the opposite stereochemistry at both C3 (3R) and C20 (20R) to that of mitragynine. Mitraciliatine is the least studied among all other minor alkaloids.66 We were able to extract a considerable amount of this alkaloid from “Red Indonesian Micro Powder” (see procedure in Materials and Methods section).
Isopaynantheine (6) is another minor alkaloid with an indole core.62,65,66 Compared to that of mitragynine, this molecule has the opposite stereochemical connectivity at both C3 (R-isomer) and C20 (R-isomer). The other difference is the replacement of the C20 ethyl moiety with a vinyl moiety. This compound is also the C3 epimer of paynantheine (2). Studies on isopaynantheine as an opioid probe are unknown in the literature. We extracted this from “Red Indonesian Micro Powder” (detailed extraction in the Materials and Methods section).
Two oxindole alkaloids corynoxine (9) and corynoxine B (10) have also been isolated from M. speciosa.44,48,67 These two molecules with an oxindole68 core differ in their stereochemistry at spiro C7 being (7S) for 9 and (7R) for 10.44 Although the biological property of these templates have not been studied extensively, total synthesis has been carried out on these two scaffolds.69 For our studies, we purchased corynoxine and corynoxine B from BOC sciences (NY, USA).
Pharmacology. Corynantheidine.
Corynantheidine has been characterized ex vivo in guinea pig ileum assays. It showed no agonist actions in these assays and has been reported to be an MOR antagonist.57 In recent years, the binding affinity at MOR and α adrenergic receptor subtype 1D (α1D) has been reported to be 118 and 41 nM, respectively.56
We reevaluated this natural product in binding assays and studied the signaling properties of this molecule at both mouse opioid receptors as well as at human opioid receptors. In our hands, corynantheidine had moderate affinity of Ki = 57 nM for the mouse mu opioid receptor (mMOR) (Table 1), about ∼4-fold higher than that of mitragynine (Ki = 230 nM)51 in the same assay. It also had moderate affinity at the mouse kappa opioid receptor (mKOR) and mDOR (Table 1). In [35S]GTPγS assays (Table 2), corynantheidine was a partial agonist (Emax = 74%) with efficacy comparable to that of mitragynine at mMOR (Emax of mitragynine = 65%), and its potency was comparable to its binding affinity. It did not signal through mKOR or mDOR and can be classified as an MOR selective agonist in this assay (Table 2).
Table 1.
Affinity Determination in Mouse Opioid Receptor Transfected Cell Lines
| affinity (Ki nM)a | |||
|---|---|---|---|
| compound | mMOR | mKOR | mDOR |
| corynantheidine | 57.1 ± 8.3 | 385.4 ± 47 | 172 ± 24.5 |
| mitraciliatine | 135.1 ± 7.7 | 101.2 ± 2.3 | 827.1 ± 85.7 |
| isopaynantheine | 91.8 ± 5.8 | 325.9 ± 31.9 | 822.9 ± 126.9 |
| corynoxine | 140.6 ± 73.9 | 3081.4 ± 1727.5 | 1273.9 ± 225.7 |
| corynoxine B | 5010 ± 388 | >10 μM | 1682 ± 260.92 |
| mitragynineb | 230 ± 47 | 231 ± 21 | 1011 ± 49 |
| 7OH mitragynineb | 37 ± 4 | 3081.4 ± 1727.5 | 1273.9 ± 225.7 |
| DAMGOb | 3.3 ± 0.43 | ||
| morphineb | 4.6 ± 1.8 | - | - |
| U50,488Hb | - | 0.73 ± 0.32 | - |
| DPDPEb | - | - | 1.39 ± 0.67 |
Competition studies were performed with the indicated natural prodcuts or compounds against 125I-IBNtxA (0.1 nM) in membranes from CHO cells stably expressing cloned mouse opioid receptors. Results are presented as nM ± SEM from three independent experiments performed in triplicate. The dash (–) means “not determined”.
Literature values from Varadi et al., 2016, 59(18), 8381–97.
Table 2.
Functional Activity Characterization Using [35S]GTPγS Assays in Opioid Transfected Cell Lines
| [35S]GTPγS functional assaya | ||||||
|---|---|---|---|---|---|---|
| mMOR | mKOR | mDOR | ||||
| compound | EC50 (nM) | Emax (%) | EC50 (nM) | Emax (%) | EC50 (nM) | Emax (%) |
| corynanthedine | 104.24 ± 4.59 | 74.13 ± 1.24 | no activity | no activity | no activity | no activity |
| mitraciliatine | 228 ± 9.6 | 47.5 ± 0.05 | 218 ± 6.4 | 98.7 ± 5 | no activity | |
| isopaynantheine | 191.8 ± 5.8 | 50 ± 5 | 282.2 ± 18 | 93.5 ± 5 | no activity | |
| corynoxine | 202.8 ± 4.4 | 136.4 ± 3.3 | no activity | no activity | no activity | |
| corynoxine B | no activity | no activity | no activity | |||
| DAMGOb | 3.4 ± 0.2 | 100 | - | - | - | - |
| U50,488Hb | - | - | 9.5 ± 1.8 | 100 | - | - |
| DPDPEb | - | - | - | - | 16.2 ± 5.1 | 100 |
Efficacy and potency data were obtained using agonist i [35S]GTPγS binding assay. Efficacy is represented as EC50 (nM) and percent maximal stimulation (Emax) relative to standard agonist DAMGO (MOR), DPDPE (DOR), or U50,488H (KOR) at 1000 nM. Results are presented as nM ± SEM from three independent experiments performed in triplicate. The dash (–) denotes not determined or not applicable. No activity = no agonism at 1000 nM drug concentration or no antagonism of 100 nM prototypic control agonist.
Literature values from Uprety et al. Elife. 2021 Feb 8;10:e56519.
The signaling of this molecule was further characterized at human opioid receptors using BRET assays.32,70 The Gi-1 activation profile at the human mu opioid receptor (hMOR) was similar to that of [35S]GTPγS assay in mMOR (Figure 2A). Corynantheidine was a selective partial agonist at MOR (Emax = 37%) in this assay but was 20 times less potent compared to [D-Ala2,N-MePhe4,Gly-ol5]-enkephalin (DAMGO). In the [35S]GTPγS assay, it was about 30-fold less potent than DAMGO. It showed no signaling through the human kappa opioid receptor (hKOR) (Figure 2C) or hDOR (Figure 2E) as well as no βarrestin-2 recruitment (Figure 2B,D,F) at any opioid subtypes consistent with the pharmacology of the mitragynine template where βarrestin-2 recruitment is not seen.43,51,71 Potential off-target labeling of this molecule was examined using radioligand assays at ∼50 CNS human receptors using the PDSP-NIMH (Table 3C). Corynantheidine was found to bind to as many as 11 targets in addition to hMOR (Ki = 339 nM) and hKOR, with the highest affinity demonstrated for hα2A (Ki = 74 nM) and hNMDA (Ki = 83 nM). The antinociceptive actions of corynantheidine, to the best of our knowledge, are unknown. To evaluate in vivo agonist activity of corynantheidine, we utilized the 55 °C warm-water tail withdrawal assay32,51 with C57BL/6J mice. Supraspinal administration (intracerebroventricular, i.c.v.) of the compound was utilized to avoid potential blood–brain barrier (BBB) permeability issues and to minimize the effects of metabolism. The compound showed a ceiling effect in antinociception assays (with a maximal 50% maximum possible effect (MPE)) consistent with the observed partial agonism seen in cell lines at mMOR in the [35S]GTPγS assays (Table 2). To probe for MOR dependent actions, we also evaluated corynantheidine in MOR KO mice, finding antinociception to be MOR dependent (Figure 2G). It is possible that the antinociception is mediated by multiple targets in addition to MOR based on the off-target labeling displayed by this molecule, notably the higher affinity for α adrenergic receptor subtype 2A (α2A) affinity over MOR. The adverse effects of corynantheidine were not investigated further because of its structural similarity to mitragynine and other congeners.
Figure 2.

G-protein and βarrestin-2 signaling of Corynantheidine at opioid receptors and dose dependent antinociception time course of Corynantheidine. Corynantheidine shows MOR selective G-protein signaling while lacking measurable βarrestin-2 recruitment. (A) Gi-1 activation measured using BRET assays at hMOR. Corynantheidine is a hMOR partial agonist. DAMGO EC50 (nM) (pEC50 ± SEM) = 3.56 (8.45 ± 0.13) nM, Emax% ± SEM = 100 ± 4.17. Corynantheidine EC50 (nM) (pEC50 ± SEM) = 67.2 (7.17 ± 0.29) nM, Emax% ± SEM = 37.2 ± 4.30. (B) No measurable βarrestin-2 recruitment was observed in BRET assays of corynantheidine at hMOR. DAMGO EC50 (nM) (pEC50 ± SEM) = 172.89 (6.76 ± 0.08) nM, Emax% ± SEM = 100 ± 3.48. Corynantheidine EC50 (nM) (pEC50 ± SEM) = not determined (n.d.), Emax% ± SEM = <20. (C) Gi-1 activation in BRET assay of corynantheidine at hKOR. U50488H EC50 (nM) (pEC50 ± SEM) = 2.8 (8.55 ± 0.21) nM, Emax% ± SEM = 100 ± 5.89. Corynantheidine EC50 (nM) (pEC50 ± SEM) = n.d., Emax% ± SEM = <20. (D) No measurable βarrestin-2 recruitment was observed in BRET assays of corynantheidine at hKOR. U50488H EC50 (nM) (pEC50 ± SEM) = 229.54 (6.64 ± 0.11) nM, Emax% ± SEM = 100 ± 4.63. Corynantheidine EC50 (nM) (pEC50 ± SEM) = n.d., Emax% ± SEM = <20. (E) Gi-1 activation in BRET assay of corynantheidine at hDOR. DPDPE EC50 (nM) (pEC50 ± SEM) = 1.23 (8.91 ± 0.28) nM, Emax% ± SEM = 100 ± 11.69. Corynantheidine EC50 (nM) (pEC50 ± SEM) = n.d., Emax% ± SEM = <20%. (F) No measurable βarrestin-2 recruitment was observed in BRET assays of corynantheidine at hDOR. DPDPE EC50 (nM) (pEC50 ± SEM) = 12.28 (7.91 ± 0.12) nM, Emax% ± SEM = 100 ± 4.08. Corynantheidine EC50 (nM) (pEC50 ± SEM) = n.d., Emax% ± SEM = <20. n.d. = not determined. Data from both Gi-1 activation and βarrestin-2 assays using human opioid receptors were normalized to Emax of the corresponding controls, DAMGO, U50,488H, and DPDPE. The data were processed in GraphPad Prism using a three-parameter logistic equation to fit the dose response curves. Mean EC50 (pEC50 ± SEM) represents the data for assays run in triplicate (n = 3). See Table S1 for values. (G) Time course of tail withdrawal antinociception of corynantheidine: Groups of C57BL/6J mice (n = 8 each group) received (i.c.v) corynantheidine, and antinociceptive response was measured at doses of 10, 30, and 100 nmol in WT mice at the indicated time points using the 55 °C tail withdrawal assay. Each point represents mean% antinociception ± SEM corynantheidine displayed potent dose dependent antinociceptive response. Attenuated effect of antinociception of corynantheidine (100 nmol, i.c.v.) was observed in MOR KO mice at 10–20 min (*p = 0.05, unpaired t test per row corrected for multiple comparisons using Holm-Sidak method).
Table 3.
Off-Target Profiling of Three Alkaloids at ~50 CNS Human Receptors through PDSP
| (a) mitraciliatine PDSP screening | |
|---|---|
| target | affinity (Ki (pEC50 ± SEM) nMa |
| hNET | 6604 (5.2 ± 0.1) |
| hMOR | 304 (6.52 ± 0.06) |
| hKOR | 73 (7.14 ± 0.07) |
| hα2A | 2153 (5.67 ± 0.09) |
| (b) Isopaynantheine PDSP Screening | |
| hSERT | 5111 (5.3 ± 0.2) |
| hDAT | 5066 (5.3 ± 0.2) |
| hMOR | 122 (6.91 ± 0.07) |
| hKOR | 27 (7.57 ± 0.07) |
| hH1 | 206 (6.7 ± 0.1) |
| hα2A | 2152 (5.7 ± 0.1) |
| (c) Corynantheidine PDSP Screening | |
| h5HT1A | 1156 (5.96 ± 0.06) |
| hSERT | 2615 (5.6 ± 0.2) |
| hDAT | 3915 (5.4 ± 0.2) |
| hMOR | 339 (6.47 ± 0.07) |
| hKOR | 2109 (5.68 ± 0.07) |
| hH2 | 5126 (5.29 ± 0.07) |
| hα1A | 397 (6.4 ± 0.06) |
| hα1B | 3304 (5.48 ± 0.06) |
| hα2A | 74 (7.1 ± 0.1) |
| hα2B | 1539 (5.81 ± 0.08) |
| hα2C | 269 (6.57 ± 0.09) |
| hSigma 2 | 1073 (6 ± 0.1) |
| hNMDA | 83 (7.08 ± 0.09) |
National Institute of Mental Health Psychoactive Drug Screening Program (NIMH-PDSP) conducted the binding affinities reported in Table 3. The methods and radioligands details used for the binding assays are available on the NIMH PDSP website and Besnard et al., 2012, 492(7428), 215–20.
Corynoxine and Corynoxine B.
We next looked at the oxindoles of corynantheidine, i.e., corynoxine and corynoxine B. In addition to Mitragyna speciosa, these alkaloids are also found in Cat’s claw.72–74 The binding affinities at hMOR and antinociception in rats have been recently reported, although functional characterization was not reported for both molecules.48 Corynoxine exhibited moderate affinity for mMOR (Ki = 140 nM, Table 1) and poor affinity for mKOR and mDOR (Ki > 1000 nM). In [35S]GTPγS functional assays (Table 2), corynoxine was similar to mitragynine with respect to its potency, but it had a higher efficacy than both mitragynine as well as DAMGO (Emax = 136%). It did not signal through mKOR or mDOR, consistent with its binding affinity at these targets.
At human opioid receptors, similar to [35S]GTPγS assays, corynoxine was a selective MOR full agonist (Figure 3A) with a potency 58-fold less than that of DAMGO. Corynoxine did not signal through other opioid receptor subtypes (Figure 3C,E) nor did it recruit βarrestin-2 at any opioid receptor subtypes (Figure 3B,D,F), similar to mitragynine and corynantheidine as we described before. In mice, corynoxine showed potent antinociception in the tail-withdrawal assay with an ED50 (and 95% C.I.) value of 3.43 (0.53–6.41) nmol using i.c.v. administration and comparable to the ED50 (and 95% C.I.) value of morphine (4.77 (1.49–28.8) nmol) (Figure 4A,B). Receptor selectivity in vivo was assessed in MOR KO mice, and corynoxine displayed MOR dependent antinociception, consistent with its in vitro activity (Figure 4A). Because most classical MOR agonists are known to lead to respiratory depression and hyperlocomotion, we evaluated this structurally unique natural product in the CLAMS (Comprehensive Laboratory Animal Monitoring System) assay.32,39 Whereas a 100 nmol, i.c.v. dose of morphine showed a significant decrease in breath rate, this dose of corynoxine showed no depression of breath rate; instead, it showed statistically significant respiratory stimulation (Figure 4C). Moreover, in contrast to morphine, this dose of corynoxine did not produce significant hyperlocomotion (Figure 4D).
Figure 3.

G-protein and βarrestin-2 signaling of corynoxine at opioid receptors. Corynoxine shows MOR selective G-protein signaling while not recruiting βarrestin-2. (A) Gi-1 activation measured using BRET assays at hMOR. Corynoxine is a full agonist at hMOR compared to DAMGO. DAMGO EC50 (nM) (pEC50 ± SEM) = 28.12 (7.55 ± 0.08) nM, Emax% ± SEM = 100 ± 2.78. Corynoxine EC50 (nM) (pEC50 ± SEM) = 1630 (5.79 ± 0.09) nM, Emax% ± SEM = 96.54 ± 4.69. (B) No measurable βarrestin-2 recruitment was observed in BRET assays of corynoxine at hMOR. DAMGO EC50 (nM) (pEC50 ± SEM) = 90.6 (7.04 ± 0.07) nM, Emax% ± SEM = 100 ± 2.29. Corynoxine EC50 (nM) (pEC50 ± SEM) = n.d. Emax% ± SEM < 20%. (C) Gi-1 activation measured using BRET assays at hKOR. U50488H EC50 (nM) (pEC50 ± SEM) = 6.93 (8.16 ± 0.06) nM, Emax% ± SEM = 100 ± 1.61. Corynoxine EC50 (nM) (pEC50 ± SEM) = n.d. Emax% ± SEM < 20%. (D) No measurable βarrestin-2 recruitment was observed in BRET assays of corynoxine at hKOR. U50488H EC50 (nM) (pEC50 ± SEM) = 48.95 (7.31 ± 0.04) nM, Emax% ± SEM = 100 ± 1.47. Corynoxine EC50 (nM) (pEC50 ± SEM) = n.d., Emax% ± SEM < 20. (E) Gi-1 activation measured using BRET assays at hDOR. DPDPE EC50 (nM) (pEC50 ± SEM) = 3.65 (8.43 ± 0.37) nM, Emax% ± SEM = 100 ± 14.51. Corynoxine EC50 (nM) (pEC50 ± SEM) = n.d. Emax% ± SEM < 20%. (F) No measurable βarrestin-2 recruitment was observed in BRET assays of corynoxine at hDOR. DPDPE EC50 (nM) (pEC50 ± SEM) = 17.69 (7.75 ± 0.17) nM, Emax% ± SEM = 100 ± 5.8. Corynoxine EC50 (nM) (pEC50 ± SEM) = n.d., Emax% ± SEM < 20. n.d. = not determined. Data from both Gi-1 activation and βarrestin-2 assays using human opioid receptors were normalized to Emax of the corresponding controls, DAMGO, U50,488H, and DPDPE. The data were processed in GraphPad Prism using a three-parameter logistic equation to fit the dose response curves. Mean EC50 (pEC50 ± SEM) represents the data for assays run in triplicate (n = 3). See Table S1 for values.
Figure 4.

Dose dependent antinociception time course, respiratory depression, and locomotor effects of corynoxine. Corynoxine shows MOR dependent antinociception. Attenuated respiratory depression and locomotor effects were observed for corynoxine at equianalgesic morphine doses. (A) Time course of tail withdrawal antinociception of corynoxine: Groups of C57BL/6J mice (n = 8 each group) received (i.c.v.) corynoxine, and antinociceptive response was evaluated at doses of 3, 10, and 30 nmol in WT mice at the indicated time points using the 55 °C tail withdrawal assay. Each point represents mean% antinociception ± SEM. Corynoxine exhibited potent potent dose dependent antinociceptive response. Reduced effect of antinociception of corynoxine (30 nmol, i.c.v.) was observed in MOR KO mice at 10–30 min (****p < 0.0001, unpaired t test per row corrected for multiple comparisons using Holm-Sidak method). (B) Antinociception time course of Morphine: Groups of C57BL/6J mice (n = 8 each group) received (i.c.v.) morphine, and antinociceptive response was evaluated at doses of 0.3, 1, 3, and 10 nmol in WT mice at the indicated time points using the 55 °C tail withdrawal assay. Each point represents mean% antinociception ± SEM. (C) Respiratory rate corynoxine: Groups of mice received either vehicle (n = 12), saline (n = 12), morphine (100 nmol, i.c.v.; n = 12), or corynoxine (100 nmol, i.c.v.; n = 12), and the measurement of breath rates was done every 20 min for 180 min. Corynoxine showed an increase in breath rates at 60 min (***p = 0.0002), 80–160 min (****p < 0.0001), and 180 min (***p = 0.001) compared to those of the vehicle, while morphine decreased breadth rates at 20–40 min (*p = 0.0387). The p values were calculated by 2-way ANOVA followed by Dunnett’s multiple-comparison test. (D) Locomotor effect of corynoxine: Groups of mice received either vehicle (n = 24), saline (n = 12), morphine (100 nmol, i.c.v.; n = 12), or corynoxine (100 nmol, i.c.v.; n = 12), and the distance traveled by charcoal for each group of mice was measured. Corynoxine showed no significant hyperlocomotion compared to that of the vehicle as determined by 2-way ANOVA followed by Dunnett’s multiple-comparison test. However, significant locomotor effect was observed for morphine at 80–100 min (****p < 0.0001), 120 min (**p = 0.0038), 140–160 min (****p < 0.0001), and 180 min (**p = 0.0093) compared to that of the vehicle. The p values were calculated by 2-way ANOVA followed by Dunnett’s multiple-comparison test.
Screening of corynoxine B showed poor binding affinity at all opioid subtypes (Ki > 1000 nM) (Table 1) and no activity in [35S]GTPγS assays (Table 2). Accordingly, this molecule was not screened in BRET assays for either G-protein or βarrestin-2 recruitment nor is it tested in mice.
Mitraciliatine.
We next screened mitraciliatine. This natural product has been described in the literature to be present in kratom,44 but to the best of our knowledge, this natural product has not been evaluated pharmacologically. Similar to other kratom natural products, this molecule had moderate mMOR (Table 1) affinity (Ki = 135 nM). To our surprise, it had similar mKOR affinity (Ki = 101 nM), in contrast to the majority of mitragynine template based compounds which tend to have higher affinity and activity at MOR.34 In [35S]GTPγS assays, it was a mixed mMOR–mKOR dual agonist (Table 2). Although its potency was similar to that of mitragynine (EC50 ∼ 200 nM), at both subtypes, it was found to be a partial agonist (Emax = 47%) at mMOR and displayed full agonism at mKOR (Emax = 98%). At mDOR, mitraciliatine had no activity (Table 2). In BRET based assays with hMOR and hKOR (Figure 5B–D), mitraciliatine differentially recruited βarrestin-2. Consistent with mitragynine template based compounds, no βarrestin-2 recruitment was seen through hMOR (Figure 5B). In contrast, mitraciliatine robustly recruited βarrestin-2 (Emax = 104%) through hKOR (Figure 5D). At hDOR, it showed no signaling at both Gi as well as βarrestin-2 (Figure 5E,F). Consistent with results utilizing rodent receptors, mitraciliatine was a partial agonist (Emax = 51%) at hMOR while displaying full agonism at hKOR. Unlike other mitragynine congeners, this compound had a cleaner off-target profile (Table 3a). It had a moderate hKOR affinity (Ki = 73 nM)/hMOR affinity (Ki = 304 nM) and labeled two other nonopioid targets: the norepinephrine transporter (hNET) (Ki = 6604 nM) and hα2A receptor (Ki = 2153 nM). When administered supraspinally (i.c.v.) to mice, mitraciliatine produced a ceiling effect in antinociception (Figure 5G), with maximal antinociception of 67% at a dose of 100 nmol, i.c.v. Testing in opioid receptor knockout mice suggested that the antinociception was MOR dependent and, surprisingly, independent of KOR (Figure 5G). When evaluated in the CLAMS assay at a 100 nmol, i.c.v. dose, mitraciliatine showed no respiratory depression or hyperlocomotion. (Figure 5H,I).
Figure 5.

G-protein and βarrestin-2 signaling, antinociception time course, respiratory depression, and locomotor effects of mitraciliatine. Mitraciliatine shows agonism at hMOR and hKOR while showing differential βarrestin-2 signaling at the same subtypes. Mitraciliatine shows MOR dependent antinociception. Attenuated respiratory depression and locomotor effects were observed for mitraciliatine at equianalgesic morphine doses. (A) Gi-1 activation measured using BRET assays at hMOR. Mitracilliatine shows G-protein signaling at hMOR while showing no βarrestin-2 signaling, compared to DAMGO. DAMGO EC50 (nM) (pEC50 ± SEM) = 3.56 (8.45 ± 0.13) nM, Emax% ± SEM = 100 ± 4.17. Mitraciliatine EC50 (nM) (pEC50 ± SEM) = 23.69 (7.62 ± 0.16) nM, Emax% ± SEM = 50.7 ± 3.22. (B) No measurable βarrestin-2 recruitment was observed in BRET assays of mitracilatine at hMOR. DAMGO EC50 (nM) (pEC50 ± SEM) = 90.6 (7.04 ± 0.07) nM, Emax% ± SEM = 100 ± 2.29. Mitraciliatine EC50 (nM) (pEC50 ± SEM) = n.d., Emax% ± SEM < 20. (C) Gi-1 activation measured using BRET assays at hKOR. Mitracilliatine shows G-protein signaling at hKOR as well as robust βarrestin-2 signaling at hKOR. U50488H EC50 (nM) (pEC50 ± SEM) = 6.93 (8.16 ± 0.06) nM, Emax% ± SEM = 100 ± 1.61. Mitraciliatine EC50 (nM) (pEC50 ± SEM) = 269.19 (6.57 ± 0.06) nM, Emax% ± SEM = 114.32 ± 2.42. (D) Significant βarrestin-2 recruitment was observed in BRET assays of mitracilatine at hKOR. U50488H EC50 (nM) (pEC50 ± SEM) = 48.95 (7.31 ± 0.04) nM, Emax% ± SEM = 100 ± 1.47. Mitraciliatine EC50 (nM) (pEC50 ± SEM) = 383.12 (6.42 ± 0.06) nM, Emax% ± SEM = 104.13 ± 2.57. (E) Gi-1 activation measured using BRET assays at hDOR. Mitraciliatine did not show G-protein or βarrestin-2 signaling at hDOR. DPDPE EC50 (nM) (pEC50 ± SEM) = 3.65 (8.43 ± 0.37) nM, Emax% ± SEM = 100 ± 14.54. Mitraciliatine EC50 (nM) (pEC50 ± SEM) = n.d., Emax% ± SEM = n.d. (F) No measurable βarrestin-2 recruitment was observed in BRET assays of mitracilatine at hDOR. DPDPE EC50 (nM) (pEC50 ± SEM) = 17.69 (7.75 ± 0.17) nM, Emax% ± SEM = 100 ± 5.8. Mitraciliatine EC50 (nM) (pEC50 ± SEM) = n.d., Emax% ± SEM < 20. n.d. = not determined. Data from both Gi-1 activation and βarrestin-2 assays using human opioid receptors were normalized to Emax of the corresponding controls, DAMGO, U50,488H, and DPDPE. The data were processed in GraphPad Prism using a three-parameter logistic equation to fit the dose response curves. Mean EC50 (pEC50 ± SEM) represents the data for assays run in triplicate (n = 3). See Table S1 for values. (G) Antinociception time course of mitraciliatine: Groups of C57BL/6J mice (n = 8 each group) received (i.c.v.) mitraciliatine, and antinociceptive response was evaluated at doses of 10, 30, and 100 nmol in WT mice at the indicated time points using the 55 °C tail withdrawal assay. Each point represents mean% antinociception ± SEM. We observed potent dose dependent antinociceptive response of mitraciliatine. Antinociception effect of mitraciliatine (100 nmol, i.c.v.) remained intact in KOR KO mice, although a statistically not significant increase in %MPE was seen. Antinociception was found attenuated in MOR KO mice at 10–30 min (***p = 0.0001, 0.001, and 0.0001 at 10, 20, and 30 min, respectively, unpaired t test per row corrected for multiple comparisons using Holm-Sidak method). (H) Respiratory rate mitraciliatine: Groups of mice received either vehicle (n = 12), saline (n = 12), morphine (100 nmol, i.c.v.; n = 12), or mitraciliatine (100 nmol, i.c.v.; n = 12), and the measurement of breath rates was done every 20 min for 180 min. Mitraciliatine showed increase in breath rates at 100 min (*p = 0.0233), 120 min (**p = 0.0017), and 140–180 min (****p < 0.0001) compared to vehicle, while morphine decreased breadth rates at 20–40 min (*p = 0.0387). The p values were calculated by 2-way ANOVA followed by Dunnett’s multiple-comparison test. (I) Locomotor effect mitraciliatine: Groups of mice received either vehicle (n = 24), saline (n = 12), morphine (100 nmol, i.c.v.; n = 12), or mitraciliatine (100 nmol, i.c.v.; n = 12), and the distance traveled by charcoal for each group of mice was measured. Mitraciliatine showed no significant hyperlocomotion compared to that of the vehicle as determined by 2-way ANOVA followed by Dunnett’s multiple-comparison test. However, significant locomotor effect was observed for morphine at 80–100 min (****p < 0.0001), 120 min (**p = 0.0038), 140–160 min (****p < 0.0001), and 180 min (**p = 0.0093) compared to that of the vehicle. The p values were calculated by 2-way ANOVA followed by Dunnett’s multiple-comparison test.
Isopaynantheine.
Isopaynantheine is the C20-vinyl analogue of mitraciliatine. Similar to mitraciliatine, it has been described in the literature to be present in kratom but has never been characterized pharmacologically. In binding assays, it was similar to mitraciliatine with mixed affinity (Table 1) at both mMOR (Ki = 92 nM) and mKOR (Ki = 325 nM). In [35S]GTPγS assays, too, it was found to act as a partial agonist at mMOR (Emax = 50%) and showed full agonism at mKOR (Table 2). The potencies were comparable to mitraciliatine. In BRET assays at human opioid receptors, isopaynantheine showed no G-protein activity at hMOR (Figure 6A), in sharp contrast to the structurally similar natural product mitraciliatine, which was a partial agonist at hMOR. No measurable activity was seen in the βarrestin-2 signaling pathway as well (Figure 6B). Because it was found to have modest affinity as well as partial agonism at mMOR, we next examined this natural product to see if it was an antagonist at hMOR. We found that isopaynanthine was a modestly potent MOR antagonist (IC50 = 1.26 μM) in comparison to diprenorphine (IC50 = 4.2 nM) in the Gi-1 pathway (Figure 6C). Similarly, it was an antagonist in the βarrestin-2 pathway as well (Figure 6D). At hKOR, it was found to be an agonist with a Emax = 80% and modest potency (EC50 = 560 nM) compared to that of U50,488h (EC50 = 7 nM) in the Gi-1 pathway (Figure 6E). In sharp contrast to mitraciliatine, it did not recruit βarrestin-2 at hKOR (Figure 6F). Similar to other alkaloids, no signaling was seen through hDOR (Figure 6G,H). Off-target screening (Table 3b) revealed it was less cleaner than mitraciliatine but far superior than mitragynine.45,56 It had a high hKOR affinity (Ki = 27 nM)/lower hMOR affinity (Ki = 122 nM) but also had a labeled H1 receptor with a 206 nM affinity (Table 3b). While hSERT (serotonin transporter), hDAT (dopamine transporters), and hα2A were also labeled by this molecule, the affinities were poorer for these targets.
Figure 6.

G-protein, βarrestin-2 signaling, and dose dependent antinociception time course of isopaynantheine. Isopaynantheine was an MOR antagonist–KOR biased agonist. Antinociception of Isopaynantheine is KOR dependent. (A) Gi-1 activation measured using BRET assays at MOR. Isopaynantheine did not show G-protein signaling at MOR compared to DAMGO. DAMGO EC50 (nM) (pEC50 ± SEM) = 31.27 (7.50 ± 0.03) nM, Emax% ± SEM = 100 ± 1.51. Isopaynantheine EC50 (nM) (pEC50 ± SEM) = n.d., Emax% ± SEM < 20. (B) No measurable βarrestin-2 recruitment was observed in BRET assays of isopaynantheine at MOR. DAMGO EC50 (nM) (pEC50 ± SEM) = 81.62 (7.09 ± 0.06) nM, Emax% ± SEM = 100 ± 2.55. Isopaynantheine EC50 (nM) (pEC50 ± SEM) = n.d., Emax% ± SEM < 20. (C) Gi-1 antagonism measured using BRET assays at MOR. Diprenorphine EC50 (nM) (pEC50 ± SEM) = 4.28 (8.37 ± 0.05) nM, Emax% ± SEM = 100 ± 2.90. Isopaynantheine EC50 (nM) (pEC50 ± SEM) = 1259 (5.9 ± 0.12) nM, Emax% ± SEM = 156.91 ± 13.49. (D) βarrestin-2 antagonism measured using BRET assays at MOR. Diprenorphine EC50 (nM) (pEC50 ± SEM) = 0.43 (9.37 ± 0.10) nM, Emax% ± SEM = 100 ± 2.47. Isopaynantheine EC50 (nM) (pEC50 ± SEM) = 1276.4 (5.89 ± 0.14), Emax% ± SEM = 106 ± 8.67. n.d. = not determined. (E) Gi-1 activation measured using BRET assays at KOR. Isopaynantheine shows G-protein signaling at KOR while showing no βarrestin-2 signaling. U50488H EC50 (nM) (pEC50 ± SEM) = 7.03 (8.15 ± 0.04) nM, Emax% ± SEM = 100 ± 1.58. Isopaynantheine EC50 (nM) (pEC50 ± SEM) = 560.4 (6.25 ± 0.066) nM, Emax% ± SEM = 80.5 ± 2.9. (F) No βarrestin-2 recruitment was observed in BRET assays of isopaynantheine at KOR. U50488H EC50 nM (pEC50 ± SEM) = 128.31 (6.89 ± 0.10) nM, Emax% ± SEM = 100 ± 4.12. Isopaynantheine EC50 (nM) (pEC50 ± SEM) = 599.29 (6.22 ± 0.43) nM, Emax% ± SEM < 20. (G) Gi-1 activation measured using BRET assays at DOR. Isopaynantheine did not show G-protein or βarrestin-2 signaling at DOR. SNC80 EC50 (nM) (pEC50 ± SEM) = 13.37 (7.87 ± 0.05) nM, Emax% ± SEM = 100 ± 1.89. Isopaynantheine EC50 (nM) (pEC50 ± SEM) = n.d., Emax% ± SEM < 20. (H) No measurable βarrestin-2 recruitment was observed in BRET assays of isopaynantheine at DOR. DPDPE EC50 (nM) (pEC50 ± SEM) = 7.76 (8.11 ± 0.05) nM, Emax% ± SEM = 100 ± 1.81. Isopaynantheine EC50 (nM) (pEC50 ± SEM) = n.d., Emax% ± SEM < 20. n.d. = not determined. Data from both Gi-1 activation and βarrestin-2 assays using human opioid receptors were normalized to Emax of the corresponding controls, DAMGO, U50,488H, DPDPE, and diprenorphine (for anatogonism). The data were processed in GraphPad Prism using a three-parameter logistic equation to fit the dose response curves. Mean EC50 (pEC50 ± SEM) represents the data for assays run in triplicate (n = 3). See Table S1 for values. (I) Antinociception time course of isopaynantheine: Groups of C57BL/ 6J mice (n = 8 each group) received (i.c.v.) isopaynantheine, and antinociceptive response was evaluated at doses of 10, 30, and 100 nmol in WT mice at the indicated time points using the 55 °C tail withdrawal assay. Each point represents mean% antinociception ± SEM. Isopaynantheine exhibited strong dose dependent antinociceptive response. Isopaynantheine (100 nmol, i.c.v.) retained the antinociceptive response in MOR KO mice. Antinociception was found reduced in KOR KO mice at 10–20 min (***p = 0.0001, *0.013 at 10 and 20 min, respectively, unpaired t test per row corrected for multiple comparisons using Holm-Sidak method). (J) Antinociception time course of U50488H: Groups of C57BL/6J mice (n = 8 each group) received (i.c.v.) U50488H, and antinociceptive response was evaluated at doses of 3, 10, and 30 nmol in WT mice at the indicated time points using the 55 °C tail withdrawal assay. Each point represents mean% antinociception ± SEM.
When assessed supraspinally, isopaynanthine showed antinociceptive activity with an ED50 (and 95% CI) value of 20.6 (6.97, 36.98 nmol) compared to that of U50,488H (ED50 = 8.8 (5.7, 13.5 nmol) (Figure 6I,J). Receptor selectivity was also assessed in vivo using MOR and KOR KO mice (Figure 6I). Antinociception was MOR independent and KOR dependent.
DISCUSSION
Kratom, a plant which grows in Southeast Asia has been under intense investigation in recent years. Earlier work prior to the 1990s and 2000s38,41,52 on kratom and its alkaloids focused on characterization with in vivo75–78 and ex vivo57,79 functional assays. In the past five years, the signaling properties of the component alkaloids have been assessed in cell lines expressing opioid receptors, as well as more detailed re-evaluation of kratom itself in animal models.39,50 Chemical characterization of individual alkaloids44 present in kratom are also being evaluated.
Kratom tea (a concoction of numerous alkaloids) and kratom alkaloid extracts50 have been described as agents with much-reduced symptoms of opioid withdrawal compared to those of traditional MOR agonists like morphine. Moreover, antiopioid withdrawal effects in subjects physically dependent on morphine with minimal adverse effects has also been observed in animal models.39 The major component in kratom, mitragynine, has been characterized pharmacologically in in vitro assays as well as in vivo assays. In cellular studies, mitragynine was found to exhibit partial agonism at MOR in amplified G-protein assays while showing no recruitment of βarrestin-2 by us and others.34,43,50,51,53,80 Mitragynine also has been proposed to be a low efficacy agonist81 in vivo and has been found to have no abuse liability on its own while also blunting the self-administration of opioid agonists like heroin in rats.82 In mice, it has also been reported to have less physical dependence than morphine alone and to block morphine withdrawal. The involvement of adrenergic receptors56,83 and serotonergic receptors45,78,84 as well as MOR itself in the biological actions of mitragynine has been recognized and remains a topic of emerging study.56
Another alkaloid, 7-hydroxy mitragynine (7OH, present in far less amounts <1%, not seen in our extracts),85 is a partial agonist at MOR, with enhanced efficacy and potency compared to those of mitragynine while also showing reduced βarrestin-2 recruitment.43,51 It is currently believed that the antinociceptive actions of mitragynine are mediated by its bioconversion to 7OH through a CYP450 dependent pathway.53 Unlike mitragynine, 7OH shows adverse effects similar to those of classical MOR agonists and is a potent MOR dependent antinociceptive agent with tolerance, dependence, and abuse potential similar to those of typical opioid agonists in mice.43,51,53,76
The semisynthetic analog mitragynine pseudoindoxyl (MP) was initially reported in the literature as a fungal metabolite of mitragynine.55 The spiro-pseudoindoxyl core of MP (Figure 1) resulted from the Lewis acidic rearrangement of 7OH. MP is also an antinociceptive agent through opioid receptors and shows G-protein partial agonism at MOR with minimal βarrestin-2 recruitment.51 Recent reports suggest that this alkaloid is a metabolite formed in vitro from 7OH.86
Recent efforts from the Filizola group have probed the binding of these three alkaloids and suggested subtle but important differences in the binding poses of these three alkaloids using computational simulations coupled with mutational validation.54
Through this work, we now report the binding and functional characterization of minor kratom alkaloids and develop a structure–activity relationship (SAR) for subtype selectivity, efficacy, and βarrestin-2 recruitment which may help us and others in the field toward additional derivatization efforts in the future as well as help us map out the individual contribution of kratom alkaloids toward its bioactivity in humans.
Similar to the related parent natural product, corynantheidine or 9-desmethoxy mitragynine shows MOR partial agonism, MOR subtype selectivity, and no recruitment of βarrestin-2, and it labels multiple nonopioid receptors while displaying MOR dependent antinociception in mice. The present affinity of corynantheidine at MOR, and higher affinity for adrenergic receptors over MOR, was comparable to recent reports by Obeng et al.,56 although in our case, we find a higher affinity at α2A instead of α1D.
The oxindole of corynantheidine, corynoxine retains the parent template’s MOR selectivity but interestingly shows higher efficacy at both mouse receptors measured using [35S]GTPγS assays as well as Gi-1 BRET assays at human receptors compared to those of mitragynine and corynantheidine. It is possible that the change of central core indole to oxindole leads to this increase in efficacy, although additional analogues need to be synthesized before this SAR can be validated. The binding affinity at MOR for corynoxine has been reported to be 16.4 nM at hMOR48 compared to 140 nM for mMOR in our hands. It is possible that the differences may be attributed to the use of different assays or to the screening with receptors from different species. It is worth noting that the functional activity at hMOR for corynoxine was also moderate and in line with the binding affinity in mMOR. Our findings of corynoxine antinociception in mice are in line with antinociception reported in rats following intravenous (IV) administration.48 Of interest, despite displaying full MOR agonism, corynoxine showed no respiratory depression or hyperlocomotion characteristic of typical MOR agonists. Exact mechanisms for this separation of adverse effects is unknown at this point and will be investigated in detail later on.
Two alkaloids mitraciliatine and isopaynantheine have never been characterized in the literature. Both natural products show unique mixed actions at MOR as well as KOR in vitro at mouse and human receptors. Mitragynine is reported to have weak KOR antagonism,51,87 to label adrenergic receptors,45,56 and to have serotonergic actions with in vivo testing.78 In contrast, mitraciliatine demonstrated KOR full agonism and MOR partial agonism at both mouse and human receptors. Mitraciliatine does not recruit βarrestin-2 at MOR but shows robust βarrestin-2 recruitment at KOR and, based on preliminary in vitro screening, appears to have higher receptor selectivity for opioid over adrenergic receptors with fewer off-target interactions. It is possible that the stereochemistry at C3 (3S in mitragynine but 3R for mitraciliatine) and C20 (20S in mitragynine but 20R for mitraciliatine) plays a key role in the SAR as well as a cleaner off-target profile for these compounds.
The signaling profile as well as receptor activity of isopaynantheine was unique as well. At mouse receptors similar to mitraciliatine, it showed MOR partial agonism and KOR full agonism. At human opioid receptors in sharp contrast to mitraciliatine, it was an MOR antagonist. At KOR, it was a high efficacy agonist. Differences were also seen in βarrestin-2 recruitment; at MOR, it was an antagonist, and at KOR, it showed reduced βarrestin-2 recruitment.
Mitraciliatine also showed no respiratory depression or hyperlocomotion, potentially due to its partial agonism in both cell lines and mice, where a ceiling effect was observed. Recently, partial agonism has been revisited as a means to develop MOR-mediated antinociception with attenuated respiratory depression.88,89
A discrepancy between the in vitro/in vivo receptor activity was observed in the present activity of mitraciliatine. In spite of full agonism at mKOR in [35S]GTPγS assays, antinociceptive actions were independent of KOR in mice and only dependent on MOR. While further study is needed, there is precedence for discrepancies of this type in the opioid field. For example, a lack of correlation between in vitro/in vivo actions exist with MP1104, which possesses pM affinity90 and agonist activity70 in functional assays at MOR but was found to lack MOR activity in vivo.20
An unexpected increase in breathing rate was observed with both corynoxine as well as mitraciliatine in the CLAMS assay. In the literature, enhanced respiration has been attributed to KOR agonism,91 serotonin,92 and adrenergic receptors;93 the exact mechanism of this respiratory stimulation remains unknown and requires more detailed examination in due course.
C20 substituent (vinyl vs ethyl) was identified as a functional switch for in vivo MOR vs KOR receptor selectivity. Mitraciliatine with C20 ethyl substituent was an MOR agonist, while isopaynantheine with a vinyl group was a KOR agonist in mice.
CONCLUSION
In conclusion, we report a thorough and complete in vitro pharmacological characterization of five kratom based minor alkaloids. Given their low abundance, it seems unlikely that these alkaloids play a major mediating role in the biological actions of kratom consumed by humans. However, these alkaloids represent novel starting points for optimizing probes to better understand opioid receptor function.
There are three major findings from this present work. First, we identify three new templates present in kratom with antinociceptive activity in mice, with corynoxine being equipotent to morphine. Second, we identify ligands with an array of pharmacological profiles, ranging from the partial opioid agonism displayed by corynantheidine and mitraciliatine and full agonism of corynoxine and KOR agonism with isopaynantheine. Finally, we identify corynoxine and mitraciliatine to be structurally unique natural products with safer, MOR dependent antinociception, and we identify isopaynantheine as the first kratom alkaloid with KOR mediated antinociceptive actions.
Our studies add to the growing chorus of reports highlighting kratom as a valuable source of opioid-active templates. Nearly ∼41 individual alkaloids in kratom still remain uncharacterized. It is hoped that kratom and its alkaloids (including indole and oxindole templates reported in this manuscript) may eventually lead to a new generation of analgesics.
MATERIALS AND METHODS
Drugs and Chemicals.
The Research Technology Branch of the National Institute on Drug Abuse (Rockville, MD) provided the opiates. Buffers and miscellaneous chemicals were purchased from Sigma-Aldrich. We purchased kratom “Red Indonesian Micro Powder” from Moon Kratom (Austin, TX). Corynoxine and corynoxine B were purchased from BOC Sciences (NY, USA). DMSO was used to dissolve all nonradioactive compounds and diluted with water for conducting assays. The assays were conducted with 1–2.5% of final concentration of DMSO.
Chemistry.
We purchased all the chemicals from Sigma-Aldrich, and they were used directly with no further purification. Flame-dried reaction flasks were used to carry out the reactions. All reactions were performed under inert atmosphere using argon. Purification of the reaction mixtures were achieved by flash column chromatography on E. Merck 230–400 mesh silica gel 60 using a Teledyne ISCO CombiFlash Rf instrument with UV detection at 280 and 254 nm. We used RediSep Rf silica gel normal phase columns. Isolated yields are reported in all the cases. A Varian 400/500 MHz NMR spectrometer was used to record the NMR spectra. All the NMR data were processed with MestReNova software. The chemical shifts are reported in parts per million (ppm) downfield of tetramethylsilane and referenced to the residual solvent peak unless otherwise noted (CDCl3 1H = 7.26, 13C = 77.3). Peak multiplicity is reported using the following abbreviations: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad. Coupling constants (J) are expressed in hertz (Hz). A Bruker Daltonics 10 T Apex Qe Fourier Transform Ion Cyclotron Resonance-Mass Spectrometer (ESI-MS) was used to record the high resolution mass spectra. The accurate masses of the molecular ion [M + H]+ are presented and matched well with the calculated value. High pressure liquid chromatography (HPLC) was carried out to determine the purity of the synthesized and isolated compounds. Instrumentation details of HPLC are provided in the Supporting Information (SI).
Isolation of Mitragynine from Mitragyna speciosa (Kratom).
Extraction of mitragynine was carried out from the dry powdered kratom leaves adopting a modification of previously reported protocol.51 Kratom powder (500 g) was refluxed in MeOH, 500 mL, for 30 min. Next, the suspension was filtered, and the alcoholic extraction process was repeated two more times (2 × 500 mL). The solvent from 3 combined extract was evaporated under reduced pressure, and the alkaloid content was dried in high vacuum. The residue was suspended in 10% aqueous AcOH (1 L) and washed several times with hexane (4 × 500 mL). Then, the aqueous layer was cooled to 0 °C in an ice bath and basified (pH ∼ 10) slowly with aqueous NaOH solution (2.5M. ∼1L). EtOAc (4 × 400 mL) was used to extract the alkaloids from the aqueous layer. The combined EtOAc layer was washed with brine (250 mL) and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure, and the residue was dried in high vacuum to obtain the crude alkaloid extract (9.8 g). This crude kratom extract was subjected to silica gel column chromatography, using 0–15% MeOH in dichloromethane to isolate mitragynine (4.7 g), paynantheine (568 mg), speciogynine (343 mg), and speciociliatine (754 mg). After removing the major alkaloid (mitragynine, paynantheine, speciogynine, and speciociliatine) fractions out, the rest of the crude material was again injected to silica gel column chromatography, using 5–20% MeOH in dichlomethane to isolate mitraciliatine (2 mg; 0.02% of total alkaloid content) and isopaynantheine (3 mg; 0.03% of total alkaloid content) as the minor alkaloids.
Isopaynantheine (6).
1H NMR of 6 matched well with the literature reported value.44 1H NMR (500 MHz, CDCl3): δ 7.88 (s, 1H), 7.27 (d, J = 1.1 Hz, 1H), 7.05 (t, J = 7.8 Hz, 1H), 6.99 (d, J = 8.2 Hz, 1H), 6.50 (d, J = 7.6 Hz, 1H), 5.37 (ddd, J = 16.8, 10.3, 8.4 Hz, 1H), 4.92 (dd, J = 17.1, 2.2 Hz, 1H), 4.85 (dd, J = 10.3, 2.1 Hz, 1H), 4.46 (s, 1H), 3.91 (d, J = 1.0 Hz, 3H), 3.76 (s, 3H), 3.68 (d, J = 1.1 Hz, 3H), 3.29–3.23 (m, 2H), 3.19 (qd, J = 7.9, 6.6, 3.7 Hz, 1H), 3.01–2.92 (m, 1H), 2.85 (dd, J = 15.5, 2.8 Hz, 1H), 2.64 (q, J = 4.4, 3.4 Hz, 2H), 2.46 (dq, J = 13.4, 8.0, 4.9 Hz, 1H), 2.37 (ddd, J = 13.0, 10.8, 2.8 Hz, 1H), 1.99 (d, J = 13.4 Hz, 1H). Purity = 98.6%
Mitraciliatine (7).
1H NMR of 7 matched well with the literature reported value.44 1H NMR (500 MHz, CDCl3): δ 7.92 (s, 1H), 7.31 (s, 1H), 7.04 (t, J = 7.8 Hz, 1H), 6.99 (dd, J = 8.1, 0.8 Hz, 1H), 6.49 (dd, J = 7.7, 0.9 Hz, 1H), 4.45 (dd, J = 5.2, 2.5 Hz, 1H), 3.90 (s, 3H), 3.76 (s, 3H), 3.69 (s, 3H), 3.26 (ddt, J = 8.1, 6.2, 2.6 Hz, 2H), 3.22–3.17 (m, 1H), 2.89–2.82 (m, 1H), 2.78 (dd, J = 11.2, 3.4 Hz, 1H), 2.48–2.38 (m, 2H), 2.25–2.13 (m, 2H), 1.99–1.93 (m, 1H), 1.34–1.27 (m, 2H), 0.78–0.75 (m, 3H). Purity = 95.1%
Chemical Charecterization of Synthesized Compounds. Methyl(E)-2-((2S,3S,12bS)-3-ethyl-8-(((trifluoromethyl)sulfonyl)oxy)1,2,3,4,6,7,12,12b-octahydroindolo[2,3-a]quinolizin-2-yl)-3-methoxyacrylate (13).
To a solution of 12 (9-hydroxymitragynine, 130 mg, 0.32 mmol) in dry dichloromethane (6 mL), N-phenylbis(trifluoromethanesulfonimide) (133 mg, 0.36 mmol) was added under argon at RT. Et3N (0.14 mL, 1 mmol) was added to the reaction mixture, and stirring was continued for 12 h. Next, the reaction mixture was concentrated and worked up with EtOAc (40 mL) and brine (2 × 30 mL) and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure, and the residue was purified in flash chromatography using 15–70% EtOAc in hexane. Desired triflate 13 (102 mg) was obtained as a white solid (yield, 61%). Only 1HNMR was recorded, because this is an intermediate compound. 1H NMR (400 MHz, CDCl3): δ 8.01 (s, 1H), 7.44 (d, J = 0.8 Hz, 1H), 7.28 (dt, J = 8.0, 0.8 Hz, 1H), 7.07 (dd, J = 8.5, 7.6 Hz, 1H), 6.98 (d, J = 7.9 Hz, 1H), 3.74 (s, 3H), 3.71 (s, 3H), 3.23–3.10 (m, 2H), 3.02 (tt, J = 17.0, 4.9 Hz, 3H), 2.91 (dd, J = 15.4, 3.6 Hz, 1H), 2.62–2.51 (m, 2H), 2.47 (dd, J = 11.7, 3.1 Hz, 1H), 1.85–1.69 (m, 2H), 1.69–1.60 (m, 1H), 1.25–1.16 (m, 1H), 0.87 (t, J = 7.3 Hz, 3H). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H28F3N2O6S 517.1620; found, 517.1611.
Corynantheidine (5).
To a solution of triflate 13 (77.5 mg, 0.15 mmol) in dry DMF (3 mL) in a sealed tube, Pd(OAc)2 (11 mg, 0.05 mmol), dppp (31 mg, 0.07 mmol), Et3N (0.4 mL, 3 mmol), and HCOOH (8 uL, 0.27 mmol) were added. The stirring was continued at 80 °C for 8 h. Next, the reaction mixture was allowed to cool to RT and diluted with EtOAc (50 mL). Regular work up was done using EtOAc and brine (15 × 4 mL). The EtOAc part was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The residue was purified by flash column chromatography using 20–70% EtOAc in hexanes. Compound 5 (40 mg, 65%) was obtained as a white amorphous solid after purification. 1H NMR of 5 matched well with the literature reported value. 1H NMR (500 MHz, CDCl3): δ 7.76 (s, 1H), 7.47 (dd, J = 7.6, 1.2 Hz, 1H), 7.44 (s, 1H), 7.29 (dt, J = 7.9, 1.0 Hz, 1H), 7.13–7.07 (m, 2H), 3.72 (s, 3H), 3.71 (s, 3H), 3.24 (dq, J = 11.4, 2.2 Hz, 1H), 3.09–2.99 (m, 4H), 2.76–2.69 (m, 1H), 2.63–2.58 (m, 1H), 2.56–2.50 (m, 2H), 1.87–1.81 (m, 1H), 1.81–1.75 (m, 1H), 1.66 (dt, J = 11.1, 3.1 Hz, 1H), 1.27–1.20 (m, 1H), 0.89 (t, J = 7.4 Hz, 3H). Purity = 98.9%
Biological Assays.
Affinity Determination Using Binding Assays.
Radioligand binding assays using [125I]IBNtxA as the radioactive ligand were used to determine the affinity (Ki) of natural products by using previosuly published protocols.90,94,95 Membranes from Chinese hamster ovary cells (CHOs) which stably express mMOR, mDOR, and mKOR were used in the assay carried out at 25 οC for 90 min. For mMOR, 50 mM potassium phosphate buffer with 5 mM MgSO4 was used, and 20 μg/μL membranes was used. For mKOR and mDOR binding, only 50 mM potassium phosphate buffer was used, and 40 μg/μL membranes was used. Following incubation, assay contents were filtered through glass fiber filters (obtained from Whatman Schleicher and Schuell, Keene, NH), and assay tubes were washed with 3 mL of ice-cold 50 mM Tris–HCl buffer at pH 7.4 thrice on a semiautomatic cell harvester. Levallorphan (8 μM) was used to determine nonspecific binding and specific binding determined by subtracting total binding from nonspecific binding. Protein concentrations were determined using the Lowry assay using BSA as control as described before.96 Affinities (Ki) were determined using the Cheng Prusoff equation: Ki = (IC50)/(1 + L), where L = (concentration of radioligand [125I[IBNtxA used in asasy)/(Kd of radioligand [125I]IBNtxA) by nonlinear regression analysis in GraphPad Prism.97,98
Functional Assays [35S]GTPγS.
To determine if the natural products behaved as full agonists or partial agonists, the [35S]GTPγS assay in the same membranes (mMOR, mKOR, and mDOR stably expressed in CHO cell lines) as those used in the binding assays were used. Assay incubation time was 60 min at 30 °C in buffer (50 mM Tris–HCl, pH 7.4, 3 mM MgCl2, 0.2 mM EGTA, and 10 mM NaCl) containing 0.05 nM [35S]GTPγS; 2 μg/mL each of protease inhibitors, namely, leupeptin, pepstatin, aprotinin, and bestatin; and 30 μM GDP.98 Following incubation, the assay tubes were filtered through glass fiber filters (obtained from Whatman Schleicher and Schuell, Keene, NH), and assay tubes were washed with 3 mL of ice-cold 50 mM Tris–HCl buffer at pH 7.4 thrice on a semiautomatic cell harvester. Following filtration, filters were placed in vials containg 3 mL of Liquiscint (obtained National Diagnostics, Atlanta, GA), and counts were measured using scintillation spectroscopy using a Tri-Carb 2900TR counter (obtained from PerkinElmer Life and Analytical Sciences). Basal activity was determined in the presence of GDP and in the absence of the tested drug. Data obtained was then normalized against appropriate controls, e.g., 1000 nM DAMGO, DPDPE, and U50,488h for mMOR, mDOR, and mKOR, respectively. Agonistic potency (EC50), antagonistic potency (IC50), and maximal stimulation (%Emax) values were calculated by nonlinear regression analysis in GraphPad Prism.
Bioluminescence Resonance Energy Transfer (BRET) Assay.70,99
To measure the Gαi1 protein mediated activation, human embryonic kidney (HEK293T) cells were cotransfected using a 1:1:1:1 DNA ratio of receptor:Gαi-Rluc:Gβ1:Gγ2-GFP2, while a 1:5 DNA ratio of receptor-Rluc:mVenus-arrestin2 was used to measure the β-arrestin2 recruitment. Transfection was performed in OptiMEM using Transit 2020 at a 2:1 ratio of Transit:micrograms (μg) of DNA. After at least 18 h, 0.05% trypsin-EDTA was added to gently detach the cells, and subsequently, the cells were plated in a plating media (DMEM supplemented with 1% dialyzed FBS) at a density of 30 000–50 000 cells per well in poly-D-lysine-coated white and clear-bottom 96-well assay plates. The next day, a white adhesive bottom seal was applied, and the culture medium was carefully decanted and replaced by 60 μL of a drug buffer (1× Hank’s balanced salt solution (HBSS) and 20 mM HEPES, pH 7.4). Following assay buffer aspiration, cells were treated with 60 μL of freshly prepared coelenterazine 400a (5 μM) final concentration for Gαi1 activation or coelenterazine h (5 μM) concentration for β-arrestin2 recruitment. After 5 min of incubation, 30 μL of indole and oxindole based alkaloids (mitraciliatine, corynoxine, corynantheidine, and isopaynantheine) was individually added and incubated for an additional 5 min. Subsequently, the plates were placed in an LB940 Mithras plate reader (Berthold Technologies) to measure BRET ratios by reading each well for 1 s. With respect to Gαi1 protein mediated activation, the BRET2 ratio was expressed as the ratio of the GFP2 emission to RLuc8 emission at 510 and 395 nm, respectively. In regard to β-arrestin2 recruitment, the BRET1 ratio was defined as the ratio of mVenus/RLuc with 485 and 530 nm. The ratio of mVenus/RLuc was calculated per well, and the net BRET ratio was calculated by subtracting the mVenus/RLuc per well from the mVenus/RLuc ratio in wells without the presence of mVenus-arrestin2. The Graphpad Prism 8 software was used to plot the net BRET ratio versus the corresponding drug concentration.
Mice.
Male C57BL/6J mice (24–34 g) were purchased from Jackson Laboratories (Bar Harbor, ME) at 7 weeks of age. MOR KO and KOR KO mice were bred in the McLaughlin laboratory at University of Florida. Progenitors of the colonies for MOR KO and KOR KO were obtained from Jackson Laboratories. All mice used throughout the manuscript were opioid naïve. All mice were maintained on a 12 h light/dark cycle with Purina rodent chow and water available ad libitum and housed in groups of five until testing. For all behavior experiments, 50% DMSO:saline was used as a vehicle to dissolve drugs.
Assessing Antinociception:
The 55 °C Warm-Water Tail-Withdrawal Assay. The 55 °C warm-water tail-withdrawal assay was conducted in C57BL/6J mice as a measure of acute thermal antinociception as described previously.51 Briefly, warm (55 °C) water in a 1.5 L heated water bath was used as the thermal nociceptive stimulus, with the latency of each mouse to withdraw its tail taken as the end point. After first determining baseline latencies (1.44 ± 0.02 s across tested subjects), mice were administered a single graded dose of the tested compound supraspinally, through the intracerebroventricular (i.c.v.) route. Following drug administration, the tail-withdrawal latency for each mouse was repeatedly determined every 10 min for 1 h or until latencies returned to the baseline values. A cutoff time of 15 s was utilized to prevent tissue damage; if the mouse failed to display a tail-withdrawal response within this maximum response, the tail was removed from the water, and the animal was assigned a maximal antinociceptive score of 100%. At each time point, antinociception was calculated by the formula: % antinociception = 100[(test latency – baseline latency)/(15 – baseline latency)]. Presentation of antinociception was utilized to account for innate variability of initial latency values between mice. ED50 antinociceptive values for compounds was calculated from peak effect observed.
Assessment of Respiratory Depression and Locomotor Effects.
Animal breathing rates (measured as breaths per minute) and spontaneous locomotive activity (measured as ambulations per minute) were measured using the automated, computer-controlled Comprehensive Lab Animal Monitoring System (CLAMS) (Columbus Instruments, Columbus, OH) as described previously.39 Mice were allowed to move freely through the closed, sealed individual cages (23.5 cm × 2.2 cm × 13 cm) during all testing, which began immediately after a 60 min habituation period. After habituation, mice were administered i.c.v. compound or vehicle and, 5 min later, confined to the CLAMS testing cages for 180 min. Pressure transducers built into each sealed CLAMS cage measured the frequency of respiration (at breaths/min) of each occupant mouse. Infrared beams located in the floor measured locomotion by counting sequential breaks of adjacent beams (as ambulations). Data were averaged over 20 min periods for the 3 h test and are expressed as % vehicle response ± SEM.
Data Analysis.
Data analysis was carried out using Graphpad Prism 8.0 software. Results for both radioligand binding assays and [35S]GTPγS are presented as nM ± SEM from n = 3 experiments performed in triplicate. Data from both Gi-1 activation and βarrestin2 assays using human opioid receptors were normalized to Emax of the corresponding agonist controls, DAMGO, U50,488H, and DPDPE. The three-parameter logistic equation using Graph Pad Prism was used to fit the dose response curves obtained. All data is presented as mean EC50 (pEC50 ± SEM) for assays run in triplicate.
Statistical analysis used either one-way or two-way ANOVA or unpaired t test wherever appropriate with p < 0.05 being considered to be significant.
Supplementary Material
ACKNOWLEDGMENTS
S.M. is supported by funds from NIH grants DA045884 and DA048379 and start-up funds from Center for Clinical Pharmacology, University of Health Sciences & Pharmacy and Washington University. Y.-X.P. is supported by NIH grants DA042888 and DA046714. This research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748 to MSKCC. The authors would like to thank Professor Bryan L. Roth and the National Institute of Mental Health’s Psychoactive Drug Screening Program for their assistance in determining binding affinities at nonopioid CNS targets. We are thankful to Dr. Mariko Kitajima (Graduate School of Pharmaceutical Sciences, Chiba University, Japan) for providing isopaynantheine, used as standard.
ABBREVIATIONS
- mMOR
mouse mu opioid receptor
- hMOR
human mu opioid receptor
- mKOR
mouse kappa opioid receptor
- hKOR
human kappa opioid receptor
- NET
norepinephrine transporter
- DAT
dopamine transporter
- SERT
serotonin transporter
- α2A
α adrenergic receptor subtype 2A
- α1D
α adrenergic receptor subtype 1D
- 7OH
7-hydroxy mitragynine
- MP
mitragynine pseudoindoxyl
- DAMGO
[D-Ala2,N-MePhe4,Gly-ol5]-enkephalin
- DCM
dichloromethane
- BBB
blood–brain barrier
- MPE
maximum possible effect
- IV
intravenous
- i.c.v.
intracerebroventricular
- CLAMS
Comprehensive Laboratory Animal Monitoring System
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.1c00149.
HPLC experimental method and HPLC traces of compounds 5-7 and functional binding data of the compounds at opioid receptors (PDF)
The authors declare the following competing financial interest(s): S.M. is a co-founder of Sparian Inc. S.M. is an inventor on patent applications related to mitragynine analogues, which may lead to royalties or other licensing revenues from future commercial products.
Contributor Information
Soumen Chakraborty, Center for Clinical Pharmacology, University of Health Sciences & Pharmacy at St. Louis and Washington University School of Medicine, St. Louis, Missouri 63110, United States; Department of Anesthesiology, Washington University School of Medicine, St. Louis, Missouri 63110, United States.
Rajendra Uprety, Department of Neurology, Memorial Sloan Kettering Cancer Center, New York, New York 10065, United States.
Amal E. Daibani, Center for Clinical Pharmacology, University of Health Sciences & Pharmacy at St. Louis and Washington University School of Medicine, St. Louis, Missouri 63110, United States; Department of Anesthesiology, Washington University School of Medicine, St. Louis, Missouri 63110, United States
Valerie L. Rouzic, Department of Neurology, Memorial Sloan Kettering Cancer Center, New York, New York 10065, United States
Amanda Hunkele, Department of Neurology, Memorial Sloan Kettering Cancer Center, New York, New York 10065, United States.
Kevin Appourchaux, Center for Clinical Pharmacology, University of Health Sciences & Pharmacy at St. Louis and Washington University School of Medicine, St. Louis, Missouri 63110, United States.
Shainnel O. Eans, Department of Pharmacodynamics, University of Florida, Gainesville, Florida 032610, United States
Nitin Nuthikattu, Center for Clinical Pharmacology, University of Health Sciences & Pharmacy at St. Louis and Washington University School of Medicine, St. Louis, Missouri 63110, United States.
Rahul Jilakara, Center for Clinical Pharmacology, University of Health Sciences & Pharmacy at St. Louis and Washington University School of Medicine, St. Louis, Missouri 63110, United States.
Lisa Thammavong, Center for Clinical Pharmacology, University of Health Sciences & Pharmacy at St. Louis and Washington University School of Medicine, St. Louis, Missouri 63110, United States.
Gavril W. Pasternak, Department of Neurology, Memorial Sloan Kettering Cancer Center, New York, New York 10065, United States.
Ying-Xian Pan, Department of Neurology, Memorial Sloan Kettering Cancer Center, New York, New York 10065, United States; Department of Anesthesiology, Rutgers New Jersey Medical School, Newark, New Jersey 07103, United States.
Jay P. McLaughlin, Department of Pharmacodynamics, University of Florida, Gainesville, Florida 032610, United States.
Tao Che, Center for Clinical Pharmacology, University of Health Sciences & Pharmacy at St. Louis and Washington University School of Medicine, St. Louis, Missouri 63110, United States; Department of Anesthesiology, Washington University School of Medicine, St. Louis, Missouri 63110, United States; Department of Pharmacology, University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, North Carolina 27599, United States.
Susruta Majumdar, Center for Clinical Pharmacology, University of Health Sciences & Pharmacy at St. Louis and Washington University School of Medicine, St. Louis, Missouri 63110, United States; Department of Anesthesiology, Washington University School of Medicine, St. Louis, Missouri 63110, United States.
REFERENCES
- (1).Pasternak GW, and Pan Y-X (2013) Mu Opioids and Their Receptors: Evolution of a Concept. Pharmacol. Rev 65 (4), 1257–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Corbett AD, Henderson G, Mcknight AT, and Paterson SJ (2006) 75 Years of Opioid Research: The Exciting but Vain Quest for the Holy Grail. Br. J. Pharmacol 147 (Suppl 1), S153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Compton WM, Jones CM, and Baldwin GT (2016) The Authors Reply. New England Journal of Medicine, 1296. [DOI] [PubMed] [Google Scholar]
- (4).Overdose Death Rates, National Institute on Drug Abuse (NIDA), https://www.drugabuse.gov/related-topics/trendsstatistics/overdose-death-rates (accessed 2019-09-23).
- (5).Baumann MH, Majumdar S, Le Rouzic V, Hunkele A, Uprety R, Huang XP, Xu J, Roth BL, Pan Y-X, and Pasternak GW (2018) Pharmacological Characterization of Novel Synthetic Opioids (NSO) Found in the Recreational Drug Marketplace. Neuropharmacology 134 (Pt A), 101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Huang XP, Che T, Mangano TJ, Le Rouzic V, Pan YX, Majumdar S, Cameron MD, Baumann MH, Pasternak GW, and Roth BL (2017) Fentanyl-Related Designer Drugs W-18 and W-15 Lack Appreciable Opioid Activity in Vitro and in vivo. JCI Insight 2, No. e97222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Faouzi A, Varga BR, and Majumdar S. (2020) Biased Opioid Ligands. In Molecules, p 4257, MDPI AG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Hübner H, Huang XP, Sassano MF, Giguère PM, Löber S, Duan D, Scherrer G, Kobilka BK, Shoichet BK, et al. (2016) Structure-Based Discovery of Opioid Analgesics with Reduced Side Effects. Nature 537 (7619), 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Brust TF, Morgenweck J, Kim SA, Rose JH, Locke JL, Schmid CL, Zhou L, Stahl EL, Cameron MD, Scarry SM, Aubé J, Jones SR, Martin TJ, and Bohn LM (2016) Biased Agonists of the Kappa Opioid Receptor Suppress Pain and Itch without Causing Sedation or Dysphoria. Sci. Signaling 9 (456), No. ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Gomes I, Ayoub MA, Fujita W, Jaeger WC, Pfleger KDG, and Devi LA (2016) G Protein-Coupled Receptor Heteromers. Annu. Rev. Pharmacol. Toxicol 56, 403–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Faouzi A, Uprety R, Gomes I, Massaly N, Keresztes AI, Le Rouzic V, Gupta A, Zhang T, Yoon HJ, Ansonoff M, Allaoa A, Pan YX, Pintar J, Morón JA, Streicher JM, Devi LA, and Majumdar S. (2020) Synthesis and Pharmacology of a Novel μ-ΔOpioid Receptor Heteromer-Selective Agonist Based on the Carfentanyl Template. J. Med. Chem 63 (22), 13618–13637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Burford NT, Traynor JR, and Alt A. (2015) Positive Allosteric Modulators of the μ-Opioid Receptor: A Novel Approach for Future Pain Medications. Br. J. Pharmacol 172 (2), 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Livingston KE, and Traynor JR (2018) Allostery at Opioid Receptors: Modulation with Small Molecule Ligands. Br. J. Pharmacol, 2846–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kandasamy R, Hillhouse TM, Livingston KE, Kochan KE, Meurice C, Eans SO, Li M-H, White AD, Roques BP, McLaughlin JP, Ingram SL, Burford NT, Alt A, and Traynor JR (2021) Positive Allosteric Modulation of the Mu-Opioid Receptor Produces Analgesia with Reduced Side Effects. Proc. Natl. Acad. Sci. U. S. A 118 (16), No. e2000017118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Majumdar S, Grinnell S, Le Rouzic V, Burgman M, Polikar L, Ansonoff M, Pintar J, Pan Y-X, and Pasternak GW (2011) Truncated G Protein-Coupled Mu Opioid Receptor MOR-1 Splice Variants Are Targets for Highly Potent Opioid Analgesics Lacking Side Effects. Proc. Natl. Acad. Sci. U. S. A 108 (49), 19778–19783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Marrone GF, Lu Z, Rossi G, Narayan A, Hunkele A, Marx S, Xu J, Pintar J, Majumdar S, Pan YX, and Pasternak GW (2016) Tetrapeptide Endomorphin Analogs Require Both Full Length and Truncated Splice Variants of the Mu Opioid Receptor Gene Oprm1 for Analgesia. ACS Chem. Neurosci 7 (12), 1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Marrone GF, Grinnell SG, Lu Z, Rossi GC, Le Rouzic V, Xu J, Majumdar S, Pan YX, and Pasternak GW (2016) Truncated Mu Opioid GPCR Variant Involvement in Opioid-Dependent and Opioid-Independent Pain Modulatory Systems within the CNS. Proc. Natl. Acad. Sci. U. S. A 113 (13), 3663–3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zaverí NT (2016) Nociceptin Opioid Receptor (NOP) as a Therapeutic Target: Progress in Translation from Preclinical Research to Clinical Utility. J. Med. Chem, 7011–7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ding H, Kiguchi N, Yasuda D, Daga PR, Polgar WE, Lu JJ, Czoty PW, Kishioka S, Zaveri NT, and Ko MC (2018) A Bifunctional Nociceptin and Mu Opioid Receptor Agonist Is Analgesic without Opioid Side Effects in Nonhuman Primates. Sci. Transl. Med 10 (456), 3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Atigari DV, Uprety R, Pasternak GW, Majumdar S, and Kivell BM (2019) MP1104, a Mixed Kappa-Delta Opioid Receptor Agonist Has Anti-Cocaine Properties with Reduced Side-Effects in Rats. Neuropharmacology 150, 217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Atigari DV, Paton KF, Uprety R, Váradi A, Alder AF, Scouller B, Miller JH, Majumdar S, and Kivell BM (2021) The Mixed Kappa and Delta Opioid Receptor Agonist, MP1104, Attenuates Chemotherapy-Induced Neuropathic Pain. Neuropharmacology 185, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ulker E, Toma W, White A, Uprety R, Majumdar S, and Damaj MI (2020) The Antinociceptive Effects of a Dual Kappa-Delta Opioid Receptor Agonist in the Mouse Formalin Test. Behav. Pharmacol 31, 174–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Gillis A, Gondin AB, Kliewer A, Sanchez J, Lim HD, Alamein C, Manandhar P, Santiago M, Fritzwanker S, Schmiedel F, Katte TA, Reekie T, Grimsey NL, Kassiou M, Kellam B, Krasel C, Halls ML, Canals M, et al. (2020) Low Intrinsic Efficacy for G Protein Activation Can Explain the Improved Side Effect Profiles of New Opioid Agonists. Sci. Signaling 13 (625), 31. [DOI] [PubMed] [Google Scholar]
- (24).Che T, and Roth BL (2021) Structural Insights Accelerate the Discovery of Opioid Alternatives. Annu. Rev. Biochem 90, 739. [DOI] [PubMed] [Google Scholar]
- (25).Bhowmik S, Galeta J, Havel VV, Nelson M, Faouzi A, Bechand B, Fiala T, Hunkele A, Kruegel AC, Ansonoff M, Pintar JE, Majumdar S, Javitch JA, and Sames D. (2021) Site Selective C-H Functionalization of Mitragyna Alkaloids Reveals a Molecular Switch for Tuning Opioid Receptor Signaling Efficacy. Nat. Commun 12, 1–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Paton KF, Atigari DV, Kaska S, Prisinzano T, and Kivell BM (2020) Strategies for Developing k Opioid Receptor Agonists for the Treatment of Pain with Fewer Side Effects. Journal of Pharmacology and Experimental Therapeutics, 332–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Parker KE, Sugiarto E, Taylor AMW, Pradhan AA, and Al-Hasani R. (2020) Pain, Motivation, Migraine, and the Microbiome: New Frontiers for Opioid Systems and Disease. Mol. Pharmacol, 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Van Rijn RM, Defriel JN, and Whistler JL Pharmacological Traits of Delta Opioid Receptors: Pitfalls or Opportunities? (2013) Psychopharmacology, pp 1–18, Springer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Massaly N, Copits BA, Wilson-Poe AR, Hipólito L, Markovic T, Yoon HJ, Liu S, Walicki MC, Bhatti DL, Sirohi S, Klaas A, Walker BM, Neve R, Cahill CM, Shoghi KI, Gereau RW, McCall JG, Morón JA, et al. (2019) Pain-Induced Negative Affect Is Mediated via Recruitment of The Nucleus Accumbens Kappa Opioid System. Neuron 102 (3), 564–573 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Mores KL, Cummins BR, Cassell RJ, and Van Rijn RM (2019) A Review of the Therapeutic Potential of Recently Developed G Protein-Biased Kappa Agonists. In Frontiers in Pharmacology, Frontiers Media S.A.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Stanczyk MA, Livingston KE, Chang L, Weinberg ZY, Puthenveedu MA, and Traynor JR (2019) The δ-Opioid Receptor Positive Allosteric Modulator BMS 986187 Is a G-ProteinBiased Allosteric Agonist. Br. J. Pharmacol 176 (11), 1649–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Uprety R, Che T, Zaidi SA, Grinnell SG, Varga BR, Faouzi A, Slocum ST, Allaoa A, Varadi A, Nelson M, Bernhard SM, Kulko E, LeRouzic V, Eans SO, Simons CA, Hunkele A, Subrath J, Majumdar S, et al. (2021) Controlling Opioid Receptor Functional Selectivity by Targeting Distinct Subpockets of the Orthosteric Site. eLife 10, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Li JWH, and Vederas JC (2009) Drug Discovery and Natural Products: End of an Era or an Endless Frontier? Science, 161–165. [DOI] [PubMed] [Google Scholar]
- (34).Chakraborty S, and Majumdar S. (2021) Natural Products for the Treatment of Pain: Chemistry and Pharmacology of Salvinorin A, Mitragynine, and Collybolide. Biochemistry 60, 1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kearney SE, Zahoránszky-Kohalmi G, Brimacombe KR, Henderson MJ, Lynch C, Zhao T, Wan KK, Itkin Z, Dillon C, Shen M, Cheff DM, Lee TD, Bougie D, Cheng K, Coussens NP, Dorjsuren D, Eastman RT, and Rohde JM (2018) Canvass: A Crowd-Sourced, Natural-Product Screening Library for Exploring Biological Space. ACS Cent. Sci 4 (12), 1727–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Newman DJ, and Cragg GM (2016) Natural Products as Sources of New Drugs from 1981 to 2014. Journal of Natural Products, 629–661. [DOI] [PubMed] [Google Scholar]
- (37).Ramanathan S, and McCurdy CR (2020) Kratom (Mitragyna Speciosa): Worldwide Issues. Curr. Opin. Psychiatry 33 (4), 312–318. [DOI] [PubMed] [Google Scholar]
- (38).Adkins JE, Boyer EW, and McCurdy CR (2011) Mitragyna Speciosa A Psychoactive Tree from Southeast Asia with Opioid Activity. Curr. Top. Med. Chem 11 (9), 1165–1175. [DOI] [PubMed] [Google Scholar]
- (39).Wilson LL, Harris HM, Eans SO, Brice-Tutt AC, Cirino TJ, Stacy HM, Simons CA, León F, Sharma A, Boyer EW, Avery BA, McLaughlin JP, and McCurdy CR (2020) Lyophilized Kratom Tea as a Therapeutic Option for Opioid Dependence. Drug Alcohol Depend. 216, 108–310. [DOI] [PubMed] [Google Scholar]
- (40).Prozialeck WC, Jivan JK, and Andurkar SV (2012) Pharmacology of Kratom: An Emerging Botanical Agent with Stimulant, Analgesic and Opioid-like Effects. J. Am. Osteopath. Assoc 112 (12), 792–799. [PubMed] [Google Scholar]
- (41).Takayama H. (2004) Chemistry and Pharmacology of Analgesic Indole Alkaloids from the Rubiaceous Plant, Mitragyna Speciosa. Chem. Pharm. Bull 52 (8), 916–928. [DOI] [PubMed] [Google Scholar]
- (42).León F, Habib E, Adkins JE, Furr EB, McCurdy CR, and Cutler SJ (2009) Phytochemical Characterization of the Leaves of Mitragyna Speciosa Grown in USA. Nat. Prod. Commun 4 (7), 907–910. [PMC free article] [PubMed] [Google Scholar]
- (43).Gutridge AM, Robins MT, Cassell RJ, Uprety R, Mores KL, Ko MJ, Pasternak GW, Majumdar S, and Rijn RM (2020) G Protein-Biased Kratom-Alkaloids and Synthetic Carfentanil-Amide Opioids as Potential Treatments for Alcohol Use Disorder. Br. J. Pharmacol 177, 1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Flores-Bocanegra L, Raja HA, Graf TN, Augustinović M, Wallace ED, Hematian S, Kellogg JJ, Todd DA, Cech NB, and Oberlies NH (2020) The Chemistry of Kratom [ Mitragyna Speciosa]: Updated Characterization Data and Methods to Elucidate Indole and Oxindole Alkaloids. J. Nat. Prod 83 (7), 2165–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Ellis CR, Racz R, Kruhlak NL, Kim MT, Zakharov AV, Southall N, Hawkins EG, Burkhart K, Strauss DG, and Stavitskaya L. (2020) Evaluating Kratom Alkaloids Using PHASE. PLoS One 15 (3), e0229646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).León F, Habib E, Adkins JE, Furr EB, Mccurdy CR, and Cutler SJ (2009) Phytochemical Characterization of the Leaves of Mitragyna Speciosa Grown in USA. Nat. Prod. Commun 4, 1934578X0900400. [PMC free article] [PubMed] [Google Scholar]
- (47).Raffa RB (2014) Kratom and Other Mitragynines: The Chemistry and Pharmacology of Opioids from Non-Opium Source, CRC Press, New York. [Google Scholar]
- (48).Chear NJ-Y, León F, Sharma A, Kanumuri SRR, Zwolinski G, Abboud KA, Singh D, Restrepo LF, Patel A, Hiranita T, Ramanathan S, Hampson AJ, McMahon LR, and McCurdy CR (2021) Exploring the Chemistry of Alkaloids from Malaysian Mitragyna Speciosa (Kratom) and the Role of Oxindoles on Human Opioid Receptors. J. Nat. Prod 84, 1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Sharma A, Kamble SH, León F, Chear NJ-Y, King TI, Berthold EC, Ramanathan S, McCurdy CR, and Avery BA (2019) Simultaneous Quantification of Ten Key Kratom Alkaloids in Mitragyna Speciosa Leaf Extracts and Commercial Products by Ultraperformance Liquid Chromatography-tandem Mass Spectrometry. Drug Test. Anal 11 (8), 1162–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Wilson LL, Chakraborty S, Eans SO, Cirino TJ, Stacy HM, Simons CA, Uprety R, Majumdar S, and McLaughlin JP (2021) Kratom Alkaloids, Natural and Semi-Synthetic, Show Less Physical Dependence and Ameliorate Opioid Withdrawal. Cell. Mol. Neurobiol 41, 1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Váradi A, Marrone GF, Palmer TC, Narayan A, Szabó MR, Le Rouzic V, Grinnell SG, Subrath JJ, Warner E, Kalra S, Hunkele A, Pagirsky J, Eans SO, Medina JM, Xu J, Pan YX, Borics A, Majumdar S, et al. (2016) Mitragynine/Corynantheidine Pseudoindoxyls As Opioid Analgesics with Mu Agonism and Delta Antagonism, Which Do Not Recruit β-Arrestin-2. J. Med. Chem 59 (18), 8381–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Takayama H. (2004) Chemistry and Pharmacology of Analgesic Indole Alkaloids from the Rubiaceous Plant, Mitragyna Speciosa. Chem. Pharm. Bull 52 (8), 916–28. [DOI] [PubMed] [Google Scholar]
- (53).Kruegel AC, Uprety R, Grinnell SG, Langreck C, Pekarskaya EA, Le Rouzic V, Ansonoff M, Gassaway MM, Pintar JE, Pasternak GW, Javitch JA, Majumdar S, and Sames D. (2019) 7-Hydroxymitragynine Is an Active Metabolite of Mitragynine and a Key Mediator of Its Analgesic Effects. ACS Cent. Sci 5 (6), 992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Zhou Y, Ramsey S, Provasi D, El Daibani A, Appourchaux K, Chakraborty S, Kapoor A, Che T, Majumdar S, and Filizola M. (2021) Predicted Mode of Binding to and Allosteric Modulation of the μ-Opioid Receptor by Kratom’s Alkaloids with Reported Antinociception in vivo. Biochemistry 60, 1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Zarembo JE, Douglas B, Valenta J, and Weisbach JA (1974) Metabolites of Mitragynine. J. Pharm. Sci 63 (9), 1407–1415. [DOI] [PubMed] [Google Scholar]
- (56).Obeng S, Kamble SH, Reeves ME, Restrepo LF, Patel A, Behnke M, Chear NJY, Ramanathan S, Sharma A, León F, Hiranita T, Avery BA, McMahon LR, and McCurdy CR (2020) Investigation of the Adrenergic and Opioid Binding Affinities, Metabolic Stability, Plasma Protein Binding Properties, and Functional Effects of Selected Indole-Based Kratom Alkaloids. J. Med. Chem 63 (1), 433–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Takayama H, Ishikawa H, Kurihara M, Kitajima M, Aimi N, Ponglux D, Koyama F, Matsumoto K, Moriyama T, Yamamoto LT, Watanabe K, Murayama T, and Horie S. (2002) Studies on the Synthesis and Opioid Agonistic Activities of Mitragynine-Related Indole Alkaloids: Discovery of Opioid Agonists Structurally Different from Other Opioid Ligands. J. Med. Chem 45 (9), 1949–1956. [DOI] [PubMed] [Google Scholar]
- (58).Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguiz RM, Huang XP, Norval S, Sassano MF, Shin AI, Webster LA, Simeons FRC, Stojanovski L, Prat A, Seidah NG, Constam DB, Bickerton GR, Read KD, Hopkins AL, et al. (2012) Automated Design of Ligands to Polypharmacological Profiles. Nature 492 (7428), 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Trager WF, Lee CM, and Beckett AH (1967) Corynantheidine-Type Alkaloids-I. Establishment of Physical Criteria for the Normal, Pseudo, Allo and Epiallo Configurations by Conformational Analysis. Tetrahedron 23 (1), 365–374. [DOI] [PubMed] [Google Scholar]
- (60).Bartlett MF, Sklar R, Taylor WI, Schlittler E, Amai RLS, Beak P, Bringi NV, and Wenkert E. (1962) Rauwolfia Alkaloids. XXXVIII.1 Stereospecific Degradations Leading to the Absolute Configurations and Structures of Ajmaline, Sarpagine and Corynantheidine. J. Am. Chem. Soc 84 (4), 622–630. [Google Scholar]
- (61).King TI, Sharma A, Kamble SH, León F, Berthold EC, Popa R, Cerlati O, Prentice BM, McMahon LR, McCurdy CR, and Avery BA (2020) Bioanalytical Method Development and Validation of Corynantheidine, a Kratom Alkaloid, Using UPLC-MS/MS, and Its Application to Preclinical Pharmacokinetic Studies. J. Pharm. Biomed. Anal, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Shellard EJ, Houghton PJ, and Resha M. (1978) The Mitragyna Species of Asia. Part XXXII. The Distribution of Alkaloids in Young Plants of Mitragyna Speciosa Korth Grown from Seed Obtained from Thailand. Planta Med. 34 (3), 253–263. [Google Scholar]
- (63).Beckett AH, Shellard EJ, and Tackie AN (2011) THE MITRAGYNA SPECIES OF GHANA. J. Pharm. Pharmacol 15 (S1), 166T–169T. [DOI] [PubMed] [Google Scholar]
- (64).Shellard EJ, and Sarpong K. (2011) The Alkaloids of the Leaves of Mitragyna Inermis (Willd.) O. Kuntze. J. Pharm. Pharmacol 21 (S1), 113S–117S. [DOI] [PubMed] [Google Scholar]
- (65).Ali Z, Demiray H, and Khan IA (2014) Isolation, Characterization, and NMR Spectroscopic Data of Indole and Oxindole Alkaloids from Mitragyna Speciosa. Tetrahedron Lett. 55 (2), 369–372. [Google Scholar]
- (66).Philipp AA, Wissenbach DK, Weber AA, Zapp J, and Maurer HH (2011) Metabolism Studies of the Kratom Alkaloids Mitraciliatine and Isopaynantheine, Diastereomers of the Main Alkaloids Mitragynine and Paynantheine, in Rat and Human Urine Using Liquid Chromatography-Linear Ion Trap-Mass Spectrometry. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci 879 (15–16), 1049–1055. [DOI] [PubMed] [Google Scholar]
- (67).Shellard EJ, Houghton PJ, and Resha M. (1978) The Mitragyna Species of Asia. Part XXXI. The Alkaloids of Mitragyna Speciosa Korth from Thailand. Planta Med. 34 (1), 26–36. [Google Scholar]
- (68).Bindra JS (1973) Oxindole Alkaloids. Alkaloids Chem. Physiol 14 (C), 83–121. [Google Scholar]
- (69).Wanner MJ, Ingemann S, van Maarseveen JH, and Hiemstra H. (2013) Total Synthesis of the Spirocyclic Oxindole Alkaloids Corynoxine, Corynoxine B, Corynoxeine, and Rhynchophylline. Eur. J. Org. Chem 2013 (6), 1100–1106. [Google Scholar]
- (70).Che T, Majumdar S, Zaidi SA, Ondachi P, McCorvy JD, Wang S, Mosier PD, Uprety R, Vardy E, Krumm BE, Han GW, Lee M-Y, Pardon E, Steyaert J, Huang X-P, Strachan RT, Tribo AR, Roth BL, et al. (2018) Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor. Cell 172 (1–2), 55–67 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Todd DA, Kellogg JJ, Wallace ED, Khin M, Flores-Bocanegra L, Tanna RS, McIntosh S, Raja HA, Graf TN, Hemby SE, Paine MF, Oberlies NH, and Cech NB (2020) Chemical Composition and Biological Effects of Kratom (Mitragyna Speciosa): In Vitro Studies with Implications for Efficacy and Drug Interactions. Sci. Rep 10 (1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Zhang Y, Liu C, Qi Y, Li S, Pan Y, and Li Y. (2015) Circulating Ultrasound-Assisted Extraction, Countercurrent Chromatography, and Liquid Chromatography for the Simultaneous Extraction, Isolation, and Analysis of the Constituents of Uncaria Tomentosa. J. Chromatogr. A 1388, 36–42. [DOI] [PubMed] [Google Scholar]
- (73).Ndagijimana A, Wang X, Pan G, Zhang F, Feng H, and Olaleye O. (2013) A Review on Indole Alkaloids Isolated from Uncaria Rhynchophylla and Their Pharmacological Studies. Fitoterapia, 35–47. [DOI] [PubMed] [Google Scholar]
- (74).Wang K, Zhou X-Y, Wang Y-Y, Li M-M, Li Y-S, Peng L-Y, Cheng X, Li Y, Wang Y-P, and Zhao Q-S (2011) Macrophyllionium and Macrophyllines A and B, Oxindole Alkaloids from Uncaria Macrophylla. J. Nat. Prod 74 (1), 12–15. [DOI] [PubMed] [Google Scholar]
- (75).Macko E, Weisbach JA, and Douglas B. (1972) Some Observations on the Pharmacology of Mitragynine. Arch. Int. Pharmacodyn. Ther 198 (1), 145–161. [PubMed] [Google Scholar]
- (76).Matsumoto K, Horie S, Ishikawa H, Takayama H, Aimi N, Ponglux D, and Watanabe K. (2004) Antinociceptive Effect of 7-Hydroxymitragynine in Mice: Discovery of an Orally Active Opioid Analgesic from the Thai Medicinal Herb Mitragyna Speciosa. Life Sci. 74 (17), 2143–2155. [DOI] [PubMed] [Google Scholar]
- (77).Matsumoto K, Hatori Y, Murayama T, Tashima K, Wongseripipatana S, Misawa K, Kitajima M, Takayama H, and Horie S. (2006) Involvement of μ-Opioid Receptors in Antinociception and Inhibition of Gastrointestinal Transit Induced by 7Hydroxymitragynine, Isolated from Thai Herbal Medicine Mitragyna Speciosa. Eur. J. Pharmacol 549 (1–3), 63–70. [DOI] [PubMed] [Google Scholar]
- (78).Matsumoto K, Mizowaki M, Suchitra T, Murakami Y, Takayama H, Sakai SI, Aimi N, and Watanabe H. (1996) Central Antinociceptive Effects of Mitragynine in Mice: Contribution of Descending Noradrenergic and Serotonergic Systems. Eur. J. Pharmacol 317 (1), 75–81. [DOI] [PubMed] [Google Scholar]
- (79).Yamamoto LT, Horie S, Takayama H, Aimi N, Sakai SI, Yano S, Shan J, Pang PKT, Ponglux D, and Watanabe K. (1999) Opioid Receptor Agonistic Characteristics of Mitragynine Pseudoindoxyl in Comparison with Mitragynine Derived from Thai Medicinal Plant Mitragyna Speciosa. Gen. Pharmacol 33 (1), 73–81. [DOI] [PubMed] [Google Scholar]
- (80).Zhou Y, Ramsey S, Provasi D, El Daibani A, Appourchaux K, Chakraborty S, Kapoor A, Che T, Majumdar S, and Filizola M. (2020) Predicted Mode of Binding to and Allosteric Modulation of the μ-Opioid Receptor by Kratom’s Alkaloids with Reported Antinociception In vivo. Biochemistry, 1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Obeng S, Wilkerson JL, León F, Reeves ME, Restrepo LF, Gamez-Jimenez LR, Patel A, Pennington AE, Taylor VA, Ho NP, Braun T, Fortner JD, Crowley ML, Williamson MR, Pallares VL, Mottinelli M, Lopera-Londoño C, and Hiranita T. (2021) Pharmacological Comparison of Mitragynine and 7-Hydroxymitragynine: In Vitro Affinity and Efficacy for Mu-Opioid Receptor and Opioid-Like Behavioral Effects in Rats. J. Pharmacol. Exp. Ther 376 (3), 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Yue K, Kopajtic TA, and Katz JL (2018) Abuse Liability of Mitragynine Assessed with a Self-Administration Procedure in Rats. Psychopharmacology (Berl). 235 (10), 2823–2829. [DOI] [PubMed] [Google Scholar]
- (83).Hiranita T, Leon F, Felix JS, Restrepo LF, Reeves ME, Pennington AE, Obeng S, Avery BA, McCurdy CR, McMahon LR, and Wilkerson JL (2019) The Effects of Mitragynine and Morphine on Schedule-Controlled Responding and Antinociception in Rats. Psychopharmacology (Berl). 236 (9), 2725–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Matsumoto K, Mizowaki M, Suchitra T, Takayama H, Sakai SI, Aimi N, and Watanabe H. (1996) Antinociceptive Action of Mitragynine in Mice: Evidence for the Involvement of Supraspinal Opioid Receptors. Life Sci. 59 (14), 1149–1155. [DOI] [PubMed] [Google Scholar]
- (85).Ponglux D, Wongseripipatana S, Takayama H, Kikuchi M, Kurihara M, Kitajima M, Aimi N, and Sakai S. (1994) A New Indole Alkaloid, 7 α-Hydroxy-7 H -Mitragynine, from Mitragyna Speciosa in Thailand. Planta Med. 60 (06), 580–581. [DOI] [PubMed] [Google Scholar]
- (86).Kamble S, León F, King TI, Berthold EC, Lopera-Londoño C, Siva Rama Raju K, Hampson A, Sharma A, Avery B, McMahon L, and McCurdy CR (2020) Metabolism of a Kratom Alkaloid Metabolite in Human Plasma Increases Its Opioid Potency and Efficacy. ACS Pharmacol. Transl. Sci 3, 1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Kruegel AC, Gassaway MM, Kapoor A, Váradi A, Majumdar S, Filizola M, Javitch JA, and Sames D. (2016) Synthetic and Receptor Signaling Explorations of the Mitragyna Alkaloids: Mitragynine as an Atypical Molecular Framework for Opioid Receptor Modulators. J. Am. Chem. Soc 138 (21), 6754–6764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Gillis A, Sreenivasan V, and Christie MJ (2020) Intrinsic Efficacy of Opioid Ligands and Its Importance for Apparent Bias, Operational Analysis, and Therapeutic Window. Mol. Pharmacol, 410–424. [DOI] [PubMed] [Google Scholar]
- (89).Cuitavi J, Hipólito L, and Canals M. (2020) The Life Cycle of the Mu-Opioid Receptor. Trends Biochem. Sci, 1. [DOI] [PubMed] [Google Scholar]
- (90).Váradi A, Marrone GF, Eans SO, Ganno ML, Subrath JJ, Le Rouzic V, Hunkele A, Pasternak GW, McLaughlin JP, and Majumdar S. (2015) Synthesis and Characterization of a Dual Kappa-Delta Opioid Receptor Agonist Analgesic Blocking Cocaine Reward Behavior. ACS Chem. Neurosci 6 (11), 1813–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Dosaka-Akita K, Tortella FC, Holaday JW, and Long JB (1993) The Kappa Opioid Agonist U-50,488H Antagonizes Respiratory Effects of Mu Opioid Receptor Agonists in Conscious Rats. J. Pharmacol. Exp. Ther 264 (2), 631–637. [DOI] [PubMed] [Google Scholar]
- (92).Hilaire G, Voituron N, Menuet C, Ichiyama RM, Subramanian HH, and Dutschmann M. (2010) The Role of Serotonin in Respiratory Function and Dysfunction. In Respiratory Physiology and Neurobiology, pp 76–88, Elsevier. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Oliveira LM, Moreira TS, Kuo FS, Mulkey DK, and Takakura AC (2016) A1- and A2-Adrenergic Receptors in the Retrotrapezoid Nucleus Differentially Regulate Breathing in Anesthetized Adult Rats. J. Neurophysiol 116 (3), 1036–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Pickett JE, Váradi A, Palmer TC, Grinnell SG, Schrock JM, Pasternak GW, Karimov RR, and Majumdar S. (2015) Mild, Pd-Catalyzed Stannylation of Radioiodination Targets. Bioorg. Med. Chem. Lett 25 (8), 1761–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Váradi A, Hosztafi S, Le Rouzic V, Tóth G, Urai Á, Noszál B, Pasternak GW, Grinnell SG, and Majumdar S. (2013) Novel 6β-Acylaminomorphinans with Analgesic Activity. Eur. J. Med. Chem 69, 786–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Lowry OH, Rosebrough NJ, Farr AL, and Randall RJ (1951) Protein Measurement with the Folin Phenol Reagent. J. Biol. Chem 193 (1), 265–275. [PubMed] [Google Scholar]
- (97).Yung-Chi C, and Prusoff WH (1973) Relationship between the Inhibition Constant (KI) and the Concentration of Inhibitor Which Causes 50 per Cent Inhibition (I50) of an Enzymatic Reaction. Biochem. Pharmacol 22 (23), 3099–3108. [DOI] [PubMed] [Google Scholar]
- (98).Chou T-C (1974) Relationships between Inhibition Constants and Fractional Inhibition in Enzyme-Catalyzed Reactions with Different Numbers of Reactants, Different Reaction Mechanisms, and Different Types and Mechanisms of Inhibition. Mol. Pharmacol 10 (2), 235. [PubMed] [Google Scholar]
- (99).Olsen RHJ, DiBerto JF, English JG, Glaudin AM, Krumm BE, Slocum ST, Che T, Gavin AC, McCorvy JD, Roth BL, and Strachan RT (2020) TRUPATH, an Open-Source Biosensor Platform for Interrogating the GPCR Transducerome. Nat. Chem. Biol 16 (8), 841–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.