

Abstract
Cancer is a genetic disease originating from the accumulation of gene mutations in a cellular subpopulation. Although many therapeutic approaches have been developed to treat cancer, recent studies have revealed an irrefutable challenge that tumors evolve defenses against some therapies. Gene therapy may prove to be the ultimate panacea for cancer by correcting the fundamental genetic errors in tumors. The engineering of nanoscale inorganic carriers of cancer therapeutics has shown promising results in the efficacious and safe delivery of nucleic acids to treat oncological diseases in small-animal models. When these nanocarriers are used for co-delivery of gene therapeutics along with auxiliary treatments, the synergistic combination of therapies often leads to an amplified health benefit. In this review, an overview of the inorganic nanomaterials developed for combinatorial therapies of gene and other treatment modalities is presented. First, the main principles of using nucleic acids as therapeutics, inorganic nanocarriers for medical applications and delivery of gene/drug payloads are introduced. Next, the utility of recently developed inorganic nanomaterials in different combinations of gene therapy with each of chemo, immune, hyperthermal, and radio therapy is examined. Finally, current challenges in the clinical translation of inorganic nanomaterial-mediated therapies are presented and outlooks for the field are provided.
Keywords: combinatorial therapy, gene delivery, inorganic nanomaterials, nanomedicine, tumors
1. Introduction
Cancer arises from the accumulation of unrepaired genetic mutations. These mutations can confer survival and proliferative advantages to cancer cells so they may outperform normal cells in resource competitions and growth. A systematic pan-cancer analysis discovered over 600 000 somatic mutations among which 127 were significantly mutated genes driving multiple tumorigenic pathways across 12 major cancer types.[1] The unchecked proliferation of cancer cells can eventually develop into malignant tumors capable of invading other organs. Thus, cancer is fundamentally a genetic disease of which development involves a series of microevolutionary events.[2] Conventional tumor treatments usually involve surgery assisted by radiotherapy and phototherapy for local tumor mass removal in combination with chemotherapeutic drugs for systemic tumor suppression.[3] However, tumor eradication is extremely challenging because surgery can only remove the bulk of a resectable tumor mass. The chances for microscopic cancer cell clusters to be missed during bulk tumor resection are high, and leftover cancer cells often develop resistance to adjuvant therapies such as chemo, radio, and photo therapies through further genetic mutation. As a result, tumor recurrence rates after conventional treatments are alarmingly high (e.g., nearly 100% for glioblastoma, 85% for ovarian cancers, and 30% for breast cancers).[4–6] New treatment modalities with higher tumor specificity and stronger potency are needed to achieve swift and effective tumor killing.
With considerable advances in cancer immunology and genomics, novel immune and genetic strategies are rapidly altering the landscape of anticancer therapy. Remarkable results from recent progress in immunotherapy development, including the identification of key immune checkpoint inhibitors and the invention of adoptive cell transfer technology, shed light on the next generation of antitumor treatments.[7] Yet, the risks of adverse systemic immune responses, unpredictable therapeutic efficacy, and high treatment costs impede the clinical translation of these immunotherapies.[8]
Manipulating the genetic behavior of cancer cells can directly address the root cause of cancer: genetic mutations. Nucleic acid-based therapeutics can silence oncogenes, restore mutated antioncogenes, and increase the recognizability of cancer cells by the body’s innate immune system. Unlike small-molecule or protein-based drugs, which require repeated infusion for long-term therapeutic effects, gene therapy offers sustained expression or silencing of endogenous proteins upon stable transfection.[9] Since each tumor has its own distinct history in accumulating defective pathways, the antitumor therapeutic effect of a single treatment modality is often limited by the heterogeneity of cancer pathogenesis.[10] To enhance antitumor effects, gene therapy can be combined with other treatment modalities to attack multiple pathways of oncogenesis at once and achieve synergistic tumor eradication effects. However, combinatorial delivery of nucleic acid and other antitumor therapies remains challenging as sophisticated and multifunctional carriers are required for this purpose.
Currently, the primary obstacles inhibiting widespread adoption of gene delivery for cancer treatment are safety concerns associated with viral vectors and low transfection efficiency of nonviral vectors. The incipient human clinical trials evaluating the efficacy of gene therapy, carried out in 1990, delivered nucleic acids to human cells using viral vectors. However, the shortcomings of these viral vectors such as limited loading capacity, risks of mutagenesis, and immunogenicity significantly limit their applicability.[11] With regards to combinatorial therapy, viral vectors are unsuitable since they can only carry nucleic acid payloads, meaning that other drugs/treatments would have to be delivered separately; an undesirable inconvenience in therapeutic administration. Consequently, the emergence of myriad nanomaterials as nonviral therapeutic carriers has significantly improved the efficiency of nonviral nucleic acid and small-molecule drug delivery.
Organic nanomaterials such as polymeric micelles and liposomes have been extensively investigated for gene/drug delivery and exhibit promising results due to their favorable biocompatibility, loading capacity, and versatility in their molecular designs. Nevertheless, organic nanomaterials often suffer from unpredictable size variations when placed in physiological environments or when loaded with therapeutic cargos. It has been reported that a 15 nm lysine dendrimer can bypass the renal glomerular filtration barrier with a size cutoff of ≈6 nm; this means that nanomaterials that are too small will be excreted from the body via the urine stream prior to performing any useful function.[12] On the other hand, nanomaterials that are too large will be recognized by the body’s systemic immune system and be marked for sequestration and removal.
Organic nanomaterials’ sizes are mostly governed by weak secondary intermolecular interactions such as hydrogen bonding, electrostatic attraction, and hydrophobic effects. Monomeric units of organic nanomaterials would have to be carefully engineered and mixed under meticulous conditions to ensure that a delicate equilibrium between multiple intermolecular interactions can be reached to form assemblies at desired sizes.[13] However, this size equilibrium is likely to be compromised when loading highly charged molecules (e.g., nucleic acids) or hydrophobic chemotherapeutics (e.g., paclitaxel), and drug loading may lead to unpredictable size outcomes. Furthermore, without rigid support, certain soft organic nanomaterials can be structurally deformed under pressure or shear stress so that they squeeze through biological pores unexpectedly and undesirably.[14]
Alternatively, inorganic nanomaterials are well-positioned to serve as nanocarriers for combinatorial therapeutics delivery. Such nanocarriers typically consist of an inorganic core and a polymeric surface coating. The physicochemical properties of inorganic cores, including size and shape, may be precisely controlled during synthesis; a scenario that lies in direct contrast to the situation concerning organic nanomaterials. Polymeric coatings can be conjugated onto the rigid inorganic cores to confer desired biocompatibility and nucleic-acid/drug-loading capacity onto the inorganic nanocarriers. With inorganic cores serving as a rigid support for polymeric coatings, combinations of nucleic acids and other types of antitumor drugs can be loaded and delivered without significantly altering the size or shapes of the nanocarriers. Furthermore, inorganic nanomaterials also enable additional treatment modalities such as hyperthermal ablation and radiotherapy enhancement due to their unique magnetic and photoelectric properties. Therefore, inorganic nanocarriers can enable combinatorial antitumor therapies by not only the simultaneous delivery of different types of therapeutics but also because of their intrinsic properties that promote the harnessing of external energy for therapeutic benefit. Figure 1 shows the structure of a representative inorganic nanomaterial carrying multiple therapeutic payloads.
Figure 1.
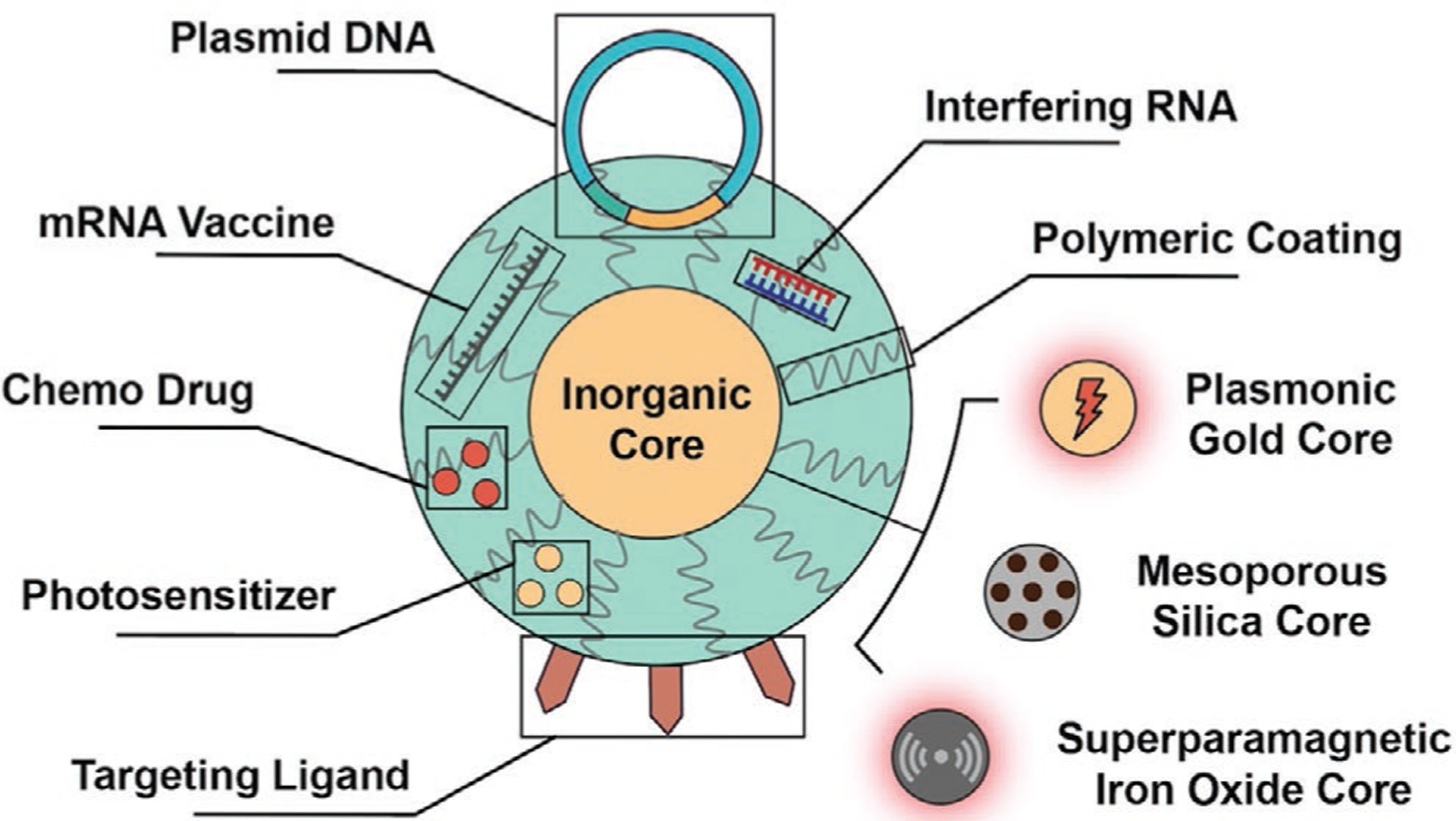
Schematic illustration of a representative inorganic nanocarrier loaded with combinatorial therapeutics.
In this article, we provide a comprehensive overview of the applications of inorganic nanomaterials in gene therapy when combined with other antitumor treatment modalities. We begin with a brief introduction of the characteristics of different types of nucleic acids and their potential for use in gene therapy. Then, we probe into the details of the criteria requisite of inorganic nanomaterials for in vivo nucleic acid delivery. Principal synthesis methods of inorganic cores and their properties are covered first. Major classes of polymeric coatings on inorganic nanomaterials serving the purposes of biocompatibility improvement, nucleic acid condensing, and controlled release are then reviewed. Insights into polymeric coating designs for nucleic acid delivery are also provided. Delivery of nucleic acids mediated by inorganic nanocarriers is then examined with an emphasis on strategies for overcoming biological barriers. Next, we review recent advances in inorganic nanomaterial-mediated gene therapy when combined with each of chemo, immune, hyperthermal, and radio therapy. The combination of gene therapy with two other therapies (trimodal therapies) are also discussed. We highlight the synergy between gene therapy and each of the treatment modalities on improving in vivo antitumor effects compared to single therapies. Outlooks on the applications of inorganic nanomaterials in combinatorial therapy are provided at the end of the review.
2. Therapeutic Nucleic Acids for Gene Therapy
Gene therapy is a treatment modality wherein nucleic acids are used to artificially modify the genetic expression of cells. With respect to cancer, considerable advances in understanding of the cellular genome has facilitated identification of the genes responsible for tumorigenesis and cancer’s resistance to certain therapies. With this genetic information, exogeneous nucleic acid sequences can be designed and introduced into cells to modulate specific pathways to achieve antitumor effects. Therapeutic nucleic acids come in different forms, including deoxyribonucleic acid (DNA), ribonucleic acid (RNA), and messenger RNA (mRNA), which are used for augmented expression of tumoricidal genes; small interfering RNA (siRNA), short hairpin RNA (shRNA), and microRNA (miRNA) are used to silence oncogene expression.[15] Additionally, certain immunostimulatory nucleic acids can induce the body’s immune system to more vigorously attack the cancer residing in the body.
Since the in vivo application of naked (i.e., unprotected) nucleic acids suffers from enzymatic degradation, low tissue uptake and short blood half-life,[16,17] shielding of nucleic acids upon their introduction into the body is essential to treatment success. Therefore, to avoid degradation from free floating enzymes that destroy exogenous nucleic acids, delivery vehicles must be engineered to safely ferry nucleic acid payloads to their desired destination. Once therapeutic nucleic acids reach cancer cells, therapeutic efficacy is usually achieved by either augmented expression of suicidal or proapoptotic genes or silencing of tumorigenic oncogenes. Therapeutic nucleic acids can alternatively be used to alter immune cells’ genetic behaviors by either immune checkpoint blockade or heightened tumor recognition to induce tumoricidal responses. The features and mechanisms of therapeutic nucleic acids are discussed in this section.
2.1. Augmented Gene Expression
Therapeutic DNAs and mRNAs are designed to encode for proteins of which overexpression can have an antitumor effect. DNAs are usually delivered as circular plasmids due to their molecular manipulability and ability for independent transcription from the host genome. Plasmids are typically around 5k base pairs and >100 nm in length when uncondensed.[18] The nucleotide (nt) sequence composing a plasmid can be readily altered through recombinant DNA technology. Reporter genes, therapeutic genes, viral-derived promoters, and origins of replication can be cut from their original sequences and incorporated into one plasmid to enable trackable, efficient tumor-specific expression of therapeutic proteins. Examples of genes commonly incorporated in plasmids for cancer treatment include the p53 antioncogene, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) gene, and herpes simplex virus-1 thymidine kinase (HSVTK) suicidal gene.[19,20] Notwithstanding their multifunctionality and effectiveness in gene therapy, application of plasmids is not without challenge. Due to their large size and negatively charged backbones, plasmids need to be condensed into supercoil formations to improve their durability during transportation (please see Section 3.7 for a more detailed discussion).[21] Another significant hurdle in plasmid delivery is that, unlike some other therapeutic nucleic acids, plasmids must overcome the nuclear membrane to gain access to the transcriptional machineries residing inside cell nuclei.
On the other hand, single-stranded mRNAs do not require nuclear entry for translation and hence only need to gain entry to the cytosol. Besides the simplified delivery process, utilizing mRNA for augmented gene expression offers several advantages over plasmid DNAs.[22] First, mRNA risks no adverse genomic integration or insertional DNA mutagenesis, as is the case with plasmid DNAs. Second, mRNA translation is dose dependent and rapid in comparison to the time required for DNA transcription and translation. Third, mRNA is less immunogenic than DNA so that it is more suitable for sensitive cells. However, mRNA is intrinsically less stable than DNA and more costly to produce at large scales.[23] Delivery of tumor antigen encoding mRNAs such as tyrosinase-related protein 2 (Trp2) mRNA and mucin 1 (MUC1) mRNA to immune cells can enhance antigen presentation and induce cytotoxic immune responses,[24,25] proving the antitumor efficacy of mRNA as a therapeutic nucleic acid.
2.2. RNA Interference (RNAi) Gene Silencing
RNAi is a natural process for mRNA degradation prior to its translation into proteins that results in gene silencing. In the cytoplasm, the RNAi pathway starts with the cleavage of long double-stranded RNA (dsRNA) containing more than 30 nt and pre-miRNA (70–100 nt) into smaller dsRNA fragments, siRNA (21–23 nt), and miRNA (19–25 nt) by a ribonuclease III-like enzyme named Dicer. Dicer-processed siRNA and miRNA then activate the RNA-induced silencing complex (RISC) and recruit the endonuclease argonaute 2 (AGO2) component.[26] The passenger strand of the siRNA is then cleaved by AGO2, and that of miRNA is simply unwound and discarded while the guide strand remains bound to the RISC for subsequent mRNA processing. It was later found that the presence of long dsRNA in cytoplasm could induce interferon (IFN)-related immune responses leading to apoptosis.[27] Therefore, exogenous siRNA and miRNA are applied to bypass the cytoplasmic presence of long dsRNA.
The siRNA guide strand binds to target mRNA with perfect sequence alignment; therefore, siRNA can achieve highly specific silencing of target genes by activating AGO2 to cleave the mRNA target. shRNA is an alternative to siRNA in specific gene silencing as shRNA behaves similarly to siRNA in RISCs.[28] On the other hand, the miRNA guide strand only requires partial complementary base pairing with mRNA so that miRNA can target an array of mRNA simultaneously. As an example, miRNA-124 has been shown to suppress the expressions of 174 genes in brain tissues at the same time.[29] Without complementary pairing, AGO2 is rarely activated so that miRNA silences mRNA by translation repression or direct degradation by exonucleases.[30] RNAi is a promising approach for antitumor gene therapy as it offers an efficient approach in specific silencing of oncogenes, mutated tumor suppressor genes, and resistance-conferring genes with minimum systemic toxicity.[31] In addition, RNAi is generally considered superior to small-molecule drugs as siRNA and miRNA can theoretically be engineered to silence any gene, including those responsible for the expression of undruggable targets.[32] Owing to their huge therapeutic potential, siRNA, shRNA, and miRNA have been extensively developed for cancer therapy.
2.3. Immunostimulatory Nucleic Acids
There are also some nucleic acids that do not participate in genetic modifications but rather directly stimulate immune responses. Single-stranded cytosine-phosphate-guanine (CpG) oligonucleotides, unmethylated CpG sequences in DNA and dsRNA polyinosinic-polycytidylic (poly I:C) have all been shown to activate innate immune responses by interaction with Toll-like receptors (TLRs). For example, CpG oligonucleotides and poly I:C are known as TLR agonists. Upon binding to TLR9 receptors on dendritic cells (DCs), CpG can activate and mature DCs, which in turn produce various cytokines such as interleukin 12 to engage T-cell immune responses.[33] Poly I:C and other viral dsRNA-mimicking analogs can interact with TLR3 receptors and induce type I IFN responses, inflammatory cytokine/chemokine production, and DC maturation.[34] Delivery of these immunostimulatory nucleic acids to tumors and immune cells can significantly enhance tumor recognition by the immune system and induce potent tumoricidal immune responses. However, due to the nonspecific nature of these nucleic acids, delivery needs to be tightly regulated to prevent undesired immune responses.
3. Inorganic Nanomaterials as Therapeutic Gene Carriers
From the advent of nanotechnology in the 1980s, marked by the invention of scanning tunneling microscopy, human knowledge in atom manipulation and nanomaterials synthesis has been advancing rapidly.[35] Nanomaterials have interfaced with the biomedical field mainly as therapeutic carriers. Inorganic nanomaterials can be tailored in size, shape, and elemental composition to meet the demands of therapeutic loading capacity, biocompatibility, prolonged circulation time, and cellular uptake.[36–39] Robust inorganic nanocarriers provide therapeutic payloads such as chemically unstable drugs, nucleic acids, and peptides with protection against hydrolysis and enzymatic degradation.[40–42] In addition, unique physical properties of certain inorganic nanomaterials offer more versatility in therapy modalities.
Common inorganic carriers are composed of noble metals (Au/Ag), iron oxide (Fe3O4), and silica (SiO2). Nanocarriers made of noble metals usually possess strong absorption in the near infrared (NIR) region of the electromagnetic spectrum and can be used for photothermal therapy and photoacoustic imaging. The superparamagnetism of iron oxide nanoparticles (IONPs) can be harnessed for magnetotransfection, contrast enhancement in magnetic resonance imaging, and magnetic hyperthermia.[43] Mesoporous silica-based nanoparticles (MSNs) have adjustable pore sizes that can accommodate loading of different types of therapeutic agents.[44] Lanthanide ion-based (e.g., Gd3+, Yb3+, and Er3+) upconversion nanomaterials have been applied as dual therapeutic-diagnostic carriers due to their ability to convert NIR radiation into higher energy visible or ultraviolet light.[45] Futhermore, hybrid inorganic nanomaterials that contain constituent elements from different types of nanocomposites such as NaGdF4:Yb3+, Er3+/Au/MSN, and IONP/MSN were also invented to enable multifunctionality.[46,47]
The aforementioned nanomaterials are typically synthesized by the bottom-up approach where precursor molecules and constituent atoms go through growth control mechanisms under specific reaction environments to form uniform ultrasmall nanomaterials (<20 nm).[48] Subsequent surface functionalization further equips these nanomaterials with desired biocompatibility, loading capacity, and targeting capabilities for nucleic acid delivery. The synthesis processes and the properties of inorganic nanocarriers are discussed in the following subsections.
3.1. Synthesis Approaches for Inorganic Nanomaterials
Typical methods for inorganic nanomaterials synthesis include hot colloidal chemistry, microemulsion, and seed-mediated synthesis. Various inorganic nanomaterials, including noble metallic nanoparticles, magnetic nanoparticles, and lanthanide-based upconversion nanoparticles benefit from being synthesized by hot colloidal chemistry techniques, as such methods allow for the precise control over key physicochemical properties of the outcoming nanomaterials.[48–51] Hot colloidal chemistry usually involves organometallic precursors, surfactant capping ligands, and solvents with high boiling points.
In hot colloidal synthesis, nanoparticles were formed following the LaMer nucleation and growth mechanism;[52] first, organometallic precursors decompose into reactive monomers via thermolysis. As thermal decomposition drives the concentration of monomers beyond the saturation limit and reaches the nucleation concentration threshold, burst nucleation occurs, and nuclei form as the new phase in the supersaturated solution. When the monomer-consuming nucleation process lowers the monomer concentration below the nucleation concentration, the remaining monomers diffuse to nucleation sites and further increase the size of nuclei. When the monomer concentration dips further, near the saturation limit, Ostwald ripening may take place and cause heterogeneous nanoparticle sizes. Proper control of the ligands capping onto the growing nuclei is essential to preventing Ostwald ripening and ensures the creation of nanoparticles with a uniform size distribution. Since high boiling point solvents are generally organic, nanoparticles produced from hot colloidal synthesis usually contain hydrophobic ligands on their surfaces.
Microemulsion is a commonly applied approach for synthesizing silica-based nanoparticles, calcium phosphate (CaP) nanoparticles, noble metallic nanoparticles, and upconversion nanoparticles.[45,53,54] Water, oil, and a surfactant are required for microemulsion synthesis. Surfactants are amphiphilic and can stabilize the oil–water interface by forming a micelle. Reactants confined in different micelles can be exchanged and reacted when two or more micelles collide. In a normal microemulsion, oil droplets are stabilized by surfactants in aqueous solution so that hydrophobic precursors are encapsulated inside the droplets. In a reverse microemulsion, polar precursors are entrapped in water droplets, which are dispersed in oil-based solutions. Due to volume constraints inside the micelles, the size of synthesized nanoparticles cannot exceed the micellar cavity. Thus, nanoparticle sizes can be controlled by tuning the water/oil/surfactant ratio to regulate the micellar size.[55] Normal microemulsions can also be combined with reverse microemulsions to create alternating polar/nonpolar/polar layers in nanocomposites.[56] In this case, hydrophilic payloads can be fully encapsulated by the organic layer and effectively separated from the outer environment by a hydrophilic barrier.
Seed-mediated synthesis is a two-step process, which separates the nucleation step from the growth process to induce epitaxial growth of noble metallic nanocrystals. Generally, seed particles are synthesized first, followed by subsequent particle growth in the presence of metal precursors, reducing agents and shape-directing reagents.[57] In the classical gold nanorod (AuNR) synthesis procedure, as an example, 3–4 nm seed gold nanoparticles (AuNPs) are produced by mixing a metal salt (HAuCl4), a capping reagent (sodium citrate), and a strong reducing agent (NaBH4). The seed AuNPs are then placed in a growth medium where amphiphilic shape-directing cetyltrimethylammonium bromide (CTAB) preferentially binds to and passivates certain facets of AuNP so that Au deposition is inhibited on those facets. A mild reducing agent (ascorbic acid) further reduces the metal salt so that the reduced elemental Au can be deposited onto the non-CTAB-passivated facets on seed AuNPs. The combination of preferential CTAB binding and continuous Au deposition results in epitaxial growth of seed AuNPs to form AuNRs, which are around 20 nm wide and hundreds of nm long.[58] The aspect ratio of AuNRs can be tuned by adjusting the concentration ratio of shape-directing reagents to metal salts.
3.2. Surface Functionalization for Improved Biocompatibility
Even though some nanoparticles such as gold, CaP, and silica-based nanoparticles are synthesized in aqueous conditions, the surface of hydrophobic nanoparticles, such as those resulting from hot colloidal chemistry, need to be functionalized with hydrophilic coatings in order to be relevant in biomedical applications. Without hydrophilic surface modifications, hydrophobic nanoparticles cannot load hydrophilic cargos (e.g., nucleic acids) and would form aggregates in aqueous environment (e.g., blood). Surface modification strategies, including micelle encapsulation, ligand exchange, and silanization, were developed to convey hydrophilicity onto nanomaterials.[48] Judicious selection of surface modification ligands can not only render nanomaterials hydrophilic but also introduce functional anchors (e.g., carboxyl, amine, and thiol groups) for further bioconjugation.
For successful delivery of therapeutic agents, nanocarriers must also possess immune stealthiness. Nanocarriers with chemically reactive surfaces often adsorb serum proteins and form protein coronae, which could lead to sequestration by the mononuclear phagocyte system (MPS). Therefore, the surface of inorganic nanocarriers needs to be passivated, typically via bioconjugation, to avoid immunorecognition. A few examples of commonly applied bioconjugation chemistries include the coupling between amine and epoxide/succinimide/epoxide, sulfhydryl and maleimide/disulfide/iodoacetyl, carboxyl and ethyl(dimethylaminopropyl) carbodiimide/N-hydroxysuccinimide, and alkyne and azide.[59] In the case of mismatching coupling groups, bifunctional linker chemistry such as amine-to-sulfhydryl succinimidyl-[(N-maleimidopropionamido) poly(ethylene glycol)n] ester crosslinkers can be applied alternatively. With bioconjugation chemistry, other functional moieties such as stabilizing polymers, charged polymers, therapeutic agents, and targeting ligands can be readily bonded with the functional anchors on inorganic nanomaterials.
Incorporation of poly(ethylene glycol) (PEG) into nanocarrier designs has been a quintessential strategy to drastically increase a nanocarrier’s serum stability and prolong blood circulation time. An outer PEG layer can shield interior surface charges and create a biochemically inert cloak for nanocarriers. As a result, PEGylation is effective in preventing the formation of a protein corona on nanocarriers and helps nanocarriers to avoid uptake by the MPS.[60] Nanocarriers conjugated with PEG can have substantially better in vivo pharmacokinetics.[61] However, it has been shown that the inertness of PEG layers also poses challenges to cellular uptake and intracellular trafficking of nanocarriers due to reduced interactions with cellular compartments, a phenomenon called the PEG dilemma.[61] Therefore, judicious selections of molecular weight and conjugation density when using PEG is essential to balancing the physiological stability and cellular uptake capacity of nanocarriers.
The strategy of transitioning from PEGylation to de-PEGylation during cellular uptake has been further proposed to solve the PEG dilemma.[62] Relying on environmental stimuli specific to tumor microenvironments and intracellular spaces, PEG can be conjugated onto nanocarriers via acid-labile or redox-sensitive linkages such as hydrazone bonds, Schiff’s base bonds, and disulfide bridges.[63–65] With these environmentally responsive linkages, PEG layers can be stripped off and nanocarriers can be better situated for cellular interaction and payload release.
3.3. Cationic Polymer Coatings for Nucleic Acid Loading
Since PEG is chemically inert and does not have the capacity for nucleic acid loading, additional functional moieties need to be conjugated onto nanomaterials for nucleic acid loading. The principle behind nucleic acid loading is based on the electrostatic attraction between the negatively charged phosphate groups on the nucleic acid backbone and the positive charge of cationic loading materials. The number of negative charges carried by nucleic acids can be easily calculated as each nt carries one negative charge on its phosphate group. Despite the fact that all nucleic acids share the anionic backbone feature, the binding between cationic materials and different types of nucleic acids can be idiosyncratic.[23] Double-stranded DNA plasmids can be effectively condensed by cationic materials via multivalent binding due to its richness in negative charges. However, large amounts of cationic materials might be required to be able to fully condense the large DNA plasmids. Since siRNAs are quite small and do not have nearly as many negative charges as DNA, lesser quantities of cationic materials but higher densities of positive charges are required to stabilize these small nucleic acid sequences. mRNA is intermediate in size but chemically unstable due to its single stranded structure. Thus, the design of mRNA nanocarriers must emphasize the cargo protection feature.
Similar to naturally occurring DNA compaction by amine-rich histone proteins, a plethora of amine-based cationic polymers including poly(l-lysine) (PLL), poly(ethyleneimine) (PEI), and poly(amido amine) (PAA) have been integrated in nonviral gene carriers due to their capacity for condensing and protecting nucleic acids. PLL was one of the first cationic and biodegradable peptides developed for in vivo use. However, its applications were limited by low transfection efficiency and inefficient endosomal escape.[66] PEI is a highly efficient transfection cationic polymer, so much so that the high molecular weight 25 kDa PEI is considered to be the gold standard cationic polymer in nonviral transfection.[67] PEI’s structure can be either linear or branched. Owing to the abundance of primary, secondary, and tertiary amines, branched PEI can usually condense nucleic acids and mediate endosomal escape via the proton sponge effect where branched PEI causes excessive influx of water and ions into endosome causing the endosomal membrane to burst open for higher transfection efficiency than its linear counterpart.[68] Nevertheless, PEI is nonbiodegradable, and its abundance of positive charges also tends to inflict severe damage on cellular organelles, especially mitochondria, thereby inducing cell death.[69] To alleviate PEI’s toxicity, PEG is usually grafted onto PEI to partially shield the positive charges.[41] Alternatively, crosslinked PEIs with bioreducible crosslinkers have also been developed so that large networks of PEI can be degraded into nontoxic low molecular weight PEIs after transfection.[69] PAA is similar to PEI as it also possesses a high density of amines in branched structures and inevitably shares PEI’s features in high transfection efficiency and cytotoxicity. Other cationic polymers such as chitosan, poly(β-amino esters) (PBAE), and poly(2-dimethylaminoethyl methacrylate) have also been exploited in nonviral gene delivery due to their low toxicity.[70–72]
3.4. Cationic Polymer Coatings for Controlled Nucleic Acid Release
Many recently developed cationic polymer coatings possess stimuli-responsive degradation mechanisms for nucleic acid release. Figure 2 shows the structural degradation of representative bioreducible polymeric coatings including disulfide-crosslinked, acid labile, and hydrolysable polymers on inorganic nanocarriers. The most commonly utilized stimulatory cue for cargo release is glutathione (GSH), a disulfide-reducing agent abundantly present in cytoplasm. GSH plays an important role in intracellular protein folding by generating active thiol sites on protein disulfide isomerase.[73] The drastic discrepancy between the intracellular (0.5 × 10−3−10 × 10−3 m) and extracellular (2 × 10−6−20 × 10−6 m) GSH concentrations makes GSH a unique intracellular cue for nucleic acid release.[74] Therefore, disulfide bonds have been incorporated into major types of nanocarriers’ polymer coatings including PEI, PLL, and PAA for GSH-sensitive nucleic acid release.
Figure 2.
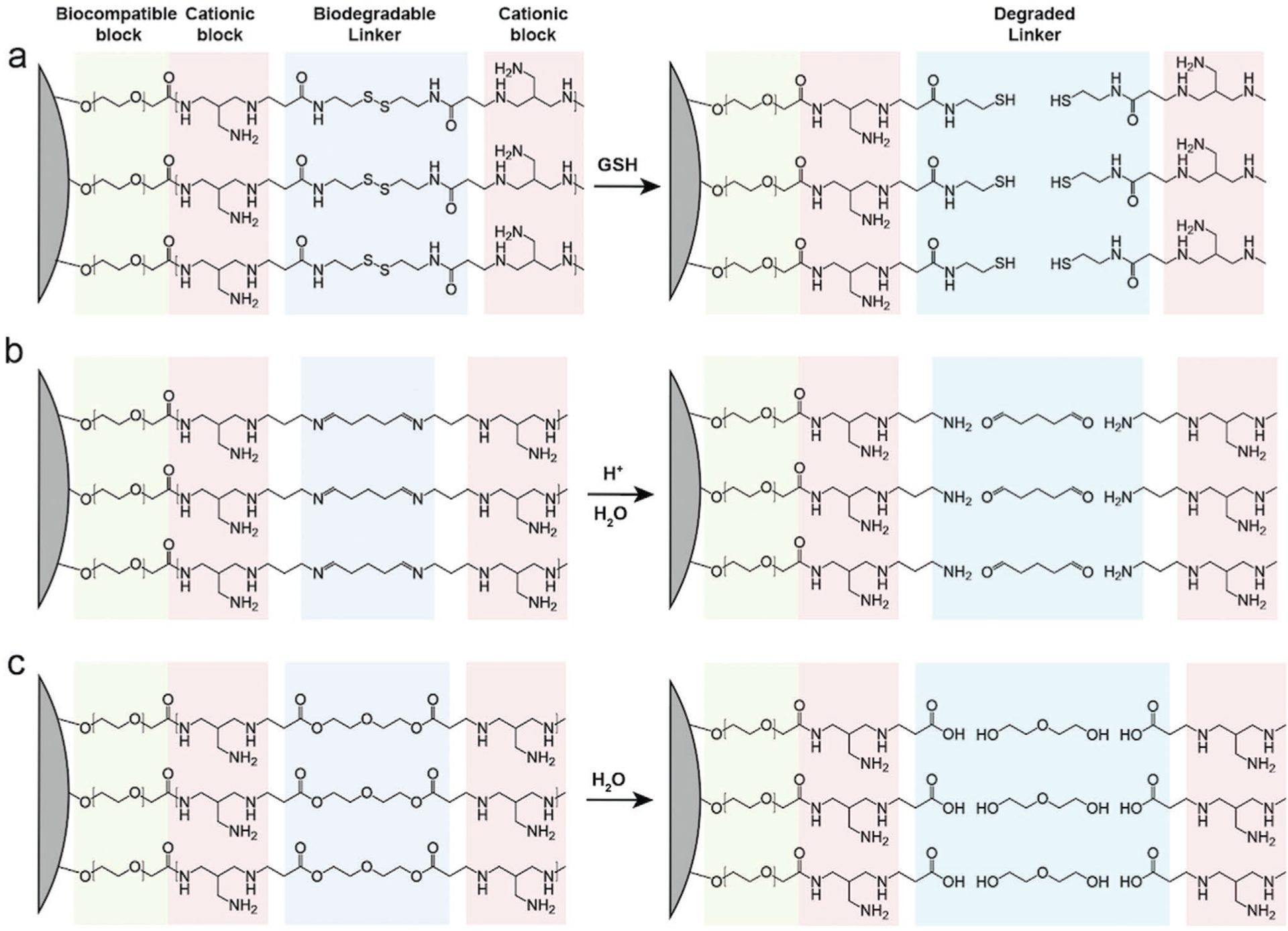
Degradation of representative biodegradable polymer coatings on inorganic cores (illustrated as gray surfaces). PEG represents the biocompatible polymer block. A simplified branched structure of PEI represents the cationic polymer block. The disulfide, imine, and beta-amino ester-containing linkers represent the GSH reducible, acid labile, and hydrolysable moieties, respectively. a) Disulfide-crosslinked cationic polymer coating cleaved by GSH. b) Imine-crosslinked cationic polymer coating degrades in acidic solution. c) Beta amino ester-crosslinked cationic polymer coating hydrolyzes in water. Straight up free molecules, change color for middle one column.
Integration of disulfide bonds into cationic polymers can be achieved via the crosslinking of oligoamines or through polymer thiolation.[75] Disulfide-containing crosslinkers such as bis(acryloyl)cystamine, dithiobis(succinimidyl propionate), and dithiodipropionic acid are commonly applied to crosslink oligoamines into a bioreducible network. For example, low molecular weight polymers such as 800 Da PEI can be effectively connected by crosslinkers into high molecular weight polycations. Alternatively, active thiol groups can be introduced onto cationic polymers by iminothiolanes so that they can form disulfide bridges with each other under oxidizing conditions. Crosslinking of thiolated PEI and PLL has been reported to be successful in transfection applications.[76,77] Using disulfide-containing crosslinkers to form polycations ensures an abundant presence of disulfide in the network, but the polymerization degree is often challenging to control so that the molecular weight of the crosslinked products can be unpredictable. On the other hand, thiolation of preformed polymers can yield relatively predictable products and conserves sufficient charge for nucleic acid condensation. Nevertheless, successful thiolation and disulfide formation might require extensive optimization with respect to reaction conditions.
Another environmental cue for intracellular nucleic acid release is acidic pH. The pH values in different biological compartments have been extensively studied. It is well established now that the pH for blood and cytosol is near 7.4, and the pH between different organelles can greatly vary (e.g., pH 8.0 in mitochondria and pH 4.5 in lysosomes).[78] Nanocarriers would experience near neutral pH when travelling in the blood circulation system or the intracellular cytosol. When going through receptor-mediated endocytosis (details provided in Section 3.6), nanocarriers inevitably encounter acidic environments associated with endo/lysosomes. Therefore, acid-labile linkers have been incorporated into cationic polymers to enable intracellularspecific release. For example, low molecular weight 1.8 kDa PEI was reacted with glutadialdehyde to form an acid-labile PEI network crosslinked by imine groups for DNA delivery.[79] In this case, acid-labile PEI was able to degrade around 1 h at pH 4.5 but needed 118 h to degrade at pH 7.4. Another example of an acid labile cationic polymer is the branched poly(ortho ester amino alcohol)s (bPOEAAs).[80] The ortho ester groups in bPOEAAs are highly sensitive to endosomal acidification and can be degraded at pH 4.0 within 3 h to release DNA.
Depending on endosomal escape propensity, nanocarriers can be subjected to various stages of acidity including pH 6.3 in early endosomes, pH 5.5 in late endosomes, and eventually pH 4.5 in lysosomes.[81] Additionally, the transition from early to late endosome can be as short as a few minutes whereas the transition from late endosomes to lysosomes can take an hour.[82] Thus, it is important to study the acid-labile polycations’ endosomal escape timeline and release profiles under different pH values so that acid-labile linkages can be cleaved at the optimal pH at the right time. It would be ideal for endosomal escape to immediately follow acid-triggered polymer degradation to avoid degradation of the released nucleic acids. Last but not least, one factor to consider for tumor delivery is that acidosis in the tumor environment produces a pH (≈6.5) value similar to that of early endosomes.[83] Acid-labile polymers should be designed to respond to pH values lower than 6.0 in order to avoid premature release in a tumor’s acidic extracellular matrix.
Besides disulfide and acid-labile linkers, hydrolysable moieties have also been integrated into polycations for controlled release of nucleic acids. An exemplary polymer for hydrolyzation is PBAE. PBAE polymers are synthesized by the reaction between amine-based monomers and diacrylate esters. PBAE can be degraded into monomeric units via the hydrolyzation of ester bonds within 5 h at physiological pH.[84] Due to simplicity in the synthesis procedures and the great versatility in choosing amine-based and diacrylate ester units, a library of 2350 combinations of different PBAE structures have been synthesized in a semiautomated system and screened for transfection efficiency.[85] Other than PBAE, ester bonds along with other hydrolysable linkages such as amide, imine, and carbonate functional groups have been incorporated into other cationic polymers such as PEI and poly(2-dimethylaminoethyl methacrylate) for controlled release of nucleic acids.[86,87] For hydrolysable linkage, it is essential for the nucleic acids to be delivered within cytoplasm before the complete hydrolytic breakdown of polycations. Therefore, comprehensive understanding of the degradation timeline of the hydrolysable polycations under different pH values and temperatures is necessary.
Since polymer crosslinking mostly relies on random reactions between crosslinkers and monomers with multivalent reactive sites, the molecular weight and the structure of the resultant products are often unpredictable. Nevertheless, the crosslinking procedure is usually simple enough so that screening over a matrix of reaction parameters is feasible. Generally, prolonged reaction duration in concentrated solutions with high crosslinker/monomer molar ratio could result in high molecular weight and hyperbranched products and vice versa. Sometimes elevated temperature could enhance the reactivity of reactants and boost the crosslinking efficiency. The acidity of the reaction environment also plays an essential role in aqueous polymer crosslinking as it could decide the protonation state and hence the reactivity of the monomers. Over-crosslinking could produce large aggregates whereas under-crosslinking could lead to low yield of desired products. Therefore, the interplay between various reaction parameters must be well understood in order to empirically identify the balanced conditions for synthesizing crosslinked polymers.
3.5. Passive Tumor Accumulation and Penetration
Even though gene therapy can take effect at relatively low dosages, achieving sufficient nucleic acid bioavailability at tumor sites remains an overarching obstacle to gene delivery. From systemic administration to arrival at intracellular destinations, nanocarriers have to go through a series of biological barriers while protecting their nucleic acid payloads. High tumor accumulation and deep tumor penetration are prerequisites for subsequent intracellular therapeutic release. Figure 3a shows the process of inorganic nanocarriers extravasating from the leaky tumor vasculature and penetrating into tumor tissue. Inorganic nanocarriers then interact with tumor cells to induce cell death.
Figure 3.

Schematic representation of tumor-targeted delivery of combinatorial therapeutics by inorganic nanocarriers. a) Passive tumor targeting. Inorganic nanocarriers extravasate from leaky tumor blood vessels into tumor tissue, and then penetrate into deep tumor tissue. b) Active tumor targeting and intracellular trafficking. After penetration into tumor tissue, inorganic nanocarriers first enter tumor cells by receptor-mediated endocytosis followed by endosomal escape and release therapeutic cargos from inorganic cores such as plasmid DNA, mRNA, interfering RNA, chemotherapeutics, and photosensitizers into the cytosol. Plasmid DNA then needs to pass through the nuclear membrane in order to be transcribed inside the nucleus. Inorganic cores and the released payloads can mediate multiple treatment modalities including gene, chemo, immune, radio, and hyperthermal therapy.
Size plays a significant role in determining the ability of a nanocarrier to traverse biological barriers. Application of nanocarriers between 10 and 200 nm in size automatically enables the enhanced permeability and retention (EPR) effect. The fact that substances smaller than 10 nm can be readily cleared out renally and those larger than 200 nm tend to be sequestered by macrophages in hepatic and spleenic tissue collectively determine the optimal size range for prolonged blood circulation.[88] Therefore, nanocarriers within the 10–200 nm range will have greater chances of encountering tumor cells. Since the vasculature network in tumors are tortuous and leaky, nanocarriers tend to passively accumulate in the tumor compartment by extravasating through these leaky vessels. Nanocarriers with sizes around 100 nm have been shown to passively accumulate in tumors most efficiently.[89] Unfortunately, the bioavailability of systemically administered nanocarriers in tumors is usually less than 5% by EPR.[62] The EPR effect can greatly vary at different locations as the pore cutoff size can be starkly different (e.g., 100 nm pore cutoff size in brain and pancreatic tumors vs >1 μm in breast tumors).
To further enhance therapeutic efficacy, nanocarriers extravasated from the leaky vasculature need to penetrate deeper into tumor tissue instead of being stagnant at the peripheral tumor margin. Notably, a series of studies have revealed the size dilemma in tumor accumulation and penetration. Although 100 nm is the optimal nanocarrier size for tumor accumulation, nanocarriers with smaller sizes around 20 nm demonstrated significantly deeper tumor penetration albeit with lower tumor accumulation.[90,91] Therefore, the respective size effects on tumor accumulation and penetration must be wisely balanced for optimal payload effect. Besides size, surface charge and the shape of nanocarriers also have profound effects on tumor accumulation and penetration, which have not been conclusively elucidated as they are usually material-dependent phenomena. Nanocarriers with cationic charges have been reported to be electrostatically attracted to the anionic and leaky tumor blood vessel walls. Attraction to tumor blood vessel walls in turn results in higher extravasation and tumor accumulation. However, the attraction between cationic nanocarriers and the anionic extracellular matrix hinders further penetration into tumor tissue.[92] Studies have also shown that extravasation favors nanocarriers with nonspherical shapes over spherical shapes for some materials and vice versa for others.[93] Therefore, well-rounded considerations on multiple physicochemical properties such as size, charge, and shape are required in order to systematically improve the EPR effect. In general, the combination of a mild positive charge and anisotropic shape should offer nanocarriers certain advantages in tumor accumulation and penetration compared to the combination of neutral/negative charge and spherical shape.[94,95]
3.6. Active Targeting for Tumor-Specific Uptake
After EPR-mediated accumulation and penetration, inorganic nanocarriers must enter the intracellular space to release their therapeutic payloads into the cytoplasm in order to achieve gene delivery. Figure 3b demonstrates the process of cellular uptake and intracellular trafficking of inorganic nanocarriers. The intracellular fate and function of different therapeutic cargos released by inorganic nanocarriers are also illustrated. There are many different modes of cellular uptake including phagocytosis, pinocytosis, macropinocytosis, and clathrin-mediated and caveolae-mediated endocytosis.[96] Inorganic nanocarriers of nucleic acids are commonly positively charged and of a size ranging between tens to hundreds of nm; due to these properties, the main cellular uptake pathways for inorganic nanocarriers are clathrin-mediated endocytosis, caveolae-mediated endocytosis, and macropinocytosis.
Clathrin-mediated endocytosis is activated by a receptor–ligand interaction. Upon ligand binding, clathrin-coated vesicles form on the plasma membrane to wrap around the ligand-bound receptors. The vesicles later become endosomes in the cytoplasm which subsequently fuse with acidic lysosomes to degrade the substances trapped inside the endosomes.[97] Another receptor-mediated endocytosis mechanism that complements clathrin-mediated endocytosis is caveolae-mediated endocytosis.[98] Caveolae are small invaginations (50–100 nm) that form on plasma membranes, which can be pinched off from the plasma membrane to form caveolar endocytic vesicles with receptor signaling. As opposed to clathrin-mediated endocytosis, caveolar endocytic vesicles do not necessarily fuse with endolysosomes; therefore, the encapsulated particles may avoid acidic degradation. Caveolae-mediated endocytosis can take in caveolae-sized particles as well as particles that are hundreds of nm in size. This phenomenon can be explained by the changeable size and shape of caveolae; these changes are regulated by receptor signal transduction. Cellular uptake via macropinocytosis is rather nonspecific. Macropinocytosis involves actin-based plasma membrane ruffling to take in large amounts of extracellular liquids and form macropinosomes.[99] Macropinosomes are leaky in nature and fuse into endolysosomes in a cell type-dependent manner. Studies have shown that particles smaller than 200 nm mostly experience clathrin-mediated endocytosis whereas those between 200 and 500 nm can enter cells via macropinocytosis.[100,101] Last but not least, the fact that cationic nanocarriers can adsorb onto the negatively charged plasma membrane also facilitates the endocytic process in general.
To exploit receptor-mediated endocytic pathways, inorganic nanocarriers are usually conjugated with targeting moieties for specific cellular uptake. Tumor cells overexpress various receptors of which expression in normal cells is tightly regulated to sustain cellular nutrient needs and control growth. As the marked difference between normal and tumor cells, the overexpressed receptors have been widely targeted for antitumor therapy. Extensively investigated receptors include human epidermal growth factor receptor 2 (HER2), epidermal growth factor receptor (EGFR), folate receptor (FAR), and transferrin receptor (TfR).[102] Nanocarriers are often conjugated with targeting moieties such as antibodies/peptides, aptamers, or targeting ligands with high binding affinity to the overexpressed receptors, which helps the nanocarrier to achieve preferential uptake by tumor cells.
Although active targeting moieties can significantly boost the uptake of nanocarriers, it is important to find the optimal density for targeting ligand conjugation. Low ligand density due to underconjugation could result in insufficient binding with the receptor. Alternatively, dense ligand-packing could enable multivalent receptor–ligand binding and improve the binding affinity, but it could also result in inefficient uptake due to steric hindrance, high competition for receptors between ligands or over-recruitment/depletion of cell receptors.[103] Moreover, overconjugation of targeting moieties could compromise a nanocarrier’s stealth characteristics and lead to opsonized sequestration. As a rough guideline, the targeting moiety packing density for sub-100 nm nanocarriers usually falls in the range of 0.1–100/100 nm2.[104] The size of different targeting moieties also needs to be taken into account. Some targeting moieties such as antibodies are significantly larger than others such as the small molecule folic acid (FA). Conjugation of small targeting ligands will not significantly affect the size or surface chemistry of nanocarriers. On the other hand, conjugation of large targeting moieties, which can be nanometers in dimensions and tens of kDa in weight, will inevitably limit the permitted packing density and alter the size of nanocarriers. Therefore, cell receptor density, nanocarrier size, targeting moiety size, and conjugation density are all to be scrutinized when designing tumor-targeting nanocarriers.
3.7. Intracellular Trafficking and Nuclear Entry
Once nanocarriers are endocytosed via receptor-mediated routes, it is essential for nanocarriers to escape from endosomes early to avoid acidic degradation and achieve efficient cargo release. Typically <2% of the internalized DNAs and RNAs can escape from endosomes while the vast majority of delivered nucleic acids are degraded prior to performing any useful therapeutic function.[105] Amine-based cationic polymers have strong pH buffering capacity and can engage the “proton sponge effect” for endosomal escape. Since amines are self-protonated under acidic pH in endolysosomes, continuous consumption of protons by cationic polymers keep the lysosomal proton pump active to infuse more protons and counterions into the lysosome. As a result, high ionic concentrations lead to an influx of water and high osmotic pressure, which eventually bursts endolysosomes open.[106] Peptides capable of lysosomal membrane destabilization can also be conjugated onto cationic polymers to further accelerate endosomal escape into the cytoplasm. During and after endosomal escape, the aforementioned intracellular cues such as low pH and GSH trigger degradation of polymer coatings on nanocarriers to release nucleic acids into cytoplasm.
Nevertheless, DNAs still need to overcome the nuclear membrane barrier in order to get transcribed before the final products of gene expression, proteins, can be created to perform the desired therapeutic function. Exogenous DNA usually enters cell nuclei either through direct diffusion during nuclear envelope break down or via nuclear pore complex (NPC)-mediated pathways. Nuclear membranes in mammalian cells break down during mitosis and provide entry for DNA.[107] It is usually observed that cells in the G2/M phases exhibit the highest DNA transfection rate.[108] When nuclear membranes are intact, the nuclear gatekeeper NPC with a pore cutoff size of only 9 nm (equivalent to only ≈300 base pairs in DNA) would prevent DNA plasmids from diffusing into nuclei.[109] However, the NPC does allow passage of molecules as large as 50 nm with the assistance of nuclear localization sequences (NLSs).[110] NLSs are typically simian virus 40-derived short amino acid sequences (i.e., peptides) that have high binding affinity to importin, a protein that mediates importation through the NPC. A series of studies have reported significantly higher nuclear uptake of NLS-conjugated plasmid DNA polyplexes compared to counterparts without NLS.[111–113] However, addition of NLSs into nanocarriers and DNA polyplexes can be challenging as compatible chemistry and optimal NLS density must be identified to achieve notable improvements in nuclear uptake. Alternatively, DNA can be designed to directly complex with NLSs. DNA nuclear targeting sequences, which are regional viral DNA sequences such as viral origins of replication and promoters, can also be directly incorporated into plasmids so that they can be transported into nuclei alone.[114,115]
Without NLS, DNA could also be ferried into cell nucleus when complexed by polycations, bypassing the entry size limit enforced by NPC. Although the exact mechanism has yet to be elucidated, it has been proposed that polycations could penetrate nuclear envelope by either electrostatic permeabilization or lipidic fusion because they are highly charged and could be adsorbed by anionic lipids after intracellular entry.[116,117] In addition, studies have also shown that polycations like PEI can facilitate nuclear transport without compromising the nuclear membrane and not interfere with transcription after nuclear entry.[118,119] However, the long term intranuclear effects of different types of polycations are still not clear. More studies on the interaction between polycations and nuclear membranes are needed to obtain insights into this issue.
4. Combinatorial Delivery of Gene–Chemo Therapeutics by Inorganic Nanoparticles
Chemotherapeutics are potent anticancer drugs widely applied in front-line clinical treatments for cancer patients. Chemotherapeutics can damage cancer cells’ DNA as well as mitotic machineries to induce apoptosis in cancer cells. An exemplary chemotherapeutic drug, doxorubicin (Dox) intercalates with DNA to prevent transcription and eventually kills cancer cells.[120] Due to its anticancer potency, hydrophilicity, and intrinsic fluorescence, Dox has been extensively studied in cancer research. Unfortunately, the application of chemotherapeutics still faces major challenges. Chemotherapy can cause severe side effects due to nonspecific toxicity to healthy tissue. Patients receiving chemotherapy often require a withdrawal period to recover from the side effects.[121] Therefore, administration dosage and frequency of chemotherapeutic treatments are limited. Moreover, cancer cells can develop multidrug resistance (MDR) either under the selection pressure of chemotherapeutic exposure or via the intrinsic expression of resistance-related genes. By activating DNA repairing and detoxification systems, cancer cells with MDR can effectively lower the intracellular concentration of chemotherapeutics and evade chemotherapeutic-induced apoptosis.[122] Thus, MDR can significantly negate the antiproliferative effects of chemotherapeutics and lead to a 90% failure rate in treating aggressive metastatic cancers.[123] Higher dosages of chemotherapeutics could potentially overcome MDR but would inflict more severe side effects.
Strategies of combinatorial delivery of chemotherapeutics and nucleic acids were developed to improve the efficacy of chemotherapy by addressing MDR. Therapeutic nucleic acids such as siRNA and DNA can silence genes responsible for MDR and thereby resensitize cancer cells to delivered chemotherapeutics. In this case, nucleic acids and chemotherapeutics can each be adjusted to doses well-tolerated by healthy tissue while retaining sufficient potency to inhibit tumor growth. Nanomaterial carriers can further enhance combinatorial therapeutic efficacy through targeted delivery. Nanomaterial-guided combinatorial therapy is a promising cancer treatment modality that may one day be the primary form of clinical remediation efforts.
4.1. MDR Gene Silencing for Chemosensitization
Tumor drug resistance often involves synergistic interactions between multiple dynamic gene expression and cascade pathways. MDR mechanisms are rather complicated and can be divided into five categories including regulated transportation of chemotherapeutics, sequestration of chemotherapeutics, heightened DNA repairing, drug inactivation and detoxification, and disruption of apoptosis.[124] Among the five categories, regulation of chemotherapeutic transportation is the most common and extensively studied resistance mechanism. Most hydrophilic chemotherapeutic drugs are protonated and carry positive charges in acidic tumor microenvironments established by the Warburg effect.[125] Denied entry of the charged chemotherapeutics by plasma membranes decreases the influx of hydrophilic chemotherapeutics into cytoplasm. Even though hydrophobic drugs can travel past plasma membranes readily by diffusion, energy-dependent transporters can expel them to lower the intracellular drug concentration.
Erroneous overexpression of the adenosine triphosphate-binding cassette (ABC) transporter, either through intrinsic mutations or chemotherapeutic stimulus, is the main cause of increased drug efflux observed in cancer cells.[126] As a member of the ABC superfamily, transmembrane P-glycoproteins (P-gp) have been known for conferring resistance against various types of chemotherapeutics. P-gp can capture hydrophobic drugs as they partition into the lipid bilayer and efficiently transport them out to the extracellular space via adenosine triphosphate hydrolysis.[127] Without drug efflux pumps, active expression of the Bcl2 antiapoptotic gene and c-myc oncogene can also confer significant drug resistance to cancer cells.[128,129]
Successful silencing of MDR genes is the premise of effective combinatorial nucleic acid and chemotherapy. The RNAi pathway, especially when exploited using siRNA, is an efficient approach to knockdown resistance-related genes due to the gene specificity and low dosage requirements associated with RNAi.[130] Nanomaterials have exhibited promising targeted-delivery of siRNAs[41,131] and chemotherapeutics.[40,132] In the case of intracellular delivery to cancer cells exhibiting MDR, nanomaterials can bypass drug efflux pumps via receptor mediated-endocytosis and escort chemotherapeutics and nucleic acids to deeper cytoplasmic and peri-nuclear regions to reach cytotoxic concentration thresholds.[133] Given its biocompatibility and high loading capacity in the pores and tunable sizes, MSNs are considered ideal nanocarriers for small-molecule drugs and large biomacromolecules.[44] MSNs modified with disulfide-linked PEI-hyaluronic acid (HA) can be applied to co-deliver Dox and Bcl2 siRNA to drug-resistant MCF7 breast cancer cells.[134] CD44 receptors are highly expressed in MDR cancer cells.[135] As a binding ligand to CD44 receptors, HA is used as the targeting ligand for certain MDR-expressing cancer cells.[136] After entry into the cytoplasm, high intracellular concentrations of GSH and hyaluronidase can degrade the MSN-PEI-HA constructs and release their payloads. The co-delivery of Dox and Bcl2 siRNA with an MSN-PEI-HA nanocarrier demonstrated increased tumor apoptosis compared to the individual delivery of Dox and Bcl2 siRNA separately. Similarly, MSNs modified with PEG-grafted-PEI were used to co-deliver P-gp siRNA and Dox to knockdown P-gp efflux pump expression and sensitize cancer cells to Dox.[137] MSNs were designed to have two different pore sizes (2–3 and 40–45 nm).[138] Dual-pore size design allows for more efficient cargo loading. In the present case, small-molecule Dox was loaded into the smaller pores and bulkier Bcl2 siRNA was fitted into the larger pores, both via electrostatic adsorption. The dual-pore MSNs showed promising drug-releasing profiles as well as significant therapeutic efficacy on HeLa cancer cells.
In addition to efficient loading of both large- and small-size therapeutic payloads, sequential release of nucleic acids and chemotherapeutics is needed to prevent the premature release of chemotherapeutics. Since loading capacity is limited on nanomaterials, chemotherapeutics released prior to gene-silencing are likely to be obviated by MDR mechanisms and significantly reduce their therapeutic effects. It has been shown that, sometimes, simultaneous delivery of nucleic acids and chemotherapeutics fails to achieve the expected synergistic anticancer effects.[139,140] It has also been validated that chemotherapeutics such as paclitaxel and temozolomide exhibit significantly stronger potency when administered hours later than interfering RNA.[141,142] When interfering RNAs and chemotherapeutics are co-delivered, rapid release of RNAs followed by delayed release of chemotherapeutics is important for optimal therapeutic efficacy.
Recently, a core–shell hierarchical MSN (H-MSN) was fabricated for sequential release of Dox and P-gp siRNA in drug-resistant MCF7-ADR tumors.[143] The small pores on the silica cores serve as the reservoirs for loading Dox. The outer organosilica shell, hybridized with PEIs and disulfide bonds, can encapsulate P-gp siRNA in its 100 nm large pores (Figure 4a). When H-MSN-Dox/siRNA is uptaken by cancer cells, the degradation of the superficial disulfide-linked organosilica shell will trigger the quick release of P-gp siRNA. Only after most of the shell is degraded and the siRNA has been released will Dox become free in the cytoplasm (Figure 4b). In a Dox cellular uptake experiment, low Dox uptake was observed in free Dox-treated MCF7-ADR cells at 2 and 24 h, indicating that active P-gp was able to expel most of the free Dox within 2 h. The H-MSN-Dox-treated MCF7-ADR cells showed decent Dox uptake at 2 h but significantly reduced at 24 h. This observation suggests that H-MSNs were only able to temporarily delay Dox efflux (Figure 4c,d). Notably, the H-MSN-Dox/siRNA-treated MCF7-ADR cells demonstrated high Dox uptake at both 2 and 24 h, meaning that P-gp siRNA was able to silence P-gp expression which was also confirmed by western blot. When applied in vivo, H-MSN-Dox/siRNA demonstrated pronounced antitumor effects by effectively inducing tumor apoptosis and inhibiting tumor growth (Figure 4e–g).
Figure 4.

Core–shell H-MSNs enable sequential release of Dox and siRNA for tumor treatment. a) Schematic illustration of mesopore surface modification with PEI for the subsequent loading of P-gp siRNA. b) Mechanism scheme demonstrating the therapeutic functions of H-MSNs in suppressing MDR of cancer cells and enhancing chemotherapeutic efficiency. c) Confocal laser scanning microscope images of MCF7-ADR cancer cells after co-incubation with free Dox, H-MSNs-Dox, and H-MSNs-Dox/siRNA for 2 and 24 h. d) Quantitative mean fluorescence intensity (MFI) analysis of Dox uptake in MCF7-ADR cancer cells after incubation for 2, 12, and 24 h by employing flow-cytometry measurement. e) Photographic images of tumors from MCF7-ADR cancer-bearing mice sacrificed after being treated with different agents for 14 days. f) Tumor masses of MCF7-ADR cancer-bearing mice sacrificed after being treated with different agents for 14 days. g) Tumor-volume changes over time after the administration of different agents until mice sacrificing on the 14th day. Reproduced with permission.[143] Copyright 2017, Elsevier.
Besides MSNs, other types of inorganic nanoparticles have been employed to co-deliver MDR-silencing interfering RNAs and chemotherapeutics. Lipid-coated calcium phosphate (LCP) nanoparticles were applied to co-deliver the chemotherapeutic docetaxel and GRP78 siRNA for chemoresistance knockdown to treat prostate cancer.[144] A hybrid Mn3O4/Fe3O4 nanoparticle was developed to co-deliver Dox and Bcl2 shRNA to drug-resistant breast cancer MCF-ADR.[145] Interestingly, an NIR-responsive, hollow gold nanoparticle (HGNP) was developed for sequential release of miRNA and Dox.[146] This study highlights HGNP’s capability for treating both cancer cells and cancer stem cells to reduce the risk of tumor recurrence. miRNA-21 (miR-21) is commonly recognized as a typical onco-miRNA that targets tumor-suppressor genes. It has been reported that miR-21 is essential to the pluripotency and drug resistance of cancer stem cells.[147] Therefore, miR-21 inhibitor (miR-21i), an miR-21-binding oligonucleotide, was selected to deactivate miR-21. HGNPs were coated with polyamidoamine (PAMAM) dendrimers by thiolation through the Au–S interaction. Dox was then attached onto HGNPs electrostatically followed by miR-21i complexation with PAMAM to create Dox-PAMAM-HGNP-miR-21i (D-P-HGNP/21i) (Figure 5a). Release of Dox was tightly controlled by HGNP-collapsing NIR laser exposure to allow the preceding release of miR-21i. The results indicate that D-PHGNP/21i with NIR exposure at 4 h (D-P-HGNP/21i-4R) was the most successful treatment in activating multiple apoptotic pathways in breast cancer cells and breast cancer stem cells in vitro (Figure 5b). When applied in vivo, D-P-HGNP/21i particles mainly accumulated in tumor tissue 4 h postadministration and displayed marked antitumor effects on breast tumor-bearing mice (Figure 5c,d). These results confirm that antitumor effects can be greatly enhanced by controlling the release timing of genetic and chemotherapeutic payloads.
Figure 5.
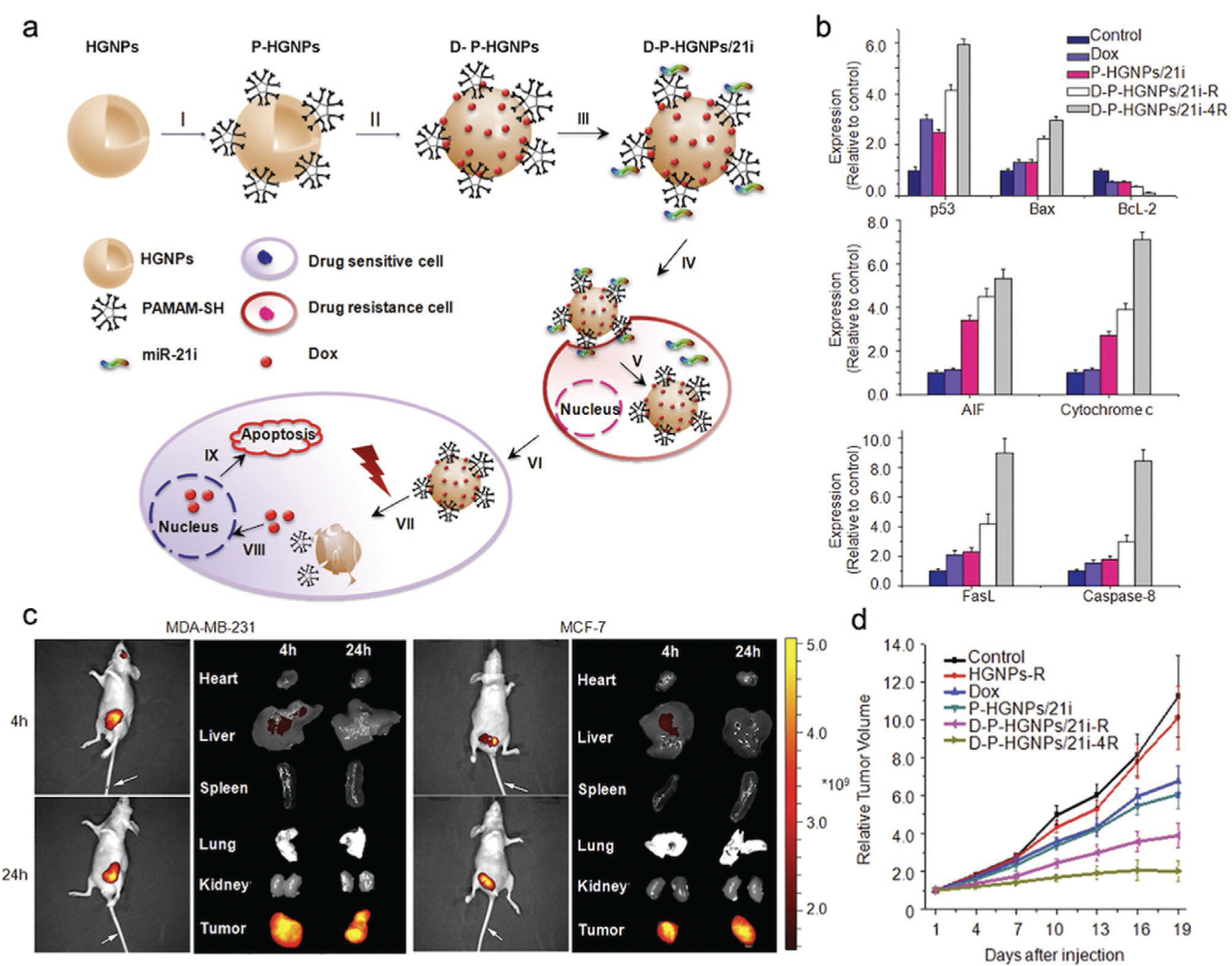
HGNPs with NIR release mechanism for co-delivery of Dox and miR-21i to breast tumors. a) Schematic illustration of HGNP-based, NIR triggered sequential miR-21i/Dox release with a precise time interval for optimal combination therapy. I–III) Formation of the HGNP co-delivery system. IV) D-P-HGNP/21i enters cells through endocytosis. V) Upon entering tumor cells, miR-21i is released first, VI) modulating the intrinsic state to a more chemosensitive state. VII) At the desired time, application of an NIR laser triggers collapse of HGNPs and a burst release of Dox, IX) activating two apoptosis signaling pathways, thereby inducing the synergistic apoptosis response. b) Quantitative results of western blotting indicate that both mitochondrial apoptosis and death receptor-related apoptosis pathways were activated by sequential therapy. c) Distribution of D-P-HGNP/21i in MDA-MB-231 and MCF7 tumor-bearing mice at 4 and 24 h postinjection with Cy5.5 labeled D-P-HGNP/21i. d) Tumor growth curves for nude mice bearing MDA-MB-231 human breast tumors. Reproduced with permission.[146] Copyright 2016, Elsevier.
4.2. Co-Delivery of Tumor-Suppressing Nucleic Acids and Chemotherapeutics
Malignant transition from normal tissue to cancerous tissue usually involves dysregulated expression of proto-oncogenes and tumor-suppressor genes. For example, malfunctioned proto-oncogene c-myc and tumor suppressor gene p53 are well studied because of their profound effects on promoting tumorigenesis. C-myc encodes for a proproliferative transcription factor responsible for malignancy development and is found to be broadly upregulated in various types of cancers.[148] On the other hand, p53, which is responsible for DNA repair and induces apoptosis, is found to be either mutated or deleted in more than half of human tumors.[149] Oncogene activation and tumor suppressor gene inhibition can further induce downstream effects such as heightened angiogenesis in tumors mediated by the regulatory cytokine vascular endothelial growth factor (VEGF).[150] Therefore, tumorigenesis drivers such as c-myc transcriptional factors and p53 proteins are ideal therapeutic targets. However, these driver proteins remain elusive as drugs due to their complex mechanisms and the involvement of many cofactors.[151] Alternatively, tumor suppressor gene-and oncogene-interfering RNAs can be exogenously applied to manipulate the behaviors of tumors on a genetic level. Instead of silencing MDR-genes mainly to enhance the antitumor efficacy of chemotherapeutics, the combination of tumor-suppressing nucleic acids and chemotherapeutics can arrest tumor growth simultaneously via independent pathways and achieve additive antitumor effects. The timing and sequence of nucleic acid and chemotherapeutic release are more flexible in this case.
Similar to the design principles concerned with loading MDR-silencing RNAs, inorganic nanoparticles are commonly modified with cationic polymers to electrostatically condense tumor-suppressing nucleic acids. For example, a Dox-embedded biodegradable silica nanoparticle (DS-Dox) modified with β-cyclodextrin-poly(glycidyl methacrylate) (CD-PGEA) was developed to condense p53 plasmids.[152] The DS-Dox core was synthesized by a simple one-pot method where Dox, disulfide-bridged silanes and tetraethyl orthosilicate (TEOS) were mixed with ammonia to allow growth of the degradable silica networks (Figure 6a). CD-PGEA was then attached onto DS-Dox by adamantine-mediated host–guest assembly for loading p53 plasmids. The release of Dox by DS-Dox-PGEA-p53 was maximized under acidic pH with dithiothreitol (DTT), indicating the dual-responsiveness of the intracellular release mechanism (Figure 6b). DS-Dox-PGEA-p53 not only displayed strong additive antitumor effects on C6 glioma-bearing mice (Figure 6c) but also potentiates mass production of biodegradable silica nanoparticles in the future. In a similar one-pot synthesis design, a selenium-based inorganic nanoparticle was developed to co-deliver c-myc siRNA and Dox.[153] Dox, vitamin C, and Na2SeO3 were first mixed together to form Dox-embedded selenium-core nanoparticles (SeNP-Dox), which were then mixed with arginine–glycine–aspartic acid peptide (RGD)-modified PAMAM to complex with c-myc siRNA. RGD-SeNP-Dox-siRNA demonstrated a marked ability for penetrating the blood brain barrier and transformed its shape in U251 glioblastoma cells’ lysosomes for precise payload release.
Figure 6.
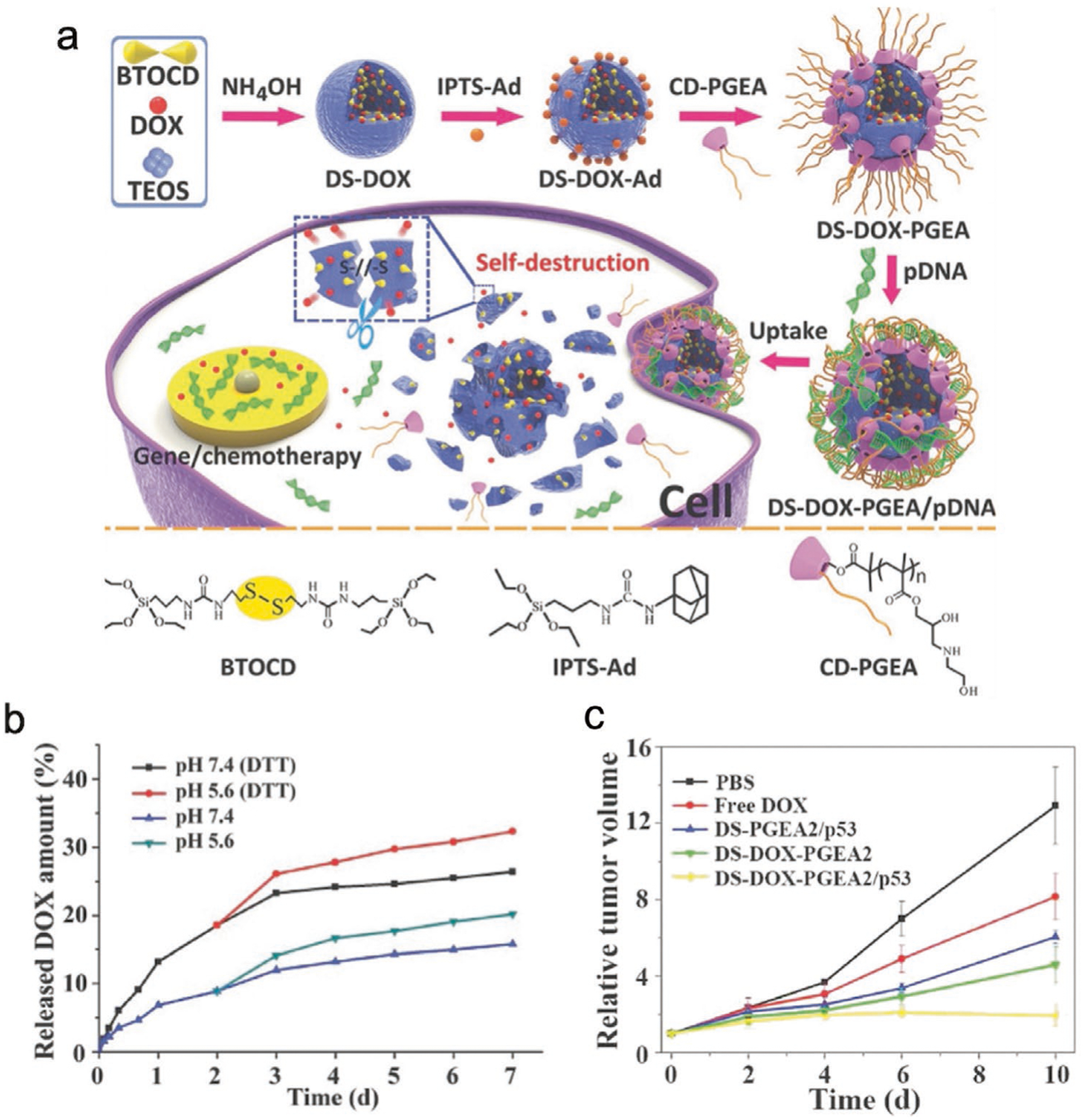
Inorganic nanoparticle-mediated co-delivery of tumor-suppressing nucleic acids and chemotherapeutics. a) Schematic illustration of the preparation of DS-Dox-PGEA-p53 and the resultant stimuli-responsive drug/gene co-delivery process. b) Dox-releasing profiles from DS-Dox-PGEA-p53 with or without DTT in PBS at 37 °C at different pH values. c) Growth curves of glioma tumors. Reproduced with permission.[152] Copyright 2017, Wiley-VCH.
Since VEGF-mediated angiogenesis contributes to tumor survival, maintenance and migration,[154] inorganic nanoparticle platforms have been incorporated into anti-VEGF therapies which can cut off the oxygen and nutrient supplies in tumors. An iron-silica hybrid magnetic MSN (Mag-MSN) modified with PEI and the targeting ligand FA was developed to co-deliver Dox and VEGF shRNA (Mag-MSN(Dox)-PEI(VEGF)-FA).[155] In this study, the specific cellular uptake of Mag-MSN(Dox)-PEI(VEGF)-FA by HeLa cells was achieved by external magnetic field-guided accumulation around HeLa cells and then FA-mediated endocytosis. The FA-magnetic dual targeting markedly increased Mag-MSN(Dox)-PEI(VEGF)-FA’s uptake by HeLa cells in vitro. Moreover, Mag-MSN(Dox)-PEI(VEGF)-FA was able to effectively suppress the secretion of VEGF from HeLa cells and discouraged human umbilical vein endothelial cells’ invasion and microtubule formation which is essential to angiogenesis. In another study, co-delivery of the chemotherapeutic gemcitabine and VEGF siRNA by LCP nanoparticles was able to inhibit the formation of a vascular endothelium in tumors and, in turn, halt the progression of nonsmall-cell lung cancer in vivo.[156] Interestingly, the authors discovered that gemcitabine could induce apoptosis in tumor-associated endothelial cells to enhance the antiangiogenesis effects of VEGF siRNA. Suppressed vasculature formation induced by VEGF siRNA would in turn increase a tumor’s permeability to gemcitabine. The additional synergy between gemcitabine and VEGF siRNA further amplified their additive antitumor effects in vivo.
5. Combinatorial Delivery of Gene–Immuno Therapeutics by Inorganic Nanomaterials
Innate and adaptive immune responses are the main defense mechanisms against aberrant cells including viral/microbial infections as well as cancerous cells. As a survival strategy, tumors actively suppress tumoricidal immune responses via immunoediting.[157] Specifically, cancer cells downregulate surface proteins recognizable to immune cells to evade immunosurveillance. Tumor-associated immune cells also secrete regulatory chemokines and cytokines to further induce immunotolerance and create an immunosuppressive tumor micro-environment. Due to its capability for tumor targeting and long-term memory responses in cases of recurrence, antitumor immunotherapy is becoming an indispensable role in treating cancers.[158]
To harness the power of the immune system and turn it against tumors, immune cell checkpoint inhibitors and immunostimulatory factors have been developed. There are more than 50 immunotherapeutic agents being assessed in clinical trials.[7] In addition, novel technologies such as adoptive cell transfer (ACT) have been developed to modulate tumoricidal immune responses. With ACT, key immune cells can be extracted and isolated from the blood and subjected to ex vivo cell engineering before infusion back into a patient’s circulatory system to elicit antitumor immunity. Specifically, DCs can be exogenously activated and loaded with tumor-specific antigens (Sipuleucel-T DCs), and T cells can be equipped with chimeric antigen receptors for specific tumor recognition (CAR-T cells).[159,160] Both CAR-T cells and Sipuleucel-T DCs have shown promising antitumor effects in clinical trials.[161] Though promising, ex vivo modification of immune cells is costly, laborious and, hence, currently unfit for widespread application.[162] Despite the recent breakthrough of CAR-T cells in treating hematological cancers, ex vivo engineered immune cells’ anticancer immunity can be greatly dampened in solid tumors due to the heterogeneity of antigen expression and the immunosuppressive mechanisms in tumor microenvironments.[163,164] Besides the challenges in ACT implementation, systemic administration of immunogenic substances such as peptide vaccines and antibodies poses the risk of an undesired global immune response and autoimmune diseases.[165,166] Heterogeneity in pathogenesis and incomprehensive understanding of the interaction between immunotherapeutic agents and immune systems also hinder the application of these agents.
Alternatively, nucleic acid-based immunotherapeutics are being developed to indirectly induce antitumor immune responses. Synthetic nucleic acids such as plasmid DNA, mRNA, and siRNA can be precisely engineered on a molecular level and easily produced.[167–169] Precise immunogenic protein expression by plasmid DNA and mRNA can elicit synergistic antitumor immune responses by targeting multiple immuno-pathways.[170–172] siRNA and miRNA can silence immunosuppressive genes and clear the path for tumoricidal immune responses.[157] With inorganic nanomaterials serving as the delivery vehicle, immunomodulatory nucleic acids can be specifically delivered to immune cells or tumor cells for immune cell activation and tumor immunoediting suppression, respectively. The combination of nanotechnology and genetic engineering points to an appealing future for antitumor immunotherapy.
5.1. Combinatorial Delivery of Nucleic Acid-Based Immunotherapeutics to Immune Cells
Nucleic acids with the capability of promoting the production of immunogenic proteins for immune cell activation and attraction or to silence key immunoinhibitory pathways are known as nucleic acid-based immunotherapeutics. Accurate delivery of immunotherapeutics into immune cells is essential for inducing potent antitumor immune responses, especially for nucleic acid-based immunotherapeutics, which require entry into the cell cytoplasm or nucleus to be functional. Research in cancer immunology has identified DCs and T cells as the primary targets for immunotherapeutic delivery.[157,161] DCs can effectively process and present antigens on their surface major histocompatibility complex. Upon migration to lymph nodes, antigen-presenting DCs can activate T cells and B cells for potent adaptive immune responses.[173] DCs can also deactivate T cells via immune checkpoint interactions to maintain self-immunotolerance.[174] As the centerpiece in orchestrating immune responses, DCs have become one of the most-wanted targets in immunotherapy.[175] Various nucleic acid-based immunotherapeutics such as signal transducer and activator of transcription 3 (STAT3) siRNA, programmed death-ligand 1 (PD-L1) siRNA, and miR-155 miRNA have been delivered to DCs to prime their antigen presentation and T cell activation functions.[176–178] These nucleic acid-based immunotherapeutics possess great potential in antitumor treatment as they can direct DC-mediated immunity to arrest tumor growth.
T cells are another important target in immunotherapy as T-cell activity decisively determines the outcomes of immunotherapy. When activated by DCs, immature T cells are transformed into effector T cells, which can in turn recognize tumor cells and induce tumor apoptosis.[179] As the executioner cells of the immune system, effector T cells are commonly incapacitated by immunosuppressive pathways such as the renowned programmed cell death protein 1 (PD1)-PDL1 and cytotoxic T-lymphocyte-associated protein 4 (CTLA4)-CD80/86 inhibitory pathways from tumor cells or immunoregulatory cells recruited in tumor microenvironments.[180] Inhibitory checkpoints expressed by T cells such as CTLA-4 and PD1 need to be blocked in order to effectively engage effector T cells.[157] Immunoregulatory cells such as tumor-associated macrophages (TAMs) and regulatory T cells are also valuable immunotherapeutic targets owing to their immunosuppressive and tumorigenic behaviors.[181,182] AuNPs capable of the specific delivery of therapeutic siRNA to CD4+/CD25+ regulatory T cells have been developed.[183]
In an immune cell activation study, an AuNR co-delivered siRNA, to inhibit the production of the immunosuppressing enzyme indoleamine 2,3-dioxygenase (IDO) and Feline McDonough Sarcoma (FMS)-like tyrosine kinase 3 ligand (Flt3-L), an essential cytokine for DC expansion and development, to DCs.[184,185] Combination of IDO-suppressing siRNA (siIDO) and Flt3-L can augment DCs’ antitumor responses by stimulating DCs’ expansion and limiting the production of T cell-suppressing IDO. AuNRs were modified with mannose (man) for DC-targeting and PEI for complexing siIDO to form man-AuNR-siIDO. Coadministration of man-AuNR-siIDO and Flt3-L to mice with Lewis lung carcinoma (LLC) showed pronounced tumor inhibition in vivo. Assessment of the DCs in the LLC-bearing mice co-treated with man-AuNR-siIDO and Flt3-L showed significant expression increase in major histocompatibility complex II, CD40 and CD80, indicating maturation in DCs. Moreover, T cells in the lymph nodes and spleen from the man-AuNR-siIDO and Flt3-L co-treated LLC-bearing mice exhibited suppressed apoptosis compared to the untreated LLC-bearing mice.[184] Taken together, the combination of man-AuNR-siIDO and Flt3-L stimulated the maturation of DCs and inhibited T cell suppression to achieve synergistic immunotherapy against LLC tumors in mice.
A novel class of CaP-based multifunctional carriers for nucleic acid-based immunotherapeutics delivery to immune cells has been reported in recent years. In particular, An asymmetric LCP nanoparticle has been developed for delivery of nucleic acids, chemo drugs, and peptides.[56] The 40 nm LCP nanoparticle is formed by coprecipitating CaCl2 and Na2HPO4 in a water-in-oil microemulsion. The CaP core is capped with an asymmetric double-layered lipid with the inner leaflet being dioleoylphosphatydic acid and the outer being PEG-modified 1,2-distearoryl-sn-glycero-3-phosphoethanolamine. LCP nanoparticles have been shown to be able to simultaneously entrap potent immunoadjuvant CpG oligonucleotides and tumor-associated antigen Trp2 peptides in its hollow core.[186] When released in DCs, LCP-delivered CpG oligonucleotides and Trp2 proteins stimulated strong immune responses against melanoma in mice. LCP’s capability of loading multiple therapeutic payloads in its hollow core and mediating quick release in endolysosomes makes LCP nanoparticles a reliable multifunctional delivery vehicle.
LCP nanoparticles were also used to co-deliver Trp2-encoding mRNA and PD-L1 siRNA to B16F10 melanoma-bearing mice (Figure 7a).[187] Trp2 is a melanoma-associated antigen. DCs transfected with Trp2 mRNA can present tumor antigens to induce specific T-cell responses against melanoma cells. In an in vivo synergistic immune response experiment, Trp2 mRNA and PD-L1 siRNA were packaged into LCP nanoparticles and subcutaneously injected into melanoma tumor-bearing mice. Twenty days later, LCP (Trp2 mRNA + PD-L1 siRNA) treated mice showed the most significant tumor growth inhibition when compared to untreated, LCP (Trp2 mRNA)-and LCP (PD-L1 siRNA)-treated mice (Figure 7b). Interestingly, the authors also demonstrated that LCP (Trp2 mRNA + PD-L1 siRNA) was able to prime cytotoxic T cells and stall melanoma’s metastasis in lymph nodes throughout the mouse body (Figure 7c,d). This result suggests that LCP (Trp2 mRNA + PD-L1 siRNA) was able to achieve global inhibition on tumor development by engaging cytotoxic immune response to suppress distant metastases.
Figure 7.
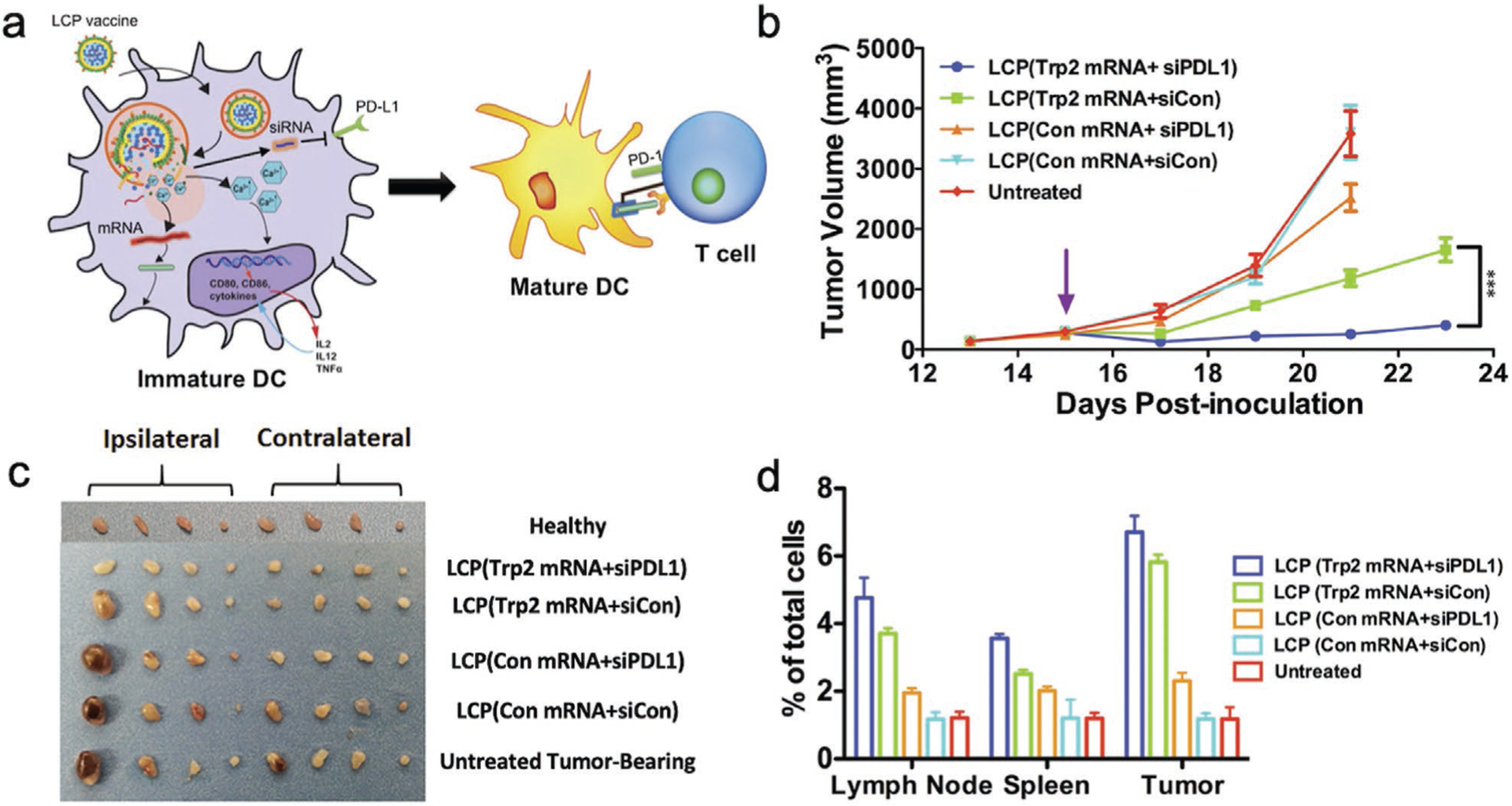
LCP co-delivers tumor antigen Trp2 mRNA and PD-L1 siRNA to treat melanoma. a) Schematic illustration of LCP (Trp2 mRNA + PD-L1 siRNA) enhancing tumor antigen presentation and silencing PD-L1 to activate T cells. b) Tumor growth inhibition study of B16F10 melanoma-bearing mice after vaccination with different LCP nanoparticles after tumors reached 300 mm3. c) Representative lymph nodes harvested from animals at the endpoint of the tumor inhibition study. From left to right: Inguinal, axillary, brachial, and sciatic lymph nodes on the ipsilateral side of the tumor, and inguinal, axillary, brachial, and sciatic lymph nodes on the contralateral side of the tumor. d) Flow cytometry analysis of CD8+ T cells generated in the lymph nodes, spleen, and tumor at the endpoint of the tumor inhibition study. Reproduced with permission.[187] Copyright 2018, Elsevier.
In a related study, MUC1 mRNA and anti-CTLA-4 monoclonal antibody (anti-CTLA-4 mAb) were packaged into LCP nanoparticles and delivered to DCs to treat 4T1 breast cancer.[188] MUC1 is a glycosylated transmembrane mucin and a well-known tumor-associated antigen.[189] CTLA-4 protein receptor is an immune checkpoint expressed by regulatory T cells and can downregulate T-cell responses once CTLA-4 binds to antigen presenting DCs (Figure 8a). Anti-CTLA-4 mAb has been developed to attenuate the CTLA-4 pathway’s inhibitory effects on T cells and was approved by the United States Food and Drug Administration (FDA) to treat melanoma.[190] When combined with anti-CTLA-4 mAb, LCP-MUC1 exhibited more than an eightfold inhibition of tumor growth when compared to placebo treatments with phosphate buffered saline, empty and free MUC1 mRNA treated mice and a more than a threefold tumor growth inhibition when compared to mice treated with only LCP-MUC1 or anti-CTLA-4 mAb (Figure 8b). The targeted delivery of LCP-MUC1 mRNA particles to DCs was able to induce interferon-γ (IFN-γ) and elicit antigen-specific cytotoxic T-cell responses in mice. Analysis of the results on the harvested tumors indicated significantly more tumor-infiltrating T cells were present in the anti-CTLA-4 mAb/LCP-MUC1 co-treated tumors than the other control groups (Figure 8c). This phenomenon suggests that tumor inhibition can be mainly attributed to the successful activation of T cells by anti-CTLA-4 mAb and LCP-MUC1.
Figure 8.

LCP delivers tumor antigen MUC1 mRNA in combination with anti-CTLA-4 mAb to treat melanoma. a) Schematic illustration of LCP (MUC1 mRNA) enhancing tumor antigen presentation and anti-CTLA-4 mAb blocking an immune checkpoint to activate T cell-mediated tumor killing. b) Tumor growth inhibition experiment. c) Flow cytometry analysis of CD8+ T cells in tumors. Adapted with permission.[188] Copyright 2018, Elsevier.
5.2. Combinatorial Delivery of Nucleic Acid-Based Immunotherapeutics to Tumor Cells
Alternative to the immune cell-targeting strategies, nucleic acids can also be delivered to tumor cells to increase their recognizability by the immune system. Tumor cells hijack immunoregulatory pathways by overexpressing complementary checkpoint proteins such as PD-L1 and CD80/86 to shut down immune cells prematurely.[7] Therefore, rendering tumors recognizable to immune cells is just as important as immune cell activation. Tumor behaviors must be reprogrammed so that either intrinsic immune inhibitory pathways are silenced or immune signaling molecules such as proinflammatory cytokines are constitutively expressed before effective antitumor immune responses can be mounted. Delivery of immunotherapeutic nucleic acids to tumor cells has been reported by a series of studies. For example, FA-modified IONPs were loaded with PD-L1 siRNA to specifically silence the immunosuppressive PD-L1 gene in gastric cancer cells to elicit T-cell responses.[191] Zinc oxide nanoparticles were able to deliver immunoadjuvant dsRNA poly I:C to melanoma tumors and induce type I IFN responses.[192] Augmented expression of immunostimulatory interleukin 12-encoding plasmids in mammary adenocarcinoma tumors via IONP-mediated magnetotransfection also demonstrated pronounced antitumor effects.[193]
To simultaneously deliver multiple immunotherapeutic nucleic acids to tumor cells, a pentapeptide conjugated AuNP (Au-CGKRK) was developed for combinatorial delivery of PD-L1 siRNA and STAT3 siRNA to melanoma cells to elicit antitumor immune responses.[194] The cationic CGKRK pentapeptide can not only complex with siRNAs but also serves as a tumor-vasculature targeting ligand for increased tumor uptake. As an important transcription factor responsible for tumor proliferation, STAT3 can upregulate PD-L1 expression on tumor cells upon its activation. PD-L1 facilitates tumor immune evasion by checkpoint interaction with T-cell programmed death receptor 1 (PD-1) and in turn inhibits the activation of cytotoxic T cells. In order to simultaneously suppress tumor growth and induce a cytotoxic T cell-mediated immune response, PD-L1 siRNA and STAT3 siRNA were cocomplexed with the positively charged CGKRK pentapeptide and administered intraperitoneally into mice with melanoma (Figure 9a). Compared to untreated, PD-L1 siRNA or STAT3 siRNA treated and Au-CGKRK only groups, melanoma-bearing mice treated with Au-CGKRK coloaded with PD-L1 siRNA and STAT3 siRNA demonstrated prominent tumor growth inhibition as well as a 75% higher survivability (Figure 9b,c).[194] The in vivo results proved that combinatorial siRNA delivery mediated by the Au-CGKRK platform can be a promising option in achieving synergistic antitumor effects.
Figure 9.

AuNP-CGKRK nanoconjugates co-deliver STAT3 siRNA and PDL1 siRNA to melanoma-tumor bearing mice. a) Schematic illustration of the synthesis of AuNP-CGKRK- STAT3/PDL1 siRNA and subsequent local administration to mice. b) Relative tumor growth inhibition profiles observed in tumor-bearing mice. Arrows underneath the x-axis indicate the time points of the five repeated intraperitoneal injections after tumor inoculation. The inset shows the average weights of the tumors excised from these differently treated mice groups. c) After the last i.p. injection on day 20 post-tumor inoculation, mice were observed for overall survival from day 21. Reproduced with permission.[194] Copyright 2018, American Chemical Society.
5.3. Other Strategies of Nucleic Acid-Based Immunotherapeutics Delivery
The presence of TAMs is considered a hallmark of tumor development because not only do TAMs facilitate tumor aggression but they also protect tumors from cytotoxic immune responses.[195] To modulate the behaviors of both tumor cells and TAMs, an AuNP was engineered to deliver VEGF siRNA to both lung adenocarcinoma cells and inflammatory tumor M2 macrophages in vivo.[196] VEGF is an essential angiogenic factor; overexpression of VEGF in both cancer cells and TAMs facilitates tumor progression, metastasis, as well as the recruitment of more TAMs.[197] By conjugating both VEGF siRNA and M2 peptides onto AuNPs, researchers were able to silence VEGF expression in both lung cancer cells and TAMs to halt their proliferation in lung tumor-bearing mice. Treated mice showed markedly suppressed tumor growth as well as decreased macrophage populations in lung tissue. This study proved that co-treating tumor cells and TAMs is a promising therapeutic strategy.
Some innovative immunotherapeutic strategies involving nucleic acid delivery to a third-party cell type have also been reported. When tumor and immune cells become challenging to target or unresponsive to direct treatments, pointing treatments at third-party cells could be beneficial. Suppressing supporting mechanisms provided by third-party cells can arrest tumor development and migration. For example, overexpression of CXCL12 by hepatic stellate cells in liver tissue facilitates the recruitment of immunosuppressive regulatory T cells.[198] Colorectal cancer cells tend to metastasize along the CXCL12 gradient and invade the liver.[199] CaP nanoparticles were developed to deliver CXCL12-trap protein encoding plasmids to hepatic stellate cells to disrupt the hepatic CXCL12 gradient and in turn halt the metastasis of colorectal cancer.[200] Another strategy involving third-party cells is to apply immunomodulatory nucleic acids first to cell types that are highly responsive to gene therapy to amplify the production of immunotherapeutic products. Minicircle DNA has been reported to be delivered to the readily transfected HEK293T cells for bispecific anti-CD3/anti-Epcam antibody production before the antibodies were applied to cancer cells.[201] By simultaneous binding to CD3 surface markers on T cells and the Epcam surface factors on SKOV3 ovarian cancer cells, the expressed bispecific antibody effectively engaged T cells in killing SKOV3 cells.
6. Inorganic Nanoparticle-Mediated Gene Therapy in Combination with Hyperthermal and Photo Therapy
Hyperthermia is a relatively noninvasive therapeutic approach aimed at tumor removal. Based on the amount of heat transferred into tumors, hyperthermia can be crudely categorized into mild and strong hyperthermia. Strong hyperthermia (over 50 °C) simply destroys cells indiscriminately and is not commonly applied.[202] Compared to strong hyperthermia, mild hyperthermia (40–45 °C) is a more feasible approach to treat tumors because mild hyperthermia can preferentially kill tumor cells while keeping healthy tissue undamaged.[203] Healthy tissue responds to mild hyperthermia by vasodilation and increased blood flow for rapid heat dissipation. It has been reported that healthy tissues, except for those in the central nervous system, can endure hyperthermia at 44 °C for one hour without being damaged.[204] On the other hand, the chaotic tumor vasculatures characterized by tortuous monolayers of endothelia can be easily damaged at elevated temperatures due to hypoxia and acidic microenvironments.[205] As a result of tumor vascular damage, decreased blood flow in turn facilitates heat retention and amplifies the tumoricidal effects caused by hyperthermia. In addition, tumor cell plasma membranes are more fluid and permeable to small-molecule and nanosized drugs when experiencing mild hyperthermia so that therapeutic efficacy of the drugs can be enhanced.[206]
Even though the selective treatment effects on tumors is clear, applications of hyperthermal ablation is not without challenges. First, it is difficult to deliver sufficient heat homogenously into the deep cores of solid tumors.[207] Thus, complete removal of bulk tumor often requires repeated and extended exposure to heat. Second, repeated heat treatments can activate a tumor’s heat shock response mechanisms, which render the tumor thermotolerant and reduces hyperthermia’s antitumor efficacy.[208] Moreover, cytoprotective heat shock responses can also impart tumors with chemoresistance and radioresistance.[209] A phototherapeutic alternative to hyperthermal ablation is photodynamic therapy (PDT) where photosensitizing chemicals react with molecular oxygen upon light activation to produce singlet oxygen to kill tumor cells.[210] Nanomaterials’ applications in conjunction with hyperthermal ablation and PDT have been widely reported due to their special photoelectronic and magnetic properties. With selective tumor targeting, inorganic nanomaterials can focus and amplify hyperthermia in the tumor region when stimulated by external energy.[211,212] In addition to serving as nanoheaters, inorganic nanomaterials can also carry photosensitizers and tumor suppressing nucleic acids such as suicidal genes and thermoresistance-silencing RNAs for combinatorial therapy. Nanomaterial-mediated photo/ablation therapies in combination with gene therapy offers a novel outlook on tumor treatments.
6.1. Magnetically Induced Hyperthermia
Magnetic-to-thermal energy transformation is achieved by subjecting superparamagnetic nanoparticles (SPNs) to an alternating magnetic field (AMF) at a specific frequency. A single-domain SPN with minimal hysteresis can generate heat via magnetization relaxation and rotational friction.[213] In an AMF, the magnetic spin of an SPN is repeatedly aligned to the external field followed by relaxation to its energetically favorable state. The energy required to flip the magnetic spin for alignment is dissipated as heat during relaxation, a phenomenon called Neél relaxation. The flipping of magnetization also exerts torque on the SPN and results in rotational friction to further generate heat, a phenomenon termed Brownian relaxation.[214] Therefore, SPNs specifically uptaken by tumors can generate localized mild hyperthermia when triggered by an AMF. Due to its high magnetic susceptibility and loading capacity, superparamagnetic iron oxide (Fe3O4) nanoparticles (SPIONs) have become the ideal nanoplatform for combinatorial magnetic hyperthermia and gene therapy.
In a magnetic Fe3O4-MSN (M-MSN) hybrid nanomaterials study, spherically shaped M-MSNs (S-M-MSNs) were compared with the rod shaped M-MSNs (R-M-MSNs) for their capability to potentiate suicidal gene magnetotransfection while inducing magnetic hyperthermia in hepatocellular carcinoma.[215] The HSVTK/ganciclovir (GCV) gene therapy system (HSVTK/GCV) was incorporated into the M-MSNs. GCV is a nontoxic prodrug which can kill tumor cells upon phosphorylation by the HSVTK protein.[216] The surface of M-MSNs was modified by PEI polymers (R/S-M-MSN-P) to condense the HSVTK-encoding plasmid, and GCV was directly adsorbed onto the mesoporous MSN surface (R/S-M-MSN-P@GCV@pHSKTK) (Figure 10a). HepG2 cells magnetotransfected with R-M-MSNP@GCV@pHSVTK and subjected to R-M-MSN-induced magnetic hyperthermia displayed the lowest cell viability, indicating that R-M-MSNs could trigger stronger synergistic killing on cancer cells than S-M-MSNs (Figure 10b). For in vivo study, the delivery of the HSVTK plasmid and GCV coloaded R-M-MSNs was guided by an external magnetic field for magnetotransfection. The treated mice were also exposed to an AMF to induce hyperthermia (Figure 10c). The R-M-MSN-P@GCV@pHSVTK treated mice displayed the most prominent tumor growth inhibition (Figure 10d). This study demonstrated that R-M-MSNs are superior to S-M-MSNs in inducing hyperthermia-gene therapy synergistic therapeutic effects against tumors, suggesting that shape could affect the performance of M-MSN materials.
Figure 10.
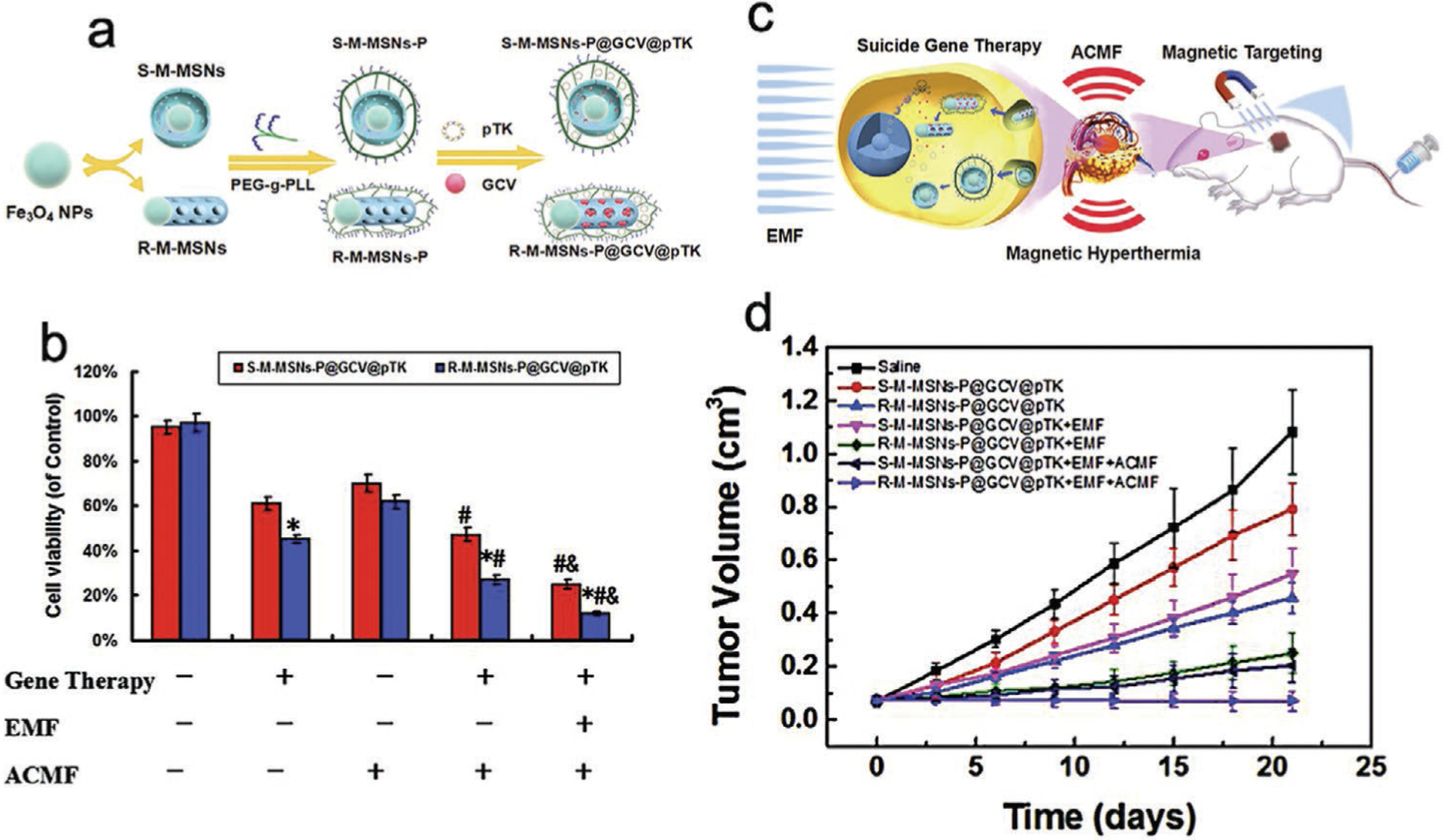
Comparison between spherical and rod-shaped M-MSNs in suicidal gene magnetotransfection and magnetic hyperthermia induction. a) Schematic of M-MSN synthesis. b) Cell viability of HepG2 cells incubated with M-MSNs-P@GCV@pHSVTK (40 μg mL−1) for 48 h with or without EMF exposure and a 20 min exposure to AMF at 24 h postincubation. *P < 0.05 versus the corresponding sphere-like MSN group, #P < 0.05 versus the corresponding gene therapy group, and &P < 0.05 versus the corresponding gene therapy + AMF group. c) Illustration of the animal study procedures. d) Tumor growth curves of HepG2 tumor-bearing mice in each group. Reproduced with permission.[215] Copyright 2018, Elsevier.
Due to their higher magnetic susceptibility than traditional Fe3O4 nanoparticles, zinc-doped iron oxide nanoparticles (ZnFe2O4) can induce magnetic hyperthermia more efficiently at lower dosages.[217] ZnFe2O4 nanoparticles loaded with heat shock response-silencing miRNA has been investigated to treat glioblastoma.[218] Cancer cells’ heat shock responses involve various pathways including upregulation of multiple oncogenes as the downstream effects of the heat shock responses; miRNA is an attractive treatment option because miRNA can silence multiple targeted pathways at once.[219] In this study, lethal-7a (let-7a) miRNA was used to simultaneously target multiple heat shock protein (HSP)-related pathways. The results demonstrated that let-7a miRNA delivered by the ZnFe2O4 nanoparticles could significantly suppress the expression of multiple HSPs responsible for thermotolerance under magnetic hyperthermia. Furthermore, the combination of hyperthermia and miRNA also downregulates the survival and proliferation pathways essential to tumor development to induce strong cytotoxicity in glioblastoma cells.
ZnFe2O4 nanoparticles has also been utilized for stem-cell therapy in conjunction with magnetic hyperthermia to treat ovarian tumors.[220] Different than the previous study, a plasmid (pHSP-TRAIL) encoding for an HSP promoter and proapoptotic TRAIL sequences were applied to mesenchymal stem cells through ZnFe2O4-mediated magnetotransfection. The expression of pHSP-TRAIL is strictly regulated by the activation of HSP genes by heat.[221] Following systemic injection into ovarian tumor-bearing mice, the ZnFe2O4-pHSP-TRAIL transfected mesenchymal stem cells were recruited and migrated to the tumor.[222] Under mild hyperthermia (41–43 °C) induced by an AMF and ZnFe2O4, the selective expression of the cytotoxic TRAIL and heat jointly caused the apoptosis of the ovarian tumors in mice.
6.2. Photothermal Hyperthermia
Photoinduced heat is another approach for hyperthermia. Photothermal hyperthermia can be better controlled spatiotemporally compared to magnetic hyperthermia because the intensity and direction of light can be easily manipulated. Nevertheless, nonspecific tissue chromophores and water-light absorption in the visible and infrared regions, respectively, hugely impair the effectiveness of light penetration.[207] Thus, the NIR region (700–1100 nm) is commonly chosen in phototherapy for deeper tissue penetration. Gold nanomaterials are usually coupled with NIR light to generate hyperthermia owing to their efficient photo-to-heat energy conversion. When interacting with the alternating electromagnetic field of light at a specific wavelength in the NIR region, gold’s free electrons absorb the optical energy to collectively oscillate at maximum amplitude, a phenomenon called surface plasmon resonance. The absorbed photoenergy is then released by the oscillating electrons in the form of heat to raise the local temperature.[223] With gold nanomaterials’ biocompatibility, modifiability, and unique optical properties, AuNPs and AuNRs have become popular choices for delivering therapeutic agents and inducing photothermal ablation therapies to treat cancers.
In a photothermal–gene combinatorial therapy study, AuNRs were utilized to deliver HSP BAG3-silencing siRNA and induce photothermal ablation to human oral squamous carcinoma Cal-27 (Figure 11a).[224] The results showed that AuNRs illuminated by an 810 nm NIR laser did induce photothermal ablation and upregulated the expression of various HSPs and BAG3. When AuNRs were loaded with BAG3 siRNA, the mRNA level of BAG3 was significantly suppressed by BAG3-silencing siRNA (Figure 11b). The co-treatment of BAG3 siRNA and phototherapy induced strong apoptosis in Cal-27 cells in vitro and demonstrated prominent tumor growth inhibition in vivo (Figure 11c). In another AuNR study, pyruvate kinase iso-enzyme type M2 (PKM2), which is a proproliferative protein overexpressed in breast cancer cells is silenced by AuNR-PKM2 siRNA.[225] With NIR heating, AuNR-PKM2 siRNA was able to reduce breast cancer cell viability to 20% in vitro.
Figure 11.
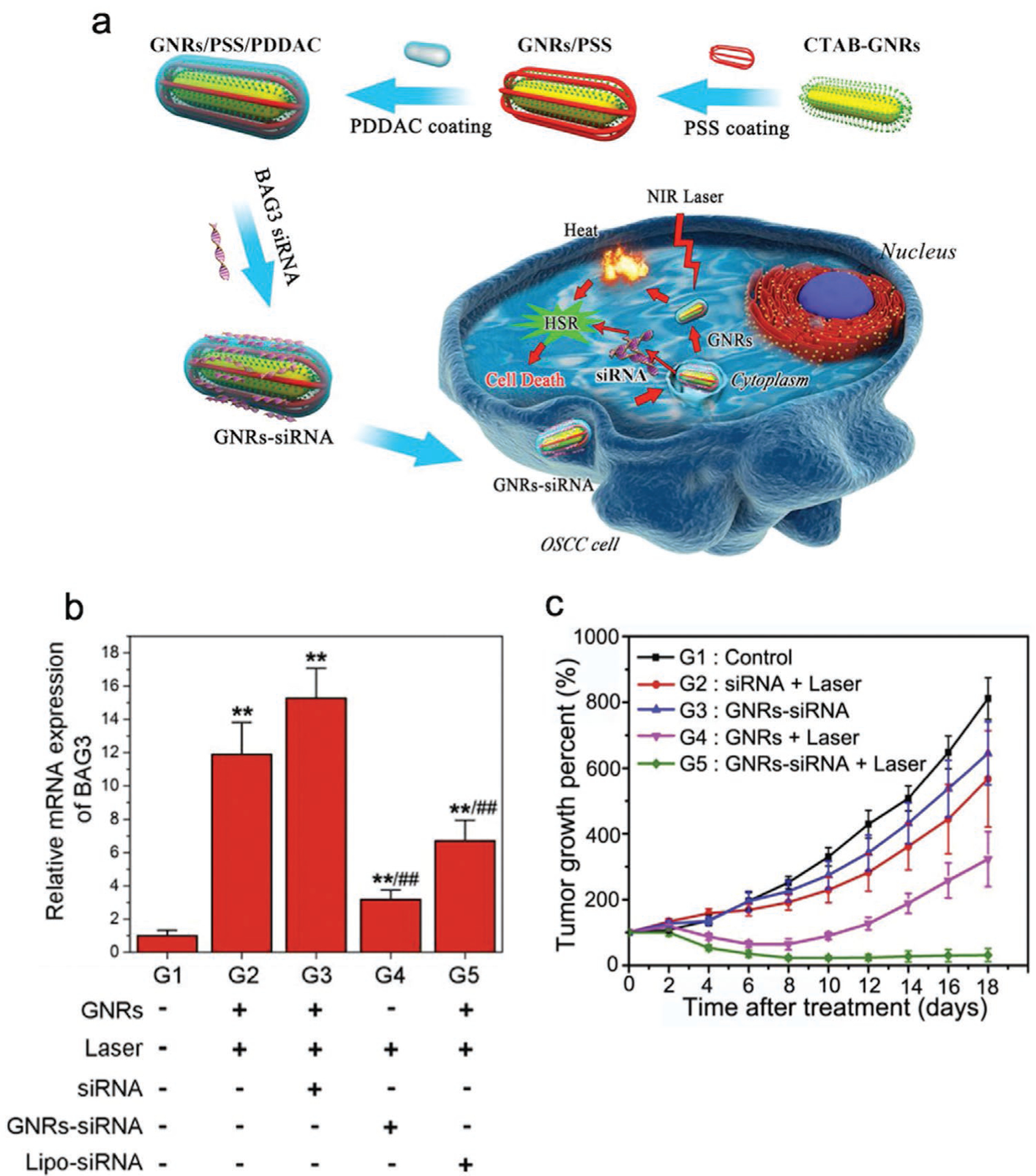
AuNRs deliver BAG3 siRNA to silence the heat shock response and mediate photothermal therapy for cancer killing. a) Schematic illustration of the design of AuNRs–siRNA in the improved photothermal therapy platform. b) Expression of BAG3 mRNA in Cal-27 cells treated with AuNRs–siRNA after laser irradiation; “+” indicates with, and “−” indicates without. c) Average growth percentage of tumors from different groups (G1–G5) in the xenograft model measured every other day after the AuNRs–siRNA and laser irradiation treatment. Reproduced with permission.[224] Copyright 2016, Elsevier.
Different from the traditional approach of condensing nucleic acids into cationic polymers, a unique design of an AuNP–DNA conjugate was reported where siRNA modified with an overhanging DNA linker can be loaded onto AuNPs by the complementary pairing between siRNA’s DNA linker siRNA and the i-motif DNA sequence on the AuNP–DNA conjugate.[226] Interestingly, the pH-responsiveness of the i-motif DNA could initiate clustering of AuNPs in acidic endolysosomes to facilitate endosomal escape and release of Plk1 siRNA. The synergistic effects between AuNP-mediated photothermal ablation and the silencing of the Plk enzyme responsible for genomic stability of cancer cells displayed promising cancer-killing profiles in vitro. Other studies also confirmed the synergistic therapeutic efficacy between the AuNP/NIR-induced photothermal ablation and the AuNP-delivered siRNA such as Bcl2 siRNA and VEGF siRNA.[227,228] The promising results from these studies validate the therapeutic potential of the combination of siRNA and photothermal ablation in antitumor treatments.
6.3. Photodynamic Therapy
Instead of relying on optically induced thermal ablation, PDT involves exogenous administration of photosensitizers, drugs that can react with molecular oxygen to produce reactive oxygen species (ROS) or singlet oxygen and in turn arrest tumor progression upon illumination.[229] Since the illumination in PDT does not result in heat generation and is well-tolerated by tissues, PDT is noninvasive and will not cause cumulative side effects.[230] As an example of photosensitizers, photosan is a chemical that has proved to be effective against head and neck cancers after systemic administration. Photosan can be converted to the photosensitizer protoporphyrin IX (PpIX) intracellularly and then activated by red light (630–640 nm) to produce cytotoxic ROS.
An LCP nanoparticle has been developed for delivery of antitumorigenic siRNAs including hypoxia induced factor and VEGF siRNA to further enhance the therapeutic efficacy of photosan-induced PDT.[231,232] In both studies, photosan and LCP-encapsulated siRNA were separately administered into human oral squamous tumor-bearing mice and showed significantly stronger synergistic antitumor effects than the mice only treated by either PDT or LCP-siRNA. Photosensitizers and siRNA have also been coloaded into a single nanoplat-form. Specifically, a NaGdF4:Yb,Er upconversion nanoparticle (UCNP) was developed to co-deliver the hydrophilic photosensitizer chorin e6 (ce6) and Plk1 siRNA (Figure 12a).[233] The results indicate that UCNPs-ce6-siPlk1 was able to effectively reduce the Plk2 mRNA level in HeLa cells and excite ce6 to produce cytotoxic singlet oxygen when subjected to NIR irradiation (980 nm) in vitro (Figure 12b,c). Ce6 and Plk2 siRNA co-delivered by UCNPs synergistically reduced the HeLa cell viability down to 20% (Figure 12d). The unique photoluminescence upconverting property grants UCNPs compatibility with deep-penetrating NIR irradiation so that PDT can be effective even for deep-seated tumors.
Figure 12.

UCNPs loaded with ce6 and Plk1 siRNA for PDT and gene combination therapy. a) Schematic showing the functionalization of UCNPs, coloading with ce6 and siRNA, and then the combined PDT and gene therapy delivered by UCNPs. b) The expression levels of Plk1 mRNA determined by quantitative real-time polymerase chain reaction. Plk1 mRNA levels were expressed as a relative index normalized against β-actin. c) The generation of singlet oxygen by measuring the fluorescence intensity changes of singlet oxygen sensor green (SOSG) dye reagent at 525 nm as a function of 980 nm light radiation time (0.8 W cm−2). d) In vitro cytotoxicity effects induced by PDT, gene therapy, or combination therapy. The combined photodynamic and gene therapy offered a significantly higher cancer cell killing effect compared to monotherapy by either gene therapy or PDT alone. Reproduced with permission.[233] Copyright 2014, Royal Society of Chemistry.
7. Inorganic Nanoparticle-Mediated Gene Therapy in Combination with Radiotherapy
Radiotherapy is an extensively applied clinical anticancer treatment as more than 50% of cancer patients are treated with radiotherapy.[234,235] Radiotherapy can be applied preoperatively to shrink tumor size to facilitate subsequent surgical removal, intraoperatively to assist tumor removal and postoperatively to reduce cancer recurrence.[236–238] Radiotherapy has also been used as a palliative approach to reduce pain from distant tumor metastases or to alleviate symptoms from unresectable tumors near critical organs and tissues.[238–240] The strong ionizing radiation from radiotherapy can penetrate deeply into tumor mass and inflicts significant damage on tumor tissue.[241] In addition, ionization of water generates ROS such as superoxide radicals, hydrogen radicals and hydrogen peroxide which can exert oxidative stress on surrounding tissues.[242] Radiotherapy-induced cytotoxicity is mainly characterized by DNA lesions including single strand breaks, double strand breaks and nucleic base modifications.[243] Cancer cells with DNA damage can halt their cell cycles at the G2/M phase while attempting DNA self-repair.[235] Irreversible apoptosis is initiated if DNA self-repair is unsuccessful.[244]
Radiotherapy also has several critical drawbacks. First, without depth restriction, ionizing radiation can damage surrounding healthy tissue and cause side effects.[245] Second, the innate cellular ability for excessive ROS quenching owing to the substantial presence of intracellular GSH can greatly impair the therapeutic efficacy of radiotherapy.[246] Last but not least, hypoxia may activate DNA repair mechanisms and render tumors radioresistant.[247] Higher doses of radiotherapy may be needed to overcome a tumor’s ROS elimination ability and radioresistance, but such dosages may also inflict harm to healthy tissue. Therefore, it is imperative to radiosensitize tumors and avoid radiotherapy overdosing.
Numerous radiosensitization strategies have been proposed over the years. For example, nanomaterials with high atomic (Z) numbers can enhance radiation energy deposition, catalyze the formation of cytotoxic compounds, and arrest cell cycles to increase a tumor’s radiosensitivity.[241] Genetic approaches can be applied to inhibit radioresistance-associated pathways, especially those related to hypoxia and DNA repair.[248,249] Nano-material-involving radiotherapy and gene therapy are rapidly advancing to help overcome obstacles in tumor treatment.
7.1. Nanomaterial-Mediated Delivery of Radioresistance-Silencing Nucleic Acids
Nucleic acids such as interfering RNAs and DNAs are potential radiosensitizers owing to their ability to modify radioresistance-associated genes. Thus, IONPs were designed to deliver apurinic endonuclease 1 (Ape1) siRNA to sensitize pediatric brain tumors and glioblastoma to radiotherapy.[250,251] Ape1 enzymes assist in repairing radiation-damaged DNA bases by cleaving the abasic sites for ligation,[252] and it has been proven to be a valuable target in oncoradiology as downregulation of Ape1 expression can increase the radiosensitivity of tumors.[253] IONPs were modified with biocompatible chitosan and PEG grafted with cationic PEI for condensation and delivery of Ape1 siRNA. Decreased Ape1 mRNA as well as suppressed abasic endonuclease activity was observed in IONP-Ape1 siRNA treated brain cancer cells in vitro. Importantly, IONP-Ape1 treatment significantly boosted tumor-associated toxicity induced by γ-ray irradiation and extended the survival period of glioblastoma tumor-bearing mice. In a similar study, human adenovirus type 5 early region 1A encoding plasmids (pE1A) were delivered by dextran-modified IONPs (SDION) to HeLa cervical tumors.[254] pE1A genes can increase cervical tumors’ radiosensitivity by upregulating the p53 antioncogene and downregulating the HER2/neu oncogene. The data showed that the tumor volume of the SDION-pE1A and X-ray radiation co-treated mice was only 50% of that in the X-ray radiation-treated mice. This study further confirms the synergy between radiotherapy and radioresistance-silencing nucleic acids.
Since nucleic acids applied in genetic approaches are also subjected to the radiative damage, premature irradiation could render radiosensitizing gene therapy ineffective. Radiotherapy should only be administered after nucleic acids have successfully radiosensitized tumors. Repeated nucleic acid administration might also be necessary after each irradiation to keep up tumors’ radiosensitivity. Therefore, it is imperative to understand the nanocarrier’s pharmacokinetic and payload releasing profile as well as the performance timescale of the radiosensitizing nucleic acids to achieve optimal treatment efficacy.
7.2. Joint Radiosensitization by Nanomaterials and Nucleic Acids
Instead of only serving as the carrier for radiosensitizing nucleic acids, certain nanomaterials possess unique photoelectric properties which can be harnessed to enhance radiotherapy’s efficacy jointly with nucleic acids. Nanomaterials composed of elements with high Z numbers can absorb radiative energy more efficiently than soft tissue due to their larger absorption coefficients associated with ionizing radiation. The absorbed radiation initiates energy cascades from high Z nanomaterials to surrounding tissues mainly through Compton scattering and the photoelectric effect.[255,256] When high Z nanomaterials are subjected to ionizing radiation, the incident photons partially transfer their energy to the weakly bound electrons upon collision and result in scattered lower-energy photons and ejected electrons, a phenomenon called Compton scattering. In the photoelectric effect, voids are created when inner-shell electron are ejected by radiation and then filled by outer-shell electrons. The energy released during the filling process releases a secondary electron (i.e., an Auger electron) along with low energy photons. The emitted electrons and scattered photons resulting from Compton scattering and the photoelectric effect effectively increase the local radiation and in turn damage tissues either by direct interaction with DNA or generation of ROS.[257] The representative high Z element in nanomaterials is gold. Besides their photoelectric properties, gold nanomaterials also exhibit catalytic ability in forming ROS as well as facilitating tumor cell cycle arrest.[258,259]
Recently, a dual-radiosensitization AuNP-based nanosystem was developed for head and neck cancer treatment.[260] STAT3 is a key transcription factor for the survival and development associated with malignant tumors; it also actively regulates the expression of hypoxia-inducible factor 1 (HIF-1) and participates in tumor adaptation to hypoxia.[261] Inhibition of STAT3 function has been reported to enhance radiosensitivity of head and neck cancers.[262,263] The anti-STAT3 decoy oligonucleotide (STAT3d ODN) can suppress STAT3 activity by selectively binding to the STAT3 protein. AuNPs were employed as both the carrier for STAT3d ODN and as high-Z radiosensitizers. Results demonstrated that STAT3d ODN and AuNPs collectively enhanced radiation-induced DNA damage on highly radioresistant cancer cells.
In another study, the combination of Gd-containing polyoxometalate-conjugated chitosan nanospheres (GdW10-CS) and HIF-1α siRNA were reported as another dual-radiosensitization nanosystem.[264] The GdW10-CS nanosystem features a multifaceted radiosensitization mechanism. GdW10-CS contains high Z elements for external radiative energy deposition and ROS generation. GdW10-CS can also deactivate GSH via W6+-triggered oxidation and allows for ROS accumulation (Figure 13a). In vitro results confirmed that the ROS accumulation in cancer cells treated by GdW10-CS and X-rays was significantly improved (Figure 13b). Additionally, the HIF-1α siRNA carried by GdW10-CS can suppress hypoxia-associated radioresistance and induce caspase-related apoptosis.[265] The in vivo results substantiated GdW10-CS’s radiosensitizing effectiveness as GdW10-CS-siRNA-treated and X-ray-irradiated mice showed significantly smaller tumor volumes than all other control groups (Figure 13c). Obvious knockdown of HIF-1α expression and DNA double strand breaks indicated by γ-H2AX were also observed in the tumor tissue harvested from the GdW10-CS-siRNA-treated and X-ray-irradiated mice (Figure 13d,e). These results collectively proved that the combination of GdW10-CS and HIF-1α siRNA can effectively sensitize tumors to radiotherapy and arrest tumor growth in vivo.
Figure 13.

Dual-radiosensitization of hypoxic tumors by GdW10CS nanoconjugates to enhance tumor killing. a) Mechanism diagram of GdW10CS-mediated ROS accumulation. b) ROS generation of BEL-7402 cells with different treatments. DCFH is a molecular probe for ROS visualization. The scale bar is 100 μm. c) Relative tumor volume curves of different groups of mice after different treatments. d) Western blot analysis of HIF-1α levels in tumors after different treatments. e) Western blot analysis of γ-H2AX levels in tumors after different treatments. Reproduced with permission.[264] Copyright 2017, American Chemical Society.
8. Inorganic Nanomaterial-Mediated Gene Therapy in Trimodal Therapy
Resistance against different antitumor treatment modalities including MDR, immune-evasion, heat shock responses, and radioresistance evidences the well-rounded defense mechanisms of cancer tissue as well as the level of complexity within tumor microenvironments. Multiple treatment modalities working in concert is necessary to overcome tumor resistance and achieve desired antitumor therapeutic efficacy. Advancement in medicinal technologies and nanotechnology has granted the emergence of multifunctional nanomaterials so that trimodal therapy can be realized by a single system. Given the conjugation compatibility and loading capacity of nanomaterials, current nanotherapeutic systems can usually carry two or three different drugs at required dosage amounts. Additional treatment modalities can be incorporated if nanomaterials possess intrinsic physical properties such as gold’s surface plasmonic resonance or iron oxide’s superparamagnetism which can be harnessed for therapeutic purposes. Under these design guidelines, the integration of chemo–gene–photothermal and chemo–gene–photodynamic trimodal therapies in a single nanoplatform have been successful.
8.1. Photothermic Nanomaterial-Mediated Delivery of Gene–Chemo Therapeutic Agents
Nanosystems combining chemotherapy and PTT have been recently extensively investigated. These systems generate photo-induced heat that can not only directly kill tumor cells but also serve as a stimulus for chemotherapeutic drug release.[266–268] With therapeutic nucleic acids introduced into these chemo–photothermal nanosystems, the tumoricidal effect can be further enhanced as a result of synergistic trimodal therapy. In a chemo–gene–photothermal nanotherapeutic system, chemotherapeutic drugs and therapeutic nucleic acids are typically carried by nanomaterials possessing photothermic properties. AuNRs have been extensively applied as both drug carriers and as photothermic nanoheaters in a series of trimodal therapy studies. For example, a rattle-structure rough nanocapsule was recently constructed for chemo–gene–photothermic trimodal therapy.[269] The nanocapsule was synthesized by the “ship in a bottle” approach. Mesoporous hollow silica nanoparticles (HSNs) with rough surfaces were first formed. Then, in situ seed-mediated growth of AuNRs occurred in the cavity of HSNs so that AuNRs were encapsulated by the HSN shells (Figure 14a). Finally, the surfaces of HSNs were modified with CD-PGEA to form Au@HSN-PGEA (AHP). The chemotherapeutic drug sorafenib (SF) was loaded into the HSN cavities and a p53 antioncogene plasmid was condensed into PGEA to form (SAHP/p53). The results showed that AHPs could effectively dissipate heat upon NIR irradiation which can be precisely controlled to trigger the temporal release of SF (Figure 14b,c). When applied in vivo, the strong photothermal effect of SAHP/p53 in combination with SF and p53 gene therapy induced the strongest tumor growth inhibition (Figure 14d). The trimodal therapy orchestrated by the AuNR-HSN nanoplatform displayed significantly stronger tumor growth inhibition effects than that caused by any combination of two of the therapies in vivo.
Figure 14.

Chemo–gene–photothermal trimodal antitumor therapy mediated by gold/silica hybrid nanocapsules. a) Schematic illustration of the preparation of SAHP and the resultant responsive drug/gene co-delivery process. b) Temperature elevation of water and AHP solutions upon irradiation at 808 nm (2 W cm−2). c) Cumulative release profile of SF from SAHPs with NIR irradiation at predetermined intervals. d) Time-dependent growth curves, average tumor weights, and representative photographs of excised tumors of mice after different treatments. Reproduced with permission.[269] Copyright 2018, American Chemical Society.
Other combinations of gene and chemotherapeutic agents that have been reported to be co-delivered by photothermic AuNRs to achieve trimodal anticancer therapy include Dox and MDR associated protein siRNA as well as Dox and immunostimulatory CpG oligonucleotides.[270,271] Notably, these AuNR-based nanotherapeutic systems all possess NIR-triggered chemo-drug release mechanism as heat provides additional energy for noncovalently linked chemo-drugs to detach from carriers. The spatiotemporal control over thermal ablation and drug release make AuNRs a centerpiece in the field of trimodal therapy. Besides AuNRs, other inorganic nanomaterials were also applied in trimodal therapy. For example, photothermic Ag2S quantum dots were grafted with MSN shells to adsorb Dox.[272] Avidin was then attached onto MSN shells to block Dox leakage and to load desthiobiotin-modified survivin antisense oligonucleotides. The synergistic effect of the Ag2S-induced photothermia, silencing of antiapoptotic survivin genes and chemotherapeutic Dox was confirmed on cervical tumor-bearing mice. Some organic materials such as polydopamine also possess strong NIR-induced photothermic properties.[273] Polydopamine thin film coated MSNs loaded with Dox and P-gp siRNA have shown promising trimodal antitumor efficacy in MCF7-ADR tumor-bearing mice.[274] When incorporated in a nanosystem, photothermic coatings similar to polydopamine can offer additional hyperthermal treatment modality to boost antitumor efficacy.
8.2. Triple Loading of Chemotherapeutics, Interfering RNAs, and Photosensitizers
For the chemo–gene–photodynamic therapy modality, three different therapeutic agents, each of which is responsible for one type of therapy, are co-loaded onto a nanocarrier. An M-MSN nanoplatform was constructed for simultaneous loading of Dox, P-gp shRNA, and photosensitizer ce6.[275] In this design, ce6 and Dox were entrapped in MSN pores followed by layer-by-layer deposition of alginate/chitosan coatings for shRNA condensation. The alginate/chitosan coating also featured pH-triggered release of ce6 and Dox in acidic environments. The trimodal therapies and controlled drug release mediated by the M-MSN-Dox-ce6-shRNA nanocomposites greatly facilitated tumor killing in mice. Polysilsesquioxane nanoparticles (PSilQ) were also designed to co-deliver the anticancer drug curcumin, PpIX and siRNA to MDA-MB-231-GFP breast cancer cells.[276] PpIX and curcumin exhibited promising synergistic anticancer efficacy in MDA-MB-231. Successful knockdown of green fluorescent protein expression indicates that PSilQ is also a reliable platform for gene therapy. Fe3O4 nanoparticles loaded with Dox, β-catenin siRNA, and anatase TiO2 as photosensitizers have also been reported to mediate trimodal therapy.[277] From these trimodal therapy studies, the effects of cancer cell killing in vitro and tumor growth inhibition in vivo caused by the chemo–gene–photodynamic combination are significantly more pronounced than that caused by any monotherapy or bimodal combinations, proving the clear benefits of adding the third type of therapy for treating tumors.
9. Summary and Outlook
The interfacing of inorganic nanomaterials and gene therapy in combination with other treatment modalities has been proven to be an effective antitumor strategy in research. To date, advanced synthesis techniques can fine tune the size, shape, and surface chemistry of inorganic nanomaterials. Polymeric surface coatings can be conjugated onto inorganic nanomaterials to improve biocompatibility, passage across biological barriers, and loading capacity for therapeutic payloads. The molecular compositions of polymeric coatings can be tailored to load different types of payloads and enable proper payload release in the presence of environmental cues. By co-delivering therapeutic nucleic acids and chemotherapeutic or immunotherapeutic agents, inorganic nanomaterial-based carriers can mediate combinatorial gene–chemotherapy or gene–immunotherapy, respectively. In addition, inorganic nanomaterials can rely on their optoelectronic or magnetic properties to harness external energy for added hyperthermal therapy and radiotherapy. When a multitude of antitumor therapies are integrated into a single inorganic nanosystem and applied to treat tumors, the besiegement from multiple pathways can lead to higher successful rates of tumor eradication. As many studies have shown, the in vivo tumor inhibition effects may be significantly boosted when tumor-bearing animals are treated with combinatorial gene therapies mediated by inorganic nanomaterials. Table 1 summarizes the previously reviewed inorganic nanocarriers’ core material types along with the therapeutic payloads they carried and the therapeutic stimuli they were subjected to. Inorganic nanocarriers’ target cancer and tumor types are also provided in the table.
Table 1.
A summary table of types of the nanomaterial and the therapeutic payloads/stimuli incorporated in the previously reviewed inorganic nanocarriers along with their target cancers/tumors. Abbreviation: ODN (oligonucleotide); AMF (alternative magnetic field); NIR (near-infrared).
| Main features of inorganic core material | Core nanomaterial | Cancer cell/tumor type | Therapeutic payload/stimulus | Section | Refs. |
|---|---|---|---|---|---|
| Silica | MSN | MCF7 | Bcl2 siRNA and DOX | 4.1 | [134] |
|
MSN | MCF7 | P-gp siRNA and DOX | 4.1 | [137] |
| MSN | HeLa | Bcl2 siRNA and DOX | 4.1 | [138] | |
| MSN | MCF7 | P-gp siRNA and DOX | 4.1 | [143] | |
| Silica NP | C6 glioma | P53 plasmid and DOX | 4.2 | [152] | |
| MSN-Ag2S | A549 and HeLa | Dox, survivin antisense OGN (db-DNA), NIR | 8.1 | [272] | |
| MSN | MCF7/ADR | Dox, P-gp siRNA, NIR | 8.1 | [274] | |
| Gold | AuNP | MCF7 and MDA-MB-231 stem cell | miR-21i and DOX | 4.1 | [146] |
|
AuNP | CD4+/25+ regulatory T cells | eGFP siRNA | 5.1 | [183] |
| AuNR | Lewis lung carcinoma (LLC) | IDO siRNA, Flt3-L | 5.1 | [184] | |
| AuNP | Melanoma | PDL1 siRNA, STAT3 siRNA | 5.2 | [194] | |
| AuNP | A549-C8 human lung adenocarcinoma | M2 peptide, VEGF siRNA | 5.3 | [196] | |
| AuNR | Cal-27 human oral squamous carcinoma | BAG3 siRNA and NIR | 6.2 | [224] | |
| AuNR | MDA-MB-231 and SUM-159 breast cancers | PKM2 siRNA and NIR | 6.2 | [225] | |
| AuNP | NIH-3T3 | Plk1 siRNA and NIR | 6.2 | [226] | |
| AuNP | PC3 prostate cancer | VEGF siRNA and NIR | 6.2 | [227] | |
| AuNP–graphene | HL-60 (human promyelocytic leukemia) | Bcl2 siRNA and NIR | 6.2 | [228] | |
| AuNP | Human epithelial carcinoma cell line A431 | STAT3 decoy ODN and linear accelerator radiation | 7.2 | [260] | |
| AuNR | PC3 tumor | Dox, MRP1 siRNA, NIR | 8.1 | [270] | |
| AuNR | Hepatocellular carcinoma cells (H22) | Dox, CpG, and NIR | 8.1 | [271] | |
| Iron oxide | Fe3O4–Mn3O4 | MCF7 | Bcl2 siRNA and DOX | 4.1 | [145] |
|
IONP | SGC-7901 gastric cancer | PD-L1 siRNA | 5.2 | [191] |
| IONP | Mammary adenocarcinoma | IL12 plasmid | 5.2 | [193] | |
| IONP | SKOV3 ovarian cancer | Anti-CD3/anti-Epcam MC.DNA | 5.3 | [201] | |
| ZnFe2O4 NP | U87 glioblastoma | Lethal-7a (let-7a) miRNA and AMF | 6.1 | [218] | |
| IONP | Glioblastoma | Ape1 siRNA and Cs-g-ray | 7.1 | [250] | |
| IONP | Pediatric brain cancer UW228 and RES196 | Ape1 siRNA and Cs-g-ray | 7.1 | [251] | |
| IONP | HeLa cervical cancer cells | E1A plasmid and X-ray | 7.1 | [254] | |
| Fe3O4–TiO2 | HeLa | Dox, beta-catenin siRNA, TiO2 NIR | 8.2 | [277] | |
| Calcium phosphate | LCP | PC-3 prostate cancer | GRP78 siRNA and docetaxel | 4.1 | [144] |
|
LCP | Nonsmall-cell lung cancer (NSCLC) | VEGF siRNA and gemacitabine | 4.2 | [156] |
| LCP | Melanoma | CpG, Trp2 peptide | 5.1 | [186] | |
| LCP | Melanoma | Trp2 mRNA, PDL1 siRNA | 5.1 | [187] | |
| LCP | Melanoma | MUC1 mRNA and CTLA4 mAb | 5.1 | [188] | |
| LCP | Colorectal cancer | CXCL12-trap plasmid | 5.3 | [200] | |
| LCP | Human oral squamous cancers | HIFa siRNA and photosan | 6.3 | [231] | |
| LCP | Head and neck squamous cell carcinoma | VEGF siRNA and photosan | 6.3 | [232] | |
| Inorganic composites | Fe3O4-MSN | HeLa | VEGF shRNA and DOX | 4.2 | [155] |
|
Fe3O4-MSN | Heptocellular carcinoma | HSVTK/GCV plasmid and AMF | 6.1 | [215] |
| Si–ZnFe2O4 NP | Ovarian cancer | HSP-TRAIL plasmid and AMF | 6.1 | [220] | |
| AuNR-MSN | HepG2 | Sorafenib, P53 plasmid, NIR | 8.1 | [269] | |
| Fe3O4–Au-MSN | MCF7 and EMT6 | Dox, P-gp shRNA, and chlorin e6 | 8.2 | [275] | |
| Biocompatible; easy surface | Selenium NP | U251 glioblastoma | c-myc siRNA and DOX | 4.2 | [153] |
| Direct cargo binding; cancer-specific toxicity | ZnO NP | Melanoma | TLR3 agonist dsRNA, Poly I:C | 5.2 | [192] |
| Convert the deep-penetrating NIR light to visible light | NaGdF4:Yb,Er UCNP | HeLa | PIk1 siRNA and chlorin e6 | 6.3 | [233] |
| Radiative energy deposition; enhanced ROS accumulation | GdW10 nanosphere | BEL-7402 tumor | HIF1a siRNA and X-ray | 7.2 | [264] |
| More degradable and higher drug loading than silica nanomaterials | Polysilsesquioxane NP | MDA-MB-231 | Curcumin, GFP siRNA, PpIX | 8.2 | [276] |
Nevertheless, the clinical translation of inorganic nanomaterial-based therapies is still in its infancy with rare clearances from clinical trials. The practical application of these nanomedicines is hindered largely by limitations on large-scale manufacturing and animal models. Mass production of nanomaterials with consistent quality and properties are prerequisites for clinical application. It is not uncommon for nanomaterials synthesized by the “bottom-up” approach in a well-controlled laboratory setting to exhibit batch-to-batch variations in their physicochemical properties such as heterogeneous sizes and shapes. Even though such inconsistencies could be negligible in small-scale syntheses and might not affect experimental outcomes, large-scale manufacturing efforts could potentially magnify the small inconsistencies and result in failed batches due to altered reaction environments and uneven mixing in large volumes. Microfluidic technologies with high throughput capabilities are currently being pursued to realize mass production of consistent inorganic nanomaterials.[278,279] Since inorganic nanomaterials often require surface functionalization to be biocompatible, the postsynthesis bioconjugation of mass-produced nanomaterials poses another obstacle with respect to the large-scale production of nanomedicines, and yet research efforts dedicated to solving this issue are scant.
Another challenge hindering the clinical translation of nanomedicines lies in the limited understanding of the interactions between nanomaterials and chosen animal models. The fate of nanomedicines in the human body has not been fully elucidated. It is well-established that nanomaterials are subject to protein corona formation, sequestration by the MPS and disintegration in acidic biological compartments.[280] However, information on the exact mechanisms of nanomaterials degradation and the metabolic routes of the dissolved nanomaterials is still lacking. More importantly, current understanding of nanomedicine pharmacokinetics is mostly based on results from in vitro investigation and small animal models. The human body is significantly larger and more complex than small animals such as mice and rats. Contrary to small animal studies, nanomedicines for clinical application would have to be applied at higher dosages, endure longer blood-circulation times, penetrate thicker biological barriers, and be equipped to recognize human-specific biomarkers to be possibly effective in human bodies. Moreover, experimental small animals are usually artificially tempered so that they are immunodeficient or genetically modified. Tumors exogenously inoculated in these small animals often fail to represent the naturally occurring pheno-types of tumors. Thus, the seemingly promising in vivo efficacy of nanomedicines tested in small animal models may not be applicable to humans. Nonhuman primates can be used as alternative experimental animals as they are closer to humans in terms of both size and genetics than small animals. But the concerns of high maintenance and animal welfare often place strict limitations on studies using primates. Findings from studies performed in tumor-bearing animal models closely resembling human tumorigenesis conditions remains as an essential yet daunting task.
In addition to addressing the hurdles of inconsistent mass-production syntheses and imperfect animal models, further technological advancements in many other aspects such as novel nucleic acid designs, artificial intelligence (AI) for nanomaterials screening and better characterization methods are also needed to push forward inorganic nanomaterial-mediated combinatorial gene therapy. Plasmid DNA sequences are being redesigned to incorporate nuclear-localization signals and tissue-specific promoters to facilitate complexation and improve transfection efficiency.[281] Sequences of siRNAs and miRNAs can also be engineered to improve chemical stability, facilitate interaction with RISCs, and minimize off-target silencing.[26] These newly designed nucleic acids could significantly boost the antitumor efficacy of gene therapy.
As a rapidly developing technology, AI has shown its powerful utilities in multiple disciplines including the field of nanomedicine. Although combinatorial therapies carried by a multifunctional nanocarrier seems to be a promising approach as the next generation antitumor treatment, the workload in developing such a system is daunting. The more complicated the system, the larger the parameter matrix and testing spaces. Not only the amount ratio but also the compatibility, synergy, and sequence of release between different components all need to be considered. Since testing of all parameter combinations in the laboratory is impractical, pre-experimental modeling and screening are becoming indispensable tools for designing therapeutic nanosystems. In recent years, AI has been applied to construct pharmacokinetic models, screen for the best combination/ratios of drugs in nanomedicines, and optimize the administration timing for maximum tumor therapeutic effects.[282,283] With parameters such as drug selection, dosage information, and nanomaterials’ properties as discretized inputs, AI can output the optimal therapy scenarios for subsequent experimental designs.[284] There is no doubt that rational application of AI will greatly reduce the redundant screening workload when developing nanosystems for combinatorial therapies.
Following the modeling and synthesis of therapeutic nanosystems, it is essential to assess nanosystems’ performance and validate their therapeutic efficacy in biological contexts. However, current analytical methods rarely accurately characterize the bio–nano interface in living organisms due to nanosystems’ complex hierarchical structures, unique physicochemical properties and highly dynamic biomolecular interactions.[285] It is not uncommon that nanosystems must be removed from biological environments, erasing their bioequilibrium signatures and prepared in a specific manner before they can be properly characterized.[286] Therefore, in situ analytical methodologies which can monitor bio–nano interfaces in real time are necessary to improve our understanding of nanosystems’ contextual behaviors in biological environments. Recent studies have reported the development of several in situ analytical techniques using nuclear magnetic resonance, fluorescence and inductively coupled plasma mass spectrometry for investigating nanosystems’ pharmacokinetics and biomolecular interactions in living organisms.[287–289] However, the resolution and sensitivity of these techniques need to be improved and their procedures simplified before they can be readily applied in clinical settings.
Last but not least, the regulatory process managed by the FDA imposes a final barrier to the clinical translation and commercialization of nanomedicines. For a new medicine to be commercialized, the review process can cost $1 billion and take more than a decade to complete.[290] As a derivative of the incipient nanotechnology, nanomedicine is still a relatively elusive concept to both researchers and regulators. The definition of nanomaterials is still in debate as the field is witnessing an unprecedented advent of novel nanomaterials along with their increasingly complicated nanoproperties. The medicinal application of nanomaterials also adds another level of uncertainty to their behaviors as the interaction between foreign materials and physiological environments is largely unknown at this point. Furthermore, the majority of existing nanomedicine guidelines are stipulated by regulator officials instead of the academics who are involved in frontier research.[291] As a result, the current FDA regulatory guidelines of nanomedicine remains in an immature state as most of them are either incomprehensive in representing nanomaterial properties or directly borrowed from the existing guidelines pertaining to bulk materials.[292] To improve current nanomedicine regulatory guidelines, extensive details of characterization and categorization of nanomedicines need to be codified. Therefore, it is imperative that governmental agencies develop a closer partnership with academics, scientists in research institutes and pharmaceutical manufacturers to bridge the gap between public perception and frontier knowledge of nanomedicine. With classification and property information of nanomaterials clearly written in the guidelines, streamlined trial procedures specific to certain types of nanomedicines are likely to be formulated to simplify the approval process and facilitate the clinical translation of nanomedicines for societal needs.
Acknowledgements
This work was supported by the National Institutes of Health Grant (NIH/NIBIB R01EB026890).
Biographies

Guanyou Lin earned his B.S. in Bioengineering from the University of Washington. He is currently pursuing his Ph.D. degree in Materials Science and Engineering at the University of Washington. His current research interest is in synthesis and application of nanoparticle systems for gene delivery and combinatorial antitumor therapies.

Richard A. Revia earned his B.S. in Biomedical Engineering from the University of Texas at Austin in 2008 and his M.S. in Electrical Engineering from Montana State University in 2013. He is currently pursuing his Ph.D. degree in Materials Science and Engineering at the University of Washington. His research interests include the development of nanoparticle platforms as contrast agents in magnetic resonance imaging and designing devices for use in tissue engineering.

Miqin Zhang is a Kyocera chair professor of Materials Science and Engineering and Neurological Surgery, and an adjunct professor in the Departments of Bioengineering, Radiology, and Orthopaedics and Sports Medicine at the University of Washington (UW). She joined the faculty of the UW in 1999 after receiving her Ph.D. from the University of California at Berkley. Her research focuses on nanomedicine for cancer diagnosis and treatment, biomaterials for tissue engineering, and biosensors for detection of chemical and biological agents.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
The ORCID identification number(s) for the author(s) of this article can be found under https://doi.org/10.1002/adfm.202007096.
References
- [1].Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MDM, Miller CA, Welch JS, Walter MJ, Wendl MC, Ley TJ, Wilson RK, Raphael BJ, Ding L, Nature 2013, 502, 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wu C-I, Wang H-Y, Ling S, Lu X, Annu. Rev. Genet 2016, 50, 347. [DOI] [PubMed] [Google Scholar]
- [3].Al-Lazikani B, Banerji U, Workman P, Nat. Biotechnol 2012, 30, 679. [DOI] [PubMed] [Google Scholar]
- [4].Colleoni M, Sun Z, Price KN, Karlsson P, Forbes JF, Thürlimann B, Gianni L, Castiglione M, Gelber RD, Coates AS, Goldhirsch A, J. Clin. Oncol 2016, 34, 927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].van Linde ME, Brahm CG, de Witt Hamer PC, Reijneveld JC, Bruynzeel AME, Vandertop WP, van de Ven PM, Wagemakers M, van der Weide HL, Enting RH, Walenkamp AME, Verheul HMW, J. Neuro-Oncol 2017, 135, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Corrado G, Salutari V, Palluzzi E, Distefano MG, Scambia G, Ferrandina G, Expert Rev. Anticancer Ther 2017, 17, 1147. [DOI] [PubMed] [Google Scholar]
- [7].Smyth MJ, Ngiow SF, Ribas A, Teng MWL, Nat. Rev. Clin. Oncol 2016, 13, 143. [DOI] [PubMed] [Google Scholar]
- [8].Lee Ventola C, Pharm. Ther 2017, 42, 514. [Google Scholar]
- [9].Dunbar CE, High KA, Joung JK, Kohn DB, Ozawa K, Sadelain M, Science 2018, 359, eaan4672. [DOI] [PubMed] [Google Scholar]
- [10].h Chen S, Lahav G, Curr. Opin. Struct. Biol 2016, 41, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Thomas CE, Ehrhardt A, Kay MA, Nat. Rev. Genet 2003, 4, 346. [DOI] [PubMed] [Google Scholar]
- [12].Ogawa M, Regino CAS, Marcelino B, Williams M, Kosaka N, Bryant LH, Choyke PL, Kobayashi H, Bioconjugate Chem 2010, 21, 955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hickey JW, Santos JL, Williford JM, Mao HQ, J. Controlled Release 2015, 219, 536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Longmire MR, Ogawa M, Choyke PL, Kobayashi H, Bioconjugate Chem 2011, 22, 993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lin G, Li L, Panwar N, Wang J, Tjin SC, Wang X, Yong KT, Coord. Chem. Rev 2018, 374, 133. [Google Scholar]
- [16].Abdelhady HG, Allen S, Davies MC, Roberts CJ, Tendler SJB, Williams PM, Nucleic Acids Res 2003, 31, 4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Huang Y, Hong J, Zheng S, Ding Y, Guo S, Zhang H, Zhang X, Du Q, Liang Z, Mol. Ther 2011, 19, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tsoi M, Do TT, Tang V, Aguilera JA, Perry CC, Milligan JR, Biophys. Chem 2010, 147, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Liu X, Wang S, Guo X, Wei F, Yin J, Zang Y, Li N, Chen D, Oncotarget 2016, 7, 18896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang K, Kievit FM, Jeon M, Silber JR, Ellenbogen RG, Zhang M, Adv. Healthcare Mater 2015, 4, 2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Catanese DJ, Fogg JM, Schrock DE, Gilbert BE, Zechiedrich L, Gene Ther 2012, 19, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Stewart MP, Langer R, Jensen KF, Chem. Rev 2018, 118, 7409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Vaughan HJ, Green JJ, Tzeng SY, Adv. Mater 2020, 32, 1901081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chen J, Li H-Y, Wang D, Zhao J-J, Guo X-Z, J. Gastroenterol. Hepatol 2011, 26, 1509. [DOI] [PubMed] [Google Scholar]
- [25].Oberli MA, Reichmuth AM, Dorkin JR, Mitchell MJ, Fenton OS, Jaklenec A, Anderson DG, Langer R, Blankschtein D, Nano Lett 2017, 17, 1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lam JKW, Chow MYT, Zhang Y, Leung SWS, Mol. Ther.–Nucleic Acids 2015, 4, e252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Barik S, J. Mol. Med 2005, 83, 764. [DOI] [PubMed] [Google Scholar]
- [28].Rao DD, Vorhies JS, Senzer N, Adv. Drug Delivery Rev 2009, 61, 746. [DOI] [PubMed] [Google Scholar]
- [29].Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM, Nature 2005, 433, 769. [DOI] [PubMed] [Google Scholar]
- [30].Huntzinger E, Izaurralde E, Nat. Rev. Genet 2011, 12, 99. [DOI] [PubMed] [Google Scholar]
- [31].Wang K, Kievit FM, Zhang M, Pharmacol. Res 2016, 114, 56. [DOI] [PubMed] [Google Scholar]
- [32].Daka A, Peer D, Adv. Drug Delivery Rev 2012, 64, 1508. [DOI] [PubMed] [Google Scholar]
- [33].Pradhan P, Qin H, Leleux JA, Gwak D, Sakamaki I, Kwak LW, Roy K, Biomaterials 2014, 35, 5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Matsumoto M, Seya T, Adv. Drug Delivery Rev 2008, 60, 805. [DOI] [PubMed] [Google Scholar]
- [35].Toumey C, Nat. Nanotechnol 2012, 7, 205. [DOI] [PubMed] [Google Scholar]
- [36].Stephen ZR, Kievit FM, Zhang M, Mater. Today 2011, 14, 330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wall MA, Harmsen S, Pal S, Zhang L, Arianna G, Lombardi JR, Drain CM, Kircher MF, Adv. Mater 2017, 29, 1605622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sun X, Zheng C, Zhang F, Yang Y, Wu G, Yu A, Guan N, J. Phys. Chem. C 2009, 113, 16002. [Google Scholar]
- [39].Chen PC, Liu G, Zhou Y, Brown KA, Chernyak N, Hedrick JL, He S, Xie Z, Lin QY, Dravid VP, O’Neill-Slawecki SA, Mirkin CA, J. Am. Chem. Soc 2015, 137, 9167. [DOI] [PubMed] [Google Scholar]
- [40].Mu Q, Lin G, Patton VK, Wang K, Press OW, Zhang M, J. Mater. Chem. B 2016, 4, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wang K, Kievit FM, Sham JG, Jeon M, Stephen ZR, Bakthavatsalam A, Park JO, Zhang M, Small 2016, 12, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wang Z, Liu G, Zheng H, Chen X, Biotechnol. Adv 2014, 32, 831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Pankhurst QA, Thanh KT, Jones SK, Dobson J, J. Phys. D: Appl. Phys 2009, 42, 224001. [Google Scholar]
- [44].Yang P, Gai S, Lin J, Chem. Soc. Rev 2012, 41, 3679. [DOI] [PubMed] [Google Scholar]
- [45].Chen G, Ågren H, Ohulchanskyy TY, Prasad PN, Chem. Soc. Rev 2015, 44, 1680. [DOI] [PubMed] [Google Scholar]
- [46].Santra S, Tapec R, Theodoropoulou N, Dobson J, Hebard A, Tan W, Langmuir 2001, 17, 2900. [Google Scholar]
- [47].Liu S, Chen G, Ohulchanskyy TY, Swihart MT, Prasad PN, Theranostics 2013, 3, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chen G, Roy I, Yang C, Prasad PN, Chem. Rev 2016, 116, 2826. [DOI] [PubMed] [Google Scholar]
- [49].Liu S, Chen G, Prasad PN, Swihart MT, Chem. Mater 2011, 23, 4098. [Google Scholar]
- [50].Xie J, Xu C, Kohler N, Hou Y, Sun S, Adv. Mater 2007, 19, 3163. [Google Scholar]
- [51].Qiu H, Yang C, Shao W, Damasco J, Wang X, Ågren H, Prasad P, Chen G, Nanomaterials 2014, 4, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lamer VK, Dinegar RH, J. Am. Chem. Soc 1950, 72, 4847. [Google Scholar]
- [53].Monguzzi A, Frigoli M, Larpent C, Tubino R, Meinardi F, Adv. Funct. Mater 2012, 22, 139. [Google Scholar]
- [54].Capek I, Adv. Colloid Interface Sci 2004, 110, 49. [DOI] [PubMed] [Google Scholar]
- [55].Das S, Jain TK, Maitra A, J. Colloid Interface Sci 2002, 252, 82. [DOI] [PubMed] [Google Scholar]
- [56].Li J, Yang Y, Huang L, J. Controlled Release 2012, 158, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Niu W, Zhang L, Xu G, Nanoscale 2013, 5, 3172. [DOI] [PubMed] [Google Scholar]
- [58].Murphy CJ, Sau TK, Gole AM, Orendorff CJ, Gao J, Gou L, Hunyadi SE, Li T, J. Phys. Chem. B 2005, 109, 13857. [DOI] [PubMed] [Google Scholar]
- [59].Veiseh O, Gunn JW, Zhang M, Adv. Drug Delivery Rev 2010, 62, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gref R, Lück M, Quellec P, Marchand M, Dellacherie E, Harnisch S, Blunk T, Müller RH, Colloids Surf., B 2000, 18, 301. [DOI] [PubMed] [Google Scholar]
- [61].Suk JS, Xu Q, Kim N, Hanes J, Ensign LM, Adv. Drug Delivery Rev 2016, 99, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Zhou Z, Liu X, Zhu D, Wang Y, Zhang Z, Zhou X, Qiu N, Chen X, Shen Y, Adv. Drug Delivery Rev 2017, 115, 115. [DOI] [PubMed] [Google Scholar]
- [63].Guan X, Guo Z, Lin L, Chen J, Tian H, Chen X, Nano Lett 2016, 16, 6823. [DOI] [PubMed] [Google Scholar]
- [64].Lai TC, Bae Y, Yoshida T, Kataoka K, Kwon GS, Pharm. Res 2010, 27, 2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Miyata K, Nishiyama N, Kataoka K, Chem. Soc. Rev 2012, 41, 2562. [DOI] [PubMed] [Google Scholar]
- [66].Mintzer MA, Simanek EE, Chem. Rev 2009, 109, 259. [DOI] [PubMed] [Google Scholar]
- [67].Kim J, Park J, Kim H, Singha K, Kim WJ, Biomaterials 2013, 34, 7168. [DOI] [PubMed] [Google Scholar]
- [68].Akinc A, Thomas M, Klibanov AM, Langer R, J. Gene Med 2005, 7, 657. [DOI] [PubMed] [Google Scholar]
- [69].Godbey WT, Wu KK, Mikos AG, Biomaterials 2001, 22, 471. [DOI] [PubMed] [Google Scholar]
- [70].Guerrero-Cázares H, Tzeng SY, Young NP, Abutaleb AO, Quiñones-Hinojosa A, Green JJ, ACS Nano 2014, 8, 5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Saranya N, Moorthi A, Saravanan S, Devi MP, Selvamurugan N, Int. J. Biol. Macromol 2011, 48, 234. [DOI] [PubMed] [Google Scholar]
- [72].Agarwal S, Zhang Y, Maji S, Greiner A, Mater. Today 2012, 15, 388. [Google Scholar]
- [73].Sevier CS, Kaiser CA, Nat. Rev. Mol. Cell Biol 2002, 3, 836. [DOI] [PubMed] [Google Scholar]
- [74].Il Kim T, Kim SW, React. Funct. Polym 2011, 71, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Oupický D, Li J, Macromol. Biosci 2014, 14, 908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Matsumoto S, Christie RJ, Nishiyama N, Miyata K, Ishii A, Oba M, Koyama H, Yamasaki Y, Kataoka K, Biomacromolecules 2009, 10, 119. [DOI] [PubMed] [Google Scholar]
- [77].Kang HC, Kang HJ, Bae YH, Biomaterials 2011, 32, 1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Asokan A, Cho MJ, J. Pharm. Sci 2002, 91, 903. [DOI] [PubMed] [Google Scholar]
- [79].Kim YH, Park JH, Lee M, Kim YH, Park TG, Kim SW, J. Controlled Release 2005, 103, 209. [DOI] [PubMed] [Google Scholar]
- [80].Wang R, Cheng J, Yan G, Wang C, Wang X, Wang J, Tang R, Polymer 2017, 123, 1. [Google Scholar]
- [81].Casey JR, Grinstein S, Orlowski J, Nat. Rev. Mol. Cell Biol 2010, 11, 50. [DOI] [PubMed] [Google Scholar]
- [82].Huotari J, Helenius A, EMBO J 2011, 30, 3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Gatenby RA, Gillies RJ, Nat. Rev. Cancer 2004, 4, 891. [DOI] [PubMed] [Google Scholar]
- [84].Lynn DM, Langer R, J. Am. Chem. Soc 2000, 122, 10761. [Google Scholar]
- [85].Anderson DG, Lynn DM, Langer R, Angew. Chem., Int. Ed 2003, 42, 3153. [DOI] [PubMed] [Google Scholar]
- [86].Funhoff AM, van Nostrum CF, Janssen APCA, Fens MHAM, Crommelin DJA, Hennink WE, Pharm. Res 2004, 21, 170. [DOI] [PubMed] [Google Scholar]
- [87].Petersen H, Merdan T, Kunath K, Fischer D, Kissel T, Bioconjugate Chem 2002, 13, 812. [DOI] [PubMed] [Google Scholar]
- [88].Steichen SD, Caldorera-Moore M, Peppas NA, Eur. J. Pharm. Sci 2013, 48, 416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Wang J, Mao W, Lock LL, Tang J, Sui M, Sun W, Cui H, Xu D, Shen Y, ACS Nano 2015, 9, 7195. [DOI] [PubMed] [Google Scholar]
- [90].Perrault SD, Walkey C, Jennings T, Fischer HC, Chan WCW, Nano Lett 2009, 9, 1909. [DOI] [PubMed] [Google Scholar]
- [91].Goodman TT, Olive PL, Pun SH, Int. J. Nanomed 2007, 2, 265. [PMC free article] [PubMed] [Google Scholar]
- [92].Carver K, Ming X, Juliano RL, Mol. Ther.–Nucleic Acids 2014, 3, e153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Li LL, Ji J, Fei R, Wang CZ, Lu Q, Zhang JR, Jiang LP, Zhu JJ, Adv. Funct. Mater 2012, 22, 2971. [Google Scholar]
- [94].Chauhan VP, Popović Z, Chen O, Cui J, Fukumura D, Bawendi MG, Jain RK, Angew. Chem., Int. Ed 2011, 50, 11417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Wang HX, Zuo ZQ, Du JZ, Wang YC, Sun R, Cao ZT, Ye XD, Wang JL, Leong KW, Wang J, Nano Today 2016, 11, 133. [Google Scholar]
- [96].El-Sayed A, Harashima H, Mol. Ther 2013, 21, 1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Rappoport JZ, Biochem. J 2008, 412, 415. [DOI] [PubMed] [Google Scholar]
- [98].Mayor S, Parton RG, Donaldson JG, Cold Spring Harbor Perspect. Biol 2014, 6, a016758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Swanson JA, Watts C, Trends Cell Biol 1995, 5, 424. [DOI] [PubMed] [Google Scholar]
- [100].Grosse S, Aron Y, Thévenot G, François D, Monsigny M, Fajac I, J. Gene Med 2005, 7, 1275. [DOI] [PubMed] [Google Scholar]
- [101].Rejman J, Oberle V, Zuhorn IS, Hoekstra D, Biochem. J 2004, 377, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Akhtar MJ, Ahamed M, Alhadlaq HA, Alrokayan SA, Kumar S, Clin. Chim. Acta 2014, 436, 78. [DOI] [PubMed] [Google Scholar]
- [103].Elias DR, Poloukhtine A, Popik V, Tsourkas A, Nanomedicine 2013, 9, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Alkilany AM, Zhu L, Weller H, Mews A, Parak WJ, Barz M, Feliu N, Adv. Drug Delivery Rev 2019, 143, 22. [DOI] [PubMed] [Google Scholar]
- [105].Stewart MP, Lorenz A, Dahlman J, Sahay G, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol 2016, 8, 465. [DOI] [PubMed] [Google Scholar]
- [106].Varkouhi AK, Scholte M, Storm G, Haisma HJ, J. Controlled Release 2011, 151, 220. [DOI] [PubMed] [Google Scholar]
- [107].Marshall ICB, Wilson KL, Trends Cell Biol 1997, 7, 69. [DOI] [PubMed] [Google Scholar]
- [108].Zhou X, Liu X, Zhao B, Liu X, Zhu D, Qiu N, Zhou Q, Piao Y, Zhou Z, Tang J, Shen Y, J. Controlled Release 2016, 234, 90. [DOI] [PubMed] [Google Scholar]
- [109].Ludtke JJ, Zhang G, Sebestyén MG, Wolff JA, J. Cell Sci 1999, 112, 2033. [DOI] [PubMed] [Google Scholar]
- [110].Van Der Aa MAEM, Mastrobattista E, Oosting RS, Hennink WE, Koning GA, Crommelin DJA, Pharm. Res 2006, 23, 447. [DOI] [PubMed] [Google Scholar]
- [111].Wang HY, Li C, Yi WJ, Sun YX, Cheng SX, Zhuo RX, Zhang XZ, Bioconjugate Chem 2011, 22, 1567. [DOI] [PubMed] [Google Scholar]
- [112].Xu Y, Liang W, Qiu Y, Cespi M, Palmieri GF, Mason AJ, Lam JKW, Mol. Pharmaceutics 2016, 13, 3141. [DOI] [PubMed] [Google Scholar]
- [113].Yin J, Meng X, Zhang S, Zhang D, Wang L, Liu C, Biomaterials 2012, 33, 7884. [DOI] [PubMed] [Google Scholar]
- [114].Ritter W, Plank C, Lausier J, Rudolph C, Zink D, Reinhardt D, Rosenecker J, J. Mol. Med 2003, 81, 708. [DOI] [PubMed] [Google Scholar]
- [115].Van Gaal EVB, Oosting RS, Van Eijk R, Bakowska M, Feyen D, Kok RJ, Hennink WE, Crommelin DJA, Mastrobattista E, Pharm. Res 2011, 28, 1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Godbey WT, Wu KK, Mikos AG, Proc. Natl. Acad. Sci. USA 1999, 96, 5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Grandinetti G, Reineke TM, Mol. Pharmaceutics 2012, 9, 2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Bieber T, Meissner W, Kostin S, Niemann A, Elsasser HP, J. Controlled Release 2002, 82, 441. [DOI] [PubMed] [Google Scholar]
- [119].Andersen H, Parhamifar L, Hunter AC, Shahin V, Moghimi SM, J. Controlled Release 2016, 244, 24. [DOI] [PubMed] [Google Scholar]
- [120].Pommier Y, Leo E, Zhang H, Marchand C, Chem. Biol 2010, 17, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Amreddy N, Babu A, Muralidharan R, Panneerselvam J, Srivastava A, Ahmed R, Mehta M, Munshi A, Ramesh R, Adv. Cancer Res 2018, 137, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Gillet JP, Gottesman MM, Methods Mol. Biol 2010, 596, 47. [DOI] [PubMed] [Google Scholar]
- [123].Chen Y, Chen H, Shi J, Mol. Pharmaceutics 2014, 11, 2495. [DOI] [PubMed] [Google Scholar]
- [124].Livney YD, Assaraf YG, Adv. Drug Delivery Rev 2013, 65, 1716. [DOI] [PubMed] [Google Scholar]
- [125].Cukierman E, Khan DR, Biochem. Pharmacol 2010, 80, 762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM, Nat. Rev. Drug Discovery 2006, 5, 219. [DOI] [PubMed] [Google Scholar]
- [127].Al-Shawi MK, Omote H, J. Bioenerg. Biomembr 2005, 37, 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Lima RT, Martins LM, Guimarães JE, Sambade C, Vasconcelos MH, Cancer Gene Ther 2004, 11, 309. [DOI] [PubMed] [Google Scholar]
- [129].Kim DY, Kim MJ, Kim HB, Lee JW, Bae JH, Kim DW, Kang CD, Kim SH, Biochim. Biophys. Acta, Mol. Basis Dis 2011, 1812, 796. [DOI] [PubMed] [Google Scholar]
- [130].Tuschl T, Borkhardt A, Mol. Interventions 2002, 2, 158. [DOI] [PubMed] [Google Scholar]
- [131].Wang K, Park JO, Zhang M, J. Gene Med 2013, 15, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Jeon M, Lin G, Stephen ZR, Kato FL, Zhang M, Adv. Ther 2019, 2, 1900081. [Google Scholar]
- [133].Gao Z, Zhang L, Sun Y, J. Controlled Release 2012, 162, 45. [DOI] [PubMed] [Google Scholar]
- [134].Zhang B, Liu Q, Liu M, Shi P, Zhu L, Zhang L, Li R, J. Biomater. Appl 2019, 33, 1382. [DOI] [PubMed] [Google Scholar]
- [135].Fang XJ, Jiang H, Zhu YQ, Zhang LY, Fan QH, Tian Y, Oncol. Rep 2014, 31, 2735. [DOI] [PubMed] [Google Scholar]
- [136].McAtee CO, Barycki JJ, Simpson MA, Adv. Cancer Res 2014, 123, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Meng H, Mai WX, Zhang H, Xue M, Xia T, Lin S, Wang X, Zhao Y, Ji Z, Zink JI, Nel AE, ACS Nano 2013, 7, 994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Lee J-H, Kang S, Ahn M, Jang H, Min D-H, Small 2018, 14, 1702564. [DOI] [PubMed] [Google Scholar]
- [139].Pacardo DB, Ligler FS, Gu Z, Nanoscale 2015, 7, 3381. [DOI] [PubMed] [Google Scholar]
- [140].Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, MacBeath G, Yaffe MB, Cell 2012, 149, 780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Jiang J, Yang SJ, Wang JC, Yang LJ, Xu ZZ, Yang T, Liu XY, Zhang Q, Eur. J. Pharm. Biopharm 2010, 76, 170. [DOI] [PubMed] [Google Scholar]
- [142].Qian X, Ren Y, Shi Z, Long L, Pu P, Sheng J, Yuan X, Kang C, Mol. Pharmaceutics 2012, 9, 2636. [DOI] [PubMed] [Google Scholar]
- [143].Sun L, Wang D, Chen Y, Wang L, Huang P, Li Y, Liu Z, Yao H, Shi J, Biomaterials 2017, 133, 219. [DOI] [PubMed] [Google Scholar]
- [144].Zhang X, He Z, Xiang L, Li L, Zhang H, Lin F, Cao H, Drug Des., Dev. Ther 2019, 13, 1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Rajendrakumar SK, Venu A, Revuri V, George Thomas R, Thirunavukkarasu GK, Zhang J, Vijayan V, Choi SY, Lee JY, Lee YK, Jeong YY, Park IK, Mol. Pharmaceutics 2019, 16, 2226. [DOI] [PubMed] [Google Scholar]
- [146].Ren Y, Wang R, Gao L, Li K, Zhou X, Guo H, Liu C, Han D, Tian J, Ye Q, Hu YT, Sun D, Yuan X, Zhang N, J. Controlled Release 2016, 228, 74. [DOI] [PubMed] [Google Scholar]
- [147].Kang HY, Stem Cell Res. Ther 2013, 4, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Hsieh AL, Dang CV, Recent Results in Cancer Research, Vol. 207, Springer, New York: 2016, pp. 73–91. [DOI] [PubMed] [Google Scholar]
- [149].Hollstein M, Sidransky D, Vogelstein B, Harris CC, Science 1991, 253, 49. [DOI] [PubMed] [Google Scholar]
- [150].Carmeliet P, Oncology 2005, 69, 4.16301830 [Google Scholar]
- [151].Dang CV, Reddy EP, Shokat KM, Soucek L, Nat. Rev. Cancer 2017, 17, 502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Zhang Q, Shen C, Zhao N, Xu F-J, Adv. Funct. Mater 2017, 27, 1606229. [Google Scholar]
- [153].Huang W, Liang Y, Sang C, Mei C, Li X, Chen T, J. Mater. Chem. B 2018, 6, 3013. [DOI] [PubMed] [Google Scholar]
- [154].Ferrara N, Arterioscler., Thromb., Vasc. Biol 2009, 29, 789. [DOI] [PubMed] [Google Scholar]
- [155].Li T, Shen X, Geng Y, Chen Z, Li L, Li S, Yang H, Wu C, Zeng H, Liu Y, ACS Appl. Mater. Interfaces 2016, 8, 13748. [DOI] [PubMed] [Google Scholar]
- [156].Zhang Y, Schwerbrock NMJ, Rogers AB, Kim WY, Huang L, Mol. Ther 2013, 21, 1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Conde J, Arnold CE, Tian F, Artzi N, Mater. Today 2016, 19, 29. [Google Scholar]
- [158].Dougan M, Dranoff G, Annu. Rev. Immunol 2009, 27, 83. [DOI] [PubMed] [Google Scholar]
- [159].Kershaw MH, Westwood JA, Darcy PK, Nat. Rev. Cancer 2013, 13, 525. [DOI] [PubMed] [Google Scholar]
- [160].Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, Xu Y, Frohlich MW, Schellhammer PF, N. Engl. J. Med 2010, 363, 411. [DOI] [PubMed] [Google Scholar]
- [161].Stewart JM, Keselowsky BG, Adv. Drug Delivery Rev 2017, 114, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Saleh T, Shojaosadati SA, Hum. Vaccines Immunother 2016, 12, 2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Saxena M, Bhardwaj N, Trends Cancer 2018, 4, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Mirzaei HR, Rodriguez A, Shepphird J, Brown CE, Badie B, Front. Immunol 2017, 8, 1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Voskens CJ, Goldinger SM, Loquai C, Robert C, Kaehler KC, Berking C, Bergmann T, Bockmeyer CL, Eigentler T, Fluck M, Garbe C, Gutzmer R, Grabbe S, Hauschild A, Hein R, Hundorfean G, Justich A, Keller U, Klein C, Mateus C, Mohr P, Paetzold S, Satzger I, Schadendorf D, Schlaeppi M, Schuler G, Schuler-Thurner B, Trefzer U, Ulrich J, Vaubel J, von Moos R, Weder P, Wilhelm T, Göppner D, Dummer R, Heinzerling LM, PLoS One 2013, 8, e53745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Reed SG, Orr MT, Fox CB, Nat. Med 2013, 19, 1597. [DOI] [PubMed] [Google Scholar]
- [167].Hardee C, Arévalo-Soliz L, Hornstein B, Zechiedrich L, Genes 2017, 8, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Sahin U, Karikó K, Türeci Ö, Nat. Rev. Drug Discovery 2014, 13, 759. [DOI] [PubMed] [Google Scholar]
- [169].Guevara ML, Jilesen Z, Stojdl D, Persano S, ACS Omega 2019, 4, 13015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Verbeke R, Lentacker I, Wayteck L, Breckpot K, Van Bockstal M, Descamps B, Vanhove C, De Smedt SC, Dewitte H, J. Controlled Release 2017, 266, 287. [DOI] [PubMed] [Google Scholar]
- [171].Dewitte H, Verbeke R, Breckpot K, De Smedt SC, Lentacker I, Nano Today 2014, 9, 743. [Google Scholar]
- [172].Andorko JI, Hess KL, Jewell CM, AAPS J 2015, 17, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Mellman I, Steinman RM, Cell 2001, 106, 255. [DOI] [PubMed] [Google Scholar]
- [174].Wykes MN, Lewin SR, Nat. Rev. Immunol 2018, 18, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Merad M, Sathe P, Helft J, Miller J, Mortha A, Annu. Rev. Immunol 2013, 31, 563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Cubillos-Ruiz JR, Baird JR, Tesone AJ, Rutkowski MR, Scarlett UK, Camposeco-Jacobs AL, Anadon-Arnillas J, Harwood NM, Korc M, Fiering SN, Sempere LF, Conejo-Garcia JR, Cancer Res 2012, 72, 1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Hobo W, Novobrantseva TI, Fredrix H, Wong J, Milstein S, Epstein-Barash H, Liu J, Schaap N, Van Der Voort R, Dolstra H, Cancer Immunol. Immunother 2013, 62, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Heo MB, Lim YT, Biomaterials 2014, 35, 590. [DOI] [PubMed] [Google Scholar]
- [179].Song W, Musetti SN, Huang L, Biomaterials 2017, 148, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Frey AB, Vaccine 2015, 33, 7393. [DOI] [PubMed] [Google Scholar]
- [181].Liakou CI, Kamat A, Tang DN, Chen H, Sun J, Troncoso P, Logothetis C, Sharma P, Proc. Natl. Acad. Sci. USA 2008, 105, 14987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, Budhu S, Ghosh A, Pink M, Tchaicha J, Douglas M, Tibbitts T, Sharma S, Proctor J, Kosmider N, White K, Stern H, Soglia J, Adams J, Palombella VJ, McGovern K, Kutok JL, Wolchok JD, Merghoub T, Nature 2016, 539, 443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Gamrad L, Rehbock C, Westendorf AM, Buer J, Barcikowski S, Hansen W, Sci. Rep 2016, 6, 28709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Zhang Y, Fu J, Shi Y, Peng S, Cai Y, Zhan X, Song N, Liu Y, Wang Z, Yu Y, Wang Y, Shi Q, Fu Y, Yuan K, Zhou N, Joshi R, Ichim TE, Min W, Int. J. Cancer 2018, 143, 2039. [DOI] [PubMed] [Google Scholar]
- [185].Xiong A, Duan L, Chen J, Fan Z, Zheng F, Tan Z, Gong F, Fang M, PLoS One 2012, 7, e46230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Xu Z, Ramishetti S, Tseng YC, Guo S, Wang Y, Huang L, J. Controlled Release 2013, 172, 259. [DOI] [PubMed] [Google Scholar]
- [187].Wang Y, Zhang L, Xu Z, Miao L, Huang L, Mol. Ther 2018, 26, 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Liu L, Wang Y, Miao L, Liu Q, Musetti S, Li J, Huang L, Mol. Ther 2018, 26, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, Mellman I, Prindiville SA, Viner JL, Weiner LM, Matrisian LM, Clin. Cancer Res 2009, 15, 5323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, Thatcher N, Wagstaff J, Zielinski C, Faulkner I, Mellstedt H, Nat. Rev. Clin. Oncol 2014, 11, 509. [DOI] [PubMed] [Google Scholar]
- [191].Luo X, Peng X, Hou J, Wu S, Shen J, Wang L, Int. J. Nanomed 2017, 12, 5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Ramani M, Mudge MC, Morris RT, Zhang Y, Warcholek SA, Hurst MN, Riviere JE, DeLong RK, Mol. Pharmaceutics 2017, 14, 614. [DOI] [PubMed] [Google Scholar]
- [193].Prijic S, Prosen L, Cemazar M, Scancar J, Romih R, Lavrencak J, Bregar VB, Coer A, Krzan M, Znidarsic A, Sersa G, Biomaterials 2012, 33, 4379. [DOI] [PubMed] [Google Scholar]
- [194].Gulla SK, Kotcherlakota R, Nimushakavi S, Nimmu NV, Khalid S, Patra CR, Chaudhuri A, ACS Omega 2018, 3, 8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Noy R, Pollard JW, Immunity 2014, 41, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Conde J, Bao C, Tan Y, Cui D, Edelman ER, Azevedo HS, Byrne HJ, Artzi N, Tian F, Adv. Funct. Mater 2015, 25, 4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Kataru RP, Jung K, Jang C, Yang H, Schwendener RA, Jung EB, Seung HH, Alitalo K, Gou YK, Blood 2009, 113, 5650. [DOI] [PubMed] [Google Scholar]
- [198].Lombardi L, Tavano F, Morelli F, Latiano TP, Di Sebastiano P, Maiello E, Crit. Rev. Oncol./Hematol 2013, 88, 696. [DOI] [PubMed] [Google Scholar]
- [199].Zeelenberg IS, Ruuls-Van Stalle L, Roos E, Cancer Res 2003, 63, 3833. [PubMed] [Google Scholar]
- [200].Goodwin TJ, Zhou Y, Musetti SN, Liu R, Huang L, Sci. Transl. Med 2016, 8, 364ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Cai J, Chen G, Jin R, Deng C, Huang S, Yuan X, Chen G, Zhao J, Wang Z, Ai H, J. Ind. Eng. Chem 2019, 76, 188. [Google Scholar]
- [202].Kim JJ, Kim JJ, Jeong C, Kim WJ, Adv. Drug Delivery Rev 2016, 98, 99. [DOI] [PubMed] [Google Scholar]
- [203].van der Zee J, Ann. Oncol 2002, 13, 1173. [DOI] [PubMed] [Google Scholar]
- [204].Fajardo LF, Cancer Res 1984, 44, 4826s. [PubMed] [Google Scholar]
- [205].Christophi C, Winkworth A, Muralihdaran V, Evans P, Surg. Oncol 1998, 7, 83. [DOI] [PubMed] [Google Scholar]
- [206].Sherlock SP, Tabakman SM, Xie L, Dai H, ACS Nano 2011, 5, 1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Beik J, Khateri M, Khosravi Z, Kamrava SK, Kooranifar S, Ghaznavi H, Shakeri-Zadeh A, Coord. Chem. Rev 2019, 387, 299. [Google Scholar]
- [208].Rybiński M, Szymańska Z, Lasota S, Gambin A, J. R. Soc., Interface 2013, 10, 20130527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Jolly C, Morimoto R, J. Natl. Cancer Inst 2000, 92, 1564. [DOI] [PubMed] [Google Scholar]
- [210].Revia RA, Zhang M, Mater. Today 2016, 19, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Yang K, Zhang S, Zhang G, Sun X, Lee ST, Liu Z, Nano Lett 2010, 10, 3318. [DOI] [PubMed] [Google Scholar]
- [212].Wang H, Mu Q, Revia R, Wang K, Tian B, Lin G, Lee W, Hong YK, Zhang M, J. Controlled Release 2018, 289, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Lee N, Yoo D, Ling D, Cho MH, Hyeon T, Cheon J, Chem. Rev 2015, 115, 10637. [DOI] [PubMed] [Google Scholar]
- [214].Hergt R, Dutz S, Zeisberger M, J. Phys.: Condens. Matter 2006, 18, 2919. [Google Scholar]
- [215].Wang Z, Chang Z, Lu M, Shao D, Yue J, Yang D, Zheng X, Li M, He K, Zhang M, Chen L, Dong W.-f., Biomaterials 2018, 154, 147. [DOI] [PubMed] [Google Scholar]
- [216].Li YF, Yuan YY, Zhang YM, Zhao N, Zhang Q, Meng FX, Gao RP, Yu BF, Zhang YH, Guo R, Wang HL, Xie J, Xu J, Qin Q, Dong XS, Mol. Med. Rep 2017, 16, 764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Jang J, Nah H, Lee J-H, Moon SH, Kim MG, Cheon J, Angew. Chem., Int. Ed 2009, 48, 1234. [DOI] [PubMed] [Google Scholar]
- [218].Yin PT, Shah BP, Lee K-B, Small 2014, 10, 4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Bartel DP, Cell 2009, 136, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Yin PT, Shah S, Pasquale NJ, Garbuzenko OB, Minko T, Lee KB, Biomaterials 2016, 81, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Rohmer S, Mainka A, Knippertz I, Hesse A, Nettelbeck DM, J. Gene Med 2008, 10, 340. [DOI] [PubMed] [Google Scholar]
- [222].Coffelt SB, Marini FC, Watson K, Zwezdaryk KJ, Dembinski JL, LaMarca HL, Tomchuck SL, Zu Bentrup KH, Danka ES, Henkle SL, Scandurro AB, Proc. Natl. Acad. Sci. USA 2009, 106, 3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Huang X, El-Sayed MA, J. Adv. Res 2010, 1, 13. [Google Scholar]
- [224].Wang BK, Yu XF, Wang JH, Bin Li Z, Li PH, Wang H, Song L, Chu PK, Li C, Biomaterials 2016, 78, 27. [DOI] [PubMed] [Google Scholar]
- [225].Shen J, Kim H-C, Mu C, Gentile E, Mai J, Wolfram J, Ji L, Ferrari M, Mao Z, Shen H, Adv. Healthcare Mater 2014, 3, 1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Son S, Nam J, Kim J, Kim S, Kim WJ, ACS Nano 2014, 8, 5574. [DOI] [PubMed] [Google Scholar]
- [227].Son S, Kim N, You DG, Yoon HY, Yhee JY, Kim K, Kwon IC, Kim SH, Theranostics 2017, 7, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Cheng FF, Chen W, Hu LH, Chen G, Miao HT, Li C, Zhu JJ, J. Mater. Chem. B 2013, 1, 4956. [DOI] [PubMed] [Google Scholar]
- [229].Dolmans DEJGJ, Fukumura D, Jain RK, Nat. Rev. Cancer 2003, 3, 380. [DOI] [PubMed] [Google Scholar]
- [230].Dolmans DEJGJ, Kadambi A, Hill JS, Waters CA, Robinson BC, Walker JP, Fukumura D, Jain RK, Cancer Res 2002, 62, 2151. [PubMed] [Google Scholar]
- [231].Chen WH, Lecaros RLG, Tseng YC, Huang L, Hsu YC, Cancer Lett 2015, 359, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Lecaros RLG, Huang L, Lee TC, Hsu YC, Mol. Ther 2016, 24, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Wang X, Liu K, Yang G, Cheng L, He L, Liu Y, Li Y, Guo L, Liu Z, Nanoscale 2014, 6, 9198. [DOI] [PubMed] [Google Scholar]
- [234].Stapleton S, Jaffray D, Milosevic M, Adv. Drug Delivery Rev 2017, 109, 119. [DOI] [PubMed] [Google Scholar]
- [235].Her S, Jaffray DA, Allen C, Adv. Drug Delivery Rev 2017, 109, 84. [DOI] [PubMed] [Google Scholar]
- [236].Nussbaum DP, Rushing CN, Lane WO, Cardona DM, Kirsch DG, Peterson BL, Blazer DG, Lancet Oncol 2016, 17, 966. [DOI] [PubMed] [Google Scholar]
- [237].Pilar A, Gupta M, Laskar SG, Laskar S, ecancermedicalscience 2017, 11, 750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Wang H, Mu X, He H, Zhang XD, Trends Pharmacol. Sci 2018, 39, 24. [DOI] [PubMed] [Google Scholar]
- [239].Murray L, Menard C, Zadeh G, Au K, Bernstein M, Millar B-A, Laperriere N, Chung C, J. Radiat. Oncol 2017, 6, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Patel SA, Qureshi MM, Sahni D, Truong MT, JAMA Dermatol 2017, 153, 1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Xie J, Gong L, Zhu S, Yong Y, Gu Z, Zhao Y, Adv. Mater 2019, 31, 1802244. [DOI] [PubMed] [Google Scholar]
- [242].Kwatra D, Venugopal A, Anant S, Transl. Cancer Res 2013, 2, 330. [Google Scholar]
- [243].Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA, Nat. Rev. Cancer 2008, 8, 193. [DOI] [PubMed] [Google Scholar]
- [244].Kastan MB, Bartek J, Nature 2004, 432, 316. [DOI] [PubMed] [Google Scholar]
- [245].Kunz-Schughart LA, Dubrovska A, Peitzsch C, Ewe A, Aigner A, Schellenburg S, Muders MH, Hampel S, Cirillo G, Iemma F, Tietze R, Alexiou C, Stephan H, Zarschler K, Vittorio O, Kavallaris M, Parak WJ, Mädler L, Pokhrel S, Biomaterials 2017, 120, 155. [DOI] [PubMed] [Google Scholar]
- [246].Ju E, Dong K, Chen Z, Liu Z, Liu C, Huang Y, Wang Z, Pu F, Ren J, Qu X, Angew. Chem., Int. Ed 2016, 55, 11467. [DOI] [PubMed] [Google Scholar]
- [247].Liu JN, Bu W, Shi J, Chem. Rev 2017, 117, 6160. [DOI] [PubMed] [Google Scholar]
- [248].Jin X, Li Q, Wu Q, Li P, Matsumoto Y, Furusawa Y, Gong L, Hao J, Dai Z, J. Radiat. Res 2011, 52, 335. [DOI] [PubMed] [Google Scholar]
- [249].Zou J, Qiao X, Ye H, Zhang Y, Xian J, Zhao H, Liu S, Cancer Biother. Radiopharm 2009, 24, 339. [DOI] [PubMed] [Google Scholar]
- [250].Kievit FM, Wang K, Ozawa T, Tarudji AW, Silber JR, Holland EC, Ellenbogen RG, Zhang M, Nanomedicine 2017, 13, 2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Kievit FM, Stephen ZR, Wang K, Dayringer CJ, Sham JG, Ellenbogen RG, Silber JR, Zhang M, Mol. Oncol 2015, 9, 1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Mol CD, Izumi T, Mitra S, Talner JA, Nature 2000, 403, 451. [DOI] [PubMed] [Google Scholar]
- [253].Bobola MS, Jankowski PP, Gross ME, Schwartz J, Finn LS, Blank A, Ellenbogen RG, Silber JR, Int. J. Cancer 2011, 129, 2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Shen L-F, Chen J, Zeng S, Zhou R-R, Zhu H, Zhong M-Z, Yao R-J, Shen H, Mol. Cancer Ther 2010, 9, 2123. [DOI] [PubMed] [Google Scholar]
- [255].Martin RF, Feinendegen LE, Int. J. Radiat. Biol 2016, 92, 617. [DOI] [PubMed] [Google Scholar]
- [256].Carter JD, Cheng NN, Qu Y, Suarez GD, Guo T, J. Phys. Chem. B 2007, 111, 11622. [DOI] [PubMed] [Google Scholar]
- [257].Goswami N, Luo Z, Yuan X, Leong DT, Xie J, Mater. Horiz 2017, 4, 817. [Google Scholar]
- [258].Roa W, Zhang X, Guo L, Shaw A, Hu X, Xiong Y, Gulavita S, Patel S, Sun X, Chen J, Moore R, Xing JZ, Nanotechnology 2009, 20, 375101. [DOI] [PubMed] [Google Scholar]
- [259].Hvolbæk B, Janssens TVW, Clausen BS, Falsig H, Christensen CH, Nørskov JK, Nano Today 2007, 2, 14. [Google Scholar]
- [260].Zhang S, Gupta S, Fitzgerald TJ, Bogdanov AA, Nanotheranostics 2018, 2, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Jung JE, Lee H, Cho I, Chung DH, Yoon S, Yang YM, Lee JW, Choi S, Park J, Ye S, Chung M, FASEB J 2005, 19, 1296. [DOI] [PubMed] [Google Scholar]
- [262].Adachi M, Cui C, Dodge CT, Bhayani MK, Lai SY, Oral Oncol 2012, 48, 1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Bonner JA, Trummell HQ, Willey CD, Plants BA, Raisch KP, Radiother. Oncol 2009, 92, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Yong Y, Zhang C, Gu Z, Du J, Guo Z, Dong X, Xie J, Zhang G, Liu X, Zhao Y, ACS Nano 2017, 11, 7164. [DOI] [PubMed] [Google Scholar]
- [265].Elser M, Borsig L, Hassa PO, Erener S, Messner S, Valovka T, Keller S, Gassmann M, Hottiger MO, Mol. Cancer Res 2008, 6, 282. [DOI] [PubMed] [Google Scholar]
- [266].Wang H, Wang K, Tian B, Revia R, Mu Q, Jeon M, Chang F-C, Zhang M, Small 2016, 12, 6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].Mu Q, Wang H, Gu X, Stephen ZR, Yen C, Chang F, Dayringer CJ, Zhang M, Adv. Healthcare Mater 2019, 8, 1801505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Wang H, Mu Q, Revia R, Wang K, Zhou X, Pauzauskie PJ, Zhou S, Zhang M, Adv. Healthcare Mater 2017, 6, 1601080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [269].Chen X, Zhang Q, Li J, Yang M, Zhao N, Xu FJ, ACS Nano 2018, 12, 5646. [DOI] [PubMed] [Google Scholar]
- [270].Yang XJ, Li XL, Chen HY, Xu JJ, ACS Appl. Bio Mater 2019, 2, 2994. [DOI] [PubMed] [Google Scholar]
- [271].Tao Y, Ju E, Liu Z, Dong K, Ren J, Qu X, Biomaterials 2014, 35, 6646. [DOI] [PubMed] [Google Scholar]
- [272].Li C, Yang XQ, Zhang MZ, Song YY, Cheng K, An J, Zhang XS, Xuan Y, Liu B, Di Zhao Y, Theranostics 2018, 8, 5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Liu Y, Ai K, Lu L, Chem. Rev 2014, 114, 5057. [DOI] [PubMed] [Google Scholar]
- [274].Cheng W, Nie J, Gao N, Liu G, Tao W, Xiao X, Jiang L, Liu Z, Zeng X, Mei L, Adv. Funct. Mater 2017, 27, 1704135. [Google Scholar]
- [275].Yang H, Chen Y, Chen Z, Geng Y, Xie X, Shen X, Li T, Li S, Wu C, Liu Y, Biomater. Sci 2017, 5, 1001. [DOI] [PubMed] [Google Scholar]
- [276].Juneja R, Lyles Z, Vadarevu H, Afonin KA, Vivero-Escoto JL, ACS Appl. Mater. Interfaces 2019, 11, 12308. [DOI] [PubMed] [Google Scholar]
- [277].Yu Q, Sun J, Zhu X, Qiu L, Xu M, Liu S, Ouyang J, Liu J, J. Mater. Chem. B 2017, 5, 6081. [DOI] [PubMed] [Google Scholar]
- [278].Lim JM, Swami A, Gilson LM, Chopra S, Choi S, Wu J, Langer R, Karnik R, Farokhzad OC, ACS Nano 2014, 8, 6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [279].Valencia PM, Farokhzad OC, Karnik R, Langer R, Nat. Nanotechnol 2012, 7, 623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [280].Feliu N, Docter D, Heine M, Del Pino P, Ashraf S, Kolosnjaj-Tabi J, Macchiarini P, Nielsen P, Alloyeau D, Gazeau F, Stauber RH, Parak WJ, Chem. Soc. Rev 2016, 45, 2440. [DOI] [PubMed] [Google Scholar]
- [281].Mak IWY, Evaniew N, Ghert M, Am. J. Transl. Res 2014, 6, 114. [PMC free article] [PubMed] [Google Scholar]
- [282].Adir O, Poley M, Chen G, Froim S, Krinsky N, Shklover J, Shainsky-Roitman J, Lammers T, Schroeder A, Adv. Mater 2020, 32, 1901989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [283].Wilson B, Km G, Nanomedicine 2020, 15, 433. [DOI] [PubMed] [Google Scholar]
- [284].Ho D, Wang P, Kee T, Nanoscale Horiz 2019, 4, 365. [DOI] [PubMed] [Google Scholar]
- [285].Wang L, Yan L, Liu J, Chen C, Zhao Y, Anal. Chem 2018, 90, 589. [DOI] [PubMed] [Google Scholar]
- [286].Lo Giudice MC, Herda LM, Polo E, Dawson KA, Nat. Commun 2016, 7, 13475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [287].Carril M, Padro D, Del Pino P, Carrillo-Carrion C, Gallego M, Parak WJ, Nat. Commun 2017, 8, 1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].Jia J, Li F, Zhou H, Bai Y, Liu S, Jiang Y, Jiang G, Yan B, Environ. Sci. Technol 2017, 51, 9334. [DOI] [PubMed] [Google Scholar]
- [289].Volkov Y, Biochem. Biophys. Res. Commun 2015, 468, 419. [DOI] [PubMed] [Google Scholar]
- [290].Bobo D, Robinson KJ, Islam J, Thurecht KJ, Corrie SR, Pharm. Res 2016, 33, 2373. [DOI] [PubMed] [Google Scholar]
- [291].Jones AAD, Mi G, Webster TJ, Trends Biotechnol 2019, 37, 117. [DOI] [PubMed] [Google Scholar]
- [292].Navya PN, Kaphle A, Srinivas SP, Bhargava SK, Rotello VM, Daima HK, Nano Convergence 2019, 6, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]