

SUMMARY
Iron overload is positively associated with diabetes risk. However, the role of iron in adipose tissue remains incompletely understood. Here, we report that transferrin receptor 1-mediated iron uptake is differentially required for distinct subtypes of adipocytes. Notably, adipocyte-specific transferrin receptor 1 deficiency substantially protects mice from high-fat diet-induced metabolic disorders. Mechanistically, low cellular iron levels have a positive impact on the health of the white adipose tissue and can restrict lipid absorption from the intestine through modulation of vesicular transport in enterocytes following high-fat diet feeding. Specific reduction of adipocyte iron by AAV-mediated overexpression of the iron exporter Ferroportin1 in adult mice effectively mimics these protective effects. In summary, our studies highlight an important role of adipocyte iron in the maintenance of systemic metabolism through an adipocyteenterocyte axis, offering an additional level of control over caloric influx into the system after feeding by regulating intestinal lipid absorption.
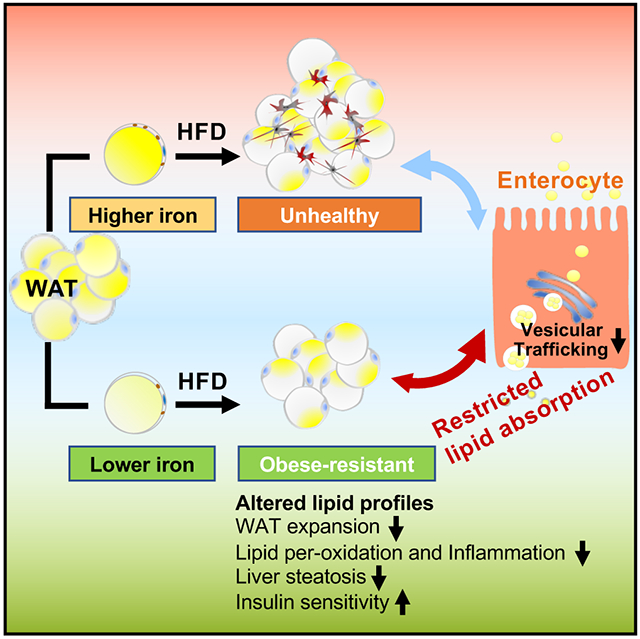
Graphical Abstract
In Brief
Zhang and colleagues reveal an important role of adipocyte iron content in the maintenance of systemic metabolic homeostasis through an adipose-gut axis. They find that lowering iron in white adipose tissue substantially protects mice from high fat diet-induced metabolic dysfunction by restricting intestinal lipid absorption.
INTRODUCTION
Obesity and type 2 diabetes have emerged as global public health issues (Mokdad et al., 2003; Saeedi et al., 2019). However, significant heterogeneity exists in the etiology of obesity and type 2 diabetes, as the underlying causes are highly complex, involving genetic, environmental and behavioral factors (Ussar et al., 2015). As chronic caloric excess is one of the main drivers of obesity and type 2 diabetes development, most studies mainly focus on dietary macronutrients, such as the types or composition of fats and carbohydrates. We know less about the role of micronutrients, especially with respect to normal cellular homeostasis of the adipocyte.
Iron is an essential trace element for humans, and there is a correlation between iron and diabetes as there is an increased frequency of diabetes during states of excess iron (Dymock et al., 1972; Simcox and McClain, 2013). Further, epidemiological studies have also suggested that overconsumption of dietary iron is associated with an increased risk of developing diabetes (Shah and Fonseca, 2011; Simcox and McClain, 2013). The role of iron in the pathogenesis of diabetes is further highlighted by the discovery that a reduction of systemic iron stores leads to an improvement of the diabetic state (Fernandez-Real et al., 2002; Gamberini et al., 2008), which fits with other findings that an iron overload can generate oxidative stress (Straub et al., 2019). As pancreatic β cells are predisposed for iron accumulation and, at the same time, extremely susceptible to oxidative damage, dysfunction and apoptosis of pancreatic islets is considered a crucial factor for the pathogenesis of diabetes under iron overload conditions (McClain et al., 2006). Nonetheless, the role of iron in regulating insulin sensitivity in specific tissues and cell types is complex and insufficiently understood.
Previous studies in our lab show that specific depletion of mitochondrial iron in adipocytes leads to a massive expansion of adipose tissue, but preserved insulin sensitivity in obesity (Kusminski et al., 2012). Studies in mouse models and humans demonstrate that iron plays a direct and causal role in determining adiponectin and leptin levels (Gabrielsen et al., 2012; Gao et al., 2015), suggesting a role of iron in regulating adipocyte function (Simcox and McClain, 2013). This is not surprising, given that iron is not only a co-factor for rate-limiting components of the electron transport chain, but also a co-factor for many other enzymes, such as catalase and lipoxygenases, along with many additional components of the iron regulatory system. However, a number of important questions remain unresolved. While high ferritin levels are positively correlated with central adiposity (Iwasaki et al., 2005), iron deficiency is also positively associated with obesity (Cepeda-Lopez et al., 2011; Pinhas-Hamiel et al., 2003). This indicates that the role of iron in obesity and diabetes is likely to be complex (Simcox and McClain, 2013). Much work also remains with respect to determining which tissues are the primary drivers of obesity and diabetic development in the context of altered iron metabolism.
To gain a better understanding of the role of iron in adipose tissue, specifically in the adipocyte, we manipulated iron levels in adipocytes and found a critical role of iron in adipocyte development, maintenance and adipose tissue and systemic physiology. Of note, limiting iron specifically in adipocytes dramatically reduced adiposity and significantly improved systemic metabolism by controlling intestinal fat absorption. Our results further indicate that these changes in adipocytes lead to secondary effects in other organs, delineating an “inter-organ communication axis” between adipocytes and the digestive tract that has so far not yet been described.
RESULTS
TFRC-mediated Iron Transport Is Required for the Survival of Certain Fat Depots, but Not Others, During Development and Maturation
White and brown adipose tissue (WAT and BAT, respectively) and beige adipose tissue are three types of adipose tissue that commonly exist in mammals. We examined the expression of the transferrin receptor 1 (Tfrc), a common receptor for iron uptake, in different fat pads. Tfrc was highly expressed in BAT relative to other adipose tissues, but it showed a decrease over the course of BAT development. In contrast, inguinal white adipose tissue (iWAT) expressed low levels of Tfrc that gradually increased over the course of development and gonadal white adipose tissue (gWAT) expressed consistently low levels of Tfrc (Figure 1A). The expression of Tfrc in fractionated adipocytes of adult mice was higher in brown than white adipocytes (Figure 1B). This pattern of Tfrc expression in different adipose tissues correlates well with their predicted demand for iron, as BAT has a much higher mitochondrial content than WAT given the former’s strong thermogenic potential.
Figure 1. The Contribution of TFRC Mediated Iron Transport during Adipocytes Development and Maturation.

(A) mRNA levels of Tfrc in different fat pads of WT mice during development. n=5.
(B) mRNA levels of Tfrc in purified adipocytes fraction and SVF from fat pads of 2-month-old WT mice. n=4.
(C) Relative mRNA levels of Tfrc in purified adipocytes fraction from fat pads of 2-month-old Tfrcfl/fl and TfrcAKO mice. n=3.
(D) DAB iron staining of BAT harvested from Tfrcfl/fl and TfrcAKO mice at indicated age. Scale bar, 50 μM. Representative of 3 biological replicates.
(E) Basal OCR of diced fat pads of 2-month-old Tfrcfl/fl and TfrcAKO mice. n=5.
(F) Photograph of fat pads harvested from Tfrcfl/fl and TfrcAKO mice at indicated age. Representative of at least 5 biological replicates.
(G) H&E staining of fat pads harvested from Tfrcfl/fl and TfrcAKO mice at indicated age. Scale bar, 100 μM. Representative of 3 biological replicates.
(H) Overview of TfrciAKO mice model.
(I) Photograph of fat pads of 2-month-old iCon and TfrciAKO mice (Dox diet: E0-2 months). Representative of at least 5 biological replicates.
(J) Photograph of fat pads of 2-month-old iCon and TfrciAKO mice (Dox diet: E13.5-2 months). Representative of at least 5 biological replicates.
(K) Photograph of fat pads of 2-month-old iCon and TfrciAKO mice (Dox diet: 3 weeks-2 months). Representative of at least 5 biological replicates.
(L) Photograph of fat pads of 4-month-old iCon and TfrciAKO mice (Dox diet: 2 months-4 months). Representative of at least 5 biological replicates.
(M) Photograph of fat pads of 2-month-old iCon and TfrciAKO mice (Dox diet: E13.5-P21). Representative of at least 5 biological replicates.
(N) Photograph of fat pads of 2-month-old Tfrcfl/fl and TfrcAKO mice (TN: E13.5-2 months). Representative of at least 5 biological replicates.
Data are presented as mean ± SD. Two-tailed Student’s t-test. *P < 0.05, **P < 0.01. Results were confirmed in at least 2 independent experiments.
To further investigate the role of iron in adipose tissue, we generated mice that lack Tfrc in adipocytes (TfrcAKO) by crossing Tfrcfl/fl mice (Chen et al., 2015) with Adipoq-Cre mice (Eguchi et al., 2011). Tfrc was successfully deleted in adipocytes (Figures 1C, 1D, and S1A). We observed no obvious compensatory upregulation of other iron transporters in Tfrc-deficient adipose tissues. However, Fpn1, an iron exporter, exhibited a significant downregulation, reflecting an overall iron-deficient status (Figures S1B-S1D). Owing to the lack of iron, both the level of mitochondria and the oxygen consumption rate (OCR) were lower in adipose tissues of TfrcAKO mice relative to their controls (Figures S1E, S1F, and 1E). Despite these changes, adipocyte-specific Tfrc disruption did not affect body weight gain or whole-body iron status of TfrcAKO mice fed a chow diet (Figures S1G-S1L). Of note, We found that BAT gradually became paler and nearly disappeared in TfrcAKO mice. iWAT displayed similar trends, but with fewer changes. No obvious size differences were observed for gWAT between TfrcAKO and control mice (Figures 1F, and S1M-S1Q). H&E staining demonstrated that BAT and iWAT histology in TfrcAKO mice is initially normal. As of 1 week of age, a significant amount of cell death and inflammatory infiltration occurred in BAT of TfrcAKO mice. We observed widespread apoptosis in Tfrc-deficient BAT and iWAT starting at 3 weeks of age, while we found no signs of apoptosis or inflammation in Tfrc-deficient gWAT (Figures 1G and S1R). These findings demonstrate that TFRC-mediated iron uptake is essential for the postnatal survival of BAT and iWAT, but not for that of gWAT.
To characterize the role of TFRC-mediated iron transport beyond adipocyte development, we established an inducible knockout mouse model to specifically ablate Tfrc in adipocytes by crossing Tfrcfl/fl mice with Adipoq-rtTA:TRE-Cre mice (TfrciAKO). We designated Tfrcfl/fl:Adipoq-rtTA littermates that lack TRE-Cre as controls (iCon). This allowed us to expose TfrciAKO mice to a doxycycline (Dox)-diet at different stages of development and thus uncouple developmental effects from effects in the mature adipocyte (Figure 1H). Given that distinguishable BAT is first observed on embryonic (E) day 15.5 of development (Schulz and Tseng, 2013) and starts to express Adipoq around the same time (Song et al., 2020), we chose E0 and E13.5 for the initiation of Dox diet. When mice were exposed to Dox diet during the prenatal period to early adulthood (i.e., from E0 - 2 months or from E13.5 – 2 months), we found that TfrciAKO mice exhibited a similar phenotype to that of non-inducible (constitutive) TfrcAKO mice, as shown by the near complete loss of the BAT and a smaller size of the iWAT (Figures 1I, 1J, S1S, and S1T). When TfrciAKO mice were exposed to a Dox-containing diet only during the postnatal period (i.e., from P21 - 2 months or from 2 months - 4 months), we observed no morphological changes of adipose tissue in TfrdiAKO mice compared to controls (Figures 1K, 1L, S1U, and S1V). When mice were exposed to a Dox-containing diet during the period of E13.5 to P21, then switched to normal chow diet, TfrciAKO mice recapitulated the phenotype observed in TfrcAKO mice (Figures 1M and S1W). We found that even exposing these mice to thermoneutrality (TN) from E13.5 to 2 months, which suppresses the thermogenic and beiging activities and puts BAT “on hold”, could not block the widespread apoptosis of BAT and iWAT in TfrcAKO mice. However, iWAT displayed fewer morphological changes at TN, reflecting a reduced need for beiging (Figures 1N, S1X, and S1Y). We observed no difference in body weights between the two groups of mice fed a Dox-containing chow diet (Figure S1Z). Taken together, these observations indicate that TFRC-mediated iron transport is indispensable, especially for BAT, but also iWAT development and maturation. However, it is not required for the development of gWAT or the maintenance of well-differentiated mature BAT, iWAT, and gWAT.
TfrcAKO Mice are Potently Protected from High-fat Diet-induced Metabolic Disorders
When mice were maintained on a chow diet, we found that circulating adiponectin levels were significantly lower in TfrcAKO mice compared to Tfrcfl/fl controls (Figure S2A). However, Tfrcfl/fl and TfrcAKO mice displayed no difference with respect to leptin levels (Figure S2B). Fasting triglyceride levels and triglyceride clearance were similar between TfrcAKO and control mice, whereas non-esterified fatty acids (NEFA) levels were lower, and cholesterol levels were slightly higher in TfrcAKO mice (Figures S2C and S2D). Additionally, we found that TfrcAKO mice exhibited comparable glucose tolerance and insulin sensitivity compared to control mice, with the exception of a slightly higher basal and fasting glucose (Figures S2E-S2H). To confirm that there was no functional brown or beige adipose tissue in TfrcAKO mice, which are primary organs for thermogenesis (Morrison et al., 2014), we tested whether TfrcAKO mice can cope with cold exposure. We observed that TfrcAKO mice were extremely cold sensitive. However, this vulnerability to acute cold stress was alleviated once food was provided or following cold acclimation (Figure S2I), which indicates that alternative thermogenic mechanisms can compensate for the lack of BAT and iWAT.
When adult mice were challenged with a high-fat diet (HFD), we found that TfrcAKO and control mice displayed comparable plasma iron parameters, but TfrcAKO mice had elevated iron storage in the liver (Figures S2J-S2N). Importantly, the body weights of the two groups began to diverge once the HFD was administered, with TfrcAKO mice gaining far less body weight (Figures 2A and 2B). Body composition analysis revealed a striking lower level of fat mass in TfrcAKO mice compared to Tfrcfl/fl controls (Figures 2C-2E). We observed no notable difference in fasting and random-fed blood glucose levels between the two groups of mice (Figure 2F). However, TfrcAKO mice were more glucose tolerant and insulin sensitive than their controls (Figures 2G-2I). Beyond these changes, we found that both triglyceride and cholesterol levels were significantly lower in TfrcAKO mice, while no differences were observed in NEFA levels and triglyceride clearance between the two groups (Figures 2J-2L and S2O). Of note, we saw that TfrcAKO mice exhibited significantly lower levels of both adiponectin and leptin compared to their controls, a very unusual scenario, as these two adipokines usually have an inverse relationship (Figures 2M and 2N). Despite their low adiponectin levels, we observed that TfrcAKO mice showed less hepatic steatosis compared to Tfrcfl/fl controls (Figures 2O and 2P), probably due to improved leptin sensitivity (Zhao et al., 2019). Also, a dramatically fewer number of crown-like structures was observed in gWAT, which however did not apply to iWAT and BAT (Figure 2Q). Notably, even upon a HFD challenge under TN, we found that TfrcAKO mice were still well protected from HFD-induced metabolic deterioration, as demonstrated by lower levels of body weight gain and fat mass, improved glucose tolerance and insulin sensitivity, less liver steatosis, as well as a much lower level of inflammation in WAT (Figures 2R-2W). Circulating adiponectin and leptin levels were lower in these TfrcAKO mice as well compared with their controls (Figures S2P and S2Q). Consistent with the low iron demand of mature adipocytes, we found that ablation of Tfrc in fully differentiated mature adipocytes had only a subtle impact in adult TfrciAKO mice fed a HFD (Figures S2R-S2U). This indicates a very low iron turnover in fully differentiated mature adipocytes, which was supported by almost undisturbed Ferritin-H expression levels (Figure S2V). When TfrciAKO mice were exposed to a Dox-containing diet at a developmental stage to efficiently reduce adipocyte iron, or when they were exposed to cold to increase the demand for iron, we observed significant differences (Figures S2W-S2Z).
Figure 2. TfrcAKO Mice Displayed Improved Metabolic Phenotype under HFD Challenge at Both Room Temperature and TN Status.

(A) Representative photograph of Tfrcfl/fl and TfrcAKO mice after 10 weeks of HFD feeding. Representative of 5 biological replicates.
(B) Body weight of Tfrcfl/fl and TfrcAKO mice following HFD feeding. n=6.
(C-F) Body mass (C), fat pads weight (D), photograph of fat pads (E), and fasting and random blood glucose (F) of Tfrcfl/fl and TfrcAKO mice after 10 weeks of HFD feeding. n=6.
(G-I) Oral glucose tolerance test (OGTT) (G), insulin tolerance test (ITT) (H), and serum insulin levels during OGTT (I) of Tfrcfl/fl and TfrcAKO mice after 10 weeks of HFD feeding. n=6.
(J-N) Fasting serum triglycerides (J), NEFA (K), cholesterol (L), and random serum adiponectin (M) and leptin (N) levels of Tfrcfl/fl and TfrcAKO mice after 10 weeks of HFD feeding. n≥6.
(O-Q) Photograph of liver (O), H&E staining of liver (P), H&E staining of fat pads (Q) harvested from Tfrcfl/fl and TfrcAKO mice after 10 weeks of HFD feeding. Scale bar, 100 μM. Representative of 3 biological replicates.
(R-T) Tfrcfl/fl and TfrcAKO mice were exposed to TN (from E13.5, and thereafter), and fed on HFD from 2-month-old under TN. Body weight (R), weight of fat pads (S), and representative photograph of fat pads (T) of Tfrcfl/fl and TfrcAKO mice were presented after 10 weeks of HFD Feeding. n=8.
(U-V) OGTT (U) and serum insulin levels during glucose tolerance test (V) of Tfrcfl/fl and TfrcAKO mice described in R-T. n=8.
(W) H&E staining of liver and fat pads of Tfrcfl/fl and TfrcAKO mice described in R-T. Scale bar, 100 μM. Representative of 3 biological replicates.
Data are presented as mean ± SD. Two-way ANOVA with Dunnett’s test (B, G, H, I, U, V) or Two-tailed Student’s t-test (C, D, F, J-N, R, S). *P < 0.05, **P < 0.01. The weight of BAT includes the weight of BAT and the WAT connected to BAT. Results were confirmed in 3-5 independent experiments.
Collectively, these data reveal a nuanced but critical role of adipocyte iron in shaping the function of adipose tissue and suggest that restriction or chelation of adipocyte iron may constitute a promising therapeutic approach to obesity and its metabolic sequelae when done at the appropriate time points.
The Improved Metabolic Phenotype of TfrcAKO Mice is not Related to Defective BAT
Given previous reports that Ucp1 knockout mice are resistant to HFD-induced obesity as they use pathways with calorically high-cost for thermogenesis (Liu et al., 2003), we investigated whether the improved metabolic phenotype of TfrcAKO mice also depends on the observed defects in BAT. We thus generated TfrcUKO mice by crossing Tfrcfl/fl mice to Ucp1-Cre mice by which Tfrc was specifically eliminated from Ucp1-expressing cells (Figure 3A). We observed that BAT of TfrcUKO mice was paler than that of Tfrcfl/fl mice (Figures 3B and 3C). When these mice were challenged with a HFD, we observed no significant differences in weight gain, plasma fasting and random-fed glucose levels, glucose tolerance, or insulin sensitivity between two groups mice (Figures 3D-3H), thus suggesting that the improved metabolic phenotype of TfrcAKO mice is not dependent on defects in BAT. TfrcAKO mice were strikingly resistant to HFD-induced obesity at both room temperature and TN, while the obesity-resistant phenotype of Ucp1 knockout mice was quickly reversed when they were exposed to TN (Feldmann et al., 2009; Liu et al., 2003). The mechanisms underlying the obesity-resistant phenotype of TfrcAKO mice are thus apparently distinct from that of Ucp1 knockout mice.
Figure 3. Specific Loss of Tfrc in BAT Does Not Protect Mice from HFD-induced Metabolic Disorder.

(A) Overview of TfrcUKO mice model.
(B) Relative mRNA levels of Tfrc in purified adipocytes fraction from BAT of 2-month-old Tfrcfl/fl and TfrcUKO mice. n=4-5.
(C) Photograph of BAT harvested from 2-month-old Tfrcfl/fl and TfrcUKO mice with 2 more months of HFD feeding. Representative of 3 biological replicates.
(D) Body weight of Tfrcfl/fl and TfrcUKO mice during HFD feeding. n=5.
(E) Fasting and random blood glucose of Tfrcfl/fl and TfrcUKO mice after 2 months of HFD feeding. n=5.
(F-H) OGTT (F), ITT (G), and serum insulin levels during OGTT (H) of Tfrcfl/fl and TfrcUKO mice after 2 months of HFD feeding. n=5.
Data are presented as mean ± SD. Two-tailed Student’s t-test (B, E) or Two-way ANOVA with Dunnett’s test (D, F-H). *P < 0.05, **P < 0.01. Results were confirmed in 2 independent experiments.
Less Intestinal Lipid Absorption Contributes to The Improved Metabolic Phenotype of TfrcAKO Mice
To ascertain the reasons for the leaner and healthier phenotypes of HFD-fed TfrcAKO mice, we first examined food intake. There was no difference in food intake between TfrcAKO and control mice (Figures 4A, 4B, S3A, and S3B). We subsequently investigated the rates of energy expenditure (EE). TfrcAKO mice exhibited even lower levels of EE and oxygen consumption relative to their controls (Figures 4C and S3C). Meanwhile, we observed no difference in the respiratory exchange rate between the two groups of mice (Figure S3D).
Figure 4. TfrcAKO Mice Show Less Intestinal Lipid Absorption compared to Tfrcfl/fl Mice.

(A-C) Realtime food intake (A), accumulated food intake (B) and energy expenditure (C) in 2-month-old Tfrcfl/fl and TfrcAKO mice following 9 weeks of HFD. n=6.
(D) Feed efficiency of Tfrcfl/fl and TfrcAKO mice during HFD feeding. n=6.
(E) Serum triglycerides levels of fasted Tfrcfl/fl and TfrcAKO mice (HFD 8 weeks) before and after refeed. n≥9.
(F) Post gavage (olive oil) serum triglycerides levels in HFD-fed Tfrcfl/fl and TfrcAKO mice pre-treated with tyloxapol. n=5.
(G-I) Daily feces weight (G), fecal energy content (H), and daily fecal calorie content (I) of Tfrcfl/fl and TfrcAKO mice after 8 weeks of HFD feeding. n≥9.
(J) Feces from HFD-fed Tfrcfl/fl and TfrcAKO mice were collected to tubes with DNA/RNA shield buffer and put at 4°C for 6 hours. Representative of 3 biological replicates.
(K-N) Radioactivity of 3H and 14C in blood and feces of HFD-fed Tfrcfl/fl and TfrcAKO mice following 3H-Triolein and 14C-Palmitate gavage. n=6.
(O-P) Oil-Red-O staining of villus of proximal small intestine of fasted Tfrcfl/fl and TfrcAKO mice (HFD 8 weeks) after 1 hour refeed (O). Scale bar, 50 μM. Quantification of the oil red signal in villus (P). Representative of 3 biological replicates.
(Q-R) Electron microscopy images for lipid droplets in enterocytes of proximal small intestine of fasted Tfrcfl/fl and TfrcAKO mice (HFD 8 weeks) after 1 hour refeed. (Q). Scale bar, 6 μM. Quantification of the vesicle area in villus (R). Representative of 3 biological replicates.
(S) Relative mRNA levels of vesicle transport related genes in small intestine of Tfrcfl/fl and TfrcAKO mice after 10 weeks of HFD feeding. n=5.
(T) Model for fat transplantation.
(U) Body weight of recipient mice (TfrcAKO) received fat from either Tfrcfl/fl or TfrcAKO mice following HFD feeding. n=5.
(V-Y) Daily food intake (V), feces weight (W), fecal energy content (X), and daily fecal calorie content (Y) of the recipient mice after 4 weeks of HFD feeding. n=5.
Data are presented as mean ± SD. Two-tailed Student’s t-test (D, E, G-I, M, N, P, R, S, V-Y) or Two-way ANOVA with Dunnett’s test (F, K, L, U). *P < 0.05, **P < 0.01. Results were confirmed in 2-4 independent experiments.
We thus hypothesized that TfrcAKO mice may absorb nutrients less efficiently. Indeed, we found that TfrcAKO have a significantly lower feed efficiency (Figure 4D) compared to Tfrcfl/fl controls. In addition, when fasted TfrcAKO mice were exposed to a HFD during refeeding, they displayed significantly lower serum triglyceride levels than their controls. Administration of tyloxapol, a lipoprotein lipase inhibitor, did not abolish this effect, indicating the lower triglyceride levels in TfrcAKO mice are not a result of lipid clearance (Figures 4E and 4F). We also measured fecal output and fecal energy content. Surprisingly, TfrcAKO mice not only discharged more feces, but also had higher fecal energy content compared to control mice (Figures 4G-4I). Notably, we observed that feces from TfrcAKO mice floated on top of the DNA/RNA Shield buffer (Figure 4J). Sudan III staining revealed more fat globules in the feces of TfrcAKO compared to control mice (Figure S3E). We then examined intestinal lipid absorption. The absorption of lipids was significantly lower in TfrcAKO mice compared to that of Tfrcfl/fl controls, as indicated by lower levels of 3H-trolein and 14C-palmitate signals in blood, whereas higher levels of these components were found in feces (Figures 4K-4N, S3F, and S3G). All of these observations argue for a low level of dietary fat absorption in TfrcAKO mice.
We then examined lipid transport. Oil-Red O staining demonstrated that a large amount of lipids accumulated in the enterocytes of the small intestine in TfrcAKO mice upon fasting and HFD-refeeding (Figures 4O, 4P, and S3H). This was further confirmed with a more detailed visualization of these structures using electron microscopy (Figures 4Q, 4R, and S3I). These observations reflect that TfrcAKO mice have slower lipid absorption on the basolateral side of enterocytes. As vesicular trafficking-mediated transcytosis is critically involved in lipid transport, we examined the expression of vesicle transport-related genes. We found that TfrcAKO mice displayed a marked lower level of these genes relative to their controls (Figure 4S), which may explain in the less lipid absorption. We furthermore observed that intestinal organoids that were co-cultured with gWAT from HFD-fed TfrcAKO mice displayed a lower expression of genes related to vesicle transport relative to their controls (Figures S3J and S3K), which indicates that the signals that control gut lipid absorption indeed arise specifically from adipose tissue. To further confirm that this regulatory signal originates from adipose tissue, we performed fat transplants (Figures 4T and S3L). We found that TfrcAKO mice that received TfrcAKO fat displayed lower body weight gain compared to mice that received Tfrcfl/fl fat, while no differences in food intake were observed (Figures 4U and 4V). Consistent with the body weight gain, we found that daily fecal output and fecal energy content were higher in mice that received TfrcAKO fat tissue compared to those that received Tfrcfl/fl fat tissue (Figures 4W-4Y). This was also validated by transplantation of TfrcAKO and Tfrcfl/fl fat tissue into FAT-ATTAC mice (Pajvani et al., 2005), a model of inducible lipodystrophy (Figures S3M-S3Q).
Collectively, these observations following the transplantation of fat tissue further support our model invoking a crosstalk between adipose tissue and the gut in the regulation of lipid uptake.
The Beneficial Effects of a Low Level of Iron in WAT
Apart from the expression of genes related to intestinal vesicular trafficking, we examined other potential factors that may contribute to the altered intestinal lipid absorption, including the length of the intestine, gastric emptying rate, gastrointestinal transit time, bile acid levels, the expression of fatty acid/cholesterol transport- and triglyceride synthesis-related genes and the expression of HIF-2α, the last of which is involved in the regulation of ceramide metabolism in the intestine and positively correlated with hepatic steatosis (Xie et al., 2017; Yen et al., 2009) (Figures S4A-S4M). We observed no obvious differences between TfrcAKO and control mice in any of these parameters.
To elucidate the underlying mechanism by which iron-deficient adipose tissue affects intestinal lipid absorption, we performed a series of multi-omic experiments. First, we performed RNA-seq on gWAT from TfrcAKO and control mice fed a HFD. Hierarchical clustering and gene ontology (GO) analysis revealed that genes related to lipid metabolism were markedly higher, while genes related to inflammation and cell proliferation were significantly lower in TfrcAKO mice relative to their controls (Figures 5A-5C). Consistent with the restricted expansion of gWAT in TfrcAKO mice, we found that mWAT also displayed a significantly lower expansion in TfrcAKO mice compared to Tfrcfl/fl controls (Figure 5D). Given the direct connection of mWAT with the serosa layer of the intestine, we performed a proteomic analysis of small extracellular vesicles (sEVs) isolated from mWAT of TfrcAKO and control mice fed a HFD. The analysis of the differential protein abundance in sEVs revealed that the most down-regulated proteins in the TfrcAKO group were related to iron transport and reactive oxygen species (ROS) production, whereas the most up-regulated proteins were related to extracellular matrix (ECM) and cell-ECM interactions (Figures 5E-5G, Table S1). In agreement with these observations, the expression of genes related to inflammation and oxidative stress were significantly lower in gWAT of TfrcAKO mice compared to their controls (Figure S5A). Extending beyond WAT, we also observed lower levels of inflammation and oxidative stress in the small intestine of TfrcAKO mice compared to Tfrcfl/fl controls (Figure S5B). Of note, F2-isoprostanes, products formed by free radical-mediated peroxidization of fatty acids, were about 20-fold lower in gWAT of TfrcAKO mice compared to that of Tfrcfl/fl controls (Figure 5H), reflecting a markedly lower lipid peroxidation. Meanwhile, triglyceride synthesis was enhanced in gWAT of TfrcAKO mice compared to Tfrcfl/fl controls (Figure 5I). These data suggest that limiting iron levels in adipocytes results in a much healthier WAT under a HFD-challenge. As mentioned above, lipid metabolism was among the most up-regulated processes in gWAT of TfrcAKO mice relative to their controls (Figure 5B). We therefore performed lipidomic profiling on gWAT and serum of TfrcAKO and control mice fed a HFD. We observed that the gWAT of TfrcAKO mice exhibited subtle changes with respect to ceramides and sphingomyelins compared to Tfrcfl/fl controls. However, dihydroceramides, for which an anti-proliferative role has been reported (Siddique et al., 2015), were significantly higher in the gWAT of TfrcAKO mice relative to that of their controls (Figure S5C, Table S2), which was consistent with restricted fat expansion. Notably, we observed that most of the detected ceramides, dihydroceramides, sphingomyelins and phospholipids were significantly lower in the serum of TfrcAKO mice compared to their controls (Figures 5J and 5K, Table S3). The serum fatty acid profile also showed lower levels of many types of fatty acids, such as myristoleic acid and palmitoleic acid (Figure 5L). We further performed proteomic profiling of the secretome of cultured adipocytes upon a short-term or long-term depletion of iron (Table S4). Cross analysis revealed a total of 44 proteins that were commonly identified with changes above two-fold. Among these proteins, Asah1, Gltp, and Glb1 are related to sphingolipid metabolism (Figures 5M and S5D). Interestingly, we found that the expression of genes related to de novo and the salvage pathway of ceramide synthesis were also significantly lower in the small intestine of TfrcAKO mice compared to Tfrcfl/fl controls (Figure S5E). This suggests that limiting iron levels in adipose tissues have profound effects on systemic lipid metabolism. Of note, when the small intestine was immersed in PBS, we observed that the diameter of the small intestine was larger in TfrcAKO mice compared to control mice (Figure 5N). H&E staining demonstrated that the density of crypts and the depth of the muscle layer were higher in TfrcAKO than control mice (Figure 5O). Taken together, the lower levels of inflammation and oxidative stress in the small intestine (Figure S5B) reflects a healthier small intestine in TfrcAKO mice challenged with a HFD. Indeed, compositional changes of the gut microbiota were also detected in HFD-fed TfrcAKO mice (Figure 5P). Overall, lowering iron in white adipocytes has multiple beneficial effects upon a HFD-feeding, all of which point to a healthier WAT.
Figure 5. The Beneficial Effects of Lowering Iron in Adipocytes upon HFD-challenge.

(A-C) Hierarchical clustering of transcriptional profiles of gWAT obtained from Tfrcfl/fl and TfrcAKO mice after 10 weeks of HFD feeding (A). Labels identify gene clusters showing enrichment GO analyses (B-C). n=3, each sample combined from 2 mice. FC, fold change.
(D) Weight of mWAT in Tfrcfl/fl and TfrcAKO mice after 10 weeks of HFD feeding. n=6. Results were confirmed in at least 3 independent experiments.
(E-G) Volcano plot depicting the fold change and significance of differential protein abundance in sEV isolated from mWAT of Tfrcfl/fl and TfrcAKO mice after 10 weeks of HFD feeding (E). GO analysis of proteins downregulated (F) and upregulated (G) more than two-fold in sEV of TfrcAKO mice. n=2, each sample combined from 3-4 mice.
(H-I) F2-Isoprostane levels (H) the expression of triglycerides synthesis related genes (I) in gWAT of Tfrcfl/fl and TfrcAKO mice after 10 weeks of HFD feeding. n=5.
(J-L) Profiling of sphingolipids (J), serum phospholipids (K), and fatty acids (L) in serum of Tfrcfl/fl and TfrcAKO mice after 10 weeks of HFD feeding. n=6.
(M) Scatter plot of differential protein levels upon fold changes in medium of fully differentiated Tfrcfl/fl and TfrcAKO adipocytes, or Adipoq-Cre adipocytes with AAV mediated specific overexpression of AAV-eGFP or AAV-Fpn1C326S. Representative of 2-3 biological replicates.
(N-O) Photograph of small intestine (N) and H&E staining of proximal small intestine (O) harvested from Tfrcfl/fl and TfrcAKO mice after 4 months of HFD feeding. Scale bar, 200 μM. Representative of at least 15 biological replicates. Results were confirmed in 3 independent experiments.
(P) Phylum level and class level comparison of gut microbiota proportional abundance in feces of Tfrcfl/fl (n=5) and TfrcAKO (n=4) mice after 2 months of HFD feeding.
Data are presented as mean ± SD. Two-tailed Student’s t-test. *P < 0.05, **P < 0.01.
Reducing Iron Levels in Adipocytes of Adult Mice Protects from HFD-induced Metabolic Deterioration
Given the low iron demand and iron turnover in well differentiated mature adipocytes, we specifically overexpressed a mutant form of the iron exporter, Fpn1C326S, in adipocytes of adult mice to determine whether specifically lowering adipocyte iron levels can protect mice from HFD-induced metabolic disorder. To this end, Adipoq-Cre mice were injected intraperitoneally (IP) with AAVs encoding Cre-dependent eGFP (Data S1) or Cre-dependent Fpn1C326S (Data S2) to mainly target the visceral fat depots in a selective manner (Figure 6A). We found that eGFP and Fpn1 C326S were successfully expressed in the gWAT of mice (Figures S6A-S6C). When these mice were challenged with a HFD, AAV-Fpn1C326S mice gained less body weight compared to AAV-eGFP mice. Body composition analysis revealed a lower fat mass in AAV-Fpn1C326S mice relative to AAV-eGFP controls. The expansion of gWAT and mWAT were most dramatically affected, whereas BAT was less affected given that we administered the virus by IP injection (Figure 6B-6E). Mice that received AAV-Fpn1C326S also displayed improved glucose tolerance and insulin sensitivity relative to AAV-eGFP controls (Figure 6F and 6G). Both triglyceride and cholesterol levels were significantly lower in the AAV-Fpn1C326S group, while no differences were observed in NEFA levels, as compared to AAV-eGFP controls (Figure 6H). In addition to the improved metabolic phenotype, H&E staining showed that mice that received AAV-Fpn1C326S exhibit less liver steatosis and displayed fewer crown-like structures in gWAT compared to AAV-eGFP controls. Unlike the constitutive deletion of Tfrc, we observed no adipocyte death or inflammatory infiltrates in iWAT and BAT of AAV-Fpn1C326S mice following IP administration of the AAVs (Figure 6I). When BAT was directly injected with AAV-Fpn1C326S to increase the local expression, widespread cell death was obvious, indicating efficient depletion of iron from brown adipocytes (Figures S6D and S6E). To examine whether reduced intestinal lipid absorption contributed to the healthier phenotype of mice receiving AAV-Fpn1C326S, we assessed their food intake and fecal output. There was no difference in food intake, whereas fecal output was significantly higher in AAV-Fpn1C326S mice compared to the AAV-eGFP group (Figure 6J and 6K). Sudan III staining demonstrated more fat globules in the feces of AAV-Fpn1C326S mice compared to AAV-eGFP controls (Figure S6F). Consistent with these observations, we observed a lower level of lipid absorption in HFD-fed AAV-Fpn1C326S mice compared to AAV-eGFP controls, as demonstrated by lower levels of 3H-trolein and 14C-palmitate signals in circulation and higher levels of these labels in feces (Figures 6L, 6M, S6G, and S6H).
Figure 6. AAV-Mediated Specific Overexpression of An Iron Exporter in Adipocytes Mitigates Metabolic Dysfunction.

(A) Overview for AAV-mediated specific overexpression of eGFP or Fpn1C326S in adipose tissue of adult mice.
(B) Body weight of Adipoq-Cre mice received either AAV-eGFP or AAV-Fpn1C326S following HFD feeding. n=6.
(C-E) Body mass (C), weight of fat pads (D), representative photograph of fat pads (E) of Adipoq-Cre mice received either AAV-eGFP or AAV-Fpn1C326S, after 4 weeks HFD feeding. n=6.
(F-H) OGTT (F), serum insulin levels during OGTT (G), fasting serum triglycerides, NEFA, and cholesterol (H) of Adipoq-Cre mice received either AAV-eGFP or AAV-Fpn1C326S, after 4 weeks HFD feeding. n=6.
(I) H&E staining of liver and fat pads of Adipoq-Cre mice received either AAV-eGFP or AAV-Fpn1C326S , after 4 weeks HFD feeding. Scale bar, 200 μM. Representative of 3 biological replicates.
(J-K) Food intake (J), fecal output (K) of Adipoq-Cre mice received either AAV-eGFP or AAV-Fpn1C326S, after 3 weeks HFD feeding. n=6.
(L-M) Radioactivity of 3H and 14C in feces following 3H-Triolein and 14C-Palmitate gavage in Adipoq-Cre mice received either AAV-eGFP or AAV-Fpn1C326S, after one week of HFD feeding. n=6.
(N-Q) Body weight (N), Body mass (O), weight of fat pads (P), representative photograph of fat pads (Q) of Adipoq-Cre mice with 2 months HFD feeding, then received either AAV-eGFP or AAV-Fpn1C326S, with continued HFD feeding for 7 weeks. n=5.
(R-T) OGTT (R), serum insulin levels during OGTT (S), fasting serum triglycerides, NEFA, and cholesterol (T) of Adipoq-Cre mice with 2 months HFD feeding, then received either AAV-eGFP or AAV-Fpn1C326S, with continued HFD feeding for 6.5 weeks. n=5.
(U-W) H&E staining of liver and fat pads (U), food intake (V), fecal output (W) of Adipoq-Cre mice with 2 months HFD feeding, then received either AAV-eGFP or AAV-Fpn1C326S , with continued HFD feeding for 7 weeks. Scale bar, 200 μM. Representative of 5 biological replicates.
Data are presented as mean ± SD. Two-way ANOVA with Dunnett’s test (B, F, G, N, R, S, V) or Two-tailed Student’s t-test (C, D, H, J-M, O, P, T, W). *P < 0.05, **P < 0.01. Results were confirmed in at least 2 independent experiments.
We subsequently examined whether lowering iron specifically in adipocytes would reverse established HFD-induced obesity. Adipoq-Cre mice were fed a HFD for 2 months and then received AAV-Fpn1C326S or AAV-eGFP by IP injection. Of note, we observed that lowering adipocyte iron levels in mice with established obesity also improved their metabolic health, as demonstrated by less body weight gain, less fat mass, improved glucose tolerance, and improved insulin sensitivity (Figures 6N-6S). We observed no obvious difference in serum triglycerides, NEFA, and cholesterol levels between AAV-Fpn1C326S and AAV-eGFP groups (Figure 6T). H&E staining showed that AAV-Fpn1C326S mice exhibited less liver steatosis and fewer crown-like structures in gWAT compared to AAV-eGFP controls (Figure 6U). We also assessed the food intake of AAV-Fpn1C326S mice, which was lower around 2-4 weeks following AAV injection, as compared to AAV-eGFP controls. Moreover, we observed that fecal output was consistently higher in AAV-Fpn1C326S group compared to the AAV-eGFP group (Figure 6V and 6W).
These observations suggest that specifically lowering iron in mature adipocytes not only protects mice from HFD-induced metabolic disorders, but it can also therapeutically alleviate the metabolic dysregulation in mice with established obesity.
DISCUSSION
As an energy storage organ, the WAT evolved to have a relatively small number of iron-containing mitochondria, which allows only an overall low respiration rate supporting its function as an energy reservoir. The BAT, in contrast, contains a high number of densely packed iron-containing mitochondria and displays extensive vascularization, which gives the tissue a brown color and a thermogenic capability (Saely et al., 2012). In this study, we show that iron is not only differentially required for distinct subtypes of adipocytes, but also selectively needed at different stages of adipocyte development. The fact that iron levels are tightly controlled in different types of adipocytes under varying conditions suggests a direct role of iron in modulating adipocyte function.
Over the past few decades, studies have shown that the adipose tissue is a metabolically dynamic endocrine organ (Kershaw and Flier, 2004; Scherer, 2006). Despite its role in storage of excess energy, the adipose tissue also exerts multiple roles in the control of metabolic homeostasis (Funcke and Scherer, 2019; Kershaw and Flier, 2004; Scherer, 2006; Straub and Scherer, 2019; Zhang and Scherer, 2018). Recent studies have shown that iron can modulate systemic metabolism through regulation of adiponectin and leptin production in adipose tissue (Gabrielsen et al., 2012; Gao et al., 2015). Manipulation of iron levels in mitochondria of adipocytes also has an impact on systemic metabolism (Kusminski et al., 2012; Kusminski and Scherer, 2019). Although the exact role of iron in obesity and diabetes development is not well defined, both animal studies and human clinical studies demonstrate that iron is critically involved in the regulation of metabolism in adipose tissue and that effects originating in adipocytes immediately affect other tissues as well (An et al., 2021). We observe that the specific deletion of Tfrc in adipocytes protects mice from HFD-induced obesity under both ambient and thermoneutral conditions. Of note, a recent study that uses a distinct Tfrcfl/fl mouse strain demonstrates a phenotype opposite of what we observed (Li et al., 2020). However, lowering adipocyte iron with alternative approach recapitulates the phenotype that we observe in our TfrcAKO mice. The most important conclusion from our studies is that the specific manipulation of adipocyte iron levels leads to a lower level of intestinal lipid absorption upon HFD feeding. Our studies lead us to suggest the existence of an inter-organ cross talk between the adipose tissue and the intestine, an axis which has not been appreciated so far.
The general involvement of adipose tissue in inter-organ crosstalk has been well recognized, especially in light of the discovery of adipokines. It is well-established that the adipose tissue cross-talks with other organs, such as the central nervous system, pancreas and liver. The identification of additional mediators, such as norepinephrine, myokines and cardiokines further adds to the complexity of this multi-directional inter-organ exchange (Romacho et al., 2014). Obesity induces fundamental changes in the gut, which have been proposed to contribute to the development of the metabolic syndrome (Konrad and Wueest, 2014). Hormones secreted by the gut, such as ghrelin, GIP and GLP-1, are also considered to have an impact on adipose tissues (Priest and Tontonoz, 2019). However, the reverse crosstalk between adipose tissue and the digestive tract remains poorly understood and has not been thoroughly examined to date. The efficient absorption of dietary fat and the storage of these lipids in adipose tissue confer a survival advantage during a famine or during times of high-energy-demand. However, adipose tissue has expansion limits (Rutkowski et al., 2015). Evolutionarily, WAT seldom reached this limit, while obesity is now widespread in industrialized countries. Overwhelming adipocytes with lipids results in a diversion of lipids to ectopic accumulation in alternate cells, which contributes to the development of lipotoxicity and the metabolic syndrome (Unger and Scherer, 2010). Here, we report a secondary mechanism that acts after food consumption to avoid a nutrient overload in the system by limiting absorption of calories. The sensor chosen by the overchallenged adipocyte for this feedback signal is a specific reduction of adipocyte iron. This is the first series of observations that involve an axis from adipose tissue to the intestine. Our model strongly supports the idea that white adipocyte iron can become rate limiting, especially under conditions of excess energy supply.
It is widely appreciated that iron is critically involved in the generation of lipid peroxidation products. Beyond that, iron is a co-factor for components of the electron transport chain, and for many proteins and enzymes involved in metabolism (Crooks et al., 2018). Although the role of iron in lipid metabolism is established, how iron regulates lipid metabolism in adipose tissue in vivo is still poorly understood. Clinical studies show that a systemic reduction of iron levels by iron chelation or phlebotomy can improve the diabetic phenotype (Fernandez-Real et al., 2002; Gamberini et al., 2008). Of note, bariatric surgery is one of the most effective weight loss approaches. Except for its long-term weight loss effects, there are many other benefits from bariatric surgery, such as prevention of type 2 diabetes (Shin, 2018). As iron deficiency is a side effect of bariatric surgery (Jauregui-Lobera, 2013), the relative lack of iron might contribute to the long-term remission of type 2 diabetes. However, systemic iron chelation or bariatric surgery leads to whole-body iron depletion, which ultimately results in iron deficiency-induced anemia. By genetically ablating Tfrc in adipocytes or using AAV-mediated specific overexpression of Fpn1C326S in adipocytes, our study shows that such cell-specific depletion of iron conveys a striking protection from metabolic disorders. This highlights the potential for the manipulation of iron levels selectively in adipocytes, rather than in the whole body, as a promising therapeutic intervention. In addition, limiting iron levels in adipocytes leads to a remarkably lower level of circulating lipids, such as ceramides. As ceramide levels show a robust correlation with co-morbidities of obesity and type 2 diabetes (Chaurasia et al., 2019), the lower levels of ceramides may also contribute to the beneficial phenotype. Notably, gut microbiota is also changed as the lack of iron in adipocytes. Though there is a well-described influence of the intestinal microbiome on systemic metabolism (Turnbaugh et al., 2009), we cannot exclude the possibility that these changes are secondary to a healthier gut or altered lipid metabolism in TfrcAKO mice.
Lowering adipocyte iron levels leads to a plethora of changes, any one of which could be the driving factor for the phenotypic changes. We suggest that the beneficial outcomes are due to a combination of multiple effects, whose underlying molecular mechanisms need to be investigated further (Figure 7). Taken together, our study indicates that the iron content of adipocytes is a promising target for therapeutic intervention. Further studies developing clinically viable approaches towards selectively lowering iron in adipocytes with all of its beneficial effects may open new avenues for the treatment of the metabolic syndrome.
Figure 7. Summary of The Effects of Lowering Adipocytes Iron on Metabolism.

Selective lowering iron in white adipocytes has multiple beneficial effects in mice upon a HFD-feeding. Adipocytes iron content can be used as a sensor of WAT to activate an adipose-gut crosstalk to regulate lipid absorption.
LIMITATIONS OF STUDY
Our study was carried out in preclinical models and awaits clinical validation. Further, we observed a fat-gut cross-talk by a number of different approaches, but we did not identify a distinct molecular mechanism that explains the means by which this cross-talk occurs. Additionally, the demand and turnover of iron is very low in well-differentiated mature adipocytes. To benefit metabolic end points, iron needs to be actively depleted from mature adipocytes, such as by overexpression of the iron exporter Fpn1. As iron is an essential trace element, targeted iron depletion needs to be controlled tightly. Lastly, our current AAV-mediated adipocyte-specific overexpression Fpn1C326S requires the help of Cre recombinase. For viable therapeutic FPNC326S overexpression in humans, a Cre-independent, but adipocyte-specific, vector design will need to be established.
STAR METHODS TEXT
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Philipp E. Scherer (Philipp.Scherer@UTSouthwestern.edu)
Material Availability
Mouse lines and plasmids generated in this study are available from the lead contact upon request.
Data and Code Availability
The accession numbers for the RNA, 16S RNA sequencing data reported in this paper are GEO: GSE144938, SRA NCBI: PRJNA718550. One of the TfrcAKO fecal sample did not generate the required number of sequencing reads, hence, was excluded from 16S analysis.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models
Mice were maintained on a 12-hour dark/light cycle and housed in groups of 3-5 with unlimited access to water and food at room temperature or as indicated below. All mice were housed in barrier facility with biosafety level 2. Only healthy mice were used for experiments. For cold exposure experiment, mouse was single-caged, and maintained at 6°C rodent chamber. For thermoneutral experiment, mice were maintained at 30°C rodent chamber. Normal chow diet, 2916, TEKLAD Diet; doxycycline (Dox) chow diet (600 mg/kg diet), S4107, Bio-serv; doxycycline 60% HFD (600mg/kg diet), S5867, Bio-serv; 60% HFD paste, S1850, Bio-Serv. 60% HFD green pellet, D12492i, Research Diets. All animal procedures were approved by the University of Texas Southwestern Medical Center Institutional Animal Care and Use Committee, animal protocol number 2015-101207G. All efforts were made to follow the Replacement, Refinement and Reduction guidelines. Tfrcfl/fl (JAX028363) (Chen et al., 2015), Adipoq-Cre (JAX028020) (Eguchi et al., 2011), Ucp1-Cre (JAX024670) (Kong et al., 2014), and TRE-Cre (JAX006234) (Perl et al., 2002) mice were obtained from Jackson Laboratory. To test the deletion of Tfrc, primers are designed on exons between the two loxp sites. The Adipoq-rtTA and FAT-ATTAC mice were generated as previously described (Pajvani et al., 2005; Sun et al., 2012). All mice were maintained on the C57BL/6J background, male mice were used for this study. All mice were used as the result of in-house matings. Tfrcfl/fl:Adipoq-Cre mice (TfrcAKO) were bred with Tfrcfl/fl mice, Tfrcfl/fl:UCP1-Cre mice (TfrcUKO) were bred with Tfrcfl/fl mice, Tfrcfl/fl:Adipoq-rtTA:TRE-Cre mice (TfrciAKO) were bred with Tfrcfl/fl:Adipoq-rtTA (iCon) mice. Littermate controls were used for experiments. For FAT-ATTAC mice, male FAT-ATTAC mice were bred with WT female mice. All FAT-ATTAC mice carried one allele of transgene. FAT-ATTAC Littermates were used as recipient of fat from Tfrcfl/fl or TfrcAKO littermates. For Doxycycline induced knockout model, doxycycline was given to all groups of mice. The age and number of mice analyzed for each experiment is detailed in the Figure legends.
Primary organoid Culture
Crypt isolation from small intestine of mice followed the protocol as previously described (McFarlane et al., 2015). Small intestines were harvested, cut open longitudinally, and placed in a 50 ml conical tube containing 30 ml ice-cold PBS with 10 mM EDTA. Intestines were incubated on ice with intermittent shaking, decanting, and replacement of buffer to get rid of villus material. Crypts were then collected and passed through 100 μm cell strainers, followed by 70 μm cell strainer twice to isolate crypts from villus debris. Thereafter, crypts were pelleted, suspended in ADMEM/F12 (Life Technologies), counted under an inverted microscope, plated to a density at 100 crypts per well (24 well plate) in a 20 μl droplet consisting of a 1:9 ratio of ADMEM/F12 and Growth Factor-Reduced Matrigel (Corning), and cultured with ENR media (basic organoid medium). Medium were changed every other day. Crypts were cultured for 12 days to form organoids. Only P0 organoids were used for experiment. For the co-culture assay, gWAT from HFD-fed Tfrcfl/fl or TfrcAKO mice were cut to small pieces in sterile dish, 50 mg gWAT of each group were put in transwell (Corning) and co-cultured overnight with organoid in ENR media supplemented with 100 μM BSA conjugated palmitate.
Primary Cell Culture
Stromal vascular fraction cells were obtained from inguinal adipose tissues from 5-week-old male Tfrcfl/fl and TfrcAKO mice. To isolate adipocytes and stromal vascular fraction cells, dissected adipose tissues were cut to small pieces and digested for about 50 min at 37°C in DMEM/F12 medium (Gibco) containing Collagenase IV (Roche, 1U/ml) and Dispase II (Sigma, 1U/ml). Digested cells were filtered through a 100 μm cell strainer to a new tube. The flow-through cells were centrifuged at 600g at 4°C for 5 min. The floated adipocytes were transferred to a new 15ml conical tube with 10 ml DMEM/F12 medium, centrifuged at 600g at 4°C for 5 min, and then transferred to 1.5 ml tube for future use. Cell pellet was re-suspended in DMEM/F12 medium contained 10% FBS, and then filtered through a 40 μm cell strainer. The flow-through cells were centrifuged at 600g at 4°C for 5 min, and re-suspended in DMEM/F12 medium contained 10% FBS, 100 U/ml Penicillin, 100 μg/ml Streptomycin (complete medium), and seeded into a 35 mm culture dish. After the cells reached confluence for 2 days, cells were exposed to the adipogenic cocktail containing dexamethasone (1 μM), insulin (5 μg/ml), isobutylmethylxanthine (0.5 mM) and rosiglitazone (1 μM) in complete medium. After 2 days, cells were maintained in complete medium containing 5 μg/ml insulin, and the medium was changed every two days.
AAV-based Fpn1C326S overexpression
The Fpn1 (Slc40a1) open reading frame (ORF) was PCR amplified from mouse intestinal cDNA as two overlapping fragments introducing a p.C326S mutation that renders the protein hepcidin-resistant (Altamura et al., 2014) and cloned into a custom pAAVK CAG-FLEX vector based on another published vector (a gift from Hongkui Zeng; Addgene #51502). As a control, an enhanced green fluorescent protein (eGFP) ORF was PCR amplified from plasmid DNA and cloned into the same pAAVK CAG-FLEX vector. Vector sequences are supplied as Data S3. The non-expressing, anti-sense eGFP or Fpn1C326S coding sequences of these AAV expression vectors are flanked by two double-lox sites in inverted orientation. Cre recombination irreversibly flips the coding sequence into the sense direction, permitting protein expression. To produce AAVs (serotype: AAV2/Rec2 or AAV2/DJ ) (Charbel Issa et al., 2013; Griffin et al., 2019), near-confluent HEK293T cells growing in 15 cm dishes were transfected with 6 μg pAAVK CAG-FLEX-eGFP/Fpn1C326S, 12 μg pHelper (Cell Biolabs Inc., San Diego, CA, USA), and 24 μg pRepCap Rec2 (a gift from Deborah Young, University of Auckland, New Zealand) or pAAV-DJ (Cell Biolabs) in DMEM supplemented with 5% FBS, 2 mM L-alanyl-L-glutamine dipeptide (GlutaMAX), 100 U/ml penicillin and 100 μg/ml streptomycin using PEI (linear, MW 25000; Polysciences Inc., Warrington, PA, USA) at a PELDNA mass ratio of 3:1. Culture supernatants were collected 3 days post-transfection and stored at 4°C and fresh DMEM supplemented with 5% FBS, 2 mM GlutaMAX, 100 U/ml penicillin and 100 μg/ml streptomycin was added. Culture supernatants and cells were collected 5 days post-transfection, added to the stored supernatants, and centrifuged to pellet the cells. AAVs were purified by an established two-phase partitioning procedure (Guo et al., 2012). The supernatants were set aside and the cells were resuspended in lysis buffer (50 mM Tris-HCl, 150 mM NaCl, and 2 mM MgCl2; pH 8.00) and lysed by three consecutive freeze-thaw cycles in liquid nitrogen and a water bath at 37°C. The cell lysates were supplemented with 50 U/ml Benzonase (Sigma-Aldrich, St. Louis, MO, USA) and 10 U/ml RNase I (ThermoFisher Scientific, Waltham, MA, USA), incubated for 30 min at 37°C, supplemented with 0.5% w/v SDS, and incubated for another 30 min at 37°C. The cell lysates were cleared from debris by centrifugation and added to the supernatants set aside earlier. To this, 500 mM NaCl and 8%w/w PEG-8000 were added, and the samples were incubated overnight at 4°C and centrifuged for 30 minutes at 4,000xg and 4°C. The pellets were resuspended in purification buffer (50 mM HEPES, 150 mM NaCI, 20 mM EDTA, 1%w/w sarkosyl; pH 8.00), an equal volume of chloroform was added, and the samples were vortexed and centrifuged for 15 min at 4,000 g. The upper aqueous phases were collected, 10%w/w PEG-8000 and 13.2%w/w (NH4)2SO4 were added, and the samples were vortexed and centrifuged for 15 min at 3,000 g with low deceleration. The lower aqueous phases were collected. Amicon Ultra-15 centrifugal filter units (100 kDa MWCO; ThermoFisher Scientific) were preconditioned with DPBS supplemented with 0.1%w/v Pluronic F-68 (ThermoFisher Scientific) and washed once with DPBS supplemented with 0.01%w/v Pluronic F-68. The collected aqueous phases washed thrice with a surplus of injection buffer (DPBS supplemented with 200 mM NaCl and 0.001 %w/v Pluronic F-68) and concentrated using the prepared filter units. Purified AAVs were aliquoted and stored at −80°C until use. To determine AAV titers, qPCR was performed according to an established protocol (Aurnhammer et al., 2012) using the following primers: 5’-GGA ACC CCT AGT GAT GGA GTT-3’ and 5’-CGG CCT CAG TGA GCG A-3’. AAVs are diluted to 1×1010/μl in injection buffer. For prevention group, two-month-old male Adipoq-Cre mice were intraperitoneally injected with 1.5×1012 AAV-eGFP/mice or 1.5×1012 AAV-Fpn1C326S/mice (Serotype: AAV2/Rec2). Mice were switched for HFD following 1 week of the injection, and were intraperitoneally injected with the same amount of virus again following 1 week of HFD. For treatment group, two-month-old male Adipoq-Cre mice were fed HFD for 2 months, and then were intraperitoneally injected with 1.5×1012 AAV-eGFP/mice or 1.5×1012 AAV-Fpn1C326S/mice (Serotype: AAV2/Rec2). For primary cultured adipocytes, stromal vascular fraction cells were obtained from inguinal adipose tissues of Adipoq-Cre mice. Cells from each mice were plated in one 10 cm dish. At day 7 of differentiation, the differentiated adipocytes were incubated with AAV-eGFP or AAV-Fpn1C326S (Serotype: AAV2/DJ) with MOI 1:2000. After 2 days of infection, infected cells were washed twice with serum and phenol red free DMEM/F12 (Gibco) medium, and then incubated in serum and phenol red free DMEM/F12 medium supplemented with 5 μg/ml insulin. Culture medium was harvested after 36 hours incubation. Medium from three 10 cm dishes was pooled together as one sample. Medium was concentrated by 3kD Ultra centrifugal filter (Amicon) for proteomic analysis.
METHOD DETAILS
Physiological assays
OGTTs were performed in mice without access to food overnight prior to administration of 2 g/kg body weight for chow diet group, 1.5 g/kg body weight for HFD group. ITTs were performed in mice without access to food for 4 hours prior to administration of 0.5 U/kg body weight insulin (Eli Lilly) for the chow diet group, 0.75 U/kg body weight for the HFD group by intraperitoneal injection. Triglyceride clearance was performed in mice without access to food for 4 hours prior to administration of 15 μl/g body weight intralipid (Sigma) by gastric gavage. 20 μl of blood was collected at the indicated time points for triglyceride measurement. After one week of HFD feeding, tyloxapol (Sigma) was used to inhibit plasma triglyceride hydrolysis, 500 mg/kg tyloxapol was injected via tail vein 30 min before olive oil gavage (10 μl olive oil/g BW). For fasting-refeeding experiments, mice were on HFD for about 2 months, and fasted for 8 hours during the light phase and refed with HFD for 30 min or 1 hour as indicated at the beginning of the successive dark phase. For 3H-Trolein and 14C-Palmitate uptake assay, mice were single caged and fed on HFD for one week. Mice were then fasted overnight, and administered with 2 μCi 3H-Triolein (PerkinElmer) together with 1 μCi 14C-Palmiate (PerkinElmer) in 200 μl 1% intralipid (Sigma) by gavage after 15 min refeeding. 10 μl of tail blood was collected at 15min, 30min, 1h, 2h, 3h, 4h, 5h, 24h, 72h following gavage, immediately added into a scintillation vial containing 5 ml 3a70B complete counting cocktail (RPI Corp, Mount Prospect, IL), and shaken vigorously to disperse. Feces were collected at 24h, 48h, 72h after gavage. Feces were immersed in 4.5 ml chloroform-methanol 2:1 mixture at 4°C overnight, and disrupted by bead vortex. The content was then mixed with 3 ml 1 M CaCl2 and centrifuge at 3000 rpm, 4°C for 30 min. The chloroform phase was air-dried completely in a fume hood, and mixed with 5 ml counting cocktail. The water–methanol phase was transferred to a scintillation vial containing 5 ml counting cocktail, and shaken vigorously to mix. All the vials were counted 5 min for 3H and 14C scintillation in a Beckman Coulter LS6500 multi-purpose scintillation counter.
Metabolic cage studies
Metabolic cage studies were performed by the UTSW Metabolic Core Facility. The mice were maintained on a 12-hour dark-light cycle at room temperature or under cold as indicated. Mice were acclimated in the metabolic chambers for 5 days before the start of the experiments. Metabolic parameters including oxygen consumption, CO2 generation, food intake and water consumption were monitored and recorded continuously using the TSE calorimetric system (TSE System). During the whole process, all transgenic mice and their littermate controls were single housed in the metabolic chambers and kept on normal chow and HFD as indicated, and water ad libitum.
Body Temperature
Mice were anesthetized, implantable electronic ID transponder IPTT-300 HTEC were embedded subcutaneously. After one week of recovery, mice were exposed at cold as indicated. Body temperature was detected by BMDS IPTT reader.
Bomb Calorimetry
Mice were single housed in a new autoclaved cage with one absorbent pad, feces were collected each day and froze in −80°C. Feces were collected for a total of 5 days. The combined feces were weighed before and after drying. Feces samples were dried for 72 hours using a LABCONCO Centrivap concentrator equipped with a LABCONCO Centrivap cold trap (−50°C) (Labconco Corporation, Kansas City, MO). Dried feces were pulverized using a multiplex bead tissue disruptor (TissueLyserII, Qiagen, Germantown, MD). Heat of combustion was determined in a 6200 Isoperibol Calorimeter Equipped with a semi-micro oxygen combustion vessel. Benzoic acid was utilized as standard (Parr Instrument Company, Moline IL).
Cold and thermoneutrality exposure
Mice were housed in 6°C cold-chamber or 30°C thermo-chamber of UTSW animal facility for cold exposure or thermoneutrality experiments. All mice were single-caged for cold exposure experiment. For acute cold exposure, mice were exposed in cold with/without food for a maximum of 6 hours with unlimited access to water. For chronic cold exposure experiments mice were exposed to the cold with unlimited access to food and water. Experiments were carried out at least for one week after cold adaption. For thermoneutrality experiment, the breeding mice were housed in thermoneutrality from E13 or earlier, the offspring were also kept at thermoneutrality.
Fat transplantation
Transplantation procedure was performed at 5-6 weeks of age. Mice were anesthetized with isoflurane. The total amount of gWAT mWAT, and iWAT from male Tfrcfl/fl and TfrcAKO mice (The weight of different fat pads were matched with the one from Tfrcfl/fl mouse) were cut to small pieces (around 25-50 mg/piece), rinsed with sterile saline, and then transplanted to TfrcAKO littermate mice or FAT-ATTAC mice (Pajvani et al., 2005), a model of inducible lipodystrophy. iWAT was transplanted subcutaneously through a small incision of the skin. Both donor and recipient gWAT were cut with wound surface, then they are stitched together by PGA synthetic absorbable suture 5.0 (Henry Schein). mWAT were stitched to the mesenterium with small incision was made to the mesenterium around the transplanted fat to stimulate the healing and vascularization. Subcutaneous injection of Carprofen every 12 hours for 2 days was used for analgesia after surgery. TfrcAKO recipient mice were put on HFD after 2 weeks of recovery. For FAT-ATTAC recipient mice, after 3 weeks of transplantation, FAT-ATTAC mice were repeatedly (every 3 days) injected with dimerizer, AP20187 (Ariad Pharmaceuticals, 0.2 μg/g BW) to ablate original adipocytes, and then fed on HFD with continued injection of dimerizer.
Gastric emptying test and gastrointestinal transit time measurement
2-month-old Tfrcfl/fl and TfrcAKO male mice were on HFD for 10 days, then mice were fasted overnight and refeed with HFD. Acetaminophen were mixed with 5% intralipid (Sigma) and administered to mice by gavage (100 mg/kg) after 15min refeeding. Tail blood was collected at 0, 15, 30, 60, 90 min following gavage. 10 μl serum were used for measurement of acetaminophen by mass spec of UTSW metabolic core. Quantification of acetaminophen was described in Mass spectrometry assay section. For gastrointestinal transit time, 2-month-old Tfrcfl/fl and TfrcAKO male mice were on white HFD paste for 2 weeks, then mice were fasted overnight and refeed with green HFD pellet. After refeed (basal time point), feces were monitored at 30 min intervals. The time for the first presence of green fecal pellet is considered as total gastrointestinal transit time.
NMR fat mass and F2-isoprostane analysis
Fat mass of was measured using a Bruker Minispec mq10 (Brucker). F2-isoprostane was measured as previously described (Milne et al., 2007).
Transmission electron microscopy
Mice were anesthetized, fixed by perfusion with a fixation buffer (0.1 mM sodium cacodylate containing 4% paraformaldehyde and 1% glutaraldehyde). The proximal small intestine was quickly harvested, transferred to 2.5% glutaraldehyde in 0.1 mM sodium cacodylate buffer and cut into small pieces, and rinsed at this buffer at room temperature for 2 hours before kept at 4°C. The tissue pieces were sent to University of Texas Southwestern Medical Center Electron Microscopy Core for subsequent sectioning and imaging processing. Electron microscopy pictures were acquired on the JEOL 1200EX transmission electron microscope (JEOL).
Oxygen-Consumption Experiments
Freshly isolated adipose tissues were weighed and cut into small pieces and incubated in a Seahorse xFe24 islet capture Fluxpak. Tissue respiration was determined by the Seahorse XF Extracellualr Flux Analyzer.
Sudan III fecal staining
Dried feces were ground to powder, take powder from about one grain of mouse feces, put it on the slide, and cycled by waterproof pen. 40 μl 36% acetic acid was added, until the powder totally soaked, then 40 μl Sudan III in 95% ETOH was added. Mixed thoroughly, then covered with a cover slide. Put the covered slide on top of the heater (100°C) until bubbling, and repeated for 3 times. The slide was quickly examined under 40 X light microscopy.
Histological analyses and immune-staining
Tissues were fixed in 10% neutralized formalin overnight, and sent to University of Texas Southwestern Medical Center Electron Microscopy Core for embedding, sectioning, and H&E staining. For Immunofluorescence staining, tissue sections were deparaffinized, heat retrieved at 96 °C for 30 min in buffer containing 10 mM Tris and 1.0 mM EDTA, pH 8.0, allowed to cool naturally, then permeabilized in 0.2% Triton X-100 for 10 min, blocked in 3% BSA (Sigma, A9418), and then incubated with primary antibodies to TOM20, 1:1000 (Santa Cruze1, SC-17764,), PLIN1, 1:1500 (Fitzgerald, 20R-PP004) , TFRC, 1:250 (Invitrogen) overnight at 4 °C. Following washes in PBST, slides were then incubated with secondary antibodies diluted in PBST containing 3% BSA for 2 hours at room temperature. Anti-guinea pig Alexa 594 1:500 (Invitrogen, A11076), Anti-mouse Alexa 488 1:500 (Invitrogen, A11001). Slides were washed in PBST, and then mounted with Prolong Anti-Fade mounting medium containing DAPI (Invitrogen, P36941). For TUNEL staining, it was performed according to the manufacturer’s protocol (Thermo Fisher, C10617). All Images were obtained using Keyence BZ-X710 microscope.
Tissue iron and serum iron measurement
About 100 mg tissues were harvested, accurately weighted and put in a polypropylene plastic tube. 1 ml of acid mixture (3M hydrochloric acid with 0.61M trichloroacetic acid) were added to tissue, and incubated at 65 °C for up to 50 hours. Vigorously vortexed for 30 min at a time for at least twice during the incubation. Spun down with high speed after fully digestion of tissues and brought to the same volume with acid mixture, mixed well and spun down. One volume of Chromagen stock (1.86 mM bathophenanthroline sulfonate with 143mM thioglycolic acid) were mixed with 5 volume saturated sodium acetate and 5 volume of ddH2O to make working chromagen solution. 10 μl of digested tissue solution or iron standard were mixed with 200 μl working chromagen solution to measure tissue iron. Serum iron parameters were measured according to manufacturer’s instructions (Pointe Scientific).
DAB iron staining
The deparaffinized tissue sections were rinsed 3 times (5 min each) in PBS, and incubated for 30min in 1% potassium ferrocyanide, 0.12M HCl. After washing with PBS, the sections were rinsed in 0.3% H2O2 in methanol for 20 min to quench the endogenous peroxidase activity. Then sections were washed with PBS and stained with DAB Quanto chromogen and substrate kit (Thermo, TA-125-QHDX). Positive iron staining is visible as a reddish brown to dark brown color. Images were obtained using Keyence BZ-X710 microscope.
Oil Red O staining
Differentiated adipocytes were fixed by 1 mL of 10% neutralized formalin for 10 min at room temperature. Following fixation, adipocytes were washed with deionized water, and then incubated with 60% isopropanol for 5 min. Cells were stained with Oil Red O working solution (2 g/L Oil red O, 60% isopropanol, 40% H2O) for 10 minutes at room temperature, and then washed by deionized water until the water was clear. For tissues, mice were perfused with 4% Paraformaldehyde, post-fixed in 4% Paraformaldehyde for another 30 min, and put in 20% sucrose at 4°C for dehydration. Tissues were embedded in optimal cutting temperature medium for section. Frozen tissue sections were then stained with oil red o stain kit according to manufacturer’s instruction (Abcam).
Gene expression analysis (qRT-PCR)
Total RNA from tissues was extracted by RNeasy Mini Kit (Qiagen) or Trizol (Invitrogen) according to manufacturer’s instructions, and reverse transcribed to cDNA using the iScript™ cDNA Synthesis Kit (Biorad, 170-8891). Gene expression differences were determined by quantitative PCR using SYBR Green PCR Master Mix (Applied Biosystems). Values were normalized to reference genes using the comparative Ct method. Primers are listed in Table S5.
Western Blotting
For western blotting, proteins were extracted from tissues in RIPA buffer, separated on SDS-PAGE gels, transferred to PVDF membranes and probed with primary antibodies. MGAT2 (Proteintech, 19514-1-AP), HIF-2α (Novus Biologicals, NB100-122SS), β-actin (Cell Signaling Technology, 4970s), Fth1 (Cell Signaling Technology, 4393s), Fpn1 (Novus Biologicals, NBP1-21502), TFRC (Invitrogen), GFP (Abeam). Samples were then stained with secondary antibodies conjugated to IRDye 800 or IRDye 680 (LI-COR Biosciences) at the dilution of 1:5000. The signal was scanned by the LI-COR Odyssey infrared imaging system.
Isolation of small extracellular vesicles from adipose tissue
Mice were perfused through the heart at the left ventricle with PBS 5 mM EDTA at a rate of 1 ml/min for 2 min to removed blood from tissues. EDTA was washed out with a second perfusion of PBS alone for 3 min. The mWAT from 4 mice were combined and digested for 60 min at 37°C in buffer containing 100 mM HEPES, 1.5% BSA, 5 mM glucose, 1 mM calcium and 1 mg/ml collagenase D. Digested samples were diluted 1:1 in PBS and 2 mM EGTA to inhibit collagenase activity for isolation of small extracellular vesicles as described before (Crewe et al., 2018).
RNA-seq analyses
Total RNA was extracted by RNeasy Mini Kit (Qiagen). Samples were run on the Agilent 2100 Bioanalyzer to determine level of degradation thus ensuring only high-quality RNA is used (RIN Score 8 or higher). mRNA is enriched using oligo(dT) beads, fragmented randomly in fragmentation buffer, followed by cDNA synthesis using random hexamers and reverse transcriptase. After first-strand synthesis, a custom second-strand synthesis buffer (Illumina) is added with dNTPs, RNase H and Escherichia coli polymerase I to generate the second strand by nick-translation. The final cDNA library is ready after a round of purification, terminal repair, A-tailing, ligation of sequencing adapters, size selection and PCR enrichment. Library concentration was first quantified using a Qubit 2.0 fluorometer (Life Technologies), and then diluted to 1 ng/μl before checking insert size on an Agilent 2100 and quantifying to greater accuracy by quantitative PCR (Q-PCR) (library activity >2 nM). Libraries are fed into HiSeq machines according to activity and expected data volume. RNA-seq was performed using the Illumina Novaseq 6000 sequencing system. STAR, HTSeq, DESeq2 softwares were used for mapping, Quantification and differential analysis. The whole process was performed at Novogene. For bacterial 16S sequencing, 2-month-old Tfrcfl/fl and TfrcAKO male mice were fed HFD for 2 months. Mice were single caged in sterile box. 3-4 grains of fresh feces were collected to DNA/RNA shield collection tube (ZYMO research). Samples were processed and sequenced by Microbiome Core of UT Southwestern Medical Center. The hypervariable region V3 & V4 of bacterial 16S rRNA gene were captured using Illumina Nextera protocol (Part # 15044223 Rev. B). A single amplicon of 460bp was amplified using 16S Forward 5'TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG and 16S Reverse Primer = 5'GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC as described in Illumina protocol. PCR product was cleaned using Agencourt AmpureXP beads from Beckman Counter Genomics. Illumina adapter and barcode sequences were ligated to amplicon in order to attach them to MiSeqDx flow cell and for multiplexing. Quality and quantity of each sequencing library was assessed using Bioanlyzer and picogreen measurements, respectively. About 6pM of pooled library was loaded onto a MiSeqDX flow cell and sequenced using PE300 (Paired end 300 bp) v3 kit. Raw fastq files were demultiplexed based on unique barcodes and assessed for quality. Samples with more than 50K QC pass sequencing reads were used for downstream 16S OTU analysis. Taxonomic classification and Operational taxonomic units (OTUs) abundance analysis was done using CLC Bio microbial genomics module (https://www.qiagenbioinformatics.com/plugins/clc-microbial-genomics-module/). Individual sample reads were annotated with Greengene database and taxonomic features were determined. Alpha and beta diversity analysis was done to understand the within and between sample diversity, respectively. Abundance data was used for numeric Principal Component Analysis (PCA) in SVS, Golden Helix Software.
Mass spectrometry analysis
Phospholipids. Adipose tissue and serum were submitted to the UTSW Metabolic Core Facility for mass spectrometry analysis. Sphingolipid profiling was conducted by liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-MS/MS), using a Nexera X2 UHPLC coupled to an LCMS-8050 (Shimadzu Scientific Instruments, Columbia, MD, USA), as previously described (Carles, 1956; Holland et al., 2011). For the analysis of phospholipid species, Folch’s lipid extraction was performed on visceral fat tissue samples (40 mg) and serum (10 μL). 20 μL of each of the following internal standard components was added to the sample; SPLASH® LIPIDOMIX® Mass Spec Standard (diluted 1:10 in MeOH), LPS 17:1 10 μM, LPG 17:1 10 μM and LPI 55 μM (Avanti Polar Lipids, Alabaster, AL). Organic extracts were dried under nitrogen stream with no heat. Samples were reconstituted in 250 μL MeOH/CH2Cl2 1:1 (v:v). Phospholipid profiling was performed using the mass spectrometric parameters and separations conditions described in the Shimadzu LC-MS/MS MRM Library for Phospholipid Profiling on a Nexera X2 UHPLC coupled to an LCMS-8060 (Shimadzu Scientific Instruments).
For free fatty acid profiling analysis in serum (10 μL), sample preparation and derivatization was carried out as previously described (Bollinger et al., 2013). Total free fatty acid distribution in serum required alkaline hydrolysis prior to free fatty acid extraction and derivatization. Briefly, lipids from 5 μL of serum were extracted using the Folch method. The final organic extract (2 mL) was hydrolyzed by adding 150 μL of 1M KOH in MeOH followed by incubation at 55°C for two hours. The reaction mixture was then neutralized by the addition of 20 μL of acetic acid and dried down under nitrogen stream at 40°C. Samples were analyzed in a Nexera UHPLC LC-40 coupled to LCMS-9030 Q-TOF mass spectrometer (Shimadzu Scientific Instruments). Free fatty acid chromatographic separation was achieved on a Nexcol C18 1.8 μm 2.1×50 mm (Shimadzu Scientific Instruments) using a gradient elution with H2O 0.1% formic acid and MeOH:MeCN 1:1 (v:v) 0.1% formic acid.
For bile acids analysis in feces, samples were dried for 72 hours using a LABCONCO Centrivap concentrator equipped with a LABCONCO Centrivap cold trap (−50°C). Dried feces (150 mg) were extracted homogenized in a borosilicate glass tube in 2.0 mL of chilled MeCN using a mechanical tissue homogenizer (Omni International, Kennesaw, GA). The homogenate was centrifuged and the supernatant was transferred to a new tube. The resulting pellet was re-extracted with 2.0 mL of chilled MeCN. Combined supernatant was dried under nitrogen stream at 40°C. Dried residues were reconstituted in 2.0 mL of MeCN. 200 μL of sample was fortified with an internal standard mixture and used for LC-MS/MS analysis. In the case of mouse serum, 25 μL of sample was quenched with 200 μL of chilled MeCN and fortified with internal standard mixture. Samples were thoroughly vortexed for 30 seconds and centrifuged to eliminate protein precipitate. Supernatant was used for LC-MS/MS analysis. A second set of mouse serum samples was diluted 10-fold with saline solution and processed as described above. Bile acid profiling was performed using the mass spectrometric parameters described in the Shimadzu LC-MS/MS Method Package for Bile Acids on a Nexera X2 UHPLC coupled to an LCMS-8060 (Shimadzu Scientific Instruments). Chromatographic separation of bile acid species was achieved on a Kinetex C18 1.7 μm 2.1x150 mm with a C18 column guard cartridge (Phenomenex, Torrance, CA) using a gradient elution with H2O 0.05% acetic acid and MeOH:MeCN 1:1 (v:v). LabSolutions V 5.82 and LabSolutions Insight V 2.0 program packages were used for data processing (Shimadzu Scientific Instruments).
For acetaminophen and related metabolites profiling, mouse serum samples (10 μL) were mixed with MeOH (90 μL) and 20 μL of internal standard solution in MeOH (10 μgr/mL). Samples were vortexed vigorously for one minute and then kept at −20°C during one hour to facilitate the precipitation of proteins. Samples were then centrifuged at 13,000 g at 4°C during 10 min and 90 μL of supernatant were transferred to a clean 1.5 mL micro-centrifuge tube. Methanolic extract was dried under a stream of nitrogen with no heat. Dried extracts were reconstituted in 100 μL of ultrapure HPLC water and transferred to polypropylene low absorption auto-sampler vials with built in 300 μL insert (Shimadzu Scientific Instruments, Columbia, MD). Calibration standards and quality control samples were prepared using blank commercial mouse Balb C serum (Innovative Research, Novi, MI) and fortified with methanolic stock acetaminophen stock solutions at known concentrations. Samples were analyzed in a LCMS-8060 triple quadrupole mass spectrometer coupled to a Nexera X2 UHPLC system (Shimadzu Scientific Instruments) using the dual ionization source in electrospray ionization mode. Analytical instrument analysis for each compound were optimized by automated flow injection analysis. During analysis the instrument operated in multiple reaction monitoring (MRM) mode. The chromatographic separation was carried out by gradient elution using a Titan C18 reverse phase column (10x2.1 cm, 2.1 cm ID, 1.9 μm; Supelco, Bellefonte, PA) maintained at 40°C. The mobile phase composed of solvent A (water 0.1% formic acid, v:v) and solvent B (MeOH:MeCN:formic acid, 50:50:0.1, v:v:v) was kept at a flow rate of 0.4 mL/min. 4 μL of sample were injected in the system. Acetaminophen was accurately quantified using a calibration curve employing true standard compound (acetaminophen certified reference standard, Cayman Chemicals, Ann Arbor, MI) and stable isotope labeled acetaminophen (acetyl-13C2, 99%; 15N, 98%, Cambridge Isotope Laboratories, Inc. Tewksbury, MA). Relative abundance of acetaminophen glucuronide, acetaminophen sulfate, cysteinylacetaminophen (Cayman chemicals), acetaminophen N-acetyl cysteinyl and acetaminophen glutathione (Santa Cruz Biotechnology, Dallas, TX) was determine by comparing their relative peak area vs acetaminophen internal standard peak area ratios.
Proteomics and Pathway Analysis
Samples were run into an SDS-PAGE gel. Proteins were excised from the gel and submitted to the UT Southwestern Proteomics Core. Samples were processed as previously described (Kalantari et al., 2016). Reactome software was used for pathway analysis.
Serum Chemistry
Insulin were measured by ELISA kit from Alpco, Leptin were measured by ELISA kit from Crystal Chem. Adiponectin was measured with mouse adiponectin ELISA kit (Millipore, Bedford, MA). Serum or plasma triglyceride and cholesterol were measured with an Infinity Triglyceride Kit and Infinity Cholesterol Kit (Thermo Electron Corporation, Vantaa, Finland), respectively. Serum NEFAs were measured with a NEFA-HR kit (Wako Diagnostics, Mountain View, CA). Blood glucose were measured by using Bayer Contour glucometers.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantifications of Oil Red O staining and vesicle area in villus of proximal small intestine were conducted by Image J. No statistical methods were used to predetermine the sample size. For in vitro assays, triplicates were used for statistical analysis, n correspond to the independent repeat. Experiments were confirmed by 3 independent experiments or as indicated. For mouse experiments, 5-8 mice were included for each experimental group. n corresponds to the number of mice in each experimental group. Critical mouse experiments were confirmed by at least 3 independent experiments. All data were expressed as the mean ± SD. We used GraphPad Prism 8.0 (GraphPad Software Inc.) or Microsoft Excel to perform the statistical analyses. Two-tailed Student’s t-test was used for comparisons between two independent groups. For multiple comparisons, One-way or Two-way ANOVA was used. P values than 0.05 was considered statistically significant. *P < 0.05, **P < 0.01. Mice were randomly-grouped based on genotype, littermates status, and ear punch numbers. All mice related experiments were performed in a blinded fashion. One of the TfrcAKO fecal sample did not generate required number of sequencing reads, hence, was excluded from 16S analysis. No other data points or samples were excluded from the study. The data were assumed to be an continuous normal distribution.
Supplementary Material
Table S1. Proteomic of sEV from mWAT of Tfrcfl/fl and TfrcAKO mice upon HFD feeding (related to Fig. 5)
Table S2. Lipidomic-Gonadal adipose tissue of Tfrcfl/fl and TfrcAKO mice upon HFD feeding related to Fig. 5)
Table S3. Lipidomic-Serum of Tfrcfl/fl and TfrcAKO mice upon HFD feeding (related to Fig. 5)
Table S4. Proteomic in medium of differentiated SVF harvested from Tfrcfl/fl, TfrcAKO, AAV-eGFP, and AAV-Fpn1C326S mice (related to Fig. 5)
Data S1. sequence of pAAVK CAG-FLEX-eGFP (related to key resources table)
Data S2. sequence of pAAVK CAG-FLEX-Slc40a1 C326S (related to key resources table)
Data S3. sequence of pAAVK CAG-FLEX (related to key resources table)
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Chicken anti-GFP | Abcam | Cat# ab13970; RRID: AB_300798 |
| Donkey anti-chicken IRDye 680RD | LI-COR Biosciences | Cat#: 926-68075; RRID:AB_10974977 |
| Donkey anti-mouse IRDye 680RD | LI-COR Biosciences | Cat#: 926-68072; RRID: AB_10953628 |
| Goat anti-guinea pig Alexa 594 | Thermo Fisher | Cat# A-11076; RRID: AB_141930 |
| Goat anti-mouse Alexa 488 | Thermo Fisher | Cat# A-11001; RRID: AB_2534069 |
| Goat anti-rabbit IRDye 800CW | LI-COR Biosciences | Cat#: 925-32211; RRID: AB_2651127 |
| Guinea pig anti-PLIN1 | Fitzgerald | Cat# 20R-PP004; RRID: AB_1288416 |
| Mouse anti-TFRC (H68.4) | Invitrogen | Cat# 13-6890; RRID: AB_86624 |
| Mouse anti-TOM20 (F10) | Santa Cruz Biotechnology | Cat# sc-17764; RRID: AB_628381 |
| Rabbit Anti-b-actin (13E5) | Cell Signaling Technology | Cat# 4970; RRID: AB_2223172 |
| Rabbit anti-Ferritin-H | Cell Signaling Technology | Cat# 4393; RRID: AB_11217441 |
| Rabbit anti-Fpn1 | Novus Biologicals | Cat# NBP1-21502; RRID: AB_2302075 |
| Rabbit anti-HIF-2a | Novus Biologicals | Cat# NB100-122SS; RRID: AB_10000872 |
| Rabbit anti-MGAT2 | Proteintech | Cat# 19514-1-AP; RRID: AB_10638917 |
| Chemicals, peptides, and recombinant proteins | ||
| 3a70B complete counting cocktail | RPI | SKU: 111154 |
| 3H-Triolein | PerkinElmer | Cat#: NEC075H050UC |
| 14C-Palmiate | PerkinElmer | Cat#: NET431001MC |
| Acetaminophen | Sigma-Aldrich | Cat#: A7085 |
| ADMEM/F12 | Gibco | Cat#: 12634010 |
| Antifade mountant with DAPI | Invitrogen | Cat#: P36941 |
| AP20187 (Dimerizer) | Ariad Pharmaceuticals | N/A |
| Bathophenanthrolinedisulfonic acid disodium salt hydrate | Sigma-Aldrich | Cat#: B1375 |
| Benzonase | Sigma-Aldrich | Cat#: E8263 |
| BSA | Sigma-Aldrich | Cat#: A8806 |
| Collagenase IV | Roche | Cat#: 11088882001 |
| Dexamethasone | Sigma-Aldrich | Cat#: D4902 |
| Dispase II | Roche | Cat#: 04942078001 |
| DMEM high glucose | Gibco | Cat#: 11965118 |
| DMEM/F12 | Gibco | Cat#: 11320082 |
| Fetal Bovine Serum | Sigma-Aldrich | Cat#: 12303C |
| Glucose | Gibco | Cat#: 15023021 |
| GlutaMax | Gibco | Cat#: 35050079 |
| Glutaraldehyde solution | Sigma-Aldrich | Cat#: G5882 |
| Growth Factor-Reduced Matrigel | Corning | Cat#: 356230 |
| HEPES | Gibco | Cat#: 15630080 |
| Insulin | Eli Lilly | Product ID: A10008415 |
| Intralipid | Sigma-Aldrich | Cat#: 6I141 |
| Iron Standard for AAS | Sigma-Aldrich | Cat#: 16596 |
| Isobutylmethylxanthine | Sigma-Aldrich | Cat#: I7018 |
| Oil Red O | Sigma-Aldrich | Cat#: O0625 |
| Palmitate | Sigma-Aldrich | Cat#: P5585 |
| Paraformaldehyde | Sigma-Aldrich | Cat#: P6148 |
| PEI linear MW25000 | Polysciences | Cat#: 23966-100 |
| Penicillin and streptomycin | Gibco | Cat#: 15140122 |
| Phenol red free DMEM/F12 | Gibco | Cat#: 21041025 |
| Pluronic F-68 | ThermoFisher | Cat#: 24040032 |
| Potassium ferrocyanide | Sigma-Aldrich | Cat#: 60279 |
| Prolong Anti-Fade mounting medium | Invitrogen | Cat#: P36941 |
| RNase I | ThermoFisher | Cat#: EN0601 |
| Rosiglitazone | Sigma-Aldrich | Cat#: R2408 |
| Sarkosyl | Sigma-Aldrich | Cat#: L9150 |
| Sodium acetate | Sigma-Aldrich | Cat#: S2889 |
| Sodium cacodylate | Sigma-Aldrich | Cat#: C0250 |
| Thioglycolic acid solution | Sigma-Aldrich | Cat#: T6750 |
| Trizol | Invitrogen | Cat#: 12034977 |
| Tyloxapol | Sigma-Aldrich | Cat#: T0307 |
| Critical commercial assays | ||
| DAB Quanto chromogen and substrate kit | ThermoFisher | Cat#: TA-125-QHDX |
| HR Series NEFA-HR(2) Color Reagent A | Wako | Cat#: 999-34691 |
| HR Series NEFA-HR(2) Color Reagent B | Wako | Cat#: 991-34891 |
| HR Series NEFA-HR(2) Color Solvent A | Wako | Cat#: 995-34791 |
| HR Series NEFA-HR(2) Color Solvent B | Wako | Cat#: 993-35191 |
| Infinity™ Cholesterol Reagent | ThermoFisher | Cat#: TR13421 |
| Infinity™ Triglycerides Reagent | ThermoFisher | Cat#: TR22421 |
| iScript™ cDNA Synthesis Kit | BIO-RAD, | Cat#: 170-8891 |
| Mouse Adiponectin Elisa Kit | Millipore | Cat#: EZMADP-60K |
| Mouse Leptin ELISA Kit | Crystal Chem | Cat#: 90030 |
| Mouse Ultrasensitive Insulin ELISA | ALPCO | Cat#: 80-INSMSU-E01 |
| Oil red O stain kit | Abcam | Cat#: ab150678 |
| Pierce BCA Protein Assay kit | ThermoFisher | Cat#: 23225 |
| RIPA buffer | Sigma-Aldrich | Cat#: R0278 |
| RNeasy Mini Kit (Qiagen) | Qiagen | Cat#: 74106 |
| Serum iron/TIBC Kit | Pointe Scientific | Cat#: I750460 |
| Sybr Green Master Mix | Applied biosystems | Cat#: A25778 |
| TUNEL staining kit | ThermoFisher | Cat#: C10617 |
| Deposited data | ||
| RNA-seq | This paper | GEO: GSE144938 |
| 16S rRNA-seq | This paper | SRA NCBI: PRJNA718550 |
| Experimental models: Organisms/strains | ||
| Mouse: Tfrcfl/fl | The Jackson Laboratory | JAX028363; RRID:IMSR_JAX:028363 |
| Mouse: Adipoq-Cre | The Jackson Laboratory | JAX028020; RRID:IMSR_JAX:028020 |
| Mouse: Ucp1-Cre | The Jackson Laboratory | JAX024670; RRID:IMSR_JAX:024670 |
| Mouse: TRE-Cre | The Jackson Laboratory | JAX006234; RRID:IMSR_JAX:006234 |
| Mouse: Adipoq-rtTA | Sun et al., 2012 | N/A |
| Mouse: FAT-ATTAC | Pajvani et al., 2005 | N/A |
| Oligonucleotides | ||
| Primers for qPCR | See Table S5 | N/A |
| Recombinant DNA | ||
| pAAVK CAG-FLEX-eGFP | See Data S1 | N/A |
| pAAVK CAG-FLEX-Fpn1C326S | See Data S2 | N/A |
| pAAVK CAG-FLEX vector | See Data S3; Gift from Hongkui Zeng | Based on Addgene plasmid #51502 |
| pHelper | Cell Biolabs | Part No: 340202 |
| pRepCap Rec2 | Gift from Deborah Young, University of Auckland, New Zealand | N/A |
| pAAV-DJ | Cell Biolabs | Part No: VPK-420-DJ |
| Software and algorithms | ||
| Adobe photoshop | Adobe | https://www.adobe.com/products/photoshop.html; RRID: SCR_014199 |
| Endnote | Endnote | http://endnote.com; RRID: SCR_014001 |
| Excel | Microsoft | https://www.microsoft.com/enus/microsoft-365/excel; RRID:SCR_016137 |
| Image J | NIH | https://imagej.nih.gov/ij/; RRID: SCR_003070 |
| PowerPoint | Microsoft | https://www.microsoft.com/enus/microsoft-365/powerpoint |
| Prism 9 | GraphPad software | https://www.graphpad.com/; RRID: SCR_002798 |
| Word | Microsoft | https://www.microsoft.com/enus/microsoft-365/word |
| Other | ||
| 1200EX Transmission Electron Microscope | JEOL | N/A |
| 60% HFD Green pellet | Research diets | D12492i |
| 60% HFD paste | Bio-serv | S1850 |
| 6200 Isoperibol Calorimeter | Parr Instrument Company | N/A |
| a Nexera X2 UHPLC | Shimadzu Scientific Instruments | N/A |
| Agilent 2100 Bioanalyzer | Agilent | N/A |
| Amicon Ultra-15 centrifugal filter unit (3KD) | Millipore | UFC9003 |
| Amicon Ultra-2 centrifugal filter unit (3KD) | Millipore | UFC200324 |
| Beckman Coulter LS6500 multi-purpose scintillation counter | Beckman | N/A |
| BMDS IPTT reader | BMDS | Model: DAS-8007-IUS |
| Bruker Minispec mq10 | Brucker | Cat#: TMTA2892161 |
| DNA/RNA shield collection tube | ZYMO research | Cat#: R1102 |
| Doxycycline 60% HFD (600mg/kg diet) | Bio-serv | S5867 |
| Doxycycline chow diet (600 mg/kg diet) | Bio-serv | S4107 |
| Glucometers | Bayer Contour | N/A |
| Illumina Novaseq 6000 sequencing system | Illumina | N/A |
| Implantable electronic ID transponder IPTT-300 HTEC | BMDS | N/A |
| Keyence BZ-X710 Fluorescence Microscope | Keyence | N/A |
| LCMS-8050 | Shimadzu Scientific Instruments | N/A |
| Normal chow diet | TEKLAD | 2916 |
| Odyssey Infrared Imager | LI-COR Biosciences | N/A |
| PGA synthetic absorbable suture 5.0 | Henry Schein | 31995 |
| Rodent cold/thermo-Chamber | Powers Scientific | N/A |
| Scintillation vial | RPI | SKU: 121000CA |
| Seahorse XF Extracellualr Flux Analyzer | Agilent | N/A |
| Seahorse xFe24 islet capture fluxpak | Agilent | Cat#: 103518-100 |
| TSE calorimetric system | TSE system | N/A |
Highlights.
TFRC expression is differentially required for distinct subtypes of adipocytes
Lowering iron in adipocytes leads to a healthier white adipose tissue (WAT)
Adipocyte iron levels activate an adipose-gut crosstalk to regulate gut lipid absorption
Lowering iron in WAT protects mice from high fat diet-induced metabolic dysfunction
ACKNOWLEDGEMENTS
We thank all the members of the Scherer laboratory for their support of this study. We also thank the University of Texas Southwestern Medical Center Metabolic Phenotyping Core Facility, Electron Microscopy Core, Proteomic core, Microbiome Research Laboratory, Animal Resource Center and the Histopathology Core. This study was supported by US National Institutes of Health grants RC2-DK118620, R01-DK55758, R01-DK099110, P01-DK088761 and P01-AG051459 to P.E.S. AHA825982 to S.Z. K01-DK125447 to Y.A.A. P.E.S. was also supported by an unrestricted research grant from the Novo Nordisk Foundation. We thank Shimadzu Scientific Instruments for the collaborative efforts in mass spectrometry technology resources.
Footnotes
DECLARATION OF INTERESTES
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altamura S, Kessler R, Grone HJ, Gretz N, Hentze MW, Galy B, and Muckenthaler MU (2014). Resistance of ferroportin to hepcidin binding causes exocrine pancreatic failure and fatal iron overload. Cell Metab 20, 359–367. [DOI] [PubMed] [Google Scholar]
- An YA, Chen S, Deng Y, Wang ZV, Funcke JB, Shah M, Shan B, Gordillo R, Yoshino J, Klein S, et al. (2021). The mitochondrial dicarboxylate carrier prevents hepatic lipotoxicity by inhibiting white adipocyte lipolysis. J Hepatol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurnhammer C, Haase M, Muether N, Hausl M, Rauschhuber C, Huber I, Nitschko H, Busch U, Sing A, Ehrhardt A, et al. (2012). Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Hum Gene Ther Methods 23, 18–28. [DOI] [PubMed] [Google Scholar]
- Bollinger JG, Naika GS, Sadilek M, and Gelb MH (2013). LC/ESI-MS/MS detection of FAs by charge reversal derivatization with more than four orders of magnitude improvement in sensitivity. J Lipid Res 54, 3523–3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carles J (1956). [Colorimetric microdetermination of phosphorus]. Bull Soc Chim Biol (Paris) 38, 255–257. [PubMed] [Google Scholar]
- Cepeda-Lopez AC, Osendarp SJ, Melse-Boonstra A, Aeberli I, Gonzalez-Salazar F, Feskens E, Villalpando S, and Zimmermann MB (2011). Sharply higher rates of iron deficiency in obese Mexican women and children are predicted by obesity-related inflammation rather than by differences in dietary iron intake. Am J Clin Nutr 93, 975–983. [DOI] [PubMed] [Google Scholar]
- Charbel Issa P, De Silva SR, Lipinski DM, Singh MS, Mouravlev A, You Q, Barnard AR, Hankins MW, During MJ, and Maclaren RE (2013). Assessment of tropism and effectiveness of new primate-derived hybrid recombinant AAV serotypes in the mouse and primate retina. PLoS One 8, e60361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaurasia B, Tippetts TS, Mayoral Monibas R, Liu J, Li Y, Wang L, Wilkerson JL, Sweeney CR, Pereira RF, Sumida DH, et al. (2019). Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science 365, 386–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AC, Donovan A, Ned-Sykes R, and Andrews NC (2015). Noncanonical role of transferrin receptor 1 is essential for intestinal homeostasis. Proc Natl Acad Sci U S A 112, 11714–11719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crewe C, Joffin N, Rutkowski JM, Kim M, Zhang F, Towler DA, Gordillo R, and Scherer PE (2018). An Endothelial-to-Adipocyte Extracellular Vesicle Axis Governed by Metabolic State. Cell 175, 695–708 e613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks DR, Maio N, Lane AN, Jarnik M, Higashi RM, Haller RG, Yang Y, Fan TW, Linehan WM, and Rouault TA (2018). Acute loss of iron-sulfur clusters results in metabolic reprogramming and generation of lipid droplets in mammalian cells. J Biol Chem 293, 8297–8311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dymock IW, Cassar J, Pyke DA, Oakley WG, and Williams R (1972). Observations on the pathogenesis, complications and treatment of diabetes in 115 cases of haemochromatosis. Am J Med 52, 203–210. [DOI] [PubMed] [Google Scholar]
- Eguchi J, Wang X, Yu S, Kershaw EE, Chiu PC, Dushay J, Estall JL, Klein U, Maratos-Flier E, and Rosen ED (2011). Transcriptional control of adipose lipid handling by IRF4. Cell Metab 13, 249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann HM, Golozoubova V, Cannon B, and Nedergaard J (2009). UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab 9, 203–209. [DOI] [PubMed] [Google Scholar]
- Fernandez-Real JM, Lopez-Bermejo A, and Ricart W (2002). Cross-talk between iron metabolism and diabetes. Diabetes 51, 2348–2354. [DOI] [PubMed] [Google Scholar]
- Funcke JB, and Scherer PE (2019). Beyond adiponectin and leptin: adipose tissue-derived mediators of inter-organ communication. J Lipid Res 60, 1648–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrielsen JS, Gao Y, Simcox JA, Huang J, Thorup D, Jones D, Cooksey RC, Gabrielsen D, Adams TD, Hunt SC, et al. (2012). Adipocyte iron regulates adiponectin and insulin sensitivity. J Clin Invest 122, 3529–3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamberini MR, De Sanctis V, and Gilli G (2008). Hypogonadism, diabetes mellitus, hypothyroidism, hypoparathyroidism: incidence and prevalence related to iron overload and chelation therapy in patients with thalassaemia major followed from 1980 to 2007 in the Ferrara Centre. Pediatr Endocrinol Rev 6 Suppl 1, 158–169. [PubMed] [Google Scholar]
- Gao Y, Li Z, Gabrielsen JS, Simcox JA, Lee SH, Jones D, Cooksey B, Stoddard G, Cefalu WT, and McClain DA (2015). Adipocyte iron regulates leptin and food intake. J Clin Invest 125, 3681–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JM, Fackelmeier B, Fong DM, Mouravlev A, Young D, and O'Carroll SJ (2019). Astrocyteselective AAV gene therapy through the endogenous GFAP promoter results in robust transduction in the rat spinal cord following injury. Gene Ther 26, 198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo P, El-Gohary Y, Prasadan K, Shiota C, Xiao X, Wiersch J, Paredes J, Tulachan S, and Gittes GK (2012). Rapid and simplified purification of recombinant adeno-associated virus. J Virol Methods 183, 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, Davis KE, Bikman BT, Halberg N, Rutkowski JM, et al. (2011). Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med 17, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki T, Nakajima A, Yoneda M, Yamada Y, Mukasa K, Fujita K, Fujisawa N, Wada K, and Terauchi Y (2005). Serum ferritin is associated with visceral fat area and subcutaneous fat area. Diabetes Care 28, 2486–2491. [DOI] [PubMed] [Google Scholar]
- Jauregui-Lobera I (2013). Iron deficiency and bariatric surgery. Nutrients 5, 1595–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalantari R, Hicks JA, Li L, Gagnon KT, Sridhara V, Lemoff A, Mirzaei H, and Corey DR (2016). Stable association of RNAi machinery is conserved between the cytoplasm and nucleus of human cells. RNA 22, 1085–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kershaw EE, and Flier JS (2004). Adipose tissue as an endocrine organ. J Clin Endocrinol Metab 89, 2548–2556. [DOI] [PubMed] [Google Scholar]
- Kong X, Banks A, Liu T, Kazak L, Rao RR, Cohen P, Wang X, Yu S, Lo JC, Tseng YH, et al. (2014). IRF4 is a key thermogenic transcriptional partner of PGC-1alpha. Cell 158, 69–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konrad D, and Wueest S (2014). The gut-adipose-liver axis in the metabolic syndrome. Physiology (Bethesda) 29, 304–313. [DOI] [PubMed] [Google Scholar]
- Kusminski CM, Holland WL, Sun K, Park J, Spurgin SB, Lin Y, Askew GR, Simcox JA, McClain DA, Li C, et al. (2012). MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat Med 18, 1539–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusminski CM, and Scherer PE (2019). Lowering ceramides to overcome diabetes. Science 365, 319–320. [DOI] [PubMed] [Google Scholar]
- Li J, Pan X, Pan G, Song Z, He Y, Zhang S, Ye X, Yang X, Xie E, Wang X, et al. (2020). Transferrin Receptor 1 Regulates Thermogenic Capacity and Cell Fate in Brown/Beige Adipocytes. Adv Sci (Weinh) 7, 1903366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Rossmeisl M, McClaine J, Riachi M, Harper ME, and Kozak LP (2003). Paradoxical resistance to diet-induced obesity in UCP1-deficient mice. J Clin Invest 111, 399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain DA, Abraham D, Rogers J, Brady R, Gault P, Ajioka R, and Kushner JP (2006). High prevalence of abnormal glucose homeostasis secondary to decreased insulin secretion in individuals with hereditary haemochromatosis. Diabetologia 49, 1661–1669. [DOI] [PubMed] [Google Scholar]
- McFarlane MR, Cantoria MJ, Linden AG, January BA, Liang G, and Engelking LJ (2015). Scap is required for sterol synthesis and crypt growth in intestinal mucosa. J Lipid Res 56, 1560–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne GL, Sanchez SC, Musiek ES, and Morrow JD (2007). Quantification of F2-isoprostanes as a biomarker of oxidative stress. Nat Protoc 2, 221–226. [DOI] [PubMed] [Google Scholar]
- Mokdad AH, Ford ES, Bowman BA, Dietz WH, Vinicor F, Bales VS, and Marks JS (2003). Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA 289, 76–79. [DOI] [PubMed] [Google Scholar]
- Morrison SF, Madden CJ, and Tupone D (2014). Central neural regulation of brown adipose tissue thermogenesis and energy expenditure. Cell Metab 19, 741–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajvani UB, Trujillo ME, Combs TP, Iyengar P, Jelicks L, Roth KA, Kitsis RN, and Scherer PE (2005). Fat apoptosis through targeted activation of caspase 8: a new mouse model of inducible and reversible lipoatrophy. Nat Med 11, 797–803. [DOI] [PubMed] [Google Scholar]
- Perl AK, Wert SE, Nagy A, Lobe CG, and Whitsett JA (2002). Early restriction of peripheral and proximal cell lineages during formation of the lung. Proc Natl Acad Sci U S A 99, 10482–10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinhas-Hamiel O, Newfield RS, Koren I, Agmon A, Lilos P, and Phillip M (2003). Greater prevalence of iron deficiency in overweight and obese children and adolescents. Int J Obes Relat Metab Disord 27, 416–418. [DOI] [PubMed] [Google Scholar]
- Priest C, and Tontonoz P (2019). Inter-organ cross-talk in metabolic syndrome. Nat Metab 1, 1177–1188. [DOI] [PubMed] [Google Scholar]
- Romacho T, Elsen M, Rohrborn D, and Eckel J (2014). Adipose tissue and its role in organ crosstalk. Acta Physiol (Oxf) 210, 733–753. [DOI] [PubMed] [Google Scholar]
- Rutkowski JM, Stern JH, and Scherer PE (2015). The cell biology of fat expansion. J Cell Biol 208, 501–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, Colagiuri S, Guariguata L, Motala AA, Ogurtsova K, et al. (2019). Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res Clin Pract 157, 107843. [DOI] [PubMed] [Google Scholar]
- Saely CH, Geiger K, and Drexel H (2012). Brown versus white adipose tissue: a mini-review. Gerontology 58, 15–23. [DOI] [PubMed] [Google Scholar]
- Scherer PE (2006). Adipose tissue: from lipid storage compartment to endocrine organ. Diabetes 55, 1537–1545. [DOI] [PubMed] [Google Scholar]
- Schulz TJ, and Tseng YH (2013). Brown adipose tissue: development, metabolism and beyond. Biochem J 453, 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SV, and Fonseca VA (2011). Iron and diabetes revisited. Diabetes Care 34, 1676–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SW (2018). Weight and Metabolic Outcomes 12 Years after Gastric Bypass. N Engl J Med 378, 95. [DOI] [PubMed] [Google Scholar]
- Siddique MM, Li Y, Chaurasia B, Kaddai VA, and Summers SA (2015). Dihydroceramides: From Bit Players to Lead Actors. J Biol Chem 290, 15371–15379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simcox JA, and McClain DA (2013). Iron and diabetes risk. Cell Metab 17, 329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song A, Dai W, Jang MJ, Medrano L, Li Z, Zhao H, Shao M, Tan J, Li A, Ning T, et al. (2020). Low- and high-thermogenic brown adipocyte subpopulations coexist in murine adipose tissue. J Clin Invest 130, 247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub LG, Efthymiou V, Grandl G, Balaz M, Challa TD, Truscello L, Horvath C, Moser C, Rachamin Y, Arnold M, et al. (2019). Antioxidants protect against diabetes by improving glucose homeostasis in mouse models of inducible insulin resistance and obesity. Diabetologia 62, 2094–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub LG, and Scherer PE (2019). Metabolic Messengers: Adiponectin. Nat Metab 1, 334–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Wernstedt Asterholm I, Kusminski CM, Bueno AC, Wang ZV, Pollard JW, Brekken RA, and Scherer PE (2012). Dichotomous effects of VEGF-A on adipose tissue dysfunction. Proc Natl Acad Sci U S A 109, 5874–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. (2009). A core gut microbiome in obese and lean twins. Nature 457, 480–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger RH, and Scherer PE (2010). Gluttony, sloth and the metabolic syndrome: a roadmap to lipotoxicity. Trends Endocrinol Metab 21, 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ussar S, Griffin NW, Bezy O, Fujisaka S, Vienberg S, Softic S, Deng L, Bry L, Gordon JI, and Kahn CR (2015). Interactions between Gut Microbiota, Host Genetics and Diet Modulate the Predisposition to Obesity and Metabolic Syndrome. Cell Metab 22, 516–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Yagai T, Luo Y, Liang X, Chen T, Wang Q, Sun D, Zhao J, Ramakrishnan SK, Sun L, et al. (2017). Activation of intestinal hypoxia-inducible factor 2alpha during obesity contributes to hepatic steatosis. Nat Med 23, 1298–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen CL, Cheong ML, Grueter C, Zhou P, Moriwaki J, Wong JS, Hubbard B, Marmor S, and Farese RV Jr. (2009). Deficiency of the intestinal enzyme acyl CoA:monoacylglycerol acyltransferase-2 protects mice from metabolic disorders induced by high-fat feeding. Nat Med 15, 442–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, and Scherer PE (2018). Adipose tissue: The dysfunctional adipocyte - a cancer cell’s best friend. Nat Rev Endocrinol 14, 132–134. [DOI] [PubMed] [Google Scholar]
- Zhao S, Zhu Y, Schultz RD, Li N, He Z, Zhang Z, Caron A, Zhu Q, Sun K, Xiong W, et al. (2019). Partial Leptin Reduction as an Insulin Sensitization and Weight Loss Strategy. Cell Metab 30, 706–719 e706. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Proteomic of sEV from mWAT of Tfrcfl/fl and TfrcAKO mice upon HFD feeding (related to Fig. 5)
Table S2. Lipidomic-Gonadal adipose tissue of Tfrcfl/fl and TfrcAKO mice upon HFD feeding related to Fig. 5)
Table S3. Lipidomic-Serum of Tfrcfl/fl and TfrcAKO mice upon HFD feeding (related to Fig. 5)
Table S4. Proteomic in medium of differentiated SVF harvested from Tfrcfl/fl, TfrcAKO, AAV-eGFP, and AAV-Fpn1C326S mice (related to Fig. 5)
Data S1. sequence of pAAVK CAG-FLEX-eGFP (related to key resources table)
Data S2. sequence of pAAVK CAG-FLEX-Slc40a1 C326S (related to key resources table)
Data S3. sequence of pAAVK CAG-FLEX (related to key resources table)
Data Availability Statement
The accession numbers for the RNA, 16S RNA sequencing data reported in this paper are GEO: GSE144938, SRA NCBI: PRJNA718550. One of the TfrcAKO fecal sample did not generate the required number of sequencing reads, hence, was excluded from 16S analysis.