

Abstract
Tissue‐resident macrophages in white adipose tissue (WAT) dynamically adapt to the metabolic changes of their microenvironment that are often induced by excess energy intake. Currently, the exact contribution of these macrophages in obesity‐driven WAT remodeling remains controversial. Here, using a transgenic CD169‐DTR mouse strain, we provide new insights into the interplay between CD169+ adipose tissue macrophages (ATMs) and their surrounding WAT microenvironment. Using targeted in vivo ATM ablation followed by transcriptional and metabolic WAT profiling, we found that ATMs protect WAT from the excessive pathological remodeling that occurs during obesity. As obesity progresses, ATMs control not only vascular integrity, adipocyte function, and lipid and metabolic derangements but also extracellular matrix accumulation and resultant fibrosis in the WAT. The protective role of ATMs during obesity‐driven WAT dysfunction supports the notion that ATMs represent friends, rather than foes, as has previously assumed.
Keywords: adipocyte hypertrophy, fibrosis, obesity‐mediated adipose tissue remodeling, tissue‐resident macrophages, vascular integrity
Subject Categories: Immunology, Metabolism
This study reveals that resident macrophages in the adipose tissue are important regulatory cells that restrain tissue dysregulation caused by obesity.

Introduction
With an estimated more than one billion obese and overweight people worldwide (Stevens et al, 2012; Kastl et al, 2020), overt metabolic dysregulation is one of the most pressing health challenges of our modern time. An important complication in obesity is insulin resistance, which can ultimately lead to type 2 diabetes mellitus (T2DM), a major cause of morbidity and mortality globally (Danaei et al, 2011). A consensus has been reached in the field that the primary underlying cause of obesity‐induced insulin resistance is chronic, low‐grade systemic inflammation originating in the white adipose tissue (WAT) (Xu et al, 2003; Hotamisligil, 2006). Obesity deregulates homeostasis in WAT not only by promoting changes in the endocrinological signals and the composition of metabolites and lipids (Shi et al, 2006; Cao, 2014; Rosen & Spiegelman, 2014) but also by inducing adipocyte hypertrophy and extensive tissue vascularization (Sun et al, 2011; Sung et al, 2013). The resulting stress responses of adipocytes and stromal cells lead to the activation and accumulation of pro‐inflammatory cells in WAT (Hotamisligil, 2006), driving an influx of Th1 CD4+ T cells, CD8+ T cells, B cells, and distinct myeloid cells, generating an inflammatory immune response that is aggravated over time as more factors and cells become involved (Lumeng et al, 2007; Nishimura et al, 2009; Winer et al, 2011; Talukdar et al, 2012; Trim et al, 2018). This results in chronic systemic production of pro‐inflammatory cytokines, which in time reduces systemic insulin sensitivity (Borst, 2004). Among other cell types (such as pre‐adipocytes/adipocytes, endothelial cells, fibroblasts, and lymphoid and myeloid cells), adipose tissue‐resident macrophages (ATMs), which represent the predominant CD45+ leukocyte fraction, “sit” in the middle of this stressed WAT microenvironment and are currently considered one of the major culprits involved in the establishment of metabolic syndrome. Based on their varied surface marker expression, tissue localization, and lipid content, ATMs represent a heterogeneous cell population (Hill et al, 2018; Jaitin et al, 2019; Silva et al, 2019; Chen & Ruedl, 2020). In individuals with obesity, ATMs accumulate by rapidly increasing their numbers in the WAT, where it is commonly believed they promote chronic, low‐grade inflammation (Weisberg et al, 2003; Xu et al, 2003). Therefore, it is generally accepted that pro‐inflammatory ATMs are linked with the development of systemic insulin resistance and other metabolic diseases (Patsouris et al, 2008), even though the complete spectrum of initiators and the pathophysiological consequences of this infiltration have yet to be defined. Recently, it was demonstrated that ATMs undergo “unconventional” metabolic activation due to their exposure to high levels of free fatty acids (FFAs) during adiposity (Kratz et al, 2014), rather than a classically activated (M1) or alternatively activated (M2) macrophage polarization (Gordon & Taylor, 2005).
One of the key factors that can affect ATMs is diet. A high‐fat diet not only augments total ATM numbers but also attracts a unique ATM subpopulation that is minimally present in WAT under steady‐state conditions (Lumeng et al, 2007; Nguyen et al, 2007). These obesity‐driven infiltrating ATMs are bone marrow (BM)‐derived (Chen & Ruedl, 2020), express markers such as CD11c, CD9, and Trem2 (Hill et al, 2018), and are frequently lipid‐laden; and the majority form classical crown‐like structures (CLS) around dead or damaged hypertrophic adipocytes (Cinti et al, 2005). Although considered to be the major initiators of the inflammatory cascade (Boutens et al, 2018), as well as one of the contributors to metabolic syndrome (Patsouris et al, 2008; Boutens & Stienstra, 2016), recent evidence highlights the role of ATMs in controlling tissue immune surveillance and therefore in maintaining a healthy adipose tissue environment via enhanced endocytic as well as lipid‐buffering capacity (Jaitin et al, 2019; Silva et al, 2019).
Here, we investigated the potential impact of different ATM subpopulations on the complex WAT cellular microenvironment during the development of obesity, in an attempt to identify the cellular mechanisms underlying high‐fat‐diet‐driven metabolic derangements. To do so, we made use of CD169‐diphtheria toxin receptor (DTR) mice, in which tissue‐resident macrophages can be depleted via diphtheria toxin (DT) injection (Purnama et al, 2014). We then performed WAT‐specific metabolome, three‐dimensional (3D) volume fluorescence imaging, and single‐cell RNA‐seq analysis to provide new insights into the interplay between ATMs, in particular the CD11c+ subpopulation, with their surrounding WAT microenvironment and their beneficial contribution to protecting WAT from pathological adipose tissue remodeling.
Results
F4/80hiMHCIIhiCD11c+ ATMs increasingly seed into epididymal WAT during obesity progression
Various distinct myeloid cells are present in epididymal WAT (eWAT). F4/80hi ATMs represent the largest fraction of CD45+ cells and can be further separated into three main subpopulations on the basis of their MHCII and CD11c expression (Chen & Ruedl, 2020). In lean mice, MHCIIlowCD11c− (subsequently referred to as MHCIIlow) and MHCIIhiCD11c− (subsequently referred to as MHCIIhi) cells are the dominant ATMs (Fig 1A); both cell types express the embryonic marker Tim4 and are only slowly replaced by BM‐derived cells (Chen & Ruedl, 2020). CD11c+ cells, which are almost undetectable in young lean mice (Fig 1A and C), only start to infiltrate eWAT with increasing age. However, huge numbers of CD11c+ cells infiltrate eWAT during the development of obesity, as clearly seen from a high‐resolution data‐driven uniform manifold approximation and projection (UMAP) analysis performed over a 16‐week period on a high‐fat diet (HFD) (Fig 1D). In fact, whereas the accumulation of MHCIIlow macrophages in the eWAT of obese mice was minimal, there was a strong correlation between obesity‐driven increasing eWAT weight and the infiltration of MHCIIhi as well as CD11c+ ATMs (Fig 1C). Under an HFD, these two F4/80hiMHCIIhi ATM subsets lost their embryonic Tim4+ signature and were swiftly replaced by BM‐derived cells (Chen & Ruedl, 2020). In obese conditions, both infiltrating ATM subsets showed a similar phenotype, lacking CD206, CD301, and CD163, but expressing higher levels of CCR2, Cx3CR1, and CD9 (Fig 1B). Interestingly, Trem2, a marker expressed on lipid‐associated macrophages (LAMs) (Jaitin et al, 2019), was detectable only on CD11c+ ATMs. Based on the UMAP analysis, we see these three ATMs as developmentally distinct subpopulations (Fig 1E). Monocytes (F4/80int Ly6Chigh MHCII−), monocyte‐derived macrophages (F4/80int Ly6ChighMHCII+, Ly6CnegMHCII+), Ly6G+ neutrophils (Fig 1C), and dendritic cells (DCs) (Chen & Ruedl, 2020) were present in lower numbers in the eWAT and minimally infiltrated this tissue during the development of obesity (Fig 1C and D). These data support that ATMs are the main leukocyte fraction in the eWAT and the three main subpopulations dynamically change in numbers and proportions during aging or progression of obesity.
Figure 1. Enhanced ATM infiltration into eWAT during high‐fat diet‐triggered obesity.
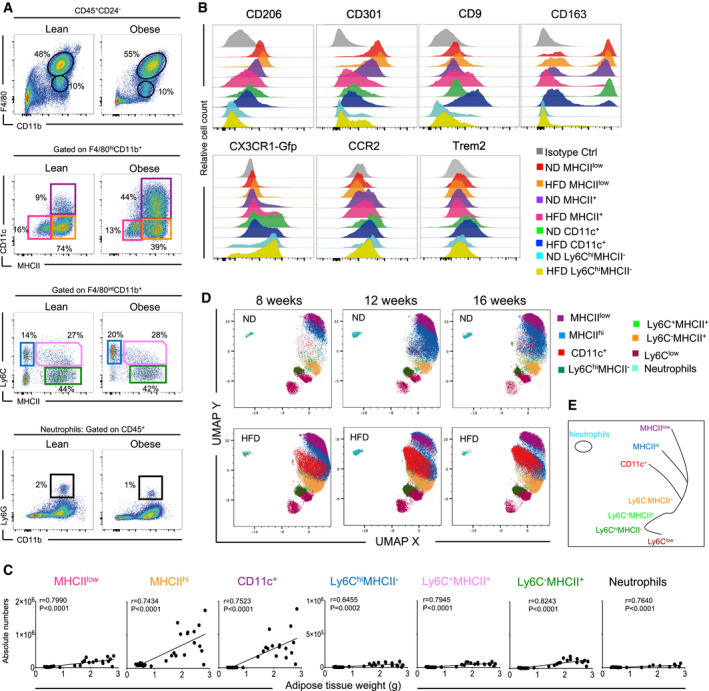
- Representative flow cytometry dot plots showing the myeloid cell landscape in eWAT of 16‐week‐old mice fed on a high‐fat diet (HFD) (right) and the corresponding lean controls (left). The upper panel shows the F4/80 versus CD11b staining profile gated by CD45+CD24−Ly6G− cells. The middle panels show the MHCII versus CD11c staining profile of F4/80hi macrophages and Ly6C versus MHCII staining profile of F4/80int cells, respectively. The lower panel shows the Ly6G versus CD11b staining profile gated by total CD45+cells (n = 8 mice).
- Representative phenotype profiling of distinct myeloid subpopulations in lean or obese (HFD 16 weeks) eWAT: CD206, CD301, CD9, CD163, Cx3CR1, CCR2, and Trem2 expression is shown in the respective colored histograms specific for each cell type (n = 3–5 mice per group). The y‐axis shows relative cell count for each population (normalized to mode).
- Pearson correlation coefficients between eWAT weight and cell numbers of MHCIIlow, MHCIIhi, CD11c+ ATMs, Ly6ChiMHCII−, Ly6ChiMHCII+, Ly6C−MHCII+, and Ly6G+ neutrophils. Significance was determined using the Pearson correlation test.
- High‐dimensional UMAP analysis shows a dynamic distribution of distinct myeloid clusters in eWAT during aging and the development of obesity. Three timepoints were analyzed (8, 12, and 16 weeks) with annotated, colored clusters based on their expression of 13 different markers (n = 3 for each timepoint).
- Illustration of global topological relationships between clusters of different myeloid cell populations in the eWAT based on the result in D.
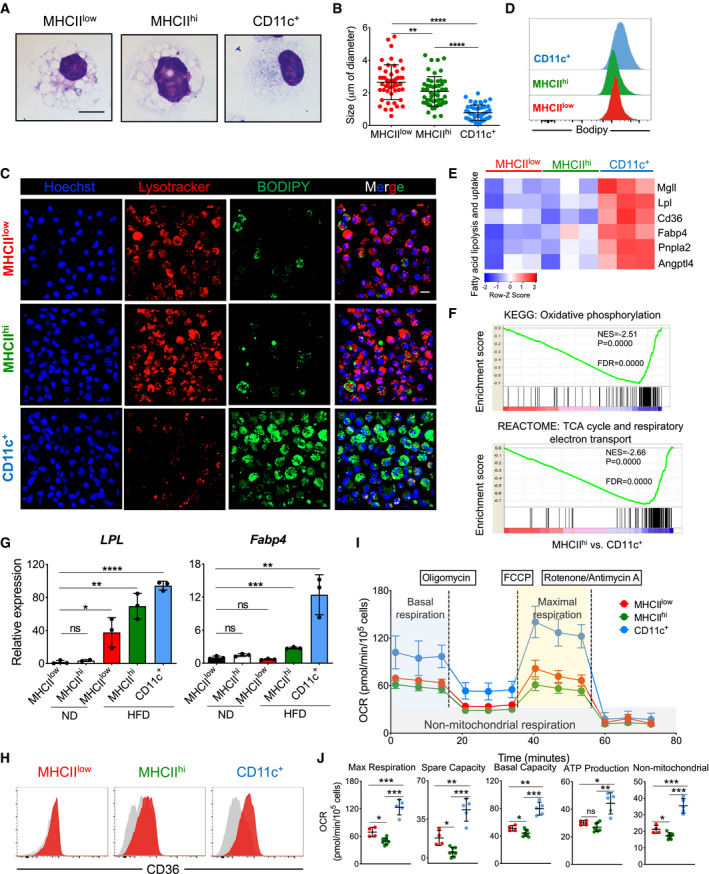
CD11c+ ATMs in obese mice possess high lipid metabolism and bioenergetic activity
To visualize and compare the morphology of different ATM subsets, we isolated MHCIIlow, MHCIIhi, and CD11c+ ATMs from pooled eWAT of mice fed on an HFD for 16 weeks. Then, we sorted and analyzed the cells in Giemsa‐stained cytospin preparations. Although all ATMs had a similar diameter of 15 µm, clear differences in vacuole size were detected among the three subpopulations, with MHCIIlow and MHCIIhi ATMs showing large cytoplasmic vacuoles, while CD11c+ ATMs exhibited significantly smaller vacuoles (Fig 2A and B). LysoTracker staining confirmed the presence of large lysosomes mainly in MHCIIlow and MHCIIhi cells, but less frequently in CD11c+ cells (Fig 2C). On the other hand, immunofluorescence and flow cytometry staining with BODIPY, a fluorescent dye commonly used to measure neutral lipid content, revealed CD11c+ cells to be the main lipid‐laden ATM fraction (Fig 2C and D).
Figure 2. Among eWAT ATM subpopulations, CD11c+ ATMs show augmented lipid and bioenergetic profiles.
-
ARepresentative Giemsa staining of sorted ATM subsets (MHCIIlow, MHCIIhi, and CD11c+) from obese (HFD 16 weeks) eWAT. Scale bar, 10 μm.
-
BQuantification of intracellular vacuole sizes in the three different ATM subsets (n = 4 mice). The average size is presented as mean ± SD. Statistical significance was determined using one‐way ANOVA test. **P < 0.01 and ****P < 0.0001.
-
CRepresentative confocal 3D images of LysoTracker‐stained (red), DAPI‐stained (blue), and BODIPY‐stained (green) ATM subsets obtained from eWAT of HFD (16 weeks) treated mice (n = 8 mice from two independent experiments). The merged figures are the result of the overlap of all three fluorochromes. Scale bar, 10 μm.
-
DRepresentative histograms showing the mean fluorescence intensity of BODIPY‐stained ATM subsets obtained from obese (HFD 16 weeks) eWAT (n = 4 mice).
-
EHeatmap showing fatty acid lipolysis‐ and uptake‐related genes differentially expressed among the eWAT ATM subsets from HFD‐treated (16 weeks) mice. Each cluster has three biological replicates. The z‐score of the gene expression profiles gives a scale to measure the differential expression.
-
FGene set enrichment analysis (GSEA) identified significant transcriptional upregulation in oxidative phosphorylation and the TCA cycle/respiratory electron transport signaling pathway in CD11c+ ATMs when compared with MHCIIhi ATMs obtained from eWAT of HFD‐treated (HFD 16 weeks) mice. Gene ontology (GO)/Kyoto Encyclopedia of Genes and Genomes (KEGG) with false discovery rate (FDR) < 0.05.
-
GqPCR validation of Lpl and Fabp4 expression in sorted ATM subsets from eWAT of ND‐ and HFD‐treated (16 weeks) mice (n = 3 of 5 pooled mice each). Data are presented as mean ± SD. *P < 0.05; **P < 0.01, ***P < 0.001; and ****P < 0.0001. ns: no significant difference.
-
HRepresentative FACS analysis of CD36 expression in ATM subsets from eWAT of HFD (16 weeks) mice (n = 4 mice).
-
I, JMito Stress assay of sorted ATM subsets (red, MHCIIlow; green, MHCIIhi; and blue, CD11c+) from 16‐week HFD obese mice showing overall oxygen consumption rate (OCR), maximal respiration, spare capacity respiration, basal respiration, ATP production, and non‐mitochondrial respiration. OCR data shown are representative of four independent experiments and expressed as mean ± SD (n = 4 MHCIIlow; n = 7 MHCIIhi; n = 5 CD11c+), with statistical significance determined using one‐way ANOVA test (*P < 0.05; **P < 0.01; and ***P < 0.001. ns: no significant difference).
We next performed RNA‐seq analysis on purified ATM subsets to determine the lipid metabolism gene expression signature. Here, we further confirmed differential lipid metabolism gene expression among the ATMs. The fatty acid transporter (Cd36) and a series of enzymes involved in lipid degradation (Lpl, Lipa, Fabp4, Mgll, PPARγ, Angptl4, and Pnpla2) were significantly and highly expressed in CD11c+ cells when compared with their expression in the other two fractions (Fig 2E–H). Moreover, gene set enrichment analysis (GSEA) showed that CD11c+ ATMs are significantly involved in TCA cycle, respiratory electron transport, and oxidative phosphorylation signaling pathways when compared to MHCIIhi ATMs (Fig 2F).
Additionally, an ex vivo bioenergetic analysis of different ATMs obtained from eWAT of obese mice clearly revealed that CD11c+ cells not only have the maximal respiration (measured in oxygen consumption rate [OCR]) but also the highest spare respiratory capacity, basal respiration, ATP production, and proton leakage among the ATMs (Fig 2I and J). The enhanced mitochondrial oxidative phosphorylation could have been fueled by the augmented fatty acid uptake observed in this obesity‐driven CD11c+ ATM subpopulation.
Overall, the expanded CD11c+ ATM population in obese mice possessed the greatest lipid metabolism activity and bioenergetic profile, suggesting possible functional polarizations between distinct ATM populations in eWAT.
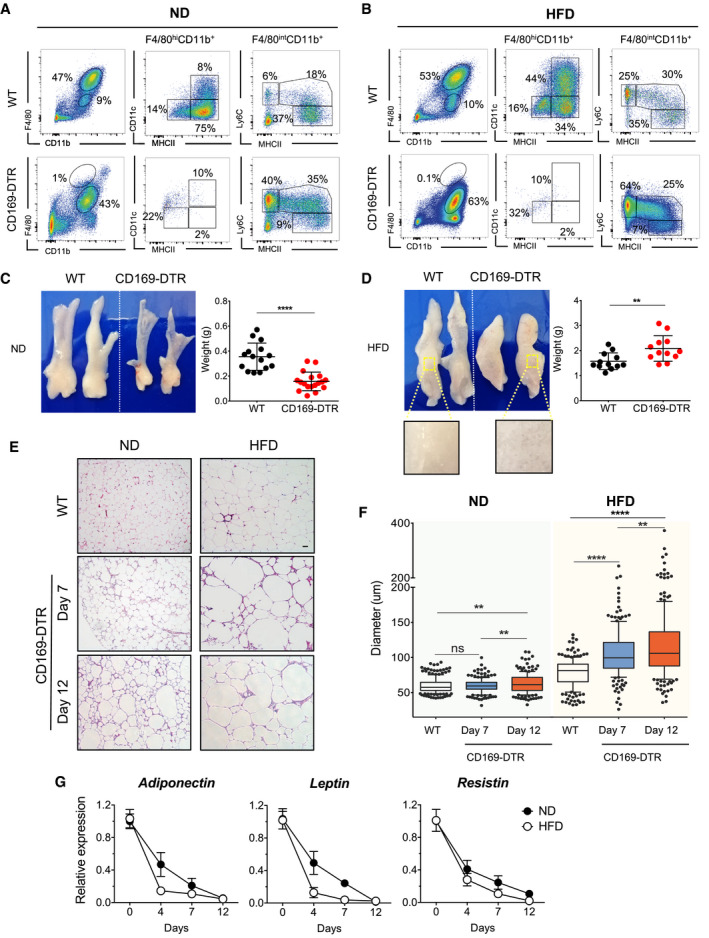
ATM depletion leads to increased hypertrophic and dysfunctional adipocytes
To investigate the role of ATMs in obesity‐driven eWAT remodeling, we exploited two different in vivo macrophage ablation mouse models: CD169‐DTR mice, which have been reported to specifically ablate tissue‐resident F4/80hi macrophages (Purnama et al, 2014; Gupta et al, 2016), and an antibody treatment specific for CSF1R, which depletes macrophages that are dependent on CSF‐1 and IL‐34 for their maintenance and survival (MacDonald et al, 2010). We recently demonstrated that CD169 expression is restricted to the three ATM subpopulations (Chen & Ruedl, 2020). The use of these two approaches clearly led to diverse depletion profiles in eWAT. CD169‐DTR mice treated with DT showed full depletion of all three F4/80hi ATM subpopulations (Fig 3A and B), whereas anti‐CFS1R antibody treatment reduced the MHCIIlow and MHChi fractions but did not affect the infiltrating CD11c+ ATMs, particularly in HFD‐treated mice (Fig EV1A). Since the macrophage niche became fully available in both lean and obese CD169‐DTR mice, some Ly6C monocytes and monocyte‐derived macrophages probably seeded the eWAT in an attempt to fill the empty space created, a phenomenon not detectable in the anti‐CSF1R antibody‐treated mice (Figs 3A and B, and EV1A).
Figure 3. ATM‐depleted CD169‐DTR mice show increased hypertrophic and dysfunctional adipocytes.
-
A, BRepresentative flow cytometric showing the specific depletion efficiency of ATM subsets from eWAT of CD169‐DTR ND‐ (A) or HFD‐treated (16 weeks) (B) mice. A representative analysis of three independent experiments is shown.
-
C, DeWAT was collected from lean (C) and obese (HFD 16 weeks) (D) WT or CD169‐DTR mice after 12 days of DT‐mediated ATM depletion. Representative pictures of dissected eWAT are shown on the left and the corresponding eWAT weights expressed as mean ± SD of more than 12 samples are shown in the right, with statistical significance calculated using Student’s t‐test (**P < 0.01 and ****P < 0.0001). The zoomed‐in area shows clear, oily lipid accumulation in the absence of ATMs (lower left part).
-
E, FAdipocyte hypertrophy is visible in the absence of ATMs. Representative images of hematoxylin and eosin staining (H&E) (E) and corresponding adipocyte diameters (F) of lean and obese (HFD 16 weeks) eWAT from WT or CD169‐DTR mice treated for 7 or 12 days with DT. Scale bar, 50 μm. Each group comprised 3–5 mice. Statistical significance was determined using one‐way ANOVA test. **P < 0.01 and ****P < 0.0001. ns: no significant difference. The box and whisker plots show the median value and 10–90 percentiles of adipocyte diameters.
-
GRelative expression of adipokines (adiponectin, leptin, and resistin) in eWAT obtained from WT or CD169‐DTR mice, as measured by qPCR after 4, 7, and 12 days of DT treatment. Black circles, ND‐fed mice and white circles, HFD‐fed (16 weeks) mice (n ≥ 5 mice per group, mean ± SD).
Figure EV1. Depletion of MHCIIlow and MHCIIhi ATM subsets does not affect eWAT functionality.
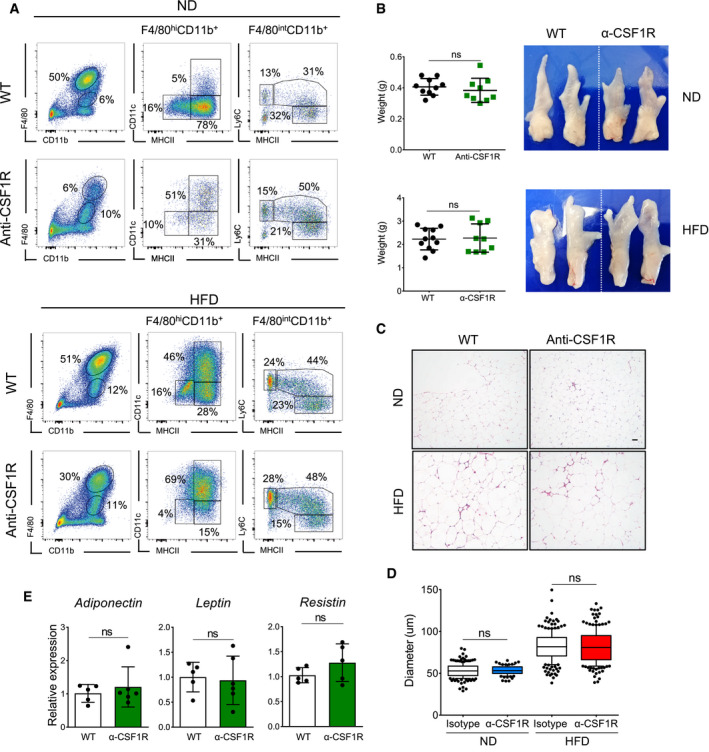
- Representative flow cytometry plots showing the specific efficiency of the depletion of ATM subsets from eWAT in anti‐CSF1R antibody‐treated lean (upper panel) and obese (lower panel, 16 weeks) mice. A representative analysis of three or four independent experiments is shown.
- eWAT was collected from lean and obese (HFD 16 weeks) anti‐CSF1R antibody‐treated mice. MHCIIlow and MHCIIhi ATM subsets were maintained at a depleted level for 12 days. Representative images of dissected eWATs are shown on the right, while on the left, the corresponding eWAT weights are expressed as mean ± SD (n = 8–12), with statistical significance calculated using Student’s t‐test. ns: no significant difference.
- Adipocyte hypertrophy is comparable between both experimental anti‐CSF1R antibody‐treated groups. Representative H&E pictures of eWAT. Scale bar, 50 μm.
- Calculated diameter of adipocytes from lean and obese (HFD 16 weeks) eWAT from WT or anti‐CSF1R antibody‐injected mice (12 days). Around 200 adipocytes from one mouse were calculated for the diameter, and 3–5 mouse were analyzed in each group. Statistical significance was determined using one‐way ANOVA test. ns: no significant difference. The box and whisker plots show the median value and 10–90 percentiles of adipocyte diameters.
- Relative expression of adipokines (adiponectin, leptin, and resistin) in eWAT obtained from obese (HFD 16 weeks) WT or anti‐CSF1R antibody‐injected mice measured by qPCR after 12 days’ antibody treatment (n = 5–6 per group, mean ± SD). Statistical significance was determined using an unpaired Student’s t‐test. ns: no significant difference.
In lean mice, 12 days’ full ablation of ATMs led to a reduction in eWAT weight (Fig 3C), which was not the case with the HFD‐treated CD169‐DTR mice, in which a significant increase in eWAT weight, but not body weight, was observed (Fig 3D and Appendix Fig S1A and B). Upon zooming in, the eWAT showed large, oily vesicles in the absence of ATMs. Such differences were not observed in the anti‐CSF1R antibody‐treated mice, since both anti‐CSF1R antibody‐injected and control mice (both lean and obese) showed comparable eWAT weights (Fig EV1B). Correlated with this, histological analysis of eWAT revealed a significant augmented adipocyte hypertrophy in the absence of all ATMs (Fig 3E and F), a phenotype not observed when the ablation was restricted to the MHCIIlow and MHCIIhi fractions (Fig EV1C and D). Under a normal chow diet (ND), adipocyte enlargement was visible only after 12 days of ATM depletion, whereas in obese CD169‐DTR mice, hypertrophy was already detectable after a 7‐day absence of ATMs when compared with the eWAT of WT controls (Fig 3E and F). Furthermore, the lack of ATMs interfered with adipocyte functionality, since the production of key adipocyte hormones, such as adiponectin, leptin, and resistin, rapidly decreased, with a more pronounced effect observed in HFD‐treated mice (Figs 3G and EV1E). Correlating with the lower levels measured in the eWAT, significant reduced levels of adiponectin and leptin were measured also in the serum of obese CD169‐DTR mice (Appendix Fig S1D). Furthermore, a slight increase of serum insulin levels was observed in CD169‐DTR mice whereas similar GTT reactivities were measured in both WT and CD169‐DTR obese mice, which indicates that type 2 diabetes was not significantly affected after this 12 days of macrophage ablation period (Appendix Fig S1C).
In summary, the complete depletion of ATMs led to major phenotypic and functional changes in adipocytes. To rule out a possible contribution of infiltrating monocytes in the observed phenotype, we crossed CCR2−/− mice with CD169‐DTR mice to obtain a CCR2−/− CD169‐DTR transgenic mouse line lacking the majority of monocytes and, if treated with DT, the ATM compartment. In the obese DT‐treated CCR2−/−CD169‐DTR mice, enhanced adipocyte hypertrophy was still observed, there was a reduction in all three adipokines, and the weight of eWAT increased (Appendix Fig S2A–D); therefore, we can conclude that the main drivers of the adipocyte dysregulation observed in CD169‐DTR mice are ATMs rather than infiltrating monocytes.
ATM depletion perturbs lipid metabolism in eWAT
Due to their transcriptional signature for lipid metabolism‐associated genes and enhanced accumulation of BODIPY dye, ATMs, in particular the CD11c+ subpopulation, are clearly actively involved in the uptake, storage, and processing of lipids (Fig 2). Therefore, we next investigated the consequences of ATM depletion on lipid metabolism in eWAT under both homeostatic and obesity conditions. To this end, following the depletion of ATMs (via DT injection in CD169‐DTR mice or anti‐CSF1R antibody injection in WT mice), fresh eWAT from lean and obese mice was collected and snap‐frozen in liquid nitrogen for further processing and analysis by mass spectrometry. Two‐dimensional principal component analysis (2D‐PCA) showed that individual samples from each group under DT treatment (WT ND, WT HFD, CD169‐DTR ND, and CD169‐DTR HFD) clustered together (Fig 4A). However, this was not clearly the case in the anti‐CSF1R antibody‐treated mice versus their corresponding WT controls (Fig 4B), indicating that the complete ablation of ATMs caused a major perturbation of lipid metabolism in eWAT under both lean and obese conditions. In particular, the glycerolipids (GL)/free fatty acid (FFA) cycle was affected in the obese eWAT when ATMs were absent, as shown in the heatmap (Fig 4C) and dot plots (Fig 4D). Fatty acids (FA) were clearly less abundant in obese eWAT of CD169‐DTR mice, whereas GLs accumulated in the obese eWAT of these mice (Fig 4D). The reduction of FFA was clearly detectable also in the serum (Appendix Fig S1D). Furthermore, a lack of ATMs downregulated the expression of several genes involved in lipid metabolism in both lean and obese eWAT. The ligand‐activated nuclear receptor central to the regulation of adipogenesis (Pparγ) and other key enzymes involved in lipolysis (Lpl) and lipogenesis (Dgat1), as well as lipid receptors/transporters (Cd36, Abca1) and lipolysis regulators (Fabp4), were all significantly downregulated in eWAT of CD169‐DTR mice when compared to WT mice. This was not observed in the anti‐CSF1R antibody‐treated mice (Fig 4E), which showed similar results to the control mice. Taken together, ATMs are involved in the control of lipid metabolism in the eWAT.
Figure 4. Perturbed lipid metabolism in eWAT in the absence of CD169+ ATMs.
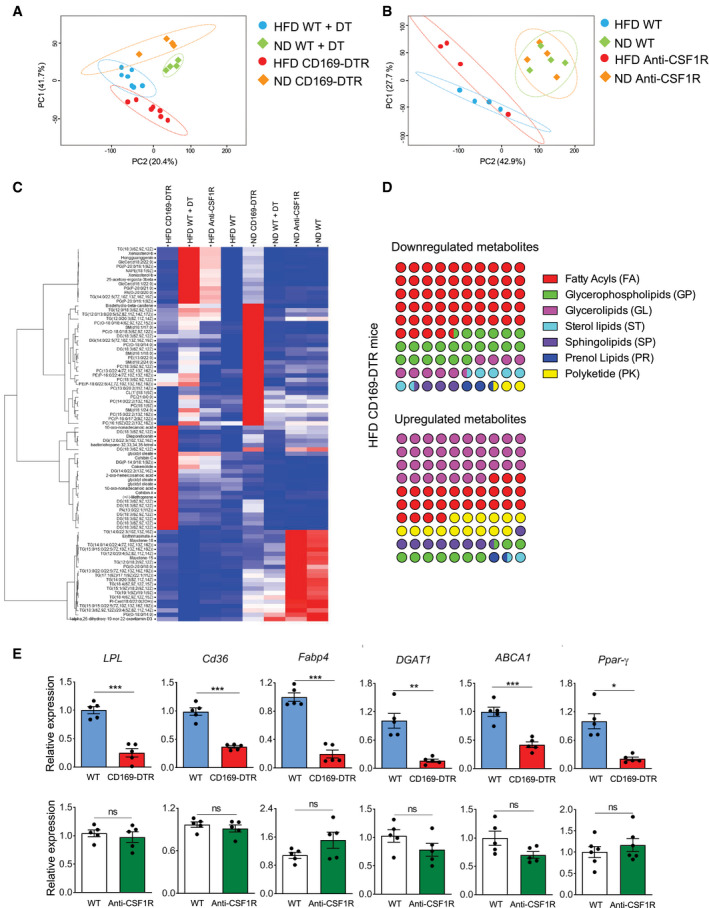
-
A, B2D PCA plots showing distinct lipid metabolite profiles of lean (square) and obese (round, HFD 16 weeks) WT (light green and light blue) and CD169‐DTR (A) or anti‐CSF1R antibody‐injected (B) (red and orange) mice. Mice were under DT or anti‐CSF1R antibody treatment for 12 days.
-
CHierarchical clustering heatmap showing the major changes in lipid metabolites in eWAT among the ND or HFD (16 weeks) CD169‐DTR or anti‐CSF1R antibody‐treated groups.
-
DDot plots showing the percentage of perturbed lipid categories (fold change >2) in HFD (16 weeks) CD169‐DTR mice. Upregulated (left) or downregulated (right) lipid metabolites between HFD (16 weeks) WT and CD169‐DTR mice were classified into their lipid categories according to the LIPID MAPS Structure Database (https://www.lipidmaps.org/data/classification/LM_classification_exp.php).
-
EqPCR showing the relative expression of genes for adipogenesis (Pparγ), lipolysis (Lpl), and lipogenesis (Dgat1), plus other lipid receptor/transporters (Cd36, Abca1) or chaperones (Fabp4) in obese (HFD 16 weeks) eWAT of CD169‐DTR (red bars) mice and DT‐injected WT controls (blue bars) (upper panel) and obese eWAT of anti‐CSF1R antibody‐injected mice (green bars) mice and isotype control‐injected WT controls (white bars) (lower panel). Mice were under DT or anti‐CSF1R antibody treatment for 12 days. (n = 5 pooled mice per group, mean ± SD). Statistical significance was determined using an unpaired Student’s t‐test. *P < 0.05, **P < 0.01, and ***P < 0.001. ns: no significant difference.
Unbiased scRNA‐seq analysis of eWAT reveals profound changes in the CD45− cell compartment in the absence of ATMs
To elucidate the mechanisms underlying the eWAT dysregulation observed in the absence of ATMs, we performed scRNA‐seq analysis of eWAT obtained from control and CD169‐DTR mice, in which the ATM pool had been maintained at a depleted level for 7 days. To specifically profile the non‐hematopoietic cell compartment in the eWAT, we separated out the CD45− and CD45+ cell fractions by excluding the high proportion of F4/80hi cells as well as Ly6Chi monocytes (Fig 5A). Pooled CD45+ and CD45− cells, in a 1:1 ratio, were then further processed for scRNA‐seq analysis. Using t‐distributed stochastic neighbor embedding (t‐SNE) analysis, we identified 23 transcriptionally distinct cell clusters present in the eWAT when the four experimental groups were combined (ND and HFD with or without ATMs) (Figs 5B and EV2A–D). Our analysis identified six main clusters of CD45− cells: two adipocyte stem cell fractions (ASC1 and ASC2), adipose‐derived multipotent mesenchymal stromal cells, including fibroblasts (stromal cells), two fractions of vascular endothelial cells (VEC1 and VEC2), and a population showing a gene signature for mesothelial‐like cells (MLCs) (Fig 5B). The ASC1 and ASC2 clusters were characterized by the expression of canonical adipocyte progenitor markers, including platelet‐derived growth factor receptors (Pdgfrα, Pdgfrβ, Gsn, Eln, Pi16, and Cebpd) (Burl et al, 2018; Merrick et al, 2019). Stromal cells expressed several collagen‐related genes (Col1a1, Col1a2, Col3a1, Col5a2, and Col6a2), fibrosis‐associated genes (Meg3, Postn, and Bgn), and Myl9 (Han et al, 2018). The VEC1 and VEC2 clusters predominantly expressed endothelial identity genes, including Pecam1, Cldn5, Cd36, Esam, Eng, ICAM‐2, Aqp1, and Aqp7 (Kalucka et al, 2020), whereas the MLCs were highly enriched for genes representing common mesothelial/epithelial markers, such as Upk3b, Msln, and Acta2 (Hepler et al, 2018) (Figs 5C and EV2E). Among the CD45+ cells, seventeen lymphoid and myeloid cell subpopulations were detectable in the eWAT, including monocytes, monocyte‐derived macrophages, dendritic cells, basophils, mast cells, neutrophils, B cells, CD4+ Th2 cells, CD4+ Th17 cells, Tregs and CD8+ T cells, NK, and NKT cells (Figs 5B and EV2A and B). We will not address CD45+ cells in detail in this work. Next, to identify the transcriptional changes in eWAT in the presence or absence of ATMs, we applied t‐SNE analysis to compare cell subpopulations obtained from eWAT of lean or obese WT and CD169‐DTR mice. As illustrated in the t‐SNE projection, an absence of ATMs in the eWAT for 1 week mainly affected the non‐hematopoietic cell compartment, while the total number of cell clusters and percentage of hematopoietic CD45+ cells remained roughly the same between the four experimental groups (Fig 5D). Ablation of ATMs in lean mice did not clearly perturb the cluster distribution of CD45− cells, whereas the scRNA‐seq analysis of obese WT and macrophage‐depleted eWATs revealed the disappearance of the three main CD45− clusters (ASC1, VEC1, and VEC2) and a pronounced increase in the stromal cell fraction displaying a fibrosis‐related signature (see later) (Fig 5D and E).
Figure 5. scRNA‐seq analysis reveals that a lack of ATMs leads to major cell perturbations in the eWAT CD45− fraction.
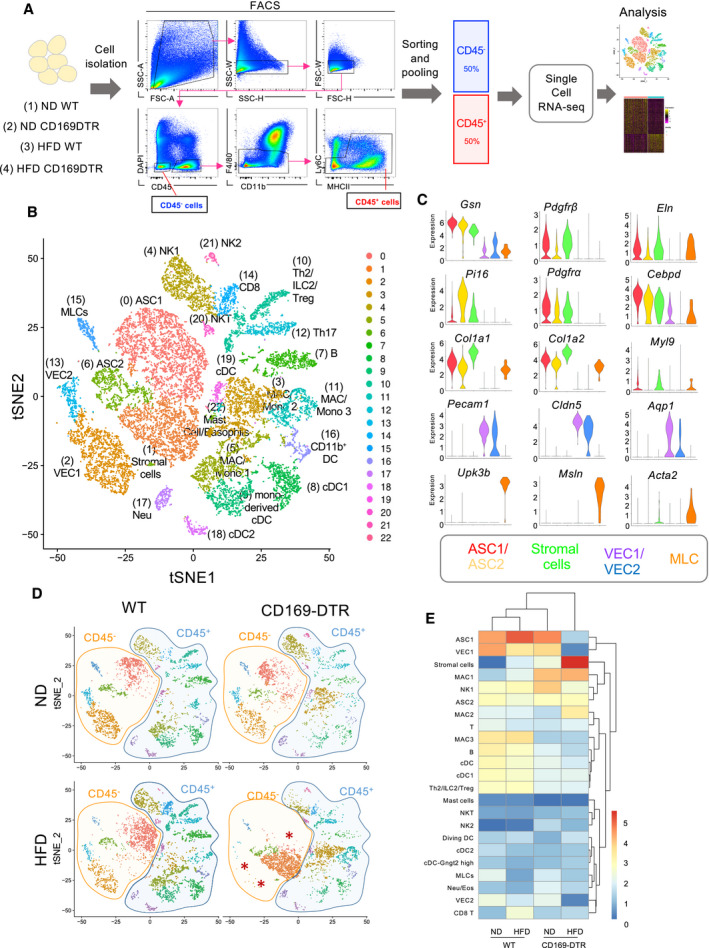
- Schematic representation of the scRNA‐seq analysis used on a 1:1 mixture of CD45− and CD45+ eWAT cells (excluding F4/80hi resident macrophages and Ly6C+ monocytes) obtained from lean/obese (HFD 16 weeks) WT and CD169‐DTR mice (7 days DT injected, n = 5–8 mice per group).
- t‐SNE projection of 18,798 pooled cells (5,032 cells from ND WT mice, 3,494 cells from ND CD169‐DTR mice, 5,096 cells from HFD (16 weeks) WT mice, and 5,176 cells from HFD (16 weeks) CD169‐DTR mice) isolated from eWAT of lean and obese WT/CD169‐DTR mice. The projection shows 23 distinct clusters of CD45− and CD45+ cell populations identified via unsupervised clustering. Six specific CD45− cell clusters were identified, including adipocyte stem cells (ASC1 and ASC2), stromal cells, vascular endothelial cells (VEC1 and VEC2), and mesothelial‐like cells (MLCs). Seventeen different CD45+ cell clusters were identified, including B and T cells, NK cells, and several myeloid subpopulations. NK: natural killer; MAC: macrophage; Mono: monocyte; DC: dendritic cell; and NKT: natural killer T.
- Violin plots depicting the expression of representative marker genes for each CD45− cell cluster: ASC1 (Gsn, Pdgfrb, Eln) (red), ASC2 (Pi16, Pdgfra, Cebpd) (yellow), stromal cells (Col1a1, Col1a2, Myl9) (green), VEC1/VEC2 (Pecam‐1, Cldn5, Aqp1) (purple and blue), and mesothelial‐like cells (MLCs) (Upk3b, Msln, Acta2) (orange).
- Four t‐SNE plots of 5,032, 3,494, 5,096, and 5,176 cells obtained from lean/obese WT and CD169‐DTR mice. Orange and blue shaded areas depict the CD45− and CD45+ cell clusters, respectively.
- Hierarchical heatmap showing the percentage (log2) of all 23 cell clusters identified in lean/obese (HFD 16 weeks) WT and CD169‐DTR mice.
Figure EV2. Dot plots of 10 marker genes for each major cell population of lean and obese eWAT related to Fig 5B .

- Marker genes of CD45+ clusters in lean eWAT.
- Marker genes of CD45+ clusters in obese (HFD 16 weeks) eWAT.
- Marker genes of CD45− clusters in lean eWAT.
- Marker genes of CD45− clusters in obese (HFD 16 weeks) eWAT. The average expression level is indicated by the color, and the percentage is indicated by the size of circle.
- Feature plots of selected maker genes of CD45− clusters related to Fig 5C. The color of the cells represents their level of expression.
Together, this scRNA‐seq analysis indicated that CD169+ ATMs are essential for maintaining the homeostasis of cell populations in eWAT, with the CD45− cell fraction in an obese microenvironment particularly perturbed in the absence of ATMs.
ATMs support vascular integrity
Although it has been suggested that distinct macrophage subpopulations occupy preferentially different niches, with MHCIIhi macrophages located adjacent to nerves and MHCIIlow macrophages residing alongside blood vessels (Chakarov et al, 2019), a recent detailed analysis of adipose tissue revealed that both these subsets of macrophages are positioned close to the vasculature and are therefore referred to as vascular‐associated macrophages (Silva et al, 2019). Here, we re‐analyzed in detail the localization of the three ATM subpopulations in both lean and obese eWAT. To test this, whole‐mount, three‐dimensional (3D) confocal imaging was employed to reconstruct a stereoscopic image of eWAT, which allowed us to visualize and calculate the distance between distinct ATMs to blood vessels or neurons. In lean mice, the two main MHCIIhi and MHCIIlow ATM subpopulations were visualized by co‐staining CD169 and MHC class II, whereas in obese mice, anti‐CD169 antibodies were combined with anti‐CD11c antibodies to delineate CD11c+ macrophages versus the remaining MHCIIhi and MHCIIlow ATMs. Blood vessels were visualized by staining the platelet endothelial cell adhesion molecule (also known as CD31), and the nerves were visualized using an antibody against a neuron‐specific class II beta tubulin (TUJ1).
We re‐confirmed that, under lean conditions, both MHCIIhi and MHCIIlow ATMs were in close proximity to blood vessels (within 1–10 µm). Similar to a previous report (Silva et al, 2019), we observed a significantly higher proportion (40%) of MHCIIhi ATMs localized less than 1 µm from the vasculature when compared to their MHCIIlow counterparts (Fig 6A–C and Movie EV1). As expected, in obese eWAT ATMs are no longer uniformly distributed throughout the tissue in the way they are under steady‐state conditions, since many ATMs, in particular CD11c+ ATMs, form classical crown‐like structures around damaged or dead adipocytes (Fig 6A and B, and Movie EV2). Our measurements also revealed that under obese conditions, most ATMs are located closely to the blood vasculature (less than 10 µm), with almost 60% of CD11c+ ATMs < 1 µm away from the vessels (Fig 6D). Notably, the majority of CD11c+ ATMs are preferentially close to small capillaries and do not reside alongside larger arteries or veins, as shown by our 3D volume fluorescence imaging analysis (Fig 6E and Movie EV3).
Figure 6. CD169+ ATMs are closely associated with the eWAT vasculature.
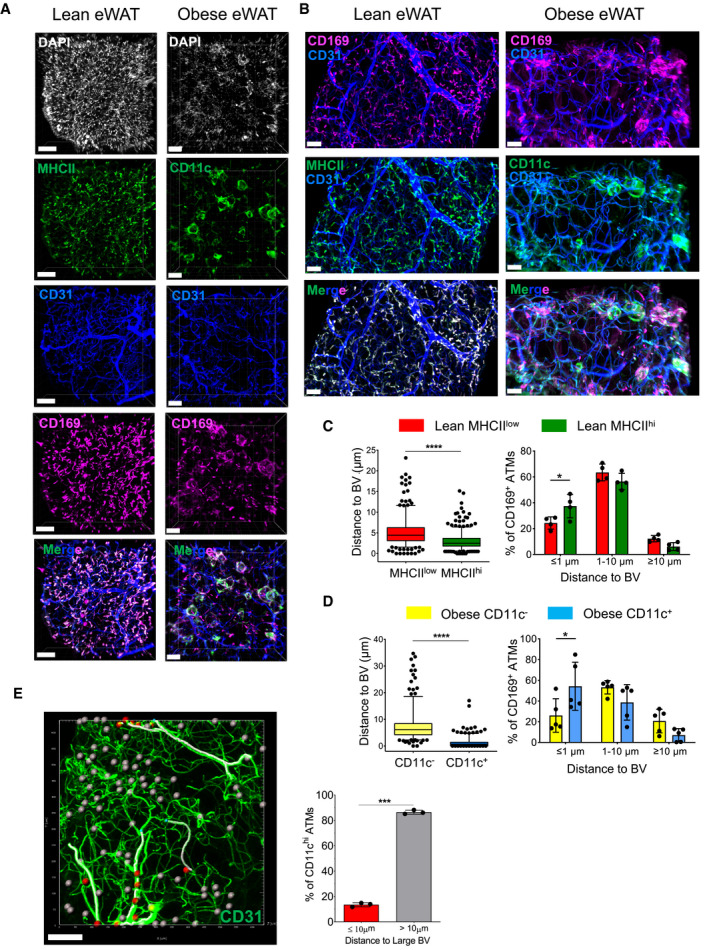
-
ARepresentative 3D fluorescence imaging of lean and obese (HFD 16 weeks) eWAT samples stained with anti‐MHCII (green in lean samples), anti‐CD11c (green in obese samples), anti‐CD31 (blue), anti‐CD169 (pink), and DAPI (white). The two images at the bottom show the images merged for the lean and obese conditions without DAPI. Scale bar: 100 µm (lean) and 70 µm (obese).
-
BHigher magnification of specific eWAT areas. Scale bar: 30 µm (lean) and 50 µm (obese).
-
C, D(left panels) Distance of different ATM subsets to blood vessels. Distances (in µm) were calculated using the Imaris software, as described in the Materials and Methods section. The box and whisker plots show the median value and 10‐90 percentiles of distance values. The right panels show the distribution of ATM subsets to blood vessels within a certain distance (≤ 1 μm, 1–10 μm, ≥ 10 μm). C: CD169+MHCIIlow and CD169+MHChi ATMs in lean eWAT. D: CD169+CD11c− and CD169+CD11c+ ATMs in obese (HFD 16 weeks) eWAT. Four mice from two independent experiments were analyzed (n = 4–5; mean ± SD). Statistical significance was determined using an unpaired Student’s t‐test. *P < 0.05 and ****P < 0.0001, bars with no asterisks showed no significant difference.
-
EModel construction based on 3D reconstructed images showing the relative distance of CD11c+ ATMs from traced large blood vessels. Blood vessels larger than 10µm diameter in the image were marked out on the image by tracing (white filaments). CD11c+ ATMs are close (< 10 µm) to the traced large blood vessels (red ellipsoids) or far (> 10 µm) (gray ellipsoids) from the traced blood vessel. Blood vessels are shown in green. Scale bar: 100 μm. The graph to the right shows the percentage of CD11chi ATMs showing a proximity of ≤ 10 or >10 µm to blood vessels quantified from 3 biological replicates from 1 experiment, and data were expressed as mean ± SD. Statistical significance was determined using an unpaired Student’s t‐test. ***P < 0.001.
Conversely, the majority of all three ATM subpopulations, in both lean and obese eWAT, were located relatively far away (> 10 µm) from neurons (Fig EV3, EV4, EV5).
Figure EV3. ATMs are not located in close proximity to nerves.
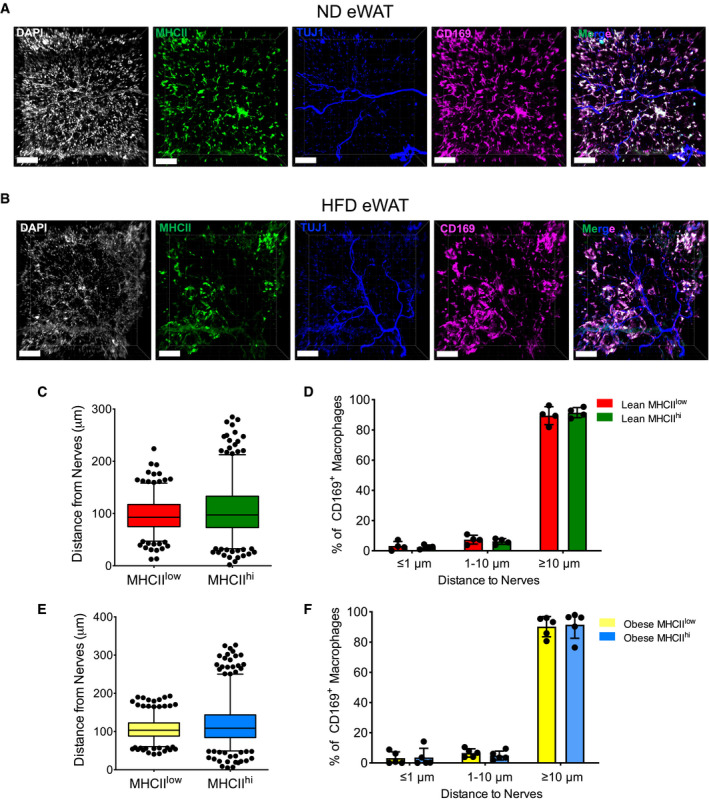
-
A, BRepresentative 3D‐reconstruction imaging showing the distance of ATM subsets to nerves in ND (A) and HFD (16 weeks) (B) eWAT (white, DAPI; green, anti‐MHCII; blue, anti‐Tuj1; and pink, CD169). The merged figure shows all three colors together without DAPI. Scale bar: 100 μm.
-
C–FStatistical analysis of the distances of different ATM subsets to nerves (C, E) and the percentage distribution of ATM subsets within a certain distance of nerves (≤ 1 μm, 1–10 μm, ≥ 10 μm) (D, F) in ND (C, D) or HFD (E, F) eWAT, as determined from 3D‐reconstruction confocal images (n = 4–5 mice from 2 independent experiments); data were expressed as mean ± SD. The box and whisker plots show the median value and 10–90 percentiles of the distance values.
Figure EV4. Depletion of MHCIIlow and MHCIIhi ATM subsets has no effect on vasculature integrity.
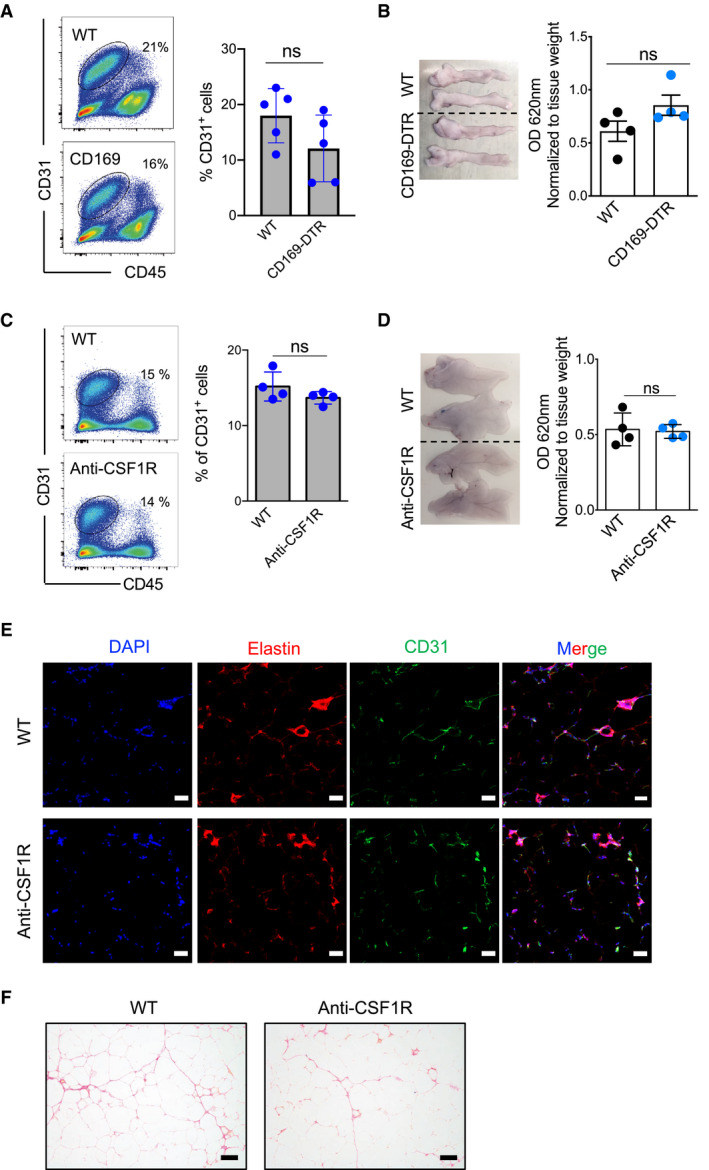
- Representative flow cytometry analysis (on the left) shows the staining profile of CD45 and CD31, and the chart (on the right) shows the percentage of CD31+CD45− fractions in the eWAT (ND 16 weeks) of five male control and five male CD169‐DTR (DT for 7 days) mice; data were expressed as mean ± SD. Statistical significance was determined using an unpaired Student’s t‐test. ns: no significant difference.
- Left: Representative Evans blue‐stained eWAT pictures obtained from WT or CD169‐DTR male mice (ND 16 weeks, DT for 7 days). Right: The chart shows the normalized (absorption values were normalized to tissue weight in grams) mean optical density ± SD (n = 4 mice per group). Statistical significance was determined using an unpaired Student’s t‐test. ns: no significant difference.
- Representative flow cytometry analysis (on the left) shows the staining profile of CD45 (x‐axis) and CD31 (y‐axis), and the chart (on the right) shows the percentage of CD31+ CD45− fractions in the eWAT (HFD 16 weeks) of 4 control and 4 anti‐CSF1R antibody‐treated mice; data were expressed as mean ± SD. Statistical significance was determined using an unpaired Student’s t‐test. ns: no significant difference.
- Left: Representative Evans blue‐stained eWAT pictures from WT or anti‐CSF1R antibody‐treated mice (HFD 16 weeks, after 12 days’ antibody treatment). Right: The chart shows the normalized (absorption values were normalized to tissue weight in grams) mean optical density ± SD (n = 4 mice per group). Statistical significance was determined using an unpaired Student’s t‐test. ns: no significant difference.
- Elastin staining showing similar ECM distribution in anti‐CSF1R antibody‐treated (HFD 16 weeks) mice compared with the ECM distribution in the control (DAPI, blue; elastin, red; and CD31, green). The final image shows the other images merged together (n = 3–5 mice per group). Scale bar: 50 μm.
- Representative picrosirius red staining showing comparable collagen expression in anti‐CSF1R antibody‐treated eWAT (HFD 16 weeks) compared with collagen expression in WT control mice (n = 3–5 mice per group). Scale bar: 100 μm.
Figure EV5. Cytokine secretion profiles in eWAT obtained from lean WT and CD169‐DTR mice (related to Fig 8).
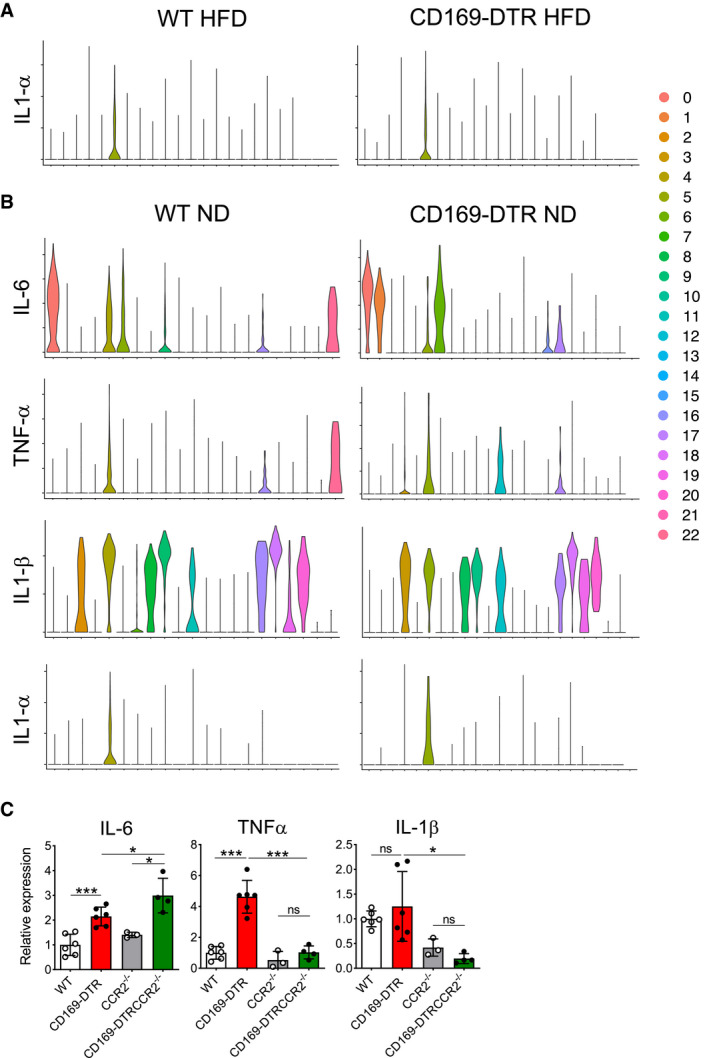
-
A, BViolin plots showing the pro‐inflammatory cytokine profile (Il‐6, Tnf‐α, Il‐1α, and Il‐1β) of all 23 cell clusters obtained from lean eWAT (lower panel). The upper panel shows the profile for Il‐1α in obese (HFD 16 weeks) eWAT.
-
CqPCR analysis showing the relative expression of Il‐6, Tnf‐α, and Il‐1β in lean eWAT collected from HFD (16 weeks) WT, CD169‐DTR, CCR2−/−, and CCR2−/− CD169‐DTR mice after 12 days of DT treatment (n = 3–6 mice per group, mean ± SD). Statistical significance was determined using a Bonferroni two‐way ANOVA test. *P < 0.05 and ***P < 0.001. ns: no significant difference.
Three‐dimensional volume fluorescence imaging of eWAT showed that after 7 days, the vascular network of DT‐treated obese CD169‐DTR mice exhibited slightly fewer branches in some areas of their eWAT compared with the vascular network in WT mice (Fig 7A and Movies EV4 and EV5). Remarkably, following cell isolation, the CD31+ cell fraction was shown to be absent in the eWAT samples obtained from obese CD169‐DTR mice. In fact, CD31+ endothelial cells, although visible in imaging, were no longer detectable by flow cytometry (Fig 7B) nor present in the scRNA‐seq analysis (Fig 5D). To rule out a possible Siglec 1 as well as human heparin‐binding EGF‐like growth factor (HBEGF, known also as DTR) expression on CD31+ cells which could lead to an undesired DT‐mediated endothelial cell deletion in CD169‐DTR mice, all 23 cell clusters obtained from our eWAT scRNA‐seq were analyzed for Siglec 1 and expression. Clearly, both VEC1 and VEC2 fractions are lacking a Siglec 1 and HBEGF gene expression; therefore, their absence is not linked to a direct DT‐mediated ablation (Appendix Fig S3A–C).
Figure 7. Vascular integrity is impaired in eWAT that lacks ATMs.
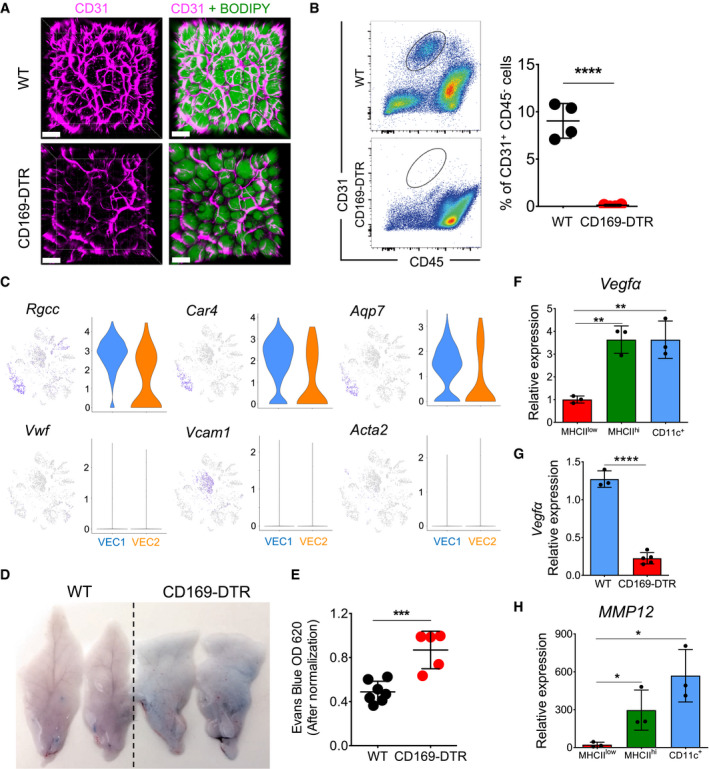
-
ARepresentative 3D fluorescence imaging of obese (HFD 16 weeks) eWAT stained with BODIPY (green) and anti‐CD31 (pink) obtained from WT and CD169‐DTR mice treated with DT over 7 days. Scale bar, 100 μm.
-
BThe lack of a CD31+ fraction after cell isolation from eWAT obtained from obese (HFD 16 weeks) CD169‐DTR mice following ablation of ATMs for 7 consecutive days. The flow cytometry analysis (on the left) shows the staining profile of CD45 (x‐axis) and CD31 (y‐axis), and the chart (on the right) shows the percentage of CD31+ CD45− fractions in the eWAT of control and CD169‐DTR mice (both n = 4); data were expressed as mean ± SD. Statistical significance was determined using an unpaired Student’s t‐test. ****P < 0.0001.
-
CViolin and feature plots of single cells in the VEC1 and VEC2 clusters showing expression of typical marker genes for capillaries or arteries (Rgcc, Car4, Acq7) and veins (Vwf, Vcam1, Acta2) in WT obese (HFD 16 weeks) eWAT.
-
D, EVascular permeability is increased in eWAT obtained from obese (HFD 16 weeks) CD169‐DTR mice. (D) Two representative images of eWAT dissected from Evans blue‐injected obese (HFD 16 weeks) WT and CD169‐DTR mice treated for 7 days with DT. Evans blue dye was extracted from eWAT using formamide, and the absorption of the extract was measured at 620 nm (the absorption values were normalized to the tissue weight in grams). (E) A chart showing the normalized mean optical density ± SD (n = 5–7 mice per group). Statistical significance was determined using an unpaired Student’s t‐test. ***P < 0.001.
-
F–HMmp12 (H) and Vegfα (F) are highly expressed in MHCIIhi and CD11c+ ATMs purified from obese (HFD 16 weeks) eWAT. qPCR analysis showed the relative expression in all three ATM subsets (n = 3), and data were expressed as mean ± SD. Statistical significance between the three ATM subpopulations was determined using one‐way ANOVA test. *P < 0.05 and **P < 0.01. (G) qPCR analysis showing the relative expression of Vegfα in obese (HFD 16 weeks) eWAT of WT (n = 3) or CD169‐DTR mice (n = 5) treated for 7 days with DT; data were expressed as mean ± SD. Statistical significance was determined using an unpaired Student’s t‐test. ****P < 0.0001.
Since the VEC1 and VEC2 fractions obtained from WT eWAT samples showed expression of the Rgcc, Car4, and Acq7 (Fig 7C) genes, which are markers for capillaries, and no expression of genes representative of large blood vessels, such as Vcam and Vwf, we speculate that the majority of CD31+ cells obtained after eWAT processing and digestion are capillary endothelial cells. In the absence of ATMs, this fraction was significantly affected and compromised in HFD‐treated mice (Fig 7B), since they were completely lost during the isolation procedure, but were minimally impacted in ND‐treated mice (Fig EV4A). To confirm the role of ATMs in mediating endothelial integrity, we monitored blood vessel permeability by intravenously injecting Evans blue into lean and obese WT and CD169‐DTR mice, in which ATM depletion was maintained for 7 consecutive days. Significant leakage of the blue dye into eWAT was observed only in obese mice, but not in lean mice lacking ATMs (Fig 7D), which was further confirmed by the optical measurement of extracted eWAT tissues (Figs 7E and Fig EV4B). As this effect was not clearly visible in anti‐CSF1R antibody‐treated mice (Fig EV4C and D), we suggest that the major macrophage fraction responsible for the observed vasculature leakiness predominately comprised CD11c+ ATMs.
One of the protective functions mediated by ATMs for vascular maintenance could be contributed by Vegfα, which encodes a known factor for vascular homeostasis (Cao, 2013), which was highly expressed under HFD conditions by MHCII+ and CD11c+ ATMs (Fig 7F). ATMs could be one of the principal sources of Vegfα since its level in eWAT was reduced in macrophage‐depleted obese CD169‐DTR mice (Fig 7G). Furthermore, matrix metalloproteinase‐12 (Mmp12), a macrophage‐secreted elastase, was abundantly expressed by CD11c+ ATMs (a 600‐fold increase under HFD conditions) when compared with its expression in other ATM subpopulations (Fig 7H). Since MMP12 regulates elastin and collagen accumulation, we hypothesized that ATMs are involved in altered elastin and collagen fiber organization in eWAT and around blood vessels, which could reflect the observed vascular instability and fragility. To test this, we co‐stained elastin with anti‐CD31 to mark the blood vessels and compared eWAT obtained from obese WT and CD169‐DTR mice. Only in the absence of ATMs, enlarged elastin deposits were detectably distributed throughout the tissue as well as in proximity to CD31+ blood vessels, a phenotype that was much less pronounced in WT obese mice (Fig 8A).
Figure 8. The absence of ATMs exacerbates eWAT fibrosis during obesity.
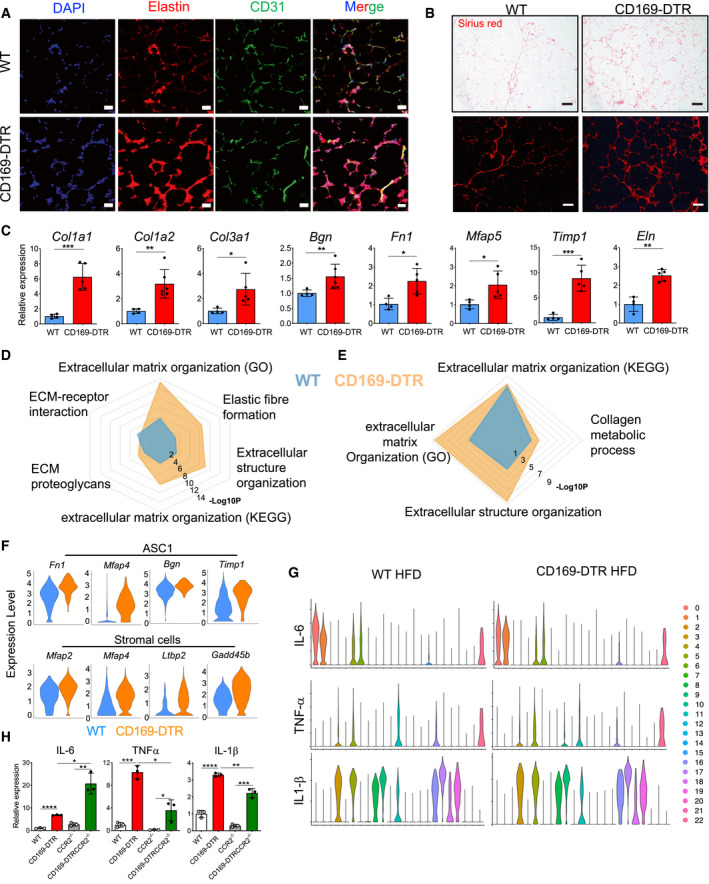
-
AElastin staining showing more pronounced ECM accumulation in eWAT of CD169‐DTR (HFD 16 weeks) when compared with ECM accumulation in the WT obese (HFD 16 weeks) control (DAPI, blue; elastin, red; and CD31, green). The fourth image in each row shows the three channels merged. Scale bar: 50 μm.
-
BPicrosirius red staining depicts excessive collagen deposits in obese (HFD 16 weeks) eWAT of CD169‐DTR mice when compared with collagen deposits in WT controls. Scale bar: 250 μm. Upper panels: brightfield microscopy and lower panels: fluorescence microscopy.
-
CqPCR analysis showing the relative expression of ECM marker genes in obese (HFD 16 weeks) eWAT of WT (blue bars) and CD169‐DTR (red bars) mice (DT treatment for 7 days). n = 4–6 mice per group, data were expressed as mean ± SD. Statistical significance was determined using an unpaired Student’s t‐test. *P < 0.05, **P < 0.01, and ***P < 0.001.
-
D, ECommon signaling pathways of single cells from clusters of ASC1 (D) and stromal cells (E) showed increased ECM remodeling pathways in HFD (16 weeks) CD169‐DTR mice when compared to WT mice.
-
FViolin plots of single cells showing upregulated ECM gene signatures in ASC1 (top, Fn1, Mfap4, Bgn, and Timp1) and stromal cell clusters (bottom, Mfap2, Mfap4, Ltbp2, and Gadd45b) in eWAT of obese (HFD 16 weeks) CD169‐DTR mice when compared with the expression of these genes in WT mice.
-
GViolin plots showing the pro‐inflammatory cytokine profile (Il‐6, Tnf‐α, and Il‐1β) of all 23 cell clusters obtained from obese (HFD 16 weeks) WT and CD169‐DTR eWAT. Cluster 0: ASC1; Cluster 1: stromal cells; Cluster 2: VEC1; Cluster 3: VEC2; Cluster 4: NK1; Cluster 5: MAC/Mono1; Cluster 6: ASC2; Cluster 7: B; Cluster 8: cDC1; Cluster 9: mono‐derived cDC; Cluster 10: Th2/ILC2/Treg; Cluster 11: MAC/Mono3; Cluster 12: Th17; Cluster 13: VEC2; Cluster 14: CD8; Cluster 15: MLCs; Cluster 16: CD11b+ DC; Cluster 17: neutrophils; Cluster 18: cDC2; Cluster 19: cDC; Cluster 20: NKT; Cluster 21: NK2; and Cluster 22: mast cells/basophils. ASC: adipocyte stem cell; VEC: vascular endothelial cell; MLC: mesothelial‐like cell; NK: natural killer; MAC: macrophage; Mono: monocyte; DC: dendritic cell; and NKT: natural killer T cells.
-
HqPCR analysis showing the relative expression of Il‐6, TnF‐α, and Il‐1β in obese (HFD 16 weeks) eWAT collected from WT, CD169‐DTR, CCR2−/−, and CCR2−/− CD169‐DTR mice (n = 3); data were expressed as mean ± SD. Statistical significance between the four experimental groups was determined using a Bonferroni two‐way ANOVA test. *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001.
Taken together, these findings suggest that during HFD‐induced eWAT remodeling, the CD11c+ ATM fraction is the fraction most closely associated with blood vessels, in particular capillaries, and supports blood vessel integrity.
ATMs hamper obesity‐induced accumulation of ECM and fibrosis
In parallel with the dysfunction of the eWAT vasculature, a lack of ATMs leads to aberrant extracellular matrix (ECM) remodeling in the obese eWAT microenvironment. In our final analyses, we thus tested whether the ECM remodeling in obese eWAT was controlled by ATMs. In the absence of ATMs, the elastin network in eWAT showed a marked difference in the organization of its matrix, with an increase in both density and thickness of its fibers (Fig 8A). Similarly, picrosirius red histology staining revealed augmented collagen depositions in eWAT of CD169‐DTR mice when compared to HFD‐fed control mice (Fig 8B). The augmented collagen and elastin deposits were only observed in eWAT of CD169‐DTR mice lacking all three ATM subpopulations and not in the anti‐CSF1R antibody‐treated mice, which were only deficient in the MHCIIlow and MHCIIhi subpopulations (Figs 8A and B, and EV4E and F). In line with the histology results, crucial ECM‐related genes, such as collagens (Col1a1, Col1a2, and Col3a1), elastin (Eln), fibronectin 1 (Fn1), proteoglycan biglycan (Bgn), and microfibrillar‐associated protein 5 (Mfap5), were increased in eWAT of obese CD169‐DTR mice (Fig 8C). Accordingly, scRNA‐seq analysis revealed a major accumulation of the stromal cell population in the eWAT of CD169‐DTR mice, which was minimally detectable in eWAT from WT obese mice (Fig 5D). The absence of macrophages affected the stromal cell cluster both quantitatively and qualitatively, as they displayed a strong “fibrotic” signature, with elevated expression of genes such as Col1a1, Col1a2, Col3a1, Postn, and Bgn (Figs 5C and EV2C and D). Moreover, when common signaling pathways from ASC1 and stromal cells were compared between WT and CD169‐DTR mice, a significant enrichment of ECM‐related pathways was observed in eWAT in the absence of ATMs (Fig 8 D and E), suggesting that the ablation of CD169+ ATMs generally promotes the enhanced remodeling of ECM in obese eWAT, which is mediated by both stromal cells and pre‐adipocytes. To explore this hypothesis, we compared the expression of ECM‐related genes in single cells of both cell types. The results showed that ECM‐related genes in both ASC1 (Fn1, Mfap4, Timp1, and Bgn) and stromal cells (Mfap2, Mfap4, Ltbp2, and Gadd45b) were upregulated in eWAT of obese CD169‐DTR mice (Fig 8F). Fibrosis is linked to tissue inflammation; therefore, pro‐inflammatory cytokine expression signatures were analyzed in all 23 cell clusters obtained from the scRNA‐seq analysis (Figs 8G and EV5A). Clearly, the expression of Il‐6 in obese eWAT was restricted to ASC1 [0], ASC2 [6], stromal cells [1], MSLs [15], and some myeloid cells (macrophages/monocytes [5] and mast cells [22]), whereas the expression of Tnf‐α, Il‐1α, and Il‐1β was mostly confined to myeloid cells (Tnf‐α: macrophages/monocytes [3, 5, 11], DCs [16], and mast cells [22]; Il‐1α: macrophages/monocytes [5]; Il‐1β: macrophages/monocytes [3, 5, 11], neutrophils [17], and DCs [8, 9, 16, 18, 19]). The comparison of lean eWAT obtained from WT and CD169‐DTR mice revealed that Il‐6 expression in particular was augmented in ASC1 (Cluster 0: Wilcoxon, P = 3.1e−15), ASC2 (Il‐6 only measurable in CD169‐DTR mice), and stromal cells (Cluster 6: Wilcoxon, P = 5.3e−06) when macrophages were depleted for 7 days (Fig EV5B), a trend that was also detectable in eWAT from obese mice although not highly significant (ASC1 (0): Wilcoxon, P = 0.024, ASC2 (1): Wilcoxon, P = 0.43, and stromal cells (6): Wilcoxon, P = 0.26) (Fig 8G). A slight increase was also observed for Tnf‐α in the macrophage/monocyte clusters. Less dysregulated were the cytokines Il‐1α and Il‐1β. The scRNA‐seq analysis was complemented by the qPCR analysis of Il‐6, Tnf‐α, and Il‐1β in the obese and lean eWAT of WT, CD169‐DTR, CCR2−/−, and CCR2−/− CD169‐DTR mice. In the absence of ATMs, all pro‐inflammatory cytokines measured were elevated in the eWAT. Tnf‐α and Il‐1β were most likely from myeloid origins, since their levels were reduced in the CCR2−/− CD169‐DTR mice, whereas Il‐6 was clearly adipocyte‐/stromal cell‐dependent since its levels were measured in both CD169‐DTR and CCR2−/− CD169‐DTR mice (Fig 8H). In obese eWATs, higher levels of pro‐inflammatory cytokines were measured when compared with their lean counterparts (Figs 8H and EV5C).
In summary, here we have shown that during HFD‐mediated eWAT remodeling, ATMs control adipocyte‐specific Il‐6 expression and prevent pathological changes such as dysregulated ECM composition, which contains the formation of tissue fibrosis.
Discussion
Immune cell infiltration in WAT during obesity is associated with local chronic inflammation and the development of metabolic syndrome, a major threat to health in developed countries. ATMs, the most abundant leukocyte fraction in WAT, are believed to be one of the main cell subpopulations involved in this metabolic dysregulation (Weisberg et al, 2003). However, this notion has recently been challenged by new evidence that indicates ATMs play a crucial role in buffering the excess fat released by lipid‐overladen adipocytes (Xu et al, 2013). A lipid receptor known as Trem2 has been identified as one of the master regulators of lipid‐associated macrophage function, helping to prevent both adipocyte hypertrophy and systemic metabolic imbalance (Jaitin et al, 2019). An evolutionarily conserved pathway was recently discovered in resident macrophages that control fat storage via PDGFcc secretion (preprint: Cox et al, 2020), which underlines their importance in maintaining the functionality of the WAT cellular microenvironment. Another study elegantly demonstrated not only the close vasculature association of ATMs and their contribution to endocytosis but also identified a clear anti‐inflammatory/detoxifying gene signature (Silva et al, 2019). A recent study also showed that ATMs perform both detrimental and beneficial functions during the progression of obesity (Coats et al, 2017). It remains an open question, therefore, as to the exact role these ATMs play: whether they cause damage to tissues or whether they have a beneficial function during the obesity–adipose tissue remodeling process. Here, by exploiting two different in vivo macrophage‐ablating mouse models (CD169‐DTR mice and anti‐CSF1R antibody treatment), we addressed this question and aimed to unravel still unappreciated macrophage‐related functions involved in the control of obesity‐driven metabolic derangements.
Recently, multicolor flow, mass cytometry analysis, and single‐cell RNA sequencing have revealed the ATMs to be a spatially and phenotypically heterogeneous cell family. During the development of obesity, several distinct subpopulations of ATMs have been identified in the WAT (Silva et al, 2019), which display different frequencies, turnover rates, and origins (Chen & Ruedl, 2020). These findings suggest that the tissue environment drives ATM heterogeneity and that each ATM subpopulation may play a unique role during either the steady‐state or obesity‐driven metabolic tissue dysregulation. Obesity not only affects the number of ATMs but also their spatial organization within the WAT. ATMs are usually uniformly distributed throughout lean WAT; however, during obesity‐driven WAT enlargement, ATMs begin to accumulate around dying adipocytes, typically forming clusters known as crown‐like structures (Cinti et al, 2005). In eWAT, we broadly identified three clearly distinct populations of tissue‐resident F4/80hi macrophages, with each population representing a separate developmental lineage. Two of these populations, the MHCIIlow and MHCIIhi macrophages, can be found in many other tissues, including the intestines, lungs, dermis, and heart (Soncin et al, 2018; Chakarov et al, 2019). The MHCIIhi resident macrophage niche was described as being close to nerve bundles and fibers in tissues such as the lungs and skin (Chakarov et al, 2019), although in the WAT, we and others have shown their proximity to the vasculature (Silva et al, 2019). MHCIIlow cells are long‐lived cells that express the embryonic‐related marker Tim‐4; they are present in various organs, including intestine (Shaw et al, 2018; Soncin et al, 2018), dermis (Sheng et al, 2015), and WAT, and are located adjacent to blood vessels (Chakarov et al, 2019; Silva et al, 2019). A unique tissue‐resident macrophage population, characterized by co‐expression of the canonical dendritic cell marker CD11c (Shaul et al, 2010), the lipid receptor Trem2 (Chakarov et al, 2019), and the tetraspanin CD9 (Hill et al, 2018), heavily infiltrates the WAT during the progression of obesity. This macrophage type is not generally found in other tissues and displays similarities with tumor‐associated macrophages that appear in the mammary glands during breast cancer progression (Franklin et al, 2014). Here, we showed that this CD11c+ obesity‐driven infiltrating ATM subpopulation is metabolically different from the other two ATM subsets. Although all three subsets are exposed to the same lipid‐rich microenvironment, CD11c+ ATMs possess some exclusive properties compared with those of the other two ATM subpopulations. Only CD11c+ ATMs are equipped, via specific receptors (e.g., CD36) and enzymes (e.g., Lpl, Fabp4, and Mgll), to take up and process lipids. However, they also show augmented fatty acid fueled oxidative phosphorylation activity and are therefore clearly functionally specialized to handle the increasing accumulation of fat during the development of obesity. Related to these properties, in obese CD169‐DTR mice lacking all three subpopulations of ATMs, we observed an increased number of hypertrophic adipocytes, a reduction in all major adipokines (e.g., adipokine, leptin, and resistin), and a marked change in the adipose tissue lipidome signature. Interestingly, in anti‐CSF1R antibody‐treated mice that lacked only MHCIIlow and MHCIIhi ATMs, this dysregulation was not clearly observed; therefore, the lipid metabolism of adipocytes is probably mainly controlled by the infiltrating CD11c+ cell fraction. A similar phenotype to the CD169‐DTR mice was found in Trem2 deficient mice, which showed not only adipocyte hypertrophy and body fat accumulation but also systemic hypercholesterolemia and glucose tolerance. This was attributed to the lipid‐associated Trem2‐expressing CD11c+ macrophages (Jaitin et al, 2019). Taken together, this evidence suggests that this particular ATM subpopulation plays a crucial and beneficial role in buffering the excess lipids accumulated during the development of obesity, and without these cells, adipocytes would become dysregulated.
To further elucidate other ATM‐related functions, we performed scRNA‐seq analysis of cells isolated from lean and obese adipose tissue, obtained from WT and CD169‐DTR mice in which the macrophage pool was ablated over 1 week. This analysis led to some important observations, such as the complete loss of the endothelial cell compartment (VEC1 and VEC2), a reduction in the pre‐adipocyte cell compartment (ASC1), and a massive increase in the stromal cell cluster in obese CD169‐DTR mice.
Based on previous studies, tissue‐resident macrophages are known to protect vessel integrity in large arteries; it has been elegantly demonstrated, for example, that aorta‐resident macrophages maintain arterial vessel tone through hyaluronan‐mediated regulation of smooth muscle collagen production (Lim et al, 2018). The control of vascular permeability by blood vessel‐associated macrophages (also called perivascular macrophages) has been described in many organs, such as brain, heart, skin, lungs, kidney, and pancreas (reviewed in Lapenna et al, (2018)), as well as in the intestine where their ablation leads to loss of VE‐Cadherin+ blood vessels and enhanced vascular leakage (De Schepper et al, 2018; Viola & Boeckxstaens, 2020).
Here, we showed that under obese conditions, the majority of CD11c+ ATMs are in close proximity to small vessels and capillaries, and their absence impacts not only the density of vascularization in some areas of the eWAT but also the vessel integrity. In fact, in CD169‐DTR mice fed on a high‐fat diet, the accelerated hypertrophic alterations were clearly linked with an impairment in the vascularization density and permeability. Factors such as Vegfα and Mmp12, which are highly expressed by infiltrating CD11c+ ATMs under obese conditions, could contribute to maintain the “fitness” of the blood vessel network during obesity‐driven adipose tissue remodeling. Dysregulated endothelial permeability and vascular leakage could be also attributed to the pro‐fibrotic microenvironment (Sun et al, 2013), which develops in the eWAT of obese CD169‐DTR mice. Crucial ECM‐related genes, including elastin, fibronectin, and various collagens, are massively upregulated in the WAT of ATM‐depleted mice. This pronounced fibrotic and ECM remodeling phenotype was also confirmed by picrosirius red staining and elastin histology, which showed large deposits of collagen and elastin in the eWAT in close proximity to its vessels. The enhanced extracellular stiffness relative to the “normal” levels observed in control mice may contribute to the observed vascular dysfunction and increased vascular leakage in CD169‐DTR mice.
As with stromal cells, adipocytes produce numerous matrix proteins necessary to maintain the structure of the tissue microenvironment. During obesity‐driven adipose tissue remodeling, the increase in the number and size of adipocytes means further tissue expansion is limited by the tight ECM framework, which subsequently undergoes fibrotic changes (Sun et al, 2011; Buechler et al, 2015). Fibrosis is often accompanied by adipocyte cell death, inflammation, and hypoxia, all factors that contribute to insulin resistance (Sun et al, 2013; Datta et al, 2018). Our detailed scRNA‐seq analysis revealed that pre‐adipocytes (ASC1) and stromal cells obtained from the eWAT of CD169‐DTR mice express augmented levels of several ECM‐ and fibrotic‐related genes, as well as IL‐6, which could explain the adipocyte dysfunction, inflammation, and increased fibrosis observed in the absence of ATMs. IL‐6 is of particular interest, since high levels of secretion of this cytokine from the WAT may contribute to metabolic complications, such as obesity‐associated hepatic insulin resistance and steatosis and, as a consequence, the development of type 2 diabetes (Allen & Febbraio, 2010; Wueest & Konrad, 2018).
Our data provide strong evidences that the presence of CD169+ ATMs in eWAT is crucial for containing excessive, pathological adipose tissue inflammation and remodeling; therefore, the role played by these cells in adipose tissue should be re‐considered. Moving away from the classical view of an inflammatory M1 macrophage type that supports the progression of obesity‐mediated WAT remodeling, CD169+ ATMs should be regarded as “amicus adipem” (Odegaard et al, 2013) and as important regulatory cells that actually restrain further tissue dysregulation caused by excess fat accumulation. These beneficial rather than detrimental properties should be considered in any eventual targeted therapeutic strategies.
Materials and Methods
Mice
C57BL/6J, CCR2−/−, and CX3CR‐1GFP (B6.129P2(Cg)‐Cx3cr1tm1Litt/J) mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). C57BL/6 CD169‐DTR mice were generated in our laboratory (Purnama et al, 2014) and crossed with CCR2−/− to obtain the final CCR2−/−CD169‐DTR mouse line. We used male mice only for these experiments. To induce obesity, mice aged 5–6 weeks were given a high‐fat diet (HFD) that comprised 60% kcal of fat (Research Diet, D12492, New Brunswick, NJ, USA) for 8, 12, or 16 weeks; age‐matched control mice were given a normal chow diet (ND).
CD169‐DTR+ and CD169‐DTR− (WT) mice were intraperitoneally (i.p.) injected with 20 ng/g DT (Sigma) every 3–4 days to maintain the depletion of CD169+ ATMs. C57BL/6J mice were i.p. injected with rat IgG2a isotype control (BioLegend) or anti‐CSF1R blocking antibody (Clone AFS98, BioXCell, West Lebanon, NH, USA), twice per week (400 μg per mouse) for 12 days. No randomization nor blinding was used during our experiments.
All mice were bred and maintained in a specific‐pathogen‐free animal facility at Nanyang Technological University (NTU), Singapore. The mice experiments were carried out in strict accordance with the recommendations of the NACLAR (National Advisory Committee for Laboratory Animal Research) and approved by the Institutional Animal Care and Use Committee (IACUC) of NTU (ARF‐ SBS/NIE A18014).
Cell isolation, flow cytometry staining, and UMAP analysis
Epididymal WAT was used in all our experiments. Adipose tissue cell isolation was performed as previously described (Chen & Ruedl, 2020). Briefly, collected eWAT was cut into small pieces and incubated in Iscove's modified Dulbecco's medium (IMDM) containing 100 µg/ml Collagenase D (Roche, Switzerland) and 20 U/ml DNase I (Life Technologies, Carlsbad, CA, USA) at 37°C for 1 h. After a short mixing for 20 s, the samples were centrifuged, cell pellets resuspended in 2% fetal bovine serum (FBS) in IMDM, filtered (100 μm), and centrifuged again at 500× g. After a short 3‐min erythrocyte lysis using 0.89% NH4CL, cells were centrifuged again at 500× g and finally resuspended in 2% FBS in PBS for flow cytometric analysis.
A single‐cell suspension was pre‐incubated with 10 μg/ml anti‐Fc receptor antibody (2.4G2) on ice for 20 min followed by another 20‐min incubation with the following antibodies: anti‐F4/80 (clone BM8; PE), anti‐CD45.2 (clone 104; PE‐cy7), anti‐CD163 (clone TNKUPJ; PerCP‐eFluor 710), anti‐CD31 (clone 390; Biotin), anti‐CD11c (clone N418; APC), anti‐MHCII (clone M5/114.15.2; eFluor 450), and BODIPYTM 493/503 (D3922) from Thermo Fisher Scientific. Anti‐CCR2 (APC, FAB5538A) and anti‐Trem2 (AF700, FAB17291N) were from R&D Systems. Anti‐ Ly‐6C (clone HK1.4; BV605), anti‐CD45 (clone 30F11; APCFire), anti‐Ly6G (clone 1A8; FITC), anti‐CD24 (clone M1/16; PE‐cy7), anti‐Tim‐4 (clone RMT4‐54; PE‐Cy7), anti‐CD206 (clone Co68C2; FITC), anti‐ MGL1/2 (clone LOM‐14, Percp‐cy5.5), anti‐CD9 (clone MZ3; FITC), anti‐CD36 (clone HM36; Percp‐cy5.5), anti‐NK1.1 (clone PK136; PE‐Cy7), anti‐B220 (clone RA3‐6B2; PE‐Cy7), and anti‐CD90.2 (clone 53‐2.1; PE‐Cy7) were from BioLegend. Anti‐CD11b (clone M1/70, BUV395) and streptavidin BUV395 were from BD Biosciences.
The cells were then centrifuged (500× g) and suspended in 100 μl 2% phosphate‐buffered saline–fetal bovine serum (PBS‐FBS) for flow cytometry (5‐laser LSRFortessa; BD Biosciences) analysis or cell sorting (FACSaria II). The data were analyzed using FlowJo software (FlowJo 10, TreeStar, Ashland, OR, USA).
For the UMAP analysis, after exclusion of dead cells using DAPI, live CD45+ Lin (B220, CD90.2, NK1.1)− cells from different groups were concatenated. UMAP analysis was performed using FlowJo software with default settings, using the Euclidean distance function with nearest neighbors of 15 and a minimum distance of 0.5 (http://arxiv.org/abs/1802.03426). For cell annotation, different cell populations were manually labeled based on their expression profiles of cell surface markers.
RNA extraction and cDNA preparation
Freshly isolated eWAT was homogenized in TRIzol (Invitrogen) solution and total RNA was extracted using an RNAsimple Total RNA Kit (DP419, TIANGEN Biotech Co., Ltd, Beijing, china), following the manufacturer’s protocol. cDNA was generated using SuperScript III Reverse Transcriptase (Cat. No. 18080093, Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s instructions.
For sorted ATMs, total RNA was isolated using an ArcturusTM PicoPureTM RNA Isolation Kit (Cat. No. KIT0214, Thermo Fisher Scientific, Waltham, MA, USA), quantified using a Quant‐iTTM RiboGreen® RNA Reagent Kit (Invitrogen), and retro‐transcribed and amplified using an Ovation® Pico WTA System V2 Kit (NuGEN Technologies, Inc., Redwood City, CA, USA), according to the manufacturers’ protocols.
Quantitative real‐time PCR
Quantitative real‐time PCR (qRT–PCR) was performed using the FAST 2X qPCR Master Mix (PrecisionFAST‐SY, Primerdesign Ltd, Cambridge, UK). Reactions were run on a real‐time qPCR system (Illumina, San Diego, CA, USA). Samples were normalized to β‐actin. The data represent the median of triplicate analyses and are displayed as a fold change from naïve wild‐type (WT) controls unless otherwise stated. The primer sequences were as follows:
β‐actin, Fwd: aaggccaaccgtgaaaagat and Rev: cctgtggtacgaccagaggcataca; adipoq, Fwd: cgattgtcagtggatctgacg and Rev: caacagtagcatcctgagccct; lep, Fwd: gagacccctgtgtcggttc and Rev: ctgcgtgtgtgaaatgtcattg; retn, Fwd: ccttgtccctgaactgctgg and Rev: tgtccagcaatttaagccaatgttc; Cd36, Fwd: gccaagctattgcgacatga and Rev: ctgcaaatgtcagaggaaaagaatc; Fabp4, Fwd: acaagctggtggtggaatgtg and Rev: cctttggctcatgcccttt; Lpl, Fwd: caacaaggtcagagccaagagaag and Rev: gttgtgttgcttgccatcctc; Dgat1, Fwd: gtgccatcgtctgcaagattc and Rev: gcatcaccacacaccaattcag; Abca1, Fwd: ccaaagagataccagcattaaggac and Rev: caggctgtccacagtagatc; Pparγ, Fwd: gtgccagtttcgatccgtaga and Rev: ggccagcatcgtgtagatga; Mmp12, Fwd: ttgatgaggcagaaacgtggac and Rev: agagaggcgaaatgtgctgg; Vegfα, Fwd: ggcctccgaaaccatgaactt and Rev: tgggaccacttggcatggtg; Col1a1, Fwd: cgcatgagccgaagctaac and Rev: ctcgggtttccacgtctcac; Col1a2, Fwd: agggtgaaactggtctccga and Rev: tctcctttagcaccgggttg; Bgn, Fwd: gcctgagttttctgcctacc and Rev: ctccgaagcccataggacag; Col3a1, Fwd: tcacccttcttcatcccact and Rev: gtcatcgcaaaggacagatcc; Eln, Fwd: gtgttggtcttccaggtgtata and Rev: ctttgactcctgtgccagt; Mfap5, Fwd: tgccgggatgagaagtttgc and Rev: aagtagttggagcgccgaag; and Timp1, Fwd: cctttgcatctctggcatctg and Rev: ggtggtctcgttgatttctggg.
Transcriptomics analysis by RNA sequencing
Total RNA was isolated from sorted cells using an ArcturusTM PicoPureTM RNA Isolation Kit and quantified using a Quant‐iTTM RiboGreen® RNA Reagent Kit, according to the manufacturers’ instructions. RNAs were analyzed using an Agilent Bioanalyzer (Agilent, Santa Clara, CA, USA) for quality assessment. cDNA libraries were prepared from a range of 18, 24.2, 68, and 100 ng total RNA starting material, using the Ovation Universal RNA‐seq system (NuGEN). The length distribution of the cDNA libraries was monitored using a DNA High Sensitivity Reagent Kit on the Agilent Bioanalyzer (Agilent, Santa Clara, CA, USA). All samples were subjected to an indexed paired‐end sequencing run of 2 × 100 bp on an Illumina NovaSeq 6000 system (Illumina). The paired‐end reads were trimmed with trim_galore1 (option: ‐q 20 –stringency 5 –paired) (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). The trimmed paired‐end reads were mapped to the mouse GRCm38/mm10 reference genome using the STAR2 (version 2.6.0a) alignment tool with multi‐sample 2‐pass mapping (Dobin et al, 2013). Mapped reads were summarized to gene level using featureCounts (Liao et al, 2014) in the Subread (Liao et al, 2013) software package (version 1.4.6‐p5) and with gene annotation from GENCODE release M19. For differentially expressed gene (DEG) analysis, DESeq2 (Love et al, 2014) was used and significant genes were identified with Benjamini–Hochberg‐adjusted P‐values < 0.05. DESeq2 analysis was carried out in R version 3.5.2. Gene ontology analysis was performed using the Gene Ontology Consortium website. Other analyses were performed using R software. For gene set enrichment analysis (GSEA), normalized data were analyzed with GSEA software, using gene set permutation, Signal2Noise, as a ranking metric and a weighted scoring scheme (all other parameters were set to default).
Single‐cell RNA sequencing (scRNA‐seq)
Cell Ranger 3.1.0 (http://10xgenomics.com) was used to process Chromium Single Cell 3’ v3 RNA‐seq output. First, “cellranger mkfastq” was used to demultiplex the sequencing samples and generate fastq files for Read1 and Read2. Second, “cellranger count” was used to align the reads to the mouse reference genome (refdata‐cellranger‐mm10‐3.0.0) using STAR (Dobin et al, 2013). Then, aligned reads were used to generate a data matrix, but only when they had valid cell barcodes and a unique molecular identifier (UMI) without PCR duplicates. Valid cell barcodes were defined based on the UMI distribution (Zheng et al, 2017). Finally, the output files for the four samples were aggregated into one gene–cell matrix using “cellranger aggr” with default parameters.
The Seurat package (version 3.1.0) (Stuart et al, 2019) was used to process Cell Ranger outputs. Cells with unique feature counts of more than 5,000 or < 200 or with > 5% mitochondrial counts were filtered. SCTransform normalization (Hafemeister & Satija, 2019) was used to remove technical variation. A total of 12,000 highly variable genes were selected and used for principal component analysis (PCA). The JackStraw resampling approach (Macosko et al, 2015) was then used to select significant principal components (PCs). t‐Distributed stochastic neighbor embedding (t‐SNE) and uniform manifold approximation and projection (UMAP) embedding were generated using significant PCs. Cell clustering was performed using the FindNeighbors and FindClusters functions with significant PCs and a resolution of 0.5. Genes differentially expressed between cell clusters were identified using FindAllMarkers with a logfc.threshold = 0.25, test.use = “bimod”, min.pct = 0.1, and min.diff.pct = ‐Inf. For each cell cluster, genes differentially expressed between experimental groups were identified using the same functions and parameters.
Lipid analysis
The tissue samples were homogenized in pre‐chilled methanol/water (1:1) using tissue homogenizing CKMix, which was preloaded with ceramic (zirconium oxide) mix beads. Chloroform was added to the homogenate, which was vortexed for 5 min and then centrifuged at 10,000 g at 4°C for 10 min. The organic layer was collected, vacuum‐dried, and resuspended in 2:1:1(v/v/v) isopropanol/acetonitrile/chloroform and mixed with a lipid internal standard. The lipid analysis was performed using ACQUITY UPLC equipment coupled with a Xevo G2‐XS QTOF/MS detector (Waters Corp. Milford, MA, USA). The chromatographic separation of all samples was performed on an ACQUITY BEH C18 column (Waters Corp.) (dimensions 100 × 2.1 mm, 1.7‐µm particle size). The column was maintained at 55°C. The mobile phase, comprising solvent A (5 mM ammonium acetate in water/acetonitrile/isopropanol (2:1:1 v/v), 20 µM phosphoric acid, and 0.05% acetic acid) and solvent B (5 mM ammonium acetate in isopropanol/acetonitrile (9:1 v/v), 0.05% acetic acid), was used to establish the required gradient. The gradient was optimized and can be described as follows: 0 min, 1% B; 2 min, 40% B; 11.5 min, 95% B; 12.0–12.50 min, 99.9% B; 12.55 min, 65% B; 12.65 min, 30% B; and 12.75–14.25 min, 1% B. Mass spectrometry analysis was conducted in positive and negative ion modes. The quadrupole time‐of‐flight mass spectrometry (QTOF/MS) acquisition parameters were capillary voltage, 2.0 to −1.5 kV; cone voltage, 25 V; source temperature, 120°C; cone gas‐flow rate, 150 l/h; desolvation temperature and flow rate, 600°C and 1,000 l/h, respectively; scan range, 50–2,000 m/z; data acquisition rate, 0.1 s; and collision energy, 6 V. The raw data were acquired using MassLynx 4.1 workstation UPLC‐QTOF/MS Acquisition software (Waters Company, Milford, MA, USA) in centroid mode. To ensure accuracy, the m/z values of all ions acquired in the QTOF/MS were real‐time adjusted using LockSpray. Leucine‐enkephalin was selected as the lock mass compound for the positive ([M + H]+ = 556.2771) and negative ion modes ([M − H]− = 554.2615). The QC sample was injected 20 times prior to formally beginning the experiment, to equilibrate the UPLC‐MS system. Further QC samples were injected at the start, every ten samples, and at the end, to monitor the reproducibility.
Cell mito stress extracellular flux assay
Sorted ATM subpopulations were washed in 2% PBS‐FBS and used according to Seahorse XF Cell Mito Stress kit instructions on a Seahorse XF96 Analyzer (Agilent Technologies, Wilmington, DE, USA) for the extracellular flux assay. Briefly, 105 cells per well were seeded into a Corning® Cell‐Tak™ adhesive coated 96‐well tissue culture plate (Agilent Technologies) and allowed to adhere for 45 min at 37°C in a non‐CO2 incubator prior to the assay. The following inhibitor concentrations were sequentially injected: 1.5 μM oligomycin, 2 μM FCCP, and 0.5 μM rotenone/antimycin A. Basal respiration was calculated from the average oxygen consumption rate (OCR) post‐rotenone/antimycin A treatment subtracted from the average OCR prior to the addition of oligomycin. Maximal respiration was calculated from the average OCR post‐rotenone/antimycin A treatment subtracted from the average OCR post‐FCCP injection. Spare respiratory capacity was calculated from the average basal OCR subtracted from the average OCR post‐FCCP treatment. Representative extracellular flux (XF) phenograms and bioenergetics plots were created or derived from recorded Cell Mito Stress assay OCR values using Wave Software (Agilent Technologies) from replicated experiments.
Giemsa staining
Sorted cells were washed twice in 2% PBS–FBS and centrifuged at 500 g. Cells (104) were immobilized on a slide following cytospin centrifugation, air‐dried, fixed in methanol for 1 min, stained in Giemsa (diluted in H2O, 1:5) for 20 min, and mounted with DPX mounting media (Sigma).
Live imaging of LysoTracker Red‐ and BODIPY‐stained sorted myeloid cells
Sorted ATM subpopulations were washed in 2% PBS‐FBS and stained with 0.5 μM LysoTracker Red (Life Technologies, Carlsbad, CA, USA), 5 μM BODIPY (Thermo Scientific, Waltham, MA, USA), and 1 μM Hoechst 33342 (Sigma) in an imaging medium of phenol red‐free Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Grand Island, NY, USA) supplemented with 10% FBS, l‐glutamate, and HEPES. Cells were incubated for 60 min at 37°C, washed twice with 2% PBS‐FBS, resuspended in pre‐warmed imaging medium, and seeded into 8‐well μ‐slides (Ibidi, Gräfelfing, Germany) at 2 × 105 cells per chamber. Seeded cells were allowed to adhere for 15 min at 37°C. Images were acquired using an inverted confocal microscope with Airyscan (LSM800), and data were processed using ZEISS ZEM microscope software.
Histology
Murine eWAT was removed and fixed in zinc‐formalin (Anatech LTD, MI USA, 170) overnight at 4°C and prepared as previously described (Kosteli et al, 2010).
Hematoxylin and eosin (H&E) staining
Paraffin‐embedded eWAT was cut into 5‐μm‐thick sections, and the sections were incubated at 40°C overnight. Dried slides were deparaffinized and re‐hydrated in xylene, 100% ethanol, 95% ethanol, and H2O. Slides were stained in Gill 3 Hematoxylin (Sigma), followed by H2O, HCl–ethanol, H2O, 1% lithium carbonate (Sigma), H2O, and Eosin Y alcoholic (Sigma). Following a rehydration process, the stained sections were air‐dried and mounted with DPX solution (Sigma).
Picrosirius red staining
Paraffin‐embedded sections were deparaffinized, re‐hydrated, and stained, first with hematoxylin followed by incubation for 1 h with picrosirius red solution (Abcam) in a humid chamber at room temperature. The stained slides were rapidly rinsed in two changes of 0.5% acetic acid solution, air‐dried, dehydrated in graded alcohol solutions, and mounted with DPX mounting solution (Sigma), before being evaluated under light microscopy.
Immunofluorescence staining
eWAT sections, 15‐μm‐thick, were incubated in ice‐cold acetone/methanol (1:1) solution for 10 min and then washed three times in 0.3% PBS–Triton X‐100 buffer for 5 min each time. Sections were blocked with mouse serum for 2 h at room temperature and stained with primary antibody in a humid chamber at 4°C overnight. Then, the slides were washed three times in 0.3% PBS–Triton X‐100 buffer and stained with fluorochrome‐conjugated secondary antibody for a further 2 h at room temperature. The stained sections were washed in 0.3% PBS–Triton X‐100 buffer and mounted with the DAPI‐based mounting medium, Dako (Sigma).
Whole‐mount immunostaining and optical clearing
eWAT samples from lean and obese mice were harvested and fixed in 4% paraformaldehyde overnight at 4°C and then washed with PBS. Samples were permeabilized using a series of methanol gradients, following which they were blocked overnight at 4°C in blocking buffer consisting of 5% dimethyl sulfoxide, 5% donkey serum, 0.3% Triton X‐100, 0.2% bovine serum albumin, and 0.01% sodium azide diluted in PBS. Samples were then incubated with primary antibodies (anti‐CD11c, clone D1V9Y, Cell Signaling Technologies; anti‐CD31, Clone 2H8, Invitrogen; anti‐MHCII, clone NIMR‐4, Abcam; anti‐CD169, clone 3D6.112; polyclonal anti‐elastin and polyclonal anti‐TUJ1, Abcam), diluted in blocking buffer, and incubated for 4 days at 4°C. The samples were then washed three times at room temperature with wash buffer consisting of 0.3% sodium chloride and 0.3% Triton X‐100 diluted in PBS. This process was repeated for the incubation with secondary antibodies (donkey anti‐rat AF488, donkey anti‐rabbit AF488, and donkey anti‐rabbit AF555, Life Technologies; goat anti‐hamster Cy3, Jackson ImmunoResearch, West Grove, PE, USA). Samples were optically cleared using solvent‐based optical clearing by immersion in a series of methanol gradients and incubation in a mixture of benzyl alcohol and benzyl benzoate. For panels involving lipid labeling using BODIPYTM 493/503 (Thermo Fisher Scientific), aqueous‐based optical clearing involving overnight immersion in RapiClear 1.52 (SunJin Labs, Hsinchu City, Taiwan) at room temperature was used.
Image acquisition, deconvolution, and image processing
Images were acquired using an Olympus FV1000 confocal microscope with a LUCPLFN 20× objective (numerical aperture: 0.45). Lasers were used for fluorophore excitation at 405, 488, 559, and 635 nm. Image deconvolution was carried out using Huygens Deconvolution Express software (Scientific Volume Imaging, Hilversum, Netherlands), and images were visualized using Imaris 9.5.0 (Bitplane).
Determination of macrophage subset distance from blood vessels and nerves
CD169+ macrophages were identified in the images using the Spots function in the Imaris software. CD169+ macrophages were categorized into either CD11chi or CD11clow subsets based on their CD11c expression within the image. The same threshold was applied across different images for both lean and adipose tissues processed within the same experiment. The Surface function in Imaris was then used to create surfaces based on CD31+ blood vessels, and the distance from CD31+ surface to CD169+ macrophages calculated using the distance transformation function. Debris within the image was filtered out using the ellipticity filter. Both Surface and Spots creation were carried out prior to performing any statistical analyses to ensure no bias during the analysis. Two images were acquired for each sample and used as technical replicates during image analysis. The number of CD169+ macrophages that were less than 1, between 1 and 10, and more than 10 µm away from the CD31+ surface was then counted, and the median distance of CD169+ macrophages from blood vessels calculated. Graphs were plotted and statistics derived using Prism 8.2.1 (GraphPad Software, La Jolla, CA, USA). The same method was carried out for nerve staining.
Measurement of vasculature leakage
Mice were intravenously (i.v.) injected with 200 µl 1% Evans blue diluted in 0.9% saline solution. Thirty minutes later, eWAT was removed and incubated in formamide at 55°C for 48 h to extract Evans blue from the tissue. The absorbance of the Evans blue mixture was subsequently measured at 620 nm, using formamide as a blank.
ELISA
Mouse Insulin ELISA (Cat # 10‐1247‐01, Mercodia, Uppsala, Sweden), mouse Adiponectin ELISA kit (Cat #: 80569, Crystal Chem, IL, USA), and mouse Leptin ELISA kit (Cat #: 90030, Crystal Chem) were used to determine insulin, adiponectin, and leptin amounts in the serum of WT and CD169‐DTR mice following the manufacturer’s instructions.
Glucose tolerance test (GTT)
GTT was performed to monitor the development of diabetes in obese WT and CD169‐DTR which ATMs were depleted for 7 days. After 6 h of fasting, the basal blood glucose levels were measured in HFD‐treated mice. A challenge of 1.5 g/kg glucose was given intraperitoneally, and the blood glucose level was checked at 15, 30, 60, 90, and 120 min post‐glucose injection.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 9 software (GraphPad Software). All values are expressed as the mean ± standard deviation (SD), as indicated in the legends. Statistical significance was determined using Student’s t‐test (two‐tailed), Welch’s t‐test, or Bonferroni two‐way ANOVA. A P‐value < 0.05 was considered to be statistically significant. *< 0.05, **< 0.01, ***< 0.001, and ****< 0.0001.
Author contributions
Conceptualization: CR; Methodology: QC, SML, YT, KL, DL, RRN, AMB, SSA, SZC and LGN Formal analysis: YT, KL, DL and QC; Bioinformatics analysis: JCT and JC; scRNA‐seq analysis: QC and SX; Writing: CR; Visualization: QC and CR; Supervision: CR; Funding acquisition: CR.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Review Process File
Acknowledgements
The authors would like to thank Monika Tetlak for mouse management, Dr. Shanshan Wu Howland and her team for scRNA‐seq, and Insight Editing London for proofreading the manuscript prior to submission. This work was supported by a Ministry of Education Tier2 grant (MOE2018‐T2‐2‐016) awarded to C.R.
EMBO reports (2021) 22: e52835.
Data availability
The accession numbers for the RNA‐seq data reported in this paper are GEO: GSE148386 and scRNA‐seq: GSE157572. The original flow cytometry data have been deposited in the NTU Open Access Data Repository (DR‐NTU) https://doi.org/10.21979/N9/9J9BWD.
References
- Allen TL, Febbraio MA (2010) IL6 as a mediator of insulin resistance: fat or fiction? Diabetologia 53: 399–402 [DOI] [PubMed] [Google Scholar]
- Borst SE (2004) The role of TNF‐alpha in insulin resistance. Endocrine 23: 177–182 [DOI] [PubMed] [Google Scholar]
- Boutens L, Hooiveld GJ, Dhingra S, Cramer RA, Netea MG, Stienstra R (2018) Unique metabolic activation of adipose tissue macrophages in obesity promotes inflammatory responses. Diabetologia 61: 942–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutens L, Stienstra R (2016) Adipose tissue macrophages: going off track during obesity. Diabetologia 59: 879–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buechler C, Krautbauer S, Eisinger K (2015) Adipose tissue fibrosis. World J Diabetes 6: 548–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burl RB, Ramseyer VD, Rondini EA, Pique‐Regi R, Lee YH, Granneman JG (2018) Deconstructing adipogenesis induced by beta3‐adrenergic receptor activation with single‐cell expression profiling. Cell Metab 28: 300–309 e304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H (2014) Adipocytokines in obesity and metabolic disease. J Endocrinol 220: T47–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y (2013) Angiogenesis and vascular functions in modulation of obesity, adipose metabolism, and insulin sensitivity. Cell Metab 18: 478–489 [DOI] [PubMed] [Google Scholar]
- Chakarov S, Lim HY, Tan L, Lim SY, See P, Lum J, Zhang X‐M, Foo S, Nakamizo S, Duan K et al (2019) Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 363: eaav0964 [DOI] [PubMed] [Google Scholar]
- Chen Q, Ruedl C (2020) Obesity retunes turnover kinetics of tissue‐resident macrophages in fat. J Leukoc Biol 107: 773–782 [DOI] [PubMed] [Google Scholar]
- Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, Wang S, Fortier M, Greenberg AS, Obin MS (2005) Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res 46: 2347–2355 [DOI] [PubMed] [Google Scholar]
- Coats BR, Schoenfelt KQ, Barbosa‐Lorenzi VC, Peris E, Cui C, Hoffman A, Zhou G, Fernandez S, Zhai L, Hall BA et al (2017) Metabolically activated adipose tissue macrophages perform detrimental and beneficial functions during diet‐induced obesity. Cell Rep 20: 3149–3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox N, Crozet L, Holtman IR, Loyer PL, Lazarov T, Mass E, Stanley ER, Glass CK, Geissmann F (2020) Diet‐regulated production of PDGFcc by macrophages controls energy storage. bioRxiv 10.1101/2020.06.15.152397 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danaei G, Finucane MM, Lu Y, Singh GM, Cowan MJ, Paciorek CJ, Lin JK, Farzadfar F, Khang Y‐H, Stevens GA et al (2011) National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country‐years and 2.7 million participants. Lancet 378: 31–40 [DOI] [PubMed] [Google Scholar]
- Datta R, Podolsky MJ, Atabai K (2018) Fat fibrosis: friend or foe? JCI Insight 3: e122289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Schepper S, Verheijden S, Aguilera‐Lizarraga J, Viola MF, Boesmans W, Stakenborg N, Voytyuk I, Schmidt I, Boeckx B, Dierckx de Casterlé I et al (2018) Self‐maintaining gut macrophages are essential for intestinal homeostasis. Cell 175: 400–415.e13 [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, Li MO (2014) The cellular and molecular origin of tumor‐associated macrophages. Science 344: 921–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S, Taylor PR (2005) Monocyte and macrophage heterogeneity. Nat Rev Immunol 5: 953–964 [DOI] [PubMed] [Google Scholar]
- Gupta P, Lai SM, Sheng J, Tetlak P, Balachander A, Claser C, Renia L, Karjalainen K, Ruedl C (2016) Tissue‐resident CD169(+) macrophages form a crucial front line against plasmodium infection. Cell Rep 16: 1749–1761 [DOI] [PubMed] [Google Scholar]
- Hafemeister C, Satija R (2019) Normalization and variance stabilization of single‐cell RNA‐seq data using regularized negative binomial regression. Genome Biol 20: 296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Wang R, Zhou Y, Fei L, Sun H, Lai S, Saadatpour A, Zhou Z, Chen H, Ye F et al (2018) Mapping the mouse cell atlas by microwell‐seq. Cell 172: 1091–1107 [DOI] [PubMed] [Google Scholar]
- Hepler C, Shan B, Zhang Q, Henry GH, Shao M, Vishvanath L, Ghaben AL, Mobley AB, Strand D, Hon GC et al (2018) Identification of functionally distinct fibro‐inflammatory and adipogenic stromal subpopulations in visceral adipose tissue of adult mice. Elife 7: e39636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DA, Lim H‐W, Kim YH, Ho WY, Foong YH, Nelson VL, Nguyen HCB, Chegireddy K, Kim J, Habertheuer A et al (2018) Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc Natl Acad Sci USA 115: E5096–E5105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS (2006) Inflammation and metabolic disorders. Nature 444: 860–867 [DOI] [PubMed] [Google Scholar]
- Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, Lundgren P, Bleriot C, Liu Z, Deczkowska A et al (2019) Lipid‐associated macrophages control metabolic homeostasis in a trem2‐dependent manner. Cell 178: 686–698.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalucka J, de Rooij LPMH, Goveia J, Rohlenova K, Dumas SJ, Meta E, Conchinha NV, Taverna F, Teuwen L‐A, Veys K et al (2020) Single‐cell transcriptome atlas of murine endothelial cells. Cell 180: 764–779.e20 [DOI] [PubMed] [Google Scholar]
- Kastl AJ Jr, Terry NA, Wu GD, Albenberg LG (2020) The structure and function of the human small intestinal microbiota: current understanding and future directions. Cell Mol Gastroenterol Hepatol 9: 33–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosteli A, Sugaru E, Haemmerle G, Martin JF, Lei J, Zechner R, Ferrante AW Jr (2010) Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. J Clin Invest 120: 3466–3479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratz M, Coats B, Hisert K, Hagman D, Mutskov V, Peris E, Schoenfelt K, Kuzma J, Larson I, Billing P et al (2014) Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab 20: 614–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapenna A, De Palma M, Lewis CE (2018) Perivascular macrophages in health and disease. Nat Rev Immunol 18: 689–702 [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W (2013) The Subread aligner: fast, accurate and scalable read mapping by seed‐and‐vote. Nucleic Acids Res 41: e108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930 [DOI] [PubMed] [Google Scholar]
- Lim HY, Lim SY, Tan CK, Thiam CH, Goh CC, Carbajo D, Chew SHS, See P, Chakarov S, Wang XN et al (2018) Hyaluronan receptor LYVE‐1‐expressing macrophages maintain arterial tone through hyaluronan‐mediated regulation of smooth muscle cell collagen. Immunity 49: 326–341 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, Deyoung SM, Bodzin JL, Saltiel AR (2007) Increased inflammatory properties of adipose tissue macrophages recruited during diet‐induced obesity. Diabetes 56: 16–23 [DOI] [PubMed] [Google Scholar]
- MacDonald KPA, Palmer JS, Cronau S, Seppanen E, Olver S, Raffelt NC, Kuns R, Pettit AR, Clouston A, Wainwright B et al (2010) An antibody against the colony‐stimulating factor 1 receptor depletes the resident subset of monocytes and tissue‐ and tumor‐associated macrophages but does not inhibit inflammation. Blood 116: 3955–3963 [DOI] [PubMed] [Google Scholar]
- Macosko E, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas A, Kamitaki N, Martersteck E et al (2015) Highly parallel genome‐wide expression profiling of individual cells using nanoliter droplets. Cell 161: 1202–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick D, Sakers A, Irgebay Z, Okada C, Calvert C, Morley MP, Percec I, Seale P (2019) Identification of a mesenchymal progenitor cell hierarchy in adipose tissue. Science 364: eaav2501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MT, Favelyukis S, Nguyen AK, Reichart D, Scott PA, Jenn A, Liu‐Bryan R, Glass CK, Neels JG, Olefsky JM (2007) A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll‐like receptors 2 and 4 and JNK‐dependent pathways. J Biol Chem 282: 35279–35292 [DOI] [PubMed] [Google Scholar]
- Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S et al (2009) CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 15: 914–920 [DOI] [PubMed] [Google Scholar]
- Odegaard JI, Ganeshan K, Chawla A (2013) Adipose tissue macrophages: amicus adipem? Cell Metab 18: 767–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG (2008) Ablation of CD11c‐positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab 8: 301–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purnama C, Ng SL, Tetlak P, Setiagani YA, Kandasamy M, Baalasubramanian S, Karjalainen K, Ruedl C (2014) Transient ablation of alveolar macrophages leads to massive pathology of influenza infection without affecting cellular adaptive immunity. Eur J Immunol 44: 2003–2012 [DOI] [PubMed] [Google Scholar]
- Rosen ED, Spiegelman BM (2014) What we talk about when we talk about fat. Cell 156: 20–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaul ME, Bennett G, Strissel KJ, Greenberg AS, Obin MS (2010) Dynamic, M2‐like remodeling phenotypes of CD11c+ adipose tissue macrophages during high‐fat diet–induced obesity in mice. Diabetes 59: 1171–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw TN, Houston SA, Wemyss K, Bridgeman HM, Barbera TA, Zangerle‐Murray T, Strangward P, Ridley AJL, Wang P, Tamoutounour S et al (2018) Tissue‐resident macrophages in the intestine are long lived and defined by Tim‐4 and CD4 expression. J Exp Med 215: 1507–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng J, Ruedl C, Karjalainen K (2015) Most tissue‐resident macrophages except microglia are derived from fetal hematopoietic stem cells. Immunity 43: 382–393 [DOI] [PubMed] [Google Scholar]
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS (2006) TLR4 links innate immunity and fatty acid‐induced insulin resistance. J Clin Invest 116: 3015–3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva HM, Báfica A, Rodrigues‐Luiz GF, Chi J, Santos PDA, Reis BS, Hoytema van Konijnenburg DP, Crane A, Arifa RDN, Martin P et al (2019) Vasculature‐associated fat macrophages readily adapt to inflammatory and metabolic challenges. J Exp Med 216: 786–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soncin I, Sheng J, Chen Q, Foo S, Duan K, Lum J, Poidinger M, Zolezzi F, Karjalainen K, Ruedl C (2018) The tumour microenvironment creates a niche for the self‐renewal of tumour‐promoting macrophages in colon adenoma. Nat Commun 9: 582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens GA, Singh GM, Lu Y, Danaei G, Lin JK, Finucane MM, Bahalim AN, McIntire RK, Gutierrez HR, Cowan M et al (2012) National, regional, and global trends in adult overweight and obesity prevalences. Popul Health Metr 10: 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, Satija R (2019) Comprehensive integration of single‐cell data. Cell 177: 1888–1902.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Kusminski CM, Scherer PE (2011) Adipose tissue remodeling and obesity. J Clin Invest 121: 2094–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Tordjman J, Clement K, Scherer PE (2013) Fibrosis and adipose tissue dysfunction. Cell Metab 18: 470–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung H‐K, Doh K‐O, Son J, Park J, Bae Y, Choi S, Nelson S, Cowling R, Nagy K, Michael I et al (2013) Adipose vascular endothelial growth factor regulates metabolic homeostasis through angiogenesis. Cell Metab 17: 61–72 [DOI] [PubMed] [Google Scholar]
- Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, Lu M, Li P, Yan Q, Zhu Y et al (2012) Neutrophils mediate insulin resistance in mice fed a high‐fat diet through secreted elastase. Nat Med 18: 1407–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trim W, Turner JE, Thompson D (2018) Parallels in immunometabolic adipose tissue dysfunction with ageing and obesity. Front Immunol 9: 169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viola MF, Boeckxstaens G (2020) Niche‐specific functional heterogeneity of intestinal resident macrophages. Gut 70: 1383–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr (2003) Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112: 1796–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, Tsui H, Wu P, Davidson MG, Alonso MN et al (2011) B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med 17: 610–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wueest S, Konrad D (2018) The role of adipocyte‐specific IL‐6‐type cytokine signaling in FFA and leptin release. Adipocyte 7: 226–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA et al (2003) Chronic inflammation in fat plays a crucial role in the development of obesity‐related insulin resistance. J Clin Invest 112: 1821–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Grijalva A, Skowronski A, van Eijk M , Serlie MJ, Ferrante AW Jr (2013) Obesity activates a program of lysosomal‐dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab 18: 816–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J et al (2017) Massively parallel digital transcriptional profiling of single cells. Nat Commun 8: 14049 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Review Process File
Data Availability Statement
The accession numbers for the RNA‐seq data reported in this paper are GEO: GSE148386 and scRNA‐seq: GSE157572. The original flow cytometry data have been deposited in the NTU Open Access Data Repository (DR‐NTU) https://doi.org/10.21979/N9/9J9BWD.