

Abstract
Predicting the tertiary structure of a protein from its primary sequence has been greatly improved by integrating deep learning and co-evolutionary analysis, as shown in CASP13 and CASP14. We describe our latest study of this idea, analyzing the efficacy of network size and co-evolution data and its performance on both natural and designed proteins. We show that a large ResNet (convolutional residual neural networks) can predict structures of correct folds for 26 out of 32 CASP13 free-modeling (FM) targets and L/5 long-range contacts with precision over 80%. When co-evolution is not used ResNet still can predict structures of correct folds for 18 CASP13 FM targets, greatly exceeding previous methods that do not use co-evolution either. Even with only primary sequence ResNet can predict structures of correct folds for all tested human-designed proteins. In addition, ResNet may fare better for the designed proteins when trained without co-evolution than with co-evolution. These results suggest that ResNet does not simply denoise co-evolution signals, but instead may learn important protein sequence-structure relationship. This has important implications on protein design and engineering especially when co-evolutionary data is unavailable.
Introduction
Elucidation of protein structure is a fundamental step toward understanding biological life and highly relevant in the development of therapeutics. In addition to low-throughput and costly experimental structure determination, computational methods are used to predict protein structure from its primary sequence. In the last years, computational structure prediction for proteins without detectable homology in PDB has been greatly advanced by the application of co-evolution data[1] and deep learning including convolutional residual networks (ResNet) implemented in RaptorX [2, 3] and lately Transformer-like network implemented in the amazing AlphaFold2. Co-evolution means two residues of the same protein mutate simultaneously. Since the proposal of ResNet for protein contact/distance and tertiary structure prediction[4–9], many groups have studied this method and contributed further improvements. At CASP13 (the thirteen Critical Assessment of Structure Prediction), AlphaFold1 demonstrated better accuracy by using a deeper ResNet[10] and performing gradient-based energy minimization on predicted distance potential. The Gong group applied ResNet to predict real-valued distance [11]. Several groups have also capitalized on ResNet’s inherent ability to predict arbitrary inter-residue relationships. Yang et al showed that ResNet may predict inter-residue orientation[12] useful for 3D model building. The Jones group showed that ResNet can accurately predict hydrogen-bonds [13].
Apart from the study of model capabilities and architecture, new ideas on input features and model training have been suggested. For example, metagenome data was used to enhance MSAs (multiple sequence alignment) for those proteins with few sequence homologs[14]. A full precision matrix generated by co-evolution analysis may provide more information than a concise co-evolution matrix[15]. In addition, MSA subsampling[10, 12], loop modification[16] and perturbation of distance matrices[10, 11] were attempted to improve ResNet training. Although these ideas have yielded promising results, RaptorX performed well in CASP13 without them and with a relatively shallow ResNet. In light of this, it will be interesting to see how much further the ResNet method can be improved by integrating these ideas, and to analyze the contribution of individual factors to further our understanding of deep learning-based structure prediction.
The success of deep learning for structure prediction is often attributed to its utilization of co-evolution information -- especially that produced by direct-coupling methods such as Evfold[17], GREMLIN[18] and CCMpred[19]. It is even suggested that the core functionality of deep learning is in enhancing the co-evolutionary signal derived from MSAs[20]. It is true that co-evolution data plays an important role, but empirically, pure co-evolution analysis alone usually fails on proteins without a very deep MSA, e.g., CASP FM targets. This presents a challenge in protein design and engineering, where it is often the case that co-evolution (and evolutionary) information is unavailable for engineered proteins. On the other hand, in nature proteins fold without knowledge of sequence homologs and thus, a method that can predict protein structure in the absence of co-evolution information should exist in principle.
These considerations motivate us to study the role of co-evolution analysis with regard to deep learning in protein structure prediction. Specifically, we study how well ResNet may perform on both human-designed and natural proteins and its dependency on the form and abundance of co-evolution signal. We evaluate its performance by long-range contact prediction and the quality of predicted structure models. We find out even when co-evolution data is omitted, ResNet may still predict structures of correct folds for 18 CASP13 FM targets, greatly exceeding a popular deep learning method RGN[21], which fails on all CASP13 FM targets. The contrast is even more pronounced for the human-designed proteins where ResNet without co-evolution may outperform its co-evolution aware counterpart (when co-evolution data is available). In fact, ResNet performs well on the human-designed proteins even with only primary sequence. Viewed holistically, our results imply that ResNet does not simply denoise co-evolution signals, but instead captures fundamental aspects of protein structure essential to deciding their folded state.
Results
Overview of the method
Our method consists of two key components. The first is a deep ResNet used to predict discrete probability distributions over distance for three backbone atom pairs (Cb-Cb, Ca-Ca and N-O) and inter-residue orientation defined in trRosetta[12]. Important for our study, our method is implemented so that it is easy to turn on and off specific input features and pairwise relationships to be predicted. The second component is an enhanced gradient-based energy minimization method that builds protein models from potentials derived from predicted distances, orientations, and backbone torsion angles. The PyRosetta[22] fast relaxation protocol is used for side-chain packing and reducing steric clashes.
The overall network architecture is similar to what we used in CASP13[7] except that the ResNet used here is larger and wider (Supplementary Fig. S1), with 100 2D convolutional layers and on average 150 filters per layer. In comparison, our ResNet in CASP13 had only 60 2D convolution layers, averaging 80 filters per layer. We use a multi-task learning strategy, implemented during CASP13 but fully tested after that, to simultaneously predict all distance and orientation matrices. Multi-task learning does not yield a clear performance gain, but it greatly reduces the time for model training and the number of deep models.
Ablation study of contact prediction accuracy on the CASP13 targets
We trained a variety of ResNet models on the 2020 Cath S35 data using the same procedure over different input features and network sizes. Under each setting, 3 ResNet models are trained and used as an ensemble for test. The results are summarized in Table 1 and see Supplementary Dataset 1 for detailed results. In Table 1, “Large” denotes a ResNet with 100 2D convolutional layers, each utilizing 150 filters on average. “Small” denotes a ResNet with 60 2D convolutional layers, each having roughly 80 filters. “All” means that all input features are used. “No coevolution” means that neither the CCMpred[19] output nor mutual information (MI) is used. “No CCMpred” means that MI is used, but not the CCMpred output. “No full CCMpred” means only the L×L co-evolution matrix is used, but not the L×L×21×21 full co-evolution matrix. “L60F150” represents a ResNet model of 60 2D convolutional layers and 150 filters per layer. “L100F80” represents a ResNet model of 100 2D convolutional layers and 80 filters per layer. The ResNet we used in CASP13 roughly corresponds to model 6 in Table 1.
Table 1.
Precision and F1 (%) of long-range contact prediction on the CASP13 targets by ResNet in different settings.
| Model Number | Network Size | Input Features | 31 CASP13 FM targets | 12 CASP13 FM/TBM targets | ||||
|---|---|---|---|---|---|---|---|---|
| Top L/5 | Top L/2 | Top L | Top L/5 | Top L/2 | Top L | |||
| F1 of long-range contact prediction | ||||||||
| 1 | Large | All | 27.8 | 44.3 | 51.8 | 30.1 | 51.3 | 60.9 |
| 2 | Large | No coevolution | 19.3 | 30.6 | 34.7 | 24.7 | 39.1 | 47.0 |
| 3 | Large | No CCMpred | 20.4 | 31.4 | 36.1 | 24.5 | 41.2 | 49.2 |
| 4 | Large | No metagenome | 25.3 | 40.5 | 47.9 | 30.7 | 52.3 | 61.7 |
| 5 | Small | All | 25.2 | 39.6 | 45.4 | 30.2 | 48.9 | 56.9 |
| 6 | Small | No full CCMpred | 22.6 | 35.9 | 41.4 | 30.4 | 47.2 | 56.1 |
| 7 | L60F150 | All | 26.5 | 41.2 | 47.6 | 29.7 | 48.7 | 58.5 |
| 8 | L100F80 | All | 27.8 | 42.1 | 48.8 | 32.1 | 52.0 | 60.2 |
| Precision of long-range contact prediction | ||||||||
| 1 | Large | All | 81.0 | 68.2 | 58.0 | 90.1 | 81.4 | 69.5 |
| 2 | Large | No coevolution | 58.2 | 47.8 | 39.1 | 76.2 | 65.0 | 54.7 |
| 3 | Large | No CCMpred | 60.8 | 49.1 | 40.6 | 76.9 | 67.9 | 56.9 |
| 4 | Large | No metagenome | 75.6 | 63.3 | 53.7 | 90.8 | 82.4 | 70.4 |
| 5 | Small | All | 74.0 | 61.4 | 51.2 | 89.8 | 78.1 | 65.1 |
| 6 | Small | No full CCMpred | 68.8 | 56.6 | 47.0 | 89.5 | 75.5 | 64.4 |
| 7 | L60F150 | All | 78.3 | 64.0 | 53.5 | 88.3 | 77.9 | 66.9 |
| 8 | L100F80 | All | 80.6 | 65.1 | 54.8 | 94.3 | 81.8 | 68.6 |
We estimate the contribution of a single factor by comparing two ResNet models in Fig. 1(a) and Table 1. Comparing models 1 and 2 suggests that without co-evolution , the F1 value on average decreases by 13.1% when CASP13 FM targets are evaluated. The minor difference between models 2 and 3 implies that MI (one type of co-evolution information) does not help much and thus, the CCMpred co-evolution matrix is very important. Comparing models 1 and 5 suggests that a large ResNet may improve F1 and precision over a small ResNet by ~4.6% and ~6.9%, respectively, on the CASP13 FM targets. The performance difference on the FM targets among models 1, 5, 7 and 8 suggests that the size of ResNet matters. Roughly the larger the network size, the better precision and F1 can be obtained. Models 7 and 8 have about 20 and 10 million parameters, respectively, but model 8 slightly outperforms model 7. This may suggest that the depth of ResNet is more important than the width (i.e., the number of filters). However, with the same depth, a narrower network may not underperform a wider network on the FM/TBM targets, e.g., model 8 vs. model 1 and model 5 vs. model 7. Comparing models 5 and 6 suggests that a full CCMpred matrix may improve F1 and precision by ~3.4% and ~4.7% on average, respectively. Comparing models 1 and 4 suggests that the small metagenome database improves F1 and precision by ~3.4% and ~5.2% on the FM targets, but slightly decreases the performance on the FM/TBM targets. In summary, the factor contributing the most to the improvement over our own CASP13 work is the network size, followed by the full CCMpred precision matrix and then, the metagenome data. We remark that these are only estimates, which may vary with respect to network size or introduction of extra features and data.
Figure 1.
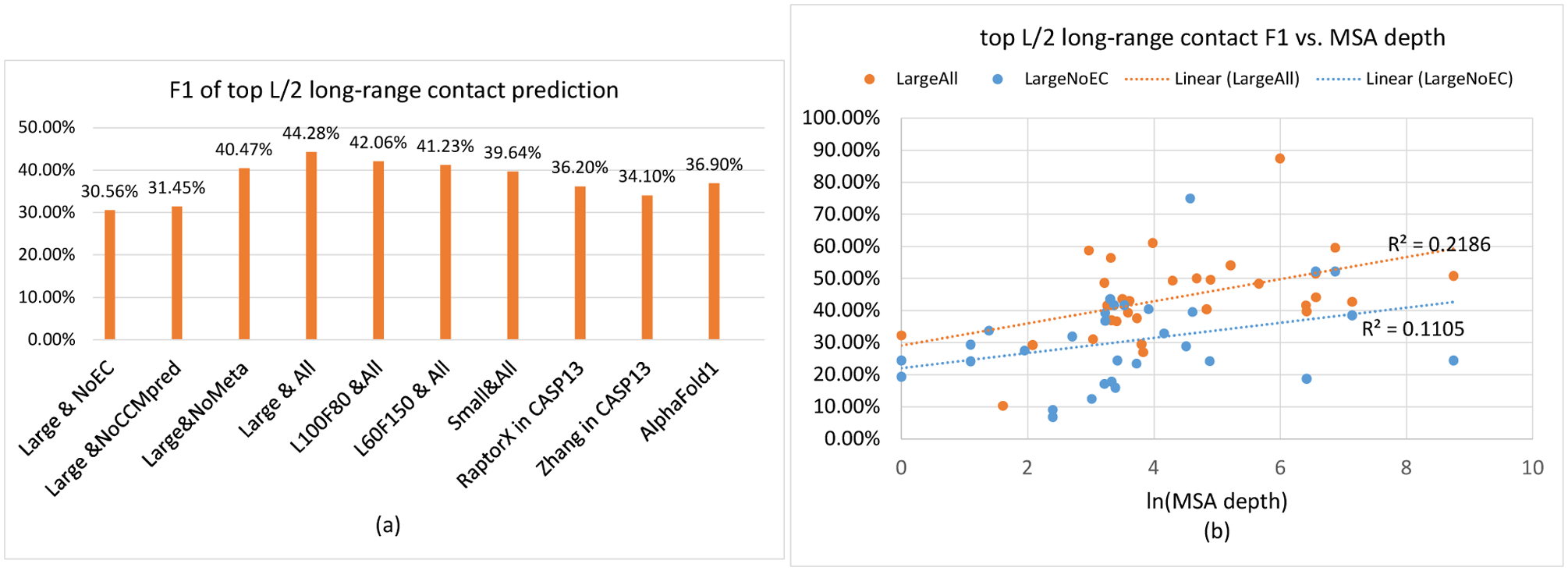
Contact prediction accuracy by various ResNet models on the 31 CASP13 FM targets. (a) F1 value of different ResNet models. As a control, the best CASP13 results are also displayed. (b) F1 value of ResNet trained with and without coevolution data vs. MSA depth. See the text for explanation of the ResNet models.
When co-evolution is not used, our top L/5, L/2 and L long-range contact precision on the 31 FM targets is 58.2%, 47.8% and 39.1%, respectively. In CASP11, where traditional neural networks utilizing direct co-evolution were employed, such as MetaPSICOV[23] and deep belief networks (e.g., DNCON[24]), the best top L/5 precision is ~27%. In CASP12, where an under-developed co-evolution aware ResNet model RaptorX-Contact[5] was tested, the best top L/5 precision is ~47%. Even in CASP13 when ResNet with co-evolution was implemented by many groups, a method with top L/5 precision of 58.2% would still be ranked among top 10. This strongly suggests that ResNet itself plays an important role in improving contact prediction and that low prediction precision prior to CASP12 cannot be simply attributed to the fact that co-evolution was not widely used.
As shown in Fig. 1(b), when co-evolution is used, there is a weak correlation between top L/2 long-range F1 and the logarithm of MSA depth (correlation coefficient=0.4675, trendline R2=0.2186). When co-evolution is not used, the correlation is weaker (correlation coefficient=0.3324, trendline R2=0.1105).
When the full information is used, our top L/2 and L contact accuracy exceeds the best CASP13 results by a good margin (Fig. 1(a), Supplementary Table S1). Although using only a very small metagenome dataset metaclust[25], our accuracy compares favorably to trRosetta[12], a ResNet method developed after CASP13 that used several large metagenome datasets.
Accuracy of predicted 3D models on the CASP13 FM targets when co-evolution is used
We test our large ResNet by generating 150 decoys per target and clustering them. Here we summarize our results and see Supplementary Dataset 2 for details. When 4 ResNet models are used as an ensemble, the average quality (TMscore) of the first and best predicted models is 0.638 and 0.659, respectively. This can be further improved. Generating 600 decoys per target may improve the first and best model quality to 0.640 and 0.675, respectively. Increasing the 2D ResNet size to 120 convolutional layers (and 170 filters per layer) may improve the first and best model quality to 0.646 and 0.673, respectively, with even only 20 decoys generated per target and the 5 lowest-energy decoys evaluated.
As shown in Fig. 2(a), our predicted structures for 24 of 32 CASP13 FM targets have TMscore higher than their respective target-training structure similarity (i.e., the highest structure similarity between one target and all training/validation proteins). For 18 targets, their predicted structures have quality at least 0.1 TMscore higher than their respective target-training structure similarity. There is almost no correlation between our model quality and the target-training structure similarity (correlation coefficient=-0.149 and trendline R2=0.0223). When the best models are considered, our method predicts structures of correct folds for 26 of the 32 FM targets. These results demonstrate that ResNets can generate novel structures, yielding model quality remarkably higher than what can be achieved by simply memorizing the training set. As shown in Fig. 2(b), there is a modest correlation between model quality and MSA depth (correlation coefficient=0.5997, trendline R2=0.3597) and our method predicts correct folds for all but two test targets with ln(MSA depth)>3.
Figure 2.
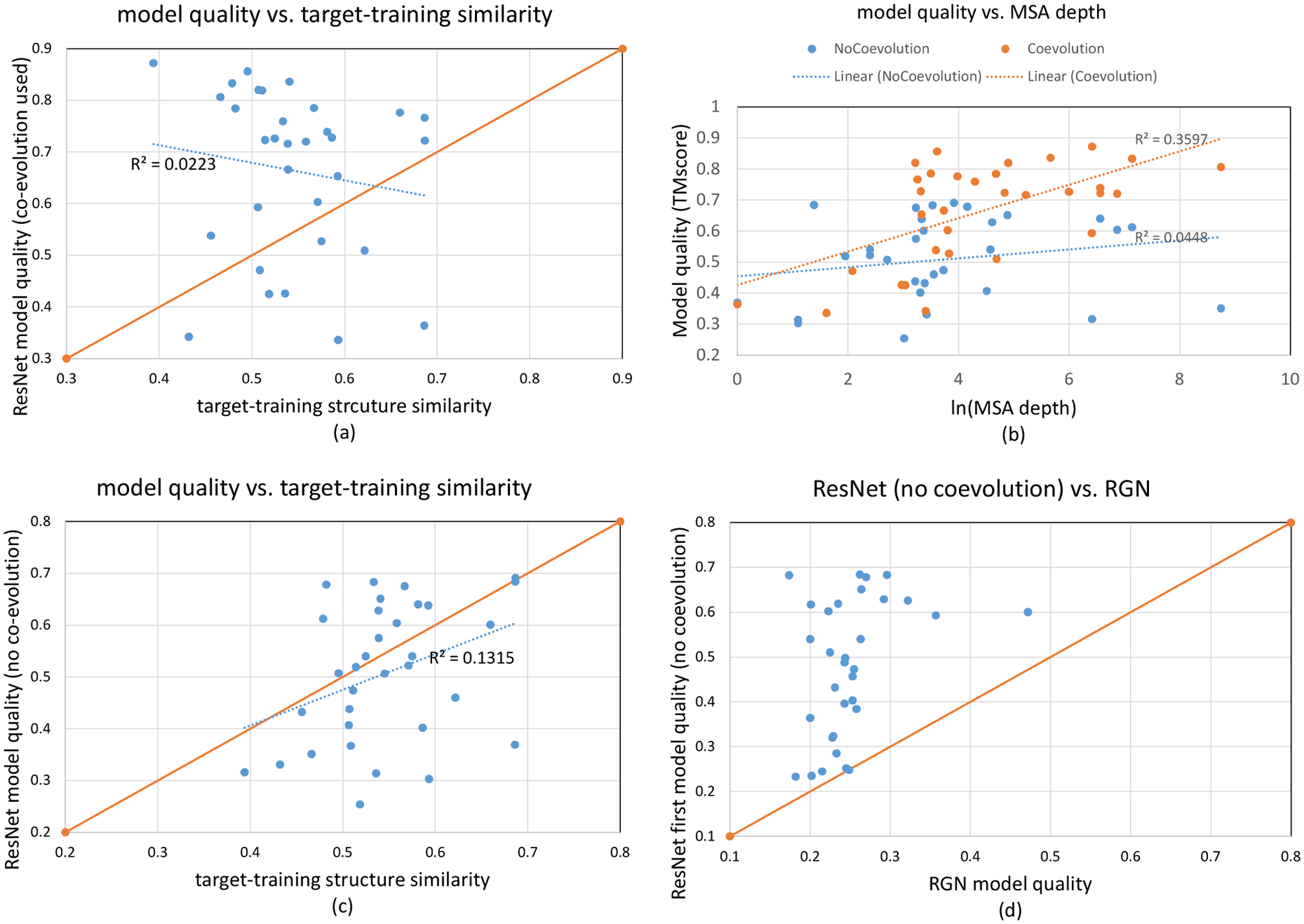
3D modeling accuracy (TMscore) on the 32 CASP13 FM targets. (a) 3D model accuracy vs. target-training structure similarity when co-evolution is used. A dot above the diagonal line indicates that the quality of our predicted 3D model is larger than the highest target-training structure similarity. (b) 3D model accuracy vs. MSA depth (i.e., Meff). When co-evoluton is used, the metagenome data is considered in calculating MSA depth. When co-evolution is not used, the metagenome data is not considered. (c) 3D model accuracy vs. target-training structure similarity when co-evolution is not used. (d) comparison between our 3D modeling accuracy when co-evolution is not used and RGN. A dot above the diagonal line indicates that our model accuracy is better than its corresponding RGN model accuracy.
Our 3D modeling accuracy compares favorably to previously reported results. For example, in CASP13 AlphaFold1 predicted structures of correct folds for 23 of 32 CASP13 FM targets with average TMscore of the first and best models being 0.583 and 0.625, respectively. The lowest-energy models produced by trRosetta on the 32 CASP13 FM targets have average TMscore 0.618. Note that trRosetta reported an average quality (0.625) of 31 (but not 32) CASP13 FM targets [12] and it used several large metagenome datasets to enhance MSAs, but we do not.
When predicted orientation is not used, the average quality of our first and best models is 0.628 and 0.648, respectively. That is, predicted orientation may improve the best model quality by ~0.01. In contrast, trRosetta reported a larger improvement (~0.03) from orientation [12]. It is not very clear why our orientation is not as useful as trRosetta’s.
Ablation study of 3D modeling accuracy
Similar to Table 1 for contact prediction, Table 2 summarizes the impact of input features and network size on 3D modeling accuracy. Table 2 lists the same ResNet models as Table 1. See Supplementary Dataset 3 for details. Comparing models 1, 5, 7 and 8 indicates that 3D modeling accuracy increases with respect to the network size. L100F80 and L60F150 have similar modeling accuracy although the former has only ~10 million parameters while the latter has ~20 million parameters, which may suggest that the network depth is more important than the network width. Comparing models 1 and 2 show that co-evolution information is very important. Comparing models 2 and 3 indicate that mutual information may improve modeling accuracy by 0.026. Comparing models 1 and 4 show that the metagenome data helps a little bit. Comparing models 5 and 6 show that the full co-evolution matrix produced by CCMpred improves modeling accuracy by 0.02–0.03.
Table 2.
The average quality (TMscore) of the first and best models generated by different ResNet models on the 32 CASP13 FM targets. To save computing time, PyRosetta relaxation is not applied in this test. In total 150 decoys per target were generated by a ResNet model and then clustered.
| Model Number | Network Size | Input Features | First model | Best model |
|---|---|---|---|---|
| 1 | Large | All | 0.627 | 0.658 |
| 2 | Large | No coevolution | 0.476 | 0.503 |
| 3 | Large | No CCMpred | 0.502 | 0.529 |
| 4 | Large | No metagenome | 0.606 | 0.632 |
| 5 | Small | All | 0.586 | 0.616 |
| 6 | Small | No full CCMpred matrix | 0.563 | 0.586 |
| 7 | L60F150 | All | 0.603 | 0.637 |
| 8 | L100F80 | All | 0.598 | 0.631 |
Accuracy of predicted 3D models when co-evolution information is not used
Here we summarize and analyze how well ResNet without co-evolution can predict protein structure for the 32 CASP13 FM targets and 21 human-designed proteins. See Supplementary Dataset 4 for details. Unless explicitly stated otherwise, we still use sequence profile (i.e., position-specific scoring matrix), which encodes evolutionary information on an individual residue basis.
Modeling the CASP13 FM targets without co-evolution.
The average quality (TMscore) of the first and best 3D models generated by our ResNet is 0.478 and 0.506, respectively. When the best predicted models are considered, our ResNet predicts correct folds for 18 out of 32 CASP13 FM targets (Fig. 2(b–c)). Fig. 2(c) shows a weak correlation between modeling accuracy and target-training structure similarity (correlation coefficient=0.363 and trendline R2=0.1315). Fig. 2(b) shows a weaker correlation between modeling accuracy and MSA depth (correlation coefficient=0.211 and trendline R2=0.0448) than when co-evolution is used, which is not unexpected.
Fig. 2(c) shows the predicted structures of 13 FM targets have quality higher than their respective target-training structure similarity. In particular, the best 3D models generated for T0950-D1 (342AAs), T0968s2-D1 (115AAs), T0969-D1 (354AAs), T0986s2-D1 (115AAs), T0987-D1 (185AAs) and T1017s2-D1 (125AAs) have TMscore 0.628, 0.651, 0.612, 0.683, 0.675, and 0.678, respectively, while their target-training structural similarity is only 0.539, 0.540, 0.478, 0.533, 0.567, and 0.482, respectively. Among the 3 FM targets with >300 residues (T0950-D1, T0969-D1 and T1000-D2), the predicted structures for T0950-D1 and T0969-D1 have correct folds and their TMscore is much higher than their respective target-training structure similarity. To understand why ResNet works well for T0969-D1 (without a similar fold in our training set) when co-evolution is not used, we visualize its predicted Cβ-Cβ distance matrices in Extended Data Fig. 1. Though less precise than when co-evolution is used, the predicted distance matrix still has a good percentage of correct long-range distances, even for pairs of residues separated by >200 residues. In summary, ResNet without co-evolution can still predict the structures of correct folds for many natural proteins.
To put our work into perspective, we compare it with a top server Robetta[26] and RGN[21]. Robetta is the best server in CASP13 that did not use deep learning. It used a combination of template-based modeling (which uses sequence profile), ab initio folding and co-evolution-based contact prediction. RGN is an end-to-end deep learning method that predicts protein structure from sequence profile but not co-evolution. We use the RGN deep model pre-trained by its authors to predict 3D models for the CASP13 targets, with sequence profile being derived from the uniref90 database dated in November 2018.
The average TMscore of the first and best models submitted by Robetta on the 32 CASP13 FM targets is 0.390 and 0.430, respectively. The average TMscore of the models by RGN is 0.251. Both Robetta and RGN underperform our method by a good margin. Furthermore, our ResNet trained without co-evolution predicts 3D models with better quality than RGN for almost all 32 FM targets (Fig. 2(d)). In fact, RGN fails to predict structures of correct folds for all the 32 test targets, while Robetta and our ResNet predict 7 and 15 correct folds, respectively, even considering only the first models. Note that when only primary sequence is used with our ResNet (trained with sequence profile), the average TMscore is only ~0.30 across all FM targets and only one has predict structures of correct fold (another one has TMscore very close to 0.5).
Modeling human-designed proteins.
It was shown that ResNet could predict structures of correct folds for 16 of the 18 in-house designed proteins[12]. Here we conduct a more comprehensive study on 21 de novo proteins designed by two groups in 2018–2020[27–29], among which 11 proteins were used to test trRosetta. None of these 21 proteins has evolutionarily related homologs in our training set (HHblits E-value<0.1). We consider two types of ResNet models. One is trained and tested with all input features including co-evolution. The other is trained and tested with all but co-evolution, mainly using input features derived from sequence profile. We build MSAs (multiple sequence alignment) by running HHblits on the 2018 uniclust30 database and the 2020 uniref30 database, respectively. No sequence homologs are found for any test proteins in the 2018 database and few are found for 9 test proteins in the 2020 database.
Table 3 summarizes the modeling accuracy. When evolutionary information is not available at all (i.e., the 2018 uniclust30 database is used), our ResNet still can predict structures of correct folds for almost all the 21 proteins no matter how ResNet is trained (Fig. 3(a) and (b)). When trained with co-evolution, ResNet performs similarly no matter whether the input features are derived from the 2018 or 2020 sequence databases. This implies that co-evolution is not very helpful possibly because there are two few sequence homologs and many of them are also human-designed proteins. Table 3 and Fig. 3 show that sequence profile (if available) helps improving modeling accuracy, especially when only the first 3D models are evaluated and ResNet is trained with only sequence profile.
Table 3.
Average modeling accuracy of deep ResNet on 21 human-designed proteins. GDT and GHA are scaled to [0, 1].
| ResNet trained with | Sequence database used to build input features | First Model | Best Model | ||||
|---|---|---|---|---|---|---|---|
| TM | GDT | GHA | TM | GDT | GHA | ||
| Co-evolution | 2018 uniclust30 | 0.650 | 0.662 | 0.479 | 0.675 | 0.686 | 0.498 |
| Co-evolution | 2020 uniref30 | 0.662 | 0.675 | 0.490 | 0.683 | 0.694 | 0.509 |
| No coevolution | 2018 uniclust30 | 0.584 | 0.598 | 0.430 | 0.676 | 0.695 | 0.509 |
| No coevolution | 2020 uniref30 | 0.654 | 0.663 | 0.495 | 0.723 | 0.738 | 0.556 |
Figure 3.
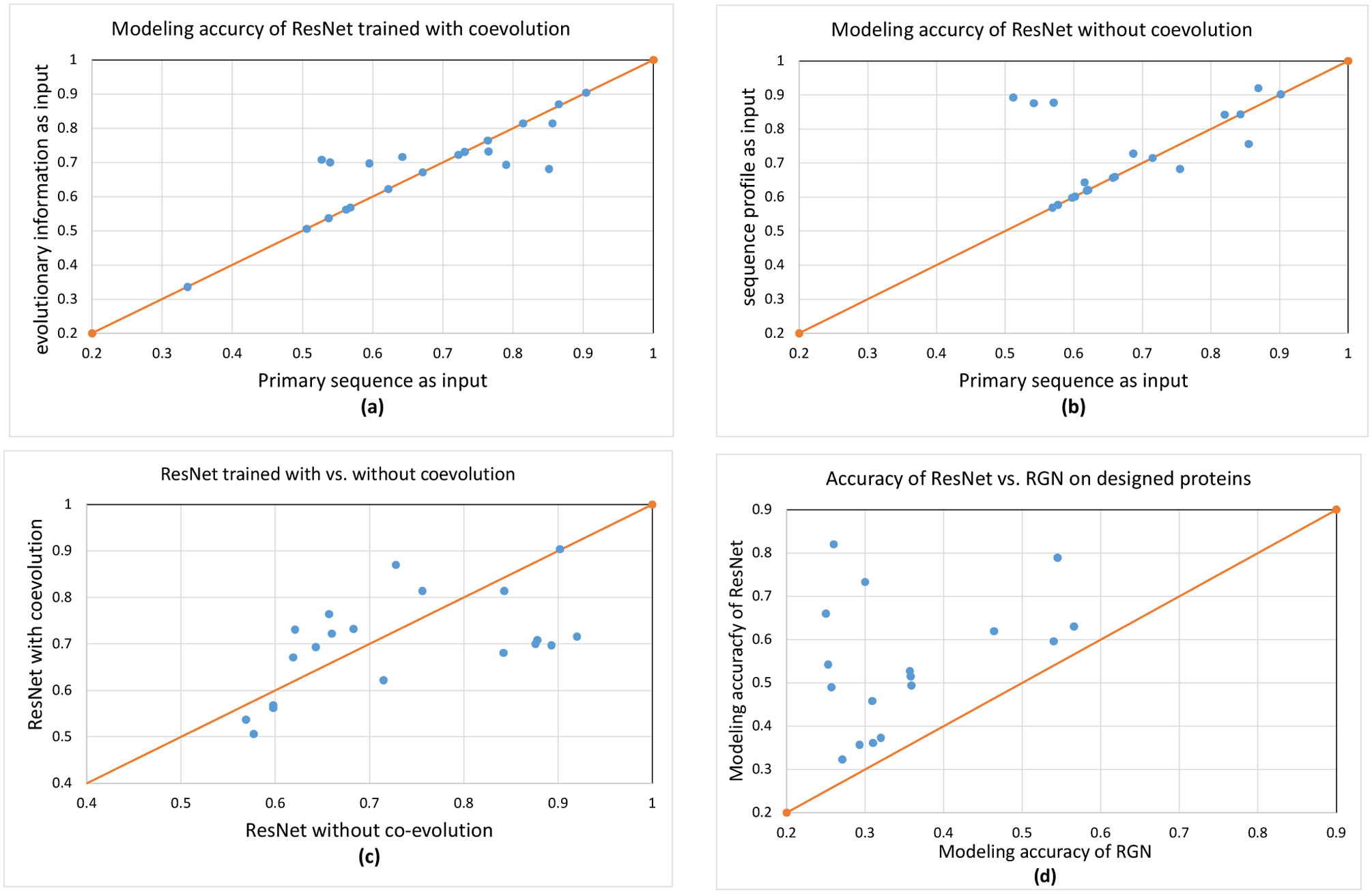
3D modeling accuracy on the human-designed proteins. (a) the best modeling accuracy of ResNet trained with co-evolution. A dot above the diagonal line indicates that a 3D model predicted with evolutionary information (coevolution and sequence profile) has higher quality. Twelve test proteins do not have any sequence homologs, so they lie on the diagonal line. (b) the best modeling accuracy of ResNet trained with only sequence profile. A dot above the diagonal line indicates that a 3D model predicted with sequence profile has higher quality. (c) the best modeling accuracy of ResNet trained with vs. without co-evolution. MSAs built from the 2020 database are used to derive input features. A dot above the diagonal line indicates that a 3D model predicted by ResNet trained with co-evolution has higher quality than corresponding 3D model predicted by ResNet trained without co-evolution. (d) ResNet vs. RGN. ResNet is trained with only sequence profile and primary sequence is used as input. A dot above the diagonal line indicates that the first model predicted by ResNet for a specific target has higher quality.
When few sequence homologs are available and the best 3D models are considered, ResNet trained without co-evolution outperforms ResNet trained with co-evolution (Fig. 3(c)). The average TMscore of the best 3D models by ResNet without and with co-evolution is 0.723 and 0.683, respectively. This may be because that when very few sequence homologs are available (and many of them are designed proteins), co-evolution information is not as reliable as sequence profile or that the patterns learned by ResNet from co-evolutionary information do not generalize well to the structural space of designed proteins.
The 21 models generated by RGN have average TMscore 0.363, much worse than ResNet trained without co-evolution and using primary sequence as input (Fig. 3(d)). In total RGN predicts correct folds for only 3 human-designed proteins while ResNet predicts 14 correct folds considering only the first models. Yang et al tested trRosetta on 11 of the 21 human-designed proteins. trRosetta predicted 3D models with average TMscore 0.661, comparable to our ResNet trained with co-evolution, which is not very surprising since trRosetta also uses a ResNet trained with co-evolution.
Performance on the CASP14 targets
In CASP14 RaptorX applied almost the same protocol to all the targets (see Methods). The average GDT of the best 3D models submitted by RaptorX for the TBM-easy, TBM-hard, FM/TBM and FM targets is 80.30, 65.35, 60.17 and 35.38, respectively. In contrast, the average GDT by Rosetta server (which used ResNet of 140 2D convolutional layers) is 78.29, 64.75, 63.77 and 33.52, respectively, and the GDT by Zhang’s QUARK server is 79.00, 64.15, 67.49 and 49.74. RaptorX, Rosetta and QUARK all used ResNet. QUARK stood out on the FM and FM/TBM targets mainly because QUARK made use of three very large metagenome databases (JGI IMG[30], BFD[31] and MGnify[32]) while RaptorX and Rosetta did not. The Baker human group used these large databases (coupled with model refinement) and outperformed all the others except AlphaFold2. These large databases have a big impact on most CASP14 FM targets and some FM/TBM targets such as the 9 test domains cut from T1044 (PDB code 6vr4), a viral protein with thousands of sequence homologs in these databases but none in UniRef. The Baker human group generated up to 6 MSAs for each target. We ran RaptorX on Baker’s MSAs only for the FM targets (due to the limit of computing resources), predicting 100 decoys from each MSA and selecting the 5 lowest-energy decoys for each target as the final predictions. The average TMscore and GDT obtained by RaptorX on the FM targets is 0.628 and 54.45, respectively, higher than what QUARK obtained in CASP14. Meanwhile, 16 of 23 predicted structures have correct fold, i.e., TMscore>0.5. This result is consistent with our previous experiment that when only primary sequence is used ResNet fails to predict structures of correct folds for most of the CASP13 FM targets
Conclusion and Discussions
We have presented our latest study of deep ResNet for protein contact/distance prediction and template-free structure prediction. We show that, although structure prediction has been significantly improved by ResNet, further improvements can still be obtained by integrating a variety of ideas. It helps by using a larger ResNet, the full co-evolution matrix, and the metagenome data, which when combined may result in measurable improvement. Additionally, gradient-based energy minimization and inter-residue orientation also help with 3D modeling. Our structure prediction pipeline is still less sophisticated than trRosetta[12], especially in using metagenome data to generate MSAs and in how to feed predicted distance/orientation restraints to PyRosetta to build 3D models.
When co-evolution is not used, ResNet trained with sequence profile can predict structures of correct folds for more than half of the CASP13 FM targets and all the human-designed proteins we tested. Further, for the human-designed proteins, sequence profile may be more useful than co-evolution when few sequence homologs are available. In the case where evolutionary information is not available (i.e., only primary sequence is available), ResNet still can predict correct folds for almost all the human-designed proteins, albeit with a loss in average model quality. These results suggest ResNet can learn important information governing protein folding from experimental protein structures and the functionality of ResNet extends beyond denoising co-evolution signals. Accordingly, ResNet can be used to quickly validate if a designed protein has a desired shape or not, avoiding expensive and time-consuming wet lab.
Although ResNet can predict structures of correct folds for human-designed proteins without using any evolutionary information, it does not work well on natural proteins, maybe because human-designed proteins usually have deeper energy wells than natural proteins. For most natural proteins, our method still needs a small number of sequence homologs to work. Nevertheless, in nature a protein folds without knowledge of its sequence homologs. It is desirable to have a method that can fold a protein without using sequence homologs. Recently, language models such as Transformer have been used to model protein sequences[33]. We are studying Transformer-like networks to determine their use in structure prediction when few homologs are present. In fact, AlphaFold2 in CASP14 has achieved an amazing result by using a Transformer-like network, directly predicting atom 3D coordinates from an MSA and training on almost the whole PDB. A Transformer-like network may infer co-evolution signal directly from MSAs without using existing direct-coupling analysis such as CCMpred and thus, avoid the errors produced by CCMpred when the MSA in question is shallow. By learning from all protein families Transformer shall have better generalization performance for shallow MSAs than CCMpred, which learns its parameters from only a single MSA. Currently our ResNet takes the CCMpred output as input and thus, inherits some errors produced by CCMpred.
Methods
Training and validation data
In CASP13 we trained deep ResNet models using PDB25, a set of representative protein chains generated by the PISCES server[34] in which no two protein chains share >25% sequence identity. Here we use Cath S35 as our training and validation proteins. Cath S35 is a representative set of CATH domains (https://www.cathdb.info/), in which any two domains share no more than 35% sequence identity. According to our previous work[6], PDB25 and Cath S40 result in similar performance on template-free modeling (FM) targets, but Cath S35 may have a slightly better performance on template-free/template-based modeling (FM/TBM) targets. The protein domains in Cath S35 on average are shorter than the protein chains in PDB25, so it may reduce GPU memory consumption by using Cath S35. For each protein domain in Cath S35, we generated their multiple sequence alignments (MSAs) by running HHblits with E-value=0.001 on the uniclust30 library dated in 2017 and then derived input features for the prediction of distance and orientation.
We downloaded two versions of Cath S35 at ftp://orengoftp.biochem.ucl.ac.uk/cath/releases/daily-release/archive/. One is dated in March 2018 and the other on January 1, 2020. They have 32140 and 32511 entries, respectively, and differ by about 2000 entries. We excluded very short domains (<25 AAs) and those with too many missing Ca and Cb atoms and then randomly split them into two non-overlapping subsets: one for training and the other for validation (1800 domains). In total we generated 6 splits, but not all of them were used to train our deep learning models due to limit of computing resources. Our results show that there is little difference between these two Cath S35 datasets in terms of both contact prediction and 3D modeling. In terms of the top L/5, L/2 and L long-range contact prediction F1 value, the ResNet models trained on the 2020 Cath data obtained 27.7%, 45.1% and 52.1% while those trained on the 2018 Cath data obtained 27.8%, 44.6% and 51.3%. In terms of the average TMscore of the predicted 3D models, the ResNet models trained on the 2020 Cath data obtained 0.629 and 0.663 for the first and best models, respectively, while those trained on the 2018 Cath data obtained 0.638 and 0.659.
Independent test data
Target-training similarity.
We use HHblits[35] with E-value=0.1 to check if there are evolutionarily related proteins in our training/validation data for a specific test target. When the E-value returned by HHblits is larger than 0.1, we say that this target has no evolutionarily related proteins in our training set. We also use TMalign[36] to calculate target-training structure similarity, which is defined as the highest structure similarity (measured TMscore) between a specific target and all our training/validation proteins. By the way, we always use the target length (i.e., the number of valid Ca atoms in an experimental structure) as the normalization constant while using TMalign to calculate structure similarity and the TMscore program to evaluate the quality of one predicted 3D model, so that TMscore returned by TMalign and the TMscore program is comparable.
CASP targets.
The CASP organizers divide the test targets into four categories: TBM-easy, TBM-hard, FM/TBM and FM. Roughly speaking, a target belongs to TBM-easy or TBM-hard if it has reliable sequence homologs in PDB (Protein Data Bank). A target belongs to FM if it does not have sequence homologs in PDB. A target belongs to FM/TBM if it may have weakly similar sequence homologs in PDB. In total there are 32 CASP13 FM targets and 13 FM/TBM targets, among which one FM target T0950 is a server-only target. T0953s1 and T0955 have very few long-range contacts, so they are not used to evaluate contact prediction. We mainly use the CASP13 FM targets to evaluate our method since some FM/TBM targets may have evolutionarily related proteins in PDB dated before CASP13. We use HHblits (with E-value=0.1) and TMalign to check sequence profile and structure similarity between the CASP13 FM targets and our training set (i.e., the two Cath S35 datasets). HHblits returns a large E-value (>10) for many of the 32 FM targets. Only two FM targets T0975 and T1015s1 have HHblits E-value less than 0.1. T0975 is related to 4ic1D with HHblits E-value=4.2E-12, and T1015s1 is related to 4iloA with HHblits E-value=0.024. Both 4ic1D and 4iloA were deposited to PDB well before 2018, so it is fair to include them in our training set. Further, the structure similarity (TMscore) between T0975 and 4ic1D is less than 0.5, so is that between T1015s1 and 4iloA. A detailed description of these FM targets is available in Supplementary Dataset 2.
De novo proteins.
Yang et al has tested trRosetta on 18 de novo proteins [12], all of which were designed by Baker group and 7 of them were deposited to PDB before 2018. We collected 35 de novo proteins (including 7 membrane proteins) designed by two research groups in the past several years and 28 of them were deposited to PDB after January 1, 2018. See Supplementary Dataset 4 for details. Meanwhile, 4 of them are designed by Kortemme group very recently [29] and the others by Baker group. Among these proteins, 21 of them have HHblits E-value>0.001 with our training proteins in the 2018 Cath S35 dataset, so we only use these 21 proteins as our test set. Their PDB codes are 6B87, 6CZG, 6CZH, 6CZI, 6CZJ, 6D0T, 6DG5, 6DG6, 6E5C, 6M6Z, 6MRR, 6MRS, 6MSP, 6NUK, 6O35, 6TJ1, 6TMSA, 6TMSG, 6VG7, 6VGA and 6VGB. To further remove potential redundancy between our training set and these test proteins, we exclude 20 proteins from the 2018 Cath S35 data so that all the 21 test proteins have HHblits E-value>0.1 with our training set. All our deep ResNet models used to predict these 21 de novo proteins are trained by this revised Cath S35 dataset. Among the 21 proteins, 11 of them have been used to evaluate trRosetta [12]: 6CZG, 6CZH, 6CZI, 6CZJ, 6D0T, 6DG5, 6DG6, 6E5C, 6MRR, 6MRS, 6MSP and 6NUK.
We run HHblits (E-value=0.001) to search through two sequence databases ( http://wwwuser.gwdg.de/~compbiol/uniclust/) released by Soding group (i.e., uniclust30 dated in August 2018 and uniref30 in February 2020 at) to see if there are any sequence homologs for the 21 test proteins. It turns out that HHblits does not find any sequence homologs in 2018 uniclust30 possibly because these 21 proteins are released since 2018. When the 2020 uniref30 database is used, HHblits finds few sequence homologs for 9 of the 21 proteins (6B87, 6CZG, 6CZH, 6CZI, 6CZJ, 6D0T, 6VG7, 6VGA and 6VGB). Further examination indicates that most of the sequence homologs are human-designed proteins or themselves.
MSA generation and input features
We run HHblits[37] with E-value=1E-3 and 1E-5 and jackhammer[38] with E-value=1E-3 and 1E-5 to detect sequence homologs and generate MSAs (multiple sequence alignment) for CASP13 FM targets. To ensure a fair comparison, we generated their MSAs using the uniclust30 database dated in October 2017 and the uniref90 database dated in March 2018. In addition, when a CASP13 target has a shallow MSA (i.e., ln(Meff)<6), we also search a small metagenome database MetaClust dated in May 2018 (https://metaclust.mmseqs.com/) to see if more sequence homologs can be found. The metagenome data is not applied to the 21 human-designed proteins.
We use the following input features: 1) primary sequence: one-hot encoding is used to represent a sequence; 2) sequence profile: it is derived from MSA and encodes evolutionary information at each residue; we also use secondary structure and solvent accessibility predicted from sequence profile; 3) co-evolution information: mutual information and CCMpred output. Meanwhile, the CCMpred output includes one L×L co-evolution matrix and one full precision matrix of dimension L×L×21×21 where L is the protein sequence length. We have also tested one covariance matrix of dimension L×L×21×21, but not observed performance gain and thus, decided not to use it. In CASP13, we only used the L×L co-evolution matrix generated by CCMpred as an input feature, but not the full precision matrix. When sequence profile is not used, we use secondary structure and solvent accessibility predicted from primary sequence. For each CASP13 target, there are 4 different MSAs generated. We run our deep ResNet on these 4 MSAs separately to predict 4 sets of distance and orientation and the calculate their average to obtain the final prediction.
For human designed proteins, only HHblits is used to search for their sequence homologs from a 2018 uniclust30 database and a 2020 uniref30 database.
MSA depth.
We use Meff to measure the depth of an MSA, i.e., the number of non-redundant (or effective) sequence homologs in the MSA. Let Sij be a binary variable indicating whether two protein sequences i and j are similar. Sij is equal to 1 if and only if the sequence identity between i and j is >70%. For a protein i, let Si denote the sum of Si1, Si2, …, Si,n where n is the number of proteins in the MSA. Then, Meff is calculated as the sum of 1/S1, 1/S2, …,1/Sn. Generally speaking, the smaller Meff a target has, the more challenging to predict its contact and distance. MSA depth of a specific target depends on the homology search tool, sequence database and E-value cutoff. In this paper, for CASP13 targets we calculate their Meff values using the MSAs generated by HHblits (E-value=0.001) on the uniclust30 database dated in October 2017.
Protein structure representation
In this work we represent only protein backbone conformation using inter-atom distance matrices and inter-residue orientation matrices. Three types of Euclidean distance matrices are used for three types of atom pairs: Ca-Ca, Cb-Cb and N-O. In addition to the inter-residue orientation defined by N, Ca and Cb atoms of two residues and implemented in trRosetta, we have studied another type of orientation formed by four Ca atoms (of 4 residues): Ca(i), Ca(i+1), Ca(j) and Ca(j+1). This Ca-based orientation includes one dihedral angle formed by two planes Ca(i)−Ca(i+1)−Ca(j) and Ca(i+1)−Ca(j)−Ca(j+1) and two angles formed by 3 atoms, i.e., Ca(i), Ca(i+1), Ca(j) and Ca(j), Ca(j+1), Ca(i). The advantage of this Ca-based orientation is that it involves 4 residues instead of only 2 residues, no special handling is needed for Glycine, and it also allows us to use those protein structures with many missing Cb atoms to train our deep models. However, our experimental results show that this Ca-based orientation slightly underperforms the orientation employed by trRosetta, so by default we employ the inter-residue orientation implemented in trRosetta.
We discretize distance into 47 bins (i.e., bin width=0.4Å): 0–2Å, 2–2.4Å, 2.4–2.8Å, 2.8–3.2Å, …, 19.6–20Å and >20Å. One special label is used to indicate an “unknown” distance when at least one of the two atoms do not have valid 3D coordinates in the structure file. For orientation, we follow the same format as presented in trRosetta[12] and discretize each angle uniformly with the bin width set to 10 degrees. A separate orientation label is used to indicate that two residues are far away from each other (when the distance of their Cb atoms >20Å) regardless of their orientation.
Deep model training
We train our deep ResNet by subsampling MSAs. For each training/validation protein, we randomly sample 40–60% of the sequence homologs from its original MSA (when it has at least 2 sequences) and then derive input features (e.g., sequence profile, predicted secondary structure and co-evolution information) from the sampled MSA. To save time, for each protein 10 sets of sampled MSAs and input features are precomputed so that our training program only needs to load them from disk. We train our deep ResNet for 20 epochs using AdamW [39] and select the model with the minimum validation loss as our final model. To examine the impact of input features and network capacity, we have trained deep ResNet models with different input features and network sizes. Under each setting, we trained up to 6 deep ResNet models and used them as an ensemble to predict distance and orientation probability distributions.
Building protein 3D models and model clustering
We build protein 3D models from our predicted distance and orientation distribution using PyRosetta[22]. Our 3D model building protocol consists of the following major steps: (1) convert predicted distance and orientation probability distribution into discrete energy potential using the idea described in our previous work[40]. We have tested both DFIRE[41] and DOPE[42] reference states and found that the DFIRE reference state is slightly better. For orientation potential, we obtain its reference state by simple counting the occurring frequency of each orientation label in our training proteins. (2) interpolate the discrete energy potential for each pair of atoms or residues to a continuous curve using the Rosetta spline function; (3) minimize the energy potential to build a 3D model by iteratively using the LBFGS algorithm implemented in PyRosetta. The starting conformation for energy minimization is sampled from the real-valued phi/psi distribution predicted by our deep learning method from sequence profile (implemented before CASP13)[6]. Since LBFGS may not converge to the global minimum, once it reaches a local minimum, we perturb all phi/psi angles by a small deviation and then apply LBFGS again to see if a lower energy status may be reached. This perturbation procedure is repeated up to three times and may slightly improve modeling accuracy. After generating an initial 3D model by LBFGS, a two-stage fast relaxation protocol is applied to it to optimize side-chain atoms and reduce steric clashes. The first stage relaxes the initial model with only the Rosetta built-in full-atom energy function (i.e., ref2015) but not our predicted distance/orientational potential. The second stage relaxes the model further using both ref2015 and our predicted potential. The 1st stage is optional and may slightly improve modeling accuracy. We found that, on average, fast relaxation improves Ca conformation by less than 0.01 TMscore.
To simulate the real prediction scenario, while building 3D models for CASP13 domains we do not use the official domain sequences. Instead we use the domain sequences determined by our own server during the CASP13 season when the experimental structures of the CASP13 targets were not available. While evaluating the quality of a 3D model, we only consider the segments of the model that overlap with the official domain definition. In calculating TMscore and GDT, we always use the official domain length as the normalization constant. Therefore, when our domain definitions deviate a lot from the official ones, our model quality score may not be good even if the models for our own domain sequences are well predicted.
We generated 150 decoys per target and then used SPICKER[43] to cluster them into up to 10 groups. The first and the best cluster centroids are used as the first and best predicted 3D models, respectively. There is only very minor decrease in modeling accuracy when the best of top 5 (instead of top 10) cluster centroids is used as the best model.
RaptorX protocol in CASP14
In CASP14 RaptorX server applied almost the same protocol to all the targets. It first ran HHblits[35] and Jackhmmer[38] to search UniRef (https://www.uniprot.org/uniref/) and a small metagenome database (https://metaclust.mmseqs.org/) to build MSAs. Then it predicted distance/orientation distributions using ResNet and built 3D models with PyRosetta[22]. RaptorX also ran HHblits to check if a test target has reasonable templates or not (E-value=1E-5). When a template was available, RaptorX fed the template, the target-template alignment and target co-evolution information to a ResNet model (trained by both template and co-evolution information) to predict distance and orientation distributions and then built template-based 3D models with PyRosetta. The target-template alignments were built by HHblits and a distance-based protein threading tool DeepThreader[9]. How useful is a template depends on its similarity with the target and the MSA depth of the target. RaptorX ranked a template-based 3D model before a template-free 3D model only when the former has energy potential at least 10% lower than the latter. In fact, for quite a few targets with good templates such as T1047s2-D2, T1065s1-D1, T1083-D1 and T1084-D1 and T1085-D3, their template-free 3D models were ranked before their template-based models. Indeed, these template-free 3D models have GDT 88.86, 88.44, 87.77, 90.84 and 82.89, respectively, better than the template-based 3D models. Our initial analysis shows that a template is helpful when it shares at least 20% sequence identity with a target which does not have many sequence homologs. A detailed analysis of this is out of scope of this paper and will be reported in another one.
Performance metrics
Following CASP official assessment, we evaluate the top L, L/2 and L/5 long-range predicted contacts by precision and F1 value where L is the protein sequence length. We say there is a contact between two residues if the Euclidean distance of their Cb atoms is no more than 8Å. A contact is long-range if the sequence separation of its involved two residues is at least 24 residues. We mainly use TMscore[44] to evaluate the quality of a predicted 3D model, which measures the geometric similarity between a predicted 3D model and its corresponding experimental structure (i.e., ground truth). It ranges from 0 to 1 and usually a 3D model is assumed to have a correct fold when its TMscore≥0.5. GDT and GHA are also widely used in the community to measure the quality of a predicted 3D model. By definition, GDT and GHA range from 0 (worst) to 100 (best), but sometimes we normalize them by 100 to obtain the same scale as TMscore. Although different, TMscore and GDT are highly correlated, so sometimes we just use one of them to measure the quality of a predicted 3D model.
Data availability
The PDB IDs of the human-designed proteins are available in Supplementary Dataset 4. The domain sequences determined by our own CASP13 server for the CASP13 targets are available in Supplementary Dataset 5. The official domain sequences of the CASP13 targets and their corresponding PDB IDs are available at the CASP13 web site https://predictioncenter.org/casp13/index.cgi .
Code availability
The source code is available at https://github.com/j3xugit/RaptorX-3DModeling/ or https://doi.org/10.5281/zenodo.4642250 and the server is available at http://raptorx.uchicago.edu/ . In addition to template-free protein structure prediction, this package also supports comparative protein structure modeling, i.e., building protein 3D models from templates by deep learning.
Extended Data
Extended Data Figure 1.
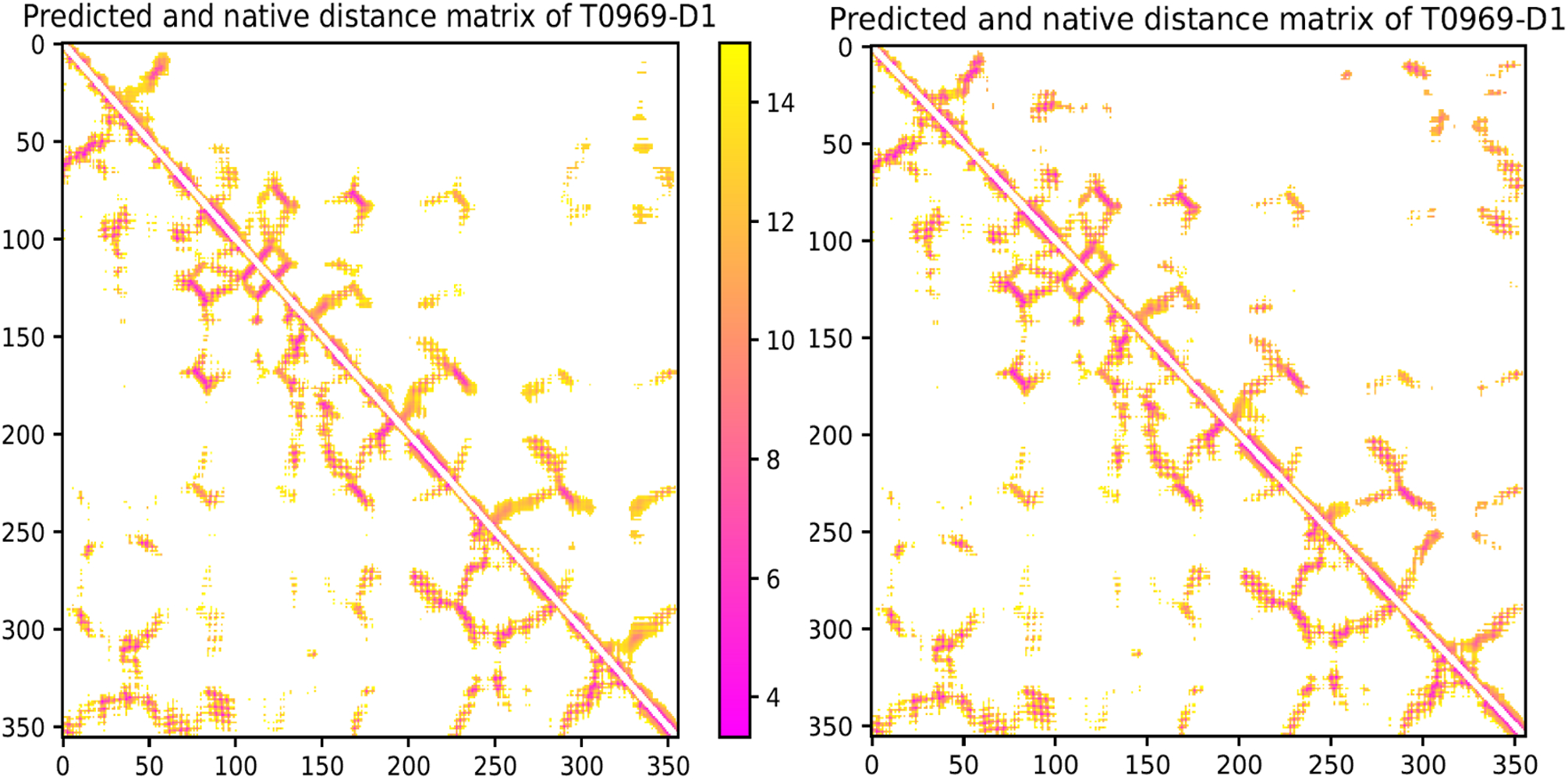
Distance matrices predicted for T0969-D1 by deep ResNet when co-evolution is not used (left) and used (right). Only distance predictions less than 15Å are displayed in color. In each picture, native distance and predicted distance are shown below and above the diagonal, respectively.
Supplementary Material
Acknowledgements
The authors are grateful to Drs. Jianyi Yang and Ivan Anishchanka for their very helpful discussions, providing trRosetta results and helping with PyRosetta. The authors are grateful to Dr. Ivan Anishchanka for providing us the MSAs built by the Baker human group for the CASP14 targets.
This work is supported by National Institutes of Health grant R01GM089753 to J.X. and National Science Foundation grant DBI1564955 to J.X. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Competing interests
The authors declare no conflict of interest.
References
- 1.De Juan D, Pazos F, and Valencia A, Emerging methods in protein co-evolution. Nature Reviews Genetics, 2013. 14(4): p. 249. [DOI] [PubMed] [Google Scholar]
- 2.Shrestha R, et al. , Assessing the accuracy of contact predictions in CASP13. Proteins-Structure Function and Bioinformatics, 2019. 87(12): p. 1058–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abriata LA, Tamo GE, and Dal Peraro M, A further leap of improvement in tertiary structure prediction in CASP13 prompts new routes for future assessments. Proteins-Structure Function and Bioinformatics, 2019. 87(12): p. 1100–1112. [DOI] [PubMed] [Google Scholar]
- 4.Wang S, et al. , Accurate de novo prediction of protein contact map by ultra-deep learning model. PLoS computational biology, 2017. 13(1): p. e1005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang S, Sun SQ, and Xu JB, Analysis of deep learning methods for blind protein contact prediction in CASP12. Proteins-Structure Function and Bioinformatics, 2018. 86: p. 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu J, Distance-based protein folding powered by deep learning. PNAS, 2019. 116(34 ): p. 16856–16865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu JB and Wang S, Analysis of distance-based protein structure prediction by deep learning in CASP13. Proteins-Structure Function and Bioinformatics, 2019. 87(12): p. 1069–1081. [DOI] [PubMed] [Google Scholar]
- 8.Wang S, et al. , Folding membrane proteins by deep transfer learning. Cell systems, 2017. 5(3): p. 202–211. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu JW, et al. , Protein threading using residue co-variation and deep learning. Bioinformatics, 2018. 34(13): p. 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Senior AW, et al. , Protein structure prediction using multiple deep neural networks in the 13th Critical Assessment of Protein Structure Prediction (CASP13). Proteins-Structure Function and Bioinformatics, 2019. 87(12): p. 1141–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ding WZ and Gong HP, Predicting the Real-Valued Inter-Residue Distances for Proteins. Advanced Science, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang JY, et al. , Improved protein structure prediction using predicted interresidue orientations. Proceedings of the National Academy of Sciences of the United States of America, 2020. 117(3): p. 1496–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greener JG, Kandathil SM, and Jones DT, Deep learning extends de novo protein modelling coverage of genomes using iteratively predicted structural constraints. Nature Communications, 2019. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ovchinnikov S, et al. , Protein structure determination using metagenome sequence data. Science, 2017. 355(6322): p. 294–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, et al. , Ensembling multiple raw coevolutionary features with deep residual neural networks for contact-map prediction in CASP13. Proteins-Structure Function and Bioinformatics, 2019. 87(12): p. 1082–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kandathil SM, Greener JG, and Jones DT, Prediction of interresidue contacts with DeepMetaPSICOV in CASP13. Proteins-Structure Function and Bioinformatics, 2019. 87(12): p. 1092–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marks DS, Hopf TA, and Sander C, Protein structure prediction from sequence variation. Nature biotechnology, 2012. 30(11): p. 1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamisetty H, Ovchinnikov S, and Baker D, Assessing the utility of coevolution-based residue–residue contact predictions in a sequence-and structure-rich era. Proceedings of the National Academy of Sciences, 2013. 110(39): p. 15674–15679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seemayer S, Gruber M, and Söding J, CCMpred—fast and precise prediction of protein residue–residue contacts from correlated mutations. Bioinformatics, 2014. 30(21): p. 3128–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y, et al. , Enhancing Evolutionary Couplings with Deep Convolutional Neural Networks. Cell Syst, 2018. 6(1): p. 65–74 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.AlQuraishi M, End-to-End Differentiable Learning of Protein Structure. Cell Systems, 2019. 8(4): p. 292–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chaudhury S, Lyskov S, and Gray JJ, PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics, 2010. 26(5): p. 689–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones DT, et al. , MetaPSICOV: combining coevolution methods for accurate prediction of contacts and long range hydrogen bonding in proteins. Bioinformatics, 2015. 31(7): p. 999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eickholt J and Cheng J, Predicting protein residue-residue contacts using deep networks and boosting. Bioinformatics, 2012. 28(23): p. 3066–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steinegger M and Soding J, Clustering huge protein sequence sets in linear time. Nature Communications, 2018. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim DE, Chivian D, and Baker D, Protein structure prediction and analysis using the Robetta server. Nucleic acids research, 2004. 32: p. W526–W531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu CF, et al. , Computational design of transmembrane pores. Nature, 2020. 585(7823): p. 129–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu PL, et al. , Accurate computational design of multipass transmembrane proteins. Science, 2018. 359(6379): p. 1042–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pan XJ, et al. , Expanding the space of protein geometries by computational design of de novo fold families. Science, 2020. 369(6507): p. 1132–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen IMA, et al. , The IMG/M data management and analysis system v.6.0: new tools and advanced capabilities. Nucleic acids research, 2021. 49(D1): p. D751–D763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steinegger M, Mirdita M, and Soding J, Protein-level assembly increases protein sequence recovery from metagenomic samples manyfold. Nature Methods, 2019. 16(7): p. 603–+. [DOI] [PubMed] [Google Scholar]
- 32.Mitchell AL, et al. , MGnify: the microbiome analysis resource in 2020. Nucleic acids research, 2020. 48(D1): p. D570–D578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rives A, et al. , Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. 2020. [DOI] [PMC free article] [PubMed]
- 34.Wang GL and Dunbrack RL, PISCES: a protein sequence culling server. Bioinformatics, 2003. 19(12): p. 1589–1591. [DOI] [PubMed] [Google Scholar]
- 35.Remmert M, et al. , HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods, 2012. 9(2): p. 173. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y and Skolnick J, TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic acids research, 2005. 33(7): p. 2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mirdita M, et al. , Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic acids research, 2017. 45(D1): p. D170–D176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson LS, Eddy SR, and Portugaly E, Hidden Markov model speed heuristic and iterative HMM search procedure. Bmc Bioinformatics, 2010. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hutter I.L.a.F., Decoupled Weight Decay Regularization. 2019, arXiv.
- 40.Zhao F and Xu J, A position-specific distance-dependent statistical potential for protein structure and functional study. Structure, 2012. 20(6): p. 1118–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou HY and Zhou YQ, Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction (vol 11, pg 2714, 2002). Protein Science, 2003. 12(9): p. 2121–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shen MY and Sali A, Statistical potential for assessment and prediction of protein structures. Protein Science, 2006. 15(11): p. 2507–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y and Skolnick J, SPICKER: A clustering approach to identify near-native protein folds. Journal of Computational Chemistry, 2004. 25(6): p. 865–871. [DOI] [PubMed] [Google Scholar]
- 44.Xu JR and Zhang Y, How significant is a protein structure similarity with TM-score=0.5? Bioinformatics, 2010. 26(7): p. 889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The PDB IDs of the human-designed proteins are available in Supplementary Dataset 4. The domain sequences determined by our own CASP13 server for the CASP13 targets are available in Supplementary Dataset 5. The official domain sequences of the CASP13 targets and their corresponding PDB IDs are available at the CASP13 web site https://predictioncenter.org/casp13/index.cgi .