

Cardiolipin (CL) is a unique glycerol-bridged dimeric phospholipid representing up to 20% of total lipids in mitochondrial membranes in cardiomyocytes. Abnormal CL metabolism is linked to heart diseases, including Barth syndrome, myocardial ischemia-reperfusion injury, and heart failure1. However, CL profiles and specific roles of CL in cardiac mitochondria remain largely obscure.
We first performed quantitative lipidomic analysis on mouse hearts at different stages and revealed a strong discrepancy in molecular compositions of CL and CL-related metabolites between embryonic and adult mouse hearts (Figure 1A). In particular, CL in embryonic hearts displayed more diverse acyl compositions, while the predominant form of CL in adult heart was tetralinoleoyl-CL, implicating that the pathways involved in CL biosynthesis and metabolism may have different functions between embryonic and adult hearts.
Figure 1. PTPMT1 is required for embryonic cardiac cardiolipin biosynthesis to regulate mitochondrial morphogenesis and heart development.
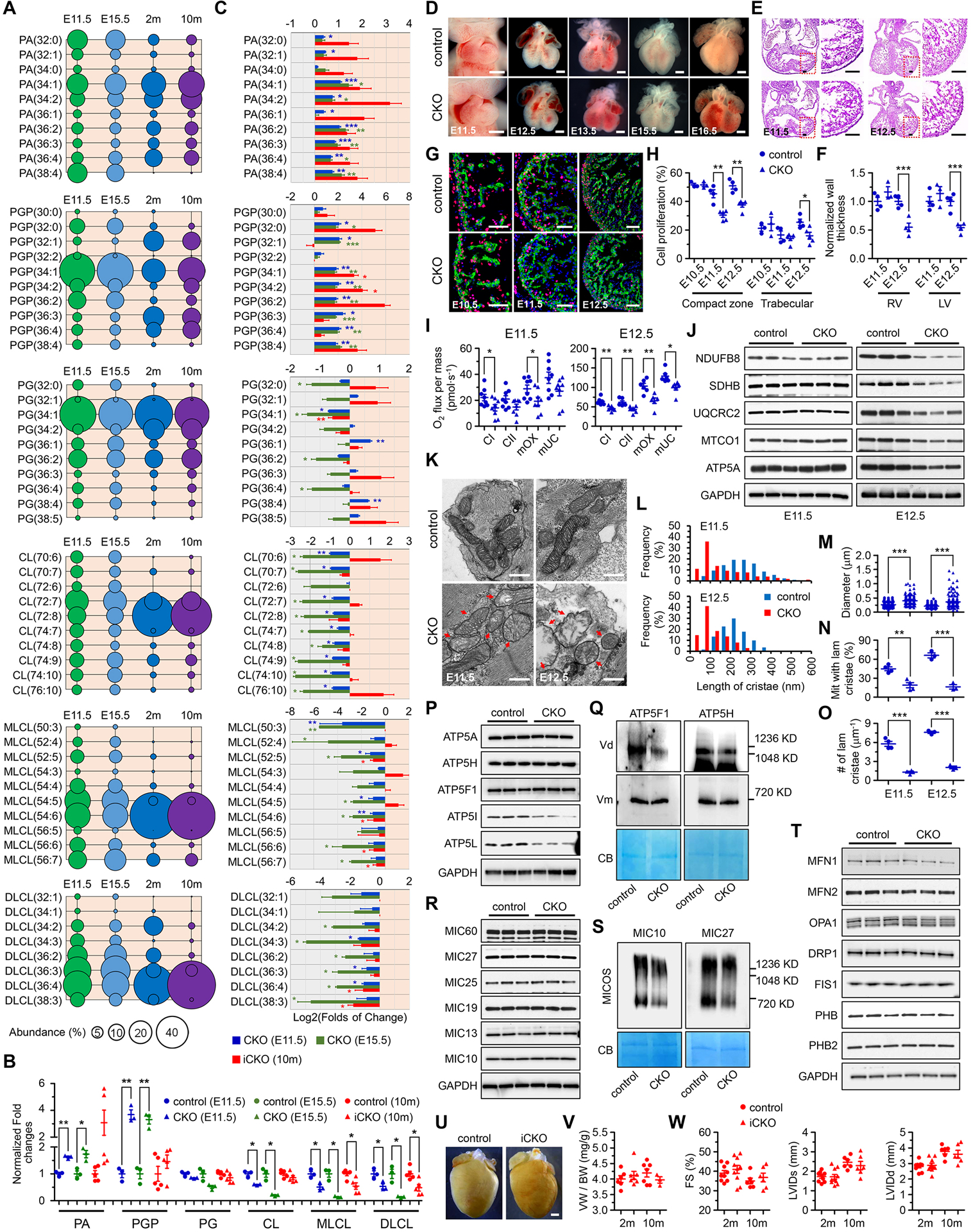
(A) Quantitative lipidomic analysis was applied to identify individual species of cardiolipin and cardiolipin-related metabolites, including phosphatidic acid (PA), phosphatidylglycerophosphate (PGP), phosphatidylglycerol (PG), cardiolipin (CL), monolyso-CL (MLCL), and dilyso-CL (DLCL) in the ventricles of control mice at embryonic day 11.5 (E11.5), E15.5, 2 months (2m), and 10m of age, respectively. The 10 most abundant species, if more than 10 species were observed, or all the species of each metabolite, were selected and further analyzed at each stage. The abundance (%) of individual species was calculated according to its total level and was represented by the area of the circle. n = 3, 3, 6, and 6, respectively. (B) The levels of total PA, PGP, PG, CL, MLCL, and DLCL in TnT-Cre-mediated PTPMT1 knockout ventricles (CKO) at E11.5 (n = 3) and E15.5 (n = 3), and in αMHC-CreER-mediated PTPMT1 knockout (iCKO) ventricles 10 months post Tamoxifen-induced gene deletion (n = 5) were analyzed and normalized to their control ventricles (n = 3, 3, and 5, respectively), respectively. Twenty-five and six ventricles with the same genotype were pooled as one sample at E11.5 and E15.5, respectively. (C) The levels of the 10 most abundant species if more than 10 species were observed, or the levels of every species of PA, PGP, PG, CL, MLCL, and DLCL in CKO and iCKO ventricles were further analyzed and normalized to their control ventricles, respectively. (D-H) Representative images of embryonic hearts (D), sections stained with hematoxylin and eosin (E), and images of EdU labeling (G) of control and CKO mice at indicated stages. Cardiac cells were co-stained with α-Actinin. Scale bar = 0.5mm, 100μm, and 50μm respectively. The thickness of left and right ventricular walls (F) of control (n = 4 at both stages) and CKO (n = 3 and 4, respectively) hearts, and the ratios of EdU-positive cardiomyocytes in ventricular compaction zone and trabecular (H) of control (n = 3, 4, and 4, respectively) and CKO (n = 3, 5, and 5, respectively) hearts were measured at indicated stages. (I) Oxygen (O2) flux representing the respiratory function of complex I (CI) and complex II (CII), the maximum OXPHOS capacity (mOX), and the maximum uncoupled capacity (mUC) were measured in control and CKO hearts by high-resolution respirometry at E11.5 and E12.5, respectively. The measurement was performed on two hearts with the same phenotype at E11.5 or one heart at E12.5 in one chamber. n = 8 per group at E11.5; n = 6 per group at E12.5. (J) Immunoblot analysis on the expression of mitochondrial OXPHOS subunits including NDUFB8 (complex I), SDHB (complex II), UQCRC2 (complex III), MTCO1 (complex IV), and ATP5A (complex V) in control and CKO embryonic hearts at E11.5 and E12.5. GAPDH was used as the loading control. (K) Representative transmission electron microscopic images of mitochondria in control and CKO cardiomyocytes at E11.5 and E12.5. Red arrows indicate the mitochondria with bubble-like cristae in CKO cardiomyocytes. Scale bar, 0.5μm. (L-O) Quantitative analysis of length of cristae (L), mitochondrial diameter in the short axis (M), the percentage of mitochondria with lamellar cristae (N), and the number of lamellar cristae along the mitochondrial long axis (O) in control and CKO cardiomyocytes at E11.5 (n = 4 per group) and E12.5 (n = 3 per group). At least 160 mitochondria were measured for each embryonic heart. (P-T) Immunoblot analysis of F1F0-ATP synthase subunits (P) including ATP5A (F1 complex subunit α), ATP5H (F0 complex subunit d), ATP5F1 (F0 complex subunit b), ATP5I (F0 complex subunit e), and ATP5L (F0 complex subunit g), MICOS complex subunits (R) including MIC60, MIC27, MIC25, MIC19, MIC13, and MIC10, as well as mitochondrial dynamics-related proteins (T) including MFN1, MFN2, OPA1, DRP1, and FIS1 and prohibitin proteins including PHB and PHB2 at E11.5. GAPDH was used as the loading control. Blue native-PAGE and immunoblot analysis of F1F0-ATP synthase complex using the antibodies against ATP5F1 and ATP5H (Q), and MICOS complex assembly using the antibodies against MIC10 and MIC27 (S). To note, the deletion of PTPMT1 reduced the formation of F1F0-ATP synthase dimers (Vd) whereas monomers (Vm) were not affected. The reduction of the MICOS complex assembly in CKO mitochondria was also observed. Each mitochondrial sample was prepared from over 50 embryonic ventricular tissues of the same genotype in 1% digitonin-containing extraction buffer, and separated by blue native-PAGE. Coomassie blue (CB) stained membranes were scanned for loading control. (U) Representative hearts of control and iCKO mice at 10 months post tamoxifen injection. Scale bar, 1mm. (V) Ratios of ventricle weight (VW) to body weight (BW) in control and iCKO mice at 2m and 10m post tamoxifen injection. n = 3–7 mice per group. (W) Echocardiographic assessment in control and iCKO mice at 2m and 10m post tamoxifen injection. n = 6–10 mice per group. All data represent mean ± SEM. Significance was determined by two-tailed, unpaired Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001 versus control.
To investigate the role of CL biosynthesis in cardiac development, we utilized TnT-Cre to generate a mouse model with cardiac-specific deletion of PTPMT1 (CKO), a mitochondrial phosphatase removing the terminal phosphate group from phosphatidylglycerophosphate (PGP) to form phosphatidylglycerol (PG)2, to interrupt CL biosynthesis in embryonic hearts. PTPMT1 deficiency indeed reduced the contents of both total CL and most of the abundantly expressed CL species in embryonic hearts at both E11.5 and E15.5, accompanied with a broad alteration in levels of total and individual CL-related metabolites (Figure 1B–C), demonstrating that PTPMT1 is required for CL biosynthesis in embryonic cardiomyocytes. All mouse protocols were approved by the Institutional Animal Care and Use Committee. The data, analytical methods, and study materials that support the findings of this study will be available to other researchers from the corresponding authors on reasonable request.
Deletion of PTPMT1 in cardiomyocytes caused abnormal cardiac development and embryonic lethality between E16.5 and E18.5. Morphological changes started in CKO hearts at E12.5 (Figure 1D), accompanied with decreased thicknesses of ventricular walls at the same stage (Figure 1E–F), which could be a consequence of defects in cardiac cell proliferation first observed in compact zone of CKO hearts at E11.5 (Figure 1G–H).
CL has been proposed to participate in regulating both mitochondrial function and structure1. We then evaluated mitochondrial respiration in permeabilized embryonic hearts by measuring oxygen consumption. Decreases in respiratory function of complex I and maximum oxidative phosphorylation capacity were observed in CKO hearts as early as E11.5 (Figure 1I). At E12.5, respiratory functions were further impaired, and changes in expression of mitochondrial complex proteins were observed in CKO hearts (Figure 1I–J).
In developing embryonic cardiomyocytes, mitochondria undergo a maturation process with increased mitochondria number and more organized lamellar cristae at later stages3. The function of CL in regulating mitochondrial morphology and ultrastructure in embryonic hearts remains unclear. We performed transmission electron microscopy analysis, and we found that membrane invagination was impaired and bubble-like inner membrane structure instead of lamellar cristae could be easily observed in CKO mitochondria at E11.5, which became more severe at E12.5 (Figure 1K). PTPMT1 deletion also altered mitochondrial diameter, percentages of mitochondria with lamellar cristae, cristae lengths, and cristae numbers in embryonic cardiomyocytes (Figure 1L–O), suggesting that PTPMT1-mediated CL biosynthesis is required for normal mitochondrial morphogenesis and cristae biogenesis in developing cardiomyocytes. Since cristae are recognized as fundamental structures to provide a sufficient area and proper spatial organization for oxidative phosphorylation and other membrane proteins in mitochondria4, abnormalities in cristae biogenesis could account for dysfunctional mitochondrial respiration in E11.5 CKO hearts.
The F1F0-ATP synthase dimers, MICOS complex, OPA1, and prohibitin proteins, have been proposed to participate in regulating mitochondrial cristae biogenesis5. ATP5I and ATP5L, two components of F1F0-ATP synthase, were downregulated in CKO hearts at E11.5. The dimerization of F1F0-ATP synthase complex was also impaired, while the assembly of complex monomer remained unaffected in CKO hearts at the same stage (Figure 1P–Q). Interestingly, deletion of PTPMT1 in cardiomyocytes impaired the assembly of MICOS complex in CKO hearts at E11.5 but did not alter the expressions of individual MICOS components (Figure 1R–S). Furthermore, we did not observe significant differences in the expressions of MFN1, MFN2, OPA1, DRP1, FIS1, and prohibitin proteins between control and CKO hearts at E11.5 (Figure 1T).
We also generated an inducible cardiac-specific Ptpmt1 knockout mouse model using αMHC-CreER. Induced deletion of PTPMT1 in adult mouse cardiomyocytes (iCKO) by tamoxifen reduced PG(34:1), the most abundant PG species, and increased PGP(34:1), the most abundant PGP species (Figure 1A, 1C), indicating that PTPMT1 is also responsible for catalyzing the conversion from PGP to PG in adult hearts. Although PTPMT1 deficiency also altered the levels of MLCL and DLCL (Figure 1B–C), the levels of total CL and individual CL species remained intriguingly unchanged (Figure 1B–C), and no morphological and functional changes were observed in iCKO hearts (Figure 1U–W).
Taken together, our results demonstrated an essential role of PTPMT1-mediated CL biosynthesis in regulating mitochondrial cristae morphogenesis in embryonic mouse cardiomyocytes and heart development. Our results also revealed a difference in not only the composition but also the metabolism of CL and CL-related metabolites between embryonic and adult hearts.
Sources of Funding
Drs J. Chen, Fang, and Patel are funded by grants from the National Heart, Lung, and Blood Institute of the US National Institutes of Health. Dr J. Chen holds an American Heart Association Endowed Chair in Cardiovascular Research. This work was also supported by the National Science Foundation of China (81970421 to KO), the Shenzhen Basic Research Foundation (JCYJ20190808174001746 to KO), the Shenzhen-Hong Kong Institute of Brain Science-Shenzhen Fundamental Research Institutions (2019SHIBS0004), the National Institutes of Health (HL091071 to HHP), and the Veterans Administration (BX001963 and BX005229 to HHP).
Conflict of Interest Disclosures:
JC consulted for and receives research funding from MyoKardia Inc.
References
- 1.Dudek J, Hartmann M, Rehling P. The role of mitochondrial cardiolipin in heart function and its implication in cardiac disease. Biochim Biophys Acta Mol Basis Dis. 2019; 1865: 810–821. [DOI] [PubMed] [Google Scholar]
- 2.Zhang J, Guan Z, Murphy AN, Wiley SE, Perkins GA, Worby CA, Engel JL, Heacock P, Nguyen OK, Wang JH, Raetz CR, Dowhan W, Dixon JE. Mitochondrial phosphatase PTPMT1 is essential for cardiolipin biosynthesis. Cell Metab. 2011; 13: 690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dorn GW, Vega RB, Kelly DP. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015; 29: 1981–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, Perales-Clemente E, Salviati L, Fernandez-Silva P, Enriquez JA, Scorrano L. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013; 155: 160–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pfanner N, Warscheid B, Wiedemann N. Mitochondrial proteins: from biogenesis to functional networks. Nat Rev Mol Cell Biol. 2019; 20: 267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]