

Abstract
Hydrogenation reactions in biology are usually carried out by enzymes with nicotinamide adenine dinucleotide (NAD(P)H) or flavin mononucleotide (FAMH2)/flavinadenine dinucleotide (FADH2) as cofactors and hydride sources. Industrial scale chemical transfer hydrogenation uses small molecules such as formic acid or alcohols (e.g. propanol) as hydride sources and transition metal complexes as catalysts. We focus here on organometallic half-sandwich RuII and OsII η6–arene complexes and RhIII and IrIII η5–Cpx complexes which catalyse hydrogenation of biomolecules such as pyruvate and quinones in aqueous media, and generate biologically important species such as H2 and H2O2. Organometallic catalysts can achieve enantioselectivity, and moreover can be active in living cells, which is surprising on account of the variety of poisons present. Such catalysts can induce reductive stress using formate as hydride source or oxidative stress by accepting hydride from NAD(P)H. In some cases, photocatalytic redox reactions can be induced by light absorption at metal or flavin centres. These artificial transformations can interfere in biochemical pathways in unusual ways, and are the basis for the design of metallodrugs with novel mechanisms of action.
Recent developments in transfer hydrogenation catalysis and photocatalysis in cancer cells by synthetic metal complexes are reviewed. They offer exciting new ways to modulate biochemical pathways for drug development and biotechnology.
Introduction
Metal complexes have long since attracted significant attention as catalysts for various chemical transformations.1–6 In most of these metal complex-catalyzed reactions, the conditions are well defined, such as concentration of catalyst and substrate, solvent, and temperature. Typically non-polar and non-aqueous solvents are used and reactions are generally carried out in the absence of any competitive reactants or catalysts.1–6 However, in spite of all the precautions, catalysts are often poisoned.7–10 On the other hand, nature very carefully uses enzymes with specific metal-ion active sites to catalyze a wide range of intracellular bio-chemical reactions essential for life.11,12 These metalloenzymes catalyse reactions in living cells in which the conditions and environment are extremely complicated.7,11,12 Metal ions such as iron, copper, zinc, and manganese are encapsulated by proteins, with engineered reaction sites which allow substrate recognition, a reactive ‘entatic/ecstatic’ state, and protection of the active metal ion against poisoning.11–13
There is interest in the design of artificial metalloenzymes involving a synergistic combination of enzymology and synthetic inorganic chemistry,14–18 but these artificial metalloenzymes are not very cost effective and generally require careful handling and storage.19 Low-molecular-weight metal complexes are attractive as mimics of metalloenzymes.7,11,19,20 However, without a large protein scaffold, small catalysts may not possess substrate specificity and intracellular nucleophiles might readily poison the active catalyst.7,11,19,20 However, recently, some progress has been achieved with transfer hydrogenation catalysis in cells by some second- and third-row transition metal low-spin d6 RuII, RhIII, IrIII and OsII complexes.21–38
Here we describe recent work on catalytic transfer hydrogenation in cells. Several enzymes are known to be involved in the reduction of NAD(P)+ to NAD(P)H, or oxidation of NAD(P)H to NAD(P)+ (Table 1).39–50 Intracellular NAD+/NADH inter-conversion has been achieved by numerous organometallic RuII, RhIII, IrIII, OsII complexes via a transfer hydrogenation pathway in the presence of a hydride donor,21–36 including catalytic reduction of pyruvate to lactate in cells,37 and in-cell photo-redox catalysis of NADH oxidation.51 We discuss these recent developments including detailed mechanisms of action, along with recent work on in-cell photo-catalytic reduction of metal complexes by flavines.20,52–55 Coenzymes NAD+/NADH play important roles in maintaining the intracellular redox balance and mitochondrial electron transport chain, and such in-cell transfer hydrogenation catalysts might find application in biotechnology and therapy, for example as catalytic anticancer drugs.
Some endogenous enzymes that transfer hydride to substrates.
| Enzyme | Reaction catalyzed | Ref. |
|---|---|---|
| Isocitrate dehydrogenase | Isocitrate + NAD+ ⇌ 2-oxoglutarate + CO2 + NADH + H+ | 39 |
| Isocitrate + NADP+ ⇌ 2-oxoglutarate + CO2 + NADPH + H+ | ||
| Oxoglutarate dehydrogenase complex | α-Ketoglutarate + NAD+ + CoA → Succinyl CoA + CO2 + NADH | 40 |
| Malate dehydrogenase | Malate + NAD+ → oxaloacetate + NADH + H+ | 41 |
| Alcohol dehydrogenases | CH3CH2OH + NAD+ → CH3CHO + NADH + H+ | 42 |
| NAD-dependent formate dehydrogenases | Formate + NAD+ ⇌ CO2 + NADH + H+ | 43 |
| NADH dehydrogenase | NADH + H+ + acceptor ⇌ NAD+ + reduced acceptor | 44 |
| NADH:ubiquinone reductase | NADH + H+ + ubiquinone ⇌ NAD+ + ubiquinol | 45 |
| Glucose dehydrogenases | Beta-d-glucose + NAD(P)+ ⇌ d-Glucono-1,5-lactone + NAD(P)H + H+ | 46 |
| Non-phosphorylating glyceraldehyde 3-phosphate dehydrogenase | Glyceraldehyde-3-phosphate + NADP+ + H2O → 3-phosphoglycerate + NADPH + H+ | 47 |
| Ferredoxin–NADP+ reductase | Reduced ferredoxin + NADP+ + H+ ⇌ oxidized ferredoxin + NADPH | 48 |
| Adrenodoxin reductase | NADPH + oxidized adrenodoxin → reduced adrenodoxin + NADP+ + H+ | 49 |
| NADPH oxidase | NADPH + 2O2 ⇌ NADP+ + 2O2− + H+ | 50 |
Enzymes that transfer hydride to substrates
Several enzymes are known to transfer hydride to NAD+ or NADP+ from a hydride source, Table 1.39–43,46–48 For example, isocitrate dehydrogenase transfers hydride to NAD+ from isocitrate,39 alcohol dehydrogenases transfer hydride from ethanol to NAD+.42 These bio-catalytic reactions are associated with the liberation of CO2 and are integral parts of cellular metabolism and the citric acid cycle.56 A few enzymes catalyse oxidation of NADH via a transfer hydrogenation, such as NADH:ubiquinone reductase which oxidizes NADH to NAD+ and transfers hydride to ubiquinone to generate ubiquinol.45 Interestingly, NADPH oxidase transfers hydride from NADPH to molecular oxygen.50 All these enzymes are important for maintaining the intracellular redox and proton balance.
Hydride sources in cells
In eukaryotic cells, the main direct sources of endogenous hydride are reduced nicotinamide adenine dinucleotide (NADH, Fig. 1), reduced nicotinamide adenine dinucleotide phosphate (NADPH, Fig. 1) and reduced flavin adenine dinucleotide (FADH2, hydroquinone form, Fig. 2).30,44,45,49,50,57–61 Their electrochemical potentials, intracellular concentrations and distributions are summarized in Table 2.
Fig. 1. Structures of NAD+/NADH and NADP+/NADPH.

Fig. 2. One- and two-electron redox reactions of flavins. The key region involved in electron/hydride transfer is highlighted in blue.

Electrochemical potential, intracellular concentration and distribution of the common hydride sources in cells.
| Hydride | Potential | Concentration | Distribution |
|---|---|---|---|
| NADH | E NAD+/NADH = −0.32 V | 100 to 200 μM | Cytosolic component: NADH shuttled from cytosol to mitochondria by malate-aspartate shuttle or glycerol 3-phosphate shuttle |
| NADPH | E NADP+/NADPH = −0.32 V | No definite range | Cytosol and mitochondria |
| FADH2 | E FAD/FADH2 = −0.22 V | No definite range | Cytosol and mitochondria |
In cells, NADH is often generated by the reduction of its oxidized form, NAD+ during metabolism and in the citric acid cycle by the enzymes isocitrate dehydrogenase, oxoglutarate dehydrogenase complex and malate dehydrogenase.39–41 Other enzymes such as alcohol dehydrogenases, NAD-dependent formate dehydrogenases (in methylotrophic yeast and bacteria) are also known to reduce NAD+ to NADH.42,43 NADH is an important hydride donor in cells and donates hydride to flavin mononucleotide (FMN) in complex I of the mitochondrial electron transport chain.57–60 NADH first binds to the hydrophilic domain of complex I which contains FMN, and then transfers two electrons to FMN, the prosthetic group of the NADH reductase,62,63 which is reduced to FMNH2. The electron acceptor is the isoalloxazine ring of FMN.64,65 NADH is oxidized to NAD+ by the enzymes NADH dehydrogenase and NADH:ubiquinone reductase, or during oxidative phosphorylation to generate ATP.44,45
Phosphorylation of NAD+ by NAD+ kinases leads to the synthesis of NADP+, which in turn is reduced to NADPH by glucose-6-phosphate dehydrogenase (G6PDH) in the first step of the pentose phosphate pathway.66 The conversion of NADP+ to NADPH is also carried out by enzymes such as isocitrate dehydrogenase (in the citric acid cycle), glucose dehydrogenases, non-phosphorylating glyceraldehyde 3-phosphate dehydrogenase (in plants, algae, and bacteria), transhydrogenases and ferredoxin–NADP+ reductase (in plants; involved in photosynthesis).39,46–48,67 NADPH is also an important hydride donor, for several biosynthetic reactions and the regeneration of glutathione.67 The enzyme adrenodoxin reductase, present in most common organisms, oxidizes NADPH to NADP+.49 In the plasma membrane and in the membranes of phagosomes, NADPH oxidase can oxidize NADPH.68,69
The other important endogenous hydride donors are FMNH2 and FADH2.61,70,71 whereas NADH and NADPH are involved only in two-electron (hydride) transfer, the flavins (FMNH2 and FADH2) can mediate both the one and two electron transfer processes as shown in the Fig. 2. Generally, during an enzymatic process, the oxidized (FMN/FAD), semiquinone (FMNH˙/FADH˙) and reduced (FMNH2/FADH2) forms undergo reversible interconversion (Fig. 2).71–76 FMNH2 donates hydride to the coenzyme Q10 (ubiquinone) and is oxidized back to FMN.76 whereas FADH2 donates hydride to complex II of the mitochondrial electron transport chain.77
The electron/H+ and hydride transfer processes for NAD(P)H and FMNH2 and enzymes are complex processes. For example, high-resolution X-ray crystallographic hydride transfer studies in the ferredoxin:NADP+ reductase (FNR) family reported by Kean et al. show the mobility of nicotinamide's C4 atom in the FNR:NADP+ complex, which results in the boat-like conformation of the nicotinamide ring. This conformational change in turn enhances hydride transfer.48
Small molecules as functional mimics of NADH have attracted significant attention for recharging cofactor-dependent enzymes, and understanding the pathways of naturally occurring biochemical reactions.78–83 For halogenation activity, tryptophan 7-halogenase needs FADH2 which is generated from the reaction of FAD with NADH by a flavin reductase.78 van Pée et al. catalytically regenerated FADH2 from FAD using the small organometallic ion [Cp*Rh(bpy)(H2O)]2+ as catalyst and formate as the electron donor.79 Sewald et al. employed the NADH mimics shown in Fig. 3 (compounds i–iv) to achieve chlorination of l-tryptophan using FAD-dependent halogenase.78 These NADH mimics take care of FADH2 regeneration from FAD.78 Scrutton and coworkers reported the excellent efficiency of the NAD(P)H mimics (iv–viii, Fig. 3) in Ene reductases catalyzed reactions.80 Compounds iv, v, vii–ix (Fig. 3) as synthetic NADH mimics for enhanced enoate reductase catalyzed reactions are reported by Hollmann et al.81 Interestingly, these mimics do not decrease the enzymatic activity or stereo-selectivity of the C C bio-reductions. Knox et al. used 1-carbamoylmethyl-3-carbamoyl-1,4-dihydropyridine (compound x in Fig. 3) as NADH mimic to activate the NAD(P)H quinone oxidoreductase 2 which finally activates cancer prodrug 5-(aziridin-1-yl)-2,4-dinitrobenzamide.82 These examples illustrate the potential for synthetic NADH mimic in bio-catalysis and biomedical applications.
Fig. 3. Structures of synthetic NADH mimics.78,80–82.

Transfer hydrogenation catalysis
Transfer hydrogenation catalysis by metal complexes involves transfer of hydride from a donor to an acceptor substrate via a metal-hydride intermediate.84–86 This reaction is well known in synthetic organic chemistry for reduction of C C, ketones (by Noyori's ruthenium arene complexes), and imines in non-aqueous media in the presence of hydride donors such as formate or isopropanol.86
Several RuII, RhIII and IrIII complexes can achieve regioselective reduction of NAD+ to 1,4-NADH in aqueous solution with sodium formate as the hydride source.87–93 In 1988, Steckhan et al. reported the regioselective reduction of NAD+ in aqueous media by [Rh(Cp*)(2,2′-bipyridine)(H2O)2]+via transfer of hydride from formate.87 They also elucidated the mechanism of NAD+ reduction by bipyridine-chelated Cp*RhIII complexes.87 Catalytic reduction of NAD+ by a series of phenanthroline-chelated RuII, RhIII and IrIII catalysts in aqueous media has been reported by Süss-Fink et al.90 Diamine Ru(ii)–arene complexes also catalyse NAD+ reduction in aqueous solution and isotope studies indicate that the formation of the Ru–H hyudride species is the rate-limiting step in the catalytic cycle.92,93
The establishment of transfer hydrogenation catalysis in aqueous solution under mild conditions provides a basis for extending the scope of catalysis to cells for the reduction of biomolecules such as NAD+ and pyruvate. This is challenging on account of the presence of numerous nucleophilic biomolecules and as well as oxidants and reductants, which might potentially poison the active catalyst.
In-cell catalytic reduction of NAD+
Coenzymes NAD+ and NADH control >400 enzymatic redox reactions which involve their inter-conversion.94–96 In cells, the conversion of NAD+ to NADH usually involves transfer hydride from a substrate to NAD+.38–43 In 2015, such reduction was achieved in living cells using arene RuII Noyori-type transfer hydrogenation catalysts (Fig. 4a) containing a chelated sulfonamidoethylenediamine ligand co-incubated with the hydride donor, formate to reduce NAD+ to NADH.21 In MeOH-d4/D2O (2 : 9 v/v) or in D2O, complexes 1–4, catalytically and regioselectively reduce NAD+ to NADH, as was evident from the 1H NMR analysis. Higher catalytic activity (higher turnover frequency, TOFs) was observed with the more electron-withdrawing sulfonamides (NbEn (4) > TfEn (3) > TsEn (2) > MsEn (1)) (Fig. 4b). Complexes 5–7 gave rise to extremely fast NAD+ reduction and the reactions were completed before the first 1H NMR spectrum could be recorded indicating that the o-terphenyl (o-terp) complexes were more catalytically active than the p-cym complexes. Aquation of the complexes (replacement of Cl by H2O, Fig. 4a) was very fast. The Ru–H intermediate was detectable by 1H NMR with a peak at −5.5 ppm and existence of the formate adduct was confirmed by the mass spectral analysis. Ru–H formation occurs in three steps (i) the initial Ru–Cl bond hydrolyses, (ii) formate binds to RuIIvia a carboxylate oxygen, and (iii) formate re-orientates to facilitate transfer of hydride to Ru(ii) with the release of CO2.
Fig. 4. (a) Structures of sulfonamidoethylenediamine RuII transfer hydrogenation catalysts 1–8.21 (b) Turnover frequencies (TOF) for NAD+ reduction in MeOH-d4/D2O (5 : 1 v/v) by complexes 1–4 determined by 1H NMR ([Ru complex]: 0.44 mM; [NAD+]: 0.88 mM; [formate]: 11.02 mM; molar ratio 1 : 2 : 25).21.

The complexes showed anticancer activity against A2780 human ovarian cancer cells with the half maximal inhibitory concentration (IC50) values in the range of 2.2–21.2 μM. When co-administrated with formate, the anticancer profile of complexes 1–7 improved (Fig. 5a). Such an effect was not observed with acetate, which is not a hydride donor (Fig. 5a). The extent of lowering of IC50 values (increase in cytotoxicity) was directly proportional to the formate concentration (Fig. 5b), suggesting a direct contribution of catalytic transfer hydrogenation to the anticancer profile. Such a direct contribution was confirmed for complex 2 which significantly decreased the intracellular NAD+/NADH ratio when cells were co-incubated with non-toxic doses of formate (Fig. 5c). Moreover, formation of coenzyme NADH induced reductive stress, a new and unusual mechanism of action for an anticancer agent, was observed. Thus this mechanism might be effective for overcoming cisplatin resistance, a major clinical problem.
Fig. 5. (a) Percentage survival of A2780 human ovarian cancer cells co-incubated with complex 2 and various concentrations of sodium acetate or sodium formate. (b) IC50 values of complex 2 when co-incubated with sodium formate and the intracellular uptake of Ru under similar conditions. (c) Linear correlation of sodium formate concentration and the NAD+/NADH ratio when co-administered with complex 2. Figure reproduced from ref. 21; J. J. Soldevila-Barreda et al., Nat. Commun., 2015, 6, 6582, published by Springer Nature.

To establish structure–activity relationships, we studied six pseudo-octahedral neutral RuII sulfonamidoethylenediamine complexes of the type [(η6-p-cym)Ru(N,N′)Cl] (9–14) (Fig. 6a).22
Fig. 6. (a) Structures of sulfonamidoethylenediamine RuII transfer hydrogenation catalysts 9–14.22 (b) Turnover frequencies (TOF) for reduction of NAD+ by formate catalyzed by complexes 9–14, determined by UV-vis spectroscopy (84 μM complex in MeOH/H2O 1 : 9 v/v, 102 mM sodium formate and 510 μM NAD+ in H2O).22.

These complexes catalyse the reduction of NAD+ to 1,4-NADH regioselectively when formate is used as the hydride donor. The catalytic activity is highly dependent on the electronic and steric effects of the N-substituent, the bulkier the substituent, the higher the rate of reduction (Fig. 6b). The rate of NAD+ reduction is dependent on pH* (deuterated solvent) and was highest between pH* 6–7.5. An increase in formate concentration increased the rate of reduction.
DFT studies indicated a mechanism involving the initial replacement of an aqua ligand by formate, followed by H− transfer to RuII and finally to NAD+. Furthermore, specific interactions between the NAD+ and the aqua complex were evident from the modelling and probably allow a pre-organisation via interaction of the aqua ligand, formate and the pyridine ring of NAD+ (Fig. 7). The complexes showed antiproliferative activity towards human ovarian cancer cells, which was further increased by 20–36% on co-administration with 2 mM sodium formate.
Fig. 7. Catalytic cycle for NAD+ reduction by the complexes 9–14 based on DFT modelling. Reproduced from ref. 22; F. Chen et al., Dalton. Trans., 2018, 47, 7178, published by The Royal Society of Chemistry.

Half-sandwich and tethered RuII complexes where the diamine ligand and the η6–arene ring are directly connected, provide control over the spatial positions of ethylenediamine substituents which results in extra stability of the complexes,97,98 are also transfer hydrogenation catalysts for reduction of ketones and imines.99,100 The neutral tethered RuII complexes [Ru(η6-Ph(CH2)3-ethylenediamine-N-R)Cl] where R is methanesulfonyl (Ms, 15), toluenesulfonyl (Ts, 16), 4-trifluoromethylbenzenesulfonyl (Tf, 17), and 4-nitrobenzenesulfonyl (Nb, 18), (Fig. 8a)23 are potent transfer hydrogenation catalysts for the reduction of NAD+ to NADH with formate as hydride donor both in aqueous solution (TOFs/h = 3.8–10) and in cancer cells. In aqueous media, the reduction can be monitored by following the absorbance of NADH at 340 nm using UV-visible spectroscopy.101 Substituents on the ethylenediamine ligand control the catalytic activity, and the turnover frequency decreases in the order Nb (18) > Tf (17) > Ts (16) > Ms (15) (Fig. 8b) again showing that more strongly electron-withdrawing groups enhance hydride transfer.
Fig. 8. (a) ORTEP diagrams of the X-ray crystal structures of tethered RuII catalysts 15–18 with sulfonyl substituents on the diamine. Figure reproduced from ref. 23; F. Chen et al., Organometallics 2018, 37, 1555, published by The American Chemical Society. (b) TOFs for complexes 15–18 for NAD+ reduction in MeOH-d4/D2O (1 : 9 v/v) determined by UV-vis spectroscopy (final concentrations: Ru complex 28 μM, NAD+ 170 μM, NaHCO2 34 mM, molar ratio 1/6/1200)).23 (c) Cell viability of A2780 cancer cells when incubated with complexes 15–18 (at equipotent 1/3 × IC50 concentrations) and various sodium formate concentrations (0, 0.5, 1.0, and 2.0 mM) for 24 h. The figure is reproduced from ref. 23; F. Chen et al., Organometallics 2018, 37, 1555, published by The American Chemical Society.

These complexes are moderately cytotoxic to human lung (A549), ovarian (A2780), breast (MCF7) and hepatocellular (HEPG2) cancer cells. Interestingly, up to 22% enhancement of cytotoxicity of the complexes was observed on co-incubation with non-toxic doses of sodium formate (0.5–2 mM) (Fig. 8c) indicating that reduction of intracellular NAD+ may contribute significantly to the anticancer activity.
Ruthenium(ii)–arene complexes with bidentate Schiff base ligands (19a, 19bFig. 9) or their reduced analogues (20a and 20b) also have anticancer activity and an ability to reduce NAD+.24 In comparison to the Schiff base complexes, the corresponding amine complexes exhibit improved anticancer activity against various human cancer cell lines, as well as higher rates of catalytic NAD+ reduction. This study shows that simple ligand modifications (reduction of an imine) can significantly alter both the biological and catalytic activities.
Fig. 9. Ruthenium arene complexes 19 and 20.24.

RhIII complexes can catalyse transfer hydrogenation for NAD+ reduction to NADH using formate as the hydride source, under biologically-relevant conditions, for example [(Cpx)Rh(N,N′)(Cl)]+ (21–30, Fig. 10a), where N,N = ethylenediamine (en), 2,2′-bipyridine (bpy), 1,10-phenanthroline (phen) or N-(2-aminoethyl)-4 (trifluoromethyl)benzenesulfonamide (TfEn), and Cpx = Cp*, 1-phenyl-2,3,4,5 tetramethylcyclopentadienyl (CpxPh) or 1-biphenyl-2,3,4,5-tetramethyl cyclopentadienyl (CpxPhPh).25
Fig. 10. (a) Rh(iii) catalysts 21–30 for the reduction of NAD+ in the presence of sodium formate as hydride source.25 (b) TOFs for reduction of NAD+ by formate, catalyzed by Cp* RhIII complexes 21, 24 and 27 in MeOH-d4/D2O (1 : 4 v/v) determined by 1H NMR (6–9 mol equiv. NAD+ and 25 mol equiv. formate).25.

The structure activity relationship showed that the N,N-chelated ligand can control the catalytic activity which decreased in the order of bpy (24) > phen (27) > en (21) as shown in the Fig. 10b. [Cp*Rh(bpy)Cl]+ (24) was the most efficient catalyst with a TOF of 37.4 ± 2 h−1 in aqueous solution. Interestingly, complexes 21–29 were able to catalytically reduce pyruvate to lactate using formate as the hydride donor. Preference for the reduction of NAD+ over pyruvate was also observed. Remarkably, when co-incubated with non-toxic doses of formate, the anticancer activity of complex 23 in A2780 cancer cells increased by ca. 50%, indicating that transfer hydrogenation may induce reductive stress in cancer cells.
Catalytic oxidation of intracellular NADH
The catalytic reduction of intracellular NAD+ to NADH in the presence of a hydride donor can lead to the alteration of intracellular redox homeostasis and cell death. The reverse process, oxidation with NADH as the hydride donor is also achievable both in aqueous solution and in cells.102–105 For example in aqueous solution, [C,N]-cyclometallated Ir(iii) complexes can convert NADH to NAD+ under weakly basic conditions.102 Also [IrIII(Cp*)(4-(1H-pyrazol-1-yl-κN2)benzoic acid-κC3)(H2O)]2SO4 catalyses the reduction of 2,3-dimethoxy-5-methyl-1,4-benzoquinone (Q0), an ubiquinone coenzyme analogue, by NADH to the corresponding reduced form (Q0H2) in aqueous solution at room temperature.103 Also [Cp*Ir(pica)Cl] where pica is picolinamidate = κ2-pyridine-2-carboxamide, catalyses the dehydrogenation of β-NADH in slightly acidic aqueous solution,104 not only to the expected β-NAD+ but also α-NAD+, nicotinamide and 1,2,5,6-tetrahydronicotinamide.104
Half-sandwich cyclopentadienyl iridium(iii) and arene ruthenium(ii) complexes [(Cpx)Ir(phen)(H2O)]2+ (31, 32) and [(arene)Ru(N,N′)Cl]+ (33–37) (Fig. 11a) can reduce ketones using NADH as the hydride source.26 Moreover, under physiological conditions, these complexes reduce pyruvate to lactate, a reduction carried out by the enzyme lactate dehydrogenase in vivo using NADH as a cofactor.106 The Ir(iii) complexes [(Cp*/CpxPh)Ir(phen)(H2O)]2+ (31, 32) behave as robust catalysts for H2 generation from NADH. Under physiologically relevant conditions (pH 7.4, 310 K), a TON of 75 after 24 h and a TOF up to 4.3 h−1 were achieved. Complex 32 almost doubled the NAD+/NADH ratio level in A2780 cancer cell lysates indicating that the complex can modulate the intracellular redox balance, and that catalytic oxidation of NADH can be achieved in cells by transfer hydrogenation catalysts.
Fig. 11. Cyclopentadienyl IrIII (31, 32) and arene RuII transfer hydrogenation catalysts (33–37) for NADH oxidation.26 Proposed mechanism for transfer of hydride from 1,4-NADH to (b) RuII and (c) IrIII complexes involving nicotinamide carbonyl coordination and arene/Cp ring slippage.26.

DFT modelling suggests that the mechanism for the oxidation of NADH to NAD+ involves transfer of hydride from NADH to the RuII/IrIII centre via formation of a six-membered transition state, by a coordination site which becomes vacant by a ring-slippage mechanism (Fig. 11b and c).107,108 Notably the formation of both Ru–H and Ir–H was detected by 1H NMR, at −7.44 ppm and −11.3 ppm, respectively.
Organoiridium(iii) complexes [(Cpxbiph)Ir(phpy)(Cl)] (38) and [(Cpxbiph)Ir(phpy)(py)]+ (39) (Fig. 12a) where Cpxbiph = biphenyltetramethylcyclopentadienyl, phpy = phenylpyridine and py = pyridine, can transfer the hydride from NADH via iridium-hydride intermediates to O2, a possible route for intracellular H2O2 generation.27 The activity of these complexes is analogous to that of NADPH oxidase which transfers hydride from NADPH to O2 in cells.50 These complexes were active against a wide range of cancer cell lines in the National Cancer Institute NCI-60 human cancer cell screen.
Fig. 12. (a) IrIII phenylpyridine transfer hydrogenation catalysts 38 and 39.27 (b) Detection of H2O2 in a solution of 39 + NADH in MeOH/H2O (3 : 7, v/v) at 310 K by Quantofix peroxide test sticks. (c) Proposed catalytic circle for the production of H2O2 by 38 and 39. Reproduced from ref. 27 with permission.

Hydride transfer from NADH to Ir(iii) was much slower for pyridine complex 39 than for chlorido complex 38, attributable to slower hydrolysis rate of 39. Importantly, H2O2 generation was also detected from the appearance of a blue colour on a H2O2 test stick in the presence of NADH (Fig. 12b) indicating transfer of hydride from NADH to O2via an Ir–H intermediate (Fig. 12c) detectable by 1H NMR.
Hydride can also be transferred from NADH to quinones by the Ir(iii) complexes [(Cp*)Ir(phen)(H2O)]2+ (40), and [(Cpxph)Ir(phen)(H2O)]2+ (41) (Fig. 13a).28 Quinones (Q) have important roles carrying electrons in the mitochondrial electron-transport chain.109,110 Quinones undergo one- or two-electron reduction to the corresponding semiquinones (QH˙) or hydroquinones (QH2), respectively (Fig. 13b). In cells, quinones are generally reduced by coenzyme NAD(P)H in the presence of either NADH ubiquinone oxidoreductase or NADPH cytochrome P-450 reductase, or NADH cytochrome b5.73,111–113 Complex 41 reduces duroquinone and menadione (vitamin K3) (Fig. 13c) with a TON of 56.6 and TOF of 12.4 h−1 (in phosphate buffer at pH 7.2) to the corresponding semiquinone radicals, which were detected by EPR.
Fig. 13. (a) IrIII TH catalysts 40 and 41 for the reduction of quinones. (b) One and two electron reduction of quinones leading to semiquinone and hydroquinone, respectively. (c) Structures of menadione and duroquinone.28 (d) Catalytic cycle for the reduction of quinones by the NADH/IrIII system. Reproduced form ref. 28 with permission.

The reduction of quinones by NADH occurs via generation of Ir–H intermediates. DFT calculations suggested two one-electron transfers, the first to form the semiquinone and a transient IrII state, which in turn transfers a second electron to a second quinone, forming the second quinone radical with the regeneration of active IrIII species (Fig. 13d). IrII is a relatively rare oxidation state, but has been reported in the literature.114–116 As a consequence, such organometallic complexes have the potential to convert the two-electron reducing power of NADH into two subsequent one-electron steps. This opens up new ways of diverting biochemical pathways in cells.
Interestingly Do et al., transferred hydride from NADH to aldehydes in PBS or cell culture media (RPMI-1640 or M199) of pH 7.4 at 37 °C, even in the presence of various biomolecules (e.g. nucleobases, amino acids, small peptides, carbohydrates) and metal ions (bio-relevant transition metals ions and alkali/alkaline-earth metal ions) using Cp* Ir(iii) complexes of pyridinecarboxamidates (42–46) (Fig. 14a).29 These complexes were active in hydrogenation of cytotoxic aldehydes responsible for various diseases.
Fig. 14. (a) IrIII TH catalysts 42–46 which reduce aldehydes in PBS and cell culture media using NADH as hydride source.29 (b) Organometallic IrIII catalysts (47, 48) for intracellular conversion of aldehydes to alcohols.30 (c) The fluorogenic BODIPY substrate (BODIPY-CHO) and its reduced form (BODIPY-OH). (d) Confocal microscope images of NIH-3T3 cells treated with (A) BODIPY-CHO (30 μM); (B) BODIPY-OH (30 μM) and (C) BODIPY-CHO (30 μM) + 47 (20 μM). Reproduced from ref. 30 with permission.

The intracellular conversion of aldehydes to alcohols in living cells can be achieved using Ir(iii) transfer hydrogenation catalysts 47 and 48 and endogenous NADH as the hydride donor (Fig. 14b).30 The reduction can be monitored in real time using a fluorogenic BODIPY-CHO substrate (Fig. 14c) and confocal microscopy (Fig. 14d). BODIPY-CHO is not highly fluorescent, but when reduced to BODIPY-OH by intracellular transfer hydrogenation, it becomes strongly fluorescent (Fig. 14c). Such biocompatible reductive chemistry may provide new biotechnological approaches and novel intracellular bio-conjugation strategies.117
Recently Liu et al. reported a mitochondria-targeting Ru(ii) complex 49 (Fig. 15a) which showed cytotoxicity against A549 cells via NADH oxidation and activation of mitochondrial membrane potential depolarization.31 Complex 49 induced overproduction of intracellular ROS possibly by transferring hydride from NADH to O2. This complex induced cell apoptosis and arrested the cell cycle at the G0/G1 phase by cyclin-dependent kinase 4/cyclin D1 inactivation.31 They also reported a new class of IrIII complexes (50–52, Fig. 15a) of N-heterocyclic carbenes (NHCs) with transfer hydrogenation ability.33 Interestingly, the TON of the NHC complex 50 was 2× times higher than the previously discussed C^N analogue, complex 38.27 This may be due to the strong electron-donating ability of the carbenes in complex 50 compared to the C^N ligand in 38, which can effectively labilize the Ir–Cl bond towards hydrolysis, the activation step of the catalysis.
Fig. 15. (a) The IrIII iminopyridine and cis-carbene transfer hydrogenation catalysts (49–52) for NADH oxidation.31,33 (b) Cyclometalated RhIII benzo[h]quinoline complexes (53–58).36.

The introduction of N,C-chelated ligands can increase the potency of Rh(iii) transfer hydrogenation catalysts for NADH oxidation as in [CpXRh(C^N)Z]0/+ (53–58), where CpX = Cp*, Cpph, or Cpbiph, C^N = benzo[h]quinoline, and Z = chloride or pyridine (Fig. 15b).36 Complex 55 was the most efficient catalyst (TON = 58 and TOF/h = 7.6 in 1.6% MeOH/98.4% phosphate buffer (5 mM, pH 7.4) over 24 h at 310 K) for NADH oxidation and moreover increased the ROS level in A549 lung cancer cells and resulting cytotoxicity. Interestingly, the chlorido complexes 53–55 showed ca. 2–4 times higher catalytic activity than their respective pyridine analogues 56–58. Such a difference in reactivity is due to the much slower hydrolysis of pyridine complexes compared to chloride analogues; hydrolysis is believed to be the activation step of the catalytic process.
Organo-osmium catalysts
Pyruvate is an important intermediate in metabolic pathways in cells.118 In a 3-step process, pyruvate is converted to acetyl-coenzyme A, which generates energy in the Krebs cycle.118,119 Hence disturbance of pyruvate metabolism would be expected to generate metabolic disorder. Chiral half-sandwich arene Os(ii) sulfonamidoethylenediamine complexes of the type [Os(arene)(TsDPEN)] where TsDPEN is N-(p-toluenesulfonyl)-1,2-diphenylethylenediamine (Fig. 16) can catalyse enantioselective reduction of pyruvate.37 These 16-electron catalysts have been synthesized as enantiomerically pure compounds by a microwave method as shown in Fig. 16.37,38
Fig. 16. Synthetic route for OsII arene sulfonamidoethylenediamine catalysts 59–65 for pyruvate reduction.

These 16e catalysts are stable as solids or in solution (including aqueous media), unlike the Ru(ii) analogues which have to be generated from 18e pre-catalysts before use. The Os(ii) complexes reduce acetophenone 3.5 × faster than the Noyori Ru catalyst37 in formic acid/triethylamine (5 : 2) azeotrope. They also reduce pyruvate to lactate in the presence of sodium formate as the hydride source via transfer hydrogenation in which Os–H is the key intermediate. The turnover frequency of catalysis is highly dependent on formate concentration compared to pyruvate concentration. Interesting, the asymmetric activity of the catalysts is retained with higher enantiomeric excess (83%).
The complexes show moderate to good antiproliferative activitiy against A2780 lung cancer cells (IC50= 4–30 μM), with no significant difference in activity between the enantiomers (R,R or S,S TsDPEN). Non-toxic concentrations of sodium formate potentiate the anticancer activity of the complexes indicating that the complexes might act as transfer hydrogenation catalysts for pyruvate reduction in cells. Lactate dehydrogenase reduces cytosolic pyruvate to l-lactate in cells.106 Remarkably, the R,R enantiomer of the complexes increased the d-lactate concentration in cells when they were co-incubated with sodium formate, suggesting that the catalysts can carry out enantioselective transfer hydrogenation of pyruvate in cells. Since cell survival was not significantly influenced by co-administration of osmium catalysts and formate to non-cancerous cells, pyruvate may be new cellular target for design of the next generation of anticancer drugs.
Photo-catalytic oxidation of NADH
Photo-catalysis can achieve novel chemical transformations with high yields of products and high reaction specificity.120–122 The stable cyclometalated luminescent Ir(iii) catalyst [Ir(ttpy)(pq)Cl]PF6 (66, Fig. 17a), containing tridentate ttpy = 4′-(p-tolyl)-2,2′:6′,2′′-terpyridine, and bidentate pq = 3-phenylisoquinoline ligands, synthesized by treating [Ir(ttpy)Cl3] with excess of 3-phenylisoquinoline in glycol under N2, can oxidise NADH photocatalytically in cells.51 DFT calculations indicated that the trans C–Cl isomer found in the crystal structure (Fig. 17b) is significantly more stable than the cis C–Cl isomer.51
Fig. 17. (a) Line structure, and (b) X-ray crystal structure of photocatalyst complex 66; the counter ion PF6− is omitted for clarity. Reproduced from ref. 51 with permission. Catalytic cycle for photo-oxidation of NADH by complex 66; (c) under normoxia, and (d) under hypoxia in the presence of Fe3+–cyt c as the terminal electron acceptor.51.

Photo-stable complex 66, has an extremely high excited-state reduction potential (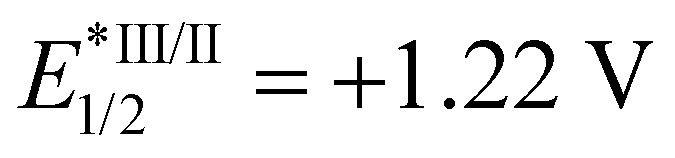 versus the saturated calomel electrode, where E1/2 is the half-wave potential), and photo-catalytically oxidises NADH to NAD+via NAD˙ radical formation with a high turnover frequency under normoxia. Molecular O2 plays an important role in regenerating the active Ir(iii) catalyst from the Ir(ii) state and is converted to the reactive oxygen species H2O2 (Fig. 17c). In DFT models, the chelated ligands ttpy and pq can π-stack with NADH, positioning the triplet-excited-state hole close to an NADH electron-donor site. Moreover, this complex catalytically reduces cytochrome c in the presence of NADH under hypoxic conditions (Fig. 17d).
versus the saturated calomel electrode, where E1/2 is the half-wave potential), and photo-catalytically oxidises NADH to NAD+via NAD˙ radical formation with a high turnover frequency under normoxia. Molecular O2 plays an important role in regenerating the active Ir(iii) catalyst from the Ir(ii) state and is converted to the reactive oxygen species H2O2 (Fig. 17c). In DFT models, the chelated ligands ttpy and pq can π-stack with NADH, positioning the triplet-excited-state hole close to an NADH electron-donor site. Moreover, this complex catalytically reduces cytochrome c in the presence of NADH under hypoxic conditions (Fig. 17d).
Interestingly, the photosensitizer complex 66 exhibited high immunogenic apoptotic phototoxicity under both normoxia and hypoxia (IC50ca. 1–8 μM), whilst having low toxicity both in the dark (IC50 17–50 μM) and towards normal MRC-5 human lung fibroblasts and LO2 human hepatocyte cells. Complex 66 localized in mitochondria (Fig. 18a), where both NADH and cytochrome c play crucial roles in electron transport, and induced intracellular NADH depletion upon light irradiation (Fig. 18b). In contrast to current photosensitizers, complex 66 generates ROS, decreases the mitochondrial membrane potential, and depletes intracellular NADH under both normoxia and hypoxia. Complex 66 also showed high photocytotoxicity on two-photon red light irradiation (760 nm, 12 J cm−2) against A549 multicellular cancer spheroids, a model for solid tumours.
Fig. 18. (a) Confocal microscopy images showing mitochondrial localization of the photosensitiser complex 66. (b) NADH concentration in A549 cells treated with complex 66 with or without photo-irradiation. Reproduced from ref. 51 with permission.

In view of the therapeutic resistance of hypoxic tumours,123–125 this catalyst might provide the basis for a new generation of hypoxia-active anticancer agents. Furthermore, its mitochondrial targeting ability has the potential to bypass nucleotide excision repair (NER), one of the factors responsible for acquired drug resistance of Pt chemotherapeutics, as NER is not involved in the repair of mitochondrial damage.126
The NAD+/NADH redox couple is emerging as the new target for next-generation anticancer drugs.21–28,31–36,51 Considering the vital role of these coenzymes in mitochondrial electron transport chain, cell metabolism, enzymatic reactions and several other biochemical pathways, changes of the intracellular NAD+/NADH ratio can ultimately lead to cell death.21–28,31–36,51 Cancer cells with their disturbed mitochondrial functions are highly sensitive to intracellular redox balance changes, especially those caused by changes of the NAD+/NADH ratio.51 Thus either intracellular NAD+ reduction or NADH oxidation by transfer hydrogenation catalysis might provide a novel way to achieve tumor targeting anticancer activity, and remains to be further investigated.
In-cell photo-catalytic reduction by flavins
Flavins (FAD or FMN), can be reduced upon light activation in the presence of a sacrificial electron donor such as EDTA.127,128 Flavin-coupled systems are widely employed to achieve both oxidation and reduction for numerous organic transformations.129 The general mechanism of such reactions involves photoactivation of flavins to an excited state which can extract electrons from a sacrificial electron donor. The photo-generated reducing equivalent can now reduce and activate various molecules, including pro-drugs.
Riboflavin (Rf) can catalyze the photo-reduction of PtIV prodrugs to active cisplatin under physiologically relevant conditions.52 The two-electron-reduced flavin, formed after accepting electrons from a sacrificial electron donor (MES; (2-(N-morpholino)ethanesulfonic acid buffer), is the active catalyst (Fig. 19).52 The active catalyst appears to form a transient adduct with the PtIV prodrug and this catalyst–substrate interaction facilitates the transfer of electrons to the PtIV centre (Fig. 19). Importantly, this chemistry can be translated from reaction flasks to cells. The Rf/67 pair shows dose-dependent light- (460 nm, 0.36 J cm−2) induced anticancer activity against PC-3 (human prostate cancer) cells, with comparable anticancer activity to cisplatin in the dark. All the four components viz., Rf, complex 67, MES and light are necessary for activity towards PC-3 cells. Cisplatin-like apoptosis was observed as the mode of cell death. Moreover, such a Rf–PtIV photocatalyst–substrate pair was effective as a photoactive anticancer agent against pancreatic cancer cells.53
Fig. 19. Photo-catalytic cycle for reduction of Pt(iv) prodrug 67 by riboflavin (Rf) upon 460 nm light irradiation. Reproduced from ref. 52; S. Alonso-de Castro et al., Chem. Sci., 2017, 8, 4619, published by The Royal Society of Chemistry.

Interestingly, flavoproteins and flavoenzymes can be used as photocatalysts to convert PtIV prodrugs to active PtII complexes by photoreduction as well as RuII prodrugs to active RuII active species by release of a monodentate ligand, just like the flavins alone do (Fig. 20).54 The flavoprotein mini singlet oxygen generator (miniSOG) and NADH oxidase (NOX) photo-catalytically reduce PtIV prodrugs in the presence of electron donors (e.g. MES, NADH) and irradiation with low doses of visible light (460 nm, 6 mW cm−2). Remarkably, NOX, in the presence of NADH as the electron donor, catalyzes PtIV activation even in the dark indicating that flavoenzymes themselves may activate PtIV pro-chemotherapeutics using endogenous NADH as the electron source.
Fig. 20. (a) Flavins and flavoproteins for the catalytic photoactivation of anticancer metal complexes and (b) their selected reactions. Reproduced from ref. 20 and 54 with permission.

Concluding remarks
Metals are in the active sites of about 40% of all enzymes.13 Achieving catalytic reactions similar to those achieved by natural metalloenzymes in cells by synthetic metal-based catalysts is highly challenging on account of the lack of features enjoyed by natural metalloenzymes, including a protein scaffold able to select out substrates from the complicated chemical environment, and a means to shield the active metal centre from inhibitors (poisons). However, even natural metalloenzymes have to cope with some degree of mis-match in substrate selection, and poisoning, which can result in their degradation. Indeed mechanisms to switch the activity of metalloenzymes on and off are vital for the regulation of biochemical pathways. Also not all natural metalloenzyme centres are well shielded by such surrounding scaffolds, e.g. Mg2+ in ribozymes. Small metal complexes can be viewed as potential metalloenzyme mimics, but equally interesting is the potential for small metal catalysts to carry out unnatural reactions in cells that might lead to new applications in biotechnology and medicine.
Here we have focused on recent advances in the design of metal complexes which catalyse transfer hydrogenation reactions. Such catalysts are widely used on a large scale in the chemicals industry. Examples are chiral tosyldiphenylethylenediamine Ru(ii) (Noyori) catalysts for the reduction of ketones and imines to alcohols and amines using e.g. isopropanol as a hydride source. The challenge is to translate such catalysis from non-aqueous solvents into the aqueous media of biological cells.
There is now a range of organometallic complexes in the general class of half-sandwich organometallic Ru(ii), Os(ii) arenes and Rh(iii), Ir(iii) cyclopentadienyl complexes which can catalyse transfer hydrogenation in aqueous media, typically using formate as hydride donor. Formate is relatively nontoxic to cells and is a natural metabolite. Importantly, complexes such as 1–30 can be tailored so as to accept hydride from formate and subsequently reduce NAD(P)+, or alternatively for e.g. complexes 31–58 to accept hydride from NAD(P)H and generate NAD(P)+ and reduce a substrate such as a quinone or O2. Moreover, complex 32 can convert NADH into a sequential one electron donor to quinones via a reduced Ir(ii) intermediate. The Os(ii) complexes 59–65 have higher catalytic efficiency than their Ru(ii) analogues and are more stable. They can achieve asymmetric hydrogenation in cells and convert pyruvate into unnatural d-lactate. The photocatalyst 66 is stable in the dark, but can extract one electron from NADH on irradiation with visible light both in solution and in cancer cells providing novel impetus in photo-activated cancer drug development with spatiotemporal control over drug activation.
Our review illustrates that metal complex-mediated transfer hydrogenation catalysis can successfully be achieved in cells. Catalytic efficiencies of such catalysts can be tuned with a judicial selection of ligand systems. Such catalysis leads to the reduction of NAD+/pyruvate or oxidation of NADH and can potentially create redox imbalance in cells or metabolic disorder – either of which is highly useful to achieve anticancer activity. Moreover, synthetic NADH mimics can also be used for certain bio-catalytic reactions in cells, and are promising for use in biomedical research on drug development. Flavins and flavoproteins can activate cancer prodrugs in a bio-friendly approach for tumour targeting cancer therapy. Thus, in-cell catalysis might provide promising catalytic anticancer drug development strategies, as is evident from the initial in vitro data. Moreover such catalysts can also transfer hydride from hydride sources to organic electrophiles such as quinones or aldehydes. This in-cell transfer hydrogenation strategy is expected to be useful in new biotechnologies and novel intracellular bio-conjugation methodology.
These promising in-cell catalyses pave the way for more detailed investigations on small metal catalysts to (i) achieve substrate specificity in cells, (ii) improve the lifetime of the catalysts (towards deactivation after a few cycles), (iii) discover other biocompatible hydride donors, (iv) determine how the distribution and compartmentalization of the catalyst in cells (e.g. amongst the cellular organelles and membranes) influences its performance, and importantly (iv) identify applications which can be validated both in vitro and in vivo.
Conflicts of interest
There are no conflicts of interest to declare.
Supplementary Material
Acknowledgments
We thank the ERC (grant no. EP/P030572/1), Wellcome Trust (grant no. 107691/Z/15/Z), Anglo American Platinum, and Royal Society (Royal Society-SERB Newton International Fellowship, NF151429, for S. B.) for their support for our research. PJS thanks all his past and present group members, and collaborators, who have contributed to the original research articles.
Biographies
Biography
Samya Banerjee .

Dr. Samya Banerjee received his PhD in 2015 from the Department of Inorganic and Physical Chemistry, Indian Institute of Science, Bangalore, under the supervision of Prof. Akhil R. Chakravarty. Subsequently he was a postdoctoral fellow at Johns Hopkins University, USA with Prof. Marc M. Greenberg and a Royal Society-SERB Newton International Fellow (2017–2019) at the University of Warwick in the laboratory of Prof. Peter J. Sadler. Now he is working as a postdoc at the Institute of Inorganic Chemistry, Georg-August-Universität Göttingen, Germany with Prof. Dr Herbert W. Roesky. He has recently been awarded an Inspire Faculty Fellowship by the government of India to develop his independent academic career. His research interests include the development of next-generation metal-based anticancer drugs with novel mechanisms of action, mechanistic studies of clustered lesions in nucleosome core particles, and radicals of heavier group 13 elements.
Biography
Peter J. Sadler .

Prof. Peter J. Sadler obtained his BA, MA and DPhil at the University of Oxford. Subsequently he was a MRC Research Fellow at the University of Cambridge and National Institute for Medical Research. From 1973–1996 he was Lecturer, Reader and Professor at Birkbeck College, University of London, and from 1996–2007 Crum Brown Chair of Chemistry, University of Edinburgh. Then he became Head of the Department of Chemistry at the University of Warwick, where he is now a Professor. He is a Fellow of the Royal Society of Chemistry (FRSC), Royal Society of Edinburgh (FRSE) and the Royal Society of London (FRS), an EPSRC RISE Fellow (Recognizing Inspirational Scientists and Engineers), a Fellow of the European Academy of Sciences, and Honorary Fellow of the Chemical Research Society of India, and the Chinese Chemical Society. His research is focused on the chemistry of metals in medicine, and in particular on organometallic and photoactivatable anticancer complexes.
References
- Deuss P. J. Barta K. From models to lignin: Transition metal catalysis for selective bond cleavage reactions. Coord. Chem. Rev. 2016;306:510–532. doi: 10.1016/j.ccr.2015.02.004. [DOI] [Google Scholar]
- Hayler J. D. Leahy D. K. Simmons E. M. A Pharmaceutical Industry Perspective on Sustainable Metal Catalysis. Organometallics. 2019;38:36–46. doi: 10.1021/acs.organomet.8b00566. [DOI] [Google Scholar]
- Gandeepan P. Ackermann Lutz. Transient Directing Groups for Transformative C–H Activation by Synergistic Metal Catalysis. Chem. 2018;4:199–222. [Google Scholar]
- Seechurn C. C. C. J. Kitching M. O. Colacot T. J. Snieckus V. Palladium-catalyzed cross-coupling: a historical contextual perspective to the 2010 Nobel prize. Angew. Chem., Int. Ed. 2012;51:5062–5085. doi: 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]
- Ruiz-Castillo P. Buchwald S. L. Application of palladium-catalyzed C–N cross-coupling reactions. Chem. Rev. 2016;116:12564–12649. doi: 10.1021/acs.chemrev.6b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas J. P. Guin S. Maiti D. Highvalent 3d metal-oxo mediated C–H halogenation: Biomimetic approaches. Coord. Chem. Rev. 2020;408:213174. doi: 10.1016/j.ccr.2019.213174. [DOI] [Google Scholar]
- Soldevila-Barreda J. J. Metzler-Nolte N. Intracellular Catalysis with Selected Metal Complexes and Metallic Nanoparticles: Advances toward the Development of Catalytic Metallodrugs. Chem. Rev. 2019;119:829–869. doi: 10.1021/acs.chemrev.8b00493. [DOI] [PubMed] [Google Scholar]
- Chernyshev V. M. Astakhov A. V. Chikunov I. E. Tyurin R. V. Eremin D. B. Ranny G. S. Khrustalev V. N. Ananikov V. P. Pd and Pt Catalyst Poisoning in the Study of Reaction Mechanisms: What Does the Mercury Test Mean for Catalysis? ACS Catal. 2019;9:2984–2995. doi: 10.1021/acscatal.8b03683. [DOI] [Google Scholar]
- Biffis A. Centomo P. Zotto A. D. Zecca M. Pd Metal Catalysts for Cross-Couplings and Related Reactions in the 21st Century: A Critical Review. Chem. Rev. 2018;118:2249–2295. doi: 10.1021/acs.chemrev.7b00443. [DOI] [PubMed] [Google Scholar]
- Kumar M. Hammond G. B. Xu B. Cationic Gold Catalyst Poisoning and Reactivation. Org. Lett. 2014;16:3452–3455. doi: 10.1021/ol501663f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Hagen W. R. Hypothesis: entatic versus ecstatic states in metalloproteins. Metallomics. 2019;11:1768–1778. doi: 10.1039/C9MT00208A. [DOI] [PubMed] [Google Scholar]; (b) Soldevila-Barreda J. J. Sadler P. J. Approaches to the design of catalytic metallodrugs. Curr. Opin. Chem. Biol. 2015;25:172–183. doi: 10.1016/j.cbpa.2015.01.024. [DOI] [PubMed] [Google Scholar]
- Pan H.-J. Huang G. Wodrich M. D. Tirani F. F. Ataka K. Shima S. Hu X. A catalytically active [Mn]-hydrogenase incorporating a non-native metal cofactor. Nat. Chem. 2019;11:669–675. doi: 10.1038/s41557-019-0266-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valasatava Y. Rosato A. Furnham N. Thornton J. M. Andreini C. To what extent do structural changes in catalytic metal sites affect enzyme function? J. Inorg. Biochem. 2018;179:40–53. doi: 10.1016/j.jinorgbio.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyster T. K. Ward T. R. Genetic Optimization of Metalloenzymes: Enhancing Enzymes for Non-Natural Reactions. Angew. Chem., Int. Ed. 2016;55:7344–7357. doi: 10.1002/anie.201508816. [DOI] [PubMed] [Google Scholar]
- Lin Y.-W. Rational design of metalloenzymes: From single to multiple active sites. Coord. Chem. Rev. 2017;336:1–27. doi: 10.1016/j.ccr.2017.01.001. [DOI] [Google Scholar]
- Nastri F. Chino M. Maglio O. Bhagi-Damodaran A. Lu Y. Lombardi A. Design and engineering of artificial oxygen-activating metalloenzymes. Chem. Soc. Rev. 2016;45:5020–5054. doi: 10.1039/C5CS00923E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reetz M. T. Directed Evolution of Artificial Metalloenzymes: A Universal Means to Tune the Selectivity of Transition Metal Catalysts? Acc. Chem. Res. 2019;52:336–344. doi: 10.1021/acs.accounts.8b00582. [DOI] [PubMed] [Google Scholar]
- Thompson Z. Cowan J. A. Artificial Metalloenzymes: Recent Developments and Innovations in Bioinorganic Catalysis. Small. 2020;16:2000392. doi: 10.1002/smll.202000392. [DOI] [PubMed] [Google Scholar]
- Ngo A. H. Bose S. Do L. H. Intracellular Chemistry: Integrating Molecular Inorganic Catalysts with Living Systems. Chem. – Eur. J. 2018;24:10584–10594. doi: 10.1002/chem.201800504. [DOI] [PubMed] [Google Scholar]
- Castro S. A. Terenzi A. Gurruchaga-Pereda J. Salassa L. Catalysis Concepts in Medicinal Inorganic Chemistry. Chem. – Eur. J. 2019;25:6651–6660. doi: 10.1002/chem.201806341. [DOI] [PubMed] [Google Scholar]
- Soldevila-Barreda J. J. Romero-Canelón I. Habtemariam A. Sadler P. J. Transfer hydrogenation catalysis in cells as a new approach to anticancer drug design. Nat. Commun. 2015;6:6582. doi: 10.1038/ncomms7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F. Soldevila-Barreda J. J. Romero-Canelón I. Coverdale J. P. C. Song J.-I. Clarkson G. J. Kasparkova J. Habtemariam A. Brabec V. Wolny J. A. Schünemann V. Sadler P. J. Effect of sulfonamidoethylenediamine substituents in RuII arene anticancer catalysts on transfer hydrogenation of coenzyme NAD+ by formate. Dalton Trans. 2018;47:7178–7189. doi: 10.1039/C8DT00438B. [DOI] [PubMed] [Google Scholar]
- Chen F. Romero-Canelón I. Soldevila-Barreda J. J. Song J.-I. Coverdale J. P. C. Clarkson G. J. Kasparkova J. Habtemariam A. Wills M. Brabec V. Sadler P. J. Transfer Hydrogenation and Antiproliferative Activity of Tethered Half-Sandwich Organoruthenium Catalysts. Organometallics. 2018;37:1555–1566. doi: 10.1021/acs.organomet.8b00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghdoost M. M. Guard J. Golbaghi G. Castonguay A. Anticancer Activity and Catalytic Potential of Ruthenium(ii)−Arene Complexes with N,O-Donor Ligands. Inorg. Chem. 2018;57:7558–7567. doi: 10.1021/acs.inorgchem.8b00346. [DOI] [PubMed] [Google Scholar]
- Soldevila-Barreda J. J. Habtemariam A. Romero-Canelón I. Sadler P. J. Half-sandwich rhodium(iii) transfer hydrogenation catalysts: Reduction of NAD+ and pyruvate, and antiproliferative activity. J. Inorg. Biochem. 2015;153:322–333. doi: 10.1016/j.jinorgbio.2015.10.008. [DOI] [PubMed] [Google Scholar]
- Betanzos-Lara S. Liu Z. Habtemariam A. Pizarro A. M. Qamar B. Sadler P. J. Organometallic Ruthenium and Iridium Transfer-Hydrogenation Catalysts Using Coenzyme NADH as a Cofactor. Angew. Chem., Int. Ed. 2012;51:3897–3900. doi: 10.1002/anie.201108175. [DOI] [PubMed] [Google Scholar]
- Liu Z. Romero-Canelon I. Qamar B. Hearn J. M. Habtemariam A. Barry N. P. E. Pizarro A. M. Clarkson G. J. Sadler P. J. The Potent Oxidant Anticancer Activity of Organoiridium Catalysts. Angew. Chem., Int. Ed. 2014;53:3941–3946. doi: 10.1002/anie.201311161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z. Deeth R. J. Butler J. S. Habtemariam A. Newton M. E. Sadler P. J. Reduction of Quinones by NADH Catalyzed by Organoiridium Complexes. Angew. Chem., Int. Ed. 2013;52:4194–4197. doi: 10.1002/anie.201300747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo A. H. Ibañez M. Do L. H. Catalytic Hydrogenation of Cytotoxic Aldehydes Using Nicotinamide Adenine Dinucleotide (NADH) in Cell Growth Media. ACS Catal. 2016;6:2637–2641. doi: 10.1021/acscatal.6b00395. [DOI] [Google Scholar]
- Bose S. Ngo A. H. Do L. H. Intracellular Transfer Hydrogenation Mediated by Unprotected Organoiridium Catalysts. J. Am. Chem. Soc. 2017;139:8792–8795. doi: 10.1021/jacs.7b03872. [DOI] [PubMed] [Google Scholar]
- Li J. Guo L. Tian Z. Zhang S. Xu Z. Han Y. Li R. Li Y. Liu Z. Half-Sandwich Iridium and Ruthenium Complexes: Effective Tracking in Cells and Anticancer Studies. Inorg. Chem. 2018;57:13552–13563. doi: 10.1021/acs.inorgchem.8b02161. [DOI] [PubMed] [Google Scholar]
- Guo L. Zhang H. Tian M. Tian Z. Xu Y. Yang Y. Peng H. Liu P. Liu Z. Electronic Effects on Reactivity and Anticancer Activity by Half-Sandwich N,N-Chelated Iridium(iii) Complexes. New J. Chem. 2018;42:16183–16192. doi: 10.1039/C8NJ03360A. [DOI] [Google Scholar]
- Wang C. Liu J. Tian Z. Tian M. Tian L. Zhao W. Liu Z. Half-Sandwich Iridium N-Heterocyclic Carbene Anticancer Complexes. Dalton Trans. 2017;46:6870–6883. doi: 10.1039/C7DT00575J. [DOI] [PubMed] [Google Scholar]
- Kong D. Guo L. Tian M. Zhang S. Tian Z. Yang H. Tian Y. Liu Z. Lysosome-targeted potent half-sandwich iridium(iii) α-diimine antitumor complexes. Appl. Organometal. Chem. 2019;33:e4633. doi: 10.1002/aoc.4633. [DOI] [Google Scholar]
- Li J. Tian M. Tian Z. Zhang S. Yan C. Shao C. Liu Z. Half-Sandwich Iridium(iii) and Ruthenium(ii) Complexes Containing P^P-Chelating Ligands: A New Class of Potent Anticancer Agents with Unusual Redox Features. Inorg. Chem. 2018;57:1705–1716. doi: 10.1021/acs.inorgchem.7b01959. [DOI] [PubMed] [Google Scholar]
- Zhang W.-Y. Bridgewater H. E. Banerjee S. Soldevila-Barreda J. J. Clarkson G. J. Shi H. Imberti C. Sadler P. J. Ligand-Controlled Reactivity and Cytotoxicity of Cyclometalated Rhodium(iii) Complexes. Eur. J. Inorg. Chem. 2020:1052–1060. doi: 10.1002/ejic.201901055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coverdale J. P. C. Romero-Canelón I. Sanchez-Cano C. Clarkson G. J. Habtemariam A. Wills M. Sadler P. J. Asymmetric Transfer Hydrogenation by Synthetic Catalysts in Cancer Cells. Nat. Chem. 2018;10:347–354. doi: 10.1038/nchem.2918. [DOI] [PubMed] [Google Scholar]
- Coverdale J. P. C. Sanchez-Cano C. Clarkson G. J. Soni R. Wills M. Sadler P. J. Easy To Synthesize, Robust Organo-osmium Asymmetric Transfer Hydrogenation Catalysts. Chem. – Eur. J. 2015;21:8043–8046. doi: 10.1002/chem.201500534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli C. Grodsky N. B. Ariyaratne N. Colman R. F. Bahnson B. J. The Crystal Structure of Porcine Mitochondrial NADP+-dependent Isocitrate Dehydrogenase Complexed with Mn2+ and Isocitrate. J. Biol. Chem. 2002;277:43454–43462. doi: 10.1074/jbc.M207306200. [DOI] [PubMed] [Google Scholar]
- Tretter L. Adam-Vizi V. Alpha-ketoglutarate dehydrogenase: a target and generator of oxidative stress. Philos. Trans. R. Soc., B. 2005;360:2335–2345. doi: 10.1098/rstb.2005.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Minárik P. Tomásková N. Kollárová M. Antalík M. Malate dehydrogenases-structure and function. Gen. Physiol. Biophys. 2002;21:257–265. [PubMed] [Google Scholar]; (b) Venkat S. Gregory C. Sturges J. Gan Q. Fan C. Studying the Lysine Acetylation of Malate Dehydrogenase. J. Mol. Biol. 2017;429:1396–1405. doi: 10.1016/j.jmb.2017.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Edenberg H. J. McClintick J. N. Alcohol dehydrogenases, aldehyde dehydrogenases and alcohol use disorders: a critical review. Alcohol.: Clin. Exp. Res. 2018;42:2281–2297. doi: 10.1111/acer.13904. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zheng Y.-G. Yin H.-H. Yu D.-F. Chen X. Tang X.-L. Zhang X.-J. Xue Y.-P. Wang Y.-J. Liu Z.-Q. Recent advances in biotechnological applications of alcohol dehydrogenases. Appl. Microbiol. Biotechnol. 2017;101:987–1001. doi: 10.1007/s00253-016-8083-6. [DOI] [PubMed] [Google Scholar]
- Popov V. O. Lamzin V. S. NAD(+)-dependent formate dehydrogenase. Biochem. J. 1994;301:625–643. doi: 10.1042/bj3010625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Kerscher S., Dröse S., Zickermann V. and Brandt U., The Three Families of Respiratory NADH Dehydrogenases. Bioenergetics. Results and Problems in Cell Differentiation ed. G. Schäfer and H. S. Penefsky, Springer, Heidelberg, 2007, vol. 45 [DOI] [PubMed] [Google Scholar]; (b) Godoy-Hernandez A. Tate D. J. McMillan D. G. G. Revealing the Membrane-Bound Catalytic Oxidation of NADH by the Drug Target Type-II NADH Dehydrogenase. Biochemistry. 2019;58:4272–4275. doi: 10.1021/acs.biochem.9b00752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikova V. G. Gladyshev G. V. Vinogradov A. D. Deactivation of mitochondrial NADH:ubiquinone oxidoreductase (respiratory complex I): Extrinsically affecting factors. Biochim. Biophys. Acta, Biomembr. 2020;1861:148207. doi: 10.1016/j.bbabio.2020.148207. [DOI] [PubMed] [Google Scholar]
- Okuda-Shimazaki J. Yoshida H. Sode K. FAD dependent glucose dehydrogenases – Discovery and engineering of representative glucose sensing enzymes. Bioelectrochemistry. 2020;132:107414. doi: 10.1016/j.bioelechem.2019.107414. [DOI] [PubMed] [Google Scholar]
- Bustos D. M. Iglesias A. A. Non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase is post-translationally phosphorylated in heterotrophic cells of wheat (Triticum aestivum) FEBS Lett. 2002;530:169–173. doi: 10.1016/S0014-5793(02)03455-5. [DOI] [PubMed] [Google Scholar]
- Kean K. M. Carpenter R. A. Pandini V. Zanetti G. Hall A. R. Faber R. Aliverti A. Karplus P. A. High-resolution studies of hydride transfer in the ferredoxin:NADP+ reductase superfamily. FEBS J. 2017;284:3302–3319. doi: 10.1111/febs.14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanukoglu I. Conservation of the Enzyme–Coenzyme Interfaces in FAD and NADP Binding Adrenodoxin Reductase—A Ubiquitous Enzyme. J. Mol. Evol. 2017;85:205–218. doi: 10.1007/s00239-017-9821-9. [DOI] [PubMed] [Google Scholar]
- Magnani F. Nenci S. Fananas E. M. Ceccon M. Romero E. Fraaije M. W. Mattevi A. Crystal structures and atomic model of NADPH oxidase. Proc. Natl. Acad. Sci. U. S. A. 2017;114:6764–6769. doi: 10.1073/pnas.1713563114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H. Banerjee S. Qiu K. Zhang P. Blacque O. Malcomson T. Paterson M. J. Clarkson G. J. Staniforth M. Stavros V. G. Gasser G. Chao H. Sadler P. J. Targeted photoredox catalysis in cancer cells. Nat. Chem. 2019;11:1041–1048. doi: 10.1038/s41557-019-0328-4. [DOI] [PubMed] [Google Scholar]
- Alonso-de Castro S. Ruggiero E. Ruiz-de-Angulo A. Rezabal E. Mareque-Rivas J. C. Lopez X. Lopez-Gallego F. Salassa L. Riboflavin as a bioorthogonal photocatalyst for the activation of a PtIV prodrug. Chem. Sci. 2017;8:4619–4625. doi: 10.1039/C7SC01109A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-de Castro S. Terenzi A. Hager S. Englinger B. Faraone A. Calvo Martínez J. Galanski M. Keppler B. K. Berger W. Salassa L. Biological activity of PtIV prodrugs triggered by riboflavin-mediated bioorthogonal photocatalysis. Sci. Rep. 2018;8:17198. doi: 10.1038/s41598-018-35655-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-de Castro S. Cortajarena A. L. Llpez-Gallego F. Salassa L. Bioorthogonal Catalytic Activation of Platinum and Ruthenium Anticancer Complexes by FAD and Flavoproteins. Angew. Chem., Int. Ed. 2018;130:3197–3201. doi: 10.1002/ange.201800288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurruchaga-Pereda J. Martínez-Martínez V. Rezabal E. Lopez X. Garino C. Mancin F. Cortajarena A. L. Salassa L. ACS Catal. 2020;10:187–196. doi: 10.1021/acscatal.9b02863. [DOI] [Google Scholar]
- Akram M. Citric Acid Cycle and Role of its Intermediates in Metabolism. Cell Biochem. Biophys. 2014;68:475–478. doi: 10.1007/s12013-013-9750-1. [DOI] [PubMed] [Google Scholar]
- Lodish H., Berk A., Zipursky S. L., et al., Molecular Cell Biology, W. H. Freeman, New York, 4th edn, 2000 [Google Scholar]
- Birrell J. A. Hirst J. Investigation of NADH Binding, Hydride Transfer, and NAD+ Dissociation during NADH Oxidation by Mitochondrial Complex I Using Modified Nicotinamide Nucleotides. Biochemistry. 2013;52:4048–4055. doi: 10.1021/bi3016873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W. Wang R.-S. Handy D. E. Loscalzo J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid. Redox Signaling. 2018;28:251–272. doi: 10.1089/ars.2017.7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradov A. D. Grivennikova V. G. Oxidation of NADH and ROS production by respiratory complex I. Biochim. Biophys. Acta. 2016;1857:863–871. doi: 10.1016/j.bbabio.2015.11.004. [DOI] [PubMed] [Google Scholar]
- Ghisla S. Massey V. Mechanisms of flavoprotein-catalyzed reactions. Eur. J. Biochem. 1989;181:1–17. doi: 10.1111/j.1432-1033.1989.tb14688.x. [DOI] [PubMed] [Google Scholar]
- Sazanov L. A. Hinchliffe P. Structure of the Hydrophilic Domain of Respiratory Complex I From Thermus Thermophilus. Science. 2006;311:1430–1436. doi: 10.1126/science.1123809. [DOI] [PubMed] [Google Scholar]
- Barker C. D. Reda T. Hirst J. The Flavoprotein Subcomplex of Complex I (NADH:Ubiquinone Oxidoreductase) from Bovine Heart Mitochondria: Insights into the Mechanisms of NADH Oxidation and NAD+ Reduction from Protein Film Voltammetry. Biochemistry. 2007;46:3454–3464. doi: 10.1021/bi061988y. [DOI] [PubMed] [Google Scholar]
- Rostas A. Einholz C. Illarionov B. Heidinger L. Said T. A. Bauss A. Fischer M. Bacher A. Weber S. Schleicher E. Long-Lived Hydrated FMN Radicals: EPR Characterization and Implications for Catalytic Variability in Flavoproteins. J. Am. Chem. Soc. 2018;140:16521–16527. doi: 10.1021/jacs.8b07544. [DOI] [PubMed] [Google Scholar]
- Sousa S. F. Sousa J. F. M. Barbosa A. C. C. Ferreira C. E. Neves R. P. P. Ribeiro A. J. M. Fernandes P. A. Ramos M. J. Improving the Biodesulfurization of Crude Oil and Derivatives: A QM/MM Investigation of the Catalytic Mechanism of NADH-FMN Oxidoreductase (DszD) J. Phys. Chem. A. 2016;120:5300–5306. doi: 10.1021/acs.jpca.6b01536. [DOI] [PubMed] [Google Scholar]
- Du W. Jiang P. Mancuso A. Stonestrom A. Brewer M. D. Minn A. J. Mak T. W. Wu M. Yang X. TAp73 enhances the pentose phosphate pathway and supports cell proliferation. Nat. Cell Biol. 2013;15:991–1000. doi: 10.1038/ncb2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaans S. K. Weusthuis R. A. van der Oost J. Kengen S. W. M. NADPH-generating systems in bacteria and archaea. Front. Microbiol. 2015;6:742. doi: 10.3389/fmicb.2015.00742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D. C. How the phagocyte NADPH oxidase regulates innate immunity. Free Radical Biol. Med. 2018;125:44–52. doi: 10.1016/j.freeradbiomed.2018.06.011. [DOI] [PubMed] [Google Scholar]
- Segal A. W. The function of the NADPH oxidase of phagocytes and its relationship to other NOXs in plants, invertebrates, and mammals. Int. J. Biochem. Cell Biol. 2008;40:604–618. doi: 10.1016/j.biocel.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavins: Photochemistry and Photobiology, E. Silva and A. M. Edwards, Royal Society of Chemistry, 2006. Print ISBN: 978-0-85404-331-6; PDF eISBN: 978-1-84755-539-7 [Google Scholar]
- Buckel W. Thauer R. K. Ferredoxin, Flavodoxin, and Anaerobic Respiration With Protons (Ech) or NAD+ (Rnf) as Electron Acceptors: A Historical Review. Front. Microbiol. 2018;9:401. doi: 10.3389/fmicb.2018.00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R. Bianchet M. A. Talalay P. Amzel L. M. The three-dimensional structure of NAD(P)H:quinone reductase, a flavoprotein involved in cancer chemoprotection and chemotherapy: mechanism of the two-electron reduction. Proc. Natl. Acad. Sci. U. S. A. 1995;92:8846–8850. doi: 10.1073/pnas.92.19.8846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyanagi T. Molecular mechanism of metabolic NAD(P)H-dependent electron-transfer systems: The role of redox cofactors. Biochim. Biophys. Acta. 2019;1860:233–258. doi: 10.1016/j.bbabio.2018.11.014. [DOI] [PubMed] [Google Scholar]
- Matsuda H. Kimura S. Iyanagi T. One-electron reduction of quinones by the neuronal nitric-oxide synthase reductase domain. Biochim. Biophys. Acta. 2000;1459:106–116. doi: 10.1016/S0005-2728(00)00117-1. [DOI] [PubMed] [Google Scholar]
- Demmer J. K. Pal Chowdhury N. Selmer T. Ermler U. Buckel W. The semiquinone swing in the bifurcating electron transferring flavoprotein/butyryl-CoA dehydrogenase complex from Clostridium difficile. Nat. Commun. 2017;8:1577. doi: 10.1038/s41467-017-01746-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sazanov L. A. A giant molecular proton pump: structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 2015;16:375–388. doi: 10.1038/nrm3997. [DOI] [PubMed] [Google Scholar]
- Vanbergen O. and Wintle G., Crash Course: Metabolism and Nutrition, Elsevier, 5th edn, 2018 [Google Scholar]
- Ismail M. Schroeder L. Frese M. Kottke T. Hollmann F. Paul C. E. Sewald N. Straightforward Regeneration of Reduced Flavin Adenine Dinucleotide Required for Enzymatic Tryptophan Halogenation. ACS Catal. 2019;9:1389–1395. doi: 10.1021/acscatal.8b04500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unversucht S. Hollmann F. Schmid A. van Pée K.-H. FADH2-Dependence of Tryptophan 7-Halogenase. Adv. Synth. Catal. 2005;347:1163–1167. doi: 10.1002/adsc.200505029. [DOI] [Google Scholar]
- Knaus T. Paul C. E. Levy C. W. de Vries S. Mutti F. G. Hollmann F. Scrutton N. S. Better than Nature: Nicotinamide Biomimetics That Outperform Natural Coenzymes. J. Am. Chem. Soc. 2016;138:1033–1039. doi: 10.1021/jacs.5b12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul C. E. Gargiulo S. Opperman D. J. Lavandera I. Gotor-Fernandez V. Gotor V. Taglieber A. Arends I. W. C. E. Hollmann F. Mimicking Nature: Synthetic Nicotinamide Cofactors for C C Bioreduction Using Enoate Reductases. Org. Lett. 2013;15:180–183. doi: 10.1021/ol303240a. [DOI] [PubMed] [Google Scholar]
- Knox R. J. Jenkins T. C. Hobbs S. M. Chen S. Melton R. G. Burke P. J. Bioactivation of 5-(Aziridin-1-yl)-2,4-dinitrobenzamide (CB 1954) by Human NAD(P)H Quinone Oxidoreductase 2: A Novel Co-substrate-mediated Antitumor Prodrug Therapy. Cancer Res. 2000;60:4179–4186. [PubMed] [Google Scholar]
- Paul C. E. Churakova E. Maurits E. Girhard M. Urlacher V. B. Hollmann F. In situ formation of H2O2 for P450 peroxygenases. Bioorg. Med. Chem. 2014;22:5692–5696. doi: 10.1016/j.bmc.2014.05.074. [DOI] [PubMed] [Google Scholar]
- Wang D. Astruc D. The Golden Age of Transfer Hydrogenation. Chem. Rev. 2015;115:6621–6686. doi: 10.1021/acs.chemrev.5b00203. [DOI] [PubMed] [Google Scholar]
- Robertson A. Matsumoto T. Ogo S. The development of aqueous transfer hydrogenation. Dalton Trans. 2011;40:10304–10310. doi: 10.1039/C1DT10544B. [DOI] [PubMed] [Google Scholar]
- Gladiali S. Alberico E. Asymmetric transfer hydrogenation: chiral ligands and applications. Chem. Soc. Rev. 2006;35:226–236. doi: 10.1039/B513396C. [DOI] [PubMed] [Google Scholar]
- Ruppert R. Herrmann S. Steckhan E. Very efficient reduction of NAD(P)+ with formate catalysed by cationic rhodium complexes. J. Chem. Soc., Chem. Commun. 1988:1150–1151. doi: 10.1039/C39880001150. [DOI] [Google Scholar]
- Lo H. C. Leiva C. Buriez O. Kerr J. B. Olmstead M. M. Fish R. H. Bioorganometallic chemistry. 13. Regioselective reduction of NAD+ models, 1-benzylnicotinamde triflate and beta-nicotinamide ribose-5′-methyl phosphate, with in situ generated [Cp*Rh(Bpy)H]+: structure–activity relationships, kinetics, and mechanistic aspects in the formation of the 1,4-NADH derivatives. Inorg. Chem. 2001;40:6705–6716. doi: 10.1021/ic010562z. [DOI] [PubMed] [Google Scholar]
- Buriez O. Kerr J. B. Fish R. H. Regioselective reduction of NAD+ models with [Cp*Rh(bpy)H]+: structure-activity relationships and mechanistic aspects in the formation of the 1,4-NADH derivatives. Angew. Chem., Int. Ed. 1999;38:1997–2000. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<1997::AID-ANIE1997>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Canivet J. Süss-Fink G. Štěpnička P. Water-soluble phenanthroline complexes of rhodium, iridium and ruthenium for the regeneration of NADH in the enzymatic reduction of ketones. Eur. J. Inorg. Chem. 2007:4736–4742. doi: 10.1002/ejic.200700505. [DOI] [Google Scholar]
- Stringer T. Melis D. R. Smith G. S. N,O-Chelating quinoline-based half-sandwich organorhodium and -iridium complexes: synthesis, antiplasmodial activity and preliminary evaluation as transfer hydrogenation catalysts for the reduction of NAD+ Dalton Trans. 2019;48:13143–13148. doi: 10.1039/C9DT02030F. [DOI] [PubMed] [Google Scholar]
- Yan Y. K. Melchart M. Habtemariam A. Peacock A. F. A. Sadler P. J. Catalysis of regioselective reduction of NAD+ by ruthenium(ii) arene complexes under biologically relevant conditions. J. Biol. Inorg. Chem. 2006;11:483–488. doi: 10.1007/s00775-006-0098-5. [DOI] [PubMed] [Google Scholar]
- Soldevila-Barreda J. J. Bruijnincx P. C. A. Habtemariam A. Clarkson G. J. Deeth R. J. Sadler P. J. Improved Catalytic Activity of Ruthenium−Arene Complexes in the Reduction of NAD+ Organometallics. 2012;31:5958–5967. doi: 10.1021/om3006307. [DOI] [Google Scholar]
- Gebicki J. Marcinek A. Zielonka J. Transient Species in the Stepwise Interconversion of NADH and NAD+ Acc. Chem. Res. 2004;37:379–386. doi: 10.1021/ar030171j. [DOI] [PubMed] [Google Scholar]
- McSkimming A. Colbran S. B. The coordination chemistry of organo-hydride donors: new prospects for efficient multi-electron reduction. Chem. Soc. Rev. 2013;42:5439–5488. doi: 10.1039/C3CS35466K. [DOI] [PubMed] [Google Scholar]
- Eisner U. Kuthan J. Chemistry of dihydropyridines. Chem. Rev. 1972;72:1–42. doi: 10.1021/cr60275a001. [DOI] [Google Scholar]
- Martins J. E. D. Clarkson G. J. Wills M. Ru(ii) Complexes of N-Alkylated TsDPEN Ligands in Asymmetric Transfer Hydrogenation of Ketones and Imines. Org. Lett. 2009;11:847–850. doi: 10.1021/ol802801p. [DOI] [PubMed] [Google Scholar]
- Hayes A. M. Morris D. J. Clarkson G. J. Wills M. A Class of Ruthenium(ii) Catalyst for Asymmetric Transfer Hydrogenations of Ketones. J. Am. Chem. Soc. 2005;127:7318–7319. doi: 10.1021/ja051486s. [DOI] [PubMed] [Google Scholar]
- Soni R. Cheung F. K. Clarkson G. J. Martins J. E. D. Graham M. A. Wills M. The importance of the N–H bond in Ru/TsDPEN complexes for asymmetric transfer hydrogenation of ketones and imines. Org. Biomol. Chem. 2011;9:3290–3294. doi: 10.1039/C1OB05208J. [DOI] [PubMed] [Google Scholar]
- Hannedouche J. Clarkson G. J. Wills M. A New Class of “Tethered” Ruthenium(ii) Catalyst for Asymmetric Transfer Hydrogenation Reactions. J. Am. Chem. Soc. 2004;126:986–987. doi: 10.1021/ja0392768. [DOI] [PubMed] [Google Scholar]
- Rover Jr. L. Fernandes J. C. B. Neto G. de O. Kubota L. T. Katekawa E. Serrano S. H. P. Study of NADH Stability Using Ultraviolet–Visible Spectrophotometric Analysis and Factorial Design. Anal. Biochem. 1998;260:50–55. doi: 10.1006/abio.1998.2656. [DOI] [PubMed] [Google Scholar]
- Maenaka Y. Suenobu T. Fukuzumi S. Efficient Catalytic Interconversion between NADH and NAD+ Accompanied by Generation and Consumption of Hydrogen with a Water-Soluble Iridium Complex at Ambient Pressure and Temperature. J. Am. Chem. Soc. 2012;134:367–374. doi: 10.1021/ja207785f. [DOI] [PubMed] [Google Scholar]
- Suenobu T. Shibata S. Fukuzumi S. Catalytic Formation of Hydrogen Peroxide from Coenzyme NADH and Dioxygen with a Water-Soluble Iridium Complex and a Ubiquinone Coenzyme Analogue. Inorg. Chem. 2016;55:7747–7754. doi: 10.1021/acs.inorgchem.6b01220. [DOI] [PubMed] [Google Scholar]
- Bucci A. Dunn S. Bellachioma G. Rodriguez G. M. Zuccaccia C. Nervi C. Macchioni A. A Single Organoiridium Complex Generating Highly Active Catalysts for both Water Oxidation and NAD+/NADH Transformations. ACS Catal. 2017;7:7788–7796. doi: 10.1021/acscatal.7b02387. [DOI] [Google Scholar]
- Ganesan V. Sivanesan D. Yoon S. Correlation between the Structure and Catalytic Activity of [Cp*Rh(Substituted Bipyridine)] Complexes for NADH Regeneration. Inorg. Chem. 2017;56:1366–1374. doi: 10.1021/acs.inorgchem.6b02474. [DOI] [PubMed] [Google Scholar]
- Buhl S. Jackson K. Y. Optimal conditions and comparison of lactate dehydrogenase catalysis of the lactate-to-pyruvate and pyruvate-to-lactate reactions in human serum at 25, 30, and 37 degrees C. Clin. Chem. 1978;24:828–831. doi: 10.1093/clinchem/24.5.828. [DOI] [PubMed] [Google Scholar]
- Jones W. D. Kuykendall V. L. Selmeczy A. D. Ring migration reactions of (C5Me5)Rh(PMe3)H2. Evidence for.eta.3 slippage and metal-to-ring hydride migration. Organometallics. 1991;10:1577–1586. doi: 10.1021/om00051a057. [DOI] [Google Scholar]
- Lo H. C. Buriez O. Kerr J. B. Fish R. H. Regioselective Reduction of NAD+ Models with [Cp*Rh(bpy)H]+: Structure–Activity Relationships and Mechanistic Aspects in the Formation of the 1,4-NADH Derivatives. Angew. Chem., Int. Ed. 1999;38:1429–1432. doi: 10.1002/(SICI)1521-3773(19990517)38:10<1429::AID-ANIE1429>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Hirst J. Energy Transduction by Respiratory Complex I-an Evaluation of Current Knowledge. Biochem. Soc. Trans. 2005;33:525–529. doi: 10.1042/BST0330525. [DOI] [PubMed] [Google Scholar]
- Yagi T. Matsuno-Yagi A. The Proton-Translocating NADH−Quinone Oxidoreductase in the Respiratory Chain: The Secret Unlocked. Biochemistry. 2003;42:2266–2274. doi: 10.1021/bi027158b. [DOI] [PubMed] [Google Scholar]
- Matsushita K. Ohnishi T. Kaback H. R. NADH-ubiquinone Oxidoreductases of the Escherichia Coli Aerobic Respiratory Chain. Biochemistry. 1987;26:7732–7737. doi: 10.1021/bi00398a029. [DOI] [PubMed] [Google Scholar]
- Bachur N. R. Gordon S. L. Gee M. V. Kon H. NADPH Cytochrome P-450 Reductase Activation of Quinone Anticancer Agents to Free Radicals. Proc. Natl. Acad. Sci. U. S. A. 1979;76:954–957. doi: 10.1073/pnas.76.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J. Towards the molecular mechanism of respiratory complex I. Biochem. J. 2010;425:327–339. doi: 10.1042/BJ20091382. [DOI] [PubMed] [Google Scholar]
- Kimura T. Ishiwata K. Kuwata S. Ikariya T. Trapping of a Doubly Unsaturated Dinuclear Iridium(ii) Sulfonylimido Complex with Phosphine and Lewis Acidic Group 11 and 12 Metals. Organometallics. 2012;31:1204–1207. doi: 10.1021/om201218j. [DOI] [Google Scholar]
- Meiners J. Scheibel M. G. Lemre-Cailleau M.-H. Mason S. A. Boeddinghaus M. B. Fässler T. F. Herdtweck E. Khusniyarov M. M. Schneider S. Square-Planar Iridium(ii) and Iridium(iii) Amido Complexes Stabilized by a PNP Pincer Ligand. Angew. Chem., Int. Ed. 2011;50:8184–8187. doi: 10.1002/anie.201102795. [DOI] [PubMed] [Google Scholar]
- Teets T. S. Cook T. R. McCarthy B. D. Nocera D. G. Redox Chemistry, Acid Reactivity, and Hydrogenation Reactions of Two-Electron Mixed Valence Diiridium and Dirhodium Complexes. Inorg. Chem. 2011;50:5223–5233. doi: 10.1021/ic2005248. [DOI] [PubMed] [Google Scholar]
- McFarland J. M. Francis M. B. Reductive Alkylation of Proteins Using Iridium Catalyzed Transfer Hydrogenation. J. Am. Chem. Soc. 2005;127:13490–13491. doi: 10.1021/ja054686c. [DOI] [PubMed] [Google Scholar]
- Haas R. Cucchi D. Smith J. Pucino V. Macdougall C. E. Mauro C. Intermediates of Metabolism: From Bystanders to Signalling Molecules. Trends Biochem. Sci. 2016;41:460–471. doi: 10.1016/j.tibs.2016.02.003. [DOI] [PubMed] [Google Scholar]
- Bhagavan N. V. and Ha C.-E., Essentials of Medical Biochemistry, 2nd edn, 2015 [Google Scholar]
- Prier C. K. Rankic D. A. MacMillan D. W. C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser F. Wenger O. S. Recent progress in the development of transition-metal based photoredox catalysts. Coord. Chem. Rev. 2020;405:213129. doi: 10.1016/j.ccr.2019.213129. [DOI] [Google Scholar]
- Romero N. A. Nicewicz D. A. Organic Photoredox Catalysis. Chem. Rev. 2016;116:10075–10166. doi: 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]
- He Z. Huang X. Wang C. Li X. Liu Y. Zhou Z. Wang S. Zhang F. Wang Z. Jacobson O. Zhu J.-J. Yu G. Dai Y. Chen X. A Catalase-Like Metal–Organic Framework Nanohybrid for O2-Evolving Synergistic Chemoradiotherapy. Angew. Chem., Int. Ed. 2019;58:8752–8756. doi: 10.1002/anie.201902612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teicher B. A. Hypoxia and drug resistance. Cancer Metastasis Rev. 1994;13:139–168. doi: 10.1007/BF00689633. [DOI] [PubMed] [Google Scholar]
- Huang C. C. Chia W. T. Chung M. F. Lin K. J. Hsiao C. W. Jin C. Lim W. H. Chen C. C. Sung H. W. An implantable depot that can generate oxygen in situ for overcoming hypoxia-induced resistance to anticancer drugs in chemotherapy. J. Am. Chem. Soc. 2016;138:5222–5225. doi: 10.1021/jacs.6b01784. [DOI] [PubMed] [Google Scholar]
- Fulda S. Galluzzi L. Kroemer G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discovery. 2010;9:447–464. doi: 10.1038/nrd3137. [DOI] [PubMed] [Google Scholar]
- Massey V. Stankovich M. Hemmerich P. Light-mediated reduction of flavoproteins with flavins as catalysts. Biochemistry. 1978;17:1–8. doi: 10.1021/bi00594a001. [DOI] [PubMed] [Google Scholar]
- Massey V. Hemmerich P. Knappe W. R. Duchstein H. J. Fenner H. Photoreduction of Flavoproteins and other Biological Compounds Catalyzed by Deazaflavins. Appendix: Photochemical Formation of Deazaflavin Dimers. Biochemistry. 1978;17:9–17. doi: 10.1021/bi00594a002. [DOI] [PubMed] [Google Scholar]
- Schmermund L., Jurkas V., Özgen F. F., Barone G. D., Büchsenschütz H. C., Winkler C. K., Schmidt S., Kourist R. and Kroutil W., Photo-Biocatalysis: Biotransformations in the Presence of Light, ACS Catal., 2019, 9, 4115–4144 [Google Scholar]