

Abstract
Background
Immune checkpoint inhibitor (ICI) therapy is highly effective in metastatic mismatch repair‐deficient (MMR‐D) colorectal cancer (CRC). In this study, we evaluated molecular and clinical predictors of ICI response in MMR‐D CRC.
Materials and Methods
Patient databases at four cancer institutions were queried. The Fisher exact test was performed to test the association of clinical and molecular markers. The Kaplan‐Meier method was used to estimate progression‐free survival (PFS) and compared by the log‐rank test. Twelve‐ and 24‐month PFS rates were compared by the Z test.
Results
A total of 60 patients with CRC with MMR‐D/microsatellite instability‐high who previously received ICIs were identified. Patients with liver metastasis had a lower overall response rate as compared with other sites of metastasis (36.4% vs. 68.7%; p = .081). Patients with MLH1/PMS2 loss had worse 1‐year and 2‐year PFS rates compared with patients with MSH2/MSH6 loss (84.2% vs. 57.8% and 78.2% vs. 54.2%, respectively; p < .001). There were improved 1‐year and 2‐year PFS rates in patients with wild‐type BRAF when compared with patients with BRAF V600E mutation (73.3% vs. 40%, and 73.3% vs. 26.7%; respectively; p < .001). Patients aged >65 had significantly worse PFS rates as compared with patients aged ≤65 (p < .001).
Conclusion
BRAF V600E mutation, MLH1 and/or PMS2 loss, as well as age >65 years and liver metastasis, may be predictive of duration of ICI response in patients with MMR‐D CRC. Larger cohorts are needed to confirm our findings.
Implications for Practice
The results of this study reveal clinically important biomarkers that potentially predict immune checkpoint inhibitor response in patients with mismatch repair‐deficient colorectal cancer.
Keywords: Colorectal cancer, Mismatch repair‐deficient, Microsatellite instability high, Immune checkpoint inhibitors, BRAF, MLH1, PMS2, MSH2, MSH6, Liver metastasis
Short abstract
This article examines the effect of molecular subsets of mismatch repair‐deficient (MMR‐D) colorectal cancer and BRAF V600E mutation status as a molecular biomarker of immune checkpoint inhibitor efficacy in patients with MMR‐D colorectal cancer.
Introduction
Colorectal cancer remains the third leading cause of cancer‐related death in the U.S., despite the significant risk reduction with screening and early diagnosis [1]. A concerning trend recently observed is the increase in advanced stage colorectal cancer in young patients, leading to a change in the recommended age of screening [2, 3]. The majority of cancer‐related deaths from colorectal cancer are due to metastasis. Although substantial progress in the management of advanced‐stage colorectal cancer has been achieved in the last decade, the 5‐year survival of patients with metastatic disease remains low.
Mismatch repair‐deficient (MMR‐D) colorectal cancer is a unique molecular subset of colorectal cancer that accounts for approximately 5% of metastatic cases [4]. Mismatch repair (MMR) deficiency is defined as the loss of expression or function in at least one of the four clinically relevant MMR genes (MLH1, PMS2, MSH2, MSH6) [5]. This functional loss results in impaired recognition and repair of DNA mismatches that occur during DNA replication [4]. This dysfunction propagates mutagenesis in the DNA, particularly in the microsatellite regions [6]. The change in the size of microsatellites results in breaks and microsatellite instability (MSI), which triggers frameshift mutations in the affected locus of DNA, creating high tumor mutation burden, also called hypermutability [7]. Approximately three‐quarters of MMR‐D colorectal cancers result from somatic mutations or silencing due to hypermethylation of the MLH1 gene [8]. Sporadic cases due to epigenetic silencing of the MMR gene(s) are associated with late‐onset disease, higher prevalence in women, and right‐sided tumors. Germline MMR gene mutations that lead to Lynch syndrome, however, are associated with younger age of onset and left‐sided colorectal cancer. In the setting of MMR‐D colorectal cancer, mutations in the BRAF gene, specifically BRAF V600E occur exclusively in sporadic cases [9]. The different molecular and clinical features of sporadic and germline MMR‐D colorectal cancer suggest these are different subtypes of which may exhibit differential response to immunotherapy [10].
Pembrolizumab, nivolumab, and nivolumab in combination with ipilimumab have demonstrated dramatic responses with durable disease control rates in patients with metastatic MMR‐D colorectal cancer [11, 12, 13]. Most recently, pembrolizumab showed a deep response as a first‐line therapy with improved 12 and 24‐month progression‐free survival (PFS) rates as compared with chemotherapy [14]. Although the results of these studies are consistent with highly promising responses, patients with de novo (intrinsic) and acquired resistance to immune checkpoint inhibitors have been reported [15]. Predictive molecular biomarkers for immune checkpoint inhibitors have not been identified in MMR‐D colorectal cancer. The objective of this multicenter study is to examine the impact of molecular subsets of MMR‐D colorectal cancer and BRAF mutation status as molecular biomarkers of immune checkpoint inhibitor efficacy in patients with MMR‐D colorectal cancer.
Materials and Methods
Patient Population
With the approval of the institutional review boards, patient databases at Winship Cancer Institute of Emory University, Mayo Clinic, Stanford University, and Vanderbilt University were screened for patients with MMR‐D colorectal cancer who were treated with immune checkpoint inhibitors between January 1, 2012, and May 1, 2019. Patients were eligible if they had biopsy‐confirmed colorectal cancer and MMR‐D/MSI‐high (MSI‐H) status analysis with immunohistochemistry or polymerase chain reaction. Patients MMR‐D/MSI‐H colorectal cancer treated with immune checkpoint inhibitors were included regardless of the choice of the immune checkpoint inhibitors (single‐agent vs. combination). Patients with liver‐only metastasis were included in the “liver metastasis” group. Patients without data regarding the loss of specific MMR protein were included in clinical and survival analysis of the general population and were excluded from the MMR protein‐specific analysis.
Data Collection
The data regarding demographic, clinical, molecular, and pathologic information of the patients included in our cohort was retrieved from institutional electronic medical records by chart review. MMR‐D status of tumor was determined based on either MMR protein immunohistochemistry (IHC) or MSI polymerase chain reaction conducted at each clinical center. The BRAF V600E mutation status was retrieved from IHC and/or next‐generation sequencing results that were available at the time of analysis. Right‐sided colon cancer was defined as any primary tumor between the cecum and transverse colon. Tumors between the descending colon and rectum were classified as left‐sided tumors. The decision for progression of disease was made by local physicians based on the clinical, laboratory, and radiologic findings. Best objective response was evaluated retrospectively by investigators using RECIST 1.1 criteria.
Statistical Analysis
The demographic, clinical, and pathological characteristics of patients were reported as frequency and percentages for categorical variables and as mean and SD for continuous variables. The loss of MLH1/PMS2 and MSH2/MSH6 was grouped by their functional dependence and rarity of PMS2 and MSH6 mutations. PFS was measured from the date of initiation of immune checkpoint inhibitor therapy to the date of disease progression or death, whichever occurred first, as documented in the electronic medical record. Patients who were alive but not progressed were censored within the analyses. Patients lost to follow‐up were censored by the last follow‐up date charted in electronic medical records. The Kaplan‐Meier method was performed to generate PFS curves and survival curves between groups were then compared by the log‐rank test. Twelve‐ and 24‐month PFS rates were compared by the Z test. The Fisher exact test was used to examine the association of clinical and molecular markers with BRAF V600E mutation and specific MMR protein loss. Cox proportional hazard model with best subset selection was used for the multivariate analysis because of the small size of the cohort and limited events in each subgroup.
Results
A total of 60 patients with MMR‐D/MSI‐H metastatic colorectal cancer who received immune checkpoint inhibitors were identified (Fig. 1; Table 1). The majority of the patients (77%) were older than 50 at the time of immune checkpoint inhibitor therapy. Most patients had right‐sided primary tumors (77%), which is consistent with the rates reported in the literature. Notably, 15 (25%) patients had no prior therapy, and the remainder of the cohort received at least one line of systemic chemotherapy prior to immune checkpoint inhibitor therapy. Forty patients (66.7%) had known BRAF V600E mutation status at the time of analysis, and nine (23%) patients with BRAF V600E mutation were identified. RAS status was known for only 25 (41.6%) patients in our cohort. Loss of specific MMR protein expression by IHC was assessed in 54 (90%) patients; among these, 34 (63%) patients had MLH1 and/or PMS2 loss, and 20 (37%) patients had MSH2 and/or MSH6 loss (Fig. 2; Table 1). Forty‐seven (78%) patients received pembrolizumab, eight (13%) patients received nivolumab, and five (8%) patients received the combination of nivolumab and ipilimumab (Fig. 2).
Figure 1.
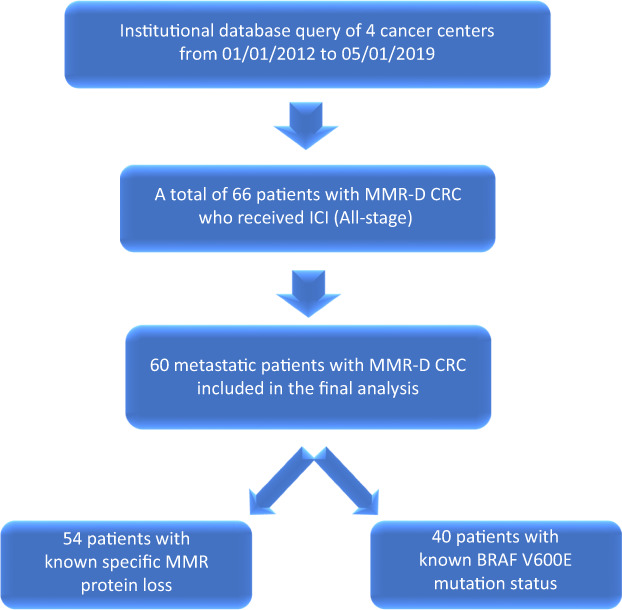
Patient flow diagram. Abbreviations: CRC, colorectal cancer; ICI, immune checkpoint inhibitor; MMR, mismatch repair; MMR‐D, mismatch repair‐deficient.
Table 1.
Baseline patient demographic and clinical characteristics
| Characteristics | No (%) |
|---|---|
| Age, years | |
| <50 | 14 (23) |
| 50–65 | 22 (37) |
| >65 | 24 (40) |
| Gender | |
| Female | 27 (45) |
| Male | 33 (55) |
| Disease stage at diagnosis | |
| II | 8 (14) |
| III | 26 (43) |
| IV | 26 (43) |
| Loss of expression | |
| MLH1/PMS2 | 30 (50) |
| PMS2 | 4 (7) |
| MSH2/MSH6 | 14 (23) |
| MSH6 | 6 (10) |
| Unknown | 6 (10) |
| Primary tumor location | |
| Left | 14 (23) |
| Right | 46 (77) |
| BRAF V600E mutation status | |
| Mutated | 9 (15) |
| Unmutated | 31 (52) |
| Unknown | 20 (33) |
| RAS mutation status | |
| Mutated | 11 (18.3) |
| Unmutated | 14 (23.3) |
| Unknown | 35 (58.3) |
| Number of prior therapies | |
| None | 15 (25) |
| 1 | 22 (36) |
| 2 | 9 (15) |
| ≥3 | 13 (21) |
| Unknown | 1 (3) |
| Agents | |
| Pembrolizumab | 47 (78) |
| Nivolumab | 8 (13) |
| Nivolumab + ipilimumab | 5 (8) |
Figure 2.
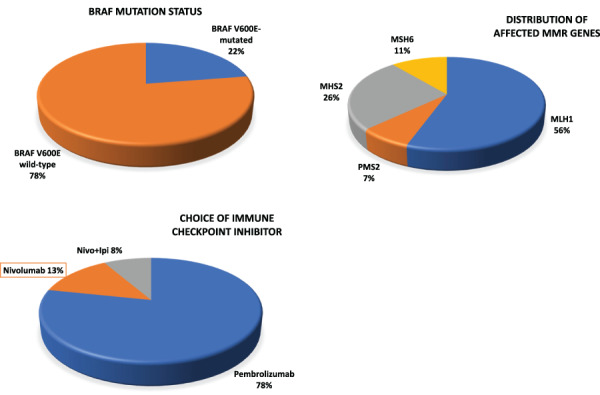
Distribution of clinical and molecular variables in the cohort of interest. Abbreviations: MMR, mismatch repair; Nivo+Ipi, nivolumab and ipilimumab.
Although it was not statistically significant, we observed more moderately and poorly differentiated tumors (62.5%) among patients with MLH1 and/or PMS2 alterations (Table 2), whereas tumor differentiation was similarly distributed in patients with MSH2 and/or MSH6 alterations. MSH2 and/or MSH6 alterations were predominantly seen in patients ≤65 (90%) and MLH1 and/or PMS2 alterations were more common among patients >65 (p < .01). More right‐sided tumors were observed in patients with MLH1 and/or PMS2 alterations as compared with those with MSH2 and/or MSH6 alterations (82.3% vs. 65%, respectively). A borderline significant association between BRAF V600E mutation and age > 65 was noted (77.8% vs. 35.5%, p = .053; Table 2). Nonsignificant differences were observed by BRAF mutation status for tumor sidedness and tumor grade (Table 2).
Table 2.
Clinical characteristics by BRAF V600E status and MMR genes
| Covariate and level | BRAF V600E mutation status | Affected MMR genes | ||||
|---|---|---|---|---|---|---|
| Not Present, n = 31 | Present, n = 9 | p value | MLH1 + PMS2, n = 34 (%) | MSH2 + MSH6, n = 20 (%) | p value | |
| Tumor grade | ||||||
| Well or moderately differentiated | 12 (40.0) | 3 (33.3) | .99 | 12 (37.5) | 9 (47.4) | .49 |
| Moderately to poorly or poorly | 18 (60.0) | 6 (66.7) | .99 | 20 (62.5) | 10 (52.6) | .49 |
| Age, years | ||||||
| ≤65 | 20 (64.5) | 2 (22.2) | .053 | 15 (44.1) | 18 (90.0) | <.001 |
| >65 | 11 (35.5) | 7 (77.8) | .053 | 19 (55.9) | 2 (10.0) | <.001 |
| Side of tumor | ||||||
| Right | 23 (74.2) | 8 (88.9) | .654 | 28 (82.3) | 13 (65.0) | .19 |
| Left | 8 (25.8) | 1 (11.1) | .654 | 6 (17.7) | 7 (35.0) | .19 |
The median follow‐up was 28.3 months for the entire cohort. We did not observe any difference for overall response rate (ORR) in patients with left‐sided tumors as compared with patients with right‐sided tumors (78.6% vs. 58.7.1%; p = .177; Table 3). Patients with liver metastasis had nearly half the response rate as compared with patients with other sites of metastasis (36.4% vs. 68.7.%; p = .081). Patients with BRAF V600E mutation exhibited a lower ORR as compared with patients with wild‐type BRAF, although this did not meet statistical significance (44.4% vs. 74.2% respectively; p = .120). There was no difference in ORR observed in patients with MLH1 and/or PMS2 loss versus MSH2 and/or MSH6 loss (70.6% vs. 60.0%; p = .425). However, 1‐year and 2‐year PFS rates favored patients who had MSH2 and/or MSH6 loss: 84.2% and 78.2% for MSH2 + MSH6 compared with 57.8% and 54.2% for MLH1 + PMS2 (p < .001; Table 4). PFS rates were significantly higher in patients with MMR‐D colorectal cancer (CRC) with wild‐type BRAF as compared with patients with BRAF V600E mutant CRC. PFS rate at 1 year for patients with BRAF V600E mutation and BRAF wild‐type was 40% and 73.3%,respectively (p < .001). A similar difference was also observed for 2‐year PFS rates (26.7% vs. 73.3%; p < .001; Fig. 3; Table 4). Patients >65 had significantly worse progression‐free outcomes as compared with patients ≤65, with a 2‐year PFS rate of 38.5% and 71.1%, respectively (Fig. 3; Table 4). Patients with liver metastasis had a statistically significant lower 2‐year PFS rate as compared with other metastatic sites (45.5% vs. 62% respectively; p = .014). No progression‐free survival outcome differences were observed based on the primary site of the tumor (left vs. right). In multivariate analysis, BRAF V600E mutation remained a statistically significant predictor of worse PFS (hazard ratio, 0.33; p = .045; supplemental online Table 1)
Table 3.
Response rates by clinical and molecular markers
| Covariate level | Best response | ||
|---|---|---|---|
| Progression of disease or stable disease, n = 22 | Partial response or complete response, n = 38 | p value | |
| Side of tumor | |||
| Right | 19 (41.3) | 27 (58.7) | .177 |
| Left | 3 (21.4) | 11 (78.6) | .177 |
| Tumor volume | |||
| Low | 10 (45.4) | 12 (54.6) | .317 |
| High | 12 (32.4) | 25 (67.6) | .317 |
| Metastatic site | |||
| Liver | 7 (63.6) | 4 (36.4) | .081 |
| Nonliver metastases | 15 (31.3) | 33 (68.7) | .081 |
| BRAF | |||
| None | 8 (25.8) | 23 (74.2) | .120 |
| Present | 5 (55.6) | 4 (44.4) | .120 |
| Age, years | |||
| ≤65 | 11 (30.6) | 25(69.4) | .229 |
| >65 | 11 (45.8) | 13 (54.2) | .229 |
| MMR genes | |||
| MLH1 + PMS2 | 10 (29.4) | 24 (70.6) | .425 |
| MSH2 + MSH6 | 8 (40.0) | 12 (60.0) | .425 |
Abbreviation: MMR, mismatch repair.
Table 4.
Twelve‐ and 24‐month PFS rates by clinical and molecular markers
| Covariate and level | 12‐month PFS rates, % | 24‐month PFS rates, % | p value |
|---|---|---|---|
| BRAF | p < .001 (for both 12‐ and 24‐month rates) | ||
| None | 73.3 | 73.3 | |
| Present | 40.0 | 26.7 | |
| MMR genes | p < .001 (for both 12‐ and 24‐month rates) | ||
| MLH1 + PMS2 | 57.8 | 54.2 | |
| MSH2 + MSH6 | 84.2 | 78.2 | |
| Age, years | p < .001 (for both 12‐ and 24‐month rates) | ||
| ≤65 | 74.2 | 71.1 | |
| >65 | 44.9 | 38.5 | |
| Metastatic site | p = .120 (for 12‐month rates) and p = .014 (for 24‐month rates) | ||
| Liver only | 54.5 | 45.5 | |
| Nonliver metastasis | 64.6 | 62.0 |
Abbreviations: MMR, mismatch repair; PFS, progression‐free survival.
Figure 3.
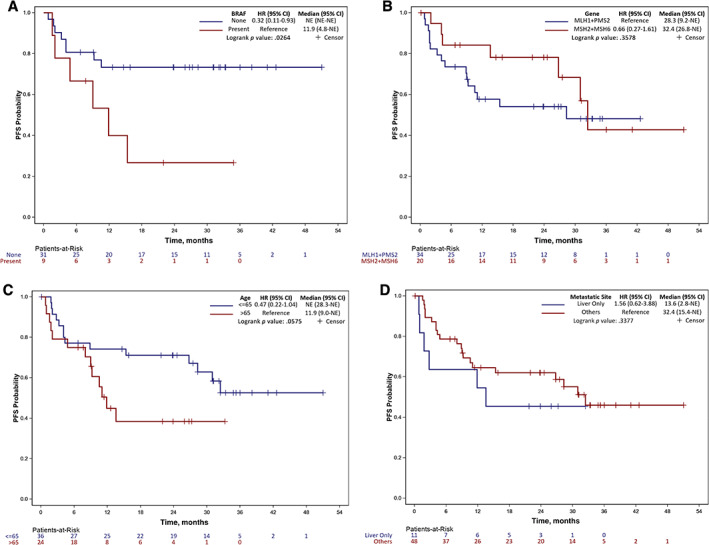
Kaplan‐Meier plots: (A) affected MMR; (B) BRAF V600 mutation status; (C) age groups; (D) metastasis site. Abbreviations: CI, confidence interval; HR, hazard ratio; NE, not estimable/not reached; PFS, progression‐free survival.
Discussion
Immune checkpoint inhibitors have led to significant improvements in survival among patients with MMR‐D colorectal cancer with sustained radiological and clinical responses [12]. Heterogeneity among patients with MMR‐D colorectal cancer exists, with variable responsiveness to immune checkpoint inhibitors, and at this time there is no biomarker of clinical response to immune checkpoint inhibitors. In our study, we identified BRAF V600E mutation and loss of expression of MLH or PMS2 proteins as a potential predictor of poor PFS rate at 1 year and 2 years in patients with MMR‐D colorectal cancer who were treated with immune checkpoint inhibitors. Patients with liver metastasis had worse clinical outcomes, which could be potential surrogates of resistance to immune checkpoint inhibitor therapy. The adverse outcomes in patients age > 65 were perhaps driven by increased BRAF V600E mutation in this subset of our cohort (Table 2).
Our findings suggest that BRAF V600E mutation may adversely affect the immune checkpoint inhibitor response in patients with MMR‐D colorectal cancer. The study of nivolumab and ipilimumab combination therapy (CheckMate 142) also investigated the effect of BRAF V600E mutation, and the response rate was found to be similar across the subgroups (55% vs. 55%) [12] The impact of BRAF mutations was also investigated in the phase II trial of single‐agent nivolumab in patients with MMR‐D colorectal cancer, and the response rate among BRAF V600E mutation carriers was 25%, whereas it was 41.4% for patients with wild‐type BRAF [13] which was deemed to be a nonsignificant difference. The KEYNOTE‐177 study investigated the potential impact of BRAF V600E mutation on survival outcomes in treatment‐naive patients, and the authors identified improved outcomes with the use of pembrolizumab as compared with chemotherapy regardless of BRAF V600E mutation [14]. In these clinical trials, however, BRAF V600E mutation was not evaluated for the durability of response among patients treated with immune checkpoint inhibitor therapy. In our study, the presence of BRAF V600E mutation correlated with worse PFS rates at 1 year and 2 years, suggesting BRAF V600E mutation may impact the durability of benefit from immune checkpoint inhibitor therapy. It is important to note that, although we observed worse outcomes with BRAF V600E mutation among patients who received immune checkpoint inhibitor therapy, patients with MMR‐D colorectal cancer with BRAF V600E mutation still had better PFS outcomes with the use of pembrolizumab as compared with chemotherapy in the KEYNOTE‐177 study [14]. This indicates that the negative predictive marker of BRAF V600E mutation should not discourage clinicians to use immune checkpoint inhibitor therapy in this subset of patients with MMR‐D colorectal cancer. Notably, consistent with previous reports from CheckMate 142 cohorts, in our study, BRAF V600E mutation did not predict ORR. These findings also suggest that biomarker analysis for immune checkpoint inhibitor response in MMR‐D colorectal cancer should evaluate the durability of response in addition to ORR.
BRAF V600E mutant MMR‐D colorectal cancer carries clinically distinct characteristics as compared with patients with Lynch syndrome [9]. BRAF mutation‐associated MMR‐D is tightly associated with the CpG island methylator phenotype in which the MLH1 gene promoter region undergoes hypermethylation that results in the silencing of this gene [10]. Most of the sporadic cases of MMR‐D colorectal cancers are linked with these molecular features. Unlike with germline MMR‐D colorectal cancer, patients harboring BRAF V600E–driven MMR‐D present later in age, with right‐sided tumors and an advanced stage at presentation [4]. In contrast, patients with germline mutations present with left‐sided colon cancer at a younger age [9]. Our findings related to clinical outcome differences in BRAF mutant MMR‐D colorectal cancer are consistent with the clinically distinct behavior of these two different MMR‐D colorectal cancer entities. Whether the direct oncogenic activity of the BRAF V600E mutation resulting in MAPK pathway activation or other BRAF V600E‐driven oncogenic pathways cause resistance to immune checkpoint inhibitors is unclear. Preclinical evidence suggests activation of the MAPK pathway, which is also activated by BRAF mutations [16], may have a significant impact on immune evasion. For example, KRAS mutations can upregulate signal transduction via BRAF and MAPK pathways and ultimately lead to immune evasion by selective conversion of T cells into regulatory T cells [17] and recruitment of myeloid‐derived suppressor cells into the tumor microenvironment [18]. Most notably, BRAF inhibitors can also have a direct effect on antitumor immunity by increasing cancer‐associated antigen expression and tumor‐reactive T cell infiltration. Collectively, these data suggest BRAF mutations may also impact immune recognition and removal of cancer cells [19, 20, 21, 22, 23].
Our study indicates that biological variation among MMR genes may influence patient survival outcomes. The role of specific MMR genes has been investigated by several studies, and the presence of mutations in distinct MMR genes confers differential risk of development of colorectal cancer [24]. For example, a cohort of 61 patients with germline PMS2 mutations evaluated the penetrance by using segregation analysis [25]. The authors reported a relatively lower risk of colorectal cancer as compared with MLH1 and MSH2 mutation carriers because of their relatively lower penetrance among monoallelic PMS2 mutation carriers [25, 26]. MSH6 mutations are also associated with a significantly lower risk of cancer development as compared with MLH1 and MSH2 mutations [26, 27], indicating significant biologic heterogeneity exists among MMR genes. Growing evidence also suggests that the mutational landscape of patients with MMR‐D colorectal cancer is highly heterogeneous [15, 28]. For example, loss of function mutations in β‐catenin, which are associated with more invasive behavior in colorectal cancer [29], appears to be more common in germline MLH1 mutations compared with other MMR genes [30]. Most notably, a recent study reported higher tumor mutation burden, a biomarker of immune checkpoint inhibitor response, in patients with colorectal cancer with loss of MSH2/MSH6 proteins compared with MLH1/PMS2 alterations [31]. The effect of this heterogeneity in the underlying cause of microsatellite instability in colorectal cancer survival outcomes with immune checkpoint inhibitors has not been studied. Perhaps hypermethylation commonly observed in patients with absent MLH1 and mutated BRAF V600E leads to silencing of expression of potentially antigenic proteins (neoantigens) [10] and limits the benefit from immune therapy. Larger cohorts are needed to better characterize the exact impact of specific MMR genes on clinical outcomes of patients with MMR‐D colorectal cancer treated with immune checkpoint inhibitors.
We identified that liver metastasis is associated with worse 2‐year PFS rates. This clinical feature of poor outcomes is consistent with data from patients with other solid tumors treated with immune checkpoint inhibitors, including in the setting of melanoma and non‐small cell lung cancer (NSCLC) [32]. The study by Tumeh et al. [32] revealed a shorter PFS rate in patients with melanoma and NSCLC with liver metastasis when they were treated with pembrolizumab. The authors identified decreased CD8+ T cell infiltration at the invasive margin in patients with liver metastasis as compared with patients with no liver metastasis. Consistently, animal studies identified decreased CD8+ T cells and increased T regulatory cells with liver metastasis along with the significantly lower expression of postactivation markers such as PD‐1, ICOS, and CTLA‐4 [33]. Our finding is in alignment with the growing evidence discussed above, and further studies are needed to better understand the underlying molecular mechanisms. Certainly, the liver has a distinct microenvironment whereby immune regulatory cells are abundant [34], and the acute phase response orchestrated by hepatic cells originates [35] by design to control the body's inherent reaction to antigenic exposure. More research is needed to uncover mechanistic facets of liver metastasis and their impact on immunotherapy response.
Our study is limited by the small size of the cohort, retrospective nature of the study, which limits data collections, and lack of overall survival data due to the heterogeneity among the lines of therapy in which immune checkpoint inhibitors were used. The data set did not also include other potential biomarkers such as tumor mutation burden and other molecular alterations including RAS status because of a lack of next‐generation–based molecular data in the majority of the patients at the time of analysis. Further prospective studies with larger cohorts are needed to confirm our findings and better understand the exact mechanisms of resistance in these subsets of patients with MMR‐D colorectal cancer.
Conclusion
Our study showed a detrimental impact of BRAF V600E mutation and MLH1/PMS2 loss on PFS outcomes of patients with MMR‐D colorectal cancer. Notably, we also identified adverse outcomes in patients with liver metastasis. Novel therapeutic approaches should be investigated, particularly for patients with BRAF V600E mutant MMR‐D colorectal cancer.
Author Contributions
Conception/design: Ibrahim Halil Sahin, Christina Wu
Provision of study material or patients: Ibrahim Halil Sahin, Yoanna Pumpalova, Mohamad B. Sonbol, Satya Das
Collection and/or assembly of data: Ibrahim Halil Sahin, Yoanna Pumpalova, Mohamad B. Sonbol, Satya Das, Subir Goyal
Data analysis and interpretation: Subir Goyal, Ibrahim Halil Sahin
Manuscript writing: Ibrahim Halil Sahin, Subir Goyal, Yoanna Pumpalova, Mohamad B. Sonbol, Satya Das, Sigurdis Haraldsdottir, Daniel Ahn, Kristen K. Ciombor, Zhengjia Chen, Amber Draper, Jordan Berlin, Tanios Bekaii‐Saab, Gregory B. Lesinski, Bassel F. El‐Rayes, Christina Wu
Final approval of manuscript: Ibrahim Halil Sahin, Subir Goyal, Yoanna Pumpalova, Mohamad B. Sonbol, Satya Das, Sigurdis Haraldsdottir, Daniel Ahn, Kristen K. Ciombor, Zhengjia Chen, Amber Draper, Jordan Berlin, Tanios Bekaii‐Saab, Gregory B. Lesinski, Bassel F. El‐Rayes, Christina Wu
Disclosures
Daniel Ahn: Eisai, Exelixis, Genentech (C/A), Bayer, Astra Zeneca (RF); Kristen K. Ciombor: C/A: Merck, Array, Foundation Medicine, Taiho, Natera, Array (C/A), Bristol‐Myers Squibb, Array, Incyte, Daiichi Sankyo, Nucana, Abbvie, Merck, Pfizer, Calithera (RF); Jordan Berlin: Bayer, Ipsen, Rafael, Seattle Genetics, EMD Serono, QED, Clovis (C/A), Dragonfly, Eli Lilly & Co, Loxo, Bayer, Pfizer, Calithera, AbbVie, Immunomedics, Incyte, Symphogen, Boston Biomedical, PsiOxus (RF), Pancreatic cancer Action Network, Novocure (Other [Data Safety Monitoring Board]); Gregory B. Lesinski: ProDa Biotech (C/A), Merck and Co., Bristol‐Myers Squibb, Boerhinger‐Ingelheim, and Vaccinex (RF [Institution]). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Table 1S Multivariate analysis with best selection method for PFS
Acknowledgments
Research reported in this publication was supported in part by the Biostatistics Shared Resource of Winship Cancer Institute of Emory University and National Institutes of Health/National Cancer Institutes under award P30CA138292. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Ethical approval and consent to participate: The approval of the institutional review boards, patient databases at Winship Cancer Institute of Emory University, Mayo Clinic, Stanford University, and Vanderbilt University were obtained (IRB00097021). Data available per request.
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer Clin 2020;70:7–30. [DOI] [PubMed] [Google Scholar]
- 2. Ahnen DJ, Wade SW, Jones WF et al. The increasing incidence of young‐onset colorectal cancer: A call to action. Mayo Clin Proc 2014;89:216–224. [DOI] [PubMed] [Google Scholar]
- 3. Liang PS, Allison J, Ladabaum U et al. Potential intended and unintended consequences of recommending initiation of colorectal cancer screening at age 45 years. Gastroenterology 2018;155:950–954. [DOI] [PubMed] [Google Scholar]
- 4. Liu B, Nicolaides NC, Markowitz S et al. Mismatch repair gene defects in sporadic colorectal cancers with microsatellite instability. Nat Genet 1995;9:48–55. [DOI] [PubMed] [Google Scholar]
- 5. Liu B, Parsons R, Papadopoulos N et al. Analysis of mismatch repair genes in hereditary non–polyposis colorectal cancer patients. Nat Med 1996;2:169–174. [DOI] [PubMed] [Google Scholar]
- 6. Parsons R, Li G‐M, Longley MJ et al. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell 1993;75:1227–1236. [DOI] [PubMed] [Google Scholar]
- 7. Strauss BS. Frameshift mutation, microsatellites and mismatch repair. Mutat Res 1999;437:195–203. [DOI] [PubMed] [Google Scholar]
- 8. Haraldsdottir S, Hampel H, Tomsic J et al. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology 2014;147:1308–1316.e1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lynch HT, Lynch JF, Shaw TG et al. HNPCC (Lynch syndrome): Differential diagnosis, molecular genetics and management ‐ A review. Hered Cancer Clin Pract 2003;1:7–18. [Google Scholar]
- 10. Weisenberger DJ, Siegmund KD, Campan M et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet 2006;38:787–793. [DOI] [PubMed] [Google Scholar]
- 11. Le DT, Uram JN, Wang H et al. PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med 2015;372:2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Overman MJ, Lonardi S, Wong KYM et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair‐deficient/microsatellite instability‐high metastatic colorectal cancer. J Clin Oncol 2018;36:773–779. [DOI] [PubMed] [Google Scholar]
- 13. Overman MJ, McDermott R, Leach JL et al. Nivolumab in patients with metastatic DNA mismatch repair‐deficient or microsatellite instability‐high colorectal cancer (CheckMate 142): An open‐label, multicentre, phase 2 study. Lancet Oncol 2017;18:1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Andre T, Shiu KK, Kim TW et al. Pembrolizumab versus chemotherapy for microsatellite instability‐high/mismatch repair deficient metastatic colorectal cancer: The phase 3 KEYNOTE‐177 study. J Clin Oncol 2020;18(suppl)LBA4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sahin IH, Akce M, Alese O et al. Immune checkpoint inhibitors for the treatment of MSI‐H/MMR‐D colorectal cancer and a perspective on resistance mechanisms. 2019;121:809–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sumimoto H, Imabayashi F, Iwata T et al. The BRAF–MAPK signaling pathway is essential for cancer‐immune evasion in human melanoma cells. J Exp Med 2006;203:1651–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zdanov S, Mandapathil M, Eid RA et al. Mutant KRAS conversion of conventional T cells into regulatory T cells. Cancer Immunol Res 2016;4:354–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liao W, Overman MJ, Boutin AT et al. KRAS‐IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell 2019;35:559–572.e557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ho PC, Meeth KM, Tsui YC et al. Immune‐based antitumor effects of braf inhibitors rely on signaling by CD40L and IFNγ. Cancer Res 2014;74:3205–3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boni A, Cogdill AP, Dang P et al. Selective BRAFV600E inhibition enhances T‐cell recognition of melanoma without affecting lymphocyte function. Cancer Res 2010;70:5213–5219. [DOI] [PubMed] [Google Scholar]
- 21. Wilmott JS, Long GV, Howle JR et al. Selective BRAF inhibitors induce marked T‐cell infiltration into human metastatic melanoma. Clin Cancer Res 2012;18:1386–1394. [DOI] [PubMed] [Google Scholar]
- 22. Knight DA, Ngiow SF, Li M et al. Host immunity contributes to the anti‐melanoma activity of BRAF inhibitors. J Clin Invest 2013;123:1371–1381. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23. Frederick DT, Piris A, Cogdill AP et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res 2013;19:1225–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Peltomäki P. Lynch syndrome genes. Fami Cancer 2005;4:227–232. [DOI] [PubMed] [Google Scholar]
- 25. Senter L, Clendenning M, Sotamaa K et al. The clinical phenotype of Lynch syndrome due to germ‐line PMS2 mutations. Gastroenterology 2008;135:419–428.e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Talseth‐Palmer BA, McPhillips M, Groombridge C et al. MSH6 and PMS2 mutation positive Australian Lynch syndrome families: Novel mutations, cancer risk and age of diagnosis of colorectal cancer. Hered Cancer Clin Pract 2010;8:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bonadona V, Bonaïti B, Olschwang S et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011;305:2304–2310. [DOI] [PubMed] [Google Scholar]
- 28. Gladbach YS, Wiegele L, Hamed M et al. Unraveling the heterogeneous mutational signature of spontaneously developing tumors in MLH1−/− mice. Cancer (Basel) 2019;11:1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ahadova A, von Knebel Doeberitz M, Bläker H et al. CTNNB1‐mutant colorectal carcinomas with immediate invasive growth: A model of interval cancers in Lynch syndrome. Fam Cancer 2016;15:579–586. [DOI] [PubMed] [Google Scholar]
- 30. Engel C, Ahadova A, Seppälä T et al. Associations of pathogenic variants in MLH1, MSH2, and MSH6 with risk of colorectal adenomas and tumors and with somatic mutations in patients with lynch syndrome. Gastroenterology 2020;158:1326–1333. [DOI] [PubMed] [Google Scholar]
- 31. Salem ME, Grothey A, Kim ES et al. Impact of MLH1, PMS2, MSH2, and MSH6 alterations on tumor mutation burden (TMB) and PD‐l1 expression in 1,057 microsatellite instability‐high (MSI‐H) tumors. J Clin Oncol 2018;36(suppl)3572a. [Google Scholar]
- 32. Tumeh PC, Hellmann MD, Hamid O et al. Liver metastasis and treatment outcome with anti‐PD‐1 monoclonal antibody in patients with melanoma and NSCLC. Cancer Immunol Res 2017;5:417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee J, Mehdizadeh S, Tsai K et al. Immunological insights into liver metastasis associated resistance to checkpoint blockade cancer immunotherapy. Am Assoc Immnol 2018;200(suppl):122.26. [Google Scholar]
- 34. Pedroza‐Gonzalez A, Verhoef C, Ijzermans JN et al. Activated tumor‐infiltrating CD4+ regulatory T cells restrain antitumor immunity in patients with primary or metastatic liver cancer. Hepatology 2013;57:183–194. [DOI] [PubMed] [Google Scholar]
- 35. He G and Karin M. NF‐κb and STAT3–Key players in liver inflammation and cancer. Cell Res 2011;21:159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Table 1S Multivariate analysis with best selection method for PFS