

Abstract
Anoctamin 1 (ANO1) is a kind of calcium-activated chloride channel involved in nerve depolarization. ANO1 inhibitors display significant analgesic activity by the local peripheral and intrathecal administration. In this study, several thiophenecarboxylic acid and benzoic acid derivatives were identified as novel ANO1 inhibitors through the shape-based virtual screening, among which the 4-arylthiophene-3-carboxylic acid analogues with the best ANO1 inhibitory activity were designed, synthesized and compound 42 (IC50 = 0.79 μmol/L) was finally obtained. Compound 42 selectively inhibited ANO1 without affecting ANO2 and intracellular Ca2+ concentration. Subsequently, the analgesic effect was investigated by intragastric administration in pain models. Compound 42 significantly attenuated allodynia which was induced by formalin and chronic constriction injury. Through homology modeling and molecular dynamics, the binding site was predicted to be located near the calcium-binding region between α6 and α8. Our study validates ANO1 inhibitors having a significant analgesic effect by intragastric administration and also provides selective molecular tools for ANO1-related research.
KEY WORDS: ANO1 (anoctamin 1, TMEM16A); Inhibitor; Synthesis; Structure–activity relationship; Analgesia
Graphical abstract
Calcium-activated chloride channel anoctamin 1 (ANO1) is a potential target for pain treatment. A novel series of 4-arylthiophene-3-carboxylic acid compounds are discovered to intensely and selectively suppress the ANO1 channel and orally attenuate allodynia which was induced by formalin and chronic constriction injury.
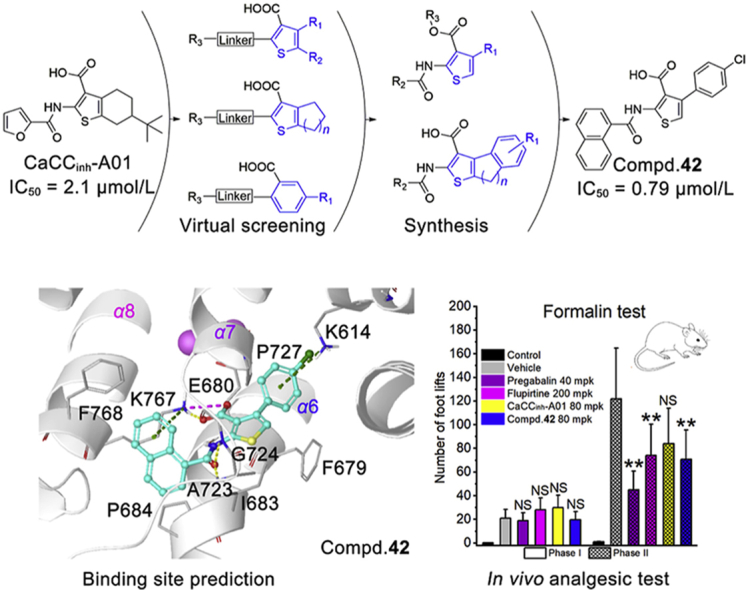
1. Introduction
Anoctamin 1 (ANO1, TMEM16A) is a kind of the calcium-activated chloride channel (CaCC) on cytomembrane, controlled by intracellular Ca2+ concentration and membrane potential1, 2, 3, 4, 5, 6, 7, 8. ANO1 is mainly expressed in airway9,10, peripheral nervous system11, gastrointestinal smooth muscle12, and some tumors13, 14, 15, which mediates and controls anion permeation to fulfill airway and exocrine gland secretion, neuronal signal transduction, and rhythmic movements of the gastrointestinal system.
In the peripheral nervous system, ANO1 is distributed in nociceptors, dorsal root ganglia (DRG), and spinal nerves, involved in nerve depolarization11,16,17. ANO1 is capable of augmenting the excitability of DRG neurons under inflammatory or neuropathic conditions and thereby aggravates inflammation- or tissue injury-induced pathological pain18,19. Knockout or inhibition of ANO1 causes significant analgesic effects. Intrathecal injection of the ANO1 inhibitor reverts the spinal nerve ligation-induced mechanical allodynia in a dose-dependent manner16. The non-selective and selective CaCC blockers reduce formalin-induced flinching behavior mainly during phase 2 of the formalin test20.
ANO1 is considered as associated with inflammation-related pain caused by the changes in temperature, ion concentration in inflamed tissues, and inflammatory factors21. The elevated temperature of inflamed tissue activates temperature-sensitive calcium channels (such as transient receptor potential channel V1, TRPV1), which activates ANO1 not only by direct interaction21,22 but also indirectly by increasing intracellular calcium ion concentrations23. Interleukin-4, an inflammatory factor in inflamed tissues or injured nerves, can promote the overexpression of ANO1, which may be related to allodynia5,16.
In the last 10 years, many ANO1 inhibitors (Fig. 1) are identified and synthesized including CaCCinh-A0124 (1), T16Ainh-A0125 (2), MONNA26 (3), AACT10 (4), Ani927 (5), and digallic acid28 (6). More recently, some clinically used drugs and natural products are found to inhibit ANO1, such as niclosamide, nitazoxanide12, flavonoids29,30, resveratrol31, etc. Among these compounds, AACT10 and Ani927 have ANO1/ANO2 selectivity (ANO2 is a homologous protein of ANO1 that functions as a CaCC in brain)32. These inhibitors were used in various pharmacodynamics studies: AACT suppresses isometric smooth muscle contraction in mouse ileum tissue10. CaCCinh-A01 and Ani9 inhibit the proliferation of ANO1 over-expressed tumor15,27. Niclosamide fully bronchodilates airways in vitro12. CaCCinh-A01, T16Ainh-A01, and MONNA attenuate formalin-induced hyperalgesia and allodynia by local peripheral and intrathecal administration16,19,20. In short, though many ANO1 inhibitors have been found, their physiological functions are varied and not fully investigated. For the discovery of potential clinical application, the biological behaviors of ANO1 and its inhibitors need to be clearly elucidated, and more importantly, structurally diverse and active inhibitors are required.
Figure 1.

Chemical structure of reported ANO1 inhibitors.
In this study, to explore new ANO1 inhibitors and potential analgesic agents, we firstly performed a scaffold virtual screening starting from the 2-arylamidothiophene skeleton shared in CaCCinh-A01 (1) and AACT (4), and a series of thiophenic acid and benzoic acid derivatives showed ANO1 inhibition. Subsequently, the 4-aryl-thiophene-3-acid selected from the screening results was used as the starting hit for optimization. The inhibitory activities were evaluated by whole-cell patch clamp on ANO1-overexpressing Fischer rat thyroid (FRT) cells. The activity preferred compound was used for analgesic evaluation in various animal pain models. The finding of this study is helpful for the understanding of the structure–activity relationships (SARs) of ANO1 inhibitors and provides a selective inhibitor for probing the biological function of the ANO1 channel.
2. Results and discussions
2.1. Shape and electrostatic similarity-based virtual screening
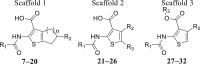
The shape and electrostatic similarity-based virtual screening query were generated from the optimized conformation of CaCCinh-A01 in OPENEYE software (Fig. 2). 300,000 compounds from the SPECS chemical library and Chinese National Compound Library of Peking University (PKU_CNCL) were screened followed by artificial selection of the molecules with Shape Tanimoto and EON index greater than 0.6. Seventy-six compounds, including thiophenic acid derivatives (Table 1, compounds 7–32), benzoic acid derivatives (Supporting Information Fig. S1, compounds S1–S13), and 37 other scaffold compounds (Fig. S1, compounds S14–S50). Their ANO1 inhibitory effects were tested using whole cell patch clamp at 100 μmol/L in ANO1-overexpression FRT cell line.
Figure 2.

Shape and electronic similarity-based virtual screening. (A) Scaffolds obtained from shape-based virtual screening; the purple surface represents the virtual screening query in OPENEYE software. (B) The general structure–activity relationships which produced by virtual screening.
Table 1.
Structure and ANO1 inhibitory activity of thiophenic acid derivatives.
 | |||||
|---|---|---|---|---|---|
| Compd. | n | R1 | R2 | R3 | Inh. (%)a |
| 1 | 2 |  |
t-Bu | ‒ | 78.3 |
| 7 | 2 |  |
t-Bu | ‒ | 82.8 |
| 8 | 2 | 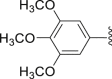 |
‒CH3 | ‒ | 25.4 |
| 9 | 2 | 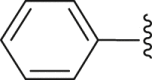 |
‒H | ‒ | 37.8 |
| 10 | 2 | 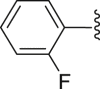 |
‒H | ‒ | 74.3 |
| 11 | 2 | 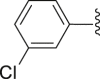 |
‒H | ‒ | 51.6 |
| 12 | 2 | 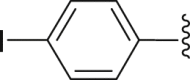 |
‒H | ‒ | 69.3 |
| 13 | 2 | 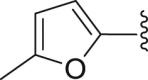 |
‒H | ‒ | 30.3 |
| 14 | 2 | 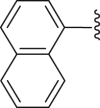 |
‒H | ‒ | 72.5 |
| 15 | 2 | 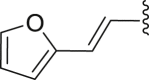 |
‒H | ‒ | 45.8 |
| 16 | 2 |  |
‒H | ‒ | 18.3 |
| 17 | 3 |  |
‒H | ‒ | 70.5 |
| 18 | 3 |  |
‒H | ‒ | 45.9 |
| 19 | 3 |  |
‒H | ‒ | 80.6 |
| 20 | 1 | 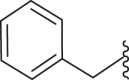 |
‒H | ‒ | 35.1 |
| 21 | ‒ | 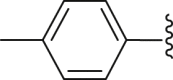 |
‒H | ‒CH3 | 33.1 |
| 22 | ‒ | 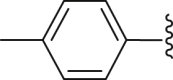 |
‒CH3 | ‒CH3 | 84.4 |
| 23 | ‒ | 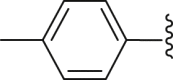 |
‒CH3 | ‒CH2CH2CH3 | 74.9 |
| 24 | ‒ | 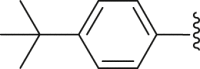 |
‒CH3 | ‒CH3 | 35.8 |
| 25 | ‒ | 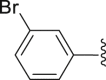 |
‒CH3 | ‒CH3 | 42.5 |
| 26 | ‒ | 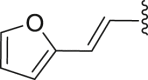 |
‒CH3 | ‒CH3 | 19.6 |
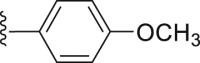
| 27 | ‒ | 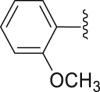 |
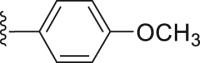 |
‒H | 33.4 |
| 28 | ‒ | 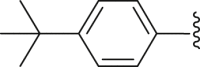 |
 |
‒H | 89.2 |
| 29 | ‒ | 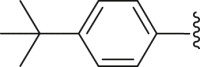 |
 |
‒CH3 | 12.4 |
| 30 | ‒ |  |
 |
‒C2H5 | 21.4 |
| 31 | ‒ |  |
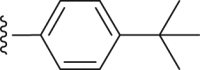 |
‒CH3 | 46.1 |
| 32 | ‒ | 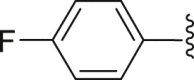 |
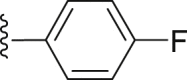 |
‒CH3 | 13.0 |
ANO1 inhibition rate of compounds at 100 μmol/L was determined using whole-cell patch clamp on ANO1-overexpressing FRT cells at +100 mV voltage. Data are represented by mean ± SEM, n = 5.
Of these 76 structurally diversified compounds, we focused on three thiophene containing scaffolds (scaffolds 1–3) which CaCCinh-A01 and AACT share (compounds 7–32, Table 1), while the ANO1 inhibitory activity of other compounds was shown in Supporting Information Table S1 due to they showed relatively low ANO1 inhibitory activity in whole-cell patch clamp.
The scaffold 1 varies in the ring size of tetrahydrobenzo[b]thiophene in CaCCinh-A01 (1). Active compounds appeared to have t-butyl substitution as R2 (1 and 7). Compounds with 2-F-phenyl (10), 4-I-phenyl (12), and 1-naphthalene (14) as R1 were more active than others. Extending the amide linker by the double bond (15) or methylene (16) is detrimental to the ANO1 inhibitory activity. Expanding the thiophene-fused aliphatic ring contributes to the enhancement of ANO1 inhibitory activity, for example, compound 17 (n = 3) is better than compound 9 (n = 2).
In scaffold 2, the moiety of tetrahydrobenzothiophene is replaced by thiophene in which positions 4′ and 5′ are substituted by alkyl or aromatic groups. Compounds 22 and 23 display comparative ANO1 inhibitory activity with CaCCinh-A01 (1), indicating the feasibility of breaking tetrahydrobenzo-ring. Alkyl substitution at R2 (22 and 23) is more favorable than non-substitution (21). Among the substituents of the R1, 4-methylphenyl (22) is superior to 4-tert-butylphenyl (24) and 3-bromophenyl (25). Extending the amide linker by the double bond is unfavorable to the ANO1 inhibitory activity.
Scaffold 3 replaces the R2 position in scaffold 2 with an aromatic group. Compound 28 with 4-(4-oxymethylbenzo)-thiophene scaffold has higher ANO1 inhibitory activity than CaCCinh-A01 (1). The 4-tert-butylphenyl substituent of R1 (28) is more active than 2-methoxyphenyl (27). Additionally, if the carboxyl group of R3 is replaced by an ester group, the ANO1 inhibitory activity declined (29–32).
Based on the preliminary SAR analysis, compound 28 with 4-arylthiophene-3-carboxylic acid backbone was selected as a hit for further optimization.
2.2. Synthesis and optimization of 4-arylthiophene-3-carboxylic acid derivatives
Based on compound 28, further optimization strategies involved three steps: firstly, 4-methoxyphenyl at thiophene 4′ position was replaced by various substituted phenyl and naphthyl groups. Then, the substructure on the carbonyl side of the amide was replaced by the preferred groups from the above virtual screening process. Finally, combined the substructures of tetrahydrobenzothiophene in CaCCinh-A01 and the aryl substituted thiophene at 4′ position in compound 28 to expand the volume of thiophene containing scaffold. ANO1 inhibitory rate was conducted by yellow fluorescence protein (YFP) fluorescence quenching assay24 in ANO1-overexpressing FRT cell line and was calculate by Eq. (1):
| Inhibition rate (%) = (Kcont − Kcomp)/Kcont × 100 | (1) |
where Kcont means the slope of negative control, Kcomp means the slope of the test compound. The ANO1 inhibitory IC50 was determined based on the inhibitory rate in 7 compound concentrations.
Compound 42 and its derivatives were synthesized by three steps (Scheme 1, Scheme 2). Firstly, ketones reacted with cyanoacetate and sulfur to form 2-aminothiophene-3-carboxylate acid ethyl ester intermediates by the Gewald reaction33. Then these intermediates are coupled with acyl chloride to form the amides derivatives. Finally, the hydrolysis reactions were conducted to form final products.
Scheme 1.

Synthetic route of 4-arylthiophene-3-carboxylic acid derivatives. Reagents and conditions: (I) step 1: ethyl cyanoacetate, morpholine, acetic acid, ethanol, 60 °C, 3 h, argon; step 2: sulfur, argon, 60 °C, 36–48 h. (II) Aryl substituted acid chloride, TEA, DCM, rt, 8 h. (III) NaOH, H2O, MeOH, THF, 60 °C, 8 h.
Scheme 2.

Synthetic route of 8H-indeno [2,1-b] thiophene-3-carboxylic acid and 4, 5-dihydronaphtho [2,1-b] thiophene-1-carboxylic acid derivatives.. Reagents and conditions: (I) step 1: ethyl cyanoacetate, ammonium acetate, toluene, acetic acid, reflux, 12 h, argon; step 2: sulfur, ethanol, argon, 60 °C, 36–48 h. (II) Aryl substituted acid chloride, TEA, DCM, rt, 8 h. (III) NaOH, H2O, MeOH, THF, 60 °C, 8 h.
2.3. SAR analysis of 4-arylthiophene-3-carboxylic acid derivatives
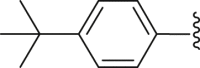
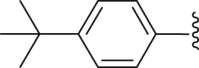
For the substituents at the R1 position (Table 2), most of the substituents are acceptable except for 4-CF3-phenyl (35) and 2,4-dichloridephenyl substitutions (38). The most potent 4-chlorophenyl (34) was selected for R2 optimization. For this, we selected the preferred fragments which showed in the previous virtual screening (such as thiophenyl, 1-naphthyl, and 4-t-bultyphenyl). Among which 1-naphthyl (42) substituted compound showed the highest ANO1 inhibitory activity (IC50 = 0.79 μmol/L). Besides, the inhibitory activity of ANO1 was slightly decreased when 2-thiophenthyl (40) and 4-methyl-1-naphthyl (46) were substituted. In summary, when R1 was substituted with 4-Cl-phenyl and R2 with 1-naphthyl, the compound showed the strongest ANO1 inhibitory activity (compound 42).
Table 2.
Structure and ANO1 inhibitory IC50 of 4-arylthiophene-3-carboxylic acid derivatives.
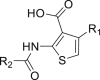 | |||
|---|---|---|---|
| Compd. | R1 | R2 | IC50 (μmol/L)a |
| 1 | – | – | 2.1 |
| 28 | 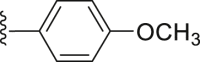 |
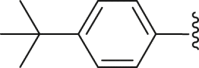 |
6.38 |
| 33 | 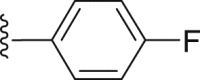 |
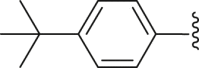 |
9.62 |
| 34 | 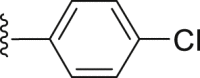 |
 |
1.34 |
| 35 | 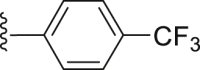 |
 |
>100 |
| 36 | 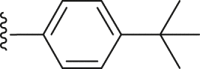 |
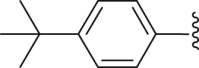 |
3.67 |
| 37 | 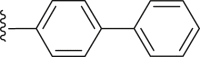 |
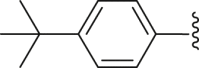 |
2.30 |
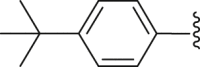
| 38 | 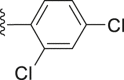 |
 |
>100 |
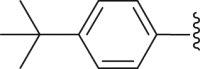
| 39 | 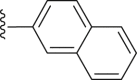 |
 |
1.29 |
| 40 | 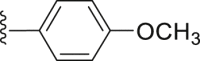 |
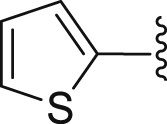 |
25.51 |
| 41 | 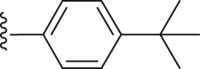 |
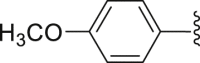 |
2.28 |
| 42 | 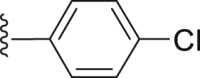 |
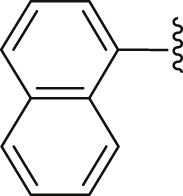 |
0.79 |
| 43 | 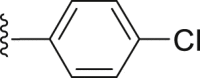 |
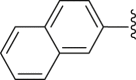 |
2.45 |
| 44 | 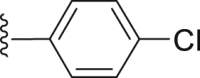 |
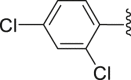 |
1.24 |
| 45 | 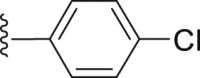 |
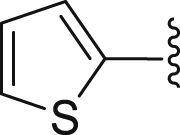 |
1.74 |
| 46 | 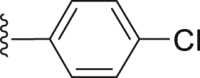 |
 |
14.50 |
IC50 value was determined by YFP fluorescence quenching assay. Data are represented by mean ± SEM, n = 3.
Compounds 47–53 combined the substructures of tetrahydrobenzothiophene in CaCCinh-A01 and the aryl substituted thiophene at 4′ position in scaffold 3 (Table 3). When the R1 was H (47), the inhibitory activity against ANO1 was higher than that of the methoxy group (48). Methyl substitution at R2 and R3 positions (42) favored ANO1 inhibitory activity. 1-Naphthyl (51) or 2-naphthyl (50) at R4 position was beneficial to ANO1 inhibition. Compound 53 was more active than compound 47, showed that the thiophene-fused a larger cycloalkane had a higher ANO1 inhibitory activity. It indicated that the flexibility between thiophene and benzene rings might favor activity. However, compounds 47–53 showed poor solubility in synthesis (only completely soluble in DMSO, slightly soluble in methanol, insoluble in water and DCM), which would cause a lot of trouble for further study, so we terminated the optimization of these scaffolds.
Table 3.
Structure and ANO1 inhibitory IC50 of compounds 47–53.
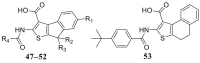 | |||||
|---|---|---|---|---|---|
| Compd. | R1 | R2 | R3 | R4 | IC50 (μmol/L)a |
| 1 | – | – | – | – | 2.1 |
| 47 | H | H | H |  |
12.59 |
| 48 | OCH3 | H | H | 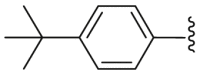 |
28.94 |
| 49 | H | H | H | 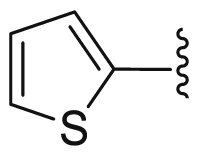 |
11.29 |
| 50 | H | H | H | 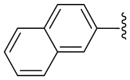 |
1.91 |
| 51 | H | H | H | 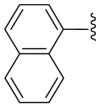 |
2.21 |
| 52 | H | CH3 | CH3 | 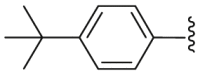 |
3.33 |
| 53 | – | – | – | – | 5.15 |
IC50 value was determined by YFP fluorescence quenching assay. Data are represented by mean ± SEM, n = 3.
Based on the SAR studies of thiophene-containing ANO1 inhibitors (both in virtual screening and structure optimization), compound 42 (IC50 = 0.79 μmol/L) was finally selected for further pharmacokinetic and pharmacodynamics study.
2.4. Electrophysiology and Ca2+ fluorescence investigations of compound 42
To verify the inhibitory activity of compound 42 on ANO1, we performed whole-cell patch clamp in ANO1-overexpressing FRT cells as previously described10. In this experiment, the ANO1 channel was activated by 1 μmol/L intracellular Ca2+ followed by changing the membrane potential +100 to −100 mV and comparing the evoked CaCC current before and after treatment. As shown in Fig. 3A, 30 μmol/L compound 42 significantly suppressed the CaCC current and was more potent than Ani9.
Figure 3.

ANO1 inhibitory activity validation. (A) CaCC current in whole-cell patch-clamp; FRT cells were clamped from the holding potential of 0 mV for 50 ms to 100 mV for 400 ms and then repolarized to −100 mV for 400 ms and subsequently returned to 0 mV. Current curve is representative of similar results obtained in two independent experiments per compound and was repeat tested more than 5 times in each independent experiment; the inhibitory rate (Inh. %) was calculated from the current under +100 mV potential; data are represented by mean ± SEM, n = 2; the concentration of compound is 30 μmol/L. (B) Ca2+ fluorescence induced by 200 μmol/L ATP under 30 μmol/L compounds concentration treated; green point derived from calcium fluorescent dye Cal 520™; scale bar, 40 μmol/L; images are representative of similar results obtained in three independent experiments per compound. (C) Quantification of Ca2+ fluorescence in 20 s; results represent mean ± SEM for the 8 of independent fields (20 μm × 20 μm) per compound shown in (B).
ANO1 is activated by intracellular Ca2+, and if the compound inhibits the Ca2+ transmembrane, it will aggravate its ANO1 inhibitory effect. Calcium fluorescent dye Cal520™ was used to evaluate the influence of compound 42 on intracellular Ca2+ concentration. Amlodipine was selected as a positive control. After 2 h pretreatment with Cal520™, compound 42 and ATP were added in turn to evoke the Ca2+ influx across cytomembrane, which showed the maximum relative fluorescence units (RFU) of the control group increased to 2.3 folds of the initial value and the RFU of amlodipine remained unchanged (Fig. 3B and C). Compound 42 almost did not affect Ca2+ influx (1.9 folds than initial value), and CaCCinh-A01 slightly suppressed Ca2+ influx (1.5 folds than initial value), whereas Ani9 hinders Ca2+ transmembrane.
2.5. ANO1/ANO2 channels selectivity of compound 42
ANO2 coexists with BK (large conductance calcium-activated potassium) and SK (small conductance calcium-activated potassium) channels in inferior olivary neurons that participate in the control of motor learning and timing34. ANO1-targeting analgesics lacking ANO1/ANO2 selectivity may cause potential central nervous system adverse effects. Thus, the ANO1/ANO2 selectivity of compound 42 was evaluated. ANO2 gene was transiently transfected into HEK293 cell, and then ANO2-mediated CaCC current was recorded by whole-cell patch clamp. The detailed method is the same as the reference35. The ANO1 and ANO2 pan-inhibitor CaCCinh-A01 was used as a negative control, and ANO1 selective inhibitor Ani9 was applied to be a positive control. The results showed that compound 42 at 30 μmol/L had little effect on ANO2 induced CaCC current. The ANO1/ANO2 selectivity of compound 42 is slightly weaker than that of Ani9 but much higher than that of CaCCinh-A01 (Fig. 4).
Figure 4.

Compound 42 performs little effect on ANO2 induced CaCC current at 30 μmol/L. (A), (B) and (C): representative traces of ANO2 currents at pCa2+ 6.0 in the pipette solution from HEK293 cells expressing ANO2; currents were recorded under control conditions and after application by 30 μmol/L compounds. HEK293 cells were depolarized from the holding potential of −40 mV to test potentials (+100 to −80 mV) by +20 mV increment for 500 ms and subsequently repolarized to −80 mV for 250 ms every 15 s; current curve is representative of similar results obtained in two independent experiments per compound and was repeat tested more than 5 times in each independent experiment. (D), (E) and (F): current–voltage relationships from the experiment showed in (A), (B) and (C); data are represented by mean ± SEM, n = 10; current values were measured at the end of each voltage step.
2.6. Stability and MDCK permeability of compound 42
We wanted to test the in vivo analgesic activity of ANO1 inhibitors by intragastric gavage (i.g.) administration rather than local peripheral and intrathecal administration as previously reported16,19,20, so compound 42 was evaluated for acid-base stability and intestinal absorbability to ensure it could be absorbed by animals.
2.6.1. Stability of compound 42 under different pH condition
The acid-base stability of compounds was tested under different pH buffer incubation by HPLC, which showed compound 42 remained stable in both acidic and alkaline environments (Table 4).
Table 4.
Compound stability under different pH conditions.
| Compd. | 5 h residual compound under different pH (%)a |
||||
|---|---|---|---|---|---|
| pH 1.0 | pH 4.0 | pH 6.8 | pH 7.4 | pH 9.0 | |
| CaCCinh-A01 | 93.8 ± 4.0 | 85.3 ± 8.1 | 94.9 ± 2.9 | 98.7 ± 0.7 | 97.6 ± 1.2 |
| 42 | 99.0 ± 4.6 | 99.4 ± 4.4 | 99.2 ± 4.9 | 99.4 ± 3.5 | 99.5 ± 0.9 |
Data are represented by the mean ± SD, n = 3.
2.6.2. MDCK permeability of compound 42
We performed the trans-well assay in the Madin–Darby canine kidney (MDCK) cell line to simulate compound absorption in the small intestine. Both compound 42 and CaCCinh-A01 passed through MDCK cells, indicating all of the two compounds could be absorbed by intestinal cells (Table 5).
Table 5.
MDCK permeability of compound 42.
Papp, permeability apparent coefficient.
F prediction, oral bioavailability prediction; data are represented by mean ± SD, n = 3.
2.7. Cytotoxicity and acute toxicity of compound 42
2.7.1. Proliferative cytotoxicity of compound 42
Sulforhodamine B (SRB) assay in Chinese Hamster Ovary (CHO) cells was conducted to examine the proliferative cytotoxicity of compound 42 incubated for 72 h (Fig. 5). All of the testing ANO1 inhibitors displayed proliferation inhibitory activity at 10 μmol/L or higher. Compound 42 and T16Ainh-A01 had a weak effect on proliferation under 1 and 3 μmol/L, while CaCCinh-A01 inhibited cell proliferation at all concentrations tested.
Figure 5.

Proliferation of CHO cells incubated with ANO1 inhibitors. Bar graph represents cytotoxicity of ANO1 inhibitors on CHO cell proliferation; n = 3; data are represented by mean ± SEM; NS, no significant, ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 compared to the 0.1% (v/v) DMSO in each group (control), t-test.
2.7.2. Acute toxicity of compound 42
The acute toxicity was carried out in 6–8 weeks aged C57BL/6 male mice which were treated with 1000 mg/kg compound 42 by i.g. administration. All mice survived and no intense acute toxic response (such as coma, tachypnea, and moribund status) was observed in 72 h. The acute toxicity test showed that compound 42 is safe at the dose of 1000 mg/kg or lower by i.g. administration.
2.8. In vivo analgesic effects of compound 42
The preliminary in vivo analgesic effects of compound 42 were investigated in the formalin test (Fig. 6A), chronic constriction injury model (Fig. 6B), hot plate test (Supporting Information Fig. S3) and writhing test (Supporting Information Fig. S4).
Figure 6.

The results of in vivo analgesic test. (A) Formalin test on rat; n = 12; the control group was injected with 50 μL saline instead of 5% (v/v) formalin. (B) Chronic constriction injury model (CCI) on rat; n = 8. Basic represents basic pain threshold before sciatic nerve ligation operation; 0 h, before corresponding compounds treated; 1 h, 1 h after the corresponding compound treated. PTI, the pain threshold rate change (%) of 1 h compared to 0 h. mpk represents mg/kg. All data in (A) and (B) are represented by the mean ± SD. Statistical significance was determined by ANOVA, ∗P < 0.05 and ∗∗P < 0.01 compared to the vehicle.
In the formalin test, phase 1 of the nociceptive response began immediately after formalin administration and then declined gradually in approximately 10 min. Phase 2 began about 15 min after formalin administration and lasted about 1 h. Pregabalin and flupirtine were selected as positive compounds. The results showed that these compounds had no significant effect on the number of foot lifts compared with the vehicle group (Fig. 6A). While, it was very encouraging that 80 mg/kg compound 42 (70 foot lifts) showed a significant analgesic effect in phase 2 compared with vehicle (121 foot lifts), which was almost as effective as flupirtine at 200 mg/kg (74 foot lifts) and pregabalin at 40 mg/kg (44 foot lifts). CaCCinh-A01 at 80 mg/kg (84 foot lifts) had no significant analgesic effect. Compound 42, as an ANO1 inhibitor, has a significant analgesic effect on phase 2 but not phase 1 in formalin test, which is consistent with the previous reports20.
The chronic constriction injury (CCI) of the sciatic nerve in the rat was used to evaluate the analgesic effects of compounds on the nerve injure-derived hyperalgesia. As shown in Fig. 6B, the threshold of paw withdraw measured by Electronic Von Frey before (basic) and after (0 h) CCI model established, and 1 h after compounds administration the threshold of paw withdraw was measured again (1 h). Pregabalin (40 mg/kg) and flupirtine (100 mg/kg) could significantly increase the threshold of paw withdraw with the pain threshold rate change (PTI, %) of 60.4 and 36.2 respectively. Treatment with compound 42 at 40 mg/kg also resulted in a significant analgesic effect (PTI of 45.2), which was more potent than 100 mg/kg flupirtine.
Based on the analgesic effects of ANO1 inhibitor compound 42 in different kinds of pain models, by i.g. administration compound 42 displayed more strength analgesic effects on chronic pain, including formalin-induced allodynia and sciatic nerve ligation-induced mechanical allodynia, especially on inflammation-related chronic pain (phase 2 in formalin test). For acute pain, however, no significant analgesic effect was observed with ANO1 inhibitors treatment in the hot plate test and the writhing test (Supporting Information Figs. S3 and S4).
2.9. Binding pose prediction of compound 42
To get insight into the binding behavior of ANO1 and its inhibitor, a lot of computer-aided drug design work has been conducted. Firstly, the human ANO1 (hANO1) homology modeling of dimer structure (Fig. 7A, Supporting Information) was constructed based on mANO1 cryo-electron microscopy structure36, 37, 38, 39, 40 using Schrödinger Prime software (sequence alignment results are shown in Supporting Information Fig. S5). Then Schrödinger SiteMap software was used to predict binding sites, and three ligand binding sites were provided (Fig. 7B). Site 1 is located outside the membrane surrounded by the terminals of α5, α6, and α9. Site 2 is adjacent to the Ca2+ binding region and consists of α6, α7, and α8. Site 3 was near the exit of the pore inside the membrane.
Figure 7.

Homology modeling and binding site prediction of hANO1 protein. (A) Dimer hANO1 homology modeling protein; the colors of left ANO1 monomer represent sequence similarity to the mANO1: dark blue, identical; cyan, similar; red, different; the color of the right monomer represents the hANO1 transmembrane helix: red, α1: E326‒D361; orange, α2: A408‒T441; yellow, α3: Y516‒M544; green, α4: T563‒E594; cyan, α5: K600‒K626; dark blue: α6: L657‒L693; purple, α7: T726‒T740; pink, α8: T752‒T770; plum, α9: R779‒F807; grey, α10: N875‒W910; black, α11: D918‒R936. The circles mean the potent ligand binding site predicted by SiteMap software. (B) The results of SiteMap prediction, the yellow surface represents hydrophobic volume. (C) and (D) Docking poses superposition of ANO1 inhibitors to site 1 (C) and site 2 (D).
Many hydrophilic arginine and glutamine residues enriched in site 3, lead to most of the hydrophobic ANO1 inhibitors were incapable to bind this area, so site 3 was ruled out first. Then reported ANO1 inhibitors (Fig. 1) and compound 42 were docked into site 1 and site 2 to evaluate whether the docking conformation conformed to the corresponding SAR. In site 1, ANO1 inhibitors docking conformation were randomly distributed in the binding pocket around K865, which indicated site 1 volume is too large for these inhibitors to form a stable docking conformation (Fig. 7C). In site 2, all inhibitors showed a very consistent binding conformation (Fig. 7D): the aromatic ring on the carbonyl side of the amide bond was embedded in the hydrophobic loop between α7 and α8, the carboxylic acid kept towards K767. As previously reported39,40, α6 and the calcium-binding region produced large conformational changes during Ca2+ induced ANO1 activation, thereby compounds binding to this region might be effective to inhibit ANO1. Thereby, site 2 is more likely to be the inhibitors binding site.
Flexible molecular docking was used to dock the CaCCinh-A01, AACT, and compound 42 into the site 2, and 30 ns molecular dynamic simulation was conducted based on docking conformation for more accurate protein‒ligand interaction pattern (Fig. 8).
Figure 8.

Interaction pattern of ANO1 inhibitors to hANO1. (A), (C), and (E): The interaction pattern of ANO1-inhibitors; yellow line, hydrogen bond; cyan line, π‒π stacking; green line, cation‒π interaction; pink line, electronic interaction. (B), (D), and (F): Interaction diagram during 10–0 ns molecular dynamic simulations. The percentage values on the interaction line represent the time proportion of this interaction over the course of the trajectory: for example, a value of 70% suggests that 70% of the simulation time the specific interaction is maintained. Values over 100% are possible as some protein residue may make multiple contacts of the same subtype with the ligand.
The carboxyl group of compound 42 and CaCCinh-A01 formed an electrostatic interaction with K767. Amide in all three compounds formed hydrogen bonds to A723 and G724 main chain. Naphthalene of compound 42 is embedded into the hydrophobic loop between α7 and α8 to form the cation‒π interaction with K767. The 4-chlorophenyl of compound 42 formed the cation‒π interaction with K614.
To verify the binding site, we mutated the hANO1 residues and constructed a transient ANO1 high expression cell line on HEK293 cells. The differences of inhibition rate of 30 μmol/L CaCCinh-A01 against wild type (69.4 ± 9.8%, average ± SD, n = 3) and mutant types were tested in whole-cell patch clamp at +100 mV potential. R321A (in site 3) and K614A (in site 2) mutations did not affect ANO1 inhibitory activity of CaCCinh-A01, and K671A (near the Ca2+ binding pocket) reduced ANO1 inhibition rate of CaCCinh-A01 (38.0 ± 13.6%, average ± SD, n = 3, P < 0.05, t-test). Whereas the mutation of K687A, K767A, and F768A in site 2 changed the electrophysiological features of CaCC current, led to unable to test ANO1 inhibition. Therefore, the uncertainty still exists to reveal the binding site without other experimental evidence such as structural biology.
3. Conclusions
Herein, we found a series of thiophenic acid and benzoic acid derivative ANO1 inhibitors by virtual screening. Among which, 4-arylthiophene-3-carboxylic acid scaffold was selected as a hit for further structural optimization, and compound 42 (IC50 = 790 nmol/L) was obtained. Activity verification showed that compound 42 could strongly inhibit the CaCC current in whole-cell patch clamp, and did not affect ANO2-induced CaCC current and weakly hinder Ca2+ transmembrane. Furthermore, the preliminary analgesic activity of compound 42 was evaluated in a variety of pain models by i.g. administration. The results showed that compound 42 had significant analgesic activity on inflammation-related pain. Finally, the binding site of compound 42 to ANO1 was predicted by homology modeling, docking, and molecular dynamics, which suggested that the binding site was close to the Ca2+ binding region. Our study is the first to show that an oral ANO1 inhibitor has a significant analgesic effect and also provides selective molecular tools for ANO1-related research.
4. Experimental
4.1. Virtual screening
4.1.1. Ligands and proteins preparation
Ligand preparation was using Schrödinger LigPrep software (2014-2 suites) with the OPLS-2005 force field and default parameters. The SPECS (http://www.specs.com) and PKU_CNCL (http://www.pkucncl.com) database were downloads from the website.
4.1.2. Shape and electronic distribution-based virtual screening
Shape and electronic distribution-based virtual screening were based on CaCCinh-A01 optimized conformation using ROCs and EON in OPENEYE software. In ROCs, the parameters were set as: “Tanimoto cutoff 0.6, rank by Shape Tanimoto” and in EON, the parameters were set as default.
4.2. Chemistry
The screened compounds were purchased from the SPECS database and PKU-CNCL. The purities of all purchased compounds were more than 90%. For the chemical synthesis, reagents and solvents were obtained from J&K Chemical (Beijing, China) without further purification. T16Ainh-A01 was purchased from TOCRIS (USA). 1H-NMR and 13C-NMR spectra were recorded on Bruker (400 MHz) instruments, using DMSO-d6 or CDCl3 as solvents. High-resolution mass spectra (HRMS) were recorded on the Bruker Apex IV FTMS mass spectrometer using ESI (electrospray ionization). The microwave-assisted reaction was performed on the CEM Discover SP microwave reactor. The purity of the final compound was determined by NMR and HPLC to be >95% (UV detection (λ = 254 nm), compound structures in smiles format and more detail can be found in Supporting Information).
4.2.1. Synthesis of CaCCinh-A01
6-(tert-Butyl)-2-(furan-2-carboxamido)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylic acid (CaCCinh-A01, 1) was synthesized according to the reported method24. Yellow powder, m.p. 205–215 °C. 1H-NMR (400 MHz, DMSO-d6) δ 13.25 (s, 1H), 12.20 (s, 1H), 8.06 (s, 1H), 7.36 (s, 1H), 6.80 (s, 1H), 3.06 (d, J = 17.0 Hz, 1H), 2.70 (d, J = 14.6 Hz, 1H), 2.45–2.34 (m, 1H), 2.03–1.94 (m, 1H), 1.43 (d, J = 5.9 Hz, 1H), 1.32–1.14 (m, 2H), 0.93 (s, 9H). 13C-NMR (101 MHz, DMSO-d6) δ 168.12, 154.62, 147.66, 146.89, 146.56, 132.12, 127.80, 117.44, 113.97, 113.20, 45.29, 33.06, 28.00, 27.84, 26.22, 24.83. HRMS (ESI-TOF+) Calcd. for C18H20NO4S [M‒H]‒ m/z: 346.1113, Found 346.1105.
4.2.2. Synthesis of compounds 33‒46
Taking compound 42 as an example: 4-chloroacetophenone 18.55 g (120 mmol), ethyl cyanoacetate 16.29 g (144 mmol), acetic acid 7.12 g (120 mmol), morpholine 23.00 g (264 mmol), and ethanol 500 mL were added to a 1 L round bottom flask. Stirred at 68 °C for 3 h under an argon atmosphere, followed by the addition of 4.91 g (144 mmol) sulfur and then stirred for 48 h. The solution was diluted with DCM and washed with brine four times. The organic phase was separated and evaporated 10 mL EA was added to re-dissolve the residue. Acidic EA was added, the precipitates were collected, washed, and then was added to 200 mL 50% ammonia and stirred for 30 min. Followed by extraction with DCM four times, the combined organic phase dried with anhydrous Na2SO4. After evaporation, 2-aminothiophene-3-carboxylate acid ethyl ester was obtained (yield 21%). 0.195 g (0.73 mmol) 2-aminothiophene-3-carboxylate acid ethyl ester was dissolved in 10 mL DCM, and 0.125 g (0.66 mmol) 1-naphthyl chloride in 10 mL DCM was added at 0 °C. The solution was stirred at room temperature for 8 h, followed by evaporation and recrystallization using DCM/petroleum ether. White powder intermediate 0.227 g (0.54 mmol) was obtained (yield 75%). 0.143 g (0.34 mmol) white powder intermediate and NaOH 0.068 g (1.7 mmol) were dissolved in THF/methanol/water solution and stirred at room temperature for 8 h, acetic acid was used to adjust pH to neutral and water was added to precipitate. 0.094 g (0.23 mmol) 42 was obtained at a yield of 69%.
4.2.2.1. 2-(4-(tert-Butyl) benzamido)-4-(4-fluorophenyl) thiophene-3-carboxylic acid (33)
White powder, m.p. 210–211 °C. Total yield 33%. 1H-NMR (400 MHz, DMSO-d6) δ 12.44 (s, 1H), 7.92 (d, J = 8.3 Hz, 2H), 7.69 (d, J = 8.3 Hz, 2H), 7.42 (dd, J = 8.4, 5.7 Hz, 2H), 7.21 (t, J = 8.8 Hz, 2H), 6.97 (s, 1H), 1.36 (s, 9H). 13C-NMR (101 MHz, DMSO-d6) δ 167.62, 163.43, 163.13, 160.71, 156.46, 149.98, 139.05, 134.11, 131.47, 131.39, 129.68, 127.47, 126.56, 116.33, 114.75, 114.54, 112.83, 35.30, 31.24. HRMS (ESI-TOF+) Calcd. for C22H19FNO3S [M‒H]‒ m/z: 396.1070, Found: 396.1070.
4.2.2.2. 2-(4-(tert-Butyl) benzamido)-4-(4-chlorophenyl) thiophene-3-carboxylic acid (34)
White powder, m.p. 245–246 °C. Total yield 30%. 1H-NMR (400 MHz, DMSO-d6) δ 12.43 (s, 1H), 7.92 (d, J = 7.7 Hz, 2H), 7.70 (d, J = 7.7 Hz, 2H), 7.43 (dd, J = 15.8, 7.5 Hz, 4H), 7.01 (s, 1H), 1.37 (s, 9H). 13C-NMR (101 MHz, DMSO-d6) δ 167.48, 163.47, 156.51, 150.03, 138.82, 136.66, 132.23, 131.33, 129.67, 127.87, 127.49, 126.61, 116.63, 112.78, 35.35, 31.28. HRMS (ESI-TOF+) Calcd. for C22H19ClNO3S [M‒H]‒ m/z: 412.0774, Found: 412.0762.
4.2.2.3. 2-(4-(tert-Butyl) benzamido)-4-(4-(trifluoromethyl) phenyl) thiophene-3-carboxylic acid (35)
White powder, m.p. 230–231 °C. Total yield 34%, 1H-NMR (400 MHz, DMSO-d6) δ 12.51 (s, 1H), 7.93 (d, J = 8.0 Hz, 2H), 7.72 (dd, J = 21.2, 8.0 Hz, 4H), 7.62 (d, J = 7.8 Hz, 2H), 7.09 (s, 1H), 1.36 (s, 9H). 13C-NMR (101 MHz, DMSO-d6) δ 167.37, 163.51, 156.53, 150.08, 142.06, 138.67, 130.34, 129.66, 127.52, 126.61, 124.72, 117.29, 112.94, 35.35, 31.27. HRMS (ESI-TOF+) m/z Calcd. for C23H19F3NO3S [M‒H]‒ m/z: 446.1038, Found: 446.1041.
4.2.2.4. 2-(4-(tert-Butyl) benzamido)-4-(4-(tert-butyl) phenyl) thiophene-3-carboxylic acid (36)
Grey powder, m.p. 220–221 °C. Total yield 33%, 1H-NMR (400 MHz, DMSO-d6) δ 12.47 (s, 1H), 7.93 (d, J = 7.9 Hz, 2H), 7.69 (d, J = 7.9 Hz, 2H), 7.40 (d, J = 7.8 Hz, 2H), 7.32 (d, J = 7.8 Hz, 2H), 6.92 (s, 1H), 1.36 (s, 9H), 1.34 (s, 9H). 13C-NMR (101 MHz, DMSO-d6) δ 167.74, 163.40, 156.44, 149.87, 149.64, 140.15, 134.88, 129.76, 129.20, 127.48, 126.57, 124.65, 116.00, 112.84, 35.32, 34.69, 31.64, 31.27. HRMS (ESI-TOF+) m/z Calcd. for C26H28NO3S [M‒H]‒ m/z: 434.1790, Found: 434.1793.
4.2.2.5. 4-([1,1′-Biphenyl]-4-yl)-2-(4-(tert-butyl) benzamido) thiophene-3-carboxylic acid (37)
White powder, m.p. 225–226 °C. Total yield 36%, 1H-NMR (400 MHz, DMSO-d6) δ 12.44 (s, 1H), 7.93 (d, J = 7.5 Hz, 2H), 7.72 (dd, J = 17.5, 8.1 Hz, 6H), 7.51 (dd, J = 17.2, 7.9 Hz, 4H), 7.41 (t, J = 7.2 Hz, 1H), 7.02 (s, 1H), 1.37 (s, 9H). 13C-NMR (101 MHz, DMSO-d6) δ 167.64, 163.49, 156.52, 149.95, 140.33, 139.77, 139.18, 136.93, 130.10, 129.73, 129.44, 127.88, 127.50, 127.03, 126.61, 126.19, 116.37, 112.87, 35.35, 31.28. HRMS (ESI-TOF+) m/z Calcd. for C28H24NO3S [M‒H]‒ m/z: 454.1477, Found: 454.1477.
4.2.2.6. 2-(4-(tert-Butyl) benzamido)-4-(2,4-dichlorophenyl) thiophene-3-carboxylic acid (38)
White powder, m.p. 225–226 °C. Yield 38%, 1H-NMR (400 MHz, DMSO-d6) δ 12.53 (s, 1H), 7.92 (d, J = 7.1 Hz, 2H), 7.68 (dd, J = 19.8, 9.0 Hz, 4H), 7.39 (d, J = 8.1 Hz, 1H), 7.10 (s, 1H), 1.37 (s, 9H). 13C-NMR (101 MHz, DMSO-d6) δ 167.28, 163.52, 156.53, 150.01, 138.49, 137.47, 131.40, 130.48, 130.14, 130.00, 129.97, 129.70, 127.51, 126.62, 117.27, 112.98, 35.36, 31.29. HRMS (ESI-TOF+) m/z Calcd. for C22H18Cl2NO3S [M‒H]‒ m/z: 446.0384, Found: 446.0378.
4.2.2.7. 2-(4-(tert-Butyl) benzamido)-4-(naphthalen-2-yl) thiophene-3-carboxylic acid (39)
White powder, m.p. 230–231 °C. Total yield 23%, 1H-NMR (400 MHz, DMSO-d6) δ 12.47 (s, 1H), 8.01–7.87 (m, 6H), 7.71 (d, J = 7.9 Hz, 2H), 7.56 (s, 3H), 7.09 (s, 1H), 1.37 (s, 9H). 13C-NMR (101 MHz, DMSO-d6) δ 167.65, 163.50, 156.50, 150.01, 140.11, 135.57, 133.06, 132.50, 129.74, 128.60, 128.30, 127.88, 127.51, 127.43, 126.83, 126.62, 126.52, 126.36, 116.63, 113.00, 35.36, 31.29. HRMS (ESI-TOF+) m/z Calcd. for C26H22NO3S [M‒H]‒ m/z: 428.1320, Found: 428.1321.
4.2.2.8. 4-(4-Methoxyphenyl)-2-(thiophene-2-carboxamido) thiophene-3-carboxylic acid (40)
White powder, m.p. 241–242 °C. Total yield 37%. 1H-NMR (400 MHz, DMSO-d6) δ 12.55 (s, 1H), 8.05 (d, J = 4.5 Hz, 1H), 7.80 (d, J = 3.0 Hz, 1H), 7.34 (s, 1H), 7.31 (d, J = 8.5 Hz, 2H), 6.94 (d, J = 8.4 Hz, 2H), 6.89 (s, 1H), 3.81 (s, 3H). 13C-NMR (101 MHz, DMSO-d6) δ 167.67, 158.86, 158.48, 139.98, 137.36, 133.81, 130.61, 130.28, 130.01, 129.25, 115.63, 113.34, 55.53. HRMS (ESI-TOF+) m/z Calcd. for C17H12NO4S2 [M‒H]‒ m/z: 358.0208, Found: 358.0207.
4.2.2.9. 4-(4-(tert-Butyl) phenyl)-2-(4-methoxybenzamido) thiophene-3-carboxylic acid (41)
White powder, m.p. 215–216 °C. Total yield 30%, 1H-NMR (400 MHz, DMSO-d6) δ 12.44 (s, 1H), 7.95 (d, J = 8.2 Hz, 2H), 7.40 (d, J = 7.6 Hz, 2H), 7.32 (d, J = 7.9 Hz, 2H), 7.21 (d, J = 8.2 Hz, 2H), 6.91 (s, 1H), 3.90 (s, 3H), 1.34 (s, 9H). 13C-NMR (101 MHz, DMSO-d6) δ 167.77, 163.34, 163.02, 150.05, 149.62, 140.10, 134.91, 129.59, 129.19, 124.65, 124.56, 115.85, 115.04, 112.64, 56.08, 34.70, 31.64. HRMS (ESI-TOF+) m/z Calcd. for C23H22NO4S [M‒H]‒ m/z: 408.1270, Found: 408.1268.
4.2.2.10. 2-(1-Naphthamido)-4-(4-chlorophenyl) thiophene-3-carboxylic acid (42)
Colorless crystal, m.p. 241–242 °C. Total yield 17%. 1H-NMR (400 MHz, DMSO-d6) δ 12.11 (s, 1H), 8.47 (d, J = 7.7 Hz, 1H), 8.24 (d, J = 8.0 Hz, 1H), 8.11 (d, J = 7.3 Hz, 1H), 8.02 (d, J = 6.8 Hz, 1H), 7.71 (dd, J = 14.0, 6.8 Hz, 3H), 7.43 (q, J = 7.6 Hz, 4H), 7.08 (s, 1H). 13C-NMR (101 MHz, DMSO-d6) δ 167.07, 165.67, 149.32, 138.85, 136.61, 133.89, 132.59, 132.27, 131.83, 131.30, 130.12, 129.10, 128.18, 127.92, 127.28, 126.75, 125.66, 125.33, 116.82, 113.44. HRMS (ESI-TOF+) m/z Calcd. for C22H13ClNO3S [M‒H]‒ m/z: 406.0305, Found: 406.0305.
4.2.2.11. 2-(2-Naphthamido)-4-(4-chlorophenyl) thiophene-3-carboxylic acid (43)
White powder, m.p. 240–241 °C. Total yield 14%, 1H-NMR (400 MHz, DMSO-d6) δ 12.94 (s, 1H), 8.61 (s, 1H), 8.16 (d, J = 6.5 Hz, 2H), 8.05 (dd, J = 18.1, 8.3 Hz, 2H), 7.70 (dt, J = 19.4, 7.0 Hz, 2H), 7.44 (s, 4H), 7.02 (s, 1H). 13C-NMR (101 MHz, DMSO-d6) δ 167.49, 163.59, 149.62, 139.02, 136.75, 135.19, 132.67, 132.16, 131.36, 129.91, 129.67, 129.46, 128.98, 128.69, 128.24, 127.84, 127.75, 123.55, 116.57, 113.81. HRMS (ESI-TOF+) m/z Calcd. for C22H13ClNO3S [M‒H]‒ m/z: 406.0305, Found: 406.0304.
4.2.2.12. 4-(4-Chlorophenyl)-2-(2,4-dichlorobenzamido)thiophene-3-carboxylic acid (44)
White powder, m.p. 239–240 °C. Total yield 24%, 1H-NMR (400 MHz, DMSO-d6) δ 12.07 (s, 1H), 7.88 (s, 2H), 7.67 (d, J = 8.3 Hz, 1H), 7.42 (dd, J = 20.3, 7.6 Hz, 4H), 7.08 (s, 1H). 13C-NMR (101 MHz, DMSO-d6) δ 166.70, 162.51, 148.18, 138.87, 137.15, 136.46, 132.63, 132.29, 132.07, 131.95, 131.24, 130.51, 128.55, 127.93, 117.18, 114.21. HRMS (ESI-TOF+) m/z Calcd. for C18H9Cl3NO3S [M‒H]‒ m/z: 423.9369, Found: 423.9368.
4.2.2.13. 4-(4-Chlorophenyl)-2-(thiophene-2-carboxamido) thiophene-3-carboxylic acid (45)
White powder, m.p. 225–226 °C. Total yield 22%. 1H-NMR (400 MHz, DMSO-d6) δ 12.40 (s, 1H), 8.05 (d, J = 4.8 Hz, 1H), 7.81 (s, 1H), 7.42 (q, J = 8.1 Hz, 4H), 7.35 (t, J = 3.7 Hz, 1H), 7.00 (s, 1H). 13C-NMR (101 MHz, DMSO-d6) δ 167.39, 158.49, 149.49, 138.83, 137.07, 136.54, 134.02, 132.25, 131.32, 130.43, 129.29, 127.87, 116.75, 113.01. HRMS (ESI-TOF+) m/z Calcd. for C16H19ClNO3S2 [M‒H]‒ m/z: 361.9712, Found: 361.9715.
4.2.2.14. 4-(4-Chlorophenyl)-2-(4-methyl-1-naphthamido) thiophene-3-carboxylic acid (46)
White powder, m.p. 250–251 °C. Total yield 24%, 1H-NMR (400 MHz, DMSO-d6) δ 12.08 (s, 1H), 8.51 (s, 1H), 8.20 (s, 1H), 7.91 (d, J = 6.8 Hz, 1H), 7.72 (s, 2H), 7.58 (d, J = 6.4 Hz, 1H), 7.43 (d, J = 8.8 Hz, 4H), 7.07 (s, 1H), 2.79 (s, 3H). 13C-NMR (101 MHz, DMSO-d6) δ 167.10, 165.79, 149.49, 139.54, 138.82, 136.64, 132.87, 132.25, 131.30, 130.25, 130.15, 127.91, 127.82, 127.23, 126.50, 126.25, 125.88, 125.24, 116.73, 113.22, 19.92. HRMS (ESI-TOF+) m/z Calcd. for C23H15ClNO3S [M‒H]‒ m/z: 420.0461, Found: 420.0454.
4.2.3. Synthesis of compounds 47‒53
Taking compound 48 as an example. Indanone 1.409 g (8.0 mmol), ethyl cyanoacetate 1.352 g (9.6 mmol), acetic acid 0.480 g (9.6 mmol), ammonium acetate 0.736 g (9.6 mmol), and toluene 100 mL were added into a round bottom flask and refluxed for 8 h. After evaporation, morpholine 0.832 g (76.8 mmol), sulfur 0.384 g (9.6 mmol), and ethanol 100 mL were added and stirred for 48 h at 80 °C under an argon atmosphere. The reaction solution was diluted with 150 mL DCM, washed with brine four times, and dried with anhydrous Na2SO4. After evaporation and column chromatography purification, brown solid immediate was obtained (yield, 35%). The following procedures were the same as compound 42. Finally, the brown powder compound 48 was obtained.
4.2.3.1. 2-(4-(tert-Butyl) benzamido)-8H-indeno [2,1-b] thiophene-3-carboxylic acid (47)
Yellowish powder, m.p. 260–261 °C. Yield 25%, 1H-NMR (400 MHz, DMSO-d6) δ 12.48 (s, 1H), 8.25 (d, J = 7.7 Hz, 1H), 7.84 (d, J = 7.9 Hz, 2H), 7.60 (d, J = 8.0 Hz, 2H), 7.46 (d, J = 7.3 Hz, 1H), 7.28 (t, J = 7.5 Hz, 1H), 7.17 (d, J = 7.4 Hz, 1H), 3.80 (s, 2H), 1.29 (s, 9H). 13C-NMR (101 MHz, DMSO-d6) δ 167.80, 163.17, 156.38, 152.32, 147.03, 140.72, 138.97, 136.13, 129.68, 127.45, 126.90, 126.53, 125.13, 124.84, 122.63, 107.92, 35.32, 34.50, 31.26. HRMS (ESI-TOF+) m/z Calcd. for C23H20NO3S [M‒H]‒ m/z: 390.1164, Found: 390.1165.
4.2.3.2. 2-(4-(tert-Butyl) benzamido)-6-methoxy-8H-indeno [2,1-b] thiophene-3-carboxylic acid (48)
Brown powder, m.p. 213–214 °C. Total yield 19%. 1H-NMR (400 MHz, DMSO-d6) δ 12.48 (s, 1H), 8.15 (d, J = 8.6 Hz, 1H), 7.89 (d, J = 8.0 Hz, 2H), 7.66 (d, J = 8.0 Hz, 2H), 7.14 (s, 1H), 6.90 (d, J = 8.5 Hz, 1H), 3.82 (s, 2H), 3.78 (s, 3H), 1.34 (s, 9H). 13C-NMR (101 MHz, DMSO-d6) δ 167.83, 163.11, 157.78, 156.39, 152.01, 148.98, 140.63, 133.79, 131.98, 129.74, 127.44, 126.57, 123.09, 112.43, 111.21, 107.74, 55.67, 35.34, 34.65, 31.29. HRMS (ESI-TOF+) m/z Calcd. for C24H22NO4S [M‒H]‒ m/z: 420.1270, Found: 420.1263.
4.2.3.3. 2-(Thiophene-2-carboxamido)-8H-indeno [2,1-b] thiophene-3-carboxylic acid (49)
Brown powder, m.p. 245–246 °C. Total yield 21%. 1H-NMR (400 MHz, DMSO-d6) δ 8.33 (d, J = 7.7 Hz, 1H), 8.04 (d, J = 4.8 Hz, 1H), 7.81 (d, J = 2.7 Hz, 1H), 7.53 (d, J = 7.3 Hz, 1H), 7.35 (t, J = 7.2 Hz, 2H), 7.23 (t, J = 7.4 Hz, 1H), 3.88 (s, 2H). 13C-NMR (101 MHz, DMSO-d6) δ 167.75, 158.24, 151.76, 147.00, 140.82, 138.96, 137.22, 136.19, 133.88, 130.27, 129.25, 126.90, 125.16, 124.84, 122.67, 108.30, 34.56. HRMS (ESI-TOF+) m/z Calcd. for C17H10NO3S2 [M‒H]‒ m/z: 340.0102 Found: 340.0104.
4.2.3.4. 2-(2-Naphthamido)-8H-indeno [2,1-b] thiophene-3-carboxylic acid (50)
Greenish powder, m.p. 260–261 °C. Total yield 14%. 1H-NMR (400 MHz, DMSO-d6) δ 12.70 (s, 1H), 8.58 (s, 1H), 8.32 (d, J = 7.5 Hz, 1H), 8.14 (s, 2H), 8.02 (dd, J = 28.5, 7.8 Hz, 2H), 7.69 (t, J = 7.1 Hz, 2H), 7.52 (d, J = 7.0 Hz, 1H), 7.35 (t, J = 7.2 Hz, 1H), 7.23 (t, J = 7.0 Hz, 1H), 3.87 (s, 2H). 13C-NMR (101 MHz, DMSO-d6) δ 167.81, 163.29, 152.24, 147.03, 140.76, 138.94, 136.31, 135.16, 132.64, 129.70, 129.65, 129.43, 128.97, 128.72, 128.23, 127.74, 126.91, 125.16, 124.85, 123.38, 122.63, 108.12, 34.52. HRMS (ESI-TOF+) m/z Calcd. for C23H14NO3S [M‒H]‒ m/z: 384.0694, Found: 384.0694.
4.2.3.5. 2-(1-Naphthamido)-8H-indeno [2,1-b] thiophene-3-carboxylic acid (51)
Yellowish powder, m.p. 265–266 °C. Total yield 17%. 1H-NMR (400 MHz, DMSO-d6) δ 12.28 (s, 1H), 8.52 (d, J = 7.9 Hz, 1H), 8.34 (d, J = 7.6 Hz, 1H), 8.20 (d, J = 8.1 Hz, 1H), 8.05 (dd, J = 25.0, 7.1 Hz, 2H), 7.68 (t, J = 7.5 Hz, 3H), 7.53 (d, J = 7.1 Hz, 1H), 7.35 (t, J = 7.3 Hz, 1H), 7.24 (t, J = 7.1 Hz, 1H), 3.88 (s, 2H). 13C-NMR (101 MHz, DMSO-d6) δ 167.47, 165.31, 151.68, 147.06, 140.83, 138.99, 136.27, 133.91, 132.57, 131.72, 130.17, 129.06, 128.12, 127.22, 126.91, 126.74, 125.60, 125.41, 125.16, 124.85, 122.69, 108.55, 34.51. HRMS (ESI-TOF+) m/z Calcd. for C23H14NO3S [M‒H]‒ m/z: 384.0694, Found: 384.0685.
4.2.3.6. 2-(4-(tert-Butyl) benzamido)-8,8-dimethyl-8H-indeno [2,1-b] thiophene-3-carboxylic acid (52)
Brown powder, m.p. 264–265 °C. Total yield 24%, 1H-NMR (400 MHz, DMSO-d6) δ 12.56 (s, 1H), 8.26 (d, J = 7.5 Hz, 1H), 7.93 (d, J = 7.7 Hz, 2H), 7.69 (d, J = 6.7 Hz, 2H), 7.54 (d, J = 7.1 Hz, 1H), 7.30 (dt, J = 26.0, 7.0 Hz, 2H), 1.52 (s, 6H), 1.36 (s, 9H). 13C-NMR (101 MHz, DMSO-d6) δ 167.80, 163.22, 157.01, 156.45, 151.83, 147.54, 136.85, 136.70, 129.76, 127.48, 127.28, 126.60, 125.79, 122.81, 122.33, 108.15, 35.34, 31.28, 27.46. HRMS (ESI-TOF+) m/z Calcd. for C25H24NO3S [M‒H]‒ m/z: 418.1477, Found: 418.1468.
4.2.3.7. 2-(4-(tert-Butyl) benzamido)-4,5-dihydronaphtho [2,1-b] thiophene-1-carboxylic acid (53)
White powder, m.p. 255–256 °C. Total yield 24%, 1H-NMR (400 MHz, CDCl3) δ 11.98 (s, 1H), 8.06 (s, 2H), 7.94 (d, J = 8.1 Hz, 2H), 7.67 (d, J = 8.1 Hz, 1H), 7.56 (d, J = 8.1 Hz, 2H), 7.20 (d, J = 7.0 Hz, 1H), 2.95 (t, J = 7.0 Hz, 2H), 2.76 (t, J = 6.9 Hz, 2H), 1.38 (s, 9H).13C-NMR (101 MHz, CDCl3) δ 171.98, 163.89, 157.61, 156.56, 135.99, 131.85, 131.33, 130.13, 129.35, 127.65, 127.36, 127.01, 126.51, 126.16, 126.10, 125.51, 77.35, 77.24, 77.04, 76.72, 35.21, 35.17, 31.13, 31.11, 30.28, 23.61, 1.04. HRMS (ESI-TOF+) m/z Calcd. for C24H22NO3S [M‒H]‒ m/z: 404.1320, Found: 404.1313.
4.3. Biological experiments
4.3.1. Cell culture and reagents
Fischer rat thyroid (FRT) cells stably co-expressing ANO1 isoform (abc) and the halide-sensitive yellow fluorescent protein (YFP)-H148Q/I152L/F46L were kindly provided by Dr. Luis J.V. Galietta (Laboratorio di Genetica Molecolare, Istituto Giannina Gaslini, Genova, Italy) and were maintained in F12 medium containing 10% (v/v) fetal bovine serum (FBS), 1% (v/v) penicillin/streptomycin (P/S). HEK-293T and CHO cells were cultured in DMEM containing 10% (v/v) FBS, 1% (v/v) P/S. The cells were cultured at 37 °C with 5% CO2 in a humidified incubator. For plate reader assays, cells were plated in black 96-well microplates (3603, Corning, USA) and assayed 24 h after plating.
FBS (10270-106, Gibo, USA), F12 medium (11765-054, Gibco), 1% (v/v) P/S (CC004, Macgene, China), DMEM (CM10013, Macgene, China). ANO1 inhibitors, CaCCinh-A01 and T16Ainh-A01 (Sigma, USA), Ani9 (Tocris, USA) were dissolved in DMSO to a stock concentration of 10 mmol/L.
4.3.2. Electrophysiology
ANO1 currents were recorded from whole cell patches at room temperature from ANO1-YFP stably co-expressing FRT cell line. The pipet (extracellular) solution contained (in mmol/L) 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 Hepes (pH 7.4). The bath (intracellular) solution A contained (in mmol/L) 146 CsCl, 2 MgCl2, 10 sucrose and 8 Hepes (adjusting pH to 7.3 with NMDG). The bath (intracellular) solution B contained (in mmol/L) 146 CsCl, 2 MgCl2, 5 CaCl2, 5 EGTA, 10 sucrose, and 8 Hepes (adjusting pH to 7.3 with NMDG). Mix the bath solution A and B (VA:VB = 892:108) to obtain the desired solution with 1 μmol/L free Ca2+ concentration. The electrical resistance of micropipettes was 3–5 MΩ. Recordings were made using the HEKA EPC10 amplifier with PatchMaster software. The method of ANO1 whole-cell patch clamp is similar to the reference10,41. ANO1 was activated by 1 μmol/L free Ca2+ in the bath solution. The protocol for stimulation consisted of 850 ms voltage steps including +100 to −100 mV (400 ms) starting from a holding potential of 0 mV (50 ms).
ANO2 currents were recorded from whole cell patches at room temperature from ANO2 transiently expressing HEK293 cell line. The protocol, pipet solution, and bath solution are the same as the reference35. HEK293 cells were depolarized from the holding potential of −40 mV to test potentials (+100 to −80 mV) by +20 mV increment for 500 ms and subsequently repolarized to −80 mV for 250 ms every 15 s.
4.3.3. YFP fluorescence quenching assay
The method of YFP fluorescence quenching assay is similar to the reference10,41. The ANO1-overexpressed FRT cell line was plated in 96-well plates and washed with PBS three times, and incubated with 60 μL test compound/PBS solution for 1 h (PBS with 0.1% (v/v) DMSO as a negative control). The fluorescence was measured in Microplate Reader (Flexstation 3, Molecular Devices) at a frequency of once per 1.7 s for 120 s (λex/λem: 514 nm/535 nm). During the measurement, baseline fluorescence data F0 was collected for 17 s before the addition of a PBS-iodide solution (PBS with 100 mmol/L NaI) containing 200 μmol/L ATP. Fluorescence−time curve was fitted by Eq. (2):
| (2) |
The slope was calculated from 5 to 6 points where the fluorescence starts to decrease after adding the PBS-iodide solution. The inhibition rate was calculated by Eq. (1) which is described in Section 2.2. IC50 was calculated from the inhibition–concentration curve fitted by the Hill equation. Data regulation and calculation were performed with Origin 8 software. Inhibition rate–concentration curves of compounds are in Supporting Information Fig. S2.
4.3.4. Intracellular Ca2+ fluorescence
HEK-293T cells were plated in 96-well plates and cultured for 24 h before changing the medium of each well into 100 μL DMEM with 10% (w/v) F127 and 10 μmol/L cal520™. After 2 h incubation, the cells were washed with PBS and incubated for 0.5 h with 100 μL compound/PBS solution at 30 μmol/L compound concentration. Cells were imaged before and 5 s after the addition of 200 μmol/L ATP containing PBS under Nikon eclipse Ti-S inverted fluorescence microscope with the FITC filter, and the data were analyzed with ImageJ software.
4.3.5. Stability under different pH conditions
The target compounds were diluted to 100 μmol/L with pH 1.0, 4.0, 6.8, 7.4, and 9.0 buffers, and the 0 h peak area of the target compounds were obtained by HPLC−UV analysis. Then each pH compound solution was placed in 37 °C water bath. The peak areas of the target compounds were obtained at 2 and 5 h. The change rate in the peak area was obtained by comparing the peak area of the target compound at different times with the peak area of 0 h. pH phosphate buffers were purchased from Shanghai Rongbai Biological Technology Ltd. (Shanghai, China).
4.3.6. MDCK cells permeability
A resistance-stable MDCK monolayer cell model was established in the transwell. A 100 μL compound solution was added to the upper chamber of the transwell, and 0.6 mL HBSS was added to the lower chamber, followed by incubation for 120 min at 37 °C and 5% CO2. Then the lower chamber liquid was detected by LC−MS (G6120B, Agilent Technologies, USA), and the compound concentration was calculated. The apparent permeability coefficient was calculated according to Eq. (3):
| (3) |
ΔQ (μg) is the compound transport amount in Δt (s), A (cm2) is the membrane area, and C0 (μg/mL) is the initial drug concentration in the upper chamber of MDCK cells.
4.3.7. Cytotoxicity
The effect of ANO1 inhibitors on cell viability was determined by the SRB (sulforhodamine B) assay. Briefly, cells were cultured on sterile slides in 96-well plates overnight at 37 °C with 5% CO2. Then cells were treated with various dosages of compounds or medium containing 0.5% (v/v) DMSO (control) for 72 h, 200 mL 10% (v/v) trichloroacetic acid (TCA) was added to each well, followed by incubation for 1 h at 4 °C in a refrigerator. After the microplate was washed five times with sterile water, evaporation in air, 100 mL of 0.4% SRB dissolved in 1% (v/v) acetic acid was added to each well. Following an additional incubation at room temperature in the dark for 15 min. After the microplate was washed five times with 1% (v/v) acetic acid, evaporation in air, add 100 mL 10 mmol/L Tris-base to each well to dissolve SRB for 30 min. Absorbance was measured at 540 nm using a microplate reader.
4.3.8. Acute toxicity
The acute toxicity was evaluated by i.g. administration of 1000 mg/kg doses of individual compound 42 into C57BL/6 mice. The C57BL/6 mice (weight, 18–22 g; age, 6–8 weeks; 7 mice per group; male) were purchased from Vital River Laboratories (Beijing, China). 5% (v/v) DMSO+10% (v/v) Glutol+85% (v/v) saline (adjust pH to 8.0 by 1 mol/L NaOH) were used for i.g. administration. Survival was monitored daily for 3 days. All animal experiments were performed in full compliance with the protocols approved by the Animal Ethics Committee of the Institute of Medicinal Biotechnology (Beijing, China).
4.4. In vivo pharmacodynamics
4.4.1. Animal and treatment
Animal welfare is in accordance with the Guide for the Care and Use of Laboratory Animals (8th ed., 2011). All animals were purchased from Shanghai Sppr bk laboratory animals Ltd. (Shanghai, China), and fed in SPF animal laboratories. All of in vivo experiments were conducted by i.g. administration.
Pregabalin, flupirtine, and morphine hydrochloride injection were obtained from Jiangsu Ehwa Pharmaceutical Group Co., Ltd. PEG400 was purchased from Weier Huagong (Nanjing, China). Administration volume was 1 mL/100 g mouse or rat weight. Most of the compounds in pharmacodynamics tests were conducted by i.g. administration, except for morphine by intraperitoneal (i.p.) administration. Formulation (take 80 mg/kg as example): 80 mg compound 42 sodium salt was dissolved in 2.0 mL DMSO (Bidepharm, Shanghai, China), then diluted in 8.0 mL 30% (v/v) PEG400/saline (Bidepharm), ultrasonic mixing. 8 mg/mL compound 42 sodium salt solution was obtained. Fluoxetine, pregabalin and CaCCinh-A01 were also prepared using that same method.
4.4.2. Formalin test
The formalin experiment with reference to the experimental method was reported by Okuda et al.42 12 SD rats (male, 220–300 g) in each group. The control group and the vehicle group were administrated with normal saline. After 30 min, 5% (v/v) formalin 50 μL was injected into the rat's left-hand foot in the vehicle group and compound group, and 50 μL normal saline was injected in the control group. Then, the number of pain responses (foot lifting) was immediately observed and recorded. 0–10 min after the formalin injection was defined as Phase 1, and 10–60 min was defined as Phase 2. SPSS statistical software was used for data analysis.
4.4.3. Chronic constriction injury model
8 SD rats (male, 6–8 weeks, 180–200 g) in each group. The rat's right hind sciatic nerve was ligation to make CCI model according to the method reported by Chaplan et al.43, 44, 45 Paw withdrawal threshold (PWT) was measured 14 days after surgery to evaluate the success of the CCI model (at least 25% lower than before the operation). The PWT was measured 1 h after compound administration. The electronic ciliary instrument used Electronic Von Frey 2390 (IITC Life Science Inc., Woodland Hills, CA, USA). The rate of pain threshold increase (PTI) was calculated as comparable data represent the analgesic effect of different compounds according to Eq. (4):
| (4) |
PWT0 h means pain withdraw threshold before compound administration; PWT1 h means pain withdraws threshold 1 h after compound administration.
4.4.4. Hot plate test
The hot plate test method is based on that previously reported by Masocha46. 10 ICR mice (female, 6–8 weeks, 18–24 g) in each group. The hot plate temperature was 60 °C; The Latency record as the time between mice contacted with a hot plate to pain response appeared (foot lifting and jumping) before (0 h) and 3 h after compound administration. The max positive effect (MPE, %) which represents the compound analgesic capability was calculated as Eq. (5):
| (5) |
Lat means latency, statistical significance was determined by a paired t-test between 0 and 3 h in each mice.
4.4.5. Writhing test
10 ICR mice (female and male mice (1:1), 6–8 weeks, 18–24 g) each group. The compounds were administered by i.g. administration, and 1 h later, 0.6% (v/v) acetic acid solution was injected by i.p. administration. The number of body writhing was observed and recorded in 15 min. The inhibition rate of compounds was calculated as Eq. (6):
| (6) |
Nvehicle means the number of writhing in the vehicle group; Ncomp means the number of writhing in compound treated group; one-way ANOVA was used for intergroup comparison and a paired t-test was used for intra-group comparison.
4.5. Binding site analysis
4.5.1. Homology modeling
The protein sequences of human ANO family (hANO1–hANO10) were downloaded from Uniprot website (https://www.uniprot.org/). mANO1 protein (PDB code: 5OYB) was downloaded from the PDB database (http://www.rcsb.org/) and used as the template. ANO family and mANO1 sequences were aligned automatically in the Schrödinger Prime software. Protein was minimized based on molecular force field (OPLS_3, 5000 steps), then 15 ns molecular dynamics simulation was made by Schrödinger Desmond software (constrain the main chain atomic coordinates, TIP3P water model, POPC membrane model, charge neutrality, adding 0.15 mol/L NaCl, NPT model, temperature 300 K, and pressure 101.325 bar). The PDB-formatted coordinate files of the hANO1 homology modeling structure were recorded in the Supporting Information Fig. S5.
4.5.2. Binding site prediction
The binding site was predicted by the Schrödinger SiteMap component (default parameters). Schrödinger Glide XP (OPLS_3 force field, no constrains) was used for molecular docking. Induced Fit Docking (standard precision, Glide XP for refinement) in Schrödinger software was used to dock ANO1 inhibitors to hANO1. Based on the binding pose, 50 ns molecular dynamics simulation was conducted by the Schrödinger Desmond software (TIP3P water model, POPC membrane model, charge neutrality, adding 0.15 mol/L NaCl, NPT model, temperature 300 K, and pressure 101.325 bar).
Acknowledgments
This research was supported by the National Key Research and Development Project (Grant No. 2019YFC1708900), the National Natural Science Foundation of China (Grant No. 21772005), National Major Scientific and Technological Special Project for Significant New Drugs Development (2019ZX09204-001, China) and Beijing Municipal Natural Science Foundation (7202088, 7172118, China). We are very appreciative of Dr. Wei Huang (Chinese Academy of Medical Sciences) and Dr. Peilin Yu (Zhejiang University, Hangzhou, China) for their help in this work.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting information to this article can be found online at https://doi.org/10.1016/j.apsb.2020.11.004.
Contributor Information
Kewei Wang, Email: wangkw@qdu.edu.cn.
Liangren Zhang, Email: liangren@bjmu.edu.cn.
Zhenming Liu, Email: zmliu@bjmu.edu.cn.
Author contributions
Yuxi Wang contributed on conceptualization, investigation, formal analysis, software, and writing—original draft. Jian Gao contributed on conceptualization, methodology, investigation, formal analysis. Song Zhao contributed on methodology, investigation, formal analysis. Yan Song contributed on methodology. Han Huang contributed on investigation. Guiwang Zhu contributed on investigation. Peili Jiao contributed on investigation. Xiangqing Xu contributed on resources. Guisen Zhang contributed on resources. Kewei Wang contributed on resources, supervision. Liangren Zhang: contributed on supervision, writing—review & editing. Zhenming Liu contributed on supervision, writing—review & editing.
Conflicts of interest
The authors have no conflicts of interest to declare.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Manoury B., Tamuleviciute A., Tammaro P. TMEM16A/Anoctamin 1 protein mediates calcium-activated chloride currents in pulmonary arterial smooth muscle cells. J Physiol. 2010;588:2305–2314. doi: 10.1113/jphysiol.2010.189506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ousingsawat J., Martins J.R., Schreiber R., Rock J.R., Harfe B.D., Kunzelmann K. Loss of TMEM16A causes a defect in epithelial Ca2+-dependent chloride transport. J Biol Chem. 2009;284:28698–28703. doi: 10.1074/jbc.M109.012120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rock J.R., O'Neal W.K., Gabriel S.E., Randell S.H., Harfe B.D., Boucher R.C. Transmembrane protein 16A (TMEM16A) is a Ca2+-regulated Cl‒ secretory channel in mouse airways. J Biol Chem. 2009;284:14875–14880. doi: 10.1074/jbc.C109.000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davis A.J., Forrest A.S., Jepps T.A., Valencik M.L., Wiwchar M., Singer C.A. Expression profile and protein translation of TMEM16A in murine smooth muscle. Am J Physiol Cell Physiol. 2010;299:948–959. doi: 10.1152/ajpcell.00018.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caputo A., Caci E., Ferrera L., Pedemonte N., Barsanti C., Sondo E. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322:590–594. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- 6.Schroeder B.C., Cheng T., Jan Y.N., Jan L.Y. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008;134:1019–1029. doi: 10.1016/j.cell.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Y.D., Cho H.W., Koo J.Y., Tak M.H., Cho Y.Y., Shim W.S. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455:1210–1236. doi: 10.1038/nature07313. [DOI] [PubMed] [Google Scholar]
- 8.Ferrera L., Caputo A., Galietta L.J.V. TMEM16A protein: A new identity for Ca2+-dependent Cl‒ channels. Physiology. 2010;25:357–363. doi: 10.1152/physiol.00030.2010. [DOI] [PubMed] [Google Scholar]
- 9.Benedetto R., Cabrita I., Schreiber R., Kunzelmann K. TMEM16A is indispensable for basal mucus secretion in airways and intestine. FASEB J. 2019;33:4502–4512. doi: 10.1096/fj.201801333RRR. [DOI] [PubMed] [Google Scholar]
- 10.Truong E.C., Phuan P.W., Reggi A.L., Ferrera L., Galietta L.J.V., Levy S.E. Substituted 2-acylaminocycloalkylthiophene-3-carboxylic acid arylamides as inhibitors of the calcium-activated chloride channel transmembrane protein 16a (TMEM16A) J Med Chem. 2017;60:4626–4635. doi: 10.1021/acs.jmedchem.7b00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu B.Y., Linley J.E., Du X.N., Zhang X., Ooi L., Zhang H.L. The acute nociceptive signals induced by bradykinin in rat sensory neurons are mediated by inhibition of M-type K+ channels and activation of Ca2+-activated Cl‒ channels. J Clin Invest. 2010;120:1240–1252. doi: 10.1172/JCI41084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miner K., Labitzke K., Liu B., Wang P., Henckels K., Gaida K. Drug repurposing: The anthelmintics niclosamide and nitazoxanide are potent TMEM16A antagonists that fully bronchodilate airways. Front Pharmacol. 2019;10:51–80. doi: 10.3389/fphar.2019.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Espinosa I., Lee C.H., Kim M.K., Rouse B.T., Subramanian S., Montgomery K. A novel monoclonal antibody against DOG1 is a sensitive and specific marker for gastrointestinal stromal tumors. Am J Surg Pathol. 2008;32:210–218. doi: 10.1097/PAS.0b013e3181238cec. [DOI] [PubMed] [Google Scholar]
- 14.Duvvuri U., Shiwarski D.J., Xiao D., Bertrand C., Huang X., Edinger R.S. TMEM16A induces MAPK and contributes directly to tumorigenesis and cancer progression. Cancer Res. 2012;72:3270–3281. doi: 10.1158/0008-5472.CAN-12-0475-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bill A., Hall M.L., Borawski J., Hodgson C., Jenkins J., Piechon P. Small molecule-facilitated degradation of ANO1 protein. J Biol Chem. 2014;289:11029–11041. doi: 10.1074/jbc.M114.549188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia G., Martinez-Rojas V.A., Oviedo N., Murbartian J. Blockade of Anoctamin-1 in injured and uninjured nerves reduces neuropathic pain. Brain Res. 2018;1696:38–48. doi: 10.1016/j.brainres.2018.06.001. [DOI] [PubMed] [Google Scholar]
- 17.Pineda-Farias J.B., Barragan-Iglesias P., Loeza-Alcocer E., Torres-Lopez J.E., Rocha-Gonzalez H.I., Perez-Severiano F. Role of anoctamin-1 and bestrophin-1 in spinal nerve ligation-induced neuropathic pain in rats. Mol Pain. 2015;11:41–56. doi: 10.1186/s12990-015-0042-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee B., Cho H., Jung J., Yang Y.D., Yang D.J., Oh U. Anoctamin 1 contributes to inflammatory and nerve-injury induced hypersensitivity. Mol Pain. 2014;10:5–15. doi: 10.1186/1744-8069-10-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deba F., Bessac B.F. Anoctamin-1 Cl‒ channels in nociception: activation by an N-aroylaminothiazole and capsaicin and inhibition by T16Ainh-A01. Mol Pain. 2015;11:55–63. doi: 10.1186/s12990-015-0061-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia G., Martinez-Rojas V.A., Rocha-Gonzalez H.I., Granados-Soto V., Murbartian J. Evidence for the participation of Ca2+-activated chloride channels in formalin-induced acute and chronic nociception. Brain Res. 2014;1579:35–44. doi: 10.1016/j.brainres.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 21.Takayama Y., Derouiche S., Maruyama K., Tominaga M. Emerging perspectives on pain management by modulation of TRP channels and ANO1. Int J Mol Sci. 2019;20:124–136. doi: 10.3390/ijms20143411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takayama Y., Uta D., Furue H., Tominaga M. Pain-enhancing mechanism through interaction between TRPV1 and anoctamin 1 in sensory neurons. Proc Natl Acad Sci U S A. 2015;112:5213–5218. doi: 10.1073/pnas.1421507112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cho H., Yang Y.D., Lee J., Lee B., Kim T., Jang Y. The calcium-activated chloride channel Anoctamin 1 acts as a heat sensor in nociceptive neurons. Nat Neurosci. 2012;15:1015–1021. doi: 10.1038/nn.3111. [DOI] [PubMed] [Google Scholar]
- 24.De La Fuente R., Namkung W., Mills A., Verkman A.S. Small-molecule screen identifies inhibitors of a human intestinal calcium-activated chloride channel. Mol Pharmacol. 2008;73:758–768. doi: 10.1124/mol.107.043208. [DOI] [PubMed] [Google Scholar]
- 25.Namkung W., Phuan P.W., Verkman A.S. TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J Biol Chem. 2011;286:2365–2374. doi: 10.1074/jbc.M110.175109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oh S.J., Hwang S.J., Jung J., Yu K., Kim J., Choi J.Y. MONNA, a potent and selective blocker for transmembrane protein with unknown function 16/Anoctamin-1. Mol Pharmacol. 2013;84:726–735. doi: 10.1124/mol.113.087502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seo Y., Lee H.K., Park J., Jeon D.K., Jo S., Jo M. Ani9, a novel potent small-molecule ANO1 inhibitor with negligible effect on ANO2. PLoS One. 2016;11:81–89. doi: 10.1371/journal.pone.0155771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Namkung W., Thiagarajah J.R., Phuan P.W., Verkman A.S. Inhibition of Ca2+-activated Cl‒ channels by gallotannins as a possible molecular basis for health benefits of red wine and green tea. FASEB J. 2010;24:4178–4186. doi: 10.1096/fj.10-160648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ji Q., Guo S., Wang X., Pang C., Zhan Y., Chen Y. Recent advances in TMEM16A: structure, function, and disease. J Cell Physiol. 2019;234:7856–7873. doi: 10.1002/jcp.27865. [DOI] [PubMed] [Google Scholar]
- 30.Seo Y., Ryu K., Park J., Jeon D.K., Jo S., Lee H.K. Inhibition of ANO1 by luteolin and its cytotoxicity in human prostate cancer PC-3 cells. PLoS One. 2017;12:534–555. doi: 10.1371/journal.pone.0174935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chai R., Chen Y., Yuan H., Wang X., Guo S., Qi J. Identification of resveratrol, an herbal compound, as an activator of the calcium-activated chloride channel, TMEM16A. J Membr Biol. 2017;250:483–492. doi: 10.1007/s00232-017-9975-9. [DOI] [PubMed] [Google Scholar]
- 32.Nishimura K., Yamamura H., Imaizumi Y. Functional expression of Ca2+-activated Cl‒ (TMEM16B) channel in rat pineal glands. J Pharmacol Sci. 2014;124:1223–1235. [Google Scholar]
- 33.Abaee M.S., Hadizadeh A., Mojtahedi M.M., Halvagar M.R. Exploring the scope of the Gewald reaction: Expansion to a four-component process. Tetrahedron Lett. 2017;58:1408–1412. [Google Scholar]
- 34.Zhang Y., Zhang Z.S., Xiao S.H., Tien J., Le S., Le T. Inferior olivary TMEM16B mediates cerebellar motor learning. Neuron. 2017;95:1103–1114. doi: 10.1016/j.neuron.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamamura H., Nishimura K., Hagihara Y., Suzuki Y., Imaizumi Y. TMEM16A and TMEM16B channel proteins generate Ca2+-activated Cl‒ current and regulate melatonin secretion in rat pineal glands. J Biol Chem. 2018;293:995–1006. doi: 10.1074/jbc.RA117.000326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paulino C., Neldner Y., Lam A.K., Kalienkova V., Brunner J.D., Schenck S. Structural basis for anion conduction in the calcium-activated chloride channel TMEM16A. Elife. 2017;6:65–75. doi: 10.7554/eLife.26232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bill A., Popa M.O., van Diepen M.T., Gutierrez A., Lilley S., Velkova M. Variomics screen identifies the re-entrant loop of the calcium-activated chloride channel ANO1 that facilitates channel activation. J Biol Chem. 2015;290:889–903. doi: 10.1074/jbc.M114.618140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brunner J.D., Lim N.K., Schenck S., Duerst A., Dutzler R. X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature. 2014;516:207–215. doi: 10.1038/nature13984. [DOI] [PubMed] [Google Scholar]
- 39.Dang S., Feng S., Tien J., Peters C.J., Bulkley D., Lolicato M. Cryo-EM structures of the TMEM16A calcium-activated chloride channel. Nature. 2017;552:426–429. doi: 10.1038/nature25024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paulino C., Kalienkova V., Lam A.K.M., Neldner Y., Dutzler R. Activation mechanism of the calcium-activated chloride channel TMEM16A revealed by cryo-EM. Nature. 2017;552:421–425. doi: 10.1038/nature24652. [DOI] [PubMed] [Google Scholar]
- 41.Seo Y., Kim J., Chang J., Kim S.S., Namkung W., Kim I. Synthesis and biological evaluation of novel Ani9 derivatives as potent and selective ANO1 inhibitors. Eur J Med Chem. 2018;160:245–255. doi: 10.1016/j.ejmech.2018.10.002. [DOI] [PubMed] [Google Scholar]
- 42.Okuda K., Sakurada C., Takahashi M., Yamada T., Sakurada T. Characterization of nociceptive responses and spinal releases of nitric oxide metabolites and glutamate evoked by different concentrations of formalin in rats. Pain. 2001;92:107–115. doi: 10.1016/s0304-3959(00)00476-0. [DOI] [PubMed] [Google Scholar]
- 43.Chaplan S.R., Bach F.W., Pogrel J.W., Chung J.M., Yaksh T.L. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 44.Bennett G.J., Xie Y.K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 45.Hargreaves K., Dubner R., Brown F., Flores C., Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 46.Masocha W., Kombian S.B., Edafiogho I.O. Evaluation of the antinociceptive activities of enaminone compounds on the formalin and hot plate tests in mice. Sci Rep. 2016;6:53–66. doi: 10.1038/srep21582. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.