

Abstract
Minimal mimics of protein conformations provide rationally designed ligands to modulate protein function. The advantage of minimal mimics is that they can be chemically synthesized and coaxed to be proteolytically resistant; a key disadvantage is that minimization of the protein binding epitope may be associated with loss of affinity and specificity. Several approaches to overcome this challenge may be envisioned, including deployment of covalent warheads and use of nonnatural residues to improve contacts with the binding surface. Herein, we describe our computational and experimental efforts to enhance the minimal protein mimics with fragments that can contact undiscovered binding pockets on Mdm2 and MdmX – two well-studied protein partners of p53.
Graphical Abstract
Mimics of folded protein domains offer rationally designed synthetic scaffolds to interfere with biomolecular complexes.1–7 The reproduction of protein binding epitopes in minimal mimics can yield proteolytically stable and cell permeable ligands but the process of downsizing protein epitopes is often coupled with a loss of some binding contacts, which leads to reduced affinity and specificity for the target surface.8, 9 Several strategies to improve the affinity of minimal protein mimics have been introduced, including the use of electrophilic warheads to covalently capture protein targets and nonnatural side chains to engage underutilized binding pockets.10, 11
Computational tools that characterize protein surfaces and predict the contribution of native and nonnatural contact residues to binding have become critical for any inhibitor discovery campaign. We recently described a computational algorithm, AlphaSpace, to enable high-resolution fragment-centric topographical mapping (FCTM) of proteins.10, 12, 13 AlphaSpace may be deployed to design nonnatural side chains to engage underutilized binding cavities and enhance binding affinity and specificity of the ligands (Figure 1). In recent studies, we have illustrated the potential of this algorithm to elaborate protein domain mimics5 with nonnatural side chains to enhance the potency of synthetic ligands.10, 14 In this paper, we systematically analyzed and compared pockets on protein surfaces, which led to the identification of secondary binding sites on Mdm2 and MdmX. The detection of cryptic pockets on Mdm proteins required introduction of two new features in AlphaSpace12: B-Score for estimating pocket targetability and ensemble β-clustering for one-to-one pocket mapping of aligned protein structures.
Figure 1.
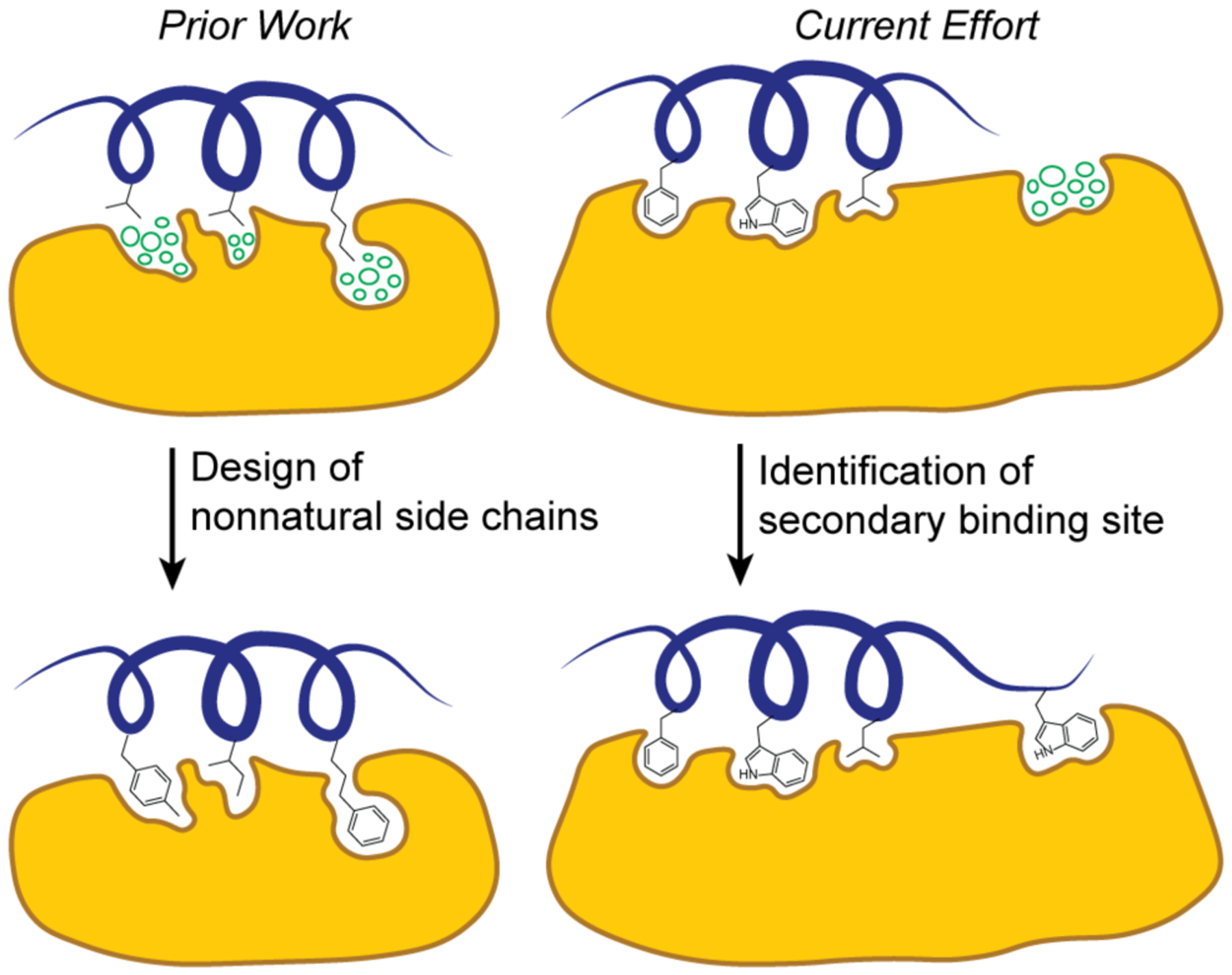
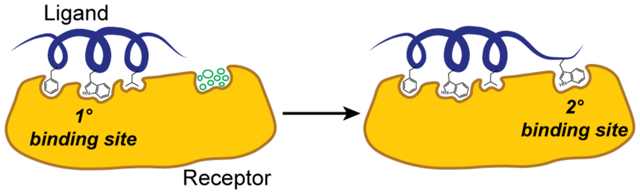
Topographic mapping of protein surfaces to improve peptidomimetic binders. Fragment-centric topographical mapping allows design of nonnatural side chains (left) and identification of secondary binding sites for peptidomimetic elaboration (right). The cluster of circles depicts unoccupied design space. Herein we describe efforts to identify a secondary binding site on Mdm2/MdmX surfaces to elaborate a p53 mimic as a dual Mdm2/MdmX ligand.
The crystal structure of the protein-protein interaction between p53 activation domain (AD) and Mdm2 has served as an inspiration for the design of small molecule and helical peptide inhibitors.15–22 The complex shows that three residues from the p53 helix – F19, W23, and L26 – engage a hydrophobic groove on Mdm2.22 Mimics of these three residues have led to clinical candidates as the canonical examples of small molecule PPI inhibitors.23, 24 Potent peptide mimics of the p53 AD sequence have also been developed and are currently in clinical trials.15 MdmX is a homolog of Mdm2, and experimental evidence indicates non-overlapping functions MDM proteins in p53 regulation.16 There has been a significant clinical interest to develop dual inhibitors of both MDM proteins to achieve optimal restoration of p53.15, 17
We analyzed the Mdm2 and MdmX surfaces with AlphaSpace to identify potential pockets distal to the primary p53 AD binding site. AlphaSpace detects concave cavities on protein surfaces and assigns pseudoatoms to optimally occupy them.12, 13 Comparison of pseudoatoms provides a quantitative difference map of protein surfaces. Our analyses of protein surfaces suggested potential secondary pockets formed by Mdm2 residues T26, M50, Y100, Y104, V108 and MdmX residues V49, M53, Y99, L102, R103 and L106 (Figure 2A). The primary site on Mdm proteins features three well-defined pockets for p53 side chain groups F19, W23, and L26 (Figure 2B). We were surprised to learn that a secondary pocket exists so close to the primary binding site in experimentally-derived structures of both proteins but had been missed in the many inhibitor design campaigns (Figure 2C); we suspect that truncation of the C-terminus of the p53 sequence to residue N29 in the initial p53-Mdm2 crystal structure focused subsequent campaigns on the primary p53 binding site. Peptide mutagenesis analysis supported the critical role of the primary binding site, indeed an NMR analysis suggests that the proceeding C-terminal region p5328–34 on its own does not meaningfully contribute to complex formation.25
Figure 2.
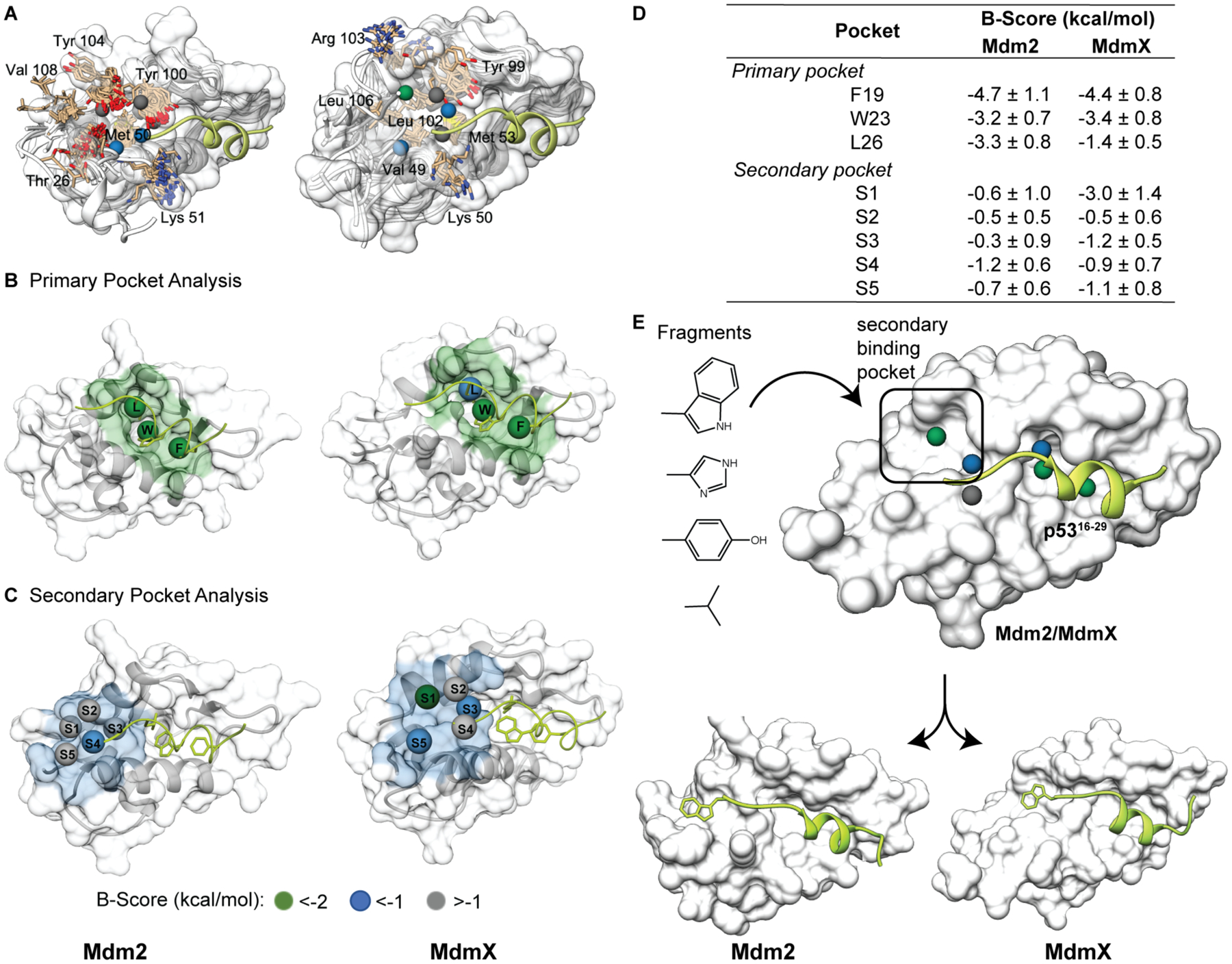
(A) AlphaSpace analysis suggests a secondary pocket on Mdm2 (left) and MdmX (right). The p53 helix is shown in green. The spheres represent the centroids of AlphaSpace-identified binding pockets. Alignment of secondary pocket residues is highlighted in tan. (B-C) Comparison of the primary and secondary pockets on Mdm2 and MdmX. The spheres represent centroids of pockets near the p53 helix. (D) AlphaSpace predicts potential enhancement in binding affinity of a ligand if a particular pocket is maximally occupied. This prediction is expressed as a B-Score. B-Scores for primary and secondary pockets for each Mdm protein are listed. (E) We performed localized docking to identify suitable natural side chain fragments (see Figure S1) for the secondary pockets. Details of the AlphaSpace analyses are included in the SI. The input structures for AlphaSpace analysis are PDB IDs 1YCR (Mdm2) and 3DAB (MdmX).
We compared the targetability of the secondary pockets vis-a-vis the primary pockets on Mdm2/X. AlphaSpace provides a B-Score for each pocket based on the binding free energies that can be gained if the particular pocket is fully occupied. B-Scores for primary and secondary pockets on Mdm2 and MdmX are shown in Figure 2D, which suggest that secondary pockets on MdmX and Mdm2 may also be targetable, and occupancy of the secondary site could confer additional affinity in concert with the primary binding region. AlphaSpace predicts higher scoring secondary pockets on MdmX than Mdm2. It should be noted that Verma and coworkers performed MD simulations with benzene probes to identify inducible pockets on the Mdm2 protein’s surface.26 Here our complementary approach analyzes concave spaces on Mdm2 and MdmX using an ensemble of static protein structures to find potentially targetable binding sites. The key difference in the two computational approaches is that AlphaSpace calculations can directly evaluate an experimentally-derived protein structure for underutilized pockets whereas extensive MD simulations with solvent probes are required to morph the protein surface for potentially inducible pockets.
We performed localized molecular docking, which allows a focused search space on the secondary binding site, to identify fragments corresponding to natural amino acid side chains that can optimally engage this region. An indole group emerged as a lead candidate from these studies (Figure 2E and Supporting Figure S1), which suggests that a C-terminal tryptophan residue may serve as the fourth hotspot on p53 to target the newly identified binding pocket.
To experimentally test the computational prediction that a simple C-terminal modification of the p53 peptide with a tryptophan residue may significantly enhance its binding affinity for Mdm2 and MdmX, we synthesized a set of peptides based on the native p53 (AD) and measured their binding affinities to Mdm2 and MdmX with a previously described competitive fluorescence polarization assay (Figure 3A and SI).27 The p53 peptide 1 (p5316–29) exhibits micromolar binding affinity for both Mdm2 and MdmX in the FP assay, while the peptide 2, in which a tryptophan residue linked by a flexible glycine residue to proline-27 is designed to contact the secondary pocket, showed an improved affinity to Mdm2 and MdmX as compared to 1. This result supports the hypothesis that the designed tryptophan residue may engage the identified secondary binding site. (Both computational and experimental mutagenesis studies suggest that the p53 C-terminal residues E-28 and N-29 do not contribute significantly to binding interactions with Mdm2.28)
Figure 3.
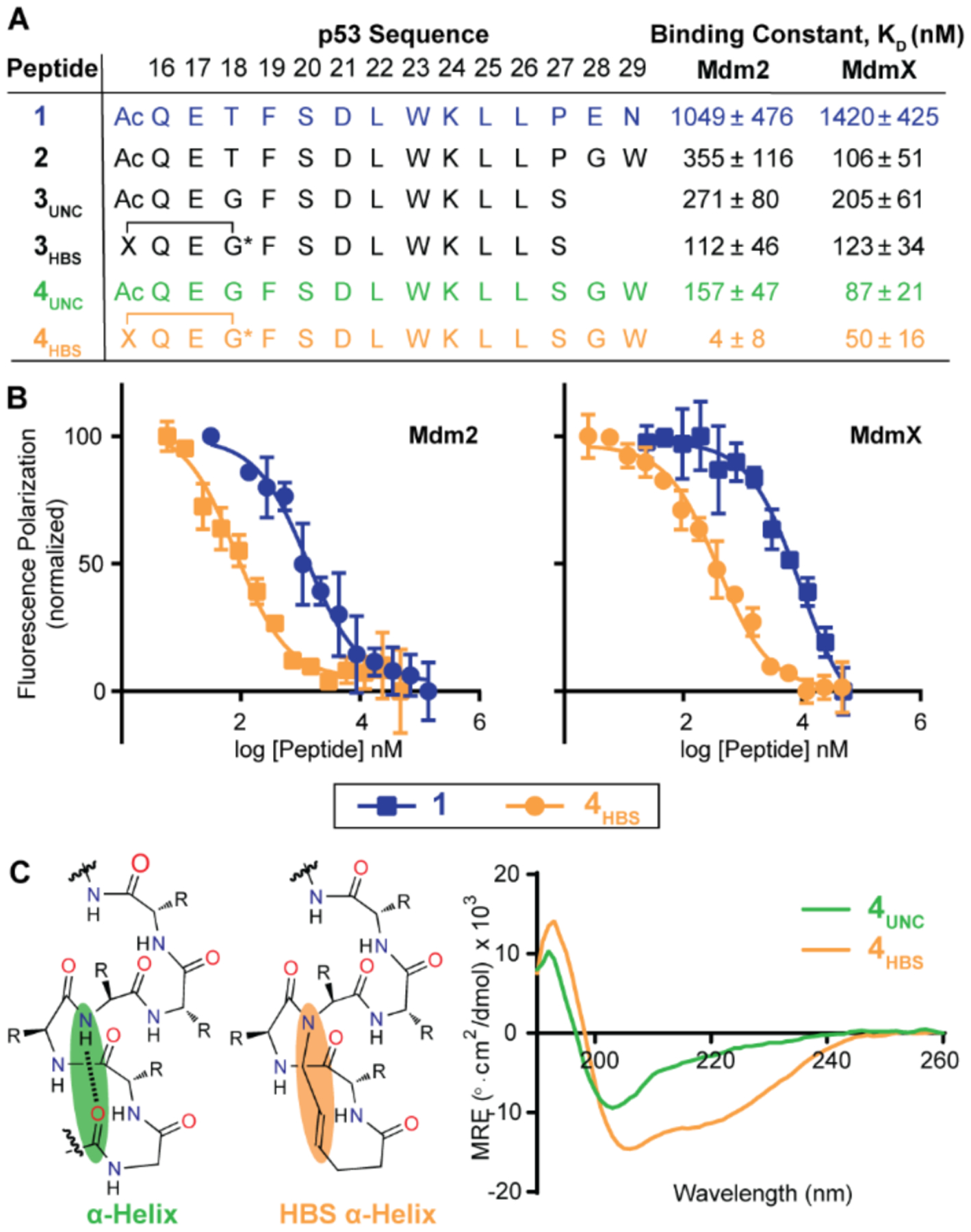
(A) The binding affinity of p53 peptides for Mdm2 and MdmX was characterized with a fluorescence polarization (FP) assay. (B) Comparison of FP binding isotherms for peptide 1 and the constrained peptide 4HBS. (C) The helical conformation of 4HBS was stabilized by the hydrogen bond surrogate approach by replacing an intrachain hydrogen bond with a carbon-carbon bond. The conformation of the constrained and unconstrained peptides were characterized by CD spectroscopy at 50 μM peptide concentration in 50 mM sodium phosphate, pH 7.4.
We next sought to determine if stabilization of the p53 sequence into an α-helical conformation would enhance its binding affinity. We have previously reported a hydrogen bond surrogate (HBS) strategy to stabilize short α-helices; HBS helices feature a covalent bond in place of the i→i+4 hydrogen bond in canonical α-helices (Figure 3C).5, 29 In prior efforts, we have also applied this strategy to mimic p53 AD and develop Mdm2 ligands.28, 30 3HBS was based on an optimized p53 sequence (3UNC) in which proline-27 was substituted with a serine residue. Prior studies have reported that the native proline residue in p53 disrupts Mdm2 binding.15, 31, 32 3HBS binds Mdm2 and MdmX with similar binding affinities, Kd = 112 ± 46 (Mdm2) and 123 ± 34 nM (MdmX). C-Terminal extension of 3HBS with glycine-tryptophan dipeptide resulted in significantly improved ligand (4HBS) for both Mdm2 and MdmX, Kd = 4 ± 8 (Mdm2) and 50 ± 16 nM (MdmX). Peptide 4UNC, which is an unconstrained analog of 4HBS, is a 40-fold weaker binder for Mdm2 but largely retains its affinity for MdmX. Circular dichroism analysis suggests that constrained peptide, 4HBS, is significantly helical as compared to its unconstrained counterpart (Figure 3C).
The secondary binding site occupancy by 4HBS was evaluated by heteronuclear single quantum coherence NMR spectroscopy (HSQC) experiment, which provides chemical shift changes of specific protein residues upon ligand binding. 15N-labeled Mdm2 and MdmX resonances were assigned as previously reported.33, 34 The results of the HSQC experiment are depicted in Figure 4. To gauge the potential of 4HBS to engage the secondary binding pocket, we compared the chemical shift perturbations (CSP) caused by 4HBS as compared to 3HBS. We compared CSP differences emanating from the addition of one equivalent of 4HBS versus 3HBS in primary and secondary pockets of 15N-labeled Mdm2 and MdmX, with the expectation that the two peptides will likely impact the primary pockets similarly but 4HBS will cause a more pronounced change in the secondary pocket. The results support our predictions and support the hypothesis that the designed tryptophan group of 4HBS binds in the secondary pocket.
Figure 4.
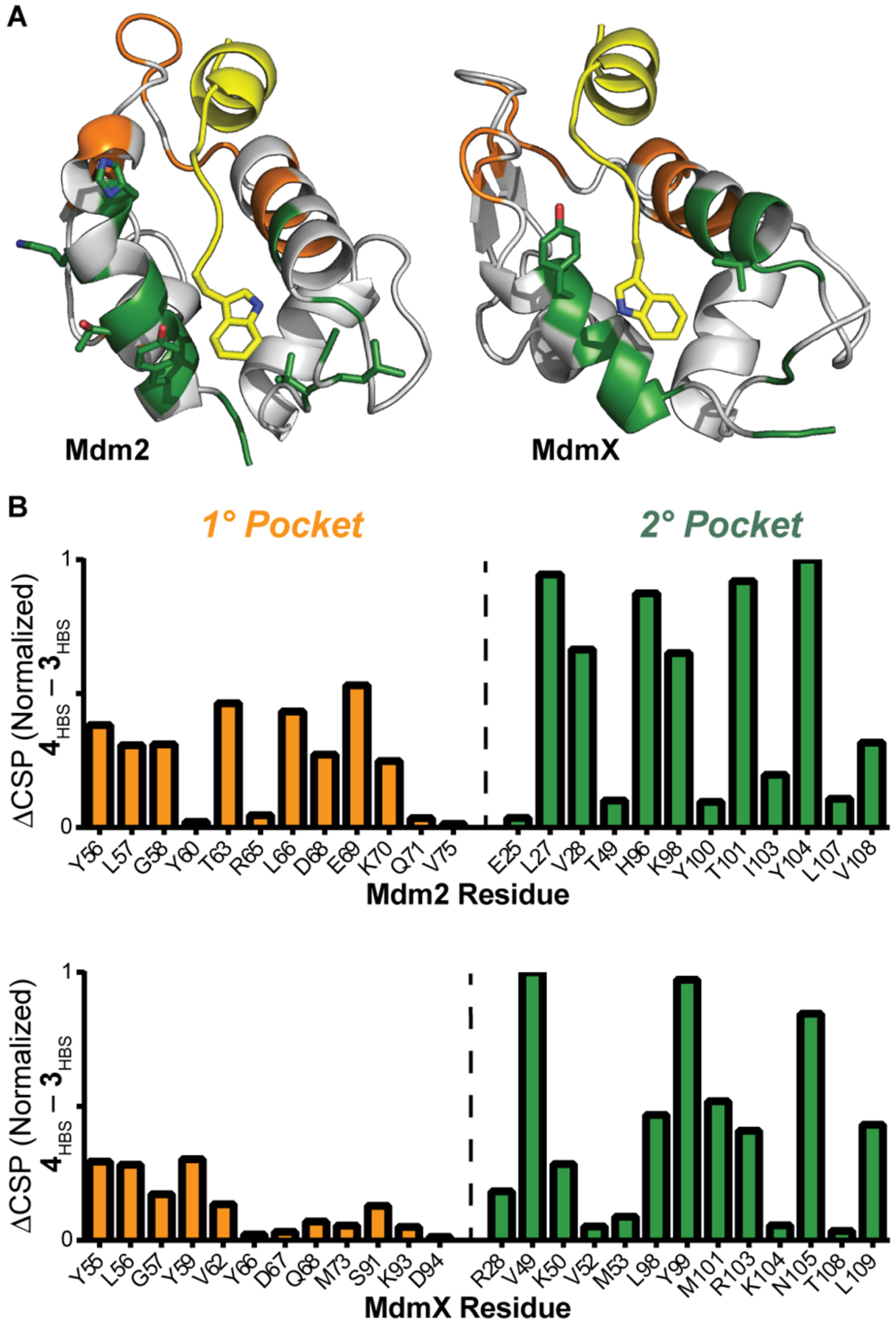
(A) Mdm2/X binding site analysis by HSQC NMR. Chemical shift changes were observed for Mdm2/X residues in the primary (orange) and secondary (green) binding pockets upon addition of 1 eq of 4HBS. (B) The difference in chemical shift perturbation caused by 4HBS and 3HBS reveals that 4HBS engages the secondary binding pocket. Full HSQC spectra are included in the Supporting Information.
In summary, our studies show the potential of topographic protein maps to rapidly identify unutilized but targetable pockets to afford optimized ligands. The studies shown here provide a benchmark for identifying new pockets on Mdm2 and MdmX – two well studied therapeutic targets. Here we focused on modifying native peptide mimics but the strategy can be readily applied to optimize small molecule and peptide ligands for Mdm2 and MdmX. We expect topographic maps of protein surfaces will complement existing approaches for fragment-screening and protein epitope mimicry to guide design of potent ligands.
Supplementary Material
ACKNOWLEDGMENT
We thank the National Institutes of Health (R35GM130333 to PSA, R35 GM127040 to YZ, and R01GM120736 to PSA and YZ) for support of this work.
Footnotes
The authors declare no competing financial interests.
Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website.
AlphaSpace analysis, and synthesis, characterization and binding analysis of peptides (PDF)
REFERENCES
- (1).Merritt HI, Sawyer N, and Arora PS (2020) Bent into shape: Folded peptides to mimic protein structure and modulate protein function, Peptide Sci 112, e24145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Pelay-Gimeno M, Glas A, Koch O, and Grossmann TN (2015) Structure-Based Design of Inhibitors of Protein–Protein Interactions: Mimicking Peptide Binding Epitopes, Angew. Chem. Int. Ed 54, 8896–8927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Horne WS, and Grossmann TN (2020) Proteomimetics as protein-inspired scaffolds with defined tertiary folding patterns, Nat. Chem 12, 331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Checco JW, Kreitler DF, Thomas NC, Belair DG, Rettko NJ, Murphy WL, Forest KT, and Gellman SH (2015) Targeting diverse protein–protein interaction interfaces with α/β-peptides derived from the Z-domain scaffold, Proc. Natl. Acad. Sci. USA 112, 4552–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sawyer N, Watkins AM, and Arora PS (2017) Protein Domain Mimics as Modulators of Protein–Protein Interactions, Acc. Chem. Res 50, 1313–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Azzarito V, Long K, Murphy NS, and Wilson AJ (2013) Inhibition of alpha-helix-mediated protein-protein interactions using designed molecules, Nat. Chem 5, 161–173. [DOI] [PubMed] [Google Scholar]
- (7).Khakshoor O, and Nowick JS (2008) Artificial beta-sheets: chemical models of beta-sheets, Curr. Opin. Chem. Biol 12, 722–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Modell AE, Blosser SL, and Arora PS (2016) Systematic Targeting of Protein-Protein Interactions, Trends Pharmacol. Sci 37, 702–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Watkins AM, and Arora PS (2015) Structure-based inhibition of protein–protein interactions, Eur. J. Med. Chem 94, 480–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Rooklin D, Modell AE, Li H, Berdan V, Arora PS, and Zhang Y (2017) Targeting Unoccupied Surfaces on Protein–Protein Interfaces, J. Am. Chem. Soc 139, 15560–15563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Berdan VY, Klauser PC, and Wang L (2021) Covalent peptides and proteins for therapeutics, Bioorg. Med. Chem 29, 115896. [DOI] [PubMed] [Google Scholar]
- (12).Katigbak J, Li H, Rooklin D, and Zhang Y (2020) AlphaSpace 2.0: Representing Concave Biomolecular Surfaces Using β-Clusters, J. Chem. Inf. Model 60, 1494–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Rooklin D, Wang C, Katigbak J, Arora PS, and Zhang Y (2015) AlphaSpace: Fragment-Centric Topographical Mapping To Target Protein–Protein Interaction Interfaces, J. Chem. Inf. Model 55, 1585–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Sadek J, Wuo MG, Rooklin D, Hauenstein A, Hong SH, Gautam A, Wu H, Zhang Y, Cesarman E, and Arora PS (2020) Modulation of virus-induced NF-κB signaling by NEMO coiled coil mimics, Nat. Commun 11, 1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To K-H, Olson KA, Kesavan K, Gangurde P, Mukherjee A, Baker T, Darlak K, Elkin C, Filipovic Z, Qureshi FZ, Cai H, Berry P, Feyfant E, Shi XE, Horstick J, Annis DA, Manning AM, Fotouhi N, Nash H, Vassilev LT, and Sawyer TK (2013) Stapled α−helical peptide drug development: A potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy, Proc. Natl. Acad. Sci. USA 110, E3445–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Biderman L, Manley JL, and Prives C (2012) Mdm2 and MdmX as Regulators of Gene Expression, Genes Cancer 3, 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Popowicz GM, Dömling A, and Holak TA (2011) The Structure-Based Design of Mdm2/Mdmx–p53 Inhibitors Gets Serious, Angew. Chem. Int. Ed 50, 2680–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Reed D, Shen Y, Shelat AA, Arnold LA, Ferreira AM, Zhu F, Mills N, Smithson DC, Regni CA, Bashford D, Cicero SA, Schulman BA, Jochemsen AG, Guy RK, and Dyer MA (2010) Identification and Characterization of the First Small Molecule Inhibitor of MDMX, J. Biol. Chem 285, 10786–10796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Li C, Pazgier M, Yuan W, Liu M, Wei G, Lu WY, and Lu W (2010) Systematic mutational analysis of peptide inhibition of the p53-MDM2/MDMX interactions, J. Mol. Biol 398, 200–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Pazgier M, Liu M, Zou G, Yuan W, Li C, Li J, Monbo J, Zella D, Tarasov SG, and Lu W (2009) Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX, Proc. Natl. Acad. Sci. USA 106, 4665–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Hu B, Gilkes DM, and Chen J (2007) Efficient p53 Activation and Apoptosis by Simultaneous Disruption of Binding to MDM2 and MDMX, Cancer Research 67, 8810–8817. [DOI] [PubMed] [Google Scholar]
- (22).Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, and Pavletich NP (1996) Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain, Science 274, 948–953. [DOI] [PubMed] [Google Scholar]
- (23).Vassilev LT (2007) MDM2 inhibitors for cancer therapy, Trends Mol Med 13, 23–31. [DOI] [PubMed] [Google Scholar]
- (24).Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, and Liu EA (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2, Science 303, 844–848. [DOI] [PubMed] [Google Scholar]
- (25).Chi S-W, Lee S-H, Kim D-H, Ahn M-J, Kim J-S, Woo J-Y, Torizawa T, Kainosho M, and Han K-H (2005) Structural Details on mdm2-p53 Interaction*, J. Biol. Chem 280, 38795–38802. [DOI] [PubMed] [Google Scholar]
- (26).Tan YS, Reeks J, Brown CJ, Thean D, Ferrer Gago FJ, Yuen TY, Goh ETL, Lee XEC, Jennings CE, Joseph TL, Lakshminarayanan R, Lane DP, Noble MEM, and Verma CS (2016) Benzene Probes in Molecular Dynamics Simulations Reveal Novel Binding Sites for Ligand Design, The J. Phys. Chem. Lett 7, 3452–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Knight SM, Umezawa N, Lee HS, Gellman SH, and Kay BK (2002) A fluorescence polarization assay for the identification of inhibitors of the p53-DM2 protein-protein interaction, Anal. Biochem 300, 230–236. [DOI] [PubMed] [Google Scholar]
- (28).Henchey LK, Porter JR, Ghosh I, and Arora PS (2010) High Specificity in Protein Recognition by Hydrogen-Bond-Surrogate alpha-Helices: Selective Inhibition of the p53/MDM2 Complex, ChemBiochem 11, 2104–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Patgiri A, Jochim AL, and Arora PS (2008) A hydrogen bond surrogate approach for stabilization of short peptide sequences in alpha-helical conformation, Acc. Chem. Res 41, 1289–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Patgiri A, Joy ST, and Arora PS (2012) Nucleation Effects in Peptide Foldamers, J. Am. Chem. Soc 134, 11495–11502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zondlo SC, Lee AE, and Zondlo NJ (2006) Determinants of specificity of MDM2 for the activation domains of p53 and p65: proline27 disrupts the MDM2-binding motif of p53, Biochemistry 45, 11945–11957. [DOI] [PubMed] [Google Scholar]
- (32).Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, and Verdine GL (2007) Reactivation of the p53 Tumor Suppressor Pathway by a Stapled p53 Peptide, J. Am. Chem. Soc 129, 2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Stoll R, Renner C, Mühlhahn P, Hansen S, Schumacher R, Hesse F, Kaluza B, Engh RA, Voelter W, and Holak TA (2000) Sequence-specific 1H, 15N, and 13C assignment of the N-terminal domain of the human oncoprotein MDM2 that binds to p53, J. Biomol. NMR 17, 91–92. [DOI] [PubMed] [Google Scholar]
- (34).Sanchez MC, Renshaw JG, Davies G, Barlow PN, and Vogtherr M (2010) MDM4 binds ligands via a mechanism in which disordered regions become structured, FEBS Lett 584, 3035–3041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.