

Abstract
Carbohydrates play essential roles in a variety of biological processes that are dictated by their structures. However, characterization of carbohydrate structures remains extremely difficult and generally unsolved. In this work, a de novo mass spectrometry-based workflow was developed to isolate and structurally elucidate oligosaccharides to provide sequence, monosaccharide compositions, and glycosidic linkage positions. The approach employs liquid chromatography–tandem mass spectrometry (LC–MS/MS)-based methods in a 3-dimensional concept: one high performance liquid chromatography-quadrupole time-of-flight mass spectrometry (HPLC-QTOF MS) analysis for oligosaccharide sequencing and two ultra high performance liquid chromatography-triple quadrupole mass spectrometry (UHPLC-QqQ MS) analyses on fractionated oligosaccharides to determine their monosaccharides and linkages compositions. The workflow was validated by applying the procedure to maltooligosaccharide standards. The approach was then used to determine the structures of oligosaccharides derived from polysaccharide standards and whole food products. The integrated LC–MS workflow will reveal the in-depth structures of oligosaccharides.
Keywords: oligosaccharide analysis, monosaccharide analysis, linkage analysis, polysaccharides, quadrupole-time-of-flight mass spectrometry, triple quadrupole mass spectrometry
Introduction
Carbohydrates have a wide range of important functions that are dictated by their specific structures. For example, human milk oligosaccharides (HMOs) are not digested by the infant’s innate metabolism. However, they are catabolized by the infant gut bacteria, thereby providing nutritional value in the form of short chain fatty acids to the infant.1,2 The specific structures of HMOs correspond uniquely to bacterial enzymes that ferment them.3−10 In human cells, oligosaccharides are also found on cell surfaces where they play important roles in cell to cell recognition and cell-binding processes.11−14 Under some conditions, gut bacteria are known to consume host cell glycans in a similarly structure-specific manner.15 In food, carbohydrates are often the largest component of the adult diet and are similarly responsible for shaping the composition of the gut microbiota.16,17
Dietary oligosaccharides such as fructooligosaccharides (FOS) and galactooligosaccharides (GOS) are termed prebiotics because they are similarly not digested by the host but are consumed by gut bacteria.18−20 These oligosaccharides are similarly selectively fermented by microbes in a structure specific manner in the large intestine.21,22 While FOS and GOS represent major industrial products, they are relatively homogeneous and are composed primarily of only a single monomer (fructose or galactose). Very many plant-based oligosaccharides may already be accessible to gut bacteria; however, we currently lack the methods to produce and characterize them. Methods for the de novo structural elucidation of oligosaccharides and polysaccharides are critically needed to develop new classes of prebiotics with potentially higher biological efficacy.
The development of methods for the complete analysis of oligosaccharides presents major challenges. A variety of techniques are typically employed to determine different aspects of their structures. Size-exclusion chromatography (SEC) is used to separate oligosaccharides from complex matrices and gives insight into the average degree of polymerization but does not elucidate the identity of the monosaccharides nor glycosidic linkages.23 Other methods including capillary electrophoresis (CE), high-performance liquid chromatography (HPLC), and high-performance anion-exchange chromatography (HPAEC) are used to separate oligosaccharides, while ultraviolet–visible (UV–vis) spectrophotometers, refractive index detectors (RID), and pulsed amperometric detectors (PAD) are used for detection.24−27 While these methods are sensitive, they lack throughput, structural information, and integration in a way that allows a deep characterization of structure. Gas chromatography (GC) is typically less used for the analysis of oligosaccharides but has long been used as the standard method for monosaccharide and linkage analysis.28,29 GC methods, however require long separation times, have low sensitivities and throughput, require extra derivatization steps in the sample preparation, focus on a limited number of monosaccharides and linkages, and yield little to no absolute quantitative information.30−32 Mass spectrometry method such as collision-induced dissociation (CID) have been employed with sodiated oligosaccharides to yield specific structural information.33−35 Newer MS techniques, such as ion mobility mass spectrometry (IM-MS) while still being developed could potentially provide anomer and sequence information. However, this and similar methods do not yet provide de novo monosaccharide nor linkage information.36−38
The impact that carbohydrates have on the microbiota has renewed interests in developing analytical methods for poly- and oligosaccharide analysis.39−43 The understanding of the role of HMOs, for example, has increased when we developed methods to obtain complete structures of large pools of compounds through LC–MS profiling. Tandem mass spectrometry (LC–MS/MS) further revealed structural information such as the degree of polymerization (DP) with composition and some sequence information when mass heterogeneity of the monomers are available.44,45 However, challenges in oligosaccharide analysis using LC–MS/MS remain. Many monosaccharides have identical masses so that CID fragmentation is insufficient for differentiating the isomers or branching positions. A hexose monosaccharide with a single mass can have several isomers which include fructose, glucose, galactose, mannose, or allose and cannot be resolved using standard LC–MS/MS workflow. Enzymatic treatments coupled with LC–MS/MS have been employed to identify the monosaccharide isomers and yield complete oligosaccharide structures.46 Enzymes used in this way were effective; however, they require prior knowledge of the monosaccharide compositions, possible linkages, and commercial availability.
We previously described a method for producing novel oligosaccharides from polysaccharides. In this report, we describe a multidimensional LC–MS/MS workflow for de novo structural elucidation of oligosaccharides derived not only from polysaccharide standards but also from a more complicated matrix such as from plant and food products. The concept involves three types of LC–MS/MS analyses representing a three-dimensional mass spectrometry (3D-MS) approach to reveal each oligosaccharide’s sequence, composition, and linkages. HPLC separation of the oligosaccharides is performed followed by a two-way split to an in-line fractionation and a high-resolution quadrupole time-of-flight mass spectrometry instrument. A 96-well plate fraction collector is used to isolate the oligosaccharides. Structural information regarding the DP is obtained through tandem MS (MS/MS). The collected fractions are subjected to a series of relatively fast methods to provide monosaccharide and linkage profiles.41,47,48 For each oligosaccharide, the method yields absolute quantitation of the monosaccharides and the relative abundance of each glycosidic linkage. Using this combinatorial approach that comprises sequence information from the QTOF MS/MS and the monosaccharide and linkage compositions from separate LC–MS/MS analyses, the in-depth oligosaccharide structures of the mixtures are elucidated.
Experimental Section
Samples and Materials
Sodium acetate, hydrogen peroxide (H2O2) (30% v/v), iron(III) sulfate pentahydrate (Fe2(SO4)3), trifluoroacetic acid (TFA) (HPLC grade), chloroform (HPLC grade), ammonium acetate, sodium hydroxide pellets (semiconductor grade 99.99% trace metals basis), ammonium hydroxide solution (NH4OH) (28–30%), 3-methyl-1-phenyl-2-pyrazoline-5-one (PMP), dichloromethane (DCM), methanol (HPLC grade), anhydrous dimethyl sulfoxide (DMSO), glacial acetic acid, d-galactose, d-mannose, d-glucose, d-allose, d-fructose, d-xylose, l-arabinose, d-xylose, l-fucose, l-rhamnose, N-acetyl-d-glucosamine (GlcNAc), N-acetyl-d-galactosamine (GalNAc), d-glucuronic acid (GlcA), and d-galacturonic acid (GalA) were purchased from Sigma-Aldrich (St. Louis, MO). Maltotriose, maltotetraose, maltopentaose, and maltohexaose were obtained from Carbosynth (Compton, UK). Galactan was purchased from Megazyme (Bray, Ireland). C18 and porous graphitized carbon (PGC) solid phase extraction (SPE) plates were purchased from Glygen (Columbia, MD). Formic acid (FA) (99.5% optima LC/MS grade) was purchased from Fisher Scientific (Hampton, NH). Acetonitrile (ACN) (HPLC grade) was purchased from Honeywell (Muskegon, MI). All purchased chemicals were used without further purification. Butternut squash was purchased from Pedrick Produce (Dixon, CA). Nanopure water was used for all experiments.
Preparation of the Maltooligosaccharides
A stock solution comprising of maltotriose, maltotetraose, maltopentaose, and maltohexaose was prepared in water at a concentration of 2 mg/mL. A 10 μL aliquot was subjected to the 3D MS workflow.
Preparation of Polysaccharide-Derived Oligosaccharides
An oxidative digestion step, Fenton’s initiation toward defined oligosaccharide groups (FITDOG), was used to generate oligosaccharides from polysaccharides.49 Briefly, a reaction solution was prepared containing 95% (v/v) 40 mM sodium acetate buffer adjusted to pH 5 with glacial acetic acid, 5% (v/v) hydrogen peroxide (30% v/v), and 65 nM iron(III) sulfate. A whole butternut squash was cut to small pieces, lyophilized, and ground to fine powder. The reaction mixture was added to 1 mg of dry galactan and 1 mg of lyophilized butternut squash powder to make a final solution of 1 mg/mL of both. The reaction was incubated at 100 °C for 1 h. The reaction was quenched by adding half of the reaction volume of cold 2 M NaOH. Glacial acetic acid was added for neutralization. A total of 30 reactions were performed on a 96-well plate.
Oligosaccharides generated from galactan were purified by 96-well plate PGC cartridges. PGC was used to trap the oligosaccharides while removing the salts. The cartridges were washed with 80% acetonitrile and 0.1% (v/v) TFA in water. The oligosaccharides were loaded and washed with five column volumes of water. The oligosaccharides were eluted with 40% acetonitrile with 0.05% (v/v) TFA in water. Samples were pooled, dried by vacuum centrifugation, and reconstituted in water prior to the de novo oligosaccharide analysis.
An additional C18 clean-up step was employed to oligosaccharides generated from butternut squash before the PGC cartridge enrichment to remove any hydrophobic components such as lipids and proteins. For the C18 cleanup, cartridges were washed with pure acetonitrile followed a water wash five times the column volumes. Analytes were loaded and eluted with two column volumes of pure water. The effluent containing the oligosaccharides was then enriched using the PGC protocol described above.
Mass Spectrometry analysis
Fractionation of Oligosaccharides
The oligosaccharide separation was performed on an Agilent 1260 Infinity II series HPLC. Inline fractionation was performed on a two-way flow splitter coupled to an Agilent 6530 QTOF mass spectrometer and a Teledyne Isco Foxy 200 96-well plate fraction collector. A 150 mm × 4.6 mm Hypercarb column from Thermo Scientific with a 5 μm particle size was used to chromatographically separate the oligosaccharides. A binary gradient consisting of solvent A: (3% (v/v) acetonitrile/water + 0.1% (v/v) formic acid) and solvent B: (90% (v/v) acetonitrile/water + 0.1% (v/v) formic acid) was used. A 45 min gradient with a flow rate of 1 mL/min was used for the separation of maltooligosaccharides 0.00–30.00 min, 8.00–10.00% B; 30.00–30.01 min; 10.00–99.00% B; 30.01–33.00 min, 99.00% B; 33.00–33.01 min, 99.00–8.00% B, 33.01–45.00 min, 8.00% B. A 120 min gradient with a flow rate of 1 mL/min was used for chromatographic separation of oligosaccharides generated from galactan and butternut squash: 0.00–90.00 min, 5.00–12.00% B; 90.00–99.01 min, 12.00–99.00% B; 90.01–110.00 min, 99.00–99.00% B; 110.00–110.01 min, 99.00–5.00% B; 110.01–120.00 min, 5.00% B.
A two-way flow splitter was used to divert 90% of the effluent to the fraction collector and the remaining 10% of the effluent to the QTOF mass spectrometer. Fractions were collected on 96-well plates at a rate of 0.5 min/fraction. The fractions were then dried to completion under vacuum centrifugation and reconstituted in 100 μL of nanopure water. A 10 μL aliquot was transferred to a separate 96-well plate for monosaccharide composition analysis, while the remaining 90 μL of solution was used for glycosidic linkage analysis. Both 96-well plates were then dried to completeness by vacuum centrifugation prior to monosaccharide and linkage analysis.
Sample was introduced into the QTOF instrument using an electrospray ionization (ESI) source operated in positive mode. The instrument was calibrated with internal calibrant ions ranging from m/z 118.086 to 2721.895. Drying gas was set to 150 °C and with a flow rate of 11 L/min. The fragment, skimmer, and Octapole 1 RF voltages were set to 75, 60, and 750 V, respectively. The collision energy was based upon the compound mass and expressed by the linear function (collision energy = 1.3(m/z) – 3.5). The QTOF data was acquired using Agilent MassHunter Workstation Data Acquisition version B.08.00. The data was then further analyzed by Agilent MassHunter Qualitative Analysis software version B.07.00.
Monosaccharide Analysis of Fractions
Monosaccharide compositional analysis performed on separated oligosaccharides used methods from Amicucci et al.47 and Xu et al.48 The separated oligosaccharides underwent a hard acid hydrolysis with 4 M TFA for 2 h at 100 °C. The released monosaccharides were dried to completion by vacuum centrifugation. A calibration curve consisting of monosaccharide standards containing d-glucose, d-galactose, d-mannose, d-fructose, d-allose, l-fucose, l-rhamnose, l-arabinose, d-xylose, d-ribose, GlcA, GalA, GlcNAc, and GalNAc were prepared in water at concentrations ranging from 0.001 to 100 μg/μL. Samples and monosaccharide standards were derivatized with 0.2 M PMP dissolved in methanol and 28% NH4OH at 70 °C for 30 min. Derivatized products were dried to completion under vacuum centrifugation and reconstituted in nanopure water. Excess PMP was removed with chloroform extraction. A 1 μL aliquot from the aqueous layer was analyzed by an Agilent 1290 Infinity II UHPLC coupled to an Agilent 6495A QqQ MS employing dynamic multiple reaction monitoring (dMRM) mode. An external standard curve was used for absolute quantitation of each monosaccharide in the fraction. The data analysis was performed using Agilent MassHunter Qualitative Analysis software version B.07.00. and Agilent MassHunter Quantitative Analysis software version B.05.02.
Linkage Analysis of Fractions
Linkage analysis performed on separated oligosaccharides used the method from Galermo et al.41 In short, permethylation was performed on separated oligosaccharides by reacting the compounds with 5 μL of 1.26 g/mL saturated aqueous NaOH and 150 μL of DMSO. A 40 μL aliquot of iodomethane was added to the mixture and reacted under argon on a shaker at room temperature for 50 min. Residual NaOH and DMSO were removed by extraction with DCM and water. The DCM layer was dried to completion by vacuum centrifugation. Permethylated oligosaccharides were hydrolyzed and derivatized in the same manner as the monosaccharide analysis. After the PMP derivatization step and the drying step, the permethylated glycosides were reconstituted in 100 μL of 70% (v/v) methanol/water. A 1 μL aliquot was analyzed on an Agilent 1290 Infinity II UHPLC coupled to an Agilent 6495A QqQ MS instrument in multiple reaction monitoring (MRM) mode. The data analysis was performed using Agilent MassHunter Qualitative Analysis software version B.07.00. and Agilent MassHunter Quantitative Analysis software version B.05.02. A pool of oligosaccharide standards was used to assign linkages.
Results and Discussion
Description of the Overall Workflow
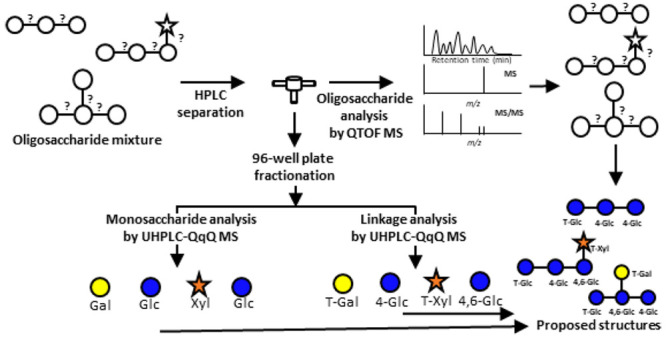
A 3D mass spectrometry workflow was developed to structurally elucidate oligosaccharides from polysaccharides using a combination of high-resolution and targeted mass spectrometry methods. This approach incorporates a nonenzymatic digestion step to generate polysaccharide-based oligosaccharides followed by three mass spectrometry analyses to obtain the in-depth structural characterization of oligosaccharides. Oligosaccharides were subjected to the workflow shown in Figure 1. The oligosaccharides are first subjected to HPLC separation using a Hypercarb PGC column. During the chromatographic run, the effluent was split two ways, one toward a QTOF MS and the other toward a 96-well plate fraction collector. The isolated oligosaccharides were characterized in the QTOF by their respective MS and MS/MS spectra. To further determine the monosaccharides and linkages of the separated oligosaccharides, collected fractions were subjected to monosaccharide analysis and linkage analysis. The workflow is the first to use a combination of three LC–MS platforms to elucidate deep structures of oligosaccharides. Furthermore, it provides several advantages, namely higher throughput in the monosaccharide and linkage determination employing a 96-well plate format.
Figure 1.

Workflow for the de novo structural elucidation of oligosaccharides using the 3D LC–MS/MS. Samples are injected in the LC–QTOF MS. The effluent is split using a flow splitter between the LC and the QTOF MS to a 96-well plate collector. The collected oligosaccharide fractions are subjected for UHPLC–QqQ MS analyses to determine monosaccharide and linkage compositions.
Workflow Validation Using Maltooligosaccharides
The workflow was demonstrated using an oligosaccharide mixture comprised of maltotriose, maltotetraose, maltopentaose, and maltohexaose. The maltooligosaccharides were chosen to model plant-based oligosaccharides derived from polysaccharides. A stock solution of the standards were pooled and subjected to the workflow. To isolate the oligosaccharides, chromatographic conditions were optimized to yield a 45 min run while still achieving base peak separation for the oligosaccharides. Complete chromatographic separation of the oligosaccharides was preferred, yet challenging to achieve, as many oligosaccharides had similar structures that tend to coelute in the more complicated mixtures described below.
The first dimension of MS results in the workflow was oligosaccharide analysis obtained through LC–QTOF MS. Two scan modes were employed on the oligosaccharides: a precursor ion scan mode to identify possible oligosaccharides based on their corresponding m/z value and a fragmentation mode, where collision-induced dissociation (CID) was employed to selected oligosaccharide ions. Fragmentation of oligosaccharide ions yielded structural information. In general, native oligosaccharide ions fragmented under CID inducing cleavages at the glycosidic bonds. Depending on the monosaccharide type (a hexose versus a pentose or a deoxyhexose), oligosaccharides can produce unique fragmentation spectra. For example, hexose, pentose, and a deoxyhexose yield losses of 162, 132, and 146 Da, respectively. The neutral mass differences and the resulting fragment ions are used to determine the monosaccharide class, some spatial arrangements, and the DP. We further note that rearrangements can occur with protonated species during CID. These rearrangements can complicate structural analysis, therefore it was helpful to identify the monosaccharide and linkages in the oligosaccharide to aid in elucidating the structure.
For the standards, the LC–QTOF MS chromatogram revealed a total of four oligosaccharides in the mixture shown in Figure 2A. The MS and MS/MS analysis revealed the m/z 505.178 as a trisaccharide, m/z 667.231 tetrasaccharide, m/z 829.283 pentasaccharide, and m/z 991.331 hexasaccharide. The compounds were split by the α and β anomer content except for the trisaccharide, which did not interact as strongly on the stationary phase. The DP of the oligosaccharides and the monosaccharide compositions (hexoses) were readily identified in the standards.
Figure 2.

Elucidated maltooligosaccharide structures obtained from 3D LC–MS/MS workflow. (A) The first dimension corresponds to base peak chromatogram (BPC) to yield the oligosaccharide profiles. (B) The second dimension revealed the absolute monosaccharides abundances in the fractions (inverted solid line graph). The third dimension produced the glycosidic linkages within the oligosaccharide (inset, pie charts).
To illustrate the details of the oligosaccharide analysis, maltohexaose was used. In the positive mode, the precursor ion corresponded to the mass of the protonated species [M + H]+ (991.331 Da, Figure 3A). Maltohexaose was subjected to CID fragmentation at a collision energy of 9.4 eV and yielded abundant fragment ions revealing the connectivity and confirming the DP (Figure 3B). The fragment ions m/z 163.062 (B1), m/z 325.113 (B2), m/z 487.166 (B3), m/z 649.212 (B4), and m/z 811.272 (B5) corresponded to one to five hexose residue(s), respectively. The observed fragment ions were consistent with the known structure of maltohexaose.
Figure 3.

(A) MS and (B) MS/MS spectra of protonated maltohexaose species [M + H]+ with assigned fragmentation obtained from the LC–QTOF MS.
The next dimension of MS in the 3D workflow yielded the monosaccharide compositional analysis. This analysis was performed on the collected fractions that were split from the LC–MS and collected in 96-well plates. A total of 76 fractions were collected and subjected to acid hydrolysis to produce monosaccharides. The released monosaccharides were then reacted in a PMP derivatization and subsequently analyzed by UHPLC–QqQ MS. The LC–MS monosaccharide analysis was performed in 10 min. The monosaccharides in the respective fractions were identified using a monosaccharide library based on retention times and MRM transitions.48 They were further quantified in absolute terms using an external calibration curve. For the standard pool, the monosaccharide results were plotted as an inverted chromatogram shown in Figure 2B. The chromatogram was constructed by plotting the absolute monosaccharide concentrations in each collected fraction with line smoothing performed to produce the figure. The compositional analysis included 14 monosaccharides; however only glucose was found as expected.
The absolute quantitation of monosaccharides was used to validate the fractionation of the oligosaccharides. Recovery analysis was performed on the LC of the maltooligosaccharide pool. LC fractions were analyzed for glucose content using the method for monosaccharide analysis. The total amounts of glucose in the fractions were determined by measuring the total glucose amounts (the prefractionated glucose amount) in the pool multiplied the molar fraction for each oligosaccharide. These values were then compared to the measured values in the fractions (fractionated glucose amount). Fractional recoveries were obtained using the absolute amounts measured in each fraction (Table 1). The glucose recoveries for maltotriose, maltotetraose, maltopentaose, and maltohexaose were determined to be 69.0%, 85.8%, 89.8%, and 82.5%, respectively. These were acceptable recoveries for all oligosaccharides with the exception of maltotriose, which may have degraded faster in the strong acid hydrolysis treatment.
Table 1. Recovery Analysis of Maltooligosaccharide Pool Based on Glucose Quantitation.
| maltooligosaccharide | degree of polymerization (DP) | collection window (min) | prefractionated glucose amount (ug) | fractionated glucose amount (ug) | glucose recovery (%) |
|---|---|---|---|---|---|
| maltotriose | 3 | 4–8 | 21.9 | 15.1 | 69.0 |
| maltotetraose | 4 | 8–12 | 16.6 | 14.2 | 85.8 |
| maltopentaose | 5 | 14–18 | 13.3 | 12.0 | 89.8 |
| maltohexaose | 6 | 21–26 | 11.1 | 9.2 | 82.5 |
| Total | N/A | N/A | 62.9 | 50.5 | 80.2 |
The third MS analysis in the workflow was used to determine the glycosidic linkages. Linkage analysis was performed by permethylating the oligosaccharides, subjecting to acid hydrolysis, and labeling with PMP. The partially methylated compounds were subjected to UHPLC–QqQ MS analysis. The separation for the UHPLC–QqQ MS analysis was performed in 15 min for each fraction. The unknown linkages in the fractions were identified using a known library based on retention times and MRM transitions.41 This analysis revealed only two types of species: a terminal glucose (t-glucose) and 4-linked glucose (4-glucose) for all fractionated oligosaccharides (Figure 2B). For the oligosaccharides, maltotriose, maltotetraose, maltopentaose, and maltohexaose were observed to contain t-glucose and 4-glucose in ratios 1.24, 0.75, 0.56, and 0.43. The expected values were 0.50, 0.33, 0.25, and 0.20, respectively. The relative abundances for observed and expected values were plotted as a function of DP (Figure S-1) and showed an over-representation of t-glucose compared to 4-glucose (Table 2). The disparity is likely due specific issues such as differences in the ionization between the various partially methylated species. Terminal monosaccharides are fully methylated, while internal linkages have at least one free hydroxide group. Permethylated species ionize better than partially methylated species. Thus, the MS method may generally overrepresent terminal linkage species over internal linkage residues.
Table 2. Degree of Polymerization (DP) versus Relative Linkage Composition of Observed and Expected Linkages for Maltooligosaccharides.
| observed
linkages |
expected
linkages |
||||||
|---|---|---|---|---|---|---|---|
| maltooligosaccharide | DP | t-glucose (%) | 4-glucose (%) | ratio (t-glucose/4-glucose) | t-glucose (%) | 4-glucose (%) | ratio (t-glucose/4-glucose) |
| maltotriose | 3 | 55.4 | 44.6 | 1.24 | 33.3 | 66.7 | 0.50 |
| maltotetraose | 4 | 42.8 | 57.2 | 0.75 | 25.0 | 75.0 | 0.33 |
| maltopentaose | 5 | 35.8 | 64.2 | 0.56 | 20.0 | 80.0 | 0.25 |
| maltohexaose | 6 | 30.2 | 69.8 | 0.43 | 16.7 | 83.3 | 0.20 |
Based on the three LC–MS/MS analyses, the structures of each maltooligosaccharides were deduced. The first QTOF MS with MS/MS yielded the DPs as 3, 4, 5, and 6. The quantitative UHPLC–QqQ MS analysis revealed that the maltooligosaccharides were solely composed of glucose, while the linkage UHPLC–QqQ MS analysis yielded 4-glucose and t-glucose were the only two linkage species observed. The maltooligosaccharide structures were determined to be composed of (1 → 4) glucose repeating units as shown in the annotated chromatogram in Figure 2A. While the α/β stereochemical information is currently not obtained with this workflow, future strategies are described below.
Validation of Structural Analysis of Oligosaccharides from Standard Polysaccharide Sources
A homopolysaccharide galactan was chemically digested using the Fenton’s initiation toward defined oligosaccharide groups (FITDOG) process described in previous publication.49 In this process, polysaccharides were subjected to oxidative cleavage with Fe3+/H2O2 conditions. The process yields distinct oligosaccharides corresponding to each polysaccharide. The resulting oligosaccharides were subjected to the workflow. The generated oligosaccharides were chromatographically separated using a 120 min run and collected into 192 fractions. The oligosaccharides were then analyzed using Q-TOF MS to determine the DP, hydrolyzed then derivatized to determine the monosaccharide compositions with UHPLC–QqQ MS analysis, and permethylated, hydrolyzed, and derivatized to determine the glycosidic linkages with UHPLC–QqQ MS analysis.
The LC–QTOF MS chromatogram of the digested galactan yielded at least seven dominant oligosaccharides (Figure 4A). The MS and MS/MS results revealed a Hex3 (6.5 min, 7.5 min), Hex4 (12.5 min, 15.0 min), Hex5 (19.5 min, 22 min), Hex6 (24.5 min, 27.5 min), Hex7 (31.5 min, 34.5 min), Hex8 (45.0 min, 47.0 min), Hex9 (65.0 min), and Hex10 (83.0 min). Over oxidation and cross-ring cleavages were observed for some oligosaccharides and were annotated with asterisks in Figure 4A. These modified oligosaccharides were sequenced by tandem MS spectra and were not analyzed further. Subsequent optimization have eliminated these side products.49 Separation of anomers (α and β at the reducing end) resulted in split peaks with the exception of Hex9 and Hex10. The CID spectra confirmed the presence of anomers and were further consistent with the proposed monosaccharide compositions (hexoses).
Figure 4.

Characterization of oligosaccharide structures derived from galactan using the 3D LC–MS. (A) Base peak chromatogram (BPC) from the QTOF MS with the proposed oligosaccharide structures. (B) Mirrored chromatogram represents the monosaccharide composition in fractions and relative linkage compositions are shown in pie charts both obtained through UHPLC–QqQ MS analysis.
The monosaccharide analysis confirmed the composition to be exclusively galactose (Figure 4B). The linkage analysis similarly yielded only two species, namely 4-galactose and t-galactose. The ratios of t-galactose and 4-galactose were consistent with the expected ratios, although the terminal linkage species were overrepresented as observed for the maltooligosaccharide (Table 3). The ratios of t-galactose and 4-galactose as a function of DP were plotted and were found to follow the trend as with the expected values (Figure S-2). The measured ratios were generally higher than expected and were significantly higher for the low DP oligosaccharides suggesting an over representation of t-galactose. As mentioned with the maltooligosaccharide example, the terminal species are more abundant than internal linkage species with the effect decreasing with higher DP. A response factor normalized to a polynomial curve may be deduced and used for future measurements if quantitation of the linkages were desired. Nonetheless, the information provided the oligosaccharide structures using the 3D MS analyses. On the basis of the information, the oligosaccharide structures were determined to be a linear (1 → 4) linked galactose oligomers as expected for galactan.
Table 3. Degree of Polymerization (DP) and Relative Linkage Composition of Observed and Expected Linkages for Fractionated Galactose Derived Oligosaccharides.
| observed
linkage |
expected
linkage |
|||||
|---|---|---|---|---|---|---|
| DP | t-galactose (%) | 4-galactose (%) | ratio (t-galactose/4-galactose) | t-galactose (%) | 4-galactose (%) | Ratio (t-galactose/4-galactose |
| 3 | 68.0 | 32.0 | 2.13 | 33.3 | 66.7 | 0.50 |
| 4 | 58.3 | 41.7 | 1.40 | 25.0 | 75.0 | 0.33 |
| 5 | 53.3 | 46.7 | 1.14 | 20.0 | 80.0 | 0.25 |
| 6 | 45.4 | 54.6 | 0.83 | 16.7 | 83.3 | 0.20 |
| 7 | 44.0 | 56.0 | 0.79 | 14.3 | 85.7 | 0.17 |
| 8 | 39.3 | 60.7 | 0.65 | 12.5 | 87.5 | 0.14 |
| 9 | 36.2 | 63.8 | 0.57 | 11.1 | 88.9 | 0.13 |
| 10 | 35.5 | 64.5 | 0.55 | 10.0 | 90.0 | 0.11 |
Determination of Oligosaccharides Produced from Food Polysaccharides
The 3D MS approach was performed on oligosaccharides produced from a plant product, namely butternut squash. The LC–QTOF chromatogram (Figure 5A) of the resulting oligosaccharides were separated and collected into a total of 96 fractions in 0.5 min increments. The mirrored chromatogram was constructed from the respective monosaccharide analysis of the corresponding fraction (Figure 5B). The linkage analysis of the same fraction was obtained and were represented in the inset pie charts. The resolution of the LC–QTOF was limited by the flow splitter between the LC and the QTOF. Chromatograms without the flow splitter were of significantly higher resolution and were used to confirm the identities of the broad peaks. The major components were composed of DPs varying from disaccharides to pentasaccharides (Table 4). As expected, a complete plant product such as butternut squash is composed of several polysaccharides. The resulting oligosaccharides yielded a small number of major products namely those composed primarily of hexose units. The monosaccharide analysis of the fractionated effluent yielded the monosaccharide compositions of the peaks (Figure 5B). Based on the combined analyses, several compounds were readily identified as those composed of starch. These oligosaccharides consisted of three, four, and five monomer units (m/z 505.180 at 11 min, m/z 667.235 at 27 min, and m/z 829.285 at 41 min). Split chromatographic peaks were observed and corresponded to the alpha and beta anomers. These compounds were comprised almost exclusively of glucose (81.7%, 90.4%, 78.1%, respectively). The coelution of other compounds decreased the relative glucose abundances. The linkage analysis of these compounds yielded 4-glucose (43%, 59%, 55%, respectively) and t-glucose (48%, 38%, 39%) (pie chart insets, Figure 5B). By combining the results from the three analyses, we conclude that the oligosaccharides were maltotriose, maltotetraose, and maltopentaose, respectively. Both the retention times and tandem MS matched the maltooligosaccharide standards, although different LC gradients were used in the separate analyses.
Figure 5.

Results from the 3D LC–MS/MS workflow for butternut squash oligosaccharides. (A) Oligosaccharide profiles from QTOF MS analysis with annotated elucidated structures. (B) Mirrored chromatogram represents the absolute monosaccharide compositions of each fraction. Linkage compositions are shown in pie charts. In the inset structures, the “X” denote undefined linkages.
Table 4. Table of Saccharides Characterized Using the De Novo Structural Analysis of Oligosaccharides Generated from Butternut Squash with Retention Time, Monosaccharide Composition, and Linkage Composition.
| monosaccharide
composition (%) |
linkage composition
(%) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| compound | m/z [M + H]+ | tR (min) | Glca | Galb | Fruc | 4-Glc | 6-Gal | 6-Glc | t-Fru | t-Gal | t-Glc |
| 2Hex | 343.116 | 6 | 43.8 | 5.1 | 51.1 | 6.7 | 0.6 | 0.6 | 1.8 | 18.5 | 71.9 |
| 3Hex | 505.180 | 11 | 81.7 | 6.8 | 11.5 | 42.5 | 0.8 | 0.8 | 0.3 | 7.3 | 48.4 |
| 3Hex | 505.180 | 19, 24 | 35.6 | 26.8 | 37.7 | 16.8 | 9.6 | 9.6 | 0.1 | 36.8 | 27.1 |
| 4Hex | 667.235 | 27 | 90.4 | 2.4 | 7.2 | 59.0 | 0.3 | 0.3 | 0.0 | 2.2 | 38.2 |
| 5Hex | 829.285 | 41 | 78.1 | 7.9 | 14.1 | 54.8 | 0.2 | 0.2 | 0.0 | 6.0 | 38.8 |
| 4Hex | 667.231 | 45 | 20.4 | 20.9 | 58.8 | 15.5 | 20.1 | 20.1 | 0.0 | 33.2 | 11.1 |
Glc, glucose.
Gal, galactose.
Fru, fructose.
Oligosaccharides with mixed monosaccharide compositions were also present. A disaccharide was found to consist of glucose (49.5%) and fructose (50.5%), while the glycosidic linkage revealed the presence of t-glucose. Fructose is a ketosugar and does not derivatize well with PMP; therefore, its linkages could not be determined. However, the results from the LC–QTOF MS showed the presence of hexose disaccharides. The structure was consistent with a disaccharide having a fructose reducing end and glucose nonreducing end (inset, Figure 5A). A trisaccharide was observed clustered at retention times 18–26 min. The oligosaccharide was composed of glucose (Glc, 35.6%), galactose (Gal, 26.8%), and fructose (Fru, 37.7%). Tandem MS yielded identical fragmentation. The multiple peaks suggested the presence of a reducing monosaccharide and the presence of anomers. Fructose does not produce anomers at the reducing end, eliminating the possibility of fructose being on the reducing end.50,51 The linkage analysis of the oligosaccharide suggested the structure as Gal-(1 → X)-Fru-(2 → 4)-Glc. The “X” corresponds to undefined linkage for the reasons written in the text below. Lastly, a tetrasaccharide at 45 min was found to consist of glucose (20.4%), galactose (20.9%), and fructose (58.8%). The linkage analysis revealed t-galactose (33.3%), and 6-glucose (20.1%). Because t-galactose (33.2%) is higher in abundance compared to t-glucose (11.1%), the t-galactose was assigned the terminal position leaving the 6-glucose to be in the internal position. The monosaccharide analysis revealed the presence of galactose and glucose residues, and linkage analysis yielded the structure Gal-(1 → 6)-Glc-(1 → X)-Fru-(2 → X)-Fru.
Limitation of 3D MS for Oligosaccharide Analysis
The concept of 3D MS provides structural information on oligosaccharides that have previously not been achievable by any single method. However, there are clear limitations to the method. Coelution of several compounds can limit the specificity of the analysis. This condition can be remedied by further optimizing the separation conditions by using a different stationary phase such as HILIC and by significantly longer chromatographic separation.
The unique structure of fructose makes it difficult to obtain linkage information. Being a keto sugar, it was difficult to label with PMP, and its sensitivity was significantly lower compared to other monosaccharides. In the partially methylated form, the monosaccharide appeared even less reactive to PMP thereby restricting the formation of labeled, partially methylated fructose. The differences in reactivity needs further investigation. However, the terminal fructose was observable and measured. Work is currently being performed to find a better labeling method for fructose.
There are additional limitations with the method serving as a reminder of the difficulties in elucidating complete structures. The α/β character of the glycosidic linkages could not be resolved using the 3D MS workflow. However, the workflow can differentiate oligosaccharide isomers by yielding differential retention times for distinct α/β linkages. For example, maltotriose with α(1 → 4) linkages and cellotriose with β(1 → 4) linkages are isomers with widely different retention times (a difference of as much as 5 min was observed on the PGC column). For this study, the anomeric character was known based on retention times and the components of the squash. However, if needed other techniques may be used and adapted to the workflow. NMR can be used on the isolated compounds. Alternatively, an LC–MS approach using enzymatic treatments that can also be applied on separated oligosaccharides.52 Enzymes can be targeted based on the elucidated monosaccharide and linkage information.
Conclusions
A 3D LC–MS/MS-based workflow was developed for the structural elucidation of oligosaccharides derived from plant polysaccharides. This workflow can be employed for pure oligosaccharides, polysaccharides, and complicated polysaccharide mixtures such as those found in plants or food. This approach utilized LC–MS with inline collection of fractions that are further probed by separate LC–MS analyses yielding the corresponding monosaccharide compositions and linkage information on the eluting oligosaccharide. Method validation was performed on maltooligosaccharides, oligosaccharides derived from the polysaccharide galactan, and a food product, butternut squash. The optimized workflow yielded separated oligosaccharides with structures. The approach can also be applied to rapidly build glycome libraries of new and novel oligosaccharides.
Acknowledgments
Funding provided by the National Institutes of Health (R01 DK124193) is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jasms.1c00133.
Plots to illustrate the effect of the linkages abundance ratios and DP for the linkages in fractionated maltooligosaccharides (Figure S-1) and oligosaccharides from galactan (Figure S-2) (PDF)
Author Present Address
# University of California Davis, Davis, Department of Chemistry, One Shields Avenue, Davis, CA 95616
Author Contributions
J.J.C. performed all experiments, analyzed samples, and produced figures and tables along with writing the manuscript. A.G.G. performed experimental design and edited the manuscript. M.J.A, E.N., G.C., and N.B. aided in experimental planning, data analysis, and editing the manuscript. Y.C. aided in galactan experiments and edited the manuscript. C.B.L. planned the overall experimental project and reviewed the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Engfer M. B.; Stahl B.; Finke B.; Sawatzki G.; Daniel H. Human Milk Oligosaccharides Are Resistant to Enzymatic Hydrolysis in the Upper Gastrointestinal Tract. Am. J. Clin. Nutr. 2000, 71 (6), 1589–1596. 10.1093/ajcn/71.6.1589. [DOI] [PubMed] [Google Scholar]
- Garrido D.; Kim J. H.; German J. B.; Raybould H. E.; Mills D. A. Oligosaccharide Binding Proteins from Bifidobacterium Longum Subsp. Infantis Reveal a Preference for Host Glycans. PLoS One 2011, 6 (3), e17315. 10.1371/journal.pone.0017315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcobal A.; Barboza M.; Froehlich J. W.; Block D. E.; German J. B.; Lebrilla C. B.; Mills D. A. Consumption of Human Milk Oligosaccharides by Gut-Related Microbes. J. Agric. Food Chem. 2010, 58 (9), 5334–5340. 10.1021/jf9044205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sela D. A.; Garrido D.; Lerno L.; Wu S.; Tan K.; Eom H. J.; Joachimiak A.; Lebrilla C. B.; Mills D. A. Bifidobacterium Longum Subsp. Infantis ATCC 15697 α-Fucosidases Are Active on Fucosylated Human Milk Oligosaccharides. Appl. Environ. Microbiol. 2012, 78 (3), 795–803. 10.1128/AEM.06762-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goubet F.; Jackson P.; Deery M. J.; Dupree P. Polysaccharide Analysis Using Carbohydrate Gel Electrophoresis. A Method to Study Plant Cell Wall Polysaccharides and Polysaccharide Hydrolases. Anal. Biochem. 2002, 300 (1), 53–63. 10.1006/abio.2001.5444. [DOI] [PubMed] [Google Scholar]
- Kobata A. Structures and Application of Oligosaccharides in Human Milk. Proc. Jpn. Acad., Ser. B 2010, 86 (7), 731–747. 10.2183/pjab.86.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newburg D. S.; Neubauer S. H. Carbohydrates in Milks: Analysis, Quantities, and Significance. Handbook of Milk Composition 1995, 273–350. 10.1016/B978-012384430-9/50015-9. [DOI] [Google Scholar]
- Wu S.; Tao N.; German J. B.; Grimm R.; Lebrilla C. B. Development of an Annotated Library of Neutral Human Milk Oligosaccharides. J. Proteome Res. 2010, 9 (8), 4138–4151. 10.1021/pr100362f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.; Grimm R.; German J. B.; Lebrilla C. B. Annotation and Structural Analysis of Sialylated Human Milk Oligosaccharides. J. Proteome Res. 2011, 10 (2), 856–868. 10.1021/pr101006u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J. C. C.; Totten S. M.; Huang J. O.; Nagshbandi S.; Kirmiz N.; Garrido D. A.; Lewis Z. T.; Wu L. D.; Smilowitz J. T.; German J. B.; Mills D. A.; Lebrilla C. B. Identification of Oligosaccharides in Feces of Breast-Fed Infants and Their Correlation with the Gut Microbial Community. Mol. Cell. Proteomics 2016, 15 (9), 2987–3002. 10.1074/mcp.M116.060665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A. Glycan-Based Interactions Involving Vertebrate Sialic-Acid-Recognizing Proteins. Nature 2007, 446 (7139), 1023–1029. 10.1038/nature05816. [DOI] [PubMed] [Google Scholar]
- Yoo S. W.; Motari M. G.; Susuki K.; Prendergast J.; Mountney A.; Hurtado A.; Schnaar R. L. Sialylation Regulates Brain Structure and Function. FASEB J. 2015, 29 (7), 3040–3053. 10.1096/fj.15-270983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleene R.; Schachner M. Glycans and Neural Cell Interactions. Nat. Rev. Neurosci. 2004, 5 (3), 195–208. 10.1038/nrn1349. [DOI] [PubMed] [Google Scholar]
- Spillmann D.; Burger M. M. Carbohydrate-Carbohydrate Interactions in Adhesion. J. Cell. Biochem. 1996, 61 (4), 562–568. 10.1002/(SICI)1097-4644(19960616)61:4<562::AID-JCB9>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Etzold S.; Kober O. I.; Mackenzie D. A.; Tailford L. E.; Gunning A. P.; Walshaw J.; Hemmings A. M.; Juge N. Structural Basis for Adaptation of Lactobacilli to Gastrointestinal Mucus. Environ. Microbiol. 2014, 16 (3), 888–903. 10.1111/1462-2920.12377. [DOI] [PubMed] [Google Scholar]
- Gehrig J. L.; Venkatesh S.; Chang H. W.; Hibberd M. C.; Kung V. L.; Cheng J.; Chen R. Y.; Subramanian S.; Cowardin C. A.; Meier M. F.; O’Donnell D.; Talcott M.; Spears L. D.; Semenkovich C. F.; Henrissat B.; Giannone R. J.; Hettich R. L.; Ilkayeva O.; Muehlbauer M.; Newgard C. B.; Sawyer C.; Head R. D.; Rodionov D. A.; Arzamasov A. A.; Leyn S. A.; Osterman A. L.; Hossain M. I.; Islam M.; Choudhury N.; Sarker S. A.; Huq S.; Mahmud I.; Mostafa I.; Mahfuz M.; Barratt M. J.; Ahmed T.; Gordon J. I. Effects of Microbiota-Directed Foods in Gnotobiotic Animals and Undernourished Children. Science (Washington, DC, U. S.) 2019, 365 (6449), eaau4732. 10.1126/science.aau4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaker B. R.; Tuncil Y. E. A Perspective on the Complexity of Dietary Fiber Structures and Their Potential Effect on the Gut Microbiota. J. Mol. Biol. 2014, 426 (23), 3838–3850. 10.1016/j.jmb.2014.07.028. [DOI] [PubMed] [Google Scholar]
- Barboza M.; Sela D. A.; Pirim C.; LoCascio R. G.; Freeman S. L.; German J. B.; Mills D. A.; Lebrilla C. B. Glycoprofiling Bifidobacterial Consumption of Galacto-Oligosaccharides by Mass Spectrometry Reveals Strain-Specific, Preferential Consumption of Glycans. Appl. Environ. Microbiol. 2009, 75 (23), 7319–7325. 10.1128/AEM.00842-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabater-Molina M.; Larqué E.; Torrella F.; Zamora S. Dietary Fructooligosaccharides and Potential Benefits on Health. J. Physiol. Biochem. 2009, 65 (3), 315–328. 10.1007/BF03180584. [DOI] [PubMed] [Google Scholar]
- Singh S. P.; Jadaun J. S.; Narnoliya L. K.; Pandey A. Prebiotic Oligosaccharides: Special Focus on Fructooligosaccharides, Its Biosynthesis and Bioactivity. Appl. Biochem. Biotechnol. 2017, 183 (2), 613–635. 10.1007/s12010-017-2605-2. [DOI] [PubMed] [Google Scholar]
- Kaplan H.; Hutkins R. W. Fermentation of Fructooligosaccharides by Lactic Acid Bacteria and Bifidobacteria. Appl. Environ. Microbiol. 2000, 66 (6), 2682–2684. 10.1128/AEM.66.6.2682-2684.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehlbaum S.; Prudence K.; Kieboom J.; Heerikhuisen M.; van den Broek T.; Schuren F. H. J.; Steinert R. E.; Raederstorff D. In Vitro Fermentation of Selected Prebiotics and Their Effects on the Composition and Activity of the Adult Gut Microbiota. Int. J. Mol. Sci. 2018, 19 (10), 3097. 10.3390/ijms19103097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler A.; Zaia J. Size-Exclusion Chromatography of Heparin Oligosaccharides at High and Low Pressure. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2006, 837 (1–2), 76–86. 10.1016/j.jchromb.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Bao Y.; Newburg D. S. Capillary Electrophoresis of Acidic Oligosaccharides from Human Milk. Electrophoresis 2008, 29 (12), 2508–2515. 10.1002/elps.200700873. [DOI] [PubMed] [Google Scholar]
- Corzo-Martínez M.; Copoví P.; Olano A.; Moreno F. J.; Montilla A. Synthesis of Prebiotic Carbohydrates Derived from Cheese Whey Permeate by a Combined Process of Isomerisation and Transgalactosylation. J. Sci. Food Agric. 2013, 93 (7), 1591–1597. 10.1002/jsfa.5929. [DOI] [PubMed] [Google Scholar]
- Füreder V.; Rodriguez-Colinas B.; Cervantes F. V.; Fernandez-Arrojo L.; Poveda A.; Jimenez-Barbero J.; Ballesteros A. O.; Plou F. J. Selective Synthesis of Galactooligosaccharides Containing β(1→3) Linkages with β-Galactosidase from Bifidobacterium Bifidum (Saphera). J. Agric. Food Chem. 2020, 68 (17), 4930–4938. 10.1021/acs.jafc.0c00997. [DOI] [PubMed] [Google Scholar]
- Benkeblia N. Fructooligosaccharides and Fructans Analysis in Plants and Food Crops. J. Chromatogr. A 2013, 1313, 54–61. 10.1016/j.chroma.2013.08.013. [DOI] [PubMed] [Google Scholar]
- Sabater C.; Prodanov M.; Olano A.; Corzo N.; Montilla A. Quantification of Prebiotics in Commercial Infant Formulas. Food Chem. 2016, 194, 6–11. 10.1016/j.foodchem.2015.07.127. [DOI] [PubMed] [Google Scholar]
- Montilla A.; Van De Lagemaat J.; Olano A.; Del Castillo M. D. Determination of Oligosaccharides by Conventional High-Resolution Gas Chromatography. Chromatographia 2006, 63 (9–10), 453–458. 10.1365/s10337-006-0770-5. [DOI] [Google Scholar]
- Biermann C. J.; McGinnis G. D.. Analysis of Carbohydrates by GLC and MS; CRC Press, 1988. [Google Scholar]
- Ruiz-Matute A. I.; Hernández-Hernández O.; Rodríguez-Sánchez S.; Sanz M. L.; Martínez-Castro I. Derivatization of Carbohydrates for GC and GC-MS Analyses. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2011, 879 (17–18), 1226–1240. 10.1016/j.jchromb.2010.11.013. [DOI] [PubMed] [Google Scholar]
- Black I.; Heiss C.; Azadi P. Comprehensive Monosaccharide Composition Analysis of Insoluble Polysaccharides by Permethylation to Produce Methyl Alditol Derivatives for Gas Chromatography/Mass Spectrometry. Anal. Chem. 2019, 91 (21), 13787–13793. 10.1021/acs.analchem.9b03239. [DOI] [PubMed] [Google Scholar]
- Hsu H. C.; Liew C. Y.; Huang S. P.; Tsai S. T.; Ni C. K. Simple Approach for De Novo Structural Identification of Mannose Trisaccharides. J. Am. Soc. Mass Spectrom. 2018, 29, 470–480. 10.1007/s13361-017-1850-5. [DOI] [PubMed] [Google Scholar]
- Hsu H. C.; Liew C. Y.; Huang S. P.; Tsai S. T.; Ni C. K. Simple Method for de Novo Structural Determination of Underivatised Glucose Oligosaccharides. Sci. Rep. 2018, 8 (1), 1–12. 10.1038/s41598-018-23903-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancilla M. T.; Wong A. W.; Voss L. R.; Lebrilla C. B. Fragmentation Reactions in the Mass Spectrometry Analysis of Neutral Oligosaccharides. Anal. Chem. 1999, 71 (15), 3206–3218. 10.1021/ac9813484. [DOI] [PubMed] [Google Scholar]
- Fasciotti M.; Sanvido G. B.; Santos V. G.; Lalli P. M.; McCullagh M.; De Sá G. F.; Daroda R. J.; Peter M. G.; Eberlin M. N. Separation of Isomeric Disaccharides by Traveling Wave Ion Mobility Mass Spectrometry Using CO2 as Drift Gas. J. Mass Spectrom. 2012, 47 (12), 1643–1647. 10.1002/jms.3089. [DOI] [PubMed] [Google Scholar]
- Hofmann J.; Hahm H. S.; Seeberger P. H.; Pagel K. Identification of Carbohydrate Anomers Using Ion Mobility-Mass Spectrometry. Nature 2015, 526 (7572), 241–244. 10.1038/nature15388. [DOI] [PubMed] [Google Scholar]
- Gray C. J.; Thomas B.; Upton R.; Migas L. G.; Eyers C. E.; Barran P. E.; Flitsch S. L. Applications of Ion Mobility Mass Spectrometry for High Throughput, High Resolution Glycan Analysis. Biochim. Biophys. Acta, Gen. Subj. 2016, 1860 (8), 1688–1709. 10.1016/j.bbagen.2016.02.003. [DOI] [PubMed] [Google Scholar]
- Amicucci M. J.; Nandita E.; Lebrilla C. B. Function without Structures: The Need for In-Depth Analysis of Dietary Carbohydrates. J. Agric. Food Chem. 2019, 67 (16), 4418–4424. 10.1021/acs.jafc.9b00720. [DOI] [PubMed] [Google Scholar]
- Amicucci M. J.; Galermo A. G.; Guerrero A.; Treves G.; Nandita E.; Kailemia M. J.; Higdon S. M.; Pozzo T.; Labavitch J. M.; Bennett A. B. A Strategy for Structural Elucidation of Polysaccharides: Elucidation of a Maize Mucilage That Harbors Diazotrophic Bacteria. Anal. Chem. 2019, 91 (20), 7254–7265. 10.1021/acs.analchem.9b00789. [DOI] [PubMed] [Google Scholar]
- Galermo A. G.; Nandita E.; Barboza M.; Amicucci M. J.; Vo T.-T. T.; Lebrilla C. B. Liquid Chromatography–Tandem Mass Spectrometry Approach for Determining Glycosidic Linkages. Anal. Chem. 2018, 90 (21), 13073–13080. 10.1021/acs.analchem.8b04124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galermo A. G.; Nandita E.; Castillo J. J.; Amicucci M. J.; Lebrilla C. B. Development of an Extensive Linkage Library for Characterization of Carbohydrates. Anal. Chem. 2019, 91 (20), 13022–13031. 10.1021/acs.analchem.9b03101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninonuevo M. R.; Park Y.; Yin H.; Zhang J.; Ward R. E.; Clowers B. H.; German J. B.; Freeman S. L.; Killeen K.; Grimm R.; Lebrilla C. B. A Strategy for Annotating the Human Milk Glycome. J. Agric. Food Chem. 2006, 54 (20), 7471–7480. 10.1021/jf0615810. [DOI] [PubMed] [Google Scholar]
- Ruhaak L. R.; Deelder A. M.; Wuhrer M. Oligosaccharide Analysis by Graphitized Carbon Liquid Chromatography-Mass Spectrometry. Anal. Bioanal. Chem. 2009, 394 (1), 163–174. 10.1007/s00216-009-2664-5. [DOI] [PubMed] [Google Scholar]
- Kailemia M. J.; Ruhaak L. R.; Lebrilla C. B.; Amster I. J. Oligosaccharide Analysis by Mass Spectrometry: A Review of Recent Developments. Anal. Chem. 2014, 86 (1), 196–212. 10.1021/ac403969n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C. W.; Tsai M. H.; Li S. T.; Tsai T. I.; Chu K. C.; Liu Y. C.; Lai M. Y.; Wu C. Y.; Tseng Y. C.; Shivatare S. S.; Wang C. H.; Chao P.; Wang S. Y.; Shih H. W.; Zeng Y. F.; You T. H.; Liao J. Y.; Tu Y. C.; Lin Y. S.; Chuang H. Y.; Chen C. L.; Tsai C. S.; Huang C. C.; Lin N. H.; Ma C.; Wu C. Y.; Wong C. H. A Common Glycan Structure on Immunoglobulin G for Enhancement of Effector Functions. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (34), 10611–10616. 10.1073/pnas.1513456112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amicucci M. J.; Galermo A. G.; Nandita E.; Vo T.-T. T.; Liu Y.; Lee M.; Xu G.; Lebrilla C. B. A Rapid-Throughput Adaptable Method for Determining the Monosaccharide Composition of Polysaccharides. Int. J. Mass Spectrom. 2019, 438, 22–28. 10.1016/j.ijms.2018.12.009. [DOI] [Google Scholar]
- Xu G.; Amicucci M. J.; Cheng Z.; Galermo A. G.; Lebrilla C. B. Revisiting Monosaccharide Analysis–Quantitation of a Comprehensive Set of Monosaccharides Using Dynamic Multiple Reaction Monitoring. Analyst 2018, 143 (1), 200–207. 10.1039/C7AN01530E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amicucci M. J.; Nandita E.; Galermo A. G.; Castillo J. J.; Chen S.; Park D.; Smilowitz J. T.; German J. B.; Mills D. A.; Lebrilla C. B. A Nonenzymatic Method for Cleaving Polysaccharides to Yield Oligosaccharides for Structural Analysis. Nat. Commun. 2020, 11 (1), 3963. 10.1038/s41467-020-17778-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogarth A. J. C. L.; Hunter D. E.; Jacobs W. A.; Garleb K. A.; Wolf B. W. Ion Chromatographic Determination of Three Fructooligosaccharide Oligomers in Prepared and Preserved Foods. J. Agric. Food Chem. 2000, 48 (11), 5326–5330. 10.1021/jf000111h. [DOI] [PubMed] [Google Scholar]
- Li J.; Hu D.; Zong W.; Lv G.; Zhao J.; Li S. Determination of Inulin-Type Fructooligosaccharides in Edible Plants by High-Performance Liquid Chromatography with Charged Aerosol Detector. J. Agric. Food Chem. 2014, 62 (31), 7707–7713. 10.1021/jf502329n. [DOI] [PubMed] [Google Scholar]
- Xu G.; Goonatilleke E.; Wongkham S.; Lebrilla C. B. Deep Structural Analysis and Quantitation of O-Linked Glycans on Cell Membrane Reveal High Abundances and Distinct Glycomic Profiles Associated with Cell Type and Stages of Differentiation. Anal. Chem. 2020, 92 (5), 3758–3768. 10.1021/acs.analchem.9b05103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.